Chapter 11 The Brain’s Dopamine Systems and Their Relevance to Restless Legs Syndrome
Synthesis and Metabolism of Dopamine
The family of catecholamines includes norepinephrine, epinephrine, and dopamine, of which dopamine is the most abundant in the central nervous system. It was not until the 1950s that dopamine was recognized as a critical neurotransmitter, and not simply an intermediate in the single biosynthetic pathway it shares with norepinephrine and epinephrine. In the first and rate-limiting step of the pathway, L-tyrosine is hydroxylated via the enzyme tyrosine hydroxylase (TH) to form L-dihydroxyphenylalanine (DOPA).1 Removal of a carboxyl group from L-DOPA by DOPA decarboxylase then produces dopamine. In subsequent steps, dopamine can then be further converted to norepinephrine and then to epinephrine by dopamine β-hydroxylase and phenylethanolamine-N-methyltrasferase, respectively (Fig. 11-1).
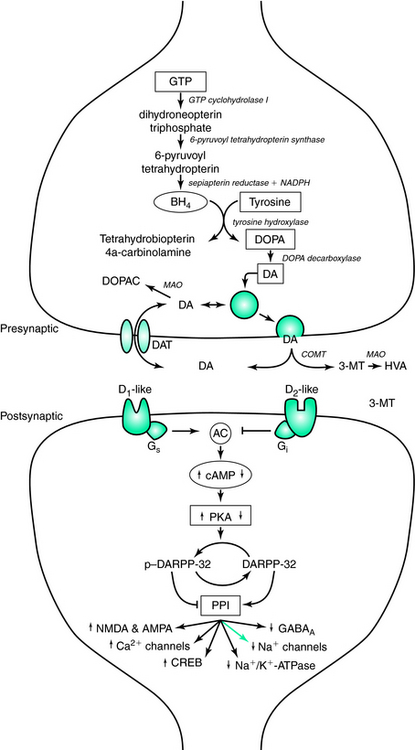
Activation of TH is the rate-limiting step in the production of dopamine, and it is under strict regulatory control by a variety of factors, including inhibitory feedback by the catecholamine products (e.g., dopamine). To convert tyrosine to L-DOPA, TH requires the binding of iron to the catalytic domain at the C terminus. Catalytic activity of TH also requires (6R)-(L-erythro-1′,2′-dihydroxypropyl)-2-amino-4-hydroxy-5, 6, 7, 8-tetrahydropteridine (6R-tetrahydrobiopterin [6RBPH4], more commonly known as tetrahydrobiopterin [BH4], a naturally occurring pteridine cofactor), to reduce the iron to the ferrous form (Fe2+). This allows the binding of the substrates (e.g., L-tyrosine and molecular oxygen) to the C terminus.2 Following a catalytic cycle, the molecular oxygen can oxidize a fraction of the iron to the ferric form, thus increasing the binding affinity for dopamine and L-DOPA. When either is bound to the regulatory domain of the N terminus, the complex is inactivated by preventing the binding of BH4. Biosynthesis of L-DOPA, and consequently dopamine, can be restored by phosphorylation of the tyrosine hydroxylase enzyme at serine 40 by cAMP-dependent protein kinase phosphorylation, thus decreasing the binding affinity for dopamine 300-fold and increasing the binding affinity for the pteridine cofactor.3,4 Meanwhile, endogenous levels of BH4 are regulated by guanosine triphosphate (GTP) cyclohydrolase activity as its synthesis is downstream of the rate-limiting GTP enzyme.5 Mutations in the GTP cyclochydrolase I gene contribute to hereditary L-DOPA responsive dsytonia,6 which manifests a dopamine-responsive circadian distribution of symptoms with greater penetrance in women, sharing these two features in common with restless legs syndrome (RLS).7,8
Once returned to the presynaptic terminal, dopamine is repackaged in synaptic vesicles via the vesicular monoamine transporter (VMAT) or metabolized to dihydroxyphenylacetic acid (DOPAC) by monoamine oxidase (MAO). Two alternative pathways are available for dopamine catabolism in the synapse, depending on whether the first step is catalyzed by MAO or catechol-O-methyltransferase (COMT). Thus, dopamine can be either deaminated to 3,4-dihydroxyphenylacetic acid (DOPAC) or methylated to 3-methoxytyramine (3-MT). In turn, deamination of 3-MT and methylation of DOPAC lead to homovanillic acid (HVA). In humans, cerebrospinal fluid levels of HVA have been used as a proxy for levels of dopaminergic activity within the brain.
Physiological Effects of Dopamine (Cellular and Subcellular)
The physiological effects of the dopaminergic system are best characterized as “neuromodulatory.” Rather than eliciting excitatory postsynaptic potentials (EPSPs) or inhibitory postsynaptic potentials (IPSPs) in a manner similar to glutamate and GABA, for example, dopamine allows the gating of inputs via alteration of membrane properties and specific ion conductances.9 This enhanced or decreased response to other inputs affects the intensity, duration, and timing of output commensurate with environmental and homeostatic demands.10–12 The multivariate control of dopamine is provided through five subtypes of seven-transmembrane domain G protein–coupled receptors (D1 through D5), which, based on similarities in pharmacology, biochemistry, and amino acid homology, are divided into two classes, D1-like (D1, D5) and D2-like (D2, D3, and D4).13 D3 demonstrates the highest affinity for endogenous dopamine, followed, in decreasing order of affinity, by D4, D2, D5, and D1.14,15 Furthermore, each receptor subtype has unique patterns of localization throughout the brain that increase the array of the behavioral effects of dopamine.
D1-like receptors activate the Gs transduction pathway stimulating the production of adenylyl cyclase, which increases the formation of cyclic adenosine monophosphate (cAMP) and ultimately increases the activity of cAMP-dependent protein kinase (PKA). PKA activates DARPP-32 (dopamine and cyclic adenosine 3′,5′-monophosphate-regulated phosphoprotein [32 kDa]) via phosphorylation, permitting phospho-DARPP-32 to then inhibit protein phosphatase-1 (PP-1). The downstream effect of decreased PP-1 activity is an increase in the phosphorylation states of assorted downstream effector proteins regulating neurotransmitter receptors and voltage-gated ion channels. Ultimately, this results in increased activity of glutamate receptors (N-methyl-D-aspartic acid [NMDA] and α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid [AMPA]), Ca2+ channels (L, N, and P types), and CREB, as well as decreased activity of GABAA receptors, Na+ channels, and Na+/K+-ATPase.16 Alternatively, D2-like receptors stimulate the Gi transduction pathway, which is negatively coupled to the production of adenylyl cyclase. Activation of D2-like receptors also leads to an increase in intracellular calcium concentrations. These two pathways can act independently, through decreased PKA and increased calcineurin, respectively, to return phospho-DARPP-32 to the inactive DARPP-32. Additionally, calcineurin activity can be stimulated by the increase in intracellular calcium concentrations caused by glutamatergic activation of NMDA receptors. Other mechanisms of action mediated by D2-like receptors include increasing K+ conductance and inhibiting Ca2+ entry via voltage-gated Ca2+ channels.
Both D1-like and D2-like receptors are found postsynaptically, exerting their effect on nondopaminergic neurons targeted by dopaminergic projections. D2-like receptors can also be localized presynaptically on the dendrites, soma, and presynaptic terminals of dopaminergic cells. The presynaptic localization of the autoreceptors enables them to provide an inhibitory feedback mechanism. The regulation in the somatodendritic region includes modulation of the firing rate of the dopaminergic cell, and in the nerve terminal autoreceptors control the synthesis and release of dopamine. In addition, dopamine appears to act on receptors present on endothelial cells lining the brain’s microvasculature promoting vasoconstriction.17 Although the exact mechanisms for the regulation of dopamine synthesis and for dopamine release remain to be elucidated, evidence does exist that these are distinct mechanisms.18 For example, in the prefrontal and cingulated cortices, activation of autoreceptors regulates the release, but not the synthesis, of dopamine.18 Low-dose effects of dopaminomimetics are mediated by autoreceptor activation, as opposed to postsynaptic receptors, due to their 10-fold higher affinity for dopamine.
Functional Anatomy of Central Dopamine Systems
In the 1960s, a group of Swedish scientists first described the nigrostriatal, mesocorticolimbic, and tuberoinfundibular dopamine neurons as giving rise to the three most conspicuous and behaviorally relevant dopamine circuits in the brain.19 Using histofluorescence and subsequently immunohistochemical identification of TH,20,21 16 unique monoaminergic cell groups were identified and given designations A1 through A16, of which dopamine has been identified as a major transmitter in a subpopulation of A2 dorsal motor vagal neurons, and in the A8 through A17 cell groups.22 There is generally marked conversation in the cellular and receptor distributions and major pathways across species with limited exceptions.23 Operationally, these groups can be characterized as long projection versus local circuit neurons with unique functions (Table 11-1).
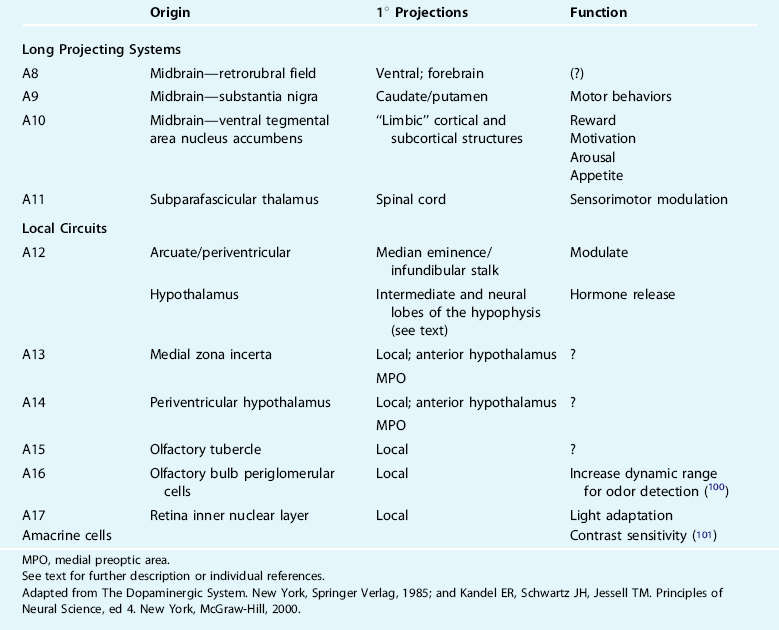
The nigrostriatal pathway originates in the midbrain, from catecholamine cell groups A8 and A9, and projects to the caudate nucleus and putamen (collectively, often referred to as the dorsal striatum). This pathway is traditionally taught to modulate voluntary (waking) movement, and its destruction or degeneration (as in Parkinson’s disease [PD]) results in impairments in the planning, initiation, and execution of movement and motor engrams.24–27 Heuristic models of this major dopaminergic circuit focus themselves on dopamine’s indirect actions (i.e., via dorsal striatal pathways to the internal segment of the globus pallidus [GPi] and substantia nigra pars reticulata [SNr]) and a series of parallel, segregated striatopallidal-thalamocortical recurrent loops centered on functionally distinct cortical regions.28,29 The anatomy of the input and output connections of the A8 and A9 neurons and associated behaviors are most often considered in isolation with the basal ganglia nuclei and the thalamus. Moreover, wakefulness has been the “default” medium through which the behavioral correlates of dopamine dysfunction are believed to play out in this major pathway. It is less well established or understood what relevance this dopaminergic system might have to the modulation of normal and pathological wake/sleep states such as RLS/periodic limb movements in sleep (RLS/PLMS) (see, however, Rye30,31).
The mesocorticolimbic pathway arises from the midbrain catecholamine cell group A10, within the ventral tegmental area, and targets the ventral striatum (nucleus accumbens), subcortical limbic nuclei such as the septum and amygdala, the hippocampus, and prefrontal cortex.32 Activation of this pathway is known to modulate various cognitive and emotive functions including reward, the psychomotor effects associated with drugs of abuse, and working memory.33–35 Disruption of this pathway is believed to modify schizophrenia, attention-deficit/hyperactivity disorder, Tourette’s syndrome, and major depression. Together, the nigrostriatal and mesocorticolimbic systems account for nearly 80% of the brain’s dopamine content.
The third major circuit, the tuberoinfundibular and tuberohypophyseal pathways, originate in the hypothalamic arcuate/periarcuate nuclei and periventricular hypothalamus (catecholamine cell groups A12 and A14, respectively). Activation of the A12 cluster modulates the release of numerous hormones in very complex ways—often in opposing ways at the cellular versus presynaptic level. Dopamine in the tuberoinfindibular system/anterior pituitary tonically inhibits release of prolactin, and luteinizing and thyroid-stimulating hormones, and promotes growth hormone release, primarily via effects on releasing hormones.36–40 The effects of dopamine on the tuberohypophyseal system include, generally, inhibitory modulation of vasopressin release and facilitation of oxytocin release.21
The little-studied A11 catecholamine cell group in the subparafascicular thalamus is the largest, likely sole, source of spinal dopamine.41–44 Within the spinal cord, these axons target the intermediolateral column (IML) housing preganglionic sympathetic neurons, dorsal horn regions related to afferent nerve processing, interneurons (e.g., Renshaw cells), and somatic motoneurons,43 where they likely dampen spinal nociceptive processing and sympathetic outflow and enhance motor output predominantly via D2-like receptor mechanisms.45,46
The axons of major projecting brain dopamine systems have a proclivity to collateralize extensively; that is, individual axons branch and innervate two or more physically, and perhaps functionally, unique regions.47–49 In addition to innervating the striatum and frontal cortex, for example, A8 through A10 neurons also target the thalamus (principally the midline, intralaminar, and reticular nuclei that modulate thalamocortical excitability), the extended amygdala, the noradrenergic locus ceruleus, and the serotonergic raphe system. Axons of individual A11 cells branch extensively to all spinal cord levels along their course in the dorsolateral funiculi, as well as to the prefrontal cortex, amygdala, and nucleus of the solitary tract.50 These arrangements are ideally suited to coordinate behaviors affected by disparate brain areas (e.g., environmental stimuli, cardiorespiratory homeostasis, and sleep/wake state) and bear remarkable resemblance to other nuclei traditionally thought of as influencing the organism’s arousal state (i.e., comprising the ascending reticular activating system).
Circadian and Homeostatic Influences on Central Dopamine Signaling
It has long been appreciated that there are ultradian rhythms in content, turnover, release and behavioral responses of many of the brain’s dopamine systems outlined above. Although we are far from a complete understanding, it is relatively safe to posit that dopamine signaling is under circadian influences that manifest as a peak in activity during the active period and a nadir in the major inactive period (i.e., sleep). This rhythmicity is retained in cultures containing hypothalamic dopamine neurons that are in close proximity to the principal circadian pacemaker (i.e., the suprachiasmatic nucleus) and less evident or absent in cultures containing forebrain or midbrain dopamine neurons.51 Circadian influences on brain dopamine systems are conveyed in part via melatonin as pinealectomy dampens rhythms in striatal dopamine content.52 There is also increasing evidence of molecular control of dopaminergic transmission by genes involved in the circadian clock.53 Mice expressing an inactive protein of the circadian-associated gene Clock, for example, exhibit increased expression and phosphorylation of TH and increased activity in A10 neurons and associated behaviors.53 The first studies to look at circadian variations in human brain monoamine content were those undertaken by Carlsson and colleagues54 on the hypothalamus in post-mortem tissue. Hypothalamic dopamine content peaks between 3 P.M and 6 P.M. and then drops continuously through the evening, reaching its nadir in early morning. The greatest variations in dopamine content in the nocturnal mouse occurs within the hypothalamus,55 peaking at midnight and then dropping significantly by 4 A.M. At least in part, this appears attributable to diurnal modulation in the expression of TH, which nadirs several hours before the major wake period and peaks in the middle of the subjective day, coincident with levels of extracellular dopamine, DOPAC, and HVA and motor activity.56–61 Expression of DAT also exhibits a diurnal rhythm that appears to lead these other rhythms in dopamine transmission, at least in the striatum.61 It remains unclear whether these rhythms are mirrored by changes in striatal receptor density58,62 and whether they might be more pronounced in limbic63 or motor64 subcircuits.
Some of the brain’s dopamine systems also appear to be under homeostatic influences, and this has been most studied in the nigrostriatal pathway. Among the brain’s dopamine systems, plasticity of the nigrostriatal pathway attending increased drives to sleep appears unique, with hypothalamic dopamine receptors, for example, being unaffected by sleep deprivation.65 Dopamine and its metabolites, as opposed to other monoamines, are increased in the striatum of rapid eye movement (REM) sleep–deprived rats.66–71 There is disagreement, on the other hand, concerning the precise receptor and regional changes that occur. Some investigators argue for no changes in the absolute receptor number,68,72 whereas others describe increases in D2-like receptor binding.73–75 Still others argue for a D1-like receptor–mediated increase in adenylate cyclase within mesocortical limbic pathways whose reversal with D1-like, as opposed to D2-like, antagonists facilitates rebound sleep.76–78
Summary: Dopamine in “State” Control and Its Relevance to RLS/PLMS
Pharmacological agents that alter neural dopaminergic signaling are some of the most prescribed and effective agents for treating sleepiness and sleep disorders such as RLS/PLMS (Table 11-2). Although this clinical experience argues that dopamine signaling is integral to the regulation of arousal state, a complete understanding is only beginning to emerge from recent scientific inquiries (see reviews30,31,79). To summarize very briefly, differential binding affinities and localizations of the various dopamine receptor subtypes likely account for the biphasic effects that dopaminomimetics have on behavioral state. Low doses promote sleep, including REM sleep, that can be antagonized by nonsedating doses of neuroleptics, suggesting a presynaptic D2-like inhibitory mechanism on terminals or somata of dopaminergic ventral tegmental area or substantia nigra pars compacta neurons. Higher doses of nonspecific dopaminergic agonists increase locomotor activity, enhance wakefulness, and suppress sleep and REM sleep, likely via D1-like postsynaptic receptors.
TABLE 11-2 Dopaminergic Drugs Used to Treat Restless Legs Syndrome are High-Affinity D3 Receptor Agonists
| Drug | Receptor Type Affinities* |
|---|---|
| Dopamine | High, with D3,4 >> D1,2,5 |
| Pergolide | High, with D3 >D2> D4 >> D1,5 |
| Pramipexole | High, with D3 > D4 > D2 >> D1,5 |
| Cabergoline | High, with D3 ∼ D2 > D5 >D4 > D1 |
| Ropinirole | Very high, with D3 >> D2, 4 >> D1,5 |
| Rotigotine | High, with D3 > D2 >> D1 |
The table shows the relative affinity of currently used dopaminergics and, for comparison, the relative affinity of dopamine for the individual molecularly defined receptors.
* Data obtained from PDSP drug database comparison: http://pdsp.cwru.edu/.
A central dopamine hypothesis (viz., a hypofunctioning) to RLS derives from the exquisite sensitivity of most patients to even low doses of dopaminomimetics. This hypothesis endures despite the lack of any compelling biological evidence of dopaminergic dysfunction in RLS patients. Genetic association studies do not point to RLS susceptibility residing in variants of proteins involved in dopamine’s synthesis or signaling80 (see, however, Desautels and colleagues81). Cerebrospinal fluid analyses of dopamine and its major metabolites have also been unrevealing.82,83 Imaging studies of the most conspicuous of brain dopamine pathways, the nigrostriatal system, have yielded inconsistent data that have been unable to differentiate between postsynaptic and presynaptic dysfunction, or a relative excess versus deficiency in the availability of synaptic dopamine.84 Decreased fluoro-dopa uptake85,86 and D2 receptor binding85,87,88 have been described by some investigators, yet these changes are small, occur in the face of normal DAT binding,88,89 and are not substantiated by another study.90 Difficult to reconcile with a significant presynaptic dysfunction in nigrostriatal terminals underlying the expression of RLS is the clinical experience that RLS (as well as PLMS) occurs more, rather than less, frequently in Parkinson’s disease, in which more than 80% of midbrain dopaminergic neurons have been lost.91–93 These findings can be reconciled if a dysfunction of dopamine signaling postsynaptically, rather than presynaptically, underlies RLS. Alternatively, these collective data may point to the primary pathophysiology residing in hypofunctioning of the little-studied A11 dopaminergic diencephalospinal pathways that, despite their small size, exert potent modulatory actions on spinal networks principally via D2-like receptors (including the D3 receptor subtype).45,94–96 One preliminary lesion study of this pathway suggests that interruption of this sole source of spinal dopamine may induce an RLS-like phenotype.97 Additional behavioral analyses and specifics concerning the synaptic, cellular, and network mechanisms involved are beginning to emerge. Dopamine and D2-like agonists, for example, depress the monosynaptic reflex amplitude in the mouse spinal cord in vitro, and at low but physiological levels of dopamine, this modulation is mediated by D3 receptors (i.e., it is absent in functional D3 receptor knock-out animals).96 Mice lacking a functional D3 receptor also exhibit a reversal of their circadian profile of TH in spinal sympathetic neurons98 and behaviorally are motorically hyperactive and manifest increased wakefulness across the rest-activity cycle.99 Given circadian nadirs of dopamine function discussed above, behavioral effects of any natural or disease related (e.g., in RLS) reductions in this dopamine circuit would therefore favor an increase in spinal cord nociceptive inputs, reflexes, and sympathetic drive and potentially a phenotype resembling RLS.
1. Nagatsu T, Levitt M, Udenfriend S. Tyrosine hydroxylase: The initial step in norepinephrine synthesis. J Biol Chem. 1964;239:2910-2917.
2. Nakashima A, Mori K, Suzuki T, et al. Dopamine inhibition of human tyrosine hydroxylase type I is controlled by the specific portion in the N-terminus of the enzyme. J Neurochem. 1999;72:2145-2153.
3. Ramsey A, Fitzpatrick P. Effects of phosphorylation of serine 40 of tyrosine hydroxylase on binding of catecholamines: Evidence for a novel regulatory mechanism. Biochemistry. 1998;37:8980-8986.
4. Ramsey A, Hillas P, Fitzpatrick P. Characterization of the active site iron in tyrosine hydroxylase redox states of the iron. J Biol Chem. 1996;271:24395-24400.
5. Nagatsu I. Tyrosine hydroxylase: Human isoforms, structure and regulation in physiology and pathology. Essays Biochem. 1995;30:15-35.
6. Bandmann O, Valente E, Holmans P, et al. Dopa-responsive dystonia: A clinical and molecular genetic study. Ann Neurol. 1998;44:649-656.
7. Nygaard T, Marsden C, Duvoisin R. Dopa-responsive dystonia. Adv Neurol. 1988;50:377-384.
8. Nygaard T. Dopa-responsive dystonia. Delineation of the clinical syndrome and clues to pathogenesis. Adv Neurol. 1993;1993:577-585.
9. Nicola S, Surmeier J, Malenka R. Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu Rev Neurosci. 2000;23:185-215.
10. Barbeau H, Rossignol S. Initiation and modulation of the locomotor pattern in the adult chronic spinal cat by noradrenergic, serotonergic and dopaminergic drugs. Brain Res. 1991;546:250-260.
11. Kiehn O, Kjaerulff O. Spatiotemporal characteristics of 5-HT and dopamine-induced rhythmic hindlimb activity in the in vitro neonatal rat. J Neurophysiol. 1996;75:1472-1482.
12. Katz P. Neurons, networks, and motor behavior. Neuron. 1996;16:245-253.
13. Missale C, Nash SR, Robinson SW, et al. Dopamine receptors: From structure to function. Physiol Rev. 1998;78:189-225.
14. Sautel F, Griffon N, Levesque D, et al. A functional test identifies dopamine agonists selective for D3 versus D2 receptors. Neuroreport. 1995;6:329-332.
15. Sokoloff P, Andrieux M, Besancon R, et al. Pharmacology of human dopamine D3 receptor expressed in a mammalian cell line: Comparison with D2 receptor. Eur J Pharmacol. 1992;225:331-337.
16. Greengard P, Allen P, Nairn A. Beyond the dopamine receptor: The DARPP-32/protein phosphatase-1 cascade. Neuron. 1999;23:435-447.
17. Krimer LS, Muly EC3rd, Williams GV, et al. Dopaminergic regulation of cerebral cortical microcirculation. Nat Neurosci. 1998;1:286-289.
18. Cooper J, Bloom F, Roth R. The Biochemical Basis of Neuropharmacology, 8. New York: Oxford University Press, 2002.
19. Dahlstrom A, Fuxe K. Evidence for the existence of monoamine-containing neurones in the central nervous system. L-Demonstration of monoamines in the cell bodies of brain stem neurones. Acta Physiol Scand Suppl. 1964;232:1-55.
20. Hökfelt T, Martensson R, Björklund A, et al. Distributional maps of tyrosine-hydroxylase-immunoreactive neurons in the rat brain. In: Björklund A, Hökfelt T, editors. Handbook of Chemical Neuroanatomy. New York: Elsevier; 1984:277-379.
21. The Dopaminergic System. New York: Springer Verlag, 1985.
22. Lindvall O, Björklund A. Neuroanatomy of central dopamine pathways: Review of recent progress. In: Kohsaka M, et al, editors. Advances in Dopamine Research. Oxford/New York: Pergamon Press; 1982:297-311.
23. Smeets W, Gonzalez A. Catecholamine systems in the brain of vertebrates: New perspectives through a comparative approach. Brain Res Brain Res Rev. 2000;33:308-379.
24. Kandel ER, Schwartz JH, Jessell TM. Principles of Neural Science, 4. New York: McGraw-Hill, 2000.
25. Grillner S, Hellgren J, Menard A, et al. Mechanisms for selection of basic motor programs—Roles for the striatum and pallidum. Trends Neurosci. 2005;28:364-370.
26. Albin R, Young A, Penney J. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366-375.
27. DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990;13:281-285.
28. Alexander GE, Crutcher MD, DeLong MR. Basal ganglia-thalamocortical circuits: Parallel substrates for motor, oculomotor, “prefrontal” and “limbic” functions. Progr Brain Res. 1990;85:119-146.
29. Alexander G. Anatomy of the basal ganglia and related motor structures. In: Watts R, Koller W, editors. Movement Disorders: Neurologic Principles and Practice. New York: McGraw-Hill; 1997:73-86.
30. Rye D. Parkinson’s disease and RLS: The dopaminergic bridge. Sleep Med. 2004;5:317-328.
31. Rye D. The two faces of Eve: Dopamine’s modulation of wakefulness and sleep. Neurology. 2004;63(suppl 3):S2-S7.
32. Williams SM, Goldman-Rakic PS. Widespread origin of the primate mesofrontal dopamine system. Cereb Cortex. 1998;8:321-345.
33. Tzschentke T, Schmidt W. Functional relationship among medial prefrontal cortex, nucleus accumbens, and ventral tegmental area in locomotion and reward. Crit Rev Neurobiol. 2000;14:131-142.
34. Fibiger H, Phillips A. Reward, motivation, cognition: Psychobiology of mesotelencephalic dopamine systems. In Bloom FE and Geiger SR (eds): Handbook of Physiology. Intrinsic Regulatory Systems of the Brain, Vol 4. Bethesda, MD: American Physiology Society. 1986:647-675.
35. Goldman-Rakic PS. Cellular basis of working memory. Neuron. 1995;14:477-485.
36. Krulich L. Central neurotransmitters and the secretion of prolactin, GH, LH and TSH. Ann Rev Physiol. 1979;41:603-615.
37. Behrends J, Prank K, Dogu E, et al. Central nervous system control of thyrotropin secretion during sleep and wakefulness. Horm Res. 1998;49:173-177.
38. Martin J. Neural regulation of growth hormone secretion. Med Clin North Am. 1978;62:327-336.
39. Benker G, Jaspers C, Hausler G, et al. Control of prolactin secretion. Klin Wochenschr. 1990;68:1157-1167.
40. Muller E. Nervous control of growth hormone secretion. Neuroendocrinology. 1973;11:338-369.
41. Björklund A, Skagerberg G. Evidence for a major spinal cord projection from the diencephalic A11 dopamine cell group in the rat using transmitter-specific fluorescent retrograde tracing. Brain Res. 1979;177:170-175.
42. Hökfelt T, Phillipson O, Goldstein M. Evidence for a dopaminergic pathway in the rat descending from the A11 cell group to the spinal cord. Acta Physiol Scand. 1979;107:393-395.
43. Skagerberg G, Björklund A, Lindvall O, et al. Origin and termination of the diencephalo-spinal dopamine system in the rat. Brain Res Bull. 1982;9:237-244.
44. Skagerberg G, Lindvall O. Organization of diencephalic dopamine neurones projecting to the spinal cord in the rat. Brain Res. 1985;342:340-351.
45. Fleetwood-Walker S, Hope P, Mitchell R. Antinociceptive actions of descending dopaminergic tracts on cat and rat dorsal horn somatosensory neurones. J Physiol (Lond). 1988;399:335-348.
46. Gladwell S, Coote J. Inhibitory and indirect excitatory effects of dopamine on sympathetic preganglionic neurones in the neonate rat spinal cord in vitro. Brain Res. 1999;818:397-407.
47. Sanchez-Gonzalez M, Garcia-Cabezas M, Rico B, et al. The primate thalamus is a key target for brain dopamine. J Neurosci. 2005;25:6076-6083.
48. Freeman A, Ciliax B, Bakay R, et al. Nigrostriatal collaterals to thalamus degenerate in parkinsonian animal models. Ann Neurol. 2001;50:321-329.
49. Gaspar P, Stepniewska I, Kaas JH. Topography and collateralization of the dopaminergic projections to motor and lateral prefrontal cortex in owl monkeys. J Comp Neurol. 1992;325:1-21.
50. Takada M. Widespread dopaminergic projections of the subparafascicular thalamic nucleus in the rat. Brain Res Bull. 1993;32:301-309.
51. Abe M, Herzog ED, Yamazaki S, et al. Circadian rhythms in isolated brain regions. J Neurosci. 2002;22:350-356.
52. Khaldy H, Leon J, Escames G, et al. Circadian rhythms of dopamine and dihydroxyphenyl acetic acid in the mouse striatum: Effects of pinealectomy and of melatonin treatment. Neuroendocrinology. 2002;75:201-208.
53. McClung C, Sidiropoulou K, Vitaterna M, et al. Regulation of dopaminergic transmission and cocaine reward by the Clock gene. Proc Natl Acad Sci U S A. 2005;102:9377-9381.
54. Carlsson A, Svennerholm L, Winblad B. Seasonal and circadian monoamine variations in human brain examined post mortem. Acta Pyschiatr Scan Suppl. 1980;280:275-285.
55. Matsumoto M, Kimura K, Fujisawa A, et al. Diurnal variations in monoamine contents in discrete brain regions of the mongolian gerbil (Meriones unguiculatus). J Neurochem. 1981;37:792-794.
56. McGeer E, McGeer P. Some characteristics of brain tyrosine hydroxylase. In: Mandel A, editor. New Concepts in Neurotransmitter Regulation. New York: Plenum Press; 1973:53-68.
57. Simon ML, George R. Diurnal variations in plasma corticosterone and growth hormone as corrlelated with regional variations in norepinephrine, dopamine and serotonin content of rat brain. Neuroendocrinology. 1975;17:125-138.
58. Bruinink A, Lichtensteiger W, Schlumpf M. Ontogeny of diurnal rhythms of central dopamine, serotonin and spirodecanone binding sites and of motor activity in the rat. Life Sci. 1983;33:31-38.
59. Smith AD, Olson RJ, Justice JBJr. Quantitative microdialysis of dopamine in the striatum: effect of circadian variation. J Neurosci Methods. 1992;44:33-41.
60. Schade R, Vick K, Sohr R, et al. Correlative circadian rhythms of cholecystokinin and dopamine content in nucleus accumbens and striatum of rat brain. Behav Brain Res. 1993;59:211-214.
61. Whittaker J, Morcol T, Patrickson J. Circadian plasticity in dopaminergic parameters in the rat substantia nigra. Soc Neurosci Abstr. 1997;23:190.
62. Watanabe S, Seeman P. D2 dopamine receptor density in rat striatum over 24 hours: Lack of detectable changes. Biol Psychiatry. 1984;19:1249-1253.
63. O’Neill RD, Fillenz M. Simultaneous monitoring of dopamine release in rat frontal cortex, nucleus accumbens and striatum: Effect of drugs, circadian changes and correlations with motor activity. Neuroscience. 1985;16:49-55.
64. Paulson PE, Robinson TE. Relationship between circadian changes in spontaneous motor activity and dorsal versus ventral striatal dopamine neurotransmission assessed with on-line microdialysis. Behav Neurosci. 1994;108:624-635.
65. Lal S, Thavundayil J, Nair NP, et al. Effect of sleep deprivation on dopamine receptor function in normal subjects. J Neural Transm. 1981;50:39-45.
66. Ghosh PK, Hrdina PD, Ling GM. Effects of REMS deprivation on striatal dopamine and acetylcholine in rats. Pharmacol Biochem Behav. 1976;4:401-405.
67. Tufik S, Lindsey CJ, Carlini EA. Does REM sleep deprivation induce a supersensitivity of dopaminergic receptors in the rat brain? Pharmacology. 1978;16:98-105.
68. Farber J, Miller JD, Crawford KA, et al. Dopamine metabolism and receptor sensitivity in rat brain after REM sleep deprivation. Pharmacol Biochem Behav. 1983;18:509-513.
69. Asakura W, Matsumoto K, Ohta H, et al. REM sleep deprivation decreases apomorphine-induced stimulation of locomotor activity but not stereotyped behavior in mice. Gen Pharmacol. 1992;23:337-341.
70. Farooqui SM, Brock JW, Zhou J. Changes in monoamines and their metabolite concentrations in REM sleep-deprived rat forebrain nuclei. Pharmacol Biochem Behav. 1996;54:385-391.
71. Lara-Lemus A, Drucker-Colin R, Mendez-Franco J, et al. Biochemical effects induced by REM sleep deprivation in naive and in D-amphetamine treated rats. Neurobiology. 1998;6:13-22.
72. Hamdi A, Brock J, Ross K, et al. Effects of rapid eye movement sleep deprivation on the properties of striatal dopaminergic system. Pharmacol Biochem Behav. 1993;46:863-866.
73. Zwicker A, Calil H. The effects of REM sleep deprivation on striatal dopamine receptor sites. Pharmacol Biochem Behav. 1986;24:809-812.
74. Nunes GJr, Tufik S, Nobrega N. Autoradiographic analysis of D1 and D2 dopaminergic receptors in rat brain after paradoxical sleep deprivation. Brain Res Bull. 1994;34:453-456.
75. Brock JW, Hamdi A, Ross K, et al. REM sleep deprivation alters dopamine D2 receptor binding in the rat frontal cortex. Pharmacol Biochem Behav. 1995;52:43-48.
76. Fadda P, Martellotta MC, De Montis MG, et al. Dopamine D1 and opioid receptor binding changes in the limbic system of sleep deprived rats. Neurochem Int. 1992;20(suppl):153S-156S.
77. Fadda P, Martellotta MC, Gessa GL, et al. Dopamine and opioids interactions in sleep deprivation. Prog Neuropsychopharmacol Biol Psychiatry. 1993;17:269-278.
78. Durán-Vázquez A, Drucker-Colín R. Differential role of dopamine receptors on motor asymmetries of nigro-striatal lesioned animals that are REM sleep deprived. Brain Res. 1997;744:171-174.
79. Rye DB, Jankovic J. Emerging views of dopamine in modulating sleep/wake state from an unlikely source: PD. Neurology. 2002;58:341-346.
80. Desautels A, Turecki G, Montplaisir J, et al. Dopaminergic neurotransmission and restless legs syndrome: A genetic association analysis. Neurology. 2001;57:1304-1306.
81. Desautels A, Turecki G, Montplaisir J, et al. Evidence for a genetic association between monoamine oxidase A and restless legs syndrome. Neurology. 2002;59:215-219.
82. Earley C, Hyland K, Allen R. CSF dopamine, serotonin, and biopterin metabolites in patients with restless legs syndrome. Mov Disord. 2001;16:144-149.
83. Stiasny-Kolster K, Moller J, Zschocke J, et al. Normal dopaminergic and serotonergic metabolites in cerebrospinal fluid and blood of restless legs syndrome patients. Mov Disord. 2004;19:192-196.
84. Wetter T, Eisensehr I, Trenkwalder C. Functional neuroimaging studies in restless legs syndrome. Sleep Med. 2004;5:401-406.
85. Turjanski N, Lees A, Brooks D. Striatal dopaminergic function in restless legs syndrome. Neurology. 1999;52:932-937.
86. Routtinen H, Partinen M, Hublin C, et al. An FDOPA PET study in patients with periodic limb movement disorders and restless legs syndrome. Neurology. 2000;54:502-504.
87. Staedt J, Stoppe G, Kogler A, et al. Nocturnal myoclonus syndrome (periodic movements in sleep) related to central dopamine D2-receptor alteration. Eur Arch Psychiatr Clin Neurosci. 1995;245:8-10.
88. Michaud M, Soucy J, Chabli A, et al. SPECT imaging of striatal pre- and postsynaptic dopaminergic status in restless legs syndrome with periodic leg movements in sleep. J Neurol. 2002;249:164-170.
89. Linke R, Eisensehr I, Wetter T, et al. Presynaptic dopaminergic function in patients with restless legs syndrome: Are there common features with early Parkinson’s disease? Mov Disord. 2004;19:1158-1162.
90. Eisensehr I, Wetter TC, Linke R, et al. Normal IPT and IBZM SPECT in drug-naive and levodopa-treated idiopathic restless legs syndrome. Neurology. 2001;57:1307-1309.
91. Bliwise D, Rye D, Dihenia B, et al. Periodic leg movements in elderly patients with parkinsonism. Sleep. 1998;21(suppl):196.
92. Wetter T, Collado-Seidel V, Pollmacher T, et al. Sleep and periodic leg movement patterns in drug-free patients with Parkinson’s disease and multiple system atrophy. Sleep. 2000;23:361-367.
93. Ondo WG, Vuong KV, Khan H, et al. Daytime sleepiness and other sleep disorders in Parkinson’s disease. Neurology. 2001;57:1392-1396.
94. Garraway S, Hochman S. Modulatory actions of serotonin, norepinephrine, dopamine, and acetylcholine in spinal cord deep dorsal horn neurons. J Neurophysiol. 2001;86:2183-2194.
95. Barriere G, Mellen N, Cazalets J. Neuromodulation of the locomotor network by dopamine in the isolated spinal cord of the newborn rat. J Neurophysiol. 2004;19:1325-1335.
96. Clemens S, Hochman S. Conversion of the modulatory actions of dopamine on spinal reflexes from depression to facilitation in D3 receptor knock-out mice. J Neurosci. 2004;24:11337-11345.
97. Ondo WG, He Y, Rajasekaran S, et al. Clinical correlates of 6-hydroxydopamine injections into A11 dopaminergic neurons in rats: A possible model for restless legs syndrome. Mov Disord. 2000;15:154-158.
98. Clemens S, Sawchuk M, Hochman S. Reversal of the circadian expression of tyrosine-hydroxylase but not nitric oxide synthase levels in the spinal cord of D3 receptor knockout mice. Neuroscience. 2005;133:353-357.
99. Hue G, Decker M, Solomon I, Rye D. Increased wakefulness and hyper-responsivity to novel environments in mice lacking functional dopamine D3 receptors. Soc Neurosci. 2003. Online publication
100. Ennis M, Zhou F, Ciombor K, et al. Dopamine D2 receptor-mediated presynaptic inhibition of olfactory nerve terminals. J Neurophysiol. 2001;86:2986-2997.
101. Witkovsky P. Dopamine and retinal function. Doc Ophthalmol. 2004;108:17-40.