Chapter 14 Spinal Cord, Dopamine, Opiates, and Restless Legs Syndrome
Restless legs syndrome (RLS) leads to abnormal limb sensations and periodic limb movements (PLMS). Due to the excellent responses to treatment with dopaminergic and opioidergic agents, an involvement of both dopamine and opioid receptors is highly likely. The spinal cord plays an essential role as the final common pathway in the generation of PLMS. Here we review anatomical and physiologic evidence contributing to PLM generation in the spinal cord and the involvement of the opioid and dopamine system. The bulk of early studies, mainly by the Lundberg group (for refs. see Schomburg1), investigated dopaminergic influence on the spinal cord in greater detail decades ago, with the interest in this research declining somewhat at the end of the last century. Modern techniques have led to a renewed interest in this subject. Two studies involving dopamine D3 receptor knockout mice (D3KO)2,3 sought to explain how impaired D3 activity could contribute to this sleep disorder. The aim of this review is to summarize neurophysiologic data regarding the role of spinal dopamine and opioids against the background of the symptomatology of RLS.
Spinal Structures
Incoming sensory afferents and the motoneuron system are interconnected by the spinal interneuronal system, which is responsible for the generation of simple reflex movements as well as complex motor patterns ranging from tonic to phasic movements and from a co-contraction of antagonistic muscles to the coordinated patterning of locomotor activity (for a review, see Schomburg1). The activity of these interneurons, like the activity of all spinal interneurons and the activity of spinal nociceptive neurons in the dorsal horn, is modulated by descending pathways with both inhibitory and excitatory components.4 During normal voluntary movements, reafferent sensory input continuously converges on the spinal circuits that are activated by these descending motor commands; this locus may serve as the anatomical correlate for the earliest suppression of reafferent sensory input5 (Fig. 14-1). Also, presynaptic inhibition of spinal afferents produced by descending commands may effectively reduce synaptic transmission at the initial synapse.6
All these interactions have to be generated quickly, and they depend on immediate motor requirements, that is, on the phase and condition (including possible external disturbances) of a movement and the position of the trunk and limbs. This so-called state-dependent modulation of neurotransmission is an important tool to adapt the spinal interneuronal systems to the requirements of performing movement in response to the existing conditions. Under conditions of disturbed descending control, such as in spasticity after a stroke, identical—or subthreshold peripheral—input becomes itself capable of inducing muscle contractions. In contrast, such as under normal conditions, reflexogenic activation is largely downregulated to prevent inappropriate reflexogenic movements from the continuously activated peripheral receptors. In this downregulated situation, the activity of individual receptor systems is generally unable to evoke reflexogenic movement. If at all, a distinct multisensorial convergence of different receptor systems is required to elicit a movement.7 During sleep, in particular during rapid eye movement (REM) sleep, state-dependent modulation is further downregulated to preclude any unwanted movements during dreaming.
Of all possible spinal structures involved, the spinal flexor reflex afferent (FRA) system and a disturbance of its supraspinal control seem to play the crucial role in the generation of PLMS. The FRA system encompasses the following features:
Dopamine in the Spinal Cord
The spinal cord is virtually devoid of dopaminergic cell bodies. The major source of the spinal dopaminergic projection into the dorsal horns and intermediolateral tracts is area A11.10 The behavioral effects of a first animal model on A11 lesion have not been convincing, most likely because the A11 neurons are spread over a comparatively large area and an isolated lesion appears to be very difficult to achieve.11 Nevertheless, an additional intriguing argument in favor of an A11 lesion as a source for RLS is its anatomical location close to the suprachiasmatic nucleus, which largely controls circadian rhythms and which could explain its predominance at night. The RLS-A11 hypothesis would require a rather selective neurodegeneration of A11 with increasing age and a concomitant reduction in the descending modulation of the dorsal horn or other related structures in the spinal cord.12
Because no RLS animal model is readily available, most data have been obtained in the course of pain studies. Dopamine may ameliorate chronic pain. In rats, intrathecal administration of the dopamine agonist apomorphine produced an analgesia that was antagonized by the prior intrathecal administration of a dopamine receptor antagonist but not by the administration of α2, serotonin, or opiate antagonists.13 Further data beyond the scope of this review confirm the antinociceptive effects of dopamine D2 receptors (for the most detailed review, see Millan10), probably via a direct inhibitory action at either the nociceptive-responsive primary afferent fiber terminals or projection neurons. An inhibitory influence of presynaptic dopamine D2 receptors on Ca2+ currents provides a potential mechanism for a presynaptic reduction of release from primary afferent fiber terminals in the dorsal horn.14 Also, L-DOPA inhibits the early flexor reflex, regardless of whether it is evoked by nociceptive or nonnociceptive afferents.15
State dependence usually depends on dopaminergic stimulation, but there are contrary cases. For example, in acute phase–dependent (during locomotion)16–19 or limb position–dependent20 reflex transmission, dopamine is not primarily involved.
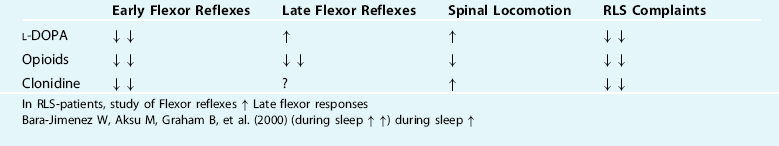
Within a medium range of spinal dopamine concentration in the spinalized and paralyzed cat, locomotor activity (“fictive locomotion”; i.e., neuronal rhythmic activity resembling locomotor pattern without real movements in paralyzed animals) can be induced. During fictive locomotion, motoneurons can develop a bistable firing characteristic, caused by their membrane potentials switching between two levels. This state is induced by L-DOPA. Depolarization during active phases was characterized by stable fixed levels and could partly be initiated by excitatory inputs and terminated by inhibitory inputs.21 Similar bistable characteristics have been postulated for RLS.22 It appears, however, unlikely that an L-DOPA–induced bistability is related to the PLM-associated firing pattern in RLS, because L-DOPA suppresses PLM instead of inducing such bursting. However, this is not a direct motoneuronal effect but has to be regarded as a consequence of the L-DOPA–induced suppression of short-latency FRA pathways (see earlier). The intricate function of the dopaminergic system in spinal motor control becomes evident when L-DOPA is applied in different dosages. Without the application of L-DOPA in the spinal animal, repeated afferent FRA stimulation evokes the “normal” short-latency flexor reflexes. After L-DOPA in a low dosage that is subthreshold for induction of rhythmic locomotor activity, the same stimulation evokes longer-lasting tonic responses. If the dopamine concentration is raised above the level that causes rhythmic locomotor activity, a spontaneous irregular motor hyperactivity occurs.18 It can be assumed that the responses to afferent stimulation during the latter stages of hyperreactivity are due to an increased responsiveness of the late FRA system. At the spinal level, L-DOPA thus heavily depresses the short-latency FRA pathways (probably its therapeutical action in RLS), whereas the transmission in the long-latency pathways, and in parallel the generation of locomotion, are facilitated (see Table 14-1). Because the depression of polysynaptic reflexes seems to be mainly mediated by D3-dopamine receptors and dopamine may cause an increase of these reflexes in the isolated spinal cord of D3KO mice, it has been assumed that an impaired D3 activity could contribute to the RLS.2
The importance of FRA for RLS has been delineated in two clinical spinal reflex studies.23,24 Here, RLS patients had significantly increased spinal cord excitability with lower threshold and greater spatial spread of the flexor reflexes, which were comparatively more prominent during sleep. Multiple late responses were seen during sleep in all patients and in some control subjects at higher thresholds. The most prominent of these responses had a very long duration and a latency range of 250 to 800 milliseconds, and because of its close temporal relation to the FRA stimulus, the authors considered it as a late, high-threshold component of the FR (FR3). This finding cannot immediately be compared with PLMS, which, as described later, decrease with depth of sleep stages. These data can, however, be modeled on the basis of the wide convergence of nociceptive and non-nociceptive flexor reflex afferents onto common interneurons in spinal reflex pathways.25,26
Relation to Sleep Stages
With increase in sleep stages, from light stage 1 sleep to deepest non-REM (NREM) sleep and even further in REM sleep, PLMS are continuously suppressed compared with a relaxed and awake state in bed.27 Nicolas and colleagues27 found that REM sleep was characterized by the shorter duration and the lowest frequency of PLMS, due most likely to the inhibition of spinal motoneurons that prevails in REM sleep. It is most likely that the system involved in REM sleep motor depression is different from the RLS generators because it is able to attenuate PLMS but does not eliminate them completely. Thus, two different systems can be assumed—one dependent on the dopamine level and responsible for PLMS, and the other responsible for suppression of movements in REM sleep and heavily dependent on GABA neurotransmission.28,29 Nevertheless, Nicolas and colleagues27 concluded from their result that during REM, the duration of sleep intervals returned to values obtained in stage 1 sleep and that these two stages shared similar patterns of electroencephalographic activity—and that a single state-dependent mechanism may be responsible for the periodicity of PLM noted both during sleep and wakefulness. It is, of course, potentially important that RLS symptoms peak at night when dopamine levels are lowest.
An altered circadian pattern in spinal dopamine synthesis was described in D3KO animals.3 Low dopamine levels (1 μmol/L) decreased the monosynaptic “stretch” reflex (MSR) amplitude in wild-type (WT) animals but increased it in D3KO animals. Higher dopamine concentrations (10 to 100 μmol/L) decreased MSR amplitudes in both groups, but always more strongly in WT mice. Like low dopamine, the D3 receptor agonists pergolide and PD 128907 reduced MSR amplitude in WT but not D3KO mice. Conversely, D3 receptor antagonists (GR 103691 and nafadotride) increased the MSR in WT but not in D3KO mice. In comparison, D2-preferring agonists bromocriptine and quinpirole depressed the MSR in both groups. Low dopamine (1 to 5 μmol/L) also depressed longer-latency (presumably polysynaptic) reflexes in WT but facilitated responses in D3KO mice. Additionally, in some experiments (e.g., during 10 μmol/L dopamine or pergolide in WT), polysynaptic reflexes were facilitated in parallel to MSR depression, demonstrating differential modulatory control of these reflex circuits. Thus, low dopamine activates D3 receptors to limit reflex excitability. The authors conclude that in D3 ligand–insensitive mice, excitatory actions are unmasked, functionally converting the modulatory action of dopamine from depression to facilitation.
Augmentation: A Paradoxical L-DOPA–Induced, Long-Latency FRA Effect
In some patients, after once-nightly treatments with carbidopa or levodopa30 but also after treatment with other dopamine agonists,31 a delayed augmentation of the RLS symptoms was observed the following day. This augmentation is a potentially serious problem for all dopaminergic treatments in RLS patients. It has been speculated that augmentation may arise as a kind of rebound or a result of a suppression of the circadian rhythm in the striatal, hypothalamic, or spinal dopaminergic system by the higher L-DOPA dosages that are usually associated with augmentation.32 However, according to experimental neurophysiological results, it seems to be more probable that the augmentation of the RLS symptoms is due to a second, delayed action of L-DOPA, which consists of facilitation of long-latency FRA pathways and of an introduction of rhythmic, partly locomotor movements or a more-or-less arrhythmic hyperactivity (cf. Table 1; review by Schomburg1). In the spinal animal, these symptoms may occur about 20 minutes to 1 hour after intravenous injection of L-DOPA (after premedication with a monaminoxidase-blocker) and could last for several hours (up to ≥8 hours, Kniffki and associates).18 More recently, we argue that in terms of receptor contribution, augmentation may be caused by a relative D1 receptor overstimulation (Paulus and Trenkwalder, 2006).33 Iron deficiency and 6-hydroxydopamine lesions in A11 nuclei differentially altered the D1, D2, and D3 receptor expression and binding capacity in the lumbar spinal cord of RLS animal model, which were accompanied by changes in locomotor activities.34
Most interestingly, A11 diencephalospinal dopaminergic neurons lack presynaptic synthesis modulating D2 autoreceptors in mice. Thus, there is no counterregulation of elevated dopamine levels in augmentation.35 The therapeutic benefit and the unwanted side effect of L-DOPA could thus be results of the two opposite effects of L-DOPA at the spinal level: a suppression of the early short-latency FRA pathways and a facilitation of the late long-latency FRA pathways. This assumption finds further support in the fact that similar augmentation of the RLS symptoms were not observed after RLS treatment with opioids and that the augmentation occurring after levodopa can be successfully treated by opioids, which are known to suppress the long-latency FRA pathways and their ability to generate rhythmic spinal motor activity36 (cf. also Table 14-1).
Opioids in the Spinal Cord
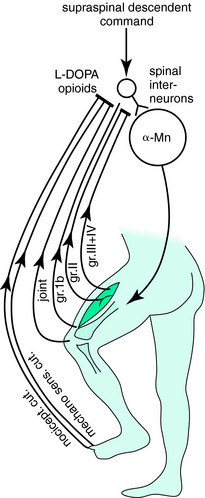
The dramatic therapeutic response to opioids in RLS patients warrants the assumption of a central role in pathophysiology. The efficacy of opiates in the relief of the symptoms is possibly partly related to the dopaminergic system.37 However, the sensitivity to opioids does not imply a crucial role of the nociceptive system in the development of RLS. Like L-DOPA, opioids do not need to have a selective antinociceptive effect at the spinal level because they suppress segmental pathways from all FRA in the same way, from group II spindle afferents, mechanosensitive skin or joint afferents, as well as from nociceptive skin and muscle afferents1,25 (Figs. 14-2 and 14-3).
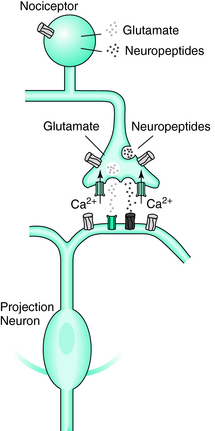
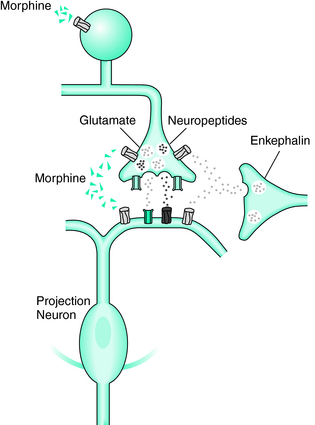
Thus, the enkephalinergic system may cooperate with the descending monoaminergic system to provide a mechanism for suppression of the relatively unspecific FRA feedback during supraspinally induced movements, which require a strictly goal-directed performance without any disturbances from the periphery.38 An intriguing interaction between both the monaminergic and the opiate system seems to be ahead in some genetic forms of RLS.39 Supraspinal motor actions of opioids are more ambiguous because they may partly increase such locomotor activity in intact animals40–43 as a result of supraspinal interactions, probably including actions on the striatum.44,45 However, at the spinal level, opioids merely depress locomotor rhythm generation36 (see also Table 14-1). Regarding the fact that locomotor rhythm generation is at least partly a function of the spinal FRA system36,46 and the above-mentioned observation of the depressing action of opioids on FRA pathways, it can be assumed that therapeutical opioidergic influence on RLS largely results from a spinal action of opioids.
Summary
We sought to bring together two separated areas of research, investigation of opioid and dopaminergic mechanisms in the spinal cord of spinalized animals and clinical features of RLS (Paulus and Schomburg, 2006).47 The most important correlation that can be drawn is a reduction of activity of the early flexor reflexes by both substances, whereas the late reflexes are facilitated by L-DOPA. The latter mechanism may contribute to the phenomenon of augmentation. The fact that the opioids reduce both may offer a rationale to the use of opioids in the treatment of augmentation.
1. Schomburg ED. Spinal sensorimotor systems and their supraspinal control. Neurosci Res. 1990;7:265-340.
2. Clemens S, Hochman S. Conversion of the modulatory actions of dopamine on spinal reflexes from depression to facilitation in D3 receptor knock-out mice. J Neurosci. 2004;24:11337-11345.
3. Clemens S, Sawchuk MA, Hochman S. Reversal of the circadian expression of tyrosine-hydroxylase but not nitric oxide synthase levels in the spinal cord of dopamine D(3) receptor knockout mice. Neuroscience. 2005;133:353-357.
4. Handwerker HO, Iggo A, Zimmermann M. Segmental and supraspinal actions on dorsal horn neurons responding to noxious and non-noxious skin stimuli. Pain. 1975;1:147-165.
5. Jankowska E, Lundberg A. Interneurones in the spinal cord. TINS. 4, 1981.
6. Seki K, Perlmutter SI, Fetz EE. Sensory input to primate spinal cord is presynaptically inhibited during voluntary movement. Nat Neurosci. 2003;6:1309-1316.
7. Lundberg A. Multisensory control of spinal reflex pathways. Prog Brain Res. 1797;50:11-28.
8. Rijsman RM, Stam CJ, Weerd AW. Abnormal H-reflexes in periodic limb movement disorder; impact on understanding the pathophysiology of the disorder. Clin Neurophysiol. 2005;116:204-210.
9. de Weerd AW, Rijsman RM, Brinkley A. Activity patterns of leg muscles in periodic limb movement disorder. J Neurol Neurosurg Psychiatry. 2004;75:317-319.
10. Millan MJ. Descending control of pain. Prog Neurobiol. 2002;66:355-474.
11. Ondo WG, He Y, Rajasekaran S, Le WD. Clinical correlates of 6-hydroxydopamine injections into A11 dopaminergic neurons in rats: a possible model for restless legs syndrome. Mov Disord. 2000;15:154-158.
12. Ellrich J, Treede RD. Convergence of nociceptive and non-nociceptive inputs onto spinal reflex pathways to the tibialis anterior muscle in humans. Acta Physiol Scand. 1998;163:391-401.
13. Jensen TS, Yaksh TL. Effects of an intrathecal dopamine agonist, apomorphine, on thermal and chemical evoked noxious responses in rats. Brain Res. 1984;296:285-293.
14. Formenti A, Martina M, Plebani A, Mancia M. Multiple modulatory effects of dopamine on calcium channel kinetics in adult rat sensory neurons. J Physiol. 1998;509(pt 2):395-409.
15. Schomburg ED, Steffens H. Comparative analysis of L-DOPA actions on nociceptive and non-nociceptive spinal reflex pathways in the cat. Neurosci Res. 1998;31:307-316.
16. Forssberg H, Grillner S, Rossignol S. Phase dependent reflex reversal during walking in chronic spinal cats. Brain Res. 1995;85:103-107.
17. Forssberg H, Grillner S, Rossignol S. Phasic gain control of reflexes from the dorsum of the paw during spinal locomotion. Brain Res. 1977;132:121-139.
18. Kniffki KD, Schomburg ED, Steffens H. Effects from fine muscle and cutaneous afferents on spinal locomotion in cats. J Physiol. 1981;319:543-554.
19. Schomburg ED, Behrends HB. The possibility of phase-dependent monosynaptic and polysynaptic is excitation to homonymous motoneurones during fictive locomotion. Brain Res. 1978;143:533-537.
20. Grillner S, Rossignol S. Contralateral reflex reversal controlled by limb position in the acute spinal cat injected with clonidine i.v. Brain Res. 1978;144:411-414.
21. Schomburg ED, Steffens H. Bistable characteristics of motoneurone activity during DOPA induced fictive locomotion in spinal cats. Neurosci Res. 1996;26:47-56.
22. Bucher SF, Steffens H. Bistable characteristics of the restless legs syndrome. Ann Neurol. 1996;26:47-56.
23. Aksu M, Bara-Jimenez W. State dependent excitability changes of spinal flexor reflex in patients with restless legs syndrome secondary to chronic renal failure. Sleep Med. 2002;3:427-430.
24. Bara-Jimenez W, Aksu M, Graham B, et al. Periodic limb movements in sleep: state-dependent excitability of the spinal flexor reflex. Neurology. 2000;54:1609-1616.
25. Schomburg ED. Restrictions on the interpretation of spinal reflex modulation in pain and analgesia research. Pain Forum. 1997;6:101-109.
26. Schomburg ED, Steffens H, Kniffki KD. Contribution of group III and IV muscle afferents to multisensorial spinal motor control in cats. Neurosci Res. 1999;33:195-206.
27. Nicolas A, Michaud M, Lavigne G, Montplaisir J. The influence of sex, age and sleep/wake state on characteristics of periodic leg movements in restless legs syndrome patients. Clin Neurophysiol. 1999;110:1168-1174.
28. Vazquez J, Baghdoyan HA. GABAA receptors inhibit acetylcholine release in cat pontine reticular formation: implications for REM sleep regulation. J Neurophysiol. 2004;92:2198-2206.
29. Xi MC, Morales FR, Chase MH. Induction of wakefulness and inhibition of active (REM) sleep by GABAergic processes in the nucleus pontis oralis. Arch Ital Biol. 2001;139:125-145.
30. Allen RP, Earley CJ. Augmentation of the restless legs syndrome with carbidopa/levodopa. Sleep. 1996;19:205-213.
31. Winkelman JW, Johnston L. Augmentation and tolerance with long-term pramipexole treatment of restless legs syndrome (RLS). Sleep Med. 2004;5:9-14.
32. Trenkwalder C, Collado Seidel V, Kazenwadel J, et al. One-year treatment with standard and sustained-release levodopa: Appropriate long-term treatment of restless legs syndrome? Mov Disord. 2003;18:1184-1189.
33. Paulus W, Trenkwalder C. Less is more: pathophysiology of dopaminergic-therapy-related augmentation in restless legs syndrome. Lancet Neurol. 2006;5:878-886.
34. Zhao H, Zhu W, Pan T, et al. Spinal cord dopamine receptor expression and function in mice with 6-OHDA lesion of the A11 nucleus and dietary iron deprivation. J Neurosci Res. 2007;85:1065-1076.
35. Pappas SS, Behrouz B, Janis KL, et al. Lack of D2 receptor mediated regulation of dopamine synthesis in A11 diencephalospinal neurons in male and female mice. Brain Res. 2008. in press
36. Schomburg ED, Steffens H. Influence of opioids and naloxone on rhythmic motor activity in spinal cats. Exp Brain Res. 1995;103:333-343.
37. Etgen T, Draganski B, Ilg C, et al. Bilateral thalamic gray matter changes in patients with restless legs syndrome. Neuroimage 2005;24:1242-1247.
38. Schmidt PF, Schomburg ED, Steffens H. Limitedly selective action of a delta-agonistic leu-enkephalin on the transmission in spinal motor reflex pathways in cats. J Physiol. 1991;442:103-126.
39. Winkelmann J, Lichtner P, Schormair P, et al. Variants in the neuronal nitric oxide synthase (nNOS, NOS1) gene are associated with restless legs syndrome. Mov Disord. 2008;23:350-358.
40. Kamerling SG, Dequick DJ, Weckman TJ, et al. Dose-related effects of ethylketazocine on nociception, behaviour and autonomic responses in the horse. J Pharm Pharmacol. 1986;38:40-45.
41. Michael-Titus A, Dourmap N, Costentin J, et al. Role of delta opioid receptors in the effects of inhibitors of enkephalin-degrading peptidases on the horizontal and vertical components of locomotion in mice. Neuropeptides. 1990;15:89-100.
42. Narita M, Takahashi Y, Takamori K, et al. Effects of kappa-agonist on the antinociception and locomotor enhancing action induced by morphine in mice. Jpn J Pharmacol. 1993;62:15-24.
43. Negri L, Noviello V, Angelucci F. Behavioural effects of deltorphins in rats. Eur J Pharmacol. 1993;209:163-168.
44. Brunello N, Volterra A, Di Giulio AM, et al. Modulation of opioid system in C57 mice after repeated treatment with morphine and naloxone: Biochemical and behavioral correlates. Life Sci. 1984;34:1669-1678.
45. Dourmap N, Michael-Titus A, Costentin J. Local enkephalins tonically modulate dopamine release in the striatum: A microdialysis study. Brain Res. 1990;524:153-155.
46. Jankowska E, Jukes MG, Lund S, et al. The effect of DOPA on the spinal cord. 5. Reciprocal organization of pathways transmitting excitatory action to alpha motoneurones of flexors and extensors. Acta Physiol Scand. 1967;70:369-388.
47. Paulus W, Schomburg ED. Dopamine and the spinal cord in restless legs syndrome: does spinal cord physiology reveal a basis for augmentation? Sleep Med Rev. 2006;10:185-196.