Chapter 29 The acute respiratory distress syndrome (ARDS)
The acute respiratory distress syndrome (ARDS) was first described in 1967 by Ashbaugh and colleagues as the ‘acute onset of tachypnea, hypoxemia and loss of compliance after a variety of stimuli’.1 Continued research examining the underlying mechanisms and management strategies is now translating into improved outcome.
DEFINITIONS
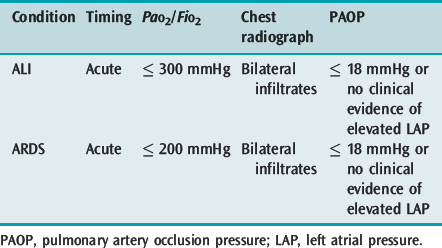
Acute lung injury (ALI) and its more severe subset ARDS describe acute hypoxaemic respiratory failure due to bilateral and diffuse alveolar damage. The most common criteria are the 1994 American–European Consensus Conference definitions2 (Table 29.1), which are broad and inclusive. However, these fail to specify an acute cause, use a PaO2/FiO2 ratio independent of respiratory support and are not specific about the radiographic criteria. The lung injury score (LIS),3 which uses a four-point score attributed to ranges of PaO2/FiO2 ratio, positive end-expiratory pressure (PEEP), respiratory system compliance and the number of quadrants involved on chest radiograph, and the Delphi definition, which demands a PaO2/FiO2 ratio ≤ 200 with 10 cmH2O PEEP, have a greater sensitivity when matched against autopsy evidence of diffuse alveolar damage.4
CHEST RADIOGRAPH AND CHEST COMPUTED TOMOGRAPHY IN ACUTE LUNG INJURY
The interpretation of the chest radiograph is central to these definitions of ALI and ARDS. However, there is considerable interobserver variability in both chest radiograph interpretation and in the definition of an infiltrate. In the American–European Consensus Conference definition the infiltrate must be bilateral and consistent with pulmonary oedema,2 whereas the LIS rates the number of quadrants with alveolar consolidation3 and the Delphi definition requires bilateral airspace disease. The intention of these descriptions is fairly clear and excludes opacity due to pleural effusion, nodules, masses, collapse and pleural thickening. However, it is desirable to improve interobserver agreement and this will require training and more specific definitions.
Chest computed tomography (CT)5 has proved extremely helpful in pathophysiological studies of ALI, has demonstrated the heterogeneity of lung inflation and is commonly used to assist clinical management. Autopsy and chest radiographs of ALI show a uniform process affecting both lungs; however, chest CT early in the course of ALI in supine patients demonstrated that there was a dorsal dependent increase in lung density, and that the ventral lung was relatively normal. In addition, CT frequently showed previously undiagnosed pneumothorax, pneumomediastinum and pleural effusion. After the second week of mechanical ventilation CT scans may demonstrate altered lung architecture and emphysematous cysts or pneumatoceles.
CT numbers or Hounsfield units can be assigned to each voxel (∼2000 alveoli in a standard 10-mm slice).5 These data can then be used to assess what proportion of a region of interest is non-aerated, poorly aerated, normally aerated or hyperinflated. Initially a single basal lung slice was studied, but it is clear that far more information can be obtained by studying the whole lung, and by using thinner slices. This allows: (1) reconstruction of the upper and lower lobes (the middle lobe is difficult to separate); (2) the same section of lung to be studied at different levels of inflation or PEEP (the lung also moves in a cephalocaudad direction with respiration); and (3) a broader picture of the lung to be obtained (lung damage is heterogeneous in ALI). However, whole-lung CT demands considerable exposure to ionising radiation, and different information, perhaps more pertinent to mechanical ventilation, is obtained from dynamic CT.
Clinical assessment of chest CT is discussed in Chapter 35, and CT findings in ALI are discussed below, in the section on clinical management.
EPIDEMIOLOGY
Estimates of the incidence and outcome from ALI and ARDS vary widely. In part this has been due to differences in the definitions used, but it also appears likely that case-mix and local factors influence outcome and incidence. Using the 1994 consensus definition the Australian incidence is 34 per 100 000 for ALI and 28 per 100 000 for ARDS;6 recent US estimates are 79 and 59 per 100 000 respectively7 – both much greater than many previous estimates. The Australian data equate to 1 in 10 non-cardiothoracic intensive care unit (ICU) patients developing ARDS, which reflects the tendency for clinicians to underestimate the incidence of ALI and ARDS.
Reported mortality rates are also influenced by the definitions used. For many years the mortality for ARDS was reported to be ∼60%; the Australian multicentre data reported mortality rates of 32% for ALI and 34% for ARDS,6 with US mortality rates a little higher at 38.5% and 41%.7 However, particular diagnostic groups such as multiple trauma have a lower mortality rate than other causes of ARDS, and patients with ALI who have chronic liver disease, non-pulmonary organ dysfunction, sepsis or age greater than 70 years (hazard ratio 2.5)8 have a higher risk of death. Consequently many factors need to be considered when assessing outcome prediction.
PULMONARY FUNCTION IN SURVIVORS
Respiratory function is most abnormal soon after discontinuation of mechanical ventilation, but usually returns towards normal by 6–12 months. Although a variety of abnormal pulmonary function tests may be found, impaired diffusing capacity is the most common. This is rarely symptomatic, but occasional patients have severe restrictive disease, and this is correlated with their cumulative LIS.9
QUALITY OF LIFE IN SURVIVORS
Compared with disease-matched ICU patients who do not develop ARDS, patients with ARDS have a more severe reduction in both pulmonary and general health-related quality of life.10 Many patients have reduction in exercise tolerance that may be attributable to associated critical-illness neuropathy and myopathy; nerve entrapment syndromes and heterotopic calcification play a role in a minority.7 Depression, anxiety and posttraumatic stress disorder are also common (20–50% of survivors).7 However, it is unclear whether neuropsychological disability is a direct consequence of the illness, or related to the associated stress. Finally, most survivors have cognitive impairments such as slowed mental processing, or impaired memory or concentration, and these correlate with the period and severity of desaturation < 90%.11 Although these data suggest that ARDS confers a specific risk of impaired quality of life, the mechanism is unclear. However, they do caution against permissive hypoxaemia as a strategy to reduce ventilator-induced lung injury (VILI).
PATIENTS AT-RISK FOR ALI AND ARDS
Clinical risk factors for the development of ALI and ARDS can be classified as either direct or indirect (Table 29.2). These identify over 80% of patients who develop ARDS; the most common risk factors are sepsis, pneumonia and aspiration of gastric contents. Multiple risk factors, low pH, chronic alcohol abuse or chronic lung disease substantially increases the incidence of ALI in at-risk patients.
Table 29.2 Clinical risk factors for acute lung injury (ALI) and acute respiratory distress syndrome (ARDS)
Rights were not granted to include this table in electronic media. Please refer to the printed book.
BIOLOGICAL MARKERS AS PREDICTORS OF ALI
In addition to identifying these clinical risk groups, there has been considerable interest in identifying possible biological markers that could be used to predict the development of ALI. Although greater specificity may be gained from sampling the epithelial lining fluid (e.g. using bronchoalveolar lavage fluid), an ideal biological marker would be more simply sampled, such as plasma. Numerous proteins, such as the cytokines interleukin (IL)-1, tumour necrosis factor-α (TNF-α) and IL-10, and von Willebrand’s factor antigen are elevated in at-risk patients and patients with ALI and ARDS; however, they are not predictive. In relatively small studies both ferritin12 and surfactant protein B13 have been shown to be predictive of ARDS. Although ferritin likely represents a non-specific oxygen free-radical response, the leakage of surfactant protein B from the alveolus into the blood is lung-specific.
PATHOGENESIS
Although it is well accepted that diffuse alveolar damage with: (1) pulmonary oedema due to damage of the alveolocapillary barrier; (2) a complex inflammatory infiltrate; and (3) surfactant dysfunction are essential components of ALI, the sequence of events is uncertain and probably depends upon the precipitating insult and host response. For example, in endotoxin-induced lung injury, hypoxaemia and reduced lung compliance occur well before recruitment of neutrophils or an increase in lung weight due to an increase in permeability.14 In addition, surfactant turnover is dramatically increased prior to these changes often thought to be typical of early ALI. Further, epithelial lining fluid sampled immediately following intubation in patients with ALI has markedly increased concentrations of type III procollagen peptide, suggestive of fibrosing alveolitis extremely early in the course of lung damage.
THE ALVEOLOCAPILLARY BARRIER
The normal lung consists of 300 million alveoli with alveolar gas separated from the pulmonary microcirculation by the extremely thin alveolocapillary barrier (0.1–0.2 μm thick). Since the endothelial pore size is 6.5–7.5 nm, and the epithelial pore size is almost one-tenth that, at 0.5–0.9 nm, the epithelium is the major barrier to protein flux.15 The surface area of the alveoli is estimated to be 50–100 m2, which is made up predominantly of alveolar type I cells, with the metabolically active type II cells accounting for ∼10% of the surface area. In turn, these cells are covered by the epithelial lining fluid, with an estimated volume of 20 ml, ∼10% of which is surfactant with the remained filtered plasma water and low-molecular-weight proteins, and a small number of cells, mainly alveolar macrophages and lymphocytes.
NEUTROPHILS IN ACUTE LUNG INJURY
Neutrophils are the most abundant cell type found in both the epithelial lining fluid (e.g. bronchoalveolar lavage fluid) and alveoli in histological specimens from early in the course of ALI. Although neutrophil migration across the endothelium or epithelium does not cause injury, when activated they release reactive oxygen species, cytokines, eicosanoids and a variety of proteases that may make an important contribution to tissue damage in ALI. Following bone marrow demargination, activated neutrophils adhere to the endothelium on their passage to the alveolus, and this may be accompanied by an early, transient leukopenia. Although neutrophils have an important role in host defence due to their bactericidal activity, there is a marked (50–1000-fold) increase in the release of cytotoxic compounds when they are activated by adherence to the endothelium, epithelium or contact with interstitial extracellular matrix proteins.16 The factors involved in adhesion of neutrophils are complex and involve the integrin family of proteins, selectins and a number of adhesion molecules.
In models of ALI, antibodies to adhesion molecules (e.g. CD11b/CD18 antibodies) ameliorate lung injury, suggesting a crucial and central role of this cell type. However, ALI occurs in neutropenic patients, and was not more common when granulocyte colony-stimulating factor was administered to patients with pneumonia.17 Clearly, other cell types play an important role, and neutrophil chemoattractants such as IL-8 must be present in the lung prior to neutrophil accumulation.
OTHER CELL TYPES INVOLVED IN ACUTE LUNG INJURY
Pulmonary endothelial cells, platelets, interstitial and alveolar macrophages and alveolar type II cells also play important roles in alveolar inflammation. Pulmonary endothelial cells express a variety of adhesion molecules and cyclooxygenase-2 (COX-2); secrete endothelin and cytokines, including IL-8;18 stimulate procoagulant activity; and ‘cross-talk’ with the alveolar macrophages and type II cells. They will be involved in generalised endothelial activation, and are subject to mechanical stress, secondary to both vascular pressure and to their close association with the alveolus. Von Willebrand’s factor antigen is synthesised by vascular endothelial cells, and although this may explain its lack of specificity for ALI, its plasma levels are a good marker of endothelial injury.19
Microvascular thrombosis is common in ALI, and contributes to pulmonary hypertension and wasted ventilation. Although platelet aggregation may contribute to ALI through release of thromboxane A2, serotonin, lysosomal enzymes and platelet-activating factor, they are less important than the other cell types.
Alveolar macrophages are the most common cell type normally found in bronchoalveolar lavage fluid, and, together with interstitial macrophages, play an important role in host defence and modulation of fibrosis. They are capable of releasing IL-6 and a host of mediators, similar to the activated neutrophil, including TNF-α and IL-8 in response to stretch,20 and may amplify lung injury. However, depletion of alveolar macrophages does not reduce neutrophil recruitment or outcome from tracheal instillation of Pseudomonas aeruginosa,21 questioning the central role of this cell type. Macrophages also release a number of factors such as transforming growth factor-α and platelet-derived growth factor that stimulate fibroblast proliferation, deposition of collagen and glycosaminoglycans, angiogenesis and lung fibrosis.
Alveolar epithelial type II cells are extremely metabolically active; they manufacture and release surfactant, control alveolar water clearance using ion pumps, express cytokines which in turn interact with surfactant production and are the progenitor of type I cells following injury. In response to both stretch and endotoxin, type II cells express IL-8 and TNF-α, with the latter cytokine augmenting Na+, and hence water, egress from the alveolus.22
CHEMOKINES IN ACUTE LUNG INJURY
The expression and secretion of chemokines (chemoattractant cytokines) at sites of inflammation are probably key proximal steps in initiating the inflammatory cascade. IL-8 appears particularly important in initiating ALI because of its ability to induce chemotaxis and activation of neutrophils. IL-8 is elevated in ALI bronchoalveolar lavage fluid within hours of the initiating insult and before recruitment of neutrophils, and in a manner that reflects subsequent morbidity and mortality. In animal models of sepsis and acid aspiration, instillation of antibodies against IL-8 prevents the recruitment of neutrophils and protects the lung. Indeed, the recruitment and retention of neutrophils require the generation and maintenance of a localised chemotactic/haptotactic gradient.23
Mediators in acute lung injury
Although many of the over 40 biologically active cytokines are implicated in ALI, TNF-α, IL-1β, IL-6 and IL-8 are the most important. However, even greater increases are found in their cognate receptors or antagonists, such as the counterregulatory cytokine IL-10, so that their biological impact is markedly reduced.24 Despite numerous studies, measurement of cytokines in blood or epithelial lining fluid has not proven predictive of the development of ALI or of mortality.
CLINICAL MANAGEMENT
MECHANICAL VENTILATION
Patients present with acute hypoxaemic respiratory failure, and an increase in the work of breathing, usually requiring mechanical ventilation (Table 29.3). The role of non-invasive ventilation in ALI is uncertain as there appears to be a greater complication rate, perhaps due to delayed intubation.25 However, non-invasive ventilation is worth considering in particular circumstances (see Chapter 33).
Table 29.3 Pathophysiology of acute lung injury (ALI) and adult respiratory distress syndrome (ARDS)
| Feature | Cause(s) |
|---|---|
| Hypoxaemia | True shunt (perfusion of non-ventilated airspaces) |
| Impaired hypoxic pulmonary vasoconstriction | |
| V/Q mismatch is a minor component | |
| ↑ Dependent densities (CT) | Surfactant dysfunction alveolar instability |
| (Collapse/consolidation) | Exaggeration of normal compression of dependent lung due to ↑ weight (↑ lung water, inflammation) |
| ↑ Elastance (↓ compliance) | Surfactant dysfunction (↑ specific elastance) |
| ↓ Lung volume (‘baby lung’) | |
| ↑ Chest wall elastance | |
| Fibrosing alveolitis (late) | |
| ↑ Minute volume requirement | ↑ Alveolar dead space (VDphys/Vt often 0.4–0.7) |
| ↑ VCO2 | |
| ↑ Work of breathing | ↑ Elastance |
| ↑ Minute volume requirement | |
| Pulmonary hypertension | Pulmonary vasoconstriction (thromboxane A2, endothelin) |
| Pulmonary microvascular thrombosis | |
| Fibrosing alveolitis | |
| Positive end-expiratory pressure |
The method and delivery of ventilatory support must take into account the pathophysiology of ALI and ARDS. For many years laboratory studies have described VILI, and the most important clinical study was the ARDS Network study where 861 ALI patients from 75 ICUs were randomised to receive a tidal volume (VT) of either 12 or 6 ml/kg predicted body weight.26 Mortality was reduced by 22% from 40% to 31% in the lower-VT group. There was a strict PEEP and FiO2 protocol, and patients were ventilated with assist-control ventilation to avoid excessive spontaneous VT. Meta-regression of five clinical studies trialling different VT strategies found that a VT < 7.7 ml/kg predicted body weight (pbw) was protective, and that above 11.2 ml/kg pbw was borderline detrimental.27 Whether this protective effect of lower VT is due to lower static distending pressure of the respiratory system is contentious; however, Hager and colleagues28 found that lower VT ventilation was protective across all quartiles of plateau pressure (Pplat) in the ARDS Network trial, and that no safe upper limit of Pplat could be identified.
AVOIDANCE OF OVERSTRETCH AND INADEQUATE RECRUITMENT
The increase in dependent lung density found on chest CT, due to non-aerated and poorly aerated lung, reduces the volume of aerated lung available for tidal inflation. Both PEEP and tidal recruitment will increase aeration of some of these airspaces, but a VT that is not reduced in proportion to the reduction in aerated lung may lead to overstretch of aerated lung parenchyma, which may lead to further diffuse alveolar damage. Studies where increased chest wall compliance led to increase airway pressure (Paw) have clearly shown that it is lung stretch, not increased Paw, that causes injury;29 consequently this has been termed volutrauma. In addition, laboratory studies have shown that repeated opening and closing of airspaces by tidal recruitment result in diffuse alveolar damage (atelectrauma). Although data from animal models support both as important mechanisms of VILI, with both resulting in alveolar inflammation and elevated alveolar cytokines (biotrauma),31 which may ‘spill’ into the systemic circulation,30 only VILI due to overstretch is supported by clinical data. CT scans performed during ARDS Network protective ventilation show that tidal inflation occurs primarily in either normally aerated or overinflated compartments, with little tidal recruitment.31 The pulmonary inflammatory response was associated with tidal overinflation.
OVERSTRETCH
The normal lung is fully inflated at a transpulmonary pressure of ∼30 cmH2O. Consequently, a maximum Pplat, the elastic distending pressure, of 30–35 cmH2O has been recommended to avoid overstretch, and the ARDS Network study targeted Pplat ≤ 30 cmH2O.28 The transpulmonary pressure may be lower than expected for a given Pplat in patients with a high chest wall elastance (e.g. obesity, abdominal compartment syndrome, postabdominal or thoracic surgery). It is also common for individual patients to show evidence of overinflation at much lower elastic distending pressures (18–26 cmH2O).32,33 Finally, inspiratory muscle effort will lower Pplat by reducing intrapleural pressure, potentially avoiding detection of an excessive transpulmonary pressure. This is particularly common when pressure support ventilation is used as a primary mode of ventilatory support: VT that would produce an unacceptably high Pplat during mechanical ventilation will produce the same volutrauma during a spontaneous or supported mode of ventilation, and should be avoided. None of these issues is easily overcome. Although placement of an oesophageal balloon (see Chapter 34) allows measurement of the transpulmonary pressure, it must be correctly placed, have an adequate occlusion pressure ratio, and measurements are preferably performed in a semisitting position in order to lift the mediastinum off the oesophagus. Similarly, static or dynamic volume–pressure curves or quantitative chest CT can be used to determine overinflation, though chest CT cannot determine overstretch.5 Consequently, unless particular expertise is available, VT limitation is currently the most practical approach.
ADEQUATE PEEP
PEEP improves PaO2 by increasing functional residual capacity, and recruiting alveoli. Because PEEP may reduce cardiac output by impairing venous return, Suter and coworkers suggested that maximum oxygen delivery (oxygen content × flow) should be used to optimise PEEP.33 Other end-points such as either titrating PEEP to a particular PaO2/FiO2 ratio or to prevent repeated opening and closing of alveoli have been suggested. The ARDS Network Study protocol28 titrates PEEP to the PaO2/FiO2 ratio, but this results in tidal overinflation in about one-third of patients.33 Concurrently there is little evidence of tidal recruitment, which may explain why a higher PEEP protocol34 did not prove beneficial. In patients at risk for ARDS prophylactic PEEP (8 cmH2O) was not protective.35
The lower inflection point of a volume–pressure curve has been used to set PEEP, because early studies suggested that this reflected recruitment of collapsed alveoli. However, in patients with ALI, recruitment occurs well above the lower inflection point, along the entire volume–pressure curve and above the upper inflection point.36,37 Concurrently, there is frequently evidence of overstretching and hyperinflation on CT scans33,38 or dynamic volume–pressure analysis.33 The amount of lung available for recruitment in ARDS is also extremely variable,39 and does not appear to differ comparing pulmonary with extrapulmonary causes.40 Although routine CT analysis has been advocated by some, it is cumbersome and has not been shown to influence outcome; non-invasive bedside alternatives are under investigation. Consequently, PEEP titration is often a compromise aiming to minimise both atelectrauma and volutrauma.41
Although a protective ventilation strategy using the ‘open-lung’ approach with PEEP levels above the lower inflection point and frequent recruitment manoeuvres did report a reduction in mortality, the treatment arm also included low VT and recruitment manoeuvres.42 Reasonable approaches to PEEP titration include: (1) the use of a scale similar to the ARDS Network protocol; (2) titration of PEEP to PaO2, aiming for a PEEP of ∼15 cmH2O; or (3) measuring elastic mechanics at the bedside. The delta-PEEP technique is a simple technique that indirectly assesses excessive stress as PEEP is changed at a constant VT33 (see Chapter 34).
Recruitment manoeuvres
It is unclear whether additional recruitment manoeuvres are of additional benefit once an adequate level of PEEP is applied. Typically a high level of continuous positive airways pressure (30–40 cmH2O) is applied for 30–40 seconds in an apnoeic patient, followed by return to a lower level of PEEP and controlled ventilation. This may be followed by a marked improvement in oxygenation. However, this is not a consistent finding, and hypotension may occur due to reduced venous return if there is inadequate fluid loading. Although a recruitment manoeuvre was applied by Amato and colleagues and associated with an improvement in outcome, they also applied a higher level of PEEP and lower VT in the protective lung strategy group.42 A number of small trials have shown improvement in oxygenation following recruitment manoeuvres; however, the largest clinical trial43 failed to show an effect. Grasso and colleagues44 found that recruitment manoeuvres were only effective early in ARDS and with lower levels of baseline PEEP, probably explaining the variable responses reported.
In addition to physical recruitment of alveoli, lung stretch above resting VT is the most powerful physiological stimulus for release of pulmonary surfactant from type II cells. This is associated with an increase in lung elastance and improved PaO2 in the isolated perfused lung,45 and is a possible explanation for the improvement in oxygenation, recruited lung volume and elastance reported with the addition of three sigh breaths in patients with ARDS.46 Similarly, in models of lung injury biologically variable or fractal VT is associated with less lung damage with lower alveolar levels of IL-8,47 improved oxygenation and lung elastance with greater surfactant release.48 Again, these data caution against monotonous low VT ventilation, and suggest that intermittent or variable lung stretch may reduce lung injury.
MODE OF VENTILATION
Non-invasive ventilation should not be routinely used in ALI and ARDS (see Chapter 33) and most patients require intubated mechanical ventilation. Following intubation controlled ventilation allows immediate reduction in the work of breathing and application of PEEP and a high FiO2. Later in the clinical course assisted or supported modes of ventilation may allow better patient–ventilator interaction (see Chapter 27), and possibly improved oxygenation through better mismatch as a result of diaphragmatic contraction.49 Withdrawal or weaning from mechanical ventilation is discussed in Chapter 27.
An advantage of assist-control ventilation (as used in the ARDS Network study) is that spontaneous effort cannot generate a greater volume than VT. Care should be taken with synchronised intermittent mandatory ventilation (SIMV), particularly if pressure support is added to SIMV, as excessive VT may occur during supported breaths. There is an increasing tendency to use pressure-controlled (PC) ventilation or pressure-regulated volume control (PRVC) as Ppk is lower than volume-controlled (VC) ventilation with a constant inspiratory flow pattern. However, the decelerating flow pattern of PC or PRVC means that most of the resistive pressure (Pres) during inspiration is dissipated by end-inspiration, which is in contrast to VC with a constant inspiratory flow pattern where Pres is dissipated at end-inspiration (see Chapter 27, Figure 27.2). Consequently, with PC and PRVC Ppk ≈ Pplat which is the same as Pplat during VC.50 In addition oxygenation, haemodynamic stability and mean airway pressure are no different between PC and VC, and a moderately sized randomised study found no difference in outcome.51 However, there may be differences in lung stress due to greater viscoelastic build up with VC.
Inverse-ratio ventilation, often together with PC, has been used in ARDS. However, when PEEPi and total PEEP are taken into account, apart from a small decrease in PaCO2, there are no advantages with inverse-ratio ventilation. Mean airway pressure is higher, with a greater risk of both haemodynamic consequences50 and regional hyperinflation.52 Consequently, an inspiratory to expiratory ratio less than 1:1 is recommended.
A number of other modes of ventilation (see Chapter 27), including airway pressure release ventilation and high-frequency oscillation, have been proposed for use in ARDS, but without new data there do not appear to be any major advantages over the optimal conventional ventilation.
TARGET BLOOD GASES
Oxygenation targets and FiO2
There must be a compromise between the major determinants of oxygenation, including the extent of poorly or non-aerated lung, hypoxic pulmonary vasoconstriction and mixed venous oxygen saturation, and the target PaO2. The association between cognitive impairment and arterial saturation (SaO2) < 90%11 suggests that SaO2 ≥ 90% and usually PaO2 > 60 mmHg is a reasonable target. Because positive-pressure ventilation may reduce cardiac output it is also important to consider tissue oxygenation.
In addition to PEEP, increased FiO2 is used to improve SaO2. However, high FiO2 may also cause tissue injury, including diffuse alveolar damage. The balance between increased airway pressure and FiO2 is unknown, but high FiO2 is generally regarded as being less damaging.53 In part this is because diffuse alveolar damage itself protects the lung against hyperoxia, perhaps through prior induction of scavengers for reactive oxygen species.54 A reasonable compromise is to start ventilation at a FiO2 of 1 and to titrate down, aiming for an FiO2 ≤ 0.6. In patients with extreme hypoxaemia, additional measures such as inhaled nitric oxide (iNO) and prone positioning may be tried, along with a lower SaO2 target.
Carbon dioxide target
Low VT strategies will result in elevations in PaCO2 unless minute ventilation is augmented by an increase in respiratory rate. The ARDS Network protocol aimed at normocapnia, with a maximum respiratory rate of 35 breaths/min, to minimise respiratory acidosis.28 This exposes the lung to more repeated tidal stretch, and may result in dynamic hyperinflation due to a shortened expiratory time.55 In addition, allowing the PaCO2 to rise above normal may not be harmful in many patients.
If hypercapnic acidosis occurs slowly, intracellular acidosis is well compensated, and the associated increase in sympathetic tone may augment cardiac output and blood pressure. Although the respiratory acidosis may worsen pulmonary hypertension and induce myocardial arrhythmias, these effects are often small, particularly if there has been time for metabolic compensation. In addition, in an ischaemia–reperfusion model of ALI, therapeutic hypercapnia reduced lung injury and apoptosis.56 However, clinical studies of permissive hypercapnia must be undertaken before therapeutic hypercapnia is considered. Hypercapnia should be avoided in patients with or at risk from raised intracranial pressure.
ADDITIONAL MEASURES TO IMPROVE OXYGENATION
PRONE POSTURE
In ∼ 70% of patients with ARDS, prone positioning will result in a significant increase in PaO2, with a modest increase in PaO2 sustained in the supine position.57 The mechanisms involved include recruitment of dorsal lung, with concurrent collapse of ventral lung; however, perfusion is more evenly distributed, leading to better matching. Although three large clinical studies58,59 did not show an improvement in mortality, post-hoc analysis suggests that mortality may be reduced in the most hypoxaemic patients.57 Although further data are awaited, prone positioning may be used as rescue therapy in life-threatening hypoxaemia.
MANIPULATION OF THE PULMONARY CIRCULATION
iNO and prostacyclin (PGI2) may be used to reduce pulmonary shunt and right ventricular afterload by reducing pulmonary artery impedance. When hypoxic pulmonary vasoconstriction is active, there is redistribution of pulmonary blood flow away from the poorly ventilated dependent areas to more normally ventilated lung, leading to an increase in PaO2. Both iNO and PGI2 are potent vasodilators, and since they are delivered as part of the gas mix (iNO) or inhaled (PGI2), they are delivered to well-ventilated lung. Both act to vasodilate the local pulmonary circulation and increase the redistribution of pulmonary blood flow away from poorly ventilated lung, reducing pulmonary shunt and improving oxygenation. Intravenous almitrine is a selective pulmonary vasoconstrictor that reinforces hypoxic pulmonary vasoconstriction, and although this may improve oxygenation alone, there is a synergistic effect with iNO.
PHARMACOLOGICAL THERAPY
Apart from improved mortality in sepsis following activated protein C,60 there are no proven pharmacological therapies for ALI or ARDS despite numerous studies. However, the lack of a protective ventilation strategy in many of these studies may have masked a drug effect.
SURFACTANT REPLACEMENT THERAPY
Surfactant dysfunction is an important and early abnormality contributing to lung damage in ALI.38,61 Pulmonary surfactant reduces surface tension, promoting alveolar stability, reducing work of breathing and lung water. In addition surfactant has important roles in lung host defence. Reactive oxygen species, phospholipases and increased protein permeability lead to inhibition of surfactant function, composition is abnormal and turnover is markedly increased. VILI is difficult to demonstrate without surfactant dysfunction.56 Consequently, there has been considerable interest in exogenous surfactant replacement therapy.
Recombinant surfactant protein C-based surfactant administered intratracheally improves oxygenation in ARDS without an improvement in mortality.62 However, as subgroup analysis appeared promising, further clinical trials are under way in direct or pulmonary causes of ARDS.
GLUCOCORTICOIDS
Glucocorticoids may have a role in ARDS through reduction of the intense inflammatory response and their potential to reduce fibroproliferation and collagen deposition, by faster degradation of fibroblast procollagen mRNA. Although a small study, with cross-over, showed a reduction in mortality,63 a study of 180 patients with persistent ARDS (of at least 7 days’ duration) found no improvement in mortality up to 180 days later, but did show an improvement in ventilator-free days and shock-free days.64 However, this was offset by neuromuscular complications, and patients enrolled at least 14 days after the onset of ARDS had increased 60- and 180-day mortality rates. These data do not support the routine use of steroids in ARDS.
KETOCONAZOLE
Ketoconazole is an antifungal drug that also inhibits thromboxane synthase and 5-lipoxygenase. However, promising results from small studies in at-risk patients have not been confirmed in a larger treatment trial.65
OTHER PHARMACOLOGIC THERAPIES
Numerous other therapies, including cytokine antagonism, non-steroidal anti-inflammatory drugs, scavengers of reactive oxygen species and lisofylline66 have been trialled without success. The complex balance of inflammation and repair in ALI, and the critical additional damage secondary to VILI, may explain these results. However, studies in less heterogeneous groups with minimisation of VILI using standardised ventilation protocols, together with a growing understanding of ALI and ARDS, offer potential pharmacological therapies.
1 Ashbaugh DG, Bigelow DB, Petty TL, et al. Acute respiratory distress in adults. Lancet. 1967;ii:319-323.
2 Bernard GR, Artigas A, Brigham KL, et al. The American–European Consensus Conference on ARDS: definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149:818-824.
3 Murray JF, Matthay MA, Luce JM, et al. An expanded definition of the adult respiratory distress syndrome. Am Rev Respir Dis. 1988;138:720-723. [Erratum, Am Rev Respir Dis 1989; 139: 1065.]
4 Ferguson ND, Frutos-Vivar F, Esteban A, et al. Acute respiratory distress syndrome: underrecognition by clinicians and diagnostic accuracy of three clinical definitions. Crit Care Med. 2005;33:2228-2234.
5 Gattinoni L, Caironi P, Pelosi P, et al. What has computed tomography taught us about the acute respiratory distress syndrome? Am J Respir Crit Care Med. 2001;164:1701-1711.
6 Bersten AD, Edibam C, Hunt T, et al. Incidence and mortality from acute lung injury and the acute respiratory distress syndrome in three Australian states. Am J Respir Crit Care Med. 2002;165:443-448.
7 Rubenfeld GD, Herridge MS. Epidemiology and outcomes of acute lung injury. Chest. 2007;131:554-562.
8 Ely EW, Wheeler AP, Thompson BT, et al. Recovery rate and prognosis in older persons who develop acute lung injury and the acute respiratory distress syndrome. Ann Intern Med. 2002;136:25-36.
9 McHugh LG, Milberg JA, Whitcomb ME, et al. Recovery of function in survivors of the acute respiratory distress syndrome. Am J Respir Crit Care Med. 1994;250:90-94.
10 Davidson TA, Caldwell ES, Curtis SR, et al. Reduced quality of life in survivors of acute respiratory distress syndrome compared with other critically ill control patients. JAMA. 1999;281:354-360.
11 Hopkins RO, Weaver LK, Pope D. Neuropsychological sequelae and impaired health status in survivors of severe acute respiratory distress syndrome. Am J Respir Crit Care. 1999;160:50-56.
12 Connelly KG, Moss M, Parsons PE, et al. Serum ferritin as a predictor of the acute respiratory distress syndrome. Am J Respir Crit Care Med. 1997;155:21-25.
13 Bersten AD, Hunt T, Nicholas TE, et al. Elevated plasma surfactant protein-B predicts development of acute respiratory distress syndrome in patients with acute respiratory failure. Am J Respir Crit Care Med. 2001;164:648-652.
14 Davidson KG, Bersten AD, Barr HA, et al. Endotoxin induces respiratory failure and increases surfactant composition and respiration independent of alveolocapillary injury in rats. Am J Respir Crit Care Med. 2002;165:1516-1525.
15 Doyle IR, Nicholas TE, Bersten AD. Partitioning lung and plasma proteins: circulating surfactant proteins as biomarkers of alveolocapillary permeability. Clin Exp Pharmacol Physiol. 1999;26:185-197.
16 Downey GP, Dong Q, Kruger J, et al. Regulation of neutrophil activation in acute lung injury. Chest. 1999;116:46S-54S.
17 Nelson S, Belknap SM, Carlson RW, et al. A randomized controlled trial of figastrim as an adjunct to antibiotics for treatment of hospitalized patients with community-acquired pneumonia. J Infect Dis. 1998;178:1075-1080.
18 Zimmeraman GA, Albertine KH, Carveth HJ, et al. Endothelial activation in ARDS. Chest. 1999;116:18S-24S.
19 Pittet JF, Mackersie RC, Martin TR, et al. Biological markers of acute lung injury: prognostic and pathogenetic significance. Am J Respir Crit Care Med. 1997;155:1187-1205.
20 Pugin J, Dunn I, Jolliet P, et al. Activation of human macrophages by mechanical ventilation in vitro. Am J Physiol. 275, 1998. L104–50
21 Cheung DO, Halsey K, Speert DP. Role of pulmonary alveolar macrophages in defense of the lung against Pseudomonas aeruginosa. Infect Immun. 2000;68:4585-4592.
22 Rezaiguia S, Garat C, Declauc C, et al. Acute bacterial pneumonia in rats increases alveolar epithelial fluid clearance by a tumor necrosis-factor-alpha dependent mechanism. J Clin Invest. 1997;99:325-335.
23 Modelska K, Pittet JF, Folkesson HG, et al. Acid-induced lung injury. Protective effect of anti-interleukin-8 pretreatment on alveolar epithelial barrier function in rabbits. Am J Respir Crit Care Med. 1999;160:1450-1456.
24 Park WY, Goodman RB, Steinberg KP, et al. Cytokine balance in the lungs of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;164:1896-1903.
25 Delclaux C, L’Her E, Alberti C, et al. Treatment of acute hypoxemic nonhypercapnic respiratory insufficiency with continuous positive airway pressure delivered by a face mask. A randomized controlled trial. JAMA. 2000;284:2352-2360.
26 Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000;342:1301-1308.
27 Moran JL, Bersten AD, Solomon PJ. Meta-analysis of controlled trials of ventilator therapy in acute lung injury and acute respiratory distress syndrome: an alternative perspective. Intens Care Med. 2005;31:227-235.
28 Hager DN, Krishnan JA, Hayden DL, et al. Tidal volume reduction in patients with acute lung injury when plateau pressures are not high. Am J Respir Crit Care Med. 2005;172:1241-1245.
29 Dreyfuss D, Saumon G. Ventilator-induced lung injury: lessons from experimental studies. Am J Respir Crit Care Med. 1998;157:294-323.
30 Chiumello D, Pristine G, Slutsky AS. Mechanical ventilation affects local and systemic cytokines in an animal model of acute respiratory distress syndrome. Am J Respir Crit Care Med. 1999;160:109-116.
31 Terragni PP, Rosboch G, Tealdi A, et al. Tidal hyperinflation during low tidal volume ventilation in acute respiratory distress syndrome. Am J Respir Crit Care Med. 2007;175:160-166.
32 Bersten AD. Measurement of overinflation by multiple linear regression analysis in patients with acute lung injury. Eur Respir J. 1998;12:526-532.
33 Suter PM, Fairley B, Isenberg MD. Optimum end-expiratory airway pressure in patients with acute pulmonary failure. N Engl J Med. 1975;292:284-289.
34 The National Heart, Lung, and Blood Institute ARDS Clinical Trials Network. Higher versus lower positive end-expiratory pressure in patients with the acute respiratory distress syndrome. N Engl J Med. 2004;351:327-336.
35 Pepe PE, Hudson LD, Carrico CJ. Early application of positive end-expiratory pressure in patients at-risk for adult respiratory distress syndrome. N Engl J Med. 1984;311:281-286.
36 Jonson B, Richard J-C, Straus R, et al. Pressure–volume curves and compliance in acute lung injury: evidence for recruitment above the lower inflection point. Am J Respir Crit Care Med. 1999;159:1172-1178.
37 Crotti S, Mascheroni D, Caironi P, et al. Recruitment and derecruitment during acute respiratory failure: a clinical study. Am J Respir Crit Care Med. 2001;164:131-140.
38 Malbouisson LM, Muller J-C, Constantin J-M, et al. Computed tomography assessment of positive end-expiratory pressure-induced alveolar recruitment in patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;163:1444-1450.
39 Gattinoni L, Caironi P, Cressoni M, et al. Lung recruitment in patients with the acute respiratory distress syndrome. N Engl J Med. 2006;354:1775-1786.
40 Thille AW, Richard J-C M, Maggiore SM, et al. Alveolar recruitment in pulmonary and extrapulmonary acute respiratory distress syndrome. Comparison using pressure–volume curve or static compliance. Anesthesiology. 2007;106:212-217.
41 Rouby JJ, Lu Q, Goldstein I. Selecting the right level of positive end-expiratory pressure in patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 2002;165:1182-1186.
42 Amato MBP, Barbas CSV, Medeiros DM, et al. Effect of a protective ventilation strategy on mortality in the acute respiratory distress syndrome. N Engl J Med. 1998;338:347-354.
43 Brower RG, Morris A, Macintyre N, et al. Effects of recruitment maneuvers in patients with acute lung injury and acute respiratory distress syndrome ventilated with high positive end-expiratory pressure. Crit Care Med. 2003;31:2592-2597.
44 Grasso S, Mascia L, Del Turco M, et al. Effects of recruiting maneuvers in patients with acute respiratory distress syndrome ventilated with protective ventilatory strategy. Anesthesiology. 2002;96:795-802.
45 Nicholas TE, Power JHT, Barr HA. The pulmonary consequences of a deep breath. Respir Physiol. 1982;49:315-324.
46 Pelosi P, Cadringher P, Bottino N, et al. Sigh in acute respiratory distress syndrome. Am J Respir Crit Care Med. 1999;159:872-880.
47 Boker A, Ruth Graham M, Walley KR, et al. Improved arterial oxygenation with biologically variable or fractal ventilation using low tidal volumes in a porcine model of acute respiratory distress syndrome. Am J Respir Crit Care Med. 2002;165:456-462.
48 Ingenito EP, Arold S, Lutchen K, et al. Effects of noisy ventilation (NV) and open lung ventilation (OLV) on lung mechanics, gas exchange, and surfactant content and properties (abstract). Am J Respir Crit Care Med. 2001;163:A483.
49 Wrigge H, Zinserling J, Neumann P, et al. Spontaneous breathing improves lung aeration in oleic acid-induced lung injury. Anesthesiology. 2003;99:376-384.
50 Lessard MR, Guerot E, Lorino H, et al. Effects of pressure-controlled with different I:E ratios versus volume-controlled ventilation on respiratory mechanics, gas exchange, and hemodynamics in patients with adult respiratory distress syndrome. Anesthesiology. 1994;80:983-991.
51 Esteban A, Alia I, Gordo F, et al. Prospective randomized trial comparing pressure-controlled ventilation and volume-controlled ventilation in ARDS. For the Spanish Lung Failure Collaborative Group. Chest. 2000;117:1690-1696.
52 Edibam C, Rutten AJ, Collins DV, et al. Effect of inspiratory flow pattern and inspiratory to expiratory ratio on nonlinear elastic behavior in patients with acute lung injury. Am J Respir Crit Care Med. 2003;167:702-707.
53 Slutsky AS. Mechanical ventilation. Chest. 1993;104:1833-1859.
54 Frank L, Yam J, Roberts RJ. The role of endotoxin in protection of adult rats from oxygen-induced lung injury. J Clin Invest. 1978;61:269-275.
55 Richard JC, Brochard L, Breton L, et al. Influence of respiratory rate on gas trapping during low volume ventilation of patients with acute lung injury. Intens Care Med. 2002;28:1078-1083.
56 Laffey JG, Tanaka M, Engelberts D, et al. Therapeutic hypercapnia reduces pulmonary and systemic injury following in vivo lung reperfusion. Am J Respir Crit Care Med. 2000;162:2287-2294.
57 Gattinoni L, Tognoni G, Pesenti A, et al. Effect of prone positioning on the survival of patients with acute respiratory failure. N Engl J Med. 2001;345:568-573.
58 Guerin C, Gaillard S, Lemasson S, et al. Effects of systematic prone positioning in hypoxemic acute respiratory failure: a randomized controlled trial. JAMA. 2004;292:2379-2387.
59 Mancebo J, Fernández R, Blanch L, et al. A multicenter trial of prolonged prone ventilation in severe acute respiratory distress syndrome. Am J Respir Crit Care Med. 2006;173:1233-1239.
60 Bernard GR, Vincent J-L, Laterre P-F, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699-709.
61 Bersten AD, Davidson K, Nicholas TE, et al. Respiratory mechanics and surfactant in the acute respiratory distress syndrome. Clin Exp Pharmacol Physiol. 1998;25:955-963.
62 Spragg RG, Lewis JF, Walmrath HD, et al. Effect of recombinant surfactant protein C-based surfactant on the acute respiratory distress syndrome. N Engl J Med. 2004;351:884-892.
63 Meduri GU, Headley AS, Golden E, et al. Effect of prolonged methylprednisolone therapy in unresolving acute respiratory distress syndrome: a randomized controlled trial. JAMA. 1998;280:159-165.
64 Steinberg KP, Hudson LD, Goodman RB, et al. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N Engl J Med. 2006;354:1671-1684.
65 Ketoconazole for early treatment of acute lung injury and acute respiratory distress syndrome. JAMA. 2000;283:1995-2002.
66 Randomized placebo-controlled trial of lisofylline for early treatment of acute lung injury and acute respiratory distress syndrome. Crit Care Med. 2002;30:1-6.