[level-membership-for-neonatal-and-perinatal-medicine-category]
9 Tetralogy of Fallot with Absent Pulmonary Valve Syndrome
I. CASE
A. Fetal echocardiography findings
1. The fetal echo reveals situs solitus of the atria, levocardia, left aortic arch, heart rate of 140 bpm.
2. Estimated fetal gestational age (GA) is 25.5 weeks.
3. There was cardiomegaly with increased cardiothoracic ratio (50%) and dextroposition of the heart.
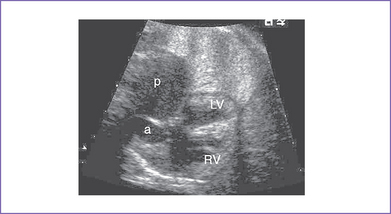
4. The four-chamber view is abnormal, with a large perimembranous subaortic ventricular septal defect (VSD), large overriding aorta, and massively dilated pulmonary artery (Fig. 9-1).
5. The pulmonary valve is dysplastic with no identifiable leaflets, small pulmonary annulus (1.7 mm), and very dilated and pulsatile main pulmonary artery and branches.
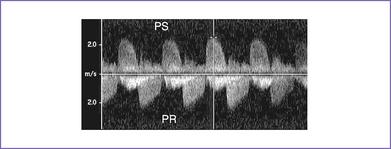
6. There is pulmonary stenosis with a peak velocity of 2.2 m/s (lower panel) and pulmonary valve regurgitation. In other words, there is to (systolic) and fro (diastolic) flow through this dysplastic valve, with severe pulmonary regurgitation (Fig. 9-2).
7. There is holosystolic tricuspid regurgitation with a dP/dt (change in pressure per change in time) of 500 mm Hg per second.
8. The foramen ovale is patent, with unrestricted right-to-left shunt.
9. Aortic valve annulus is 6 mm.
10. A ductus arteriosus cannot be identified.
11. The pulmonary venous drainage is normal.
12. Tei index (myocardial performance index).
b. Left ventricle (LV) Tei index is 0.36.
13. The umbilical artery flow pattern is normal.
14. Ductus venosus flow is abnormal, with a wave reversal (3 mm).
D. Fetal management and counseling
1. Management: Diagnosis of tetralogy of Fallot (TOF) should prompt referral for the following:
a. Thorough anatomic examination by ultrasound.
2. Follow-up included serial antenatal studies at weekly intervals.
a. Cardiac function should be monitored by Tei index, by dP/dt if sufficient tricuspid regurgitation develops (3 m/s), and by tissue Doppler (if available).
b. Development of congestive heart failure (CHF) can indicate RV dysfunction secondary to severe pulmonary regurgitation. Severe AV or semilunar valve regurgitation, particularly if it influences filling of the other ventricle, can be very poorly tolerated. Thus, Ebstein’s anomaly and absent pulmonary valves can cause CHF.
E. Delivery
1. If the fetus remains well compensated, delivery should be at term in a tertiary care center.
2. Depending upon the severity of the pathology, which can include degree of cardiomegaly and degree of pulmonary artery dilation, the baby might require ventilatory support after birth as a result of compression of the tracheobronchial tree and coexisting tracheomalacia and bronchiomalacia.
F. Neonatal management
a. Medical management of affected babies includes supporting them from a respiratory standpoint.
a. Early surgical treatment is advocated to reduce the effect of the compression by the aneurysmal pulmonary artery on the underdeveloped tracheobronchial tree.
G. Follow-up
1. When a homograft is placed in the RV–pulmonary artery conduit, the patient will require reintervention(s) to enlarge the outflow or replace the homograft over the lifetime.
2. Branch pulmonary stenosis might require postoperative balloon angioplasty, stent, or surgical repair to normalize blood flow to both lungs and to reduce the RV systolic pressure.
3. Varying levels of activity limitation may be indicated.
4. Subacute bacterial endocarditis (SBE) prophylaxis should be used as indicated.
5. Arrhythmias, particularly ventricular tachycardia, might develop later and cause sudden death. Holter monitoring, antiarrhythmic agents, and pacemaker therapy may be indicated.
I. Outcome of this case
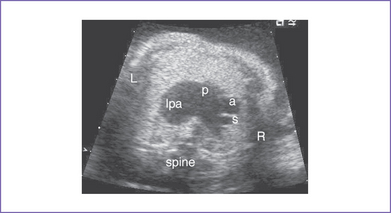
1. The baby was delivered at 36 weeks with emergency cesarean section for fetal distress (Fig. 9-3).
2. Apgar scores were 1 at 1 minute and 2 at 5 minutes.
3. The baby required immediate intubation and ventilation with full inotropic support (milrinone and dopamine) and correction of acidosis.
4. High ventilator settings (high positive end-expiratory pressure [PEEP] and relatively low peak inspiratory pressure [PIP]) were required.
5. The baby had emergency cardiac surgery within the first 24 hours of life but could not be weaned off bypass.
II. YOUR HANDY REFERENCE
A. Prevalence
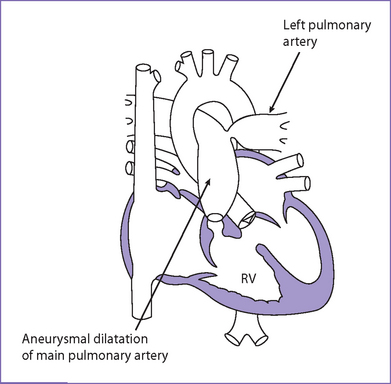
1. Tetralogy of Fallot with absent pulmonary valve syndrome (TOF/APVS) is characterized by a restrictive pulmonary valve annulus, absent or rudimentary pulmonary valve leaflets allowing moderate to severe pulmonary regurgitation, aortic override of a VSD, and RV hypertrophy. The main and branch pulmonary arteries are usually large and often aneurysmal (Fig. 9-4).
2. The incidence of this group of defects is estimated at 3% to 6% of all patients with TOF, with an equal distribution among male and female patients.
3. The fetal series of Allan and colleagues (1986) studied a total group of 2136 cases of congenital heart diseases diagnosed prenatally.
a. This series included a total of 125 cases (5.8%) of TOF diagnosed prenatally. Of the 125 cases, 18 were with TOF/APVS (14.4 %).
B. Outcome
1. Most surgical series divide the patients into two groups: early treatment and late treatment.
a. Those who require surgery in the first year of life have a higher mortality (10%-50%) than those who require surgery after 1 year of age (0%-10%).
b. Babies operated on early are likely to be those with neonatal respiratory problems that necessitate early intervention.
2. In a fetal series by Razavi and Sharland (2003), prenatal diagnosis was associated with an even worse outcome. Of 20 fetuses with APVS, there were six terminations of pregnancy (30%), three intrauterine deaths (15%), five neonatal deaths (25%), three infant deaths (15%), and three patients who did not die (15%). Ten of the 11 live-born infants required early ventilation.
3. These data about the prognosis of APVS allow expecting parents to receive information that includes total mortality and morbidity rather than information related solely to surgery.
4. The outcome of APVS diagnosed prenatally appears poor.
a. The high morbidity and mortality is in part due to the cardiovascular disease, which is associated prenatally with the evolution of hydrops in some cases.
b. There may be an ascertainment bias, with marked cardiomegaly and branch pulmonary dilation detected more often than “milder” cases.
c. A study in 2002 by Moon-Grady and colleagues documented hydrops fetalis in four of five fetuses with APVS, all of whom died.
d. After birth, the combined cardiovascular pathology and lung issues can make medical management extremely difficult.
C. Differential diagnosis
1. Abnormal four-chamber view due to RV dilation with or without systolic dysfunction.
2. Significant dilation of RVOT (usually aneurysmal) and branch pulmonary arteries, with narrowing at the level of the pulmonary valve annulus.
3. Lack of pulmonary valve tissue (or only remnants of pulmonary valve tissue may be seen).
D. Pathophysiology
1. APVS is a rare congenital cardiac malformation.
a. The pulmonary valve leaflets are either absent or rudimentary and the annulus is stenotic.
b. A massive aneurysmal dilation of the pulmonary arteries is typically present.
c. The pulmonary arteries compress the larger airways, resulting in airway obstruction and respiratory difficulties postnatally.
2. In some patients, the ductus arteriosus is absent and the aneurysmal dilation of the pulmonary arteries is more severe.
3. APVS is usually associated with tetralogy of Fallot, but it has also been described with an intact ventricular septum.
4. Because the annular stenosis is only moderate, an initial bidirectional shunt can become a predominantly left-to-right shunt beyond the newborn period as the pulmonary vascular resistance falls.
5. An embryologic scheme explaining the genesis of this syndrome has been postulated.
a. Consecutive intrauterine cardiac failure is most probably prevented by prenatal closure of the ductus arteriosus.
b. Pulmonary artery aneurysm, dilation of the RVOT, and many other coexisting deformities can be explained by a cascade of hemodynamic sequelae started by this ductus closure in utero.
c. This theory cannot explain the morphologic and hemodynamic effects seen in patients who have APVS but a patent ductus arteriosus.
6. The high incidence of intrauterine death leads one to question whether right-sided cardiac failure in itself may be an even more important determinant of poor outcome than previously appreciated.
7. In 1983, Hiraishi and colleagues found higher RV end-diastolic volume and lower RV ejection fraction angiographically in postnatal patients with poor outcome, and others have suggested a role for RV dysfunction in infants who do not survive with or without operation.
8. It has also been suggested that in utero pulmonary insufficiency results in impaired diastolic RV filling and leads to dilation and decreased compliance of the RV.
a. This might also influence the left heart, resulting in an ability to redistribute flow to at least the healthier of the ventricles.
b. The effect on the left heart, in turn, might result in high venous pressure and contribute to hydrops seen in these fetuses.
c. The condition of a volume-overloaded RV may be exacerbated by the absence of the patent ductus arteriosus.
d. It is also possible that in the uncommon patient with TOF/APVS and a patent ductus arteriosus, a further exacerbation is caused by increased left-to-right shunting at the level of the ductus.
III. TAKE-HOME MESSAGE
B. Prognosis
1. Perinatal death occurs in more than 60% of cases and is usually associated with hydrops fetalis, the presence of other malformations, or both.
2. The relatively poor survival rate is due to the high rate of terminations, associated genetic anomalies, and bronchomalacia.
3. Bronchomalacia is present in the overwhelming majority of cases featuring cardiomegaly and marked branch pulmonary dilation.
4. The association with major chromosomal anomalies or 22q11 microdeletion is consistent with previous findings. Even in the absence of extracardiac malformations, investigation for 22q11 deletion in cases of conotruncal cardiac abnormalities is recommended.
Allan LD, Sharland GK. Prognosis in fetal tetralogy of Fallot. Pediatr Cardiol. 1992;13(1):1-4.
Allan LD, Sharland GK, Milburn A, et al. Prospective diagnosis of 1,006 consecutive cases of congenital heart disease in the fetus. J Am Coll Cardiol. 1994;23(6):1452-1458.
Emmanoulides GC, Thanopoulos B, Siassi B, Fishbein M. “Agenesis” of ductus arteriosus associated with the syndrome of tetralogy of Fallot and absent pulmonary valve. Am J Cardiol. 1976;37(3):403-409.
Fouron JC, Sahn DJ, Bender R, et al. Prenatal diagnosis and circulatory characteristics in tetralogy of Fallot with absent pulmonary valve. Am J Cardiol. 1989;64(8):547-549.
Hiraishi S, Bargeron LM, Isabel-Jones JB, et al. Ventricular and pulmonary artery volumes in patients with absent pulmonary valve. Factors affecting the natural course. Circulation. 1983;67(1):183-190.
Moon-Grady AJ, Tacy TA, Brook MM, et al. Value of clinical and echocardiographic features in predicting outcome in the fetus, infant, and child with tetralogy of Fallot with absent pulmonary valve complex. Am J Cardiol. 2002;89(11):1280-1285.
Razavi RS, Sharland GK, Simpson JM. Prenatal diagnosis by echocardiogram and outcome of absent pulmonary valve syndrome. Am J Cardiol. 2003;91(4):429-432.
Volpe P, Paladini D, Marasini M, et al. Characteristics, associations and outcome of absent pulmonary valve syndrome in the fetus. Ultrasound Obstet Gynecol. 2004;24(6):623-628.
Zach M, Beitzke A, Singer H, et al. The syndrome of absent pulmonary valve and ventricular septal defect—anatomical features and embryological implications. Basic Res Cardiol. 1979;74(1):54-68.
[/level-membership-for-neonatal-and-perinatal-medicine-category][not-level-membership-for-neonatal-and-perinatal-medicine-category]
9 Tetralogy of Fallot with Absent Pulmonary Valve Syndrome
I. CASE
A. Fetal echocardiography findings
1. The fetal echo reveals situs solitus of the atria, levocardia, left aortic arch, heart rate of 140 bpm.
2. Estimated fetal gestational age (GA) is 25.5 weeks.
3. There was cardiomegaly with increased cardiothoracic ratio (50%) and dextroposition of the heart.
4. The four-chamber view is abnormal, with a large perimembranous subaortic ventricular septal defect (VSD), large overriding aorta, and massively dilated pulmonary artery (Fig. 9-1).
5. The pulmonary valve is dysplastic with no identifiable leaflets, small pulmonary annulus (1.7 mm), and very dilated and pulsatile main pulmonary artery and branches.
6. There is pulmonary stenosis with a peak velocity of 2.2 m/s (lower panel) and pulmonary valve regurgitation. In other words, there is to (systolic) and fro (diastolic) flow through this dysplastic valve, with severe pulmonary regurgitation (Fig. 9-2).
7. There is holosystolic tricuspid regurgitation with a dP/dt (change in pressure per change in time) of 500 mm Hg per second.
8. The foramen ovale is patent, with unrestricted right-to-left shunt.
9. Aortic valve annulus is 6 mm.
10. A ductus arteriosus cannot be identified.
11. The pulmonary venous drainage is normal.
12. Tei index (myocardial performance index).
b. Left ventricle (LV) Tei index is 0.36.
13. The umbilical artery flow pattern is normal.
14. Ductus venosus flow is abnormal, with a wave reversal (3 mm).
D. Fetal management and counseling
1. Management: Diagnosis of tetralogy of Fallot (TOF) should prompt referral for the following:
a. Thorough anatomic examination by ultrasound.
2. Follow-up included serial antenatal studies at weekly intervals.
a. Cardiac function should be monitored by Tei index, by dP/dt if sufficient tricuspid regurgitation develops (3 m/s), and by tissue Doppler (if available).
b. Development of congestive heart failure (CHF) can indicate RV dysfunction secondary to severe pulmonary regurgitation. Severe AV or semilunar valve regurgitation, particularly if it influences filling of the other ventricle, can be very poorly tolerated. Thus, Ebstein’s anomaly and absent pulmonary valves can cause CHF.
E. Delivery
1. If the fetus remains well compensated, delivery should be at term in a tertiary care center.
2. Depending upon the severity of the pathology, which can include degree of cardiomegaly and degree of pulmonary artery dilation, the baby might require ventilatory support after birth as a result of compression of the tracheobronchial tree and coexisting tracheomalacia and bronchiomalacia.
F. Neonatal management
a. Medical management of affected babies includes supporting them from a respiratory standpoint.
[/not-level-membership-for-neonatal-and-perinatal-medicine-category]
