17 Common Arterial Trunk or Truncus Arteriosus
I. CASE
A. Fetal echocardiography findings
1. Fetal echocardiography reveals situs solitus of the atria, normal cardiothoracic ratio (0.33), and abnormal cardiac axis of 80 degrees (oriented more toward the left).
2. The heart rate is normal at 143 bpm.
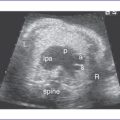
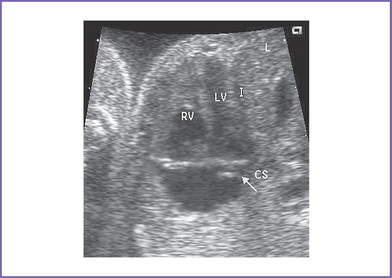
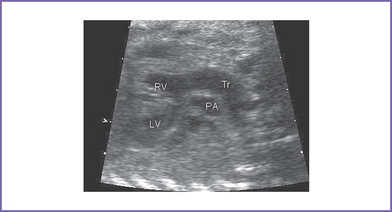
3. The four-chamber view is normal, with balanced ventricles and dilation of the coronary sinus (Fig. 17-1).
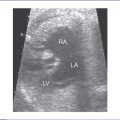
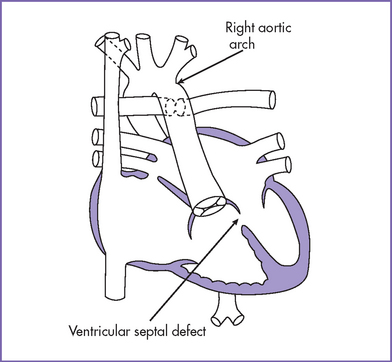
4. There is a single arterial trunk with a quadricuspid, dysplastic truncal valve.
a. Mild stenosis (Doppler velocity: 1.3 m/s).
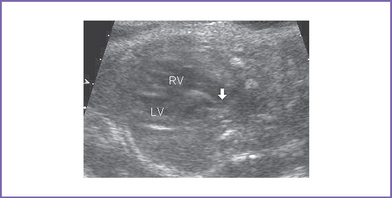
b. Mild truncal valve regurgitation arising from the heart and overriding the crest of the ventricular septum (Fig. 17-2).
5. In the parasternal long axis view of the left ventricle (LV), a moderate to large outlet ventricular septal defect (VSD) is seen.
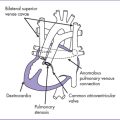
6. Two good-sized branched pulmonary arteries arise from the posterior and leftward aspect of the single arterial trunk (Edwards type II) (Fig. 17-3).
7. The aortic arch is right sided.
8. Myocardial function indices are normal. The right ventricle (RV) and LV Tei indices (myocardial performance index) were normal using the truncal valve.
D. Fetal management and counseling
a. Amniocentesis was done with fetal karyotype and fluorescent in situ hybridization (FISH) for 22q11.2 microdeletion.
b. Results showed normal male karyotype, and FISH was positive for 22q11.2 deletion.
2. Amniocentesis: Diagnosis of tetralogy of Fallot (TOF) should prompt referral for the following:
a. Thorough anatomic examination by ultrasound.
3. Follow-up included serial antenatal studies at 4-week intervals.
a. Degree of truncal valve stenosis and regurgitation and ventricular function were reassessed.
b. Development of hydrops fetalis secondary to congestive heart failure (CHF) is unlikely (approximately 5% risk for all types of congenital heart disease (CHD)), but it could occur if there is progressive truncal valve regurgitation or stenosis.
c. Check for aortic interruption or other associated cardiac anomalies.
E. Delivery
1. In classic truncus without any signs of heart failure, aortic interruption, or other associated malformation, delivery should be as close to term as possible and in an institution with at least neonatal intensive care support.
a. These babies are at high risk for significant transient hypocalcemia.
b. There is also a slight chance the baby will go into heart failure with or without myocardial ischemia (after birth the pulmonary vascular resistance falls and steals from the coronary arteries).
2. Individual management decisions must take into account the level of neonatal support, results of genetic evaluation, parents’ compliance, and the distance from specialized cardiac care.
F. Neonatal management
a. Cyanosis might not be noted immediately at birth.
b. A grade 2/6 systolic murmur with a systolic click is present along the lower sternal border.
c. A high-pitched diastolic murmur of truncal regurgitation might not be present at birth.
d. Signs of CHF can develop within days to weeks of birth as the pulmonary vascular resistance falls.
e. The clinical picture in most infants is that of a large left-to-right shunt with intracardiac mixing, the latter of which results in mild cyanosis.
a. Vigorous anticongestive measures with digitalis and diuretics could be required.
b. Close observation is necessary for evidence of significant diastolic steal, particularly affecting the coronary artery perfusion.
c. Metabolic acidosis, hypoglycemia, hypocalcemia should be treated.
d. Administration of oxygen is usually contraindicated in this disease in the absence of significant pulmonary pathology because it decreases the pulmonary vascular resistance, which would increase pulmonary blood flow further.
e. Given the presence of 22q11.2 microdeletion, the infant will need assessment of T-cell subsets and serum calcium levels. Affected infants often require calcium supplements. Given the potential for immunocompromise, CMV-negative and irradiated blood is critical.
a. Complete correction of common arterial trunk of this type is usually performed in the first 4 to 6 weeks of life. Waiting later than this places the baby at risk for significant pulmonary hypertension.
b. Correction includes closing the VSD, leaving the LV in continuity with the truncal valve, and placing a valved RV–to–pulmonary artery conduit (usually a homograft). The surgeon should attempt to repair the truncal valve, if necessary.
c. The surgical mortality ranges from 2% to 5%.
d. Timing of the operation varies by institution but is generally performed electively at 1 to 3 months of age. Earlier corrective repair may be necessary in a baby with severe heart failure or coronary artery steal.
G. Follow-up
1. Subacute bacterial endocarditis (SBE) prophylaxis is required for life after surgery.
2. For residual hemodynamic abnormalities, anticongestive medications and some restriction of activities may be required.
3. The patient must not engage in competitive sports.
4. For progressive truncal valve regurgitation or stenosis, valve replacement may be needed.
5. RV–to–pulmonary artery conduit replacement will be necessary as the child grows. Cardiac catheterization with intervention may be performed to prolong the interval to reoperation.
6. Atrial and ventricular arrhythmias are possible.
7. Renal dysfunction is possible, given the presence of a single kidney.
8. The 22q11.2 deletion may be associated with:
a. Hypoparathyroidism, necessitating treatment with calcium supplements, at least in early infancy.
b. T-cell subset abnormalities, which can place the infant at risk for certain types of infections.
c. Cleft palate and abnormalities of the pharyngeal muscles, resulting in speech pathology.
I. Outcome of this case
1. The baby was born with good weight; Apgar scores were 5 at 1 minute and 8 at 5 minutes.
2. The baby was noted to be mildly cyanotic at birth (pulse oximeter reading was 87%) in the lower extremities.
3. Postnatal echocardiography confirmed the diagnosis of common arterial trunk type II, with a right arch and mild truncal valve stenosis and regurgitation.
4. The systolic velocity across the truncal valve was 2 m/s, with mild regurgitation.
5. The baby had corrective surgery at 2 months of age (valved homograft conduit from RV to pulmonary artery).
II. YOUR HANDY REFERENCE
A. Common arterial trunk or truncus arteriosus
a. Common arterial trunk accounts for 1% of all congenital heart disease.
b. Prenatal diagnosis of common arterial trunk is not commonly reported, probably because the four-chamber view can appear normal.
a. The prenatal diagnosis of common arterial trunk is often made secondary to recognition of other extracardiac defects or abnormal karyotype.
b. Overall, the prognosis of common arterial trunk diagnosed prenatally remains poor despite much improved surgical, prenatal, and perioperative intensive care. This information is important when counseling families.
c. Risk stratification shows that low birth weight, the need for truncal valve replacement, interruption of the aortic arch, and coronary artery anomalies were associated with less favorable outcomes.
d. Risk factors for death after repair include arch obstruction, truncal valve regurgitation, coronary anomalies, and late repair.
e. Long-term follow-up of these patients showed a need for reintervention in up to 50% by 4 years following initial repair.
f. Late survival rates after repair in infancy were 90% at 5 years, 85% at 10 years, and 83% at 15 years.
3. Associated syndromes and extracardiac anomalies.
a. Microdeletion of chromosome 22q11 and DiGeorge’s syndrome in 30% to 50% of cases.
b. Chromosomal anomalies in up to 14% of fetuses with truncus arteriosus, including trisomies 7, 9, and 18 and tetraploidy.
c. A relatively high incidence of extracardiac malformations: 25% to 37%.
d. Paladini and colleagues (1996) showed that 18.3% of fetuses with conotruncal defects had an abnormal karyotype.
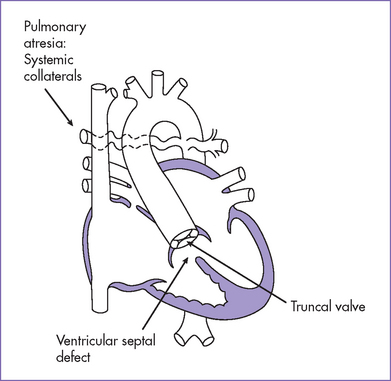
a. In TOF with pulmonary atresia, a patent pulmonary valve is not seen. However, the branch pulmonary arteries can often be seen and are supplied retrogradely from the arterial duct or from collateral arteries from the descending aorta.
b. In truncus arteriosus, the pulmonary arteries are of good size. This is in contrast to the often hypoplastic pulmonary arteries in TOF with severe pulmonary stenosis or pulmonary atresia. The aortic valve is usually thin and not dysplastic in TOF with pulmonary atresia compared to the often dysplastic, mildly regurgitant truncal valve.
c. Most truncus arteriosus is not associated with a patent ductus arteriosus, with the exception of truncus arteriosus with an interrupted aortic arch.
5. Clues to fetal sonographic diagnosis.
b. Large-outlet VSD with an overriding truncal valve.
c. Single dysplastic truncal valve, which may be regurgitant, stenotic, or both.
d. Single large great artery, the arterial trunk, from which the brachiocephalic vessels and branch pulmonary arteries arise.
e. Associated cardiac defects include:
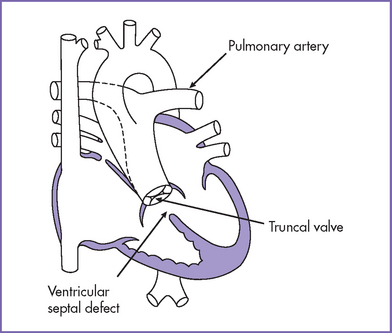
a. Common arterial trunk is characterized by a single great artery arising from the heart, which gives rise to the coronary, pulmonary, and systemic circulations.
c. There is a single arterial trunk (with a truncal valve) from the heart to the pulmonary, systemic, and coronary circulations. A large malalignment VSD is almost always present. It might or might not involve the membranous septum but it always involves the outlet septum.
d. In most cases, the truncal valve overrides the ventricular septum by 50% or less, with more commitment to the LV. Occasionally, the valve is largely committed to the RV, which can contribute to the risks of the surgery.
e. Coronary artery abnormalities (stenotic coronary ostia, abnormal branching and course) are rare but can contribute to the surgical mortality. The branch pulmonary arteries individually or through a common trunk can arise within the sinus of the common arterial trunk and be quite close to the coronary arteries.
f. Increased pulmonary blood flow is present in all types of truncus arteriosus.
 p/
p/ s).
s).g. Severe truncal valve regurgitation exacerbates the low diastolic aortic pressure and can compromise coronary perfusion. This, along with the high myocardial oxygen demand, makes the patient prone to myocardial ischemia.
7. Immediate postnatal management for patients without prenatal diagnosis of common arterial trunk.
a. Truncus arteriosus usually manifests in the neonatal period. Symptoms depend on the degree of left-to-right shunt and the truncal valve regurgitation. Mild cyanosis (88%-92% saturation) with a single second heart sound and a systolic ejection click should suggest truncus arteriosus.
b. Onset of symptoms of CHF is determined by the rate at which the pulmonary vascular resistance falls and the degree of pulmonary shunting tolerated by the left heart.
c. The presence of significant truncal valve regurgitation usually worsens the degree of heart failure because it contributes to the ventricular volume load and potentially causes coronary artery steal.
d. Some centers advocate repair at presentation, and others repair electively at 1 to 3 months of age depending on signs and symptoms of CHF.
a. Children who have undergone common arterial trunk repair will require reoperation for replacement of the RV–to–pulmonary artery conduit as they grow.
b. The actuarial survival for hospital survivors (for the postnatally diagnosed cases) is 83% at 15 years.
c. Moderate to severe truncal valve insufficiency before repair has been identified as an independent risk factor for poorer long-term survival. The need for truncal valve replacement increases with increasing age and the severity of the anatomy. Of those with truncal regurgitation, the need for repair in infancy increases the risk of valve replacement from 5% to 37% at 10 years of follow-up.
d. Long-term results are expected to continue to improve with earlier repairs and the ongoing use of cryopreserved homografts rather than Dacron-housed porcine-valved conduits in the RV outflow tract.
e. The functional status of the vast majority of reported survivors is excellent (New York Heart Association [NYHA] class I).
B. Deletion 22q11.2 syndrome
1. Deletion 22q11 is among the most common genetic syndromes, with an estimated prevalence of 1 per 2000 in the general population.
2. This deletion can manifest with a variety of phenotypes: DiGeorge’s syndrome, Shprintzen’s syndrome, or velocardiofacial syndrome; conotruncal anomaly face (or Takao syndrome); and isolated outflow tract defects of the heart including TOF, common arterial trunk, and interrupted aortic arch. A collective acronym, CATCH-22 (cardiac defect, abnormal face, thymic hypoplasia, cleft palate, hypocalcemia, and 22q11 deletion) has been proposed for these differing presentations.
3. Although each presentation is very different, they represent points along the continuum of the same genetic disorder and therefore require the same genetic counseling and medical management.
4. Deletion of 22q11 is among the most clinically variable syndromes, and more than 180 features are associated with the deletion. Physical manifestations have been reported that involve every organ system, and a well-defined behavioral and psychiatric profile has been described. Studies have shown that this variability is independent of genotype.
5. The clinical presentation can be widely different even within a single family.
6. Some physicians equate the diagnosis of DiGeorge’s syndrome with deletion 22q11. This is not true, because the findings that define the DiGeorge sequence—hypoparathyroidism, absent thymus, and congenital conotruncal heart disease—are etiologically heterogeneous and have been associated with a number of causes, including a variety of cytogenetic rearrangements (e.g., deletion 10p, deletion 17p) in infants of diabetic mothers, in Zellweger syndrome, and in peroxisomal disorders.
7. FISH testing for deletion 22q11 is essentially 100% accurate. If a patient tests negative for the deletion by FISH, then it is safe to assume that he or she does not have velocardiofacial syndrome or Shprintzen’s syndrome. However, there are other causes of the DiGeorge phenotype, including other cytogenetic abnormalities (e.g., 10q13 deletion) and nongenetic causes.
8. Not every patient with a congenital heart disease should undergo FISH testing for the deletion. Such indiscriminant testing is a waste of resources, because not every type of congenital heart disease is associated with deletion 22q11. For example, Ebstein’s anomaly and transposition of the great vessels are not typically seen in deletion 22q11.
9. There are two common misconceptions about the outcome of 22q11.2 deletions.
a. One misconception is that patients who have the identical deletion may have different disorders that have been associated with deletions of 22q11. In fact, the various terms applied to patients with 22q11.2 deletions merely represent nosologic differences, not different diagnoses. All of these different terms actually refer to the same disorder. Moreover, the phenotype develops over time, and so the clinical presentation can change dramatically over time.
b. The other misconception is that it is possible to have this syndrome without having the deletion. There is no convincing evidence to date that this is true.
III. TAKE-HOME MESSAGE
A. Diagnosis
1. The prenatal diagnosis of truncus arteriosus is often made secondary to the recognition of extracardiac anomalies that are common in this condition.
2. Common arterial trunk may also be identified where there is a more leftward cardiac axis at assessment of the four-chamber view.
3. Assessment for 22q11.2 deletion should be considered in every affected fetus and infant.
4. If a patient tests negative for the deletion by FISH, then it is safe to assume that he or she does not have velocardiofacial or Shprintzen’s syndrome. However, there are other causes of the DiGeorge phenotype.
B. Prognosis
1. Progression of the lesion during pregnancy with truncal stenosis or regurgitation can cause hydrops fetalis and fetal demise and also contributes to the mortality and morbidity of affected infants after birth.
2. Truncal valve anomalies can cause myocardial dysfunction, resulting in postnatal death before surgery.
Allan LD, Sharland GK, Milburn A, et al. Prospective diagnosis of 1,006 consecutive cases of congenital heart disease in the fetus. J Am Coll Cardiol. 1994;23(6):1452-1458.
Collett RW, Edwards JE: Persistent truncus arteriosus: a classification according to anatomic types. Surg Clin North Am.. 1949;29:1245-1270.
Duke C, Sharland GK, Jones AM, Simpson JM. Echocardiographic features and outcome of truncus arteriosus diagnosed during fetal life. Am J Cardiol. 2001;88(12):1379-1384.
Hutson MR, Kirby ML. Neural crest and cardiovascular development: A 20-year perspective. Birth Defects Res Part C Embryo Today. 2003;69(1):2-13.
McElhinney DB, Rajasinghe HA, Mora BN, et al. Reinterventions after repair of common arterial trunk in neonates and young infants. J Am Coll Cardiol. 2000;35(5):1317-1322.
Paladini D, Rustico M, Todros T, et al. Conotruncal anomalies in prenatal life. Ultrasound Obstet Gynecol. 1996;8(4):241-246.
Rajasinghe HA, McElhinney DB, Reddy VM, et al. Long-term follow-up of truncus arteriosus repaired in infancy: A twenty-year experience. J Thorac Cardiovasc Surg. 1997;113:869-878.
Van Praagh R. Truncus arteriosus: what is it really and how should it be classified? Eur J Cardiothorac Surg. 1984;1(2):65-70.
Volpe P, Paladini D, Marasini M, et al. Common arterial trunk in the fetus: Characteristics, associations, and outcome in a multicentre series of 23 cases. Heart. 2003;89(12):1437-1441.