[level-membership-for-neurosurgery-category]
Chapter 193 Surgical Resection of Sacral Tumors
Anatomy
Osseous and Ligamentous Structures
Because the lumbosacral and sacroiliac joints transmit the entire weight of the body to the hip bones and lower limbs, these joints and their supporting ligaments must be very strong. The strong dorsal ligamentous complex includes the interosseous ligaments and the dorsal sacroiliac ligaments. The very stout interosseous ligaments connect the sacral tuberosities to the overhanging bone of the iliac tuberosities and represent the single strongest ligaments binding the sacrum to the ilium. The dorsal sacroiliac ligaments are divided into deep (short) and superficial (long) parts. The deeper ligaments connect the sacral and ilial tuberosities and are composed of horizontally oriented fibers; the more superficial ligaments are oriented vertically and stretch from the posterior superior iliac spine to the tubercles of the lateral sacral crest. The caudal portions of the superficial dorsal sacroiliac ligaments blend with the sacrotuberous ligaments.
Neural Anatomy
Both the sympathetic and parasympathetic components of the autonomic nervous system have an intimate relationship with the sacrum. The sacral sympathetic trunk, continuous with the lumbar sympathetic trunk, descends against the ventral surface of the sacrum, converging in front of the coccyx to form the unpaired ganglion impar. Three or four sacral trunk ganglionic enlargements are found on each side of the midline, just medial to the ventral sacral foramina. No white rami communicantes are present in this region; however, the postsynaptic gray rami communicantes from each ganglion join the corresponding sacral or coccygeal nerves for distribution to sweat glands, blood vessels, and erector pilori muscles. In addition, the sacral sympathetic trunks provide fine branches to the superior hypogastric plexus. The superior hypogastric plexus is the caudal continuation of the periaortic sympathetic plexus; it lies on the anterior surface of the fifth lumbar vertebra and upper sacrum in the retroperitoneal tissue. Fibers of the superior hypogastric plexus diverge into right and left hypogastric nerves opposite the first sacral vertebra. The term hypogastric nerve may be a misnomer, because the structure is really a narrow plexus of fibers. The hypogastric nerves represent the principal sympathetic inputs to the inferior hypogastric plexus.
Diagnosis
Clinical Presentation
Sacral tumors are rare and difficult to diagnose at an early stage.1 The major reasons for this delay include the unique capacity of the osseous sacrum and sacral canal to allow neoplastic expansion without causing significant symptoms and the often nonspecific nature of complaints when they do arise. Tumors that originate within the sacral canal can erode or invade the walls of the sacrum and/or expand cephalad within the spinal canal. Tumor can also enter the pelvis via the ventral sacral foramina. A slow-growing, regionally expansive neoplasm can attain a large size without causing symptoms early in the course of the illness. Aggressive, rapidly growing tumors are more likely to cause mechanical instability and/or neurologic compromise earlier during the disease course.
The earliest presenting symptom in patients with sacral tumors is pain located in the lower back or sacrococcygeal region.1–3 Referred pain to the leg or buttock can occur secondary to irritation of the first sacral root or iliolumbar trunk.3 The early presentation of sacral lesions is therefore very similar to that of lumbar spondylosis. By the time a sacral lesion is diagnosed, some patients have been treated and occasionally even operated on for suspected lumbar intervertebral disc pathology.
Radiologic Evaluation
A sacral tumor can be easily overlooked on standard radiographs. The curved shape of the sacrum, its position within the pelvic girdle, and overlying bowel gas are common sources of obscuration. Destructive changes must be advanced before they become evident on plain radiographs.4 Adequate imaging should display the entire sacrum and coccyx on lateral views, and the sacrum should be visualized en face on anteroposterior views. A malignant process is suggested when lytic lesions without sharply defined borders are seen. Well-defined sclerotic margins, reflecting reactive changes in the surrounding bone, imply the presence of a benign or chronic process.
CT and MRI more readily allow the detection, characterization, and staging of sacral tumors.4,5 CT has the advantage of providing excellent bony detail and showing tumor matrix calcification. CT is also useful to image the abdomen for evidence of visceral involvement. Major advantages of MRI studies include the detailed depiction of associated soft tissue masses and the ability to assess the anatomy in multiple planes. The rostral extent of sacral involvement, which is particularly critical to surgical planning, is best appreciated on a midsagittal view.
In addition to the bone scan, CT scans of the chest, abdomen, and pelvis are warranted to rule out metastatic pathology. Intravenous pyelography and/or barium enema may be indicated in evaluating sacral tumors with significant pelvic invasion as well as for colorectal tumors that invade the sacrum.6 Angiography is useful in defining the vascularity of sacral tumors and for preoperative tumor embolization, especially in the case of highly vascular lesions such as giant cell tumor or aneurysmal bone cyst.7,8
Biopsy
The percutaneous CT-guided biopsy involves minimal risk and is currently the method of choice. The site of entry and the trajectory of the needle should be carefully selected so that they can easily be included within the margins of any subsequent resection. If possible, the puncture site should be located in the midline posteriorly. It may be worthwhile to introduce a small droplet of sterile permanent ink into the needle tract to tattoo the skin for later identification.
Pathology
Sacral tumors are categorized as those that originate from the neural elements or their supporting tissues, those arising from bone, and those that metastasize from distal sites or are the result of direct invasion from adjacent pelvic structures. The most common sacral tumors are metastatic, and the most common primary sacral tumor is chordoma.2 Some non-neoplastic entities, including developmental cysts and inflammatory conditions, mimic sacral tumors on imaging studies. A broad differential diagnosis of the various lesions encountered in this area is provided in Table 193-1.
TABLE 193-1 Differential Diagnosis of Sacral and Presacral Lesions
| Congenital Lesions |
| Posterior sacral meningocele, meningomyelocele, and lipomyelomeningocele |
| Developmental cysts |
| Dermoid and epidermoid cysts |
| Retrorectal tailgut cysts |
| Enteric duplication cysts |
| Anterior sacral meningocele |
| Lateral meningocele |
| Occult intrasacral meningocele |
| Perineural (Tarlov’s) cysts |
| Tumors |
| Primitive Neuroectodermal Tumors |
| Teratoma |
| Hamartoma |
| Chordoma |
| Inflammatory Lesions |
| Osteomyelitis |
| Abscess (pelvic abscess, perirectal abscess) |
| Neurogenic Lesions |
| Schwannoma and neurofibroma |
| Ependymoma |
| Ganglioneuroma |
| Neuroblastoma |
| Osseous Lesions |
| Bone island |
| Osteoid osteoma and osteoblastoma |
| Osteochondroma |
| Hemangioma |
| Aneurysmal bone cyst |
| Giant cell tumor |
| Chondrosarcoma |
| Osteosarcoma |
| Ewing’s sarcoma |
| Paget’s disease (monostotic) |
| Metastatic Lesions |
| Hematogenous spread (lung, breast, prostate, kidney, lymphoma) |
| Locally invasive lesions (colorectal and gynecologic malignancies, sarcoma) |
| Miscellaneous Lesions |
| Carcinoid tumor |
| Lymphoma |
| Solitary plasmacytoma, multiple myeloma |
| Meningioma |
| Hemangiopericytoma |
Congenital Tumors
Sacrococcygeal Teratoma
A teratoma is a lesion containing tissue from all three germ layers, represented by either well-differentiated or immature elements. Skin, teeth, central nervous system tissue, and respiratory and alimentary mucosa may be found within these tumors. Sacrococcygeal teratomas are the most common sacral tumor in neonates. In contrast, these tumors are rare in adults.9 They develop during intrauterine growth and can grow large enough to cause dystocia.10 The diagnosis is often made on prenatal ultrasonography.11 After birth they manifest as an exophytic mass located between the anus and coccyx, covered by normal skin. Presacral and sometimes combined pre- and postsacral (dumbbell-shaped) lesions also occur.
Chordoma
Chordoma is the most common primary bone tumor of the sacrum.12 Sacral chordomas occur almost twice as often in men compared with women and are uncommon in persons younger than 40 years. The most common presenting symptom is pain in the lower back or sciatic region.3 Chordomas can reach a very large size before constipation (from rectal compression) or lower extremity paresis (due to sacral plexus involvement) occurs.
Chordomas are considered congenital because they are thought to arise from notochordal remnants. Chordoma is characterized microscopically by the appearance of vacuolated “physaliferous” cells. The midline location of these tumors also relates to this proposed etiology.13
Chordomas are typically slow growing but locally aggressive. Significant extracompartmental growth is often seen by the time of diagnosis. Most sacral chordomas manifest as surgical stage IB with anterior extension into the pelvis.14 The tumor often displaces but does not invade the rectum, because the tough periosteum and presacral fascia resist the transgression of disease. Metastasis is usually a late event.
The usual CT appearance consists of lytic bone destruction in addition to a disproportionately large soft tissue mass. Calcification is present in 30% to 70% of cases.5 Unlike most bone tumors, chordomas can show reduced uptake or normal distribution of isotope on bone scan.
En bloc excision is the treatment of choice for sacral chordomas.2,12,13 The extent of surgical resection has been found to play a major role in determining the length of disease-free survival.13 Although a distinct capsule is often seen within the soft tissues, a radical wide posterior margin of the gluteal muscles should be employed to reduce the risk for local recurrence.15 The margins of chordoma within bone are often indistinct. Surgical resection should extend at least one sacral segment beyond the area of gross disease.2
The value of radiotherapy as primary or adjuvant treatment for chordoma has been debated.12 Supplementary radiotherapy may be a useful adjunct to surgical care, but it is not sufficient as stand-alone therapy. In a clinical series spanning 40 years, York and colleagues2 reported that the addition of radiation therapy significantly prolonged the disease-free interval for patients undergoing subtotal resection (2.12 years versus 8 months). Others have suggested that radiotherapy is of limited value in most cases.13
Currently, chemotherapy does not play a role for the treatment of chordomas, although the use of imatinib mesylate is being investigated.16
Neurogenic Tumors
Schwannoma and Neurofibroma
Sacral schwannomas are much more common than neurofibromas. They grow within the sacral canal and only rarely expand through the anterior sacral foramina into the presacral space.17,18 A complete resection of these benign tumors is potentially curative because, with the exception of plexiform neurofibromas, they do not infiltrate beyond their capsular envelope. The surgical approach depends on the size and location of the tumor, especially the degree of intraspinal and presacral extension. The majority of lesions coming to neurosurgical attention are largely confined to the sacral canal and can be resected completely using a posterior approach. This approach allows direct visualization of the relationship between the sacral nerve roots and the tumor. If there is a small presacral component, limited access to this region may be obtained by transforaminal resection. Tumors with a large presacral component should often be removed through an anterior transabdominal approach. Giant intrasacral schwannomas have been described for which radical sacral resection was performed. 17,18
Ependymoma
Sacral ependymomas are most commonly the benign myxopapillary type. They arise from ependymal cell clusters within the terminal filum and expand the sacral canal.19 Rare extradural sacrococcygeal ependymomas have been reported, including subcutaneous, presacral, and intrasacral varieties.20
Patients with sacral region ependymomas typically present with pain in either a lower back or sciatic distribution. By the time of diagnosis, which averages 2 to 3 years after the onset of symptoms, many patients exhibit some form of cauda equina syndrome.3 As with other slow-growing sacral tumors that tend to be diagnosed at an advanced stage, ependymomas may be associated with extensive bony destruction and a large soft tissue mass.19
A complete en bloc resection is advisable in order to prevent local recurrence or cerebrospinal fluid dissemination. Intradural lesions can be resected completely via a posterior approach.20 Intraoperative somatosensory-evoked potentials are useful, because dissection of tumor from the roots of the cauda equina can be difficult. The approach to extradural lesions depends on tumor location. Radiotherapy may be useful in cases of subtotal removal or recurrence.
Ganglioneuroma
Ganglioneuromas are rare slow-growing tumors composed of sympathetic ganglion cells.21 They are thought to represent the benign counterpart of malignant neuroblastomas. Ganglioneuromas can arise anywhere from the base of the skull to the pelvis. Like neuroblastomas, most ganglioneuromas arise in the abdomen, predominantly from the adrenal gland. The small percentages of pelvic tumors likely arise from sacral extensions of the sympathetic chain. As tumor expands within the pelvis, it can extend through a sacral foramen into the epidural space, causing sacral nerve root compression. The treatment is complete surgical removal.
Primary Osseous Tumors
Less than 10% of all primary bone tumors occur in the spine, with the exception of osteoblastoma, which has a 40% incidence of vertebral involvement.22 The incidence of sacral involvement among osseous tumors varies considerably. Bone tumors are a histologically diverse group of neoplasms. Some lesions are of low biological activity, such as osteoid osteoma, osteoblastoma,22,23 and aneurysmal bone cyst.24 High-grade lesions include chondrosarcoma and osteosarcoma. Although giant cell tumors are histologically benign, they are locally invasive and have a high risk for recurrence.25
Aneurysmal Bone Cysts
There is much debate over the best treatment for spinal aneurysmal bone cysts. Among the treatment modalities, complete surgical excision of the lesion remains the most effective, with the lowest rate of recurrence.26,27 Alternatively, these lesions are often treated by intralesional curettage and bone grafting. The disadvantage of this technique is that it is often associated with greater blood loss as well as high recurrence rates ranging from 20% to 60%.28,29 Selective embolization has been shown to avoid excessive bleeding when used with surgery and has also proved to be an effective sole treatment in cases that are difficult to treat surgically. Several less-invasive methods have been studied for the treatment of aneurysmal bone cysts including particulate embolization, intralesional injection of alcoholic zein, radiotherapy alone,30 and radiotherapy combined with surgery.31 Radiotherapy can be used as an alternative or adjunct to surgery when aneurysmal bone cysts are located in sites where an adequate resection cannot be done without resulting in significant morbidity or poor cosmetic results.
Osteoid Osteoma and Osteoblastoma
Osteoid osteomas and osteoblastomas are bone-producing lesions that often occur in long bones but may also be found in the spine. Although these tumors have a predilection for posterior elements of the mobile spine, they tend to involve the body of the sacrum.22 These lesions are similar histologically to each other, containing osteoblasts that produce osteoid and woven bone. Osteoid osteomas are small, self-limited, and benign, whereas osteoblastomas are often larger, are more aggressive, and can become malignant.23
Osteoid osteomas and osteoblastomas are common causes of painful scoliosis in children and adolescents. The most common presenting symptom is neck or back pain that often responds to aspirin or nonsteroidal anti-inflammatory drugs (NSAIDs). Chemotherapy has played a minimal role in the treatment of these lesions and has only been used in selected patients with recurrent aggressive lesions or in patients with surgically inaccessible lesions. Surgery becomes the primary treatment for osteoid osteoma and osteoblastoma as the pain becomes more severe and less responsive to medication. Complete resection is often curative for these lesions. Incomplete resections are more common in osteoblastomas than in osteoid osteomas because osteoblastomas are larger and more commonly involve extraosseous tissue. Osteoblastomas recur in 10% to 15% of cases,32 and the recurrence rate of osteoid osteomas is 4.5%.33
Giant Cell Tumor
Giant cell tumors of the spinal column have a predilection for the thoracolumbar and sacral regions. It is the second most common primary sacral tumor after chordoma. Patients with spinal involvement usually present in the third and forth decades of life. Giant cell tumors are believed to arise from mononuclear cells of macrophage origin. Histologically, they are multinucleated giant cells and macrophages that can contain areas resembling aneurismal bone cysts, and care should be taken when interpreting bone biopsy samples to avoid an erroneous diagnosis. Although they are histologically benign, approximately 5% to 10% of giant cell tumors undergo malignant degeneration and assume a more aggressive course.34
Clinical management of giant cell tumors is difficult given their high propensity for recurrence. En bloc resection with wide margins is the gold standard for treating these lesions.35 Unfortunately, these tumors often grow to a large size before initial diagnosis, and extension of the tumor into the spinal canal and adjacent soft tissues is not uncommon. Therefore, subtotal resection in concert with adjuvant chemotherapy, arterial embolization, and/or radiosurgery is the typical treatment when en bloc resection is not feasible.
Arterial embolization is a critical adjuvant therapy that can increase the safety of surgery as well as improve progression-free survival. Preoperative embolization is a useful tool that minimizes intraoperative blood loss during the resection of these highly vascularized tumors. Embolization may be especially useful to limit bleeding during intralesional resections. Interestingly, arterial embolization may also be useful as a primary treatment modality.8
Chondroma and Chondrosarcoma
Complete surgical resection with negative margins is the primary treatment for chondromas. Less than 10% of patients experience tumor recurrence after complete resection.36 If incomplete resection is performed, residual tumor is at risk for sarcomatous degeneration. Because chondromas are radioresistant lesions, radiosurgery currently does not have a significant role in the treatment of these tumors.
Chondrosarcomas are slow-growing tumors that commonly manifest with focal pain or neurologic deficits. Although chondrosarcomas can be divided into multiple pathologic subgroups, the most important characteristic with respect to clinical outcome is the World Health Organization (WHO) grade. Grade I chondrosarcomas have a 90% 10-year survival rate compared to 30% to 40% for high-grade lesions.37
Regardless of WHO tumor grade, the gold standard for treating chondrosarcomas is gross total resection with negative margins. The literature suggests that en bloc resection is associated with long-term recurrence-free survival. En bloc resection can result in recurrence rates of 20% or less.38 In cases of tumor recurrence, repeat resection can lead to improved survival. There is no well-defined role for radiation or chemotherapy for these lesions.
Osteosarcoma
In a study of osteosarcoma, including 15 patients with sacral lesions,39 a poorer prognosis was reported in patients with large tumors, metastases, and incomplete tumor resection. Patients with sacral osteosarcoma should be treated with a combination of chemotherapy and at least marginal excision for those with surgically accessible tumors.40 Postoperative radiotherapy may be beneficial.39
In a recent study of osteosarcoma, including 15 patients with sacral lesions, a poorer prognosis was reported in patients with large tumors, metastases, and incomplete tumor resection. Patients with sacral osteosarcoma should be treated with a combination of chemotherapy and at least marginal excision for those with surgically accessible tumors.40 Postoperative radiotherapy may be beneficial.39
Ewing’s Sarcoma
Ewing’s sarcoma is a primary bone tumor rarely found in the spine. Ewing’s sarcoma of the spine typically occurs at a young age, and the sacrum is the most common location in the spinal axis. The etiology of this tumor is unclear, although there is evidence that mesenchymal stem cell progenitors might play a role.41 Pain is the usual presenting symptom, and neurologic deficit is common.
The current treatment regimen for Ewing’s sarcoma involves surgery, chemotherapy, and radiation therapy. Patients with localized disease have been reported to have 5-year relapse-free or event-free survival rates of 59% to 76%.42–44 Chemotherapy plays a central role in the treatment of Ewing’s sarcoma. Current protocols typically use a combination of vincristine, cyclophosphamide, and doxorubicin.45
The addition of surgical resection and/or radiation to chemotherapy can improve outcomes for patients with Ewing’s sarcoma. There is some evidence that en bloc resection leads to improved local control.46 Radiation therapy plays an important role in the treatment of Ewing’s sarcoma, which is a relatively radiosensitive tumor compared to other sarcomas.
Metastatic Tumors
Metastatic tumors are by far the most common malignant neoplasms affecting the sacrum.47 Most result from hematogenous spread. Although the diagnosis of primary sacral neoplasms is often delayed, metastatic involvement of the sacrum is usually suspected relatively early because the rapidly progressive and locally invasive nature of these tumors often causes significant low back pain or radicular symptoms affecting the lower extremities.3
Metastatic involvement of the sacrum usually signifies advanced disease. The goal of treatment in these patients is palliation, which is most often accomplished with local radiotherapy and sometimes chemotherapy. Such tumors are considered for operation only under certain circumstances.47,48 For example, palliative surgical decompression of neural or pelvic structures is occasionally indicated. Spinal-pelvic stabilization may be considered in carefully selected patients with severe pain or loss of the ability to ambulate due to a pathologic fracture or instability.48
In addition to hematogenous metastases from distal sites, the sacrum is often involved by local spread of tumors arising within the pelvic viscera. Locally advanced adenocarcinoma of the rectum is a condition for which the techniques of sacral resection may be a worthwhile consideration, both at primary presentation and in patients with recurrent disease.49,50 Midline posterior tumors adherent to or invading the distal sacrum may be resectable for cure with an extended abdominoperineal resection, including the sacrum. Occasionally, total pelvic exenteration (including a portion of the sacrum) is necessary.49 Careful patient selection is paramount in ensuring a favorable outcome from these aggressive procedures.50
Surgical Treatment
Indications
The overall clinical status of the patient, the anatomic extent of the tumor, and an appraisal of the biological behavior of the tumor are major factors in determining the goals of surgery. Sacral tumors may be classified as benign tumors, low-grade locally aggressive tumors, and high-grade malignancies. Benign encapsulated tumors can be treated with lesional resection. Primary bony tumors such as chordoma or chondrosarcoma should be treated with en bloc excision, including a circumferential margin of uninvolved tissue, where possible, to effect cure.2,12 The surgical goals are similar for some primary localized high-grade malignancies such as osteosarcoma and advanced rectal cancer with sacral invasion.39 Palliative surgical debulking to preserve or restore neurologic function and reconstruction of spinopelvic junction for painful instability is a consideration for selected patients with metastatic sacral tumors.48
Functional and Biomechanical Considerations
Several detailed studies address the functional aspects of sacral amputations.13,51–54 Amputations distal to S3 generally leads to limited clinical deficits. Patients can develop reduced perineal sensation and a decline in sexual function, but sphincter function is usually preserved. The highest variability in functional results is seen for transverse resections of S2–3 (including removal of one to all four roots of S2–3).52 Although there is seldom any relevant motor deficit, many patients have saddle anesthesia and a significant reduction in sphincter control. Section of the S1 roots can result in clinically relevant motor deficits (walking with external support) and almost uniformly results in total loss of sphincter control and sexual ability. Unilateral resection of sacral roots leads to unilateral deficits in strength and sensitivity; however, sphincter control may be either preserved or only partially compromised.51,54 No matter the level of resection, damage to the lumbosacral trunks or sciatic nerves can cause serious postoperative motor and sensory deficits. Damage to the parasympathetic and sympathetic plexi alone can result in problems with sexual ability in men.
In the resection of sacral tumors, sacroiliac stability is not greatly affected if the sacroiliac joints are left intact.55 Although the sacrospinous and sacrotuberous ligaments are often transected in lower sacral amputations, the strong dorsal ligamentous complex is preserved.
Gunterberg and colleagues studied cadaveric pelves to evaluate pelvic strength after major amputations of the sacrum.55 Resection of one third of the sacroiliac joint and the associated ligamentous structures resulted in a 30% decline in the strength of the pelvic ring. Resections between S1 and S2 caused loss of stability of about 50%. In all of these experiments, the load to failure far exceeded physiologic loads. These authors concluded that weight bearing was safe for patients after sacral resection, as long as at least 50% of the sacroiliac joint (corresponding to at least the upper half of the S1 segment) remained intact.
Some partial sacrectomies involve sacroiliac joint resection on only one side. Without reconstruction, the patient can experience postoperative pain associated with proximal migration of the pelvis. It is generally recommended that lumbopelvic fixation be performed in such cases, unless the contralateral joint is completely intact and there is no anterior pelvic deficiency.56
The problem of fatigue fractures as a complication of high sacral amputation appears to be limited. Bergh and colleagues found that only 6 of 18 patients with high sacral amputations (through or above the S1–2 disc) developed fractures, and only 1 of them had permanently disabling pain.13
Preoperative Planning
Appropriate preoperative planning requires a keen appreciation of the anatomic relationships of the sacral region, familiarity with the advantages and limitations of the different exposures, and a clear sense of the surgical objective. Preoperative CT and MRI studies should be performed above and below the tumor site to define its margins and determine the relationship of the tumor with its surrounding structures. Preoperative angiography and embolization are a worthwhile consideration for many lesions, most notably for giant cell tumor.8 If the tumor mass extends well beyond the osseous confines and invades surrounding visceral structures, preoperative intravenous pyelography or a barium enema may be helpful. In cases in which resection of the bladder wall or rectum is planned, diverting colostomies or diverting urinary procedures may be necessary. These must be planned and considered in advance of the surgical procedure.6
Surgical Approaches
Transperineal Approach
The patient is placed in the Kraske position (flexed prone), and a midline incision extending rostrally from the coccygeal region is carried out. Access to the presacral space is afforded by division of the anococcygeal ligament. The caudal sacrum may be osteotomized after transection of several soft tissue attachments, including the gluteus maximus, the sacrotuberous and sacrospinous ligaments, and the piriformis muscle. The transperineal approach is discussed in detail in the section entitled Low Sacral Resection.
Anterior Approach
Wide access to the ventral sacrum is best obtained by a transabdominal route.54 This approach is often combined with a staged or simultaneous posterior approach to achieve a high sacral amputation or total sacrectomy. In these circumstances, the ventral exposure is used to gain vascular control, dissect the rectum and other pelvic contents from the tumor, and complete the anterior osteotomy. The posterior approach is used to dissect and transect the dural contents and to complete the sacral amputation.
Synchronous Abdominosacral Approach
Localio and colleagues popularized a one-stage abdominosacral approach with the patient in the lateral decubitus position.57 Although they used a retroperitoneal exposure ventrally, others have employed a transabdominal route.58 The sacrum is exposed through a separate posterior incision, and division of the anococcygeal ligament allows access to the presacral space. By developing the plane behind the rectum, an effort is made to meet with the abdominal incision. The internal iliac artery and vein can be temporarily occluded while the lateral muscular and ligamentous attachments onto the sacrum and sacroiliac joint are transected at the level of intended amputation. The gluteus maximus, piriformis and coccygeus muscles, as well as the sacrotuberous, sacrospinous, and posterior sacroiliac ligaments, are divided bilaterally. The sacroiliac joint and sacrum can then be osteotomized under direct vision both dorsally and ventrally. Although this technique can be used to simultaneously expose the sacrum anteriorly and posteriorly, it is more difficult to expose both of them well. In addition, it is difficult to carry out complex soft-tissue reconstruction and spinopelvic fixation techniques with the patient positioned laterally. We reserve this type of exposure for cases requiring hemisacrectomy.
Lateral Approach to the Sacroiliac Joint
A combined anteroposterior approach to the sacroiliac joint is useful for the en bloc excision of malignant tumors that involve not only the sacroiliac joint itself but also the lateral sacral ala and medial iliac wing.59 Chondrosarcoma is notorious for its often eccentric location within the upper sacrum and typically involves the sacroiliac joint. The technique of en bloc resection for tumor localized to the sacroiliac joint is discussed later.
Sacral Resection
The techniques of sacrectomy were popularized by Stener.60 Sacral resections may be categorized into two groups: those used for midline tumors (typically chordoma, giant cell tumor, and locally invasive rectal carcinoma) and those used for eccentric lesions (e.g., chondrosarcoma of the sacroiliac joint).
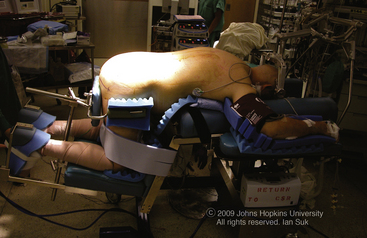
The Andrews table is recommended for patients undergoing sacral resection, particularly for patients undergoing an en bloc sacrectomy. The Mayfield headholder is recommended to limit pressure on the face and orbits (Fig. 193-1). This can potentially decrease the risk of facial decubitus ulcers and postoperative blindness.
The midline group includes low, middle, and high sacral amputations, total sacrectomy, and hemicorporectomy. Because of the significant functional consequences of nerve root sacrifice, we define the type of sacral amputation not by the level of the osteotomy but by the level of nerve root sacrifice. Low sacral amputation involves sacrifice of at least one S4 nerve root (or any level below S4). Midsacral amputation involves the sacrifice of at least one S3 nerve root. High sacral amputation requires sacrifice of at least one S2 nerve root. All of the sacrococcygeal nerve roots are lost during total sacrectomy, but the L5 nerve roots and the lumbosacral trunks should be preserved. Progressively higher sacrectomy is technically more challenging and involves greater blood loss, potential for instability, risk for complications, and loss of sphincter control and sexual ability. In hemicorporectomy, or translumbar amputation, the bony pelvis, pelvic contents, lower extremities, and external genitalia are removed following disarticulation of the lumbar spine and transection of the thecal sac.61 The lateral group of sacral resections includes hemisacrectomy and en bloc excision of the sacroiliac joint.
Low Sacral Resection
Low sacral amputation via a combined posterior and transperineal exposure was described by McCarty and colleagues in 1952.62 This approach provides access to the low presacral and retrorectal space and allows complete resection of small tumors of the middle and distal sacrum. More complicated cases, such as for patients with recurrent tumor or rectal involvement, require staged anterior and posterior procedures. Because the low sacral amputation osteotomy is performed below the level of the sacroiliac joint, it is not inherently destabilizing.
The patient is placed in a Kraske (flexed prone) position on the padded bolsters of an Andrews table to allow the abdomen to hang free and minimize compression of the inferior vena cava. After careful skin preparation and draping, a midline incision is made extending from the region of the lumbosacral junction to the coccyx. The dorsal exposure should be tailored to incorporate any biopsy incision as well as the underlying tract. The erector spinae muscles are usually dissected subperiosteally and retracted laterally. However, if preoperative imaging studies reveal tumor extending dorsally out of the sacrum, a layer of sacrospinalis musculature and fat should be left to cover the involved regions. Depending on the extent of soft tissue involvement, it may also be advantageous to transect the gluteal maximus several centimeters from its origin, leaving a cuff of gluteal musculature attached to the sacrum laterally. The lateral sacrococcygeal attachments including the sacrotuberous and sacrospinous ligaments and the coccygeus and piriformis muscles are then identified and transected close to their insertions. The pudendal and sciatic nerves as well as the gluteal arteries are carefully identified and preserved. The inferior edge of the sacroiliac joint is cleared of soft tissue using a Cobb elevator.
Division of the anococcygeal ligament allows entry into the presacral space. The rectum is gently mobilized away from the tumor surface by blunt finger dissection. A limited sacral laminectomy, immediately rostral to the level of intended amputation, allows direct visualization of the nerve roots. For example, if the intent is to amputate the sacrum at the bony S3 level with preservation of the third sacral nerve roots, a laminectomy from S1 to S3 is performed. The filum terminale externa is transected and the nerve roots below S3 are doubly ligated and transected within the sacral canal. An osteotomy of the sacral body is then performed using a high-speed drill with a diamond bur. The pelvic structures are protected during the osteotomy by maintaining a finger in the presacral space. This maneuver also provides tactile sensation to help guide the osteotomy (Fig. 193-2).
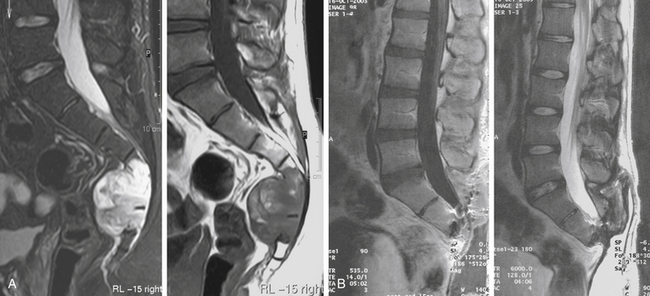
A large cavity results from this resection, and diligent hemostasis is essential. Suction drains are placed within the cavity, and the wound is reapproximated in a layered fashion. A plastic surgeon is often enlisted to mobilize local soft tissue flaps to fill the large defect and to facilitate a tensionless closure (Fig. 193-3).
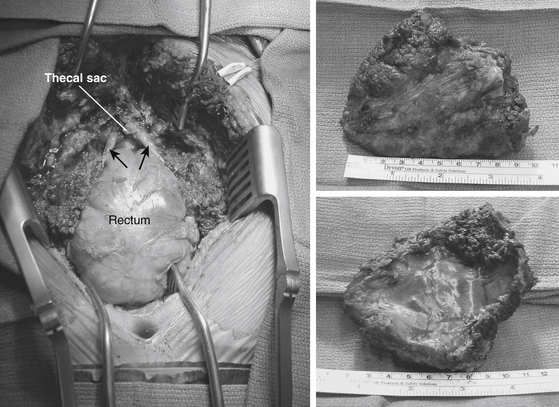
FIGURE 193-3 Postoperative dorsal view of the patient in Figure 193-2 following en bloc removal of the tumor. Seen are bilaterally preserved S3 roots (arrows), posterior rectal wall, and ligated end of the thecal sac. The last two images show a postoperative surgical specimen that was excised en bloc.
Middle and High Sacral Resection
Sacral amputation above S3 using a staged anterior and posterior approach was initially reported by Bowers.63 A transpelvic vertical rectus abdominis myocutaneous (VRAM) flap for reconstructing large sacral defects has significantly reduced problems with wound breakdown following these aggressive resections.64 Alternatively, gluteal rotational flaps can be used to fill the surgical defect and promote optimal wound healing.
Rostral dissection with mobilization of the common iliac vessels is often required for high sacral amputations. The ventral sacral foramina serve as the best landmarks to guide the ventral sacral osteotomy, although the level may also be confirmed using intraoperative radiographs or fluoroscopy. Selecting an area approximately one sacral level above the region of the planned osteotomy, the periosteum is incised in a transverse fashion and reflected downward. The sympathetic trunks are unavoidably cut, along with the hypogastric plexus in the case of very high sacral amputations. If the planned osteotomy incorporates any of the ventral sacral foramina, the sacral nerves exiting at that level are first dissected out and preserved. It is not possible to spare any sacral nerves if the osteotomy must cross the body of S1 above the foramina. The sacral alae are dissected laterally and the lumbosacral nerve trunks are exposed and freed. The nerve trunks are mobilized laterally during the osteotomy.
If the rectum is to be included with the specimen, the superior rectal vessels are identified and ligated and the rectosigmoid junction is mobilized in preparation for division with a mechanical stapler. The bowel is transected and the middle rectal vessels are ligated and divided. The stapled stumps of bowel may be oversewn to decrease the risk for wound soilage. Tumor involvement of other pelvic organs or the endopelvic fascia might require pelvic exenteration if the surgery has curative intent.49
The ventral osteotomy cuts are palpated by introducing a finger presacrally via the perineal exposure. The osteotomy is performed in two stages, each beginning in the midline and extending through the lateral sacroiliac joint to exit at the greater sacroiliac notch. Once the specimen is freed, the sacral roots, which have already been sacrificed within the spinal canal, are divided just proximal to their connections with the sciatic nerve.
The gluteal muscles may be reapproximated to each other or to bone. Suction drains are placed deep in the wound and tunneled to remote exit sites. The large sacral defect is closed using the VRAM flap, which is retrieved from the pelvis and sutured into place in a layered fashion. As an alternative, a microvascular free flap reconstruction may be performed. Free flap reconstruction is challenging in the sacral area because of difficult access to adequate recipient vessels.65 Gluteal rotation flaps may also be used, although they may be a less reliable option.64 Patients with rectal resections are again turned to the supine position, the celiotomy is reopened, and a colostomy is completed.
Total Sacrectomy
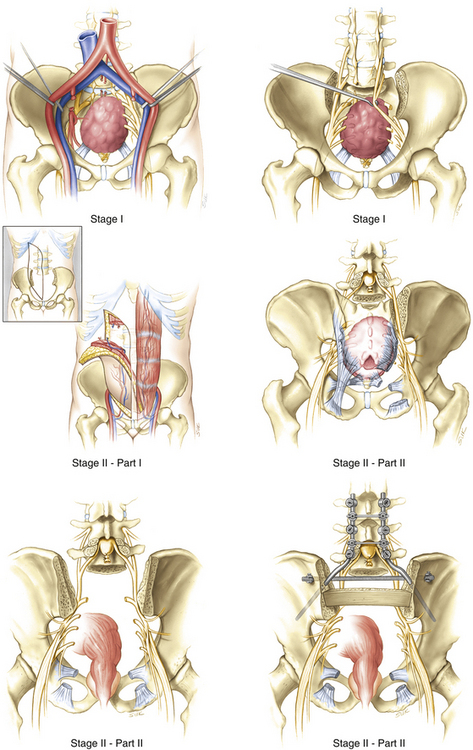
Few cases of total sacrectomy have been reported in the literature.66–70 Although it is not possible to spare any of the sacral roots during this procedure, careful preservation of the L4 and L5 nerve roots allows patients to ambulate postoperatively.
The technique is essentially the same as that discussed for high sacral amputation, with some exceptions (Fig. 193-4). During the anterior approach, bilateral ventral osteotomies along the entire length of the sacroiliac joints are performed with the lumbar nerve roots and lumbosacral trunks protected medially. Instead of a transverse osteotomy through the upper sacrum, an L5–S1 discectomy is performed. Finally, the S1–3 ventral nerve roots are transected at their foramina, if they can be visualized. During the second stage of the procedure, an L5 laminectomy and bilateral L5–S1 foraminotomies expose the L5 nerve roots, which are preserved. The posterior iliac crests are removed with Leksell rongeurs, and a high-speed drill is used to complete the sacroiliac osteotomies from the posterior approach. The thecal sac is then ligated below the level of the L5 nerve roots, and the L5–S1 discectomy is completed. The sciatic notches are exposed bilaterally and the S1–5 roots are transected. Following division of the posterior ligamentous attachments, the sacrum can be removed in toto. Lumbopelvic fixation is required. As described for high sacral amputation, resection of the rectum may be incorporated and the VRAM flap is routinely used to close the sacral defect.64
En Bloc Resection of Tumors of the Sacroiliac Joint
With the superior gluteal vessels mobilized and the neural structures protected, a Gigli saw is passed through the greater sciatic notch and the iliac osteotomy is performed. The anterior structures are retracted medially, and the sacrum is osteotomized in a posteroanterior direction, beginning just lateral to the upper three dorsal foramina. We prefer to use a high-speed drill to perform the osteotomies rather than an osteotome, because of improved hemostasis and finer control. A diamond bur does not tend to entrain adjacent soft tissues. After completing the osteotomies, the entire specimen can be removed en bloc.
Osseous bleeding is controlled with bone wax, and soft tissue hemostasis is obtained. Suction drains are placed within the resection cavity. The gluteus maximus can sometimes be reapproximated to the midline fascia. Anteriorly, the closure is performed in layers. The aponeurosis of the abdominal muscles is reattached to the soft tissue cuff left on the iliac crest during the exposure.
Hemisacrectomy
Hemisacrectomy generally involves the unilateral removal of the sacroiliac joint and a portion of the ilium along with the hemisacrum. It may be performed as part of a more extensive internal or external hemipelvectomy procedure.71 The approach is essentially an extension of the lateral approach to the sacroiliac joint. It involves combined simultaneous retroperitoneal and posterior exposures with the patient placed in the lateral decubitus position. Dorsal and ventral osteotomies can thus be performed under direct vision. Resection of all the sacral nerves on one side results in expected deficits in sensitivity and strength. However, sphincter function may be normal or only partially compromised.
Single-Stage Total Sacrectomy
En bloc total sacrectomy performed in a single-stage posterior approach has been described.72 Patient positioning and exposure of the posterior sacrum and lower lumbar spine is the same as discussed previously. A laminectomy of L5 and S1 is performed to identify the L5 and S1 nerve roots bilaterally. Lateral iliac osteotomies are performed to expose the L5 nerve roots and mobilize them from their origin off the thecal sac to the sciatic notch. An L5–S1 discectomy is performed. The thecal sac below the L5 nerve roots is divided and ligated with 0-silk suture. With careful retraction of the L5 nerve roots rostrally, resection of the posterior longitudinal ligament, annulus, and anterior longitudinal ligament is performed using a Leksell rongeur and Kerrison punches. Care must be taken to protect the iliac vessels and middle sacral artery and vein while the sacrum is disconnected ventrally.
Spinopelvic Stabilization
Partial sacrectomy involving more than 50% of the sacroiliac joint on each side is an indication for spinopelvic stabilization.55 Pedicle screws provide rigid stabilization and can be used for short segment fixation across the lumbosacral junction.73 The S1 pedicle is larger than the lumbar pedicles and often can be fitted with 7- to 8-mm diameter screws. Additional bony purchase can be obtained if the screw penetrates the anterior S1 cortex. Medially directed S1 pedicle screws that are cross-linked and attached to rods create a triangulation effect, which greatly increases torsional stability and resists pullout.74–78 Triangulation with an oblique orientation also interferes less with the superjacent facet joint and allows more purchase with a longer screw.77
A method to enhance sacral fixation with screws is to place an additional pair of laterally directed bone screws into the sacral alae below the S1 level. This has a biomechanical advantage over a single pair of S1 pedicle screws.75 However, the bone of the ala is usually of low density, and purchase may be tenuous. Moreover, the risk for neurovascular injury from laterally directed screws in this region must be kept in mind.
S2 (or lower level) pedicle screws are often of little use because the pedicles are very short. However, the S2 iliac screw technique can provide stronger purchase compared to traditional S2 pedicle screw techniques.79
A simple method of sacropelvic fixation involves the placement of long, variable-angle bone screws obliquely across the sacroiliac joint into the iliac bones. A tripod effect may be gained by combining the sacroiliac fixation with additional sacral fixation points.80
Salehi and colleagues have described a novel reconstructive technique for spinopelvic stabilization after resection of metastatic sacral tumors involving a modification of the transiliac bar.48 They used a cage and ilial bolt system for fixation of the spine to the ilium and connected rods that had been affixed to the pedicle screws with satisfactory results.
Reconstruction after Total Sacrectomy
In the case of total sacrectomy, there is no bone for sacral fixation. Iliac fixation is the only procedure to stabilize the spinopelvic junction. A number of different constructs for reconstruction following total sacrectomy have been described in the literature.6,66–70,81 Some of the early methods involved the placement of transverse sacral bars66 or Steinmann pins67 to connect the posterior iliac wings. These devices were then connected to the spinal instrumentation, which consisted of Harrington rods66 or Cotrel-Dubousset rods.67 The major disadvantage of these constructs relates to the soft bone of the posterior ilium, which does not provide firm fixation. Additionally, these methods provide poor rotational stability. Hook-and-rod systems can accidentally disengage from the transverse sacral bars or Steinmann pins.
An alternative form of reconstruction for large sacral defects is the implantation of a custom-made prosthesis81 designed to fit the individual shape of the pelvis and accommodate for the amount of resection performed. The major disadvantage is that it is impossible to make adjustments during surgery.
Galveston Technique
Allen and Ferguson from Galveston, Texas, were the first to describe a technique involving the insertion of an angled distal limb of a spinal fixation rod into the posterior iliac bones, just above the sciatic notch.82 The Galveston technique has since become the benchmark for other spinopelvic fixation systems.75
In the biomechanical testing of 10 different lumbosacral instrumentation techniques in a bovine model, McCord and colleagues75 found that the most effective construct entailed medially directed S1 pedicle screws and an iliac purchase in the Galveston-type fashion. The key to the strength of the Galveston construct is explained by the concept of the lumbosacral pivot point.75 This point is located at the intersection of the osteoligamentous column in the sagittal plane and the lumbosacral intervertebral disc in the transverse plane. It represents the axis of rotation at the lumbosacral junction. The iliac rod in the Galveston technique extends anterior to the lumbosacral pivot point, providing a long lever arm within the ilium to counteract flexion moments exerted by the lumbar spine.
Custom bending and insertion of the Galveston rod requires some technical skill to achieve the correct position with the rod remaining intracortical. Tube benders are used to create an initial sacroiliac bend of approximately 60 degrees, and then a table vise is used to stabilize the distal ilial segment of the rod while an approximately 110-degree lumbosacral bend is created.10
A technically simpler alternative to the Galveston rod is a lumbopelvic construct using iliac screws. Iliac screws are placed independently of the spinal rod, with the two subsequently linked together. Double iliac screw fixation14 involves the placement of long 7- to 8-mm-diameter threaded screws into the iliac wings and linked to the spinal rods independently. The rods may be connected with a cross-linked bar to better counteract rotational force.
Extensive bone grafting is essential to obtain a fusion. In addition to placing autogenous and allogenic corticocancellous bone extending from the transverse processes to the ilium bilaterally, we place an allogenic tibial or fibular strut graft to close the space between the two ilia and help facilitate fusion of the entire defect (Fig. 193-5).83

Postoperative Care and Complication Avoidance
The highest incidence of wound infection occurs in patients who have received preoperative radiation therapy.73 We routinely use soft tissue reconstructive techniques such as the transpelvic VRAM flap or a microvascular free flap to help reduce the risk for wound complications in patients who require high sacral resection or total sacrectomy.64
Early failure of the lumbosacral instrumentation (i.e., within 6 to 12 weeks) is usually due to screw pullout. This is most commonly caused by repetitive stress at the bone–metal interface. Early failure of the construct may be prevented by using multiple sites of fixation to best distribute the forces at the bone–implant junction. In some cases, bony fusion still occurs even in the context of instrument failure. Patients with evidence of instrumentation failure should undergo diligent radiologic follow-up. Delaying revision surgery is reasonable until it is certain that it is required to achieve an acceptable result. Reoperation is recommended if evidence of progressive deformity or painful pseudoarthrosis is observed. Revision surgery should address the cause of failure. For example, metal fatigue fracture suggests poor load sharing in the original construct, and new fixation points may need to be added. Screw pullout suggests the need for larger-diameter or longer bone screws to obtain a solid bony purchase on additional cortices. The application of methyl methacrylate may enhance screw fixation in the setting of poor-quality bone.
Allen B.L.Jr., Ferguson R.L. The Galveston technique for L rod instrumentation of the scoliotic spine. Spine. 1982;7:276-284.
Broaddus W.C., Grady M.S., Delashaw J.B.Jr., et al. Preoperative superselective arteriolar embolization: a new approach to enhance resectability of spinal tumors. Neurosurgery. 1990;27:755-759.
Carlson G.D., Abitbol J.J., Anderson D.R., et al. Screw fixation in the human sacrum. An in vitro study of the biomechanics of fixation. Spine. 1992;17(Suppl 6):196-203.
Casali P.G., Messina A., Stachiotti S., et al. Imatinib mesylate in chordoma. Cancer. 2004;101:2086-2097.
Disler D.G., Miklic D. Imaging findings in tumors of the sacrum. AJR Am J Roentgenol. 1999;173:1699-1706.
Enneking W.F. A system of staging musculoskeletal neoplasms. Clin Orthop Relat Res. 1986;204:9-24.
Feldenzer J.A., McGauley J.L., McGillicuddy J.E. Sacral and presacral tumors: problems in diagnosis and management. Neurosurgery. 1989;25:884-891.
Fourney D.R., Fuller G.N., Gokaslan Z.L. Intraspinal extradural myxopapillary ependymoma of the sacrum arising from the filum terminale externa. Case report. J Neurosurg. 2000;93(Suppl 2):322-326.
Gokaslan Z.L., Romsdahl M.M., Kroll S.S., et al. Total sacrectomy and Galveston L-rod reconstruction for malignant neoplasms. J Neurosurg. 1997;87:781-787.
Gunterberg B., Romanus B., Stener B. Pelvic strength after major amputation of the sacrum. An experimental study. Acta Orthop Scand. 1976;47:635-642.
Hasegawa K., Homma T., Hirano T., et al. Margin-free spondylectomy for extended malignant spine tumors: surgical technique and outcome of 13 cases. Spine. 2007;31:142.
Hung S.J., Chen H.C., Wei F.C. Free flaps for reconstruction of the lower back and sacral area. Microsurgery. 2000;20:72-76.
Llauger J., Palmer J., Amores S., et al. Primary tumors of the sacrum: diagnostic imaging. AJR Am J Roentgenol. 2000;174:417-424.
McCord D.H., Cunningham B.W., Shono Y., et al. Biomechanical analysis of lumbosacral fixation. Spine. 1992;17(Suppl 8):235-243.
McLoughlin G.S., Sciubba D.M., Suk I., et al. En bloc total sacrectomy performed in a single stage through a posterior approach. Neurosurgery. 2008;63:115-120.
Miles W.K., Chang D.W., Kroll S.S., et al. Reconstruction of large sacral defects following total sacrectomy. Plast Reconstr Surg. 2000;105:2387-2394.
O’Brien J.R., Yu W.D., Bhatnager R., et al. An anatomic study of the S2 iliac technique for lumbopelvic screw placement. Spine. 2009;34:439-442.
Ohata N., Ozaki T., Kunisada T., et al. Extended total sacrectomy and reconstruction for sacral tumor. Spine. 2004;29:E123-E126.
Payer M. Neurological manifestations of sacral tumors. Neurosurg Focus. 2003;15(2):E1.
Schuck A., Ahrens S., von Schorlemer I., et al. Radiotherapy in Ewing tumors of the vertebrae: treatment results and local relapse analysis of the CESS 81/86 and EICESS 92 trials. Int J Radiat Oncol Biol Phys. 2005;63:1562-1567.
Todd L.T.Jr., Yaszemski M.J., Currier B.L., et al. Bowel and bladder function after major sacral resection. Clin Orthop Relat Res. 2002;397:36-39.
Weaver J.M., Flynn M.B. Hemicorporectomy. J Surg Oncol. 2000;73:117-124.
Wuisman P., Lieshout O., Sugihara S., et al. Total sacrectomy and reconstruction: oncologic and functional outcome. Clin Orthop Relat Res. 2000;381:192-203.
York J.E., Kaczaraj A., Abi-Said D., et al. Sacral chordoma: 40-year experience at a major cancer center. Neurosurgery. 1999;44:74-80.
Zhang H.Y., Thongtrangan I., Balabhadra R.S., et al. Surgical techniques for total sacrectomy and spinopelvic reconstruction. Neurosurg Focus. 15(2), 2003. Article 5
1. Feldenzer J.A., McGauley J.L., McGillicuddy J.E. Sacral and presacral tumors: problems in diagnosis and management. Neurosurgery. 1989;25:884-891.
2. York J.E., Kaczaraj A., Abi-Said D., et al. Sacral chordoma: 40-year experience at a major cancer center. Neurosurgery. 1999;44:74-80.
3. Payer M. Neurological manifestations of sacral tumors. Neurosurg Focus. 2003;15(2):E1.
4. Disler D.G., Miklic D. Imaging findings in tumors of the sacrum. AJR Am J Roentgenol. 1999;173:1699-1706.
5. Llauger J., Palmer J., Amores S., et al. Primary tumors of the sacrum: diagnostic imaging. AJR Am J Roentgenol. 2000;174:417-424.
6. Zhang H.Y., Thongtrangan I., Balabhadra R.S., et al. Surgical techniques for total sacrectomy and spinopelvic reconstruction. Neurosurg Focus. 15(2), 2003. Article 5
7. Broaddus W.C., Grady M.S., Delashaw J.B.Jr, et al. Preoperative superselective arteriolar embolization: a new approach to enhance resectability of spinal tumors. Neurosurgery. 1990;27:755-759.
8. Lin P.P., Guzel V.B., Moura M.F., et al. Long-term follow-up of patients with giant cell tumor of the sacrum treated with selective arterial embolization. Cancer. 2002;95:1317-1325.
9. Rescorla F.J., Sawin R.S., Coran A.G., et al. Long-term outcome for infants and children with sacrococcygeal teratoma: a report from the Children’s Cancer Group. J Pediatr Surg. 1998;33:171-176.
10. De Backer A., Erpicum P., Philippe P., et al. Sacrococcygeal teratoma: results of a retrospective multicentric study in Belgium and Luxembourg. Eur J Pediatr Surg. 2001;11:182-185.
11. Chisholm C.A., Heider A.L., Kuller J.A., et al. Prenatal diagnosis and perinatal management of fetal sacrococcygeal teratoma. Am J Perinatol. 1999;16:47-50.
12. Fourney D.R., Gokaslan Z.L. Current management of sacral chordoma. Neurosurg Focus. 15(2), 2003. Article 9
13. Bergh P., Kindblom L.G., Gunterberg B., et al. Prognostic factors in chordoma of the sacrum and mobile spine: a study of 39 patients. Cancer. 2000;88:2122-2134.
14. Enneking W.F. A system of staging musculoskeletal neoplasms. Clin Orthop Relat Res. 1986;204:9-24.
15. Ishii K., Chiba K., Watanabe M., et al. Local recurrence after S2–3 sacrectomy in sacral chordoma. Report of four cases. J Neurosurg. 2002;97(Suppl 1):98-101.
16. Casali P.G., Messina A., Stachiotti S., et al. Imatinib mesylate in chordoma. Cancer. 2004;101:2086-2097.
17. Dominguez J., Lobato R.D., Ramos A., et al. Giant intrasacral schwannomas: report of six cases. Acta Neurochir (Wien). 1997;139:954-959.
18. Takeyama M., Koshino T., Nakazawa A., et al. Giant intrasacral cellular schwannoma treated with high sacral amputation. Spine. 2001;26:E216-E219.
19. Ginsberg L.E., Williams D.W., Stanton C. Intrasacral myxopapillary ependymoma. Neuroradiology. 1994;36:56-58.
20. Fourney D.R., Fuller G.N., Gokaslan Z.L. Intraspinal extradural myxopapillary ependymoma of the sacrum arising from the filum terminale externa. Case report. J Neurosurg. 2000;93(Suppl 2):322-326.
21. Marmor E., Fourney D.R., Rhines L.D., et al. Sacrococcygeal ganglioneuroma. J Spinal Disord Tech. 2002;15:265-268.
22. Boriani S., Capanna R., Donati D., et al. Osteoblastoma of the spine. Clin Orthop Relat Res. 1992;278:37-45.
23. Biagini R., Orsini U., Demitri S., et al. Osteoid osteoma and osteoblastoma of the sacrum. Orthopedics. 2001;24:1061-1064.
24. Papagelopoulos P.J., Choudhury S.N., Frassica F.J., et al. Treatment of aneurysmal bone cysts of the pelvis and sacrum. J Bone Joint Surg Am. 2001;83:1674-1681.
25. Turcotte R.E., Sim F.H., Unni K.K. Giant cell tumor of the sacrum. Clin Orthop. 1993;291:215-221.
26. Cottalorda J., Bourelle S. Modern concepts of primary aneurysmal bone cyst. Arch Orthop Trauma Surg. 2007;127:105-114.
27. Liu J.K., Brockmeyer D.L., Dailey A.T., et al. Surgical management of aneurysmal bone cysts of the spine. Neurosurg Focus. 2003;15:E4.
28. Szendroi M., Cser I., Konya A., et al. Aneurysmal bone cyst. A review of 52 primary and 16 secondary cases. Arch Orthop Trauma Surg. 1992;111:318-322.
29. Gladden M.L.Jr, Gillingham B.L., Hennrikus W., et al. Aneurysmal bone cyst of the first cervical vertebrae in a child treated with percutaneous intralesional injection of calcitonin and methylprednisolone. A case report. Spine. 2000;25:527-530.
30. Bush C.H., Drane W.E. Treatment of an aneurysmal bone cyst of the spine by radionuclide ablation. AJNR Am J Neuroradiol. 2000;21:592-594.
31. De Cristofaro R., Biagini R., Boriani S., et al. Selective arterial embolization in the treatment of aneurysmal bone cyst and angioma of bone. Skeletal Radiol. 1992;21:523-527.
32. Lucas D.R., Unni K.K., McLeod R.A., et al. Osteoblastoma: clinicopathologic study of 306 cases. Hum Pathol. 1994;25:117-134.
33. Lundeen M.A., Herring J.A. Osteoid-osteoma of the spine: sclerosis in two levels. A case report. J Bone Joint Surg Am. 1980;62:476-478.
34. Gille O., Soderlund C., Berge J., et al. Triple total cervical vertebrectomy for giant cell tumor: case report. Spine. 2005;30:E272-E275.
35. Abdelwahab I.F., Camins M.B., Hermann G., et al. Giant cell tumour of the seventh cervical vertebra. Can Assoc Radiol J. 1995;46:454-457.
36. York J., Berk R., Fuller G., et al. Chondrosarcoma of the spine:1954-1997. J Neurosurg. 1999;90:73-78.
37. Hasegawa K., Homma T., Hirano T., et al. Margin-free spondylectomy for extended malignant spine tumors: surgical technique and outcome of 13 cases. Spine. 2007;31:142.
38. Normand A.N., Ballo M.T., Yasko A.W. Palliative radiation therapy for advanced chondrosarcoma. Proc Conn Tiss Oncol Soc. 2006;12:745.
39. Ozaki T., Flege S., Liljenqvist U., et al. Osteosarcoma of the spine: experience of the Cooperative Osteosarcoma Study Group. Cancer. 2002;94:1069-1077.
40. Spiegel D.A., Richardson W.J., Scully S.P., et al. Long-term survival following total sacrectomy with reconstruction for the treatment of primary osteosarcoma of the sacrum. A case report. J Bone Joint Surg Am. 1999;81:848-855.
41. Grier H.E., Krailo M.D., Tarbell N.J., et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348:694-701.
42. Paulussen M., Ahrens S., Braun-Munzinger G., et al. EICESS 92 (European Intergroup Cooperative Ewing’s Sarcoma Study): preliminary results. Klin Padiatr. 1999;211:276-283.
43. Bernstein M., Kovar H., Paulussen M., et al. Ewing’s sarcoma family of tumors: current management. Oncologist. 2006;11:503-519.
44. Paulussen M., Craft A.W., Lewis A., et al. Results of the EICESS-92 study: two randomized trials of Ewing’s sarcoma treatment—cylophosphamide compared with ifosfamide in standard-risk patients and assessment of benefit of etoposide added to standard treatment in high-risk patients. J Clin Oncol. 2008;26:4385-4393.
45. Wagner L.M., McAllister N., Goldsby R.E., et al. Temozolomide and intravenous irinotecan for treatment of advanced Ewing sarcoma. Pediatr Blood Cancer. 2007;48:132-139.
46. Schuck A., Ahrens S., von Schorlemer I., et al. Radiotherapy in Ewing tumors of the vertebrae: treatment results and local relapse analysis of the CESS 81/86 and EICESS 92 trials. Int J Radiat Oncol Biol Phys. 2005;63:1562-1567.
47. Ozdemir M.H., Gurkan I., Yildiz Y., et al. Surgical treatment of malignant tumours of the sacrum. Eur J Surg Oncol. 1999;25:44-49.
48. Salehi S.A., McCafferty R.R., Karahalios D., et al. Neural function preservation and early mobilization after resection of metastatic sacral tumors and lumbosacropelvic junction reconstruction: report of three cases. J Neurosurg. 2002;97(Suppl 1):88-93.
49. Crowe P.J., Temple W.J., Lopez M.J., et al. Pelvic exenteration for advanced pelvic malignancy. Semin Surg Oncol. 1999;17:152-160.
50. Moffat F.L.Jr, Falk R.E. Radical surgery for extensive rectal cancer: is it worthwhile? Recent Results Cancer Res. 1998;146:71-83.
51. Gunterberg B., Norlen L., Stener B., et al. Neurological evaluation after resection of the sacrum. Invest Urol. 1975;13:183-188.
52. Biagini R., Ruggieri P., Mercuri M., et al. Neurologic deficit after resection of the sacrum. Chir Organi Mov. 1997;82:357-372.
53. Nakai S., Yoshizawa H., Kobayashi S., et al. Anorectal and bladder function after sacrifice of the sacral nerves. Spine. 2000;25:2234-2239.
54. Todd L.T.Jr, Yaszemski M.J., Currier B.L., et al. Bowel and bladder function after major sacral resection. Clin Orthop Relat Res. 2002;397:36-39.
55. Gunterberg B., Romanus B., Stener B. Pelvic strength after major amputation of the sacrum. An experimental study. Acta Orthop Scand. 1976;47:635-642.
56. Bridwell K.H. Management of tumors at the lumbosacral junction. In: Margulies J.Y., Floman Y., Farcy J.P.C., Neuwirth M.G. Lumbosacral and Spinopelvic Fixation. Philadelphia: Lippincott-Raven; 1996:109-122.
57. Localio S.A., Eng K., Ranson J.H. Abdominosacral approach for retrorectal tumors. Ann Surg. 1980;191:555-560.
58. Huth J.F., Dawson E.G., Eilber F.R. Abdominosacral resection for malignant tumors of the sacrum. Am J Surg. 1984;148:157-161.
59. McDonald J., Lane J.M. Surgical approaches to the sacroiliac joint. In: Sundaresan N., Schmidek H.H., Schiller A.L., Rosenthal D.I. Tumors of the Spine: Diagnosis and Clinical Management. Philadelphia: WB Saunders; 1996:426-431.
60. Stener B., Gunterberg B. High amputation of the sacrum for extirpation of tumors. Principles and technique. Spine. 1978;3:351-366.
61. Weaver J.M., Flynn M.B. Hemicorporectomy. J Surg Oncol. 2000;73:117-124.
62. McCarty C.S., Waugh J.M., Mayo C.W., et al. The surgical treatment of presacral tumors: a combined problem. Proc Staff Meet Mayo Clin. 1952;27:73-84.
63. Bowers R. Giant cell tumor of the sacrum. A case report. Ann Surg. 1948;1:1164-1172.
64. Miles W.K., Chang D.W., Kroll S.S., et al. Reconstruction of large sacral defects following total sacrectomy. Plast Reconstr Surg. 2000;105:2387-2394.
65. Hung S.J., Chen H.C., Wei F.C. Free flaps for reconstruction of the lower back and sacral area. Microsurgery. 2000;20:72-76.
66. Shikata J., Yamamuro T., Kotoura Y., et al. Total sacrectomy and reconstruction for primary tumors. Report of two cases. J Bone Joint Surg Am. 1988;70:122-125.
67. Santi M.D., Mitsunaga M.M., Lockett J.L. Total sacrectomy for a giant sacral schwannoma: a case report. Clin Orthop Relat Res. 1993;294:285-289.
68. Wuisman P., Lieshout O., Sugihara S., et al. Total sacrectomy and reconstruction: oncologic and functional outcome. Clin Orthop Relat Res. 2000;381:192-203.
69. Doita M., Harada T., Iguchi T., et al. Total sacrectomy and reconstruction for sacral tumors. Spine. 2003;28:E296-E301.
70. Ohata N., Ozaki T., Kunisada T., et al. Extended total sacrectomy and reconstruction for sacral tumor. Spine. 2004;29:E123-E126.
71. Karakousis C.P., Emrich L.J., Driscoll D.L. Variants of hemipelvectomy and their complications. Am J Surg. 1989;158:404-408.
72. McLoughlin G.S., Sciubba D.M., Suk I., et al. En bloc total sacrectomy performed in a single stage through a posterior approach. Neurosurgery. 2008;63:115-120.
73. Fourney D.R., Abi-Said D., Lang F.F., et al. Use of pedicle screw fixation in the management of malignant spinal disease: experience in 100 consecutive procedures. J Neurosurg. 2001;94(Suppl 1):25-37.
74. Carson W.L., Duffield R.C., Arendt M., et al. Internal forces and moments in transpedicular spine instrumentation: the effect of pedicle screw angle and transfixation-the 4R-4bar linkage concept. Spine. 1990;15:893-901.
75. McCord D.H., Cunningham B.W., Shono Y., et al. Biomechanical analysis of lumbosacral fixation. Spine. 1992;17(Suppl 8):235-243.
76. Carlson G.D., Abitbol J.J., Anderson D.R., et al. Screw fixation in the human sacrum. An in vitro study of the biomechanics of fixation. Spine. 1992;17(Suppl 6):196-203.
77. Smith D.A., Kumar R., Cahill D.W. Sacral lesions. In: Benzel E.C., editor. Spine Surgery: Techniques, Complication Avoidance, and Management. New York: Churchill Livingstone; 1999:741-758.
78. Krag M.H., Beynnon B.D., Pope M.H., et al. Depth of insertion of transpedicular vertebral screws into human vertebrae: effect upon screw-vertebra interface strength. J Spinal Disord. 1988;1:287-294.
79. O’Brien J.R., Yu W.D., Bhatnager R., et al. An anatomic study of the S2 iliac technique for lumbopelvic screw placement. Spine. 2009;34:439-442.
80. Baldwin N.G., Benzel E.C. Sacral fixation using iliac instrumentation and a variable angle screw device. J Neurosurg. 1994;81:313-316.
81. Wuisman P., Lieshout O., van Dijk M., et al. Reconstruction after total en bloc sacrectomy for osteosarcoma using a custom-made prosthesis: a technical note. Spine. 2001;26:431-439.
82. Allen B.L.Jr, Ferguson R.L. The Galveston technique for L rod instrumentation of the scoliotic spine. Spine. 1982;7:276-284.
83. Gokaslan Z.L., Romsdahl M.M., Kroll S.S., et al. Total sacrectomy and Galveston L-rod reconstruction for malignant neoplasms. J Neurosurg. 1997;87:781-787.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
Chapter 193 Surgical Resection of Sacral Tumors
Anatomy
Osseous and Ligamentous Structures
Because the lumbosacral and sacroiliac joints transmit the entire weight of the body to the hip bones and lower limbs, these joints and their supporting ligaments must be very strong. The strong dorsal ligamentous complex includes the interosseous ligaments and the dorsal sacroiliac ligaments. The very stout interosseous ligaments connect the sacral tuberosities to the overhanging bone of the iliac tuberosities and represent the single strongest ligaments binding the sacrum to the ilium. The dorsal sacroiliac ligaments are divided into deep (short) and superficial (long) parts. The deeper ligaments connect the sacral and ilial tuberosities and are composed of horizontally oriented fibers; the more superficial ligaments are oriented vertically and stretch from the posterior superior iliac spine to the tubercles of the lateral sacral crest. The caudal portions of the superficial dorsal sacroiliac ligaments blend with the sacrotuberous ligaments.
Neural Anatomy
Both the sympathetic and parasympathetic components of the autonomic nervous system have an intimate relationship with the sacrum. The sacral sympathetic trunk, continuous with the lumbar sympathetic trunk, descends against the ventral surface of the sacrum, converging in front of the coccyx to form the unpaired ganglion impar. Three or four sacral trunk ganglionic enlargements are found on each side of the midline, just medial to the ventral sacral foramina. No white rami communicantes are present in this region; however, the postsynaptic gray rami communicantes from each ganglion join the corresponding sacral or coccygeal nerves for distribution to sweat glands, blood vessels, and erector pilori muscles. In addition, the sacral sympathetic trunks provide fine branches to the superior hypogastric plexus. The superior hypogastric plexus is the caudal continuation of the periaortic sympathetic plexus; it lies on the anterior surface of the fifth lumbar vertebra and upper sacrum in the retroperitoneal tissue. Fibers of the superior hypogastric plexus diverge into right and left hypogastric nerves opposite the first sacral vertebra. The term hypogastric nerve may be a misnomer, because the structure is really a narrow plexus of fibers. The hypogastric nerves represent the principal sympathetic inputs to the inferior hypogastric plexus.
Diagnosis
Clinical Presentation
Sacral tumors are rare and difficult to diagnose at an early stage.1 The major reasons for this delay include the unique capacity of the osseous sacrum and sacral canal to allow neoplastic expansion without causing significant symptoms and the often nonspecific nature of complaints when they do arise. Tumors that originate within the sacral canal can erode or invade the walls of the sacrum and/or expand cephalad within the spinal canal. Tumor can also enter the pelvis via the ventral sacral foramina. A slow-growing, regionally expansive neoplasm can attain a large size without causing symptoms early in the course of the illness. Aggressive, rapidly growing tumors are more likely to cause mechanical instability and/or neurologic compromise earlier during the disease course.
The earliest presenting symptom in patients with sacral tumors is pain located in the lower back or sacrococcygeal region.1–3 Referred pain to the leg or buttock can occur secondary to irritation of the first sacral root or iliolumbar trunk.3 The early presentation of sacral lesions is therefore very similar to that of lumbar spondylosis. By the time a sacral lesion is diagnosed, some patients have been treated and occasionally even operated on for suspected lumbar intervertebral disc pathology.
Radiologic Evaluation
A sacral tumor can be easily overlooked on standard radiographs. The curved shape of the sacrum, its position within the pelvic girdle, and overlying bowel gas are common sources of obscuration. Destructive changes must be advanced before they become evident on plain radiographs.4 Adequate imaging should display the entire sacrum and coccyx on lateral views, and the sacrum should be visualized en face on anteroposterior views. A malignant process is suggested when lytic lesions without sharply defined borders are seen. Well-defined sclerotic margins, reflecting reactive changes in the surrounding bone, imply the presence of a benign or chronic process.
CT and MRI more readily allow the detection, characterization, and staging of sacral tumors.4,5 CT has the advantage of providing excellent bony detail and showing tumor matrix calcification. CT is also useful to image the abdomen for evidence of visceral involvement. Major advantages of MRI studies include the detailed depiction of associated soft tissue masses and the ability to assess the anatomy in multiple planes. The rostral extent of sacral involvement, which is particularly critical to surgical planning, is best appreciated on a midsagittal view.
In addition to the bone scan, CT scans of the chest, abdomen, and pelvis are warranted to rule out metastatic pathology. Intravenous pyelography and/or barium enema may be indicated in evaluating sacral tumors with significant pelvic invasion as well as for colorectal tumors that invade the sacrum.6 Angiography is useful in defining the vascularity of sacral tumors and for preoperative tumor embolization, especially in the case of highly vascular lesions such as giant cell tumor or aneurysmal bone cyst.7,8
Biopsy
The percutaneous CT-guided biopsy involves minimal risk and is currently the method of choice. The site of entry and the trajectory of the needle should be carefully selected so that they can easily be included within the margins of any subsequent resection. If possible, the puncture site should be located in the midline posteriorly. It may be worthwhile to introduce a small droplet of sterile permanent ink into the needle tract to tattoo the skin for later identification.
Pathology
Sacral tumors are categorized as those that originate from the neural elements or their supporting tissues, those arising from bone, and those that metastasize from distal sites or are the result of direct invasion from adjacent pelvic structures. The most common sacral tumors are metastatic, and the most common primary sacral tumor is chordoma.2 Some non-neoplastic entities, including developmental cysts and inflammatory conditions, mimic sacral tumors on imaging studies. A broad differential diagnosis of the various lesions encountered in this area is provided in Table 193-1.
TABLE 193-1 Differential Diagnosis of Sacral and Presacral Lesions
| Congenital Lesions |
| Posterior sacral meningocele, meningomyelocele, and lipomyelomeningocele |
| Developmental cysts |
| Dermoid and epidermoid cysts |
| Retrorectal tailgut cysts |
| Enteric duplication cysts |
| Anterior sacral meningocele |
| Lateral meningocele |
| Occult intrasacral meningocele |
| Perineural (Tarlov’s) cysts |
| Tumors |
| Primitive Neuroectodermal Tumors |
| Teratoma |
| Hamartoma |
| Chordoma |
| Inflammatory Lesions |
| Osteomyelitis |
| Abscess (pelvic abscess, perirectal abscess) |
| Neurogenic Lesions |
| Schwannoma and neurofibroma |
| Ependymoma |
| Ganglioneuroma |
| Neuroblastoma |
| Osseous Lesions |
| Bone island |
| Osteoid osteoma and osteoblastoma |
| Osteochondroma |
| Hemangioma |
| Aneurysmal bone cyst |
| Giant cell tumor |
| Chondrosarcoma |
| Osteosarcoma |
| Ewing’s sarcoma |
| Paget’s disease (monostotic) |
| Metastatic Lesions |
| Hematogenous spread (lung, breast, prostate, kidney, lymphoma) |
| Locally invasive lesions (colorectal and gynecologic malignancies, sarcoma) |
| Miscellaneous Lesions |
| Carcinoid tumor |
| Lymphoma |
| Solitary plasmacytoma, multiple myeloma |
| Meningioma |
| Hemangiopericytoma |
Congenital Tumors
Sacrococcygeal Teratoma
A teratoma is a lesion containing tissue from all three germ layers, represented by either well-differentiated or immature elements. Skin, teeth, central nervous system tissue, and respiratory and alimentary mucosa may be found within these tumors. Sacrococcygeal teratomas are the most common sacral tumor in neonates. In contrast, these tumors are rare in adults.9 They develop during intrauterine growth and can grow large enough to cause dystocia.10 The diagnosis is often made on prenatal ultrasonography.11 After birth they manifest as an exophytic mass located between the anus and coccyx, covered by normal skin. Presacral and sometimes combined pre- and postsacral (dumbbell-shaped) lesions also occur.
Chordoma
Chordoma is the most common primary bone tumor of the sacrum.12 Sacral chordomas occur almost twice as often in men compared with women and are uncommon in persons younger than 40 years. The most common presenting symptom is pain in the lower back or sciatic region.3 Chordomas can reach a very large size before constipation (from rectal compression) or lower extremity paresis (due to sacral plexus involvement) occurs.
Chordomas are considered congenital because they are thought to arise from notochordal remnants. Chordoma is characterized microscopically by the appearance of vacuolated “physaliferous” cells. The midline location of these tumors also relates to this proposed etiology.13
Chordomas are typically slow growing but locally aggressive. Significant extracompartmental growth is often seen by the time of diagnosis. Most sacral chordomas manifest as surgical stage IB with anterior extension into the pelvis.14 The tumor often displaces but does not invade the rectum, because the tough periosteum and presacral fascia resist the transgression of disease. Metastasis is usually a late event.
The usual CT appearance consists of lytic bone destruction in addition to a disproportionately large soft tissue mass. Calcification is present in 30% to 70% of cases.5 Unlike most bone tumors, chordomas can show reduced uptake or normal distribution of isotope on bone scan.
En bloc excision is the treatment of choice for sacral chordomas.2,12,13 The extent of surgical resection has been found to play a major role in determining the length of disease-free survival.13 Although a distinct capsule is often seen within the soft tissues, a radical wide posterior margin of the gluteal muscles should be employed to reduce the risk for local recurrence.15 The margins of chordoma within bone are often indistinct. Surgical resection should extend at least one sacral segment beyond the area of gross disease.2
The value of radiotherapy as primary or adjuvant treatment for chordoma has been debated.12 Supplementary radiotherapy may be a useful adjunct to surgical care, but it is not sufficient as stand-alone therapy. In a clinical series spanning 40 years, York and colleagues2 reported that the addition of radiation therapy significantly prolonged the disease-free interval for patients undergoing subtotal resection (2.12 years versus 8 months). Others have suggested that radiotherapy is of limited value in most cases.13
Currently, chemotherapy does not play a role for the treatment of chordomas, although the use of imatinib mesylate is being investigated.16
Neurogenic Tumors
Schwannoma and Neurofibroma
Sacral schwannomas are much more common than neurofibromas. They grow within the sacral canal and only rarely expand through the anterior sacral foramina into the presacral space.17,18 A complete resection of these benign tumors is potentially curative because, with the exception of plexiform neurofibromas, they do not infiltrate beyond their capsular envelope. The surgical approach depends on the size and location of the tumor, especially the degree of intraspinal and presacral extension. The majority of lesions coming to neurosurgical attention are largely confined to the sacral canal and can be resected completely using a posterior approach. This approach allows direct visualization of the relationship between the sacral nerve roots and the tumor. If there is a small presacral component, limited access to this region may be obtained by transforaminal resection. Tumors with a large presacral component should often be removed through an anterior transabdominal approach. Giant intrasacral schwannomas have been described for which radical sacral resection was performed. 17,18
Ependymoma
Sacral ependymomas are most commonly the benign myxopapillary type. They arise from ependymal cell clusters within the terminal filum and expand the sacral canal.19 Rare extradural sacrococcygeal ependymomas have been reported, including subcutaneous, presacral, and intrasacral varieties.20
Patients with sacral region ependymomas typically present with pain in either a lower back or sciatic distribution. By the time of diagnosis, which averages 2 to 3 years after the onset of symptoms, many patients exhibit some form of cauda equina syndrome.3 As with other slow-growing sacral tumors that tend to be diagnosed at an advanced stage, ependymomas may be associated with extensive bony destruction and a large soft tissue mass.19
A complete en bloc resection is advisable in order to prevent local recurrence or cerebrospinal fluid dissemination. Intradural lesions can be resected completely via a posterior approach.20 Intraoperative somatosensory-evoked potentials are useful, because dissection of tumor from the roots of the cauda equina can be difficult. The approach to extradural lesions depends on tumor location. Radiotherapy may be useful in cases of subtotal removal or recurrence.
Ganglioneuroma
Ganglioneuromas are rare slow-growing tumors composed of sympathetic ganglion cells.21 They are thought to represent the benign counterpart of malignant neuroblastomas. Ganglioneuromas can arise anywhere from the base of the skull to the pelvis. Like neuroblastomas, most ganglioneuromas arise in the abdomen, predominantly from the adrenal gland. The small percentages of pelvic tumors likely arise from sacral extensions of the sympathetic chain. As tumor expands within the pelvis, it can extend through a sacral foramen into the epidural space, causing sacral nerve root compression. The treatment is complete surgical removal.
Primary Osseous Tumors
Less than 10% of all primary bone tumors occur in the spine, with the exception of osteoblastoma, which has a 40% incidence of vertebral involvement.22 The incidence of sacral involvement among osseous tumors varies considerably. Bone tumors are a histologically diverse group of neoplasms. Some lesions are of low biological activity, such as osteoid osteoma, osteoblastoma,22,23 and aneurysmal bone cyst.24 High-grade lesions include chondrosarcoma and osteosarcoma. Although giant cell tumors are histologically benign, they are locally invasive and have a high risk for recurrence.25
Aneurysmal Bone Cysts
There is much debate over the best treatment for spinal aneurysmal bone cysts. Among the treatment modalities, complete surgical excision of the lesion remains the most effective, with the lowest rate of recurrence.26,27 Alternatively, these lesions are often treated by intralesional curettage and bone grafting. The disadvantage of this technique is that it is often associated with greater blood loss as well as high recurrence rates ranging from 20% to 60%.28,29 Selective embolization has been shown to avoid excessive bleeding when used with surgery and has also proved to be an effective sole treatment in cases that are difficult to treat surgically. Several less-invasive methods have been studied for the treatment of aneurysmal bone cysts including particulate embolization, intralesional injection of alcoholic zein, radiotherapy alone,30 and radiotherapy combined with surgery.31 Radiotherapy can be used as an alternative or adjunct to surgery when aneurysmal bone cysts are located in sites where an adequate resection cannot be done without resulting in significant morbidity or poor cosmetic results.
Osteoid Osteoma and Osteoblastoma
Osteoid osteomas and osteoblastomas are bone-producing lesions that often occur in long bones but may also be found in the spine. Although these tumors have a predilection for posterior elements of the mobile spine, they tend to involve the body of the sacrum.22 These lesions are similar histologically to each other, containing osteoblasts that produce osteoid and woven bone. Osteoid osteomas are small, self-limited, and benign, whereas osteoblastomas are often larger, are more aggressive, and can become malignant.23
Osteoid osteomas and osteoblastomas are common causes of painful scoliosis in children and adolescents. The most common presenting symptom is neck or back pain that often responds to aspirin or nonsteroidal anti-inflammatory drugs (NSAIDs). Chemotherapy has played a minimal role in the treatment of these lesions and has only been used in selected patients with recurrent aggressive lesions or in patients with surgically inaccessible lesions. Surgery becomes the primary treatment for osteoid osteoma and osteoblastoma as the pain becomes more severe and less responsive to medication. Complete resection is often curative for these lesions. Incomplete resections are more common in osteoblastomas than in osteoid osteomas because osteoblastomas are larger and more commonly involve extraosseous tissue. Osteoblastomas recur in 10% to 15% of cases,32 and the recurrence rate of osteoid osteomas is 4.5%.33
Giant Cell Tumor
Giant cell tumors of the spinal column have a predilection for the thoracolumbar and sacral regions. It is the second most common primary sacral tumor after chordoma. Patients with spinal involvement usually present in the third and forth decades of life. Giant cell tumors are believed to arise from mononuclear cells of macrophage origin. Histologically, they are multinucleated giant cells and macrophages that can contain areas resembling aneurismal bone cysts, and care should be taken when interpreting bone biopsy samples to avoid an erroneous diagnosis. Although they are histologically benign, approximately 5% to 10% of giant cell tumors undergo malignant degeneration and assume a more aggressive course.34
Clinical management of giant cell tumors is difficult given their high propensity for recurrence. En bloc resection with wide margins is the gold standard for treating these lesions.35 Unfortunately, these tumors often grow to a large size before initial diagnosis, and extension of the tumor into the spinal canal and adjacent soft tissues is not uncommon. Therefore, subtotal resection in concert with adjuvant chemotherapy, arterial embolization, and/or radiosurgery is the typical treatment when en bloc resection is not feasible.
Arterial embolization is a critical adjuvant therapy that can increase the safety of surgery as well as improve progression-free survival. Preoperative embolization is a useful tool that minimizes intraoperative blood loss during the resection of these highly vascularized tumors. Embolization may be especially useful to limit bleeding during intralesional resections. Interestingly, arterial embolization may also be useful as a primary treatment modality.8
Chondroma and Chondrosarcoma
Complete surgical resection with negative margins is the primary treatment for chondromas. Less than 10% of patients experience tumor recurrence after complete resection.36 If incomplete resection is performed, residual tumor is at risk for sarcomatous degeneration. Because chondromas are radioresistant lesions, radiosurgery currently does not have a significant role in the treatment of these tumors.
Chondrosarcomas are slow-growing tumors that commonly manifest with focal pain or neurologic deficits. Although chondrosarcomas can be divided into multiple pathologic subgroups, the most important characteristic with respect to clinical outcome is the World Health Organization (WHO) grade. Grade I chondrosarcomas have a 90% 10-year survival rate compared to 30% to 40% for high-grade lesions.37
Regardless of WHO tumor grade, the gold standard for treating chondrosarcomas is gross total resection with negative margins. The literature suggests that en bloc resection is associated with long-term recurrence-free survival. En bloc resection can result in recurrence rates of 20% or less.38 In cases of tumor recurrence, repeat resection can lead to improved survival. There is no well-defined role for radiation or chemotherapy for these lesions.
