Chapter 133 Surgical Management of Severe Closed Head Injury in Adults
Additional text and figurees are available on Expert Consult
Traumatic brain injury (TBI) is defined as damage to the brain resulting from external mechanical forces. Head injury is the term used to define any alteration in mental or physical functioning related to a blow to the head. All TBIs are head injuries, but the latter is a broader term because it also refers to damage to structures other than the brain, such as the scalp and skull. However, the terms head injury and brain injury are often used interchangeably. The most widely used method for the assessment and stratification of TBI severity is the Glasgow coma scale (GCS), which categorizes patients according to the degree of impairment of consciousness, estimated from their verbal, motor, and eye-opening responses to external stimuli.1 Following this classification system, a severe head injury is defined by an initial postresuscitation GCS score between 3 and 8 points or by deterioration to a GCS score of 8 or fewer points after injury.2
Severe TBI represents a serious medical and scientific challenge. One of the central concepts that emerged from research is that all cerebral damage from TBI does not occur at the moment of the impact but rather evolves over the ensuing hours and days. Focused protocol-driven management, including rapid resuscitation and early intubation in the field, direct transport to a major trauma center, and improved critical care management in the hospital with intracranial pressure (ICP) monitoring and immediate evacuation of mass lesions, has cut mortality in severe TBI from as much as 50% in the 1970s to 15% to 25% in the 2000s. However, this tremendous advance seems to have plateaued in the last decade despite extensive experimental and clinical research about the cellular and molecular mechanisms involved in the progression of traumatic brain damage. Interventions using drugs to reduce the effects of mediators of injury have been encouraging in animal models, but clinical TBI trials have been ubiquitously unsuccessful so far. It is naïve to expect that severely head-injured patients, each showing a huge combination of different neuropathologic post-traumatic lesions at different stages of the disease, can all benefit from a single kind of treatment.3
TBI treatment is a team effort integrated by surgeons and nonsurgeons. The role played by neurosurgeons in the integral treatment of severely head-injured patients remains critical, especially regarding the appropriate therapeutic decisions to be made in advance to the progression of secondary brain damage. For a long time, the surgical management of TBI has been based on clinical anecdotal practice. The new paradigm of evidence-based medicine has broadened to the establishment of scientific surgical and medical guidelines, which should help neurosurgeons make the better decisions using a strong scientific background, in addition to their personal anecdotal experience.4,5
This chapter focuses on the physiopathology and surgical treatment of severe TBI. Besides providing the latest update on the evidence-based medical literature, a major objective of this chapter is to present a comprehensive review of the fundamental structural and functional properties of brain tissue, which is essential to understand the mechanisms underlying traumatic brain damage. The expanding advance of neurosciences in the last decades has produced outstanding insights in the intrinsic, dynamic mechanisms of cell damage set in motion with a head trauma. We firmly believe that succinct knowledge of these basic mechanisms can help neurosurgeons involved in the care of severely head-injured patients with better assessment of the complex clinical and diagnostic information obtained from each case. Furthermore, this knowledge represents the keystone for identifying potential future therapeutic targets for TBI. In the online version of this chapter, a section focusing on the neuroscience of TBI is provided.
Epidemiology
TBI represents a serious health problem and constitutes a major cause of death and severe disability in young and adult people in Western countries. TBI afflicts 1.4 million people yearly in the United States, resulting in 50,000 deaths and 235,000 hospitalizations.6 TBI is the number one cause of coma and the leading cause of brain damage in children and young adults. The incidence (number of new cases) of head injury is approximately 300 per 100,000 people per year (0.3% of the population), with a mortality of 25 per 100,000 people in the United States. Approximately 500,000 head injuries requiring admission to a hospital occur annually in the United States, and 50,000 people with these injuries die before reaching a medical facility. Of the 450,000 initial survivors, 80% sustain minor injuries (GCS score 13-15 points), 10% have moderate injuries (GCS score 9-12 points), and 10% have severe brain injuries (GCS score 3-8 points).6 Around 100,000 patients per year may require surgical management of a post-traumatic intracranial hematoma in the United States alone.4 Roughly 50,000 head-injured patients per year will have some form of permanent disability.7
Classification Schemes for TBI
The heterogeneity of TBI is considered one of the most significant barriers to finding effective therapeutic interventions. In addition, the lack of a valid classification system for TBI that is able to identify subgroups of patients who may survive with a reasonable neurologic outcome and thus may benefit from specific therapies contributes to this therapeutic failure. A great heterogeneity of pathoanatomic lesions can be observed in neuroradiologic studies of patients classified under the same “severe TBI” category; thus, it is difficult to conceive a general therapy able to target such variability of traumatic lesions.8 To review the most common types and patterns of injury associated with TBI and develop a classification scheme that would cluster TBI patients into groups based on their major types of injury, the National Institute of Neurological Disorders and Stroke sponsored a workshop on classification of TBI for targeted therapies in October 2007.2 On the premise that TBIs with similar pathoanatomic features are likely to share common physiopathologic mechanisms, a major conclusion from the workshop was that a new, multidimensional classification system for TBI should be developed and used in future TBI trials.
TBI Classification Systems for Targeted Therapies
Different classification systems for TBI have been used depending on the categories of the data chosen for the research. Head injuries have more often been distinguished using symptom classifications, which differentiate the patients according to the severity of their symptoms. However, pathoanatomic classifications could be more useful because they provide an accurate description of the lesions that should be treated. Finally, prognostic classifications describe the combination of factors associated with the patient’s outcome. Major disadvantages of classification systems are that the optimal time to classify patients is unknown and depends on the purpose of classification. Furthermore, personnel administering classification systems need to be trained to classify consistently and correctly. In general, the complexity of the physiologic derangement is the most important factor to take into account for triage, whereas the type of injury is of predominant importance when classifying patients regarding the selection of treatment. However, outcome-based systems of classification depend less on acute physiologic changes and more on functional capacity and permanent disability.2
Classification Schemes by Injury Severity
The most commonly used severity scale for TBI is the GCS, originally designed by Teasdale and Jennett to describe unconsciousness after brain injury (Table 133-1A).1 This scale includes assessment of eye opening, best verbal response, and best motor response. The total score, ranging from 3 to 15 points, should be ascertained after resuscitation, because the neurologic examination can be markedly altered by hypotension or hypoxia. The GCS is the most commonly used neurologic injury severity scale for adults because of its high interobserver reliability and generally good capability of predicting the outcome. However, the main disadvantages of the GCS system are that summation of assessment scores results in loss of information, because multiple combinations of eye, motor, and verbal scores can equal the same total score, and that pupillary reaction and lateralization of the exam are not included. Furthermore, the reliability of this score is questionable in the setting of sedative and paralytics use and the optimal timing of measurement is unclear, with a change in GCS score being more relevant than a single measure. TBI patients are typically divided into three subgroups based on the total score: severe TBI (GCS score 3-8 points), moderate TBI (GCS score 9-12 points), and mild TBI (GCS score 13-15 points). Most severely head-injured patients are in a coma, which is defined by the International Coma Data Bank as the inability to obey commands, utter words, or open eyes.9

Classification Schemes by Pathoanatomic Findings
Schemes of classification by pathoanatomic findings describe the location and type of abnormalities to be targeted by treatment. The majority of severely head-injured patients have more than one type of injury when classified in this way, such as scalp lacerations, skull fractures, intracranial hematomas, traumatic subarachnoid hemorrhage (SAH), intraventricular hemorrhage, cerebral contusions, and diffuse patterns of injury such as diffuse axonal injury (DAI), diffuse brain swelling, or brain ischemia. Each of these entities can be further described by their location, extent, multiplicity, and so on. Gennarelli and Thibault were among the first authors to incorporate both GCS and computed tomography (CT) findings to the evaluation of the outcome after TBI, differentiating patients on the basis of the type of lesion showed by CT (focal or diffuse), their GCS score, and the length of their coma.10 They found that subdural hematomas (SDHs) and diffuse injuries associated with a coma for more than 24 hours had the highest mortality. Lobato et al., after a thorough evaluation of CT lesions displayed by patients with severe TBI, also concluded that SDHs, in addition to multiple brain contusions, were associated with the highest mortality.11
In 1991, Marshall et al. described a CT classification system based on information gathered from severe TBI patients in the Traumatic Coma Data Bank (TCDB).7 This scheme is based on the appearance of perimesencephalic cisterns, the presence of midline shift, and the presence of focal masses, and it classifies CT findings into six groups (Table 133-1B). Among the different pathoanatomic classification schemes proposed, this has been shown to be the most successful in predicting both risk of increased ICP and outcome in adults. Diffuse injury type IV was associated with the highest mortality. Maas et al. evaluated Marshall et al.’s classification system in comparison with the use of individual CT findings for prognosis after TBI and observed some discrepancies in classification, with up to 30% of patients classified as having diffuse injury type IV being reported without a midline shift greater than 5 mm (the basic criterion of diffuse injury type IV).12
Table 133-1B Classification by Pathoanatomic Findings (Marshall’s Computed Tomography Classification7)
| Type | Radiologic Findings on CT |
|---|---|
| Diffuse type I (no visible pathology) | No visible intracranial pathology seen on CT scan. |
| Diffuse type II (no swelling and no shift) | Cisterns present with midline shift < 5 mm and/or lesion densities present. No high- or mixed-density lesion > 25 ml. May include bone fragments and foreign bodies. |
| Diffuse type III (swelling) | Cisterns compressed or absent with midline shift = 0-5 mm. No high- or mixed-density lesion > 25 ml. |
| Diffuse type IV (shift) | Midline shift > 5 mm. No high- or mixed-density lesion > 25 ml. |
| Type V (evacuated) | Any lesion surgically evacuated. |
| Type VI (nonevacuated) | High- or mixed-density lesion > 25 ml not surgically evacuated. |
Classification Schemes by Biomechanical Mechanism
Injuries can be classified according to whether the head is struck or strikes an object (contact or impact loading) and/or the brain moves within the skull (noncontact or “inertial” loading). The magnitude and direction of the combined loading forces may predict the type and severity of injury.10 There is considerable but not perfect correlation between physical mechanism of injury and pathoanatomic injury type. For instance, most focal injuries, such as skull fractures, brain contusions, and epidural hematomas (EDHs), result from impact loading, whereas inertial loading generally causes more diffuse injuries, such as concussion, SDHs, and DAI. However, in clinical practice, mechanistic classification is difficult to apply because of the uncertainty about the loading conditions involved in the TBI.
Classification Schemes by Pathophysiology
Pathophysiology classification schemes include biochemical, metabolic, neurophysiologic, and/or genetic dynamic processes set in motion by the injury. One widely used physiopathologic scheme in TBI was introduced by Adams et al., who differentiated between primary injury, or unavoidable, immediate-impact parenchymal damage, and secondary injury, or potentially avoidable damage.13 However, the importance given to secondary insults has gained recognition, and since the 1990s, understanding and prevention of delayed damage have received priority. Secondary lesions are defined as those set in motion by the impact but not pathologically or clinically manifested until minutes, hours, or days after the injury. In addition to secondary ischemic injury, probably the most relevant factor influencing a worse outcome, secondary damage can be caused by neurotransmitter dysfunction, cellular and extracellular edema, blood–brain barrier (BBB) disruption, mitochondrial damage, genetic alterations such as apoptotic induction, and so on.14
Classification Schemes by Prognostic Modeling
Outcome after TBI is multifactorial and best assessed with a multidisciplinary approach. Outcome mainly depends on severity of injury, comorbid conditions, preinjury status, social factors, and patient age. The ideal TBI classification system should be able to select patients who potentially could benefit from a considered therapy from a clinical, pathoanatomic, physiopathologic, or prognostic perspective. Traditionally, the Glasgow outcome scale (GOS) has been used to categorize the long-term prognosis of patients after TBI, and the GOS score has been dichotomized into 3 to 5 points versus 1 to 2 points (Table 133-1C). Although the 5-point GOS score has been criticized for lacking sensitivity and for having a ceiling effect, this is still the most widely used scale for classifying outcome after TBI. Most TBI patients fall into the extremes of the scale, good recovery or death, and the scheme has undergone several revisions and was extended to an 8-point scale to address some of these limitations. Both the GOS and the extended GOS have been extensively used and have been shown to have validity and reliability in measurement of outcome by trained staff. Both scales are also correlated with other measurements of disability, mental status, and neurobehavioral functioning. Recent work from the International Mission for Prognosis and Clinical Trial has resulted in the development of three valid prognostic models of increasing complexity, a task particularly relevant to identifying subgroups of patients likely to benefit from a specific therapy in phase III trials.8
Table 133-1C Classification by Prognostic Modeling (Glasgow Outcome Scale)
| Score | Features |
|---|---|
| 1 | Dead |
| 2 | Vegetative state (unable to interact with environment, unresponsive) |
| 3 | Severely disabled (able to follow commands, unable to live independently) |
| 4 | Moderately disabled (able to live independently, unable to return to work/social activities) |
| 5 | Good recovery (patient has resumed most normal activities) |
Biomechanics of TBI
TBI is generally the result not of a direct impact to the skull but of inertial linear and rotational acceleration/deceleration (A/D) forces acting on brain tissue and causing structural damage and functional disturbances on the different types of brain cells. This fact was discovered early in the history of TBI research by Denny-Brown and Russell, who observed that the loss of consciousness (LOC) associated with concussion was difficult to inflict when the movement of head during the application of the impact was restricted.15 Nervous tissue strains induced by both linear and rotational forces create spatiotemporal deformation gradients (Fig. 133-1). Because the brain is a viscoelastic organ with little internal structural support, it poorly tolerates such forces. Gray matter closest to the surface of the brain is most susceptible to linear A/D forces. In contrast, the deeper cerebral white matter axons and gray matter of deep nuclei, in addition to the axonal tracts within the brain stem, are more vulnerable to rotational A/D forces. In the early 1940s, the effects of A/D forces on the brain were assessed on a physical gelatin model resembling gross brain’s physical properties, such as its volume, mass, and deformability.16 These studies were the first to describe how shear strains were caused by rotational A/D forces (also known as impulsive loadings) (Fig. 133-2). One decade later, the effects of A/D forces on brain tissue were analyzed by exhaustive postmortem neuropathologic studies performed on the brains of patients who had died from lethal road accidents.17 These studies showed that the predominant microscopic finding was diffuse degeneration of white matter characterized by the development of axoplasmic “retraction balls” at the sites of axonal discontinuity, without obvious damage to the cortex. The term that Strich used to define this damage was “shearing injury,” because he thought that traumatic damage to white matter was caused by the shearing of nerve fibers at the moment of injury.17 Clinicopathological studies undertaken by Adams et al.13 endorsed Strich’s views, and Jennet and Plum considered this to be the most common cause of the vegetative state (VS) observed in many severely head-injured patients.9 In patients with shorter survival times (less than 6 weeks following the injury), the specific microscopic pattern of axonal degeneration consisted of the presence of retraction balls, interpreted as end swellings of proximal and distal segments of severed axons, which axoplasm had extruded.18,19
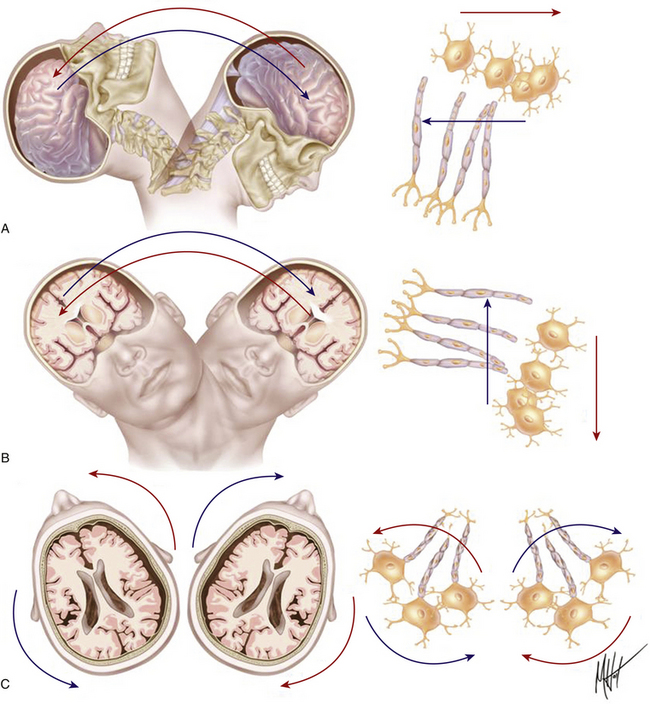
FIGURE 133-1 Biomechanics of traumatic head injury I. Classic model of axonal shear injury caused by rotational A/D forces. Impulsive inertial loadings cause damage to axons grouped in bundles within white matter without direct impact to the skull. The severity of axonal injury depends on the course followed by the fibers and the predominant spatial plane of action of the rotational A/D force: sagittal (A), lateral/coronal (B), or axial (C). Different tissue densities of gray and white matter determine differences in the inertial displacement experienced by both tissues, causing maximal stress on a definite segment of the axons. Rotational A/D forces in the lateral plane cause the severest damage to the long axonal tracts coursing along the brain stem.18,20
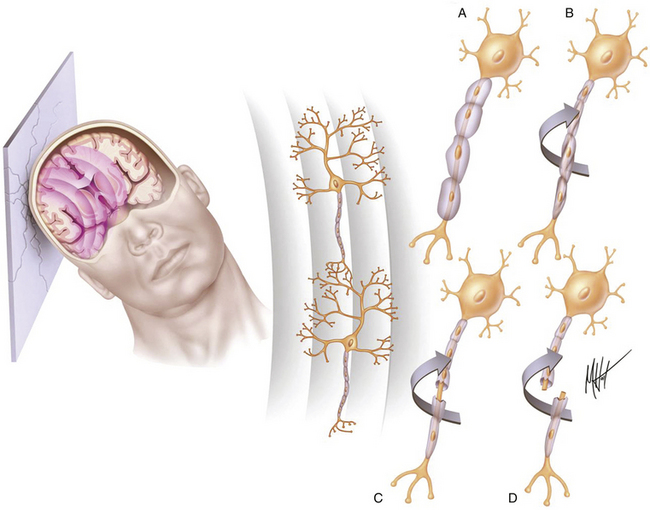
FIGURE 133-2 Biomechanics of traumatic head injury II. Classic model of spatiotemporal deformation gradient caused by A/D forces. Impact loadings inflicted on the freely moving head initiate a deformation wave that travels in the viscoelastic brain tissue, creating a spatiotemporal gradient.15,16 The major structural lesion is characterized by a stretching injury of the axolemma of scattered neurons, which cause development of multifocal swellings (A) and blocking of normal axonal transport (B), but the maximal degree of deformation may cause direct tearing (C) or breaking (D) of axons.35
Diffuse brain injury usually depends on inertial loadings as a result of rapid head rotational motions of the brain, resulting from unrestricted head movement in the instant of injury.20 Rapid A/D head movements deform white matter, inducing dynamic shear and tensile and compressive strains within the tissue (Figs. 133-1 and 133-2). A significant theoretical insight regarding the biomechanical effects of A/D forces on brain structure was made by Ommaya and Gennarelli in 1974.21 These authors proposed a “centripetal model” of severity of structural damage known as the Ommaya-Gennarelli model of TBI, which was based on experimental studies performed in subhuman primates.20,21 This probably represents the most influential biomechanical model in the field of head injury, because it explains the damage caused by A/D forces, which are most common in traffic accidents. According to this model, the mechanical strain injury operates in the brain tissue in a “centripetal sequence,” in which the higher the magnitude of the injuring forces, the deeper the brain functional and structural damage observed. The sequence of damage begins at the surface of the brain and progressively affects deeper structures as the A/D forces increase in intensity. This model uses the principle that the severity of injury is mainly determined by the direction of force, with rotational forces causing the severest injuries. On the basis of the centripetal progression of injuries, the model introduced three critical predictions: (1) when the degree of trauma is sufficient to cause LOC, cortical and subcortical structures are primarily affected and show more severe damage than that found in the rostral brain stem; (2) damage to the rostral brain stem does not occur without severer damage of cortical and subcortical structures, since the mesencephalon is the deepest structure and the last one to suffer the effects of trauma; and (3) cognitive symptoms such as confusion and disturbance of memory can occur after TBI without LOC; however, the reverse cannot occur.
The Ommaya-Gennarelli centripetal model challenged the sustained view that the main mechanism producing traumatic unconsciousness after TBI was an isolated brain stem injury. The primary brain stem damage theory was supported in the 1950s and 1960s, when the ascending arousal system (AAS) was described by Moruzzi and Magoun.22 Experimental models of TBI on subhuman primates in which the head of an animal was exposed to A/D forces using accelerator devices showed that nonimpact A/D caused concussion and axonal injury only when the moving head was allowed to rotate, while linear accelerations induced gross structural damage but not DAI.20,21 These findings were later confirmed in clinical studies, in which A/D loadings to the head in the coronal plane were observed to cause the severest injuries in humans subjected to motor vehicle accidents. Although early studies suggested that the primary site of brain injury in patients presenting with transient LOC and post-traumatic cognitive deficits was the brain stem, overwhelming neuropathologic evidence showed that most mild and moderate TBI caused damage to widespread areas of the cortex and the subcortical white matter.13,23 Recent magnetic resonance imaging (MRI) studies, in which the assessment of DAI was performed during the first month after injury, confirmed the Ommaya-Gennarelli model.24
Biomechanical theories involving linear and rotational A/D loadings are considered by many authors to be incomplete and may not adequately explain injury to deeper cerebral structures in the absence of injury to more superficial cerebral structures. Early biomechanical work suggested that brain injuries occurred principally from pressure gradients developed within the brain at the time of the impact (Fig. 133-2), but more recent research has shown that head motions occurring at the moment of injury result not only in pressure waves but also in intracranial brain deformations, which are thought to be a primary cause of the post-traumatic brain lesions. The stereotactic theory considers brain tissue to be a spherical, deformable structure composed of concentric planes with the same density in which skull vibrations generate secondary pressure waves that propagate as a spherical wave front. The spherical nature of this wave front is able to focus its energy on deeper cerebral structures without an associated injury at the brain surface.
Historical Studies on TBI: Pioneering Research By Ramón y Cajal
The Nobel Prize winner Ramón y Cajal (1852-1934) was a distinguished pioneer in the scientific study of the histologic consequences of traumatic injuries on brain tissue (Fig. 133-3A).25 He performed experimental studies on the brains of dogs and cats after severing the brain tissue with a scalpel and then studied the effect of the injury at different times up to 1 month following the injury. Ramón y Cajal described for first time the phenomenon of preservation after traumatic injury, which consists of the necrosis of cell elements distal to the lesion while these elements display perfect morphologic integrity. He also described for the first time the terminal clubs and hypertrophic stumps developing at the proximal endings of the surviving axons (retraction balls) and at the distal stumps of the preserved detached axonal segments (Fig. 133-3C).26 Retraction balls were evident in the damaged tissue 24 hours after injury, and some thin axonal appendices were seen expanding out from each ball in the direction previously followed by the axon. In addition, 6 to 8 hours after injury, he observed varicosities along the fibers proximally to the retraction balls. Retraction balls and varicosities were observable during approximately the first week after the injury and then underwent a process of degeneration. Later, and nearer the cell bodies, new buds formed (late retraction balls). These persisted for 2 to 3 weeks and then disappeared, with some perhaps entering into the formation of the last living axon collateral, which became markedly hypertrophied.25
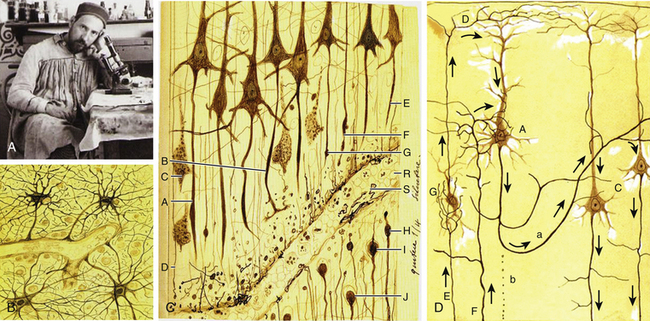
Therefore, the cortical pyramidal cells, which are those emitting long axons, were transformed after the traumatic injury into cells with short axons and hypertrophied collaterals (Fig. 133-3D). Ramón y Cajal proposed that the collateral hypertrophy is a compensatory phenomenon similar to the effects of an arterial ligature. Nevertheless, he did not know the fate reserved for the pyramidal cells with short axons. Months after the injury, he could not observe retraction balls or varicose fibers near the lesion; rather, he saw only smooth well-stained axons presenting all the signs of being normal conductors. He also observed that central tracts were incapable of regeneration and that although they had the property of producing new fibers, once the reconstructive movement is initiated, for unknown reasons the nerve sprouts lose their energy and suspend their growth. Nevertheless, he described neoformations (sprouting) in the pyramidal axons that were maximal in the second or third day after injury. However, in a few days these neoformative axon structures disappeared, and after 15 days, they were absent. In addition, he described the morphologic reactive changes in glial cells after the injury, consisting of the loss of capillary–neuroglial relationships (dislocation of perivascular astrocytes) and free migration of these cells to distant regions.25
Neuropathology of TBI
In 1990, Adams summarized the neuropathologic postmortem findings obtained from one of the largest series of TBI patients providing a full postmortem examination. These series were part of the Glasgow database, which consisted of 635 fatal closed head injuries gathered between 1968 and 1982 in Glasgow, including 434 patients with a detailed neuropathologic study.18 One third of these patients had “talked” at some time after the injury.27 The first major conclusion from this work was that severe brain damage can be caused without anything striking the head or the head striking anything. The second conclusion was that DAI represents the most important factor influencing the outcome in anyone who sustains a nonmissile head injury followed by secondary brain ischemia, which represented the second prognostic factor in importance after TBI.
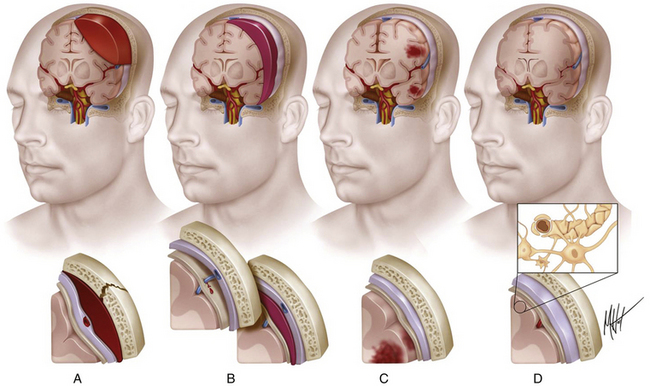
According to the pathologic classification introduced by Gennarelli in 1987,28 two major types of post-traumatic lesions could be differentiated in Adam’s series18: focal lesions, which are large enough to be identified by the naked eye, including cerebral contusions and intracranial hematomas, and diffuse injuries, which are all that cannot be macroscopically identified, including DAI, diffuse brain swelling, secondary hypoxic/ischemic brain damage, and diffuse vascular injury (Figs. 133-4 and 133-5). Overall, cerebral lesions caused by TBI can be viewed as a spectrum of injuries ranging from purely focal to purely diffuse, all brought about by A/D forces causing mechanical distortion to the head and nervous tissue. Focal brain injuries are usually produced when the stationary skull is struck by a moving object with relatively small mass, such as a stick or baseball bat. Focal brain injuries are characterized by a central area of structural disruption, surrounded by an area of primary traumatic damage without destruction of brain tissue, and finally enclosed by a peripheral tertiary zone of potential delayed insult associated with ischemia and edema.
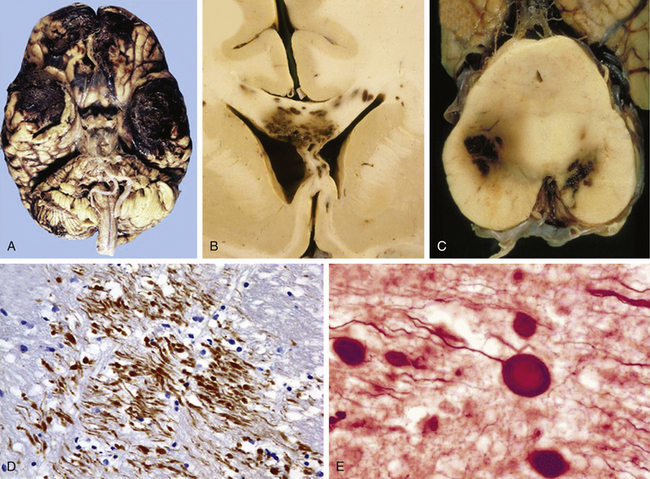
The importance of diffuse brain lesions caused by TBI was first emphasized by Symmonds as the cause of initial LOC and the frequent, long, continued disturbances of consciousness, often followed by residual symptoms in patients who were rendered unconscious at the moment of injury.14a Later experimental work by Denny-Brown and Russell15 and more recent work by Gennarelli et al.20 linked this type of damage to the prolonged unconsciousness occurring in almost half of severely head-injured patients in the absence of intracranial mass lesion. Diffuse injury is difficult to define during life, because much of it can only be recognized microscopically. Even in postmortem studies, it can hardly be recognized unless the brain is properly fixed prior to dissection. Nevertheless, this type of injury should be suspected in patients in a coma who do not present focal lesions.
Intracranial Hemorrhages
Intracranial hemorrhagic lesions represent the most common cause of clinical deterioration and death in patients who experience a lucid interval and usually develop in patients sustaining a skull fracture.27 If the lucid interval is longer than 24 hours, the hematoma is said to be “delayed.” This occurs in 30% of head-injured patients with intracranial bleeding due to the development of a new hematoma after an initial normal CT scan. The three most important intracranial hemorrhagic lesions caused by TBI are EDHs, acute SDHs, and ICHs (Fig. 133-4A-C). EDHs corresponded to the 8% of lesions observed in the Glasgow database, whereas “pure” SDHs were diagnosed in 13% and ICHs in 15% of lesions. Nevertheless, a higher rate of 26% of patients presenting with SDHs associated with ICHs was observed in the context of a “burst lobe.” This term describes a massive lobar lesion showing the coexistence of cerebral contusions, blood in the subdural space, and a hematoma in white matter deep to contusions (Fig. 133-5A). In a study performed in 1987 comparing 100 fatal head injuries with cerebral hematoma and 100 without cerebral hematoma, the cases with hematoma were observed to affect predominantly older patients.29 Structural factors such as parenchymal atrophy and weaker holding of intracerebral small-caliber vessels may probably account for this observation. Other less frequent types of focal brain injuries are traumatic lesions of the corpus callosum, usually associated with DAI; pontomedullary rents or tears in the brain stem at the junction between the pons and the medulla, often associated with a ring fracture of the base of the skull; lesions of the hypothalamus and the pituitary stalk; and lesions of the cranial nerves, with loss of sense of smell sometimes occurring after head injury because of avulsion of the olfactory nerves. For a detailed description of intracranial hematomas, refer to the section on surgical treatment of TBI.
Cerebral Contusions: Major Gray Matter Focal Lesions After TBI
Brain contusions represent frank disruptions of brain tissue, which are typically observed at the apex of cerebral gyri, appearing as coalescent petechial hemorrhages or streaks of hemorrhage with an eventual progression of bleeding into adjacent white matter (Figs. 133-4C and 133-5A).30 Brain contusions and lacerations are considered primary TBI lesions that are observed at the poles and orbital surface of the frontal lobes in up to 80% of autopsy studies performed on patients dying after the trauma (Fig. 133-5A). These are the brain areas undergoing the strongest inertial impact against the rough and irregular prominences of the inner surface of the skull following abrupt A/D forces. Contusions are basically impact-related lesions occurring with short-duration and high-acceleration impacts.20 Both brain contusions and lacerations are always accompanied by a certain degree of edema and by SAH, while intraventricular hemorrhage accompanies DAI in most cases. Cerebral lacerations represent destructive post-traumatic lesions in which disruption of cerebral tissue necessarily occurs, usually involving both the cortex and the subcortical area. Direct lacerations are caused by penetrating craniocerebral wounds, but indirect lacerations resulting from severe A/D shearing forces can be also observed.
Comprehensive studies performed by Lindenberg and Freytag in the 1950s34 and by Oprescu in the 1960s30 identified some specific types of contusions in the brains of head-injured patients dying a short time after the injury, such as contusions beneath skull fractures, coup contusions at the site of impacts (Fig. 133-4C), contrecoup contusions in regions distant to but not always opposite the impact site31 (Fig. 133-5A), herniation contusions, and finally, gliding contusions,32 which correspond to bilateral, focal, post-traumatic hemorrhages in the cortex and subcortical white matter of the superomedial parasagittal margins of cerebral hemispheres.30 Gliding contusions are thought to be caused by rotational A/D forces and are usually associated with DAI and small post-traumatic hematomas in the basal ganglia.
The complex dynamic sequence of the damage in cerebral contusions is not well understood. Early studies by Scheinker suggested that the cerebral vessels are primarily injured by the TBI and the parenchymatous tissue develop a progressive secondary damage.33 An initial vasodilatation followed by precapillary paralysis could be the triggering disturbance, causing endothelial leaking with blood extravasation that may lead to the development of true ICHs. In addition, the development of vascular thrombosis produces areas of necrosis that may become hemorrhagic, with a combination of necrotic and hemorrhagic brain tissue.30 The classic study of Lindenberg and Freytag identified the presence of two components in cerebral contusions: (1) the central area (core), in which cells undergo necrosis as the primary consequence of mechanical vascular and parenchymal injury, and (2) the peripheral area (rim), in which cellular swelling is observed.34 In contusive areas, neuronal bodies show vacuolar degeneration with chromatolysis and their axons show a varicose degeneration, a type of lesion originally described by a Ramón y Cajal in 1928 and later reported by Strich.13,17,25 Glial alterations are constantly seen with hypertrophy, swelling of astrocytes, and the development of reactive hypertrophic microgliosis or “microglial stars.” Up to 14 days after TBI, axonal retraction balls generally develop, rapidly followed by microglial and astrocytic reaction. From then on, a progressive wallerian degeneration of axons is clearly visualized.35
Physiopathology of Brain Contusions: The Osmotic Model of Progression
A rapid, massive type of cerebral edema associated with contusions may develop progressively within 12 to 72 hours after trauma, determining a marked high ICP and the shift of brain tissue, which may result in a delayed neurologic deterioration known as the “talk and deteriorate” syndrome.36,37 In addition, a “benign” slow form of delayed pericontusional edema, which rarely causes ICP elevation, is typically seen on T2-weighted MRI in white matter several days after injury.38 Contrary to the widely sustained view that increased cerebrovascular permeability is responsible for the development of contusion edema, some immunohistochemical and MRI studies have observed that vasogenic edema does not develop in contusions until 48 hours after injury, predominantly in white matter surrounding the lesion.39 Therefore, another mechanism should account for the cases in which rapid and massive contusion-associated cerebral edema develops.
In the central area of contusion, the cellular elements uniformly undergo mechanical disintegration and homogenization. This process can create a rapid increase in osmolality in the core of the contusion that attracts a large amount of water. Recent MRI–diffusion-weighted imaging (DWI) studies have observed that a large amount of edema fluid accumulates in the necrotic core of brain contusions up to 72 hours after trauma, resulting in rapid expansion of the extracellular space detected by an increased value of the apparent diffusion coefficient.38 Samples of necrotic brain tissue during surgical evacuation of cerebral contusions have revealed very high osmolality within the necrotic core of cerebral contusions when compared with the healthy brain tissue.40 Similar findings were observed in brain contusions induced in experimental TBI models. The total concentrations of Na+, K+, and Cl− are not significantly altered within the tissue, and the accumulation of metabolic intermediate osmoles (idiogenic osmoles)—which are produced in pathologic enzymatic activation, such as catabolism of proteins and lipids and deoxyribonucleic acid (DNA) fragmentation—might account for the hyperosmolar gradient created within contusions. An elevation in the brain tissue content of idiogenic osmoles associated with the formation of edema has been demonstrated in cerebral ischemia and in the early development of post-traumatic diffuse brain swelling in experimental TBI models.41
Natural History and Surgical Treatment of Cerebral Contusions
Despite intensive medical therapy, the elevated ICP caused by early massive edema associated with post-traumatic cerebral contusions is often uncontrollable and fatal. An explanation to this therapeutic failure would be that extremely high osmolality values (greater than 380 mOsm) reached within brain contusions make these lesions resistant to hyperosmotic diuretic therapy. Consequently, surgical treatment may be the best option for the efficient control of ICP. Nevertheless, indications for surgical intervention for severe cerebral contusions remain controversial.4 A recently published retrospective review of 98 patients harboring cerebral contusions who initially received conservative treatment showed that 45% of brain contusions displayed a significant progression on follow-up CT and 20% required surgical intervention.42 Contusion progression most likely happened in the first 2 days after injury, and no patient with an initial GCS score of 15 points or an initial contusion size of less than 14 ml required delayed evacuation.
The effects of surgical excision of the necrotic brain tissue in patients with severe cerebral contusions has been investigated in the Japan Neurotrauma Data Bank, in which a total of 1002 severe TBI patients were included.43 From 182 patients presenting with severe cerebral contusions, 66% were treated conservatively, whereas 34% underwent surgery. In most surgical cases, excision of necrotic tissue and clots, in addition to decompressive craniectomy, was performed. A poorer outcome was observed in the group treated conservatively 6 months after injury, which showed a double mortality rate (48% vs. 23%). These data support the usefulness of surgical treatment for cerebral contusions to prevent clinical deterioration and death, especially in patients with GCS scores of 9 points or better at the time of admission (patients who talk and deteriorate).
Basic Concepts of DAI
The term diffuse axonal injury, also known as diffuse white matter shearing injury, was coined by Gennarelli et al. to describe the scattered injury of white matter fibers observed throughout the brains of animals and humans who suffered TBI.20 DAI was observed in 30% of the cases included in the Glasgow database, typically in cases of head injury involving a pure inertial A/D noncontact loading of the head.18,44,45 Patients who sustain severe DAI are unconscious from the moment of the impact and do not experience a lucid interval, remaining unconscious, vegetative, or at least severely disabled until death. DAI has been observed in 80% of patients suffering from a VS. In the past, these severe alterations of consciousness after TBI were considered the exclusive effect of brain stem injury due to dysfunction of the fibers of the AAS, but although brain stem damage occurs in many cases, it is always accompanied by evidence of DAI. In patients recovering from a coma, DAI contributes to the functional neurologic disturbances usually present, such as spasticity, intellectual decline, and unmodulated behavior patterns.
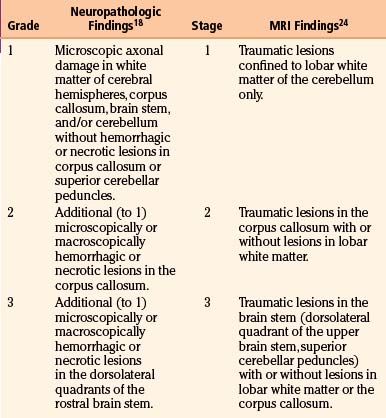
Axonal damage is usually caused by forces occurring in less than 50 msec, as seen in automobile crashes, in which the brain tissue behaves as a stiff mass transmitting tensile elongation to axons that cause damage to their cytoskeleton. The first description of specific diffuse brain lesions in patients in a permanent VS after TBI until death was provided in 1977 by Adams et al., who identified a classic clinicopathologic pattern consisting of a triad of hemorrhagic lesions in white matter, corpus callosum, and rostral brain stem typically observed in the severest form of DAI.13 According to the severity of the axonal damage, Adams et al. proposed a classification of DAI in three categories of severity (Fig. 133-5B and C and Table 133-2).46 Nevertheless, some degree of diffuse axonal damage is likely to be present in all patients with severe head injuries and is almost universally present in fatal TBI.24
The fundamental microscopic, neuropathologic finding of DAI is the development of globular expansions developed at the proximal and distal stumps of severed axons known as axonal retraction balls, which were first described by Ramón y Cajal in 1911 in models of traumatic injury after causing injury to the cerebral cortex or spinal with a surgical knife (Fig. 133-3C and D).26 Nearly half a century later, Strich described the same neuropathologic finding in the brains of patients in a permanent VS after TBI and stressed that the disruption of white matter fibers was caused by shearing forces.17 For a long time, it was believed that tensile forces on axons caused a physical disruption of these structures, splitting them into proximal and distal segments, and that this disruption produced discharging of the axoplasm content to form a swollen proximal end known as a retraction ball or end bulb (Figs. 133-1, 133-2, and 133-5D and E).25 However, traumatic primary axotomy only occurs with the strongest shearing forces, and most axons exposed to A/D forces do not tear or break at the moment of the injury; rather, they undergo dynamic, progressive, structural and functional alterations that potentially may lead to a secondary axotomy without necessarily associating it to physical disruption of the axolemma.23 Recent evidence suggests that axons are far more resistant to stretching than previous studies indicated and human neurons in culture are able to tolerate tensile strains up to 65% of their original length without undergoing axotomy.
Diagnosis of DAI
The diagnosis of DAI can only be confirmed on postmortem studies using immunohistochemical methods such as antibodies against the beta-amyloid precursor protein (β-APP) or against the 68 KD neurofilament subunit. The β-APP is a protein normally transported along the axon that accumulates at the axonal beads after DAI and can be immunohistochemically detected, serving as an accurate marker for impaired axonal transport.23,47 In TBI, β-APP accumulates before disruption of the axolemma and apparition of axonal retraction balls occur, suggesting that axonal transport failure is an early event (eFig 133-16 A). Using these immunohistochemical methods, DAI is detected in nearly 100% of moderately and severely head-injured patients.
It is accepted today that patients in whom the CT scan discloses focal petechial hemorrhages represent only the severest forms of DAI and that MRI can show may more lesions than CT in a large number of these patients. Shearing injuries are displayed on MRI studies as multiple, small, ovoid lesions of 5 to 15 mm with their long axis paralleling the direction of the affected axons, most frequently found in the corpus callosum and in parasagittal regions of the brain.24 Intracranial hematomas can be observed associated with DAI, as well as brain swelling, in 75% of cases. The most recent MRI studies confirmed the three-stage classification scheme of DAI severity proposed by Adams et al., with only the severest stage III associated with a poor outcome (Fig. 133-5B and C and Table 133-2).24,48
Neuroradiologic Diagnostic Methods in TBI
CT Scanning
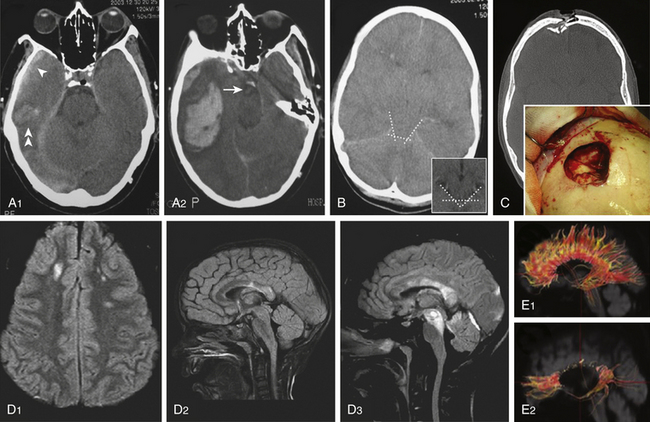
Cranial CT scanning is the imaging modality of choice for evaluation of a head-injured patient. It allows rapid assessment of the location, extent, and type of intracranial lesions caused by the head injury. With the lateral scout view and CT bone windows, skull fractures usually are well visualized, thus obviating the need for skull radiographic studies. Three major findings should be evaluated on a head-injured patient’s CT: (1) presence of intracranial hematomas, (2) appearance of basal cisterns at the midbrain level, and (3) presence of traumatic SAH. The status of the basal cisterns is related to the outcome. Basal cisterns can be open (all limbs are open), partially closed (one or two limbs are obliterated), or completely closed (all limbs are obliterated) (Fig. 133-6B). Compressed or absent basal cisterns indicate a threefold risk of raised ICP. Mortality is increased twofold in the presence of traumatic SAH, a sign that, when observed at the basal cisterns, carries a positive predictive value of unfavorable outcome of approximately 70%.4
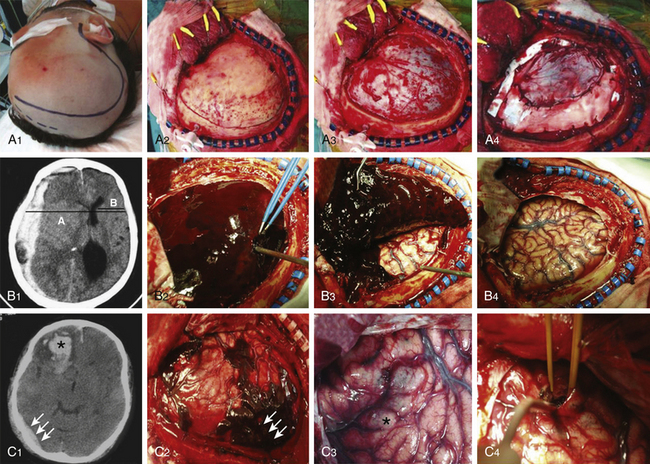
Approximately 10% of initial head CT scans in patients with severe TBI do not show abnormalities, and significant new lesions associated with increased ICP may develop in 40% of patients with an initially normal head CT scan. A CT scan should be repeated, especially if the first scan was obtained within a few hours of injury or if the patient has the potential of developing surgical lesions such as cerebral contusions, before clinical signs of neurologic deterioration are apparent (Fig. 133-6 A1 and A2). A delayed or progressive brain injury, defined as new or progressive lesions seen on a follow-up CT scan, is observed in 45% of a large series of patients with closed head injury. McBride et al., based on a review of 154 consecutive patients with closed head injury requiring surgical intervention, recommended a repeat CT scan within 4 to 8 hours of the initial scan in all patients with an abnormal initial scan, given that nearly 50% of patients received surgical intervention based on the findings of the follow-up CT scan.49 Risk factors of developing delayed cerebral insults are the clinical severity of the head injury as defined by the initial GCS score, the need for cardiopulmonary resuscitation at the accident site, the presence of an acute SDH on the first CT scan, and the presence of coagulopathy on admission. Of those patients with delayed brain injury, 55% had abnormal clotting studies, whereas only 9% of those patients whose follow-up CT scans did not worsen had abnormal coagulation studies.
Magnetic Resonance Imaging
MRI is recommended in post-traumatic coma unexplained by CT scan, both for diagnosis and for assessing prognosis. MRI is a better prognostic tool for TBI than CT scanning because it can detect DAI and brain stem lesions. Use of MRI in the acute phase was initially restricted by practical issues, such as availability, compatibility with life-support equipment, and resource capacity. These issues have been addressed; consequently, an increasing number of studies have reported on the use of MRI in severe TBI. It has been observed that 85% of severely head-injured patients showing brain stem injury on MRI have an unfavorable outcome (death, VS, or severe disability) compared to only 55% of patients without brain stem lesions.48
Table 133-3 summarizes the most important characteristics and findings obtained with MRI sequences after TBI. A conventional T2-weighted MRI sequence is useful but limited in detecting lesions within the periventricular area or the cerebral cortex due to the presence of cerebrospinal fluid (CSF) nearby. The MRI–fluid-attenuated inversion recovery (FLAIR) sequence is sensitive in evaluating nonhemorrhagic lesions on white matter because it reduces or suppresses the signal produced by CSF, thereby detecting a larger number of lesions (Fig 133-6 D). Injured white matter volume within 2 weeks of injury (total DAI volume) has been observed to be correlated with patient outcome. MRI–DWI is more sensitive to DAI and can see alterations 3 hours after injury because it evaluates the altered local water diffusion. It distinguishes cytotoxic from vasogenic edema. Timing of MRI is important, because in DAI regions there is an evolution from restricted diffusion (cytotoxic edema) to unrestricted diffusion (vasogenic edema).50
Table 133-3 MRI Techniques and Traumatic Axonal Injury24,48,50,51
| MRI Technique | Features |
|---|---|
| Conventional Imaging | |
| T2 weighted | Limited in detecting periventricular/cortical lesions due to the presence of CSF nearby. |
| FLAIR | Very sensitive to nonhemorrhagic lesions. It suppresses the signal produced by CSF and detects a larger number of lesions than does T2-weighted MRI (ischemic TAI = hypersignal). |
| T2 weighted gradient echo | Very sensitive to paramagnetic blood breakdown products and thus to microhemorrhages (hemorrhagic TAI = hyposignal). |
| More Sensitive Sequences | |
| DWI | Detects alterations >3 hours after injury. DWI quantifies the overall restriction to water diffusion (ADC). There is an evolution from decreased ADC or restricted diffusion (cytotoxic edema) to increased ADC or unrestricted diffusion (vasogenic edema) in TAI lesions. |
| DTI and DTI tractography | Quantifies the directionality of water diffusion in three-dimensional space, providing in vivo information on white matter integrity. Water diffuses more freely in the direction of white matter fibers (FA). TAI is associated with FA reduction, which could be due to misalignment of fibers, edema, fiber disruption, or axonal degeneration. |
| Proton-MRS | Detects alterations since the injury. Proton-MRS assesses neurochemical alterations in the following metabolites: Detects alterations since the injury. Proton-MRS assesses neurochemical alterations in the following metabolites: • Cr, found in glia and neurons. Its level is believed to be stable and serves as a point of reference to calculate metabolic ratios. • NAA, an amino acid present in neurons. NAA reflects the status of neuronal tissue (lower NAA/Cr ratio in TAI). • Cho, component of cell membranes. Cho reflects glial proliferation or membrane breakdown (higher Cho/Cr ratio in TAI). • Lactate, a marker of anaerobic metabolism and ischemia (higher levels are seldom found in head-injured patients). |
ADC, apparent diffusion coefficient; Cho, choline; Cr, creatine; FA, fractional anisotropy; NAA, N-acetylaspartate, TAI, traumatic axonal injury.
T2-weighted, FLAIR, and gradient echo MRI sequences underestimate axonal injury. The novel technique of diffusion tensor imaging (DTI) has evolved in recent years as a valuable complementary tool to investigate DAI. DTI characterizes the directionality of water diffusion in a three-dimensional space, thus providing information on the physiologic status of particular axon bundles, a form of noninvasive in vivo tract tracing known as DTI fiber tractography (Fig 133-6 E).51 It evaluates the interruption of axolemmal transport and axonal leakage, being capable of detecting alterations 3 hours after injury. This technique has shown good correlation with the gold standard β-APP immunohistochemical method for detection of DAI in experimental models of TBI. Two pilot studies, one performed on 9 chronic TBI patients51 and the other on 12 head-injured patients within the first week after injury,52 showed post-traumatic damage in white matter tracts, such as the corpus callosum, fornix, and peduncular projections that were correlated with the outcome, and supported this technique as a promising diagnostic and prognostic tool (Fig 133-6 E).
Magnetic resonance spectroscopy (MRS) is a method used for assessment of neurochemical alterations since the moment of the injury. Proton-MRS is the commonly used method in vivo, in which a reduction in the N-acetylaspartate/creatine ratio and an increase in the choline/creatine ratio are the most characteristic detectable findings after TBI. These changes reflect, respectively, the degree of neuronal loss/membrane breakdown and the degree of glial proliferation.41 In addition, the MRS analysis of the biochemical profile in cerebral cortex and white matter of adult TBI patients studied 7 days after injury found that glutamate (Glu)/Glu+glutamine and Glu/choline ratios were significantly elevated in patients showing long-term outcomes of 4 or fewer points on the GOS 6 to 12 months after injury.53
Surgical Treatment of TBI
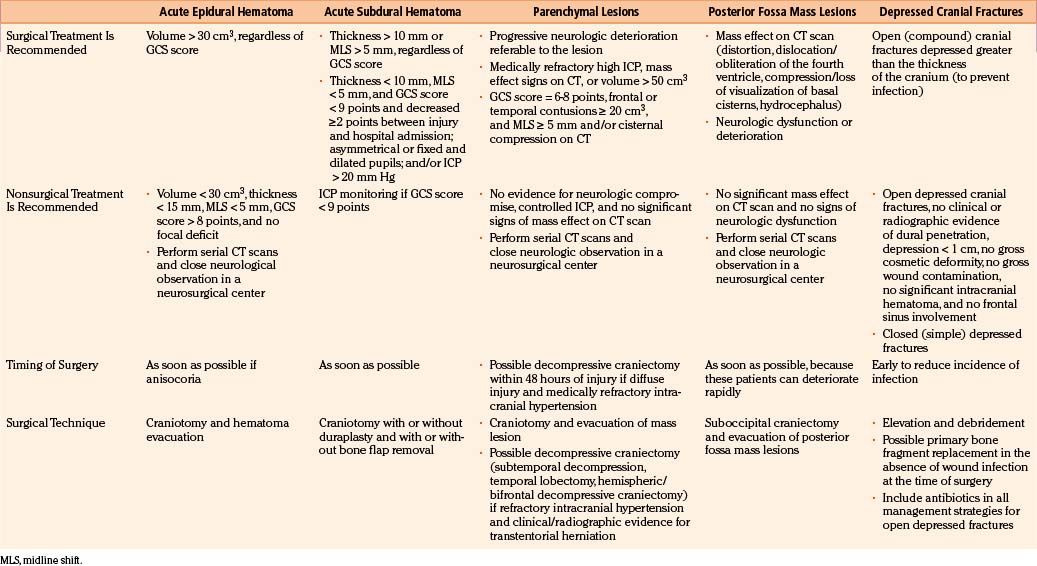
TBI treatment is an integrated team effort by surgeons and nonsurgeons. Probably the most important aspects of TBI care are nonoperative; however, the role played by neurosurgeons in the integral treatment of TBI patients remains critical, especially in making the appropriate decisions regarding whether to operate and, if operating, when to perform surgical procedures in advance to the progression of secondary damage.54 Surgical management of TBI has long been based on clinical anecdotal practice, assuming that any intracranial mass lesion in a patient with progressive neurologic deterioration should be evacuated as soon as possible by craniotomy. The new paradigm of evidence-based medicine has extend to the establishment of scientific guidelines for the surgical treatment of TBI, published in 2006, which should help neurosurgeons make the most appropriate decisions by using a strong scientific background, in addition to personal anecdotal experience.4,5 These guidelines were performed after a throughout analysis of the English-language TBI literature published since the use of CT scan. The appropriate articles were classified according to their scientific rigor in three classes of evidence with three associated levels of recommendation. No level I or II recommendation is provided by the surgical guidelines because of the lack of scientific class I or II evidence, mainly due to the ethical impossibility of carrying out prospective, randomized surgical trials. Consequently, guidelines can only formulate level III recommendations, reflecting unclear clinical certainty. As in all areas of clinical medicine, the optimal decision regarding treatment for an individual patient should not rigidly fall within a given recommendation of the guidelines but depend on a synthesis of the established knowledge based on guidelines and refined by the specific findings of the patient and the clinical judgment of the treating physician. A summary of the principal recommendations for surgical treatment of TBI lesions, together with the fundamental findings provided by the articles used for the elaboration of the guidelines, is provided in Table 133-4.
Surgical Treatment of Depressed Skull Fractures
Depressed skull fractures can be classified in two major types: (1) simple cranial depressed fractures, which have no galeal disruption and are only surgically managed if the extent of depression equals or exceeds the thickness of adjacent intact bone or if there is an associated intracranial hematoma with mass effect that requires evacuation, and (2) compound depressed cranial fractures, which have galeal disruption and an overlying scalp laceration in continuity with the fracture site and are generally treated with debridement and surgical elevation.4 Some compound depressed cranial fractures may be treated nonoperatively if there is no clinical or radiographic evidence of dural penetration, significant intracranial hematoma, depression greater than 1 cm, frontal sinus involvement, gross deformity, wound infection, pneumocephalus, or gross wound contamination.55 Several studies have also shown that the rate of postoperative infections is not increased by primary bone fragment replacement.56 There is no difference in the rate of postoperative infection between patients undergoing surgical repair of the fracture within 8 hours and those treated after that time. Anticonvulsants should be administered prophylactically to these patients, who are associated with a 20% or greater risk of developing seizures. Recommendations for the surgical treatment of depressed skull fractures, according to the surgical TBI guidelines, are listed in Table 133-4.
If the paranasal sinuses are involved by a depressed fracture, the goals of treatment are correction of the cosmetic deformity and prevention of a CSF fistula, regardless of whether the fracture is simple or compound. Depressed fractures of the anterior wall of the sinus do not result in a CSF fistula unless there is also a fracture of the posterior wall and a tear in the adjacent dura. A bicoronal scalp is fashioned, the pericranium is elevated as a separate layer, and the depressed fragments are elevated. Then the sinus mucosa is exenterated to prevent the formation of a mucocele, using a small drill if necessary. Next, the residual cavity can be filled with temporalis muscle and fat (Fig. 133-6 C). A pericranial graft can then be reflected over the sinus and sewn to the dura to further isolate it from the intracranial contents. The bone fragments comprising the anterior wall of the sinus are wired together and secured to the skull. The bone fragments of the posterior wall do not need to be replaced.
Post-Traumatic Intracranial Hematomas: Surgical Versus Nonoperative Management
The formation of intracranial hematomas after a head injury is a common occurrence. Among the 746 severely head-injured victims included in the TCDB, 37% of patients underwent surgery for removal of an EDH, SDH, or intracerebral hematoma (ICH).57 To detect hematomas at an early stage, it is essential to identify the risk factors that increase the changes that a given patient will be harboring an intracranial hematoma. They consist of the presence of a skull fracture on radiograph, any neurologic dysfunction, a seizure or severe headaches and vomiting, and a decrease in the level of consciousness. Patients with blood coagulation disorders should be observed particularly carefully for progressive enlargement of hematomas, and the clotting disorders must be vigorously treated. Overall, nonoperative management of SDHs and EDHs should be considered only if the patient is fully conscious, the extra-axial mass lesion is the single dominant lesion (i.e., there are no multiple contusions or potentially significant contralateral mass lesions, which may be preventing midline shift), and there are no features of mass effect, such as midline shift greater than 3 mm, or basal cistern effacement. Finally, there are situations in which the dismal clinical status of the patient warrants a nonoperative course despite the presence of a radiographic surgical lesion. Such cases include the adult patient who is moribund and flaccid with a GCS score of 3 points, nonreactive and dilated pupils, and no spontaneous respirations.58 Similarly, in patients older than 75 years of age with a GCS score of 5 or fewer points, a nonoperative course usually should be taken, given their consistently poor outcome with or without surgery.
High ICP and ICP Monitoring in TBI
The brain is housed in a rigid cavity and has a limited ability to compensate any volume occupation caused by intracranial hemorrhages or the development of swelling. Clinical examinations are unreliable unless the patient has progressed to brain stem herniation. The most accurate way of assessing ICP is to insert a monitor to allow the clinician to anticipate irreversible brain damage by a prompt medical or surgical intervention. There is substantial evidence that ICP monitoring may improve outcome, because it helps in earlier detection of intracranial mass lesions and guides therapies to control intracranial hypertension. About two thirds of patients with severe head injury have no significant mass lesion on the initial CT scan, but at least 80% demonstrate high ICP.59 It is generally accepted that severely head-injured patients (GCS score 3-8 points) with abnormal scans should undergo ICP monitoring. Effaced cisterns are associated with high ICP in more than 70% of cases, and mandatory ICP monitoring is advocated in such cases. The prevalence of postoperative intracranial hypertension is extremely high (greater than 75%) in patients who were in a coma before removal of an intracranial hematoma. In such patients, therefore, an ICP monitoring device should be inserted at the end of the procedure.
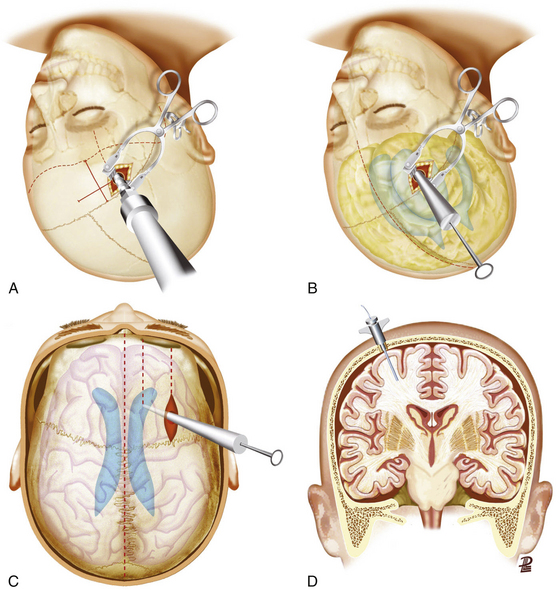
The gold standard method for measuring ICP is an intraventricular device connected to a fluid-coupled catheter with an external strain gauge (ventriculostomy catheter), because it not only monitors ICP but also provides CSF drainage to lower ICP (Fig. 133-7A-C). Nevertheless, many centers tend to use intraparenchymal probes, especially when the ventricular size is small and it is expected to need a high number of passes. This device should be inserted through a bur hole and skin incision separated from the craniotomy. A twist drill is usually made just anterior to the coronal suture in line with the pupil. Following the opening of the dura with a blunt stylet, the transducer tip is directly placed in the brain parenchyma at a distance of 2 to 3 cm into the brain parenchyma (Fig. 133-7D).
Surgical Timing in Post-Traumatic Lesions
In general, the decision regarding whether or not to proceed with surgical evacuation of a post-traumatic hematoma depends on the patient’s clinical condition, neurologic status, and imaging findings. Clinical features indicating high ICP or any impairment to the level of consciousness, oculomotor paresis ipsilateral to a sizable hematoma, bradycardia, severe headache, repeated vomiting, and/or arterial hypertension. CT features suggesting raised ICP are shift of brain structures, obliteration of all subarachnoid CSF spaces, dilatation of the lateral ventricle contralateral to the hematoma, and hematomas of large size. A prompt surgical evacuation is essential for masses producing brain stem compression because of the risk of irreversible ischemic brain damage. In these cases, the sooner the decompression is achieved, the greater the possibility of good recovery. There is clear evidence that delayed evacuation of an EDH/SDH causing significant midline shift once clinical deterioration is present is associated with much poorer results, especially if the evacuation is later than 2 to 4 hours of the onset of the coma.60
Greater controversy exists over when to evacuate parenchymal brain contusions and traumatic ICHs and whether such removal is helpful in controlling ICP and improving outcome.61 Miller et al., in their experience with more than 200 severely head-injured patients, contusions, and traumatic hematomas associated with the highest incidence of poor ICP control despite their surgical removal, recommended an initial nonoperative management for such lesions.61 Other authors believe that early surgical removal of larger hematomas and cerebral contusions provides early control of ICP and helps prevent the cascade of secondary damage. Temporal lobe hematomas are especially likely to cause brain stem compression at low ICP with little midline shift and should be evacuated sooner than other locations. An initial conservative approach is warranted, however, when eloquent cortex is involved. Similarly, intracranial hematomas confined to deep white matter or the basal ganglia are best managed without surgery.
Acute EDH
An acute EDH develops between the dura mater and the inner table of the skull and is reported in 2.5% to 5% of head-injured patients (Fig. 133-4A). EDHs adopt a lens-shaped or biconvex morphology as the periosteal layer of dura is stripped from the inner table of the skull by the bleeding. The classic and most lethal type of EDH is caused by a skull fracture crossing the groove in the temporal bone containing the middle meningeal artery. These hematomas generally accumulate rapidly, although a delayed stepwise progression over hours may also occur. EDHs may have also a venous origin, either from diploic veins or from bridging veins torn along their course into venous sinuses. These lower pressure hematomas develop slowly and are often self-limited, but no reliable imaging signs can be used to assist in their prediction, and this subtype should be considered dangerous. Some EDHs accumulate from rebleeding, accounting for up to 30% of all EDHs in some series.62 Dural stripping by the blow itself has been postulated to be the alternative presumed mechanism. Epidural collections arising solely from skull fracture bleeding are most often small and most frequently do not expand in the absence of damage to an adjacent sinus or coagulopathy. Nevertheless, nonoperative management based on anticipated stability should be supported by serial imaging.
The “classic” lucid interval consisting of a brief LOC caused by the blow per se followed by an awaken period and late deterioration due to rising ICP and mass effect (patients who “talk and die”) only occur in a minority of cases, approximately 15% to 30%.63 Between 20% and 55% of the patients are comatose on admission or immediately before surgery.62,64 Although most EDHs are diagnosed on the initial CT scan, the clinician must remain alert to the possibility of an EDH that develops or rapidly enlarges after an apparently negative scan. A vertex hematoma may be missed on a routine axial CT scan, but a coronal reconstruction should identify the lesion.
EDHs are not usually associated with underlying parenchymal damage, so their early surgical evacuation is highly successful. Global mortality of a surgically treated EDH is approximately 10%.62,64 The surgery is usually an emergency, especially in patients in a coma at the time of the diagnosis, as the duration from time of injury to treatment is an important determinant of prognosis. Other factors in determining the outcome are age, depth of coma, degree of midline shift, and size of hematoma. The mortality rate and outcome after evacuation of an EDH is directly related to the level of consciousness before surgery, ranging from 0% for patients conscious at the time of operation to 27% for patients presenting with the classic lucid interval and reaching 50% for patients who never regain consciousness after the impact.
In addition, neurosurgeons should bear in mind the possibility of a delayed EDH, which accumulates after an initial normal CT scan. These may occur in 4% to 30% of EDHs series and be associated with a slightly higher morbidity and mortality because many patients developing such lesions are diagnosed and operated on after the apparition of neurologic deterioration.62 Mechanisms involved in their origin include correction of hypotension or hypovolemia, pseudoaneurysm, or coagulopathy.
Management
There are no class I or II evidence-based guidelines for the specific management of EDHs, and treatment is based on the patient’s GCS score, pupillary exam, comorbidities, CT findings, and age and the surgeon’s personal experience.4 An expanding temporal EDH in a deteriorating patient is a surgical emergency, whereas a small frontal EDH in an awake, alert, and talking patient can usually be managed conservatively. Chen et al. analyzed factors associated with delayed neurologic deterioration and the need for surgical evacuation in initially nonoperated cases and found that volumes greater than 30 ml, clot thickness greater than 15 mm, or midline shift greater than 5 mm on the initial CT scan was statistically associated with failed indication of conservative treatment.65 When clinical observation and serial imaging are the chosen procedure, the patient must be admitted to an intensive care unit and the first follow-up CT scan should be obtained within 6 to 8 hours after TBI. No enlargement of an EDH has been observed later than 36 hours. For the recommended surgical options versus conservative treatment, surgical techniques, timing, and outcome, refer to Table 133-4 and Fig. 133-8.
Surgical Technique for Evacuation
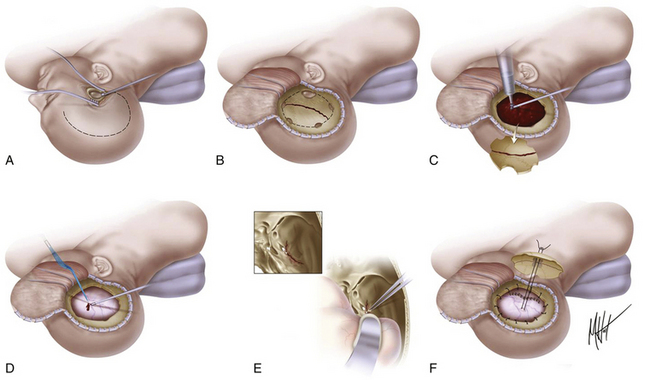
The scalp incision and craniotomy are based over the hematoma. It is crucial to achieve adequate exposure through the craniotomy to visualize and control the epidural bleeding source. A standard frontotemporal craniotomy is usually the most appropriate. The patient should be positioned supine with the head turned almost 90 degrees to the appropriate side, supported on a donut or with the Mayfield three-pin headholder. The skin incision is started about 1 cm in front of the tragus at the zygomatic arch and then curved backward and upward above the auricle to reach the midline in a question mark shape, from the parietal area toward the frontal midline, ending if possible behind the hairline. In a patient who is herniated or whose condition is deteriorating from an intracranial hematoma, the temporal end of the incision is rapidly opened, and a bur hole is placed. This opening is then extended with rongeurs to form a limited temporal craniectomy of about 3 cm in diameter. In EDHs that extend to the floor of the temporal fossa, the surgeon must ensure visual access across the floor of the middle fossa as far as the foramen spinosum (Fig. 133-8). When the bone flap is being turned, the surgeon should be aware of the location of the middle meningeal artery and be prepared to rapidly arrest bleeding from it. After removal of the bone flap, the hematoma is removed with suction, irrigation, and cup forceps. In most cases, the bleeding point corresponds to the torn middle meningeal artery or one of its main branches lying in the dura mater, and coagulation can be easily obtained with bipolar electrocautery. In some patients, the bleeding may originate directly from the foramen spinosum; in such cases, the hemorrhage can be arrested by plugging the foramen spinosum with a mixture of cotton wool and bone wax. After removal of hematoma, a small opening in the dura can be made to rule out the presence of an underlying SDH. The dura is then tacked up to the perimeter of the craniotomy using 4-0 silk sutures at 2.5-cm intervals in a circumferential fashion, to eliminate as much as possible the epidural space that has formed with the expansion of the hematoma. The bone flap is reapplied to the bone edges; if it is large, a tacking central suture is placed through two holes drilled in the center of the bone flap to tent up the dura mater centrally and minimizing the dead space. A drain is usually placed under the galeal layer. The superficial temporal fascia and then the galea are approximated using 2-0 Dexon or a similar suture. Skin staples (or silk sutures) are placed on the skin margins, the wound is dressed, and a head bandage is placed.
Acute SDH
SDHs constitute bleeding collections, which localize between the inner surface of the dura mater and the pial surface of the brain (Fig. 133-4B). Acute SDHs are those diagnosed within 14 days of the head trauma. The virtual space between the inner leaf of the dura mater and the arachnoid membrane (subdural space) is traversed by numerous, small, draining veins that bring venous blood from the brain to the dural sinus system. These veins can be damaged with minimal head trauma, particularly in elderly individuals with cerebral atrophy in whom the veins are subjected to considerable movement after head injury. Acute SDH is diagnosed on a CT scan as an extracerebral, hyperdense, crescentic (crescent shaped) collection between the dura and the brain parenchyma. Acute SDHs typically arise from torn bridging veins but may also arise from bleeding from a contused/lacerated cortex or from a torn cortical blood vessel. In most cases, the collection lies over the lateral surface of the hemispheres. In a few cases, the hematoma lies in an unusual location, for example, in the interhemispheric fissure. Subdural bleeding is usually under low pressure, and it typically tamponades early unless there is a defect in coagulation. Acute subdural bleeding is particularly dangerous in patients who take anticoagulants for vascular thrombotic disease, because continued venous leakage over several hours can cause a mass large enough to produce herniation.
Acute SDHs occur in approximately 11% of head-injured patients overall and in 20% to 30% of those severely injured.7 The mean age of presentation is between 30 and 50 years.66 SDHs are associated with a overall mortality rate of 50% to 90%, the highest of all traumatic intracranial injuries; this rate is more than four times higher in the patients older than 65 years.67 This poor outcome results largely from associated parenchymal injuries such as contusions, swelling, and DAI. At the time of diagnosis, 60% to 80% of patients are in a coma. The mass of blood accumulates rapidly, causing underlying brain edema and herniation. Mortality rates for patients belonging to the TCDB cohort with an acute SDH requiring evacuation ranged from 40% to 65%.57,66,68 No patient older than 75 years who preoperatively showed extensor or flaccid posturing to pain or had unilateral or bilateral fixed and dilated pupils made a good recovery.69
A lucid interval as typically described for EDHs has been described in 10% to 40% of SDH patients before admission. The physiopathology of fluctuations in consciousness associated with SDHs is not clear, but it is thought in some cases to be the reflection of increased ICP during plateau waves, with the patient moving in and out of diencephalic or uncal herniation. Given the breakdown in the BBB along the margin of the hematoma, fluid shifts may also contribute to these fluctuations. Approximately 50% of patients with an SDH have associated lesions, including cerebral contusions, ICHs, and brain lacerations, usually located in the frontal and temporal lobes.66,67,70
Depending on the etiology of the bleeding, three major types of acute SDHs have been described. The first type of SDH results from tearing of a bridging vein between the surface of the brain and one of the main draining venous sinuses. Under the effect of A/D forces, the maximal strain occurs in the shortest bridging veins oriented in the plane of the motion and angled in the direction of the motion. SDHs are rarely reported in the occipital region, supporting results that the largest strains are found for the parietally, centrally, or frontally located bridging veins for all impact directions.71 The second type of SDH is associated with laceration of the brain, usually at the temporal pole, in which there is a mixture of ICHs/intracerebral contusions and SDHs, a condition known as “burst temporal lobe.” The final type of SDH is caused by bleeding of a small cortical artery that may have been injured by an overlying fracture of the skull.
Management
There are no class I or II evidence-based guidelines for the specific management of SDHs, and treatment is based on the patient’s GCS score, pupillary exam, comorbidities, CT findings, age, and refractory high ICP and surgeon’s personal experience (Table 133-4).4 Different surgical techniques have been advocated for the evacuation of an SDH; the most commonly used are twist drill/bur hole trephination, craniotomy with or without dural grafting, and large decompressive hemicraniectomy with or without dural grafting. It is reasonable to leave out the bone flap in patients who appear to be actively swelling during surgery, have not had good medical control of ICP, or need other surgical procedures. For the recommended surgical options versus conservative treatment, surgical techniques, timing of the procedure, and outcome, refer to Table 133-4 and Figs. 133-9 and 133-10.
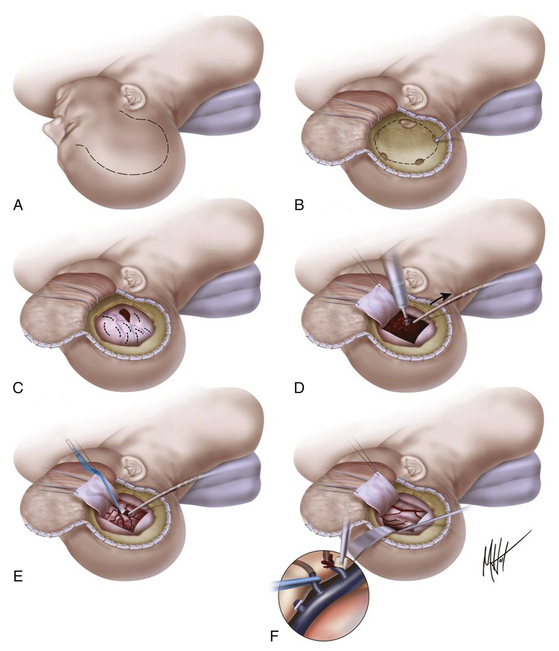
Surgical Technique for Evacuation
Most SDHs are best addressed through a large frontotemporoparietal craniotomy that provides access to the frontal and temporal poles and the area along the vertex to detect hemorrhage from bridging veins draining into the sagittal sinus (Figs. 133-9 and 133-10). The craniotomy is started with a temporal bur hole placed just above the root of the zygomatic arch, followed by the opening of the keyhole at the floor of the frontal fossae and the placement of some bur holes in the parietal and frontal regions along the line of craniotomy. After a generous U-shaped dural opening, the clotted blood is carefully removed by irrigation, gentle suction, and dissection with cup forceps until the brain surface is clear and the location of the bleeding points can be ascertained. A bleeding point may be a bridging vein or an artery on the surface of the brain that can be coagulated with the bipolar forceps, or a hemorrhage may be coming from the margins of a ragged brain laceration, which would necessitate removing a small amount of necrotic brain tissue. Areas of cerebral contusion more than 1 to 2 cm in size with irreparably damaged brain, appearing purplish and mottled, should be removed with gentle aspiration (Figs. 133-9 and 133-10). Hemorrhage from draining veins along the sagittal sinus can be troublesome. Occasionally, a bridging vein is clearly avulsed, and it can be easily cauterized with bipolar diathermy. However, when bleeding comes directly from the sinus wall, the use of bipolar diathermy is contraindicated because it only enlarges the opening. In these situations, it is best to gently tamponade the defect with oxycellulose (Surgicel) or analogous hemostatic agent or to plug it with muscle. Small amounts of clot that cannot be visualized adequately or require undue brain retraction for exposure should be left undisturbed.
In patients with signs of uncal herniation before SDH evacuation, gentle upward retraction or irrigation on the undersurface of the temporal lobe may help ease the herniated brain out of the tentorial hiatus after removing the hematoma. Effective reduction usually is accompanied by a flow of CSF as the tentorial edge is visualized. If the brain is swollen and tense and reversal of herniation is unsuccessful with gentle elevation, this maneuver should not be attempted forcibly, because it is likely only to produce more brain distortion. The dura is closed watertight, either primarily or with a graft, using either autologous pericranium/fascia lata or allogenic grafts (bovine pericardium). Some surgeons do not consider watertight closure to be essential. Dural tacking sutures are then placed around the craniotomy margin and centrally to the bone flap to prevent a postoperative EDH. The bone flap is replaced and secured with nonabsorbable sutures or cranial plates. Whenever there is significant or anticipated intracerebral swelling, a dural substitute is placed loosely over the swollen brain and sewn to the dural margins. In these situations, the bone flap is not replaced. A subgaleal drain may be placed and brought out through a separate incision. The scalp is closed in a two-layer closure with 2-0 Vicryl for the galea and staples (or silk) for the skin.
Surgical Treatment of Traumatic Parenchymal Lesions
Almost all patients with severe head injury, and about 25% of those with moderate injuries, demonstrate surface contusions and small ICHs on the initial CT scan (Fig. 133-4C).11,57 However, only 25% of trauma craniotomies are performed primarily for space-occupying contusions or ICHs. Traditionally, traumatic intraparenchymal lesions have been considered the paradigm of nonoperative treatment, despite the general assumption that surgery improves the associated mass effect arising from such lesions because of the frequent difficult recognition of the lesion margins and the potential risk of damaging the surrounding healthy brain tissue. The surgical decision is usually based on the perceived risk of delayed neurologic deterioration as estimated by the neurosurgeon. Although the ICP value remains a critical factor in the decision, a significant percentage of patients may suffer late deterioration without intracranial hypertension. There are no prospective studies addressing the efficacy of surgical treatment for specific independent factors influencing the outcome; therefore, surgical recommendations remain based mainly on two data: the degree of neurologic deterioration prior to the intervention and the ability of surgery to lower intracranial hypertension.72 For recommended indications, timing, and methods of surgery according to the surgical guidelines, see Table 133-4.
Cerebral contusion is roughly defined as bruising of brain tissue. The majority of hemorrhagic contusions occur most frequently in the frontal and temporal lobes, due to the brain impacting against the skull, whereas large hematomas are usually located at deeper areas such as in the basal ganglia, the latter frequently associated with significant parenchymal injury elsewhere. Both type of lesions are seen after severe A/D forces and represent an extreme end of the spectrum of DAI. Intraventricular hemorrhages also occur but are uncommon, and as with deeper brain stem or basal ganglia hemorrhages, they are overwhelmingly accompanied by a brain contusion and/or diffuse SAH. Direct brain injury by contusion, laceration, or tearing can lead to parenchymal hematomas, but the patient must live enough after impact and have enough blood pressure and low enough ICP to acquire any sizable hemorrhage. Gyral contusions may coalesce into areas of frank hematoma. Other hematomas may evolve from injured arterial or venous vessels due to shear forces in any region of the brain.32,44
Management of Post-traumatic Intraparenchymal Lesions
There are no class I or II evidence-based guidelines for the specific management of post-traumatic intraparenchymal lesions. ICHs and contusions generally do not require surgical evacuation unless there is significant mass effect or intracranial hypertension.4 Consequently, most surgical decisions are based on the progressive clinical deterioration, CT signs of mass effect progression—such as midline shift, clot thickness, and volume and patency of the basal cisterns—or uncontrollable rise in ICP. Because parenchymal lesions tend to evolve70,73 and because timing of surgery with respect to the occurrence of neurologic deterioration clearly affects outcome, much effort has been directed at defining patients at risk of progressive neurologic compromise as a result of their traumatic injuries.74 However, the precise characteristics of the subpopulation of patients with traumatic parenchymal lesions that will benefit from surgical intervention is not clearly defined. In patients with cerebral contusions and hematomas, perilesional swelling and enlargement of bleeding are the norms rather than the exceptions, and at least 25% of patients show clinical or radiologic worsening in the first 2 to 3 days after injury. Accordingly, CT scans should be repeated at least daily over the first 3 days after injury, unless ICP is monitored and remains low. Although steroids have not proved beneficial in head injury management overall,5 this treatment might improve the surrounding pericontusional inflammation. On average, nonevacuated parenchymal hemorrhagic lesions reabsorb in 4 to 6 weeks by macrophage phagocytosis and gliosis. For the recommended surgical options, techniques, timing, and outcome, refer to Table 133-4 and Figs. 133-10 and 133-11.
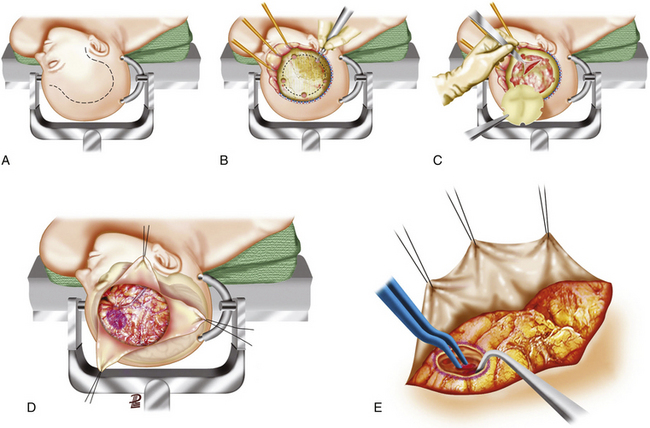
Delayed Traumatic Intracerebral Hematoma
The term delayed traumatic intracerebral hematoma (DTICH) is classically used to describe any post-traumatic intracerebral hyperdense lesion developing after admission to hospital in a part of the brain that seemed to be normal on the initial admission CT scan. These lesions only represent approximately 1.5% of all evacuated traumatic ICHs and 5% to 7% of severely head-injured patients, most appearing within 48 to 72 hours of injury.75 The etiology is often unknown, but some factors associated with DTICH development are increased vessel fragility in elderly patients,75 coagulation disturbances due to anticoagulants or platelet inhibitors, secondary systemic insults and previous decompressive craniectomy. Ultra early scanning may miss small hemorrhages in evolution, giving the false security of a negative CT scan. Some authors have noted that DTICHs occur in areas of contusion on initial, high-resolution CT scan. Indeed, the majority of studies show that patients who develop clinically relevant DTICHs have abnormal initial CT scans; therefore, it is essential that patients with initially abnormal scans undergo intensive monitoring and serial imaging to ensure early diagnosis of such delayed lesions. The hallmark of DTICHs is progressive deteriorating clinical status with seizures, progressive focal neurologic deficit, or decreasing level of consciousness.75 More than 70% of the patients showing radiologic deterioration are admitted in a clinical condition of mild–moderate TBI.76
Surgical Technique for Evacuation
Standard craniotomy with evacuation of the parenchymal hemorrhagic lesions is often effective in amelioration of brain shift and reduction of ICP. The location of the flap to be used depends on the location of the hematoma. The craniotomy can be much smaller than that used for evacuation of SDHs. After the dura is opened, a limited cortical incision is made with bipolar diathermy through the already traumatized or noneloquent brain (Figs. 133-10C and 133-11). Using gentle suction and bipolar diathermy, the contused and hemorrhagic areas are removed. Gentle retraction of the surrounding brain adjacent to the cavity is created with a handheld malleable retractor. If the hematoma is at a considerable depth from the cortical surface according to the CT scan, ultrasound can be helpful for finding its precise location. In eloquent areas, less aggressive removal of injured parenchyma should be performed. After bipolar coagulation of obvious bleeding vessels, placement of cotton balls soaked in half-strength hydrogen peroxide into the hematoma cavity is effective in achieving hemostasis. This tactic is not recommended, however, if the ventricle has been entered. After removal of the cotton balls, the hematoma bed is lined with oxycellulose (Surgicel), followed by routine closure. If there is significant swelling of the brain, a frontal or temporal “polectomy” can be carried out to minimize the pressure effects of postoperative brain swelling and the dura should be left open or closed with a dural graft sutured to its margins. In this situation, the bone flap should also be left out (Figs. 133-10C and 133-11).
Posterior Fossa Hematomas
Posterior fossa hematomas are a rare type of post-traumatic lesion, occurring in less than 3% of head injuries. Overall, they represent 1% to 13% of EDHs, 0.5% to 2.5% of SDHs, and 1.7% of intraparenchymal hematomas.77 The vast majority correspond to posterior fossa EDHs most often caused by lateral sinus injury and almost always seen in patients sustaining a direct occipital trauma that fractures of the occipital bone. They are bilateral in about one third of cases. Clival EDHs are an even rarer variant of the uncommon posterior fossa EDH that may occur by dissection from the cranial vertebra junction due to hyperflexion or extension cervical injuries or by basilar clival fractures. It is important to recognize these lesions early, because patients can undergo rapid clinical deterioration due to the limited size of the posterior fossa. It also is important to identify an occipital fracture even in the absence of a hematoma because of the possibility of delayed development of an EDH. The availability of rapid CT imaging has substantially reduced the mortality from about 25% in older series77 to near 5% in more modern series.
Management
Compressive lesions of posterior fossa usually present with headache, nausea, and vomiting, followed by LOC. They may also cause cerebellar signs and eye movement disorders. From 10% to 50% of posterior fossa EDHs are associated with hydrocephalus. Because of the rapid and life-threatening nature of neurologic deterioration secondary to expanding posterior fossa mass lesions, most patients with a posterior fossa EDH are treated surgically. Outcome is favorable in 95% of patients with an admission GCS score above 8 points, whereas it is poor in 80% of patients scoring fewer than 8 points (Table 133-4).4,78 Neurologically intact patients with a posterior fossa lesion and no CT evidence for mass effect (compression of cisterns, distortion of the fourth ventricle, and hydrocephalus) have been successfully managed nonoperatively with close observation and serial imaging.78 The best approach is unclear in patients with a traumatic posterior fossa mass lesion who do not present with a neurologic deficit but who have radiologic evidence for mass effect. In Table 133-4, several prognostic factors that adversely affect outcome regardless of the management are provided.
Surgical Technique for Evacuation
Placement of a frontal ventriculostomy is recommended before surgery for any posterior fossa traumatic lesion in view of the risk of an obstructive hydrocephalus (Fig. 133-7). To avoid the rare possibility of upward herniation after placement of the ventriculostomy, a slow CSF drainage should be allowed before craniotomy. For optimal and more comfortable control of all posterior fossa structures and to avoid the risk of air embolism, we recommend the park bench position (Fig. 133-12). The head is fixed in a Mayfield three-point device and then flexed forward to expose the occipital region, taking care of avoiding undue obstruction of the jugular veins. The bed is raised so that the head is slightly above the level of the heart. An inverted-Y skin incision provides good exposure of the supraoccipital and suboccipital compartments, allowing the surgeon to an adequate control of both lateral sinuses and the torcular herophili. Furthermore, this type of incision preserves the blood supply of the scalp flap and makes the harvest of a galea graft easier, allowing better watertight closure of the dura and reducing the risk of a postoperative CSF fistula or wound infection. Using a high-speed drill, multiple bur holes are placed above and below the transverse sinus, and a combined suprasuboccipital craniotomy is performed with the help of Leksell rongeurs or a high-speed drill, exposing widely and bilaterally the posterior fossa. Bone removal should include the rim of the foramen magnum inferiorly. It is advisable to leave a narrow bone rim over the lateral sinuses for the final stage of the craniotomy, which minimizes the risk of injury to the dural sinuses (Fig. 133-12).
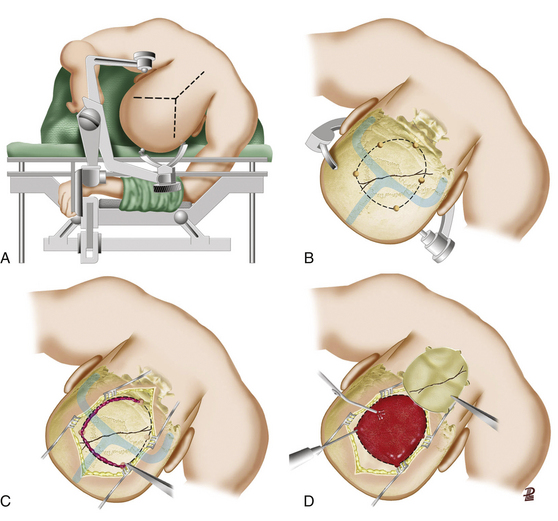
If a posterior fossa EDH is present, the dura will already be stripped from the inner table of the overlying bone by the clot. The EDH is then gently removed with the help of cut forceps and suction (Fig. 133-12). Any bleeding over the venous sinuses should be meticulously controlled, using Surgicel or a “muscle patch.” Sinus ligation should be only made in the nondominant transverse sinus. After hemostasis over the dural surface is achieved, multiple dural tacking sutures are placed along the craniotomy margin at 20-mm intervals. The bone flap is then secured using cranial miniplates. The wound is closed in layers using 2-0 Dexon sutured to the muscle layer and the subcutaneous tissues.
In a posterior fossa SDH, major bleeding should be anticipated and large-bore lines should be placed, with crossmatched blood available in the operating room. Following the suboccipital craniotomy, the dura is opened in a cruciate manner. Gentle aspiration and washing of the underlying clot are then performed, followed by exploration of the edge of the transverse sinus superiorly for identification and control of the bleeding points from the sinus or bridging veins over the superior cerebellar surface. If the underlying cerebellar surface appears contused, the damaged tissue is also aspirated and careful hemostasis is obtained with bipolar diathermy. In cases of cerebellar swelling, a dural substitute graft is used to achieve wide decompression; the posterior arch of the atlas can also be removed as part of the bony decompression.
Prognosis of Severe Head Injury
The strongest predictors of poor outcome after severe closed head injury, as documented by the TCDB cohort, included patient age, postresuscitation GCS score, pupil reactivity, proportion of time ICP is more than 20 mm Hg, presence of hypotension, and initial CT findings.57,68 Recently, a prognostic model of early death risk within the first 48 hours after the head trauma was published, and it identified similar independent predictors of early death, such as age older than 65 years, flaccidity, decerebration, nonreactive bilaterally mydriasis, shock, and diffuse injury type III to VI on Marshall et al.’s CT classification.58
• Influence of age. Age is a variable with a 70% positive predictive value for TBI outcome. An increasingly worse outcome is associated continuously and stepwise with increasing age. A meta-analysis including 5600 patients identified an essentially linear correlation of age and outcome after severe TBI.79 Around 90% of patients of the TCDB older than 55 years were severely disabled, vegetative, or dead at 6 months after injury, whereas good recovery at 6 months after injury was seen in approximately 30% of patients between 16 and 35 years old.57 In a more recent study of 600 patients with severe TBI, 56% of those younger than 20 recovered to a GOS score of 4 to 5 points, but this number fell to 39% between age 20 and 59 and to only 5% among those older than 60 years.80
• Influence of GCS score. Concerning the postresuscitation GCS score, only 7% of patients with an initial GCS score of 3 points reached a good or moderate outcome, in contrast to 77% of patients scoring 8 points.57 According to the Brain Trauma Foundation, a reasonably good indicator of outcome is the patient’s motor response to noxious stimulation, with an abnormal flexor (decortication posture), extensor (decerebration posture), or flaccid response after severe TBI predicting an outcome of fewer than 4 points on the GOS in every reported series.5 A motor response of flexion 6 hours after injury is associated with 60% mortality, and this rate increases to 80% mortality in patients with extensor or flaccid responses. Unfortunately, the European Brain Injury Consortium found that the motor score was untestable in nearly 30% of 1005 patients at the time of admission, due to prehospital medications and management with intubation.81
• Influence of pupillary response. Absent pupillary response is a variable associated with a 70% predictive positive value of an outcome of fewer than 4 points on the GOS, according to the analysis of the Brain Trauma Foundation.5 In patients with normal pupils after resuscitation and throughout their hospital course, 8.5% were left in a VS or died, compared to 75% to 80% of VSs or deaths observed in those with both pupils showing abnormal responses at some point after injury.57 Bilateral absence of pupillary and/or oculocephalic responses at any point of the evolution predicts an outcome of fewer than 4 points in the GOS, and this sign 6 hours after injury predicts 95% mortality.
• Influence of secondary insults. Secondary injuries such as episodes of hypotension/hypoxia associated with the TBI constitute independent predictors of poor outcome: a systolic blood pressure less than 90 mm Hg has a 67% positive predictive value for an outcome of fewer than 4 points on the GOS and a 79% value when combined with hypoxia. Hypotension, defined as a systolic blood pressure of 90 mm Hg or less, occurred in almost 35% of TCDB patients and was associated with an 85% increase in mortality compared with those who did not sustain a hypotensive episode.67 A single episode of hypotension (less than 90 mm Hg) is associated with a doubling of mortality and a significant increase of morbidity.
• Influence of CT findings. Class I and strong class II studies identified several CT signs associated with a positive predictive value of 70% of an outcome of fewer than 4 points on the GOS, such as mass effect with compression of basal cisterns, midline shift greater than 1.5 cm, blood within basal cisterns, and extensive post-traumatic SAH.82
• Influence of surgical treatment. In the TCDB patients undergoing evacuation of a traumatic hematoma, a favorable outcome was seen in 23% (47% in patients with an EDH, 27% in those with an ICH, and 14% in those with an SDH), whereas the overall postoperative mortality rate was 39%.57 Although the results of surgical management vary with the type of hematoma, the principal factor influencing the outcome is the patient’s level of consciousness before the operation. Comatose patients, in general, have a mortality rate 5 to 10 times higher than that of noncomatose patients (30% to 60% vs. 0% to 5%). To be precise, patients with an admission GCS score from 3 to 5 points have a mortality of 75% with an acute SDH, of 50% with an ICH, and of 36% with an EDH. Other factors that adversely affect outcome are a delay of more than 4 hours in instituting decompression and the severity of secondary insults in the postoperative period. A strong advocacy exists for a preemptive approach in diagnosis and for evacuating intracranial hematomas while the patient remains conscious.
Acknowledgments
We want to express our appreciation to the medical illustrators Matthew Holt and Scott Barrows for their superb artistic work in the elaboration of Figs. 133-1, 133-2, 133-4, 133-8, and 133-9, as well as to Pere Lluis Leon, the professional scientific illustrator who elaborated Figs. 133-7, 133-11, and 133-12.
Abbott N.J., Ronnback L., Hansson E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat Rev Neurosci. 2006;7:41-53.
Adams J.H.. Brain damage in fatal non-missile head injury in man, Braakman R., editor, Handbook of Clinical Neurology, Head Injury, 1990;57(Vol. 13):43-63 Vol 13(57). Amsterdam: Elsevier Science Publishers BV
Amiry-Moghaddam M., Ottersen O.P. The molecular basis of water transport in the brain. Nat Rev Neurosci. 2003;4:991-1001.
Bloch O., Manley G.T. The role of aquaporin-4 in cerebral water transport and edema. Neurosurg Focus. 2007;22:E3.
Brain Trauma Foundation. Guidelines for the management of severe traumatic brain injury. J Neurotrauma. 2007;24:S1-S106. 3rd ed.
Buki A., Povlishock J.T. All roads lead to disconnection? Traumatic axonal injury revisited. Acta Neurochir (Wien). 2006;148:181-193.
Bullock M.R., Chesnut R., Ghajar J., et al. Guidelines for the surgical management of traumatic brain injury. Neurosurgery. 2006;58(S2):1-62.
Ramón y Cajal S. Degeneration and regeneration of the nervous system. London: Oxford University Press; 1928.
Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6:889-898.
Farkas O., Lifshitz J., Povlishock J. Mechanoporation induced by diffuse traumatic brain injury: an irreversible or reversible response to injury? J Neurosci. 2006;26:3130-3140.
Floyd C.L., Lyeth B.G. Astroglia: important mediators of traumatic brain injury. Prog Brain Res. 2007;161:61-79.
Gaetz M. The neurophysiology of brain injury. Clin Neurophysiol. 2004;115:4-18.
Gennarelli T.A., Thibault L.E., Adams J.H., et al. Diffuse axonal injury and traumatic coma in the primate. Ann Neurol. 1982;12:564-574.
Kilinc D., Gallo G., Barbee K.A. Mechanically-induced membrane poration causes axonal beading and localized cytoskeletal damage. Exp Neurol. 2008;212:422-430.
Marmarou A. A review of progress in understanding the pathophysiology and treatment of brain edema. Neurosurg Focus. 2007;22:E1.
Myer D.J., Gurkoff G.G., Lee S.M., et al. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain. 2006;129:2761-2772.
Ommaya A.K., Gennarelli T.A. Cerebral concussion and traumatic unconsciousness: correlation of experimental and clinical observations of blunt head injuries. Brain. 1974;97:633-654.
Pascual J.M., Solivera J., Prieto R., et al. Time course of early metabolic changes following traumatic brain injury in rats as detected by 1H NMR spectroscopy. J Neurotrauma. 2007;24:944-959.
Posner J.B., Saper C.B., Schiff N.D., Plum F. Plum and Posner’s Diagnosis of Stupor and Coma, 4th ed. Oxford University Press; 2007.
Raisman G. Myelin inhibitors: does NO mean GO? Nat Rev Neurosci. 2004;5:157-161.
Rolls A., Shechter R., Schwartz M. The bright side of the glial scar in CNS repair. Nat Rev Neurosci. 2009;10:235-241.
Saatman K.E., Duhaime A.-C., Bullock R., et al. Classification of traumatic brain injury for targeted therapies. J Neurotrauma. 2008;25:719-738.
Sahuquillo J., Poca M.A. Diffuse axonal injury after head trauma. A review. Adv Tech Stand Neurosurg. 2002;27:23-86.
Servadei F., Compagnone C., Sahuquillo J. The role of surgery in traumatic brain injury. Curr Opin Crit Care. 2007;13:163-168.
Silver J., Miller J.H. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146-156.
Neuroscience of TBI: Structural and Functional Properties of Brain Tissue Determining the Mechanisms of TBI
Mechanisms of Neurovascular Regulation Coupled with Brain Functions: The Role of the BBB
BBB Function
The cerebral microvasculature is characterized by the conformation of a highly regulated barrier known as the BBB. The BBB blocks the free passage of molecules between the blood and the brain, contrary to the majority of capillary blood vessels in the body, which present leaky interendothelial cell junctions that allow the passage of many circulating molecules. Early experimental studies showed that trypan blue did not enter brain tissue when injected into the blood due to the BBB; however, when injected in the ventricles, it spread into the brain.83 The BBB is characterized by extremely low permeability to water-soluble molecules due to the presence of tight intercellular junctions, the lack of endothelial fenestrations, and the near absence of endothelial pinocytotic vesicles, the latter factor resulting in a low transcellular flux.84 The major function of the BBB is to strictly control the microenvironment of brain cells and to preserve its ionic homeostasis. The junctional complex that exists between endothelial cells, formed by tight and adherens junctions, prevents molecules from paracellular (between cells) diffusion down the concentration gradient, thus forcing most molecular traffic to take a transcellular route across the BBB83 (eFig. 133-13 A and B). These junctions even restrict the movement of small ions such as Na+ and Cl−, and only small gaseous molecules (O2 and CO2) and small lipophilic agents such as barbiturates and ethanol can diffuse freely through the lipid membranes.
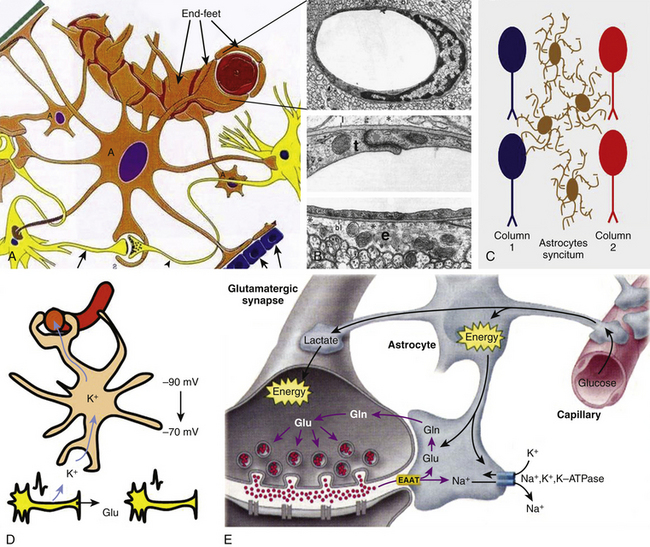
The BBB has a fundamental role in the coupling of precisely regulated local cerebral blood flow (CBF) to neural activity by maintaining the adequate extracellular microenvironment for reliable neuronal signaling.83 Perivascular end feet of astrocytic glia embracing brain capillaries contribute to the induction and regulation of the BBB.85 There is now strong evidence that these close cell–cell associations between astrocytes and brain capillaries mediate the induction of specific features of the barrier phenotype in the capillary endothelium, leading to the formation of tighter junctions—physical barriers—as well as the expression and polarized location of transporters, including the glucose carrier GLUT1.86
Most cerebral pathologic conditions and injuries caused to the brain to induce a change in BBB permeability and consequent functional and metabolic cerebral alterations derived from unrestricted permeability to water-soluble components of blood. Astrocytes and endothelial cells are able to secrete a range of chemical agents (adenosine; adenosine triphosphate, or ATP; bradykinin; prostaglandins; etc.) and able to modulate BBB function (e.g., by opening BBB tight junctions in situations such as inflammation, contributing to the development of brain edema). In addition, these cells can modulate BBB properties by inducing the upregulation or downregulation of specific endothelial transporters. Astrocytic aquaporin-4 (AQP4) is upregulated in brain edema triggered by BBB breakdown as an adaptive mechanism that may help clear the accumulating extracellular fluid.87,88
Cerebral Metabolism and Local Blood Flow Coupling
The brain requires large amounts of oxygen and glucose to remain functional. If CBF is interrupted, brain function ceases within seconds and irreversible damage to the brain’s cellular constituents ensues within minutes.89 In contrast, when neural activity increases within a region, cerebral metabolism increases to meet the energetic demands and the local CBF and cerebral metabolic rate for glucose each increase about 50% in the active areas.90 Complex cerebrovascular control mechanisms, overall known as cerebrovascular autoregulation, maintain stable cerebral perfusion despite changes in arterial pressure, ensuring that active brain regions receive an adequate amount of blood. In addition, a mechanism of functional hyperemia allows the delicate regulation of the local CBF according to the functional activity of the different brain regions so that when the activity of a considered definite brain region increases, flow to that region also increases.90 Autoregulation is achieved through tight control of cerebral vascular resistance and depends on an intact and functional BBB.
Cerebral perfusion pressure (CPP) is defined as the difference between mean arterial pressure and ICP. The brain is susceptible to ischemic injury, and in the setting of TBI, it is vital to maintain CPP and oxygenation. The lowest CBF levels are observed in diffuse injuries accompanied by significant edema, as well as large SDHs, and when the patient has experience hypotension. Hypotension and hypoxia are seen in more than a third of TBI patients and represent two of the most potent predictors of mortality. Indeed, 90% of patients who die from TBI demonstrate cerebral ischemic changes.
Pathologic events such as TBI and ischemic injury are characterized by some brain areas showing parallel reductions of local CBF and metabolism while others undergo the “uncoupling” between local CBF and metabolism. In some cases, disproportionately high local blood flow in relation to metabolism is observed (reactive hyperemia), while others are characterized by disproportionately high glucose consumption in relation to reduced local CBF (ischemic or traumatic penumbra). In the “penumbral” border around the ischemic core focal lesions, such as SDHs or cerebral contusions, glucose consumption has been shown to be increased for long periods.89 The nature of the local stimulus to pathologic hyperemia has eluded investigators to date, but it can accentuate the bulk effect associated with compartmental swelling in the brain.
Functional and Metabolic Coupling between Neurons and Glial Cells
Functional–metabolic coupling is carried out in the brain with glial cells, which cooperate with neurons through a fine regulation of local blood flow and energy supply to active synapses.91 A growing body of evidence indicates that neurons, glia, and cerebral blood vessels are organized into integrated functional glioneurovascular units, which regulate the cerebral microcirculation and the homeostasis of the brain microenvironment (eFig. 133-13 A and C).92 In these units, the glial cells signal and support the function of a particular neuronal population by interacting with local segments of the microvasculature and regulating the increased local CBF and glucose supply coupled to the heterogeneous dynamic activation of these neurons.93 In 1994, Haydon made the seminal observation that astrocytes respond to neuronal activity by releasing molecules, which have active modulatory roles in intercellular communication, such as ATP and Glu.92 This functional gliotransmission might modulate the strength of inhibitory and excitatory synaptic transmission by activating receptors in neurons.94,95
For a long time, astrocytes were regarded as electrically silent cells that did not participate in information processing in the central nervous system (CNS). However, in the 1960s, clear evidence that astrocytes are excitable cells that possess ion channels and can be depolarized, though to a much lesser extent than neurons, was provided by Kuffler and colleagues.96,97 Neuronal activity leads to transient increases in the extracellular K+ concentration ([K+]o), which if uncorrected would produce hyperactivity; therefore, the clearance of K+ from the extracellular space is considered as one of the most important functions of astrocytes for correct neuronal activity. Astrocytes play a key role in the spatial buffering of K+ by taking up and releasing excess extracellular K+ into the adjacent capillaries through their perivascular end feet (eFig. 133-13 D).97,98 Astrocyte end feet are provided with a specific type of K+ channels (inwardly rectifying the Kir4.1 K+ channel), which allow the preferred passage of K+ at negative membrane potentials. Astrocytes adjacent to neuronal active circuits experienced a change in their membrane resting potential coupled to the neuronal depolarization, which is caused by a K+ current driven from the extracellular space into the cells, sensing in this way the activity of individual neurons.83,97 Kir4.1 channels are colocalized with AQP4 channels at the astrocyte end feet ensheathing the capillaries, and this polarized enrichment suggests that both channels participate in the buffering of the K+ accumulated within the extracellular space of the synapses after neuronal depolarization.99
An additional key function of astrocytes is the removal of neurotransmitters that are released by active neurons. During normal neuronal activity, extracellular Glu in the synaptic cleft must be rapidly cleared to optimize the signal-to-noise ratio, as well as to prevent neuronal damage from excitotoxicity. Excess Glu or γ-aminobutyric acid (GABA) released at the synapses is uptaken by specific active transport systems located primarily in the perisynaptic astrocyte end feet (EAAT1 and EAAT2) in an active process linked to the influx of Na+ and water (eFig. 133-14 B).100 These neurotransmitters are then transformed within astrocytes in inactive molecules, such as glutamine, which are transferred to neurons for the synthesis of new transmitters in a metabolic cycle known as the Glu–GABA–glutamine cycle.100 This cycle is also involved in the supply of energy to neurons. Function of Glu and GABA transporters in astrocytes is linked to the activation of Na+/K+ adenosine triphosphatases (ATPases) that stimulate the anaerobic glycolysis in these cells and the generation of lactate that can be shuttled to active neurons to meet their energy needs (astrocyte–neuron lactate shuttle) (eFig. 133-13 E).91,101 Finally, astrocytes can detect the level of Glu-dependent synaptic activity and then adjust the local CBF through variations in the tone of microvessels mediated by release of prostanoids at the astrocytic perivascular end feet.
eFIGURE 133-14 Traumatic brain edema mechanisms. A, Sequence of cell mechanisms for progression of diffuse brain edema in an experimental model of diffuse head injury in rats.115 Head impact causes a transitory disruption of the BBB with an associated predominant vasogenic edema, followed by a long-lasting phase of predominant cellular edema mainly due to swelling of the astrocyte end feet that embrace capillaries. B, A major mechanism that contributes to the post-traumatic cellular edema or neurotoxic edema in glial cells is the uptake of Glu by astrocytes.126,128 Post-traumatic massive neuronal depolarization increases the concentration of Glu in the extracellular space. Astrocytes play a key role in recovering the composition of the extracellular space by the active uptake of Glu, a process that causes a net ion gain followed by the osmotic-driven water entrance through the AQP4 channel. C, Schematic illustration showing the expression pattern of AQP4 at the BBB. The concentration of AQP4 is high in the perivascular end feet membrane domains that face the vessels. The bottom panel shows strong AQP4 immunogold labeling in the perivascular membrane of an astrocytic end foot. D, Brain sections of AQP4+/+ and AQP4−/− (lacking AQP4) knockout mice after middle cerebral artery occlusion. Note the increased swelling and hemispheric enlargement in the AQP4+/+ mouse brain section (top) in comparison with a significantly reduced enlargement of the ischemic area in the AQP4−/− mice (bottom). LC, lumen capillary; E, endothelium.
(C, From Amiry-Moghaddam M, Ottersen OP. The molecular basis of water transport in the brain. Nat Rev Neurosci 2003;4:991-1001. D, From Manley GT, Fujimura M, Ma T, et al: Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med 2000;6:159-163.)
Following severe TBI, lactate is accumulated in the extracellular space. In rats subjected to a focal TBI model and treated with exogenous lactate, this energy substrate is actively uptaken and metabolized at the injury site, with the animals showing improvement in cognitive tests.102 Nevertheless, metabolic benefits from lactate solution administration has not been confirmed in recent experiments performed in a severe diffuse TBI model.103 Failure of exogenous lactate to improve the derangement of cerebral energy status after severe diffuse TBI might be caused by cerebral depletion of nicotinamide adenine dinucleotide, an essential coenzyme for the pathways metabolizing lactate.
Consciousness Impairment in TBI
Neurophysiologic Systems Supporting Consciousness
Consciousness can be defined as the functional brain state of full awareness of the self and of the relationship to the environment. Consciousness has two major components: (1) the content, which includes the sum of all types of cognitive representations (visual, auditive, proprioceptive, etc.) of the external and internal environment, as well as the associated affective responses, and (2) the arousal, which represents the level of alertness and behavioral responsiveness that in a normal individual is typically linked to wakefulness/sleep cycles.9 The cerebral cortex acts like a massively parallel processor that breaks down the components of sensory experience into an array of abstractions that are analyzed independently and in parallel during normal conscious experience. Since the 1950s, it has been known that the cerebral cortex is structurally arranged in columns about 0.3 to 0.5 mm wide in which the nerve cells located in successive layers along a line perpendicular to the pial surface share incoming signals in a vertically integrated manner, all processing information of a similar category (eFig. 133-13 C).104 Neurons of each column send information to one another and to columns in higher-order association areas. In this way, columns of neurons are able to extract progressively more complex and more abstract information from an incoming stimulus.
Cortical or subcortical focal lesions usually cause alterations in the content of consciousness characterized by the specific loss of some category of representations that is termed fractional LOC. There are also cases of multifocal brain damage that cause cessation brain function as a whole, creating a severe cognitive impairment to produce a global LOC. Diffuse brain lesions and damage to the functional systems involved in the arousal cause a reduced level of consciousness, whose deepest state is known as the coma. Coma, from the Greek word meaning “deep sleep or trance,” is a state of unresponsiveness in which the patient lies with eyes closed and cannot be aroused to respond appropriately to stimuli.9
Ascending Arousal System
In the late 19th century, consciousness was considered a brain property that is the result of the total activity of brain hemispheres, as it was proposed by Hughlings-Jackson.104a However, in the beginning of the 20th century, solid evidence of the relationship between an altered prolonged sleep state and the damage to focal gray matter regions located in the midbrain and within the hypothalamus was obtained from patients suffering from encephalitis lethargica.105 Subsequent experimental studies of transecting lesions placed sequentially at different levels of the brain stem in cats demonstrated that lesions below the midpontine level resulted in a permanent wakeful state, whereas lesions rostral to the pons forward produced electroencephalogram slowing and behavioral unresponsiveness.106 The seminal experimental work by Moruzzi and Magoun clearly showed that the paramedian reticular formation of the midbrain contained critical neural networks to forebrain arousal.22
More recent studies showed that the arousal projections to the forebrain originated from several independent and well-defined populations of neurons located at the mesopontine tegmentum. The major source of mesopontine afferents is a collection of cholinergic neurons arranged in two large clusters projecting through the paramedian midbrain reticular formation and the thalamus to specific cortical regions.9 In addition, several groups of monoamine, serotoninergic, and dopaminergic brain stem neurons, whose axons project through the hypothalamus to the cerebral cortex, contribute significantly to the arousal of the cerebral cortex.107 Finally, two groups of histaminergic and orexin-containing neurons located at the hypothalamus contribute to the AAS, firing fastest during wakefulness and slowing during both slow-wave and rapid eye movement sleep.
Mechanisms of Structural Coma in TBI
Post-traumatic parenchymal brain injuries capable of producing transitory or permanent alterations in patient’s consciousness may fall into one of the following categories: concussion, contusions, intracranial hemorrhages, diffuse brain swelling, and DAI. The term concussion refers to a transient mental alteration suffered by many patients after TBI who have usually presented with transient LOC. The hallmarks of concussion are amnesia and confusion, sometimes accompanied by headache, visual disturbances, and/or dizziness.108 The mechanism of LOC with a blow to the head is not completely understood. Physiologically, the concussion causes abrupt neuronal depolarization and release of excitatory neurotransmitters. Brief LOC, which in humans is usually not associated with any changes on a CT or MRI scan, may be due to the shearing forces transiently applied to the AAS at the mesodiencephalic junction. Experimental models of head acceleration performed in nonhuman primates by Gennarelli et al. showed that rotational acceleration in the sagittal plane typically produced only a brief LOC, whereas acceleration from the lateral direction caused mainly a prolonged and severe coma.20 Because the long axis of the brain stem is located at an angle of about 80 degrees with respect to the long axis of the forebrain, the long tracts connecting the forebrain with the brain stem take an abrupt turn at the mesodiencephalic junction, which determines that transverse shearing forces focus on this area and results in axonal injury to the tracts connecting the brain stem and the forebrain.
High ICP after TBI
In the 19th century, the predominant explanation regarding the causes of decreased level of consciousness due to expanding supratentorial lesions was diffuse impairment of cortical functions. Cushing was the first neurosurgeon to propose that the increase in ICP caused an impairment of CBF. He showed on experimental models that pressure waves generated within the supratentorial compartment were translated to the brain stem, which reactively induced an increase in blood pressure and fall in heart rate (the Cushing reflex).109 The Monro-Kellie doctrine hypothesizes that because the contents of the skull are not compressible and are within an unyielding case of bone, the sum of the volumes of brain, CSF, and intracranial blood is constant at all times.110 Intracranial contents encased by a nondistensible cage correspond to approximately 87% of brain tissue (of which 77% is water), 9% of CSF, 4% of blood volume, and a negligible volume occupied by the meninges. A corollary of this doctrine is that the same restrictions apply to each compartment (right vs. left supratentorial space, infratentorial space, or spinal subarachnoid space).
In a normal brain, increases in the size of a growing mass lesion can be compensated for by the displacement of an equal volume of CSF from the compartment. As the mass grows, there is less CSF to be displaced; hence, the compliance of the intracranial contents decreases as the size of the compressive lesion increases. Then a critical point is reached in which a small further increase in volume can produce a large increase in compartmental pressure. The two factors playing important roles in the response of the brain to increased ICP are the compliance in the intracranial system (the ability to accommodate further increases of volume without undergoing an exponential rise in ICP) and the rate of change in ICP. Loss of compliance in cases of diffuse brain edema causing high ICP can lead to the development of plateau waves, which correspond to large, sustained increases in ICP approaching the mean arterial blood pressure and occurring at intervals between 15 and 30 minutes. These waves are thought to be due to systemic vasomotor rhythms, but they may produce paroxysmal symptoms through dramatic increases of ICP and even cause herniations.
Brain Herniation Patterns Caused by Intracranial Mass Lesions
Seven major patterns of brain shift due to expansion of intracranial masses have been characterized, five of them usually due to supratentorial lesions (falcine, lateral diencephalic, uncal, central transtentorial, and rostrocaudal brain stem deterioration) and two mostly seen associated with infratentorial lesions (tonsillar and upward brain stem herniation).9 Unilateral hemispheric lesions such as basal ganglia hematomas may cause falcine herniation followed by lateral diencephalic displacement, in which a lateral displacement of the calcified pineal gland greater than 9 to 10 mm is associated with coma. Post-traumatic lesions compressing a temporal lobe or expanding at this level can cause rapid uncal herniation, a key clinical sign of which is the ipsilateral fixed pupil dilatation due to dorsal compression of the oculomotor nerve by the uncus. It is usually followed by impairment of the level of consciousness due to distortion of the AAS and hemiparesis due to compression of the cerebral peduncle by the uncus. The paresis may be either contralateral to the herniation, if the advancing uncus impinges on the adjacent cerebral peduncle, or ipsilateral, if the uncus pushes the midbrain and the opposite cerebral peduncle is compressed against the incisural edge of the Kernohan notch.111 Central herniation is clinically characterized by a progressive impairment of alertness, memory defect, and torpid drowsiness that progresses to stupor and coma. It is produced by compression and functional impairment of the diencephalic structures from an expanding mass lesion, usually deeply seated within the brain parenchyma. Rostrocaudal brain stem deterioration is a type of herniation syndrome observed when downward displacement of the midbrain or pons stretches the medial perforating branches of the basilar artery and causes necrosis and extravasation of blood, with the development of slit-like brain stem hemorrhages named Duret hemorrhages.
DAI: The Major White Matter Diffuse Lesion After TBI
Axonal Transport: Physiologic Mechanisms
Structural proteins and neurotransmitters must be shipped from the neuron cell bodies, where they are synthesized, to their axon terminals. Under physiologic conditions, there is a continuous bulk movement of axoplasm along the axon, a process known as axoplasmic flow (eFig. 133-15 A).112 Microscopic direct visualization of the movement of single vesicles along the living giant axons from the giant squid have revealed that the axonal particles are actively transported in a saltatory fashion along linear tracks of microtubules aligned with the main axis of the axon. Two components of movement can be differentiated as part of axonal transport: anterograde transport, used by cytosolic and cytoskeletal proteins and organelles to move toward axon (and dendrites) terminals, and retrograde transport, used by membrane-packing organelle material to return to the cell body for recycling or degradation (eFig. 133-15 A and B).112 In addition, two components of anterograde transport were identified: slow axonal transport at a rate of 1 to 2 mm/day, corresponding to the movement of soluble and structural proteins such as tubulin or neurofilaments, and fast axonal transport at a rate of up to 400 mm/day, representing the movement of membrane-enclosed organelles such as mitochondria and vesicles packing neurotransmitters.
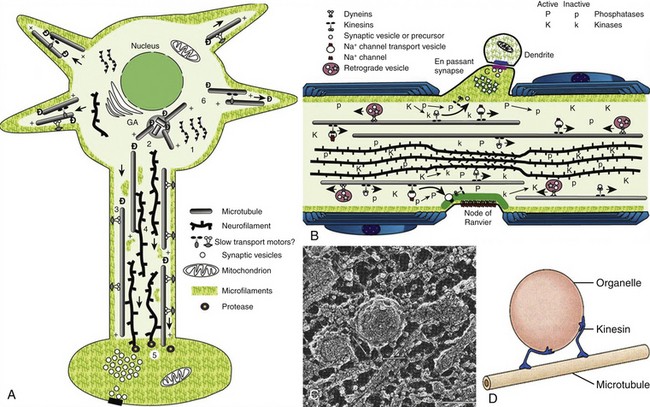
Organelles such as mitochondria and vesicle-bound transmitters moved along the axon by mechanochemical enzymes or “motors” attached to microtubules. These molecular motors are a family of ATPases with binding sites for specific organelles or molecules that hydrolyze ATP and use the energy obtained to carry organelles along the microtubules in a saltatory, step-by-step way (eFig. 133-15 C and D).113 Anterograde axonal transport is powered by kinesins, a family of microtubule-based molecular motors that moves organelles toward the plus end of microtubules. Retrograde transport of recycled membrane-bound organelles is powered by dynein, a motor enzyme that moves organelles toward the minus end of microtubules (eFig. 133-15 B-D).113 Microtubules constitute hollow tubes 24 nm in diameter and up to 100 μm in length formed typically by 13 protofilaments, each of which is formed by a linear arrangement of alpha- and beta-tubulin dimmers. Microtubules are highly dynamic structures that provide a stationary track on which specific organelles move by means of molecular motors.114 Microtubules are formed by nucleation at the microtubule-organizing center within the neuronal body and then are released for migration into the axon or dendrites, moving in a polymerized form by a mechanism involving microtubule sliding. The neurofilaments, another basic component of the cytoskeleton, are thought to move as discrete cytologic structures along the axon in association with the microtubules.
The coherent movement of neurofilaments and microtubules along the axon supports the “structural hypothesis” for axonal transport, which claims that proteins move as part of or in association with a discrete cytologic structure and not as isolated molecules. The limiting factor determining the movement of cytologic particles along the axon is the number of elements that can interact directly with transport motors, so transported material must be package appropriately to be moved. In other words, membrane-associated proteins move as membrane-bound organelles (vesicles, mitochondria, etc.), whereas tubulin and microtubule-associated proteins move as microtubules and neurofilaments move separately.114
Dynamic Process of Traumatic Axonal Injury
DAI is characterized by blocking of normal axonal transport, either with or without rupture of axon morphologic integrity, followed by apparition of focal axonal swellings or beads, which are the hallmark of this pathology (eFig. 133-16).44 Axonal beading has been observed in both postmortem human studies and in vivo experimental injury models.20 The term traumatic axonal injury refers to the specific mechanical type of axonal damage caused by TBI, which represents the physiopathologic expression of DAI.20,115 Gennarelli and Thibault consider this type of damage to be a dynamic process, not an event, and they proposed several stages of axonal damage increasing in severity, from a reversible increased axonal permeability and transitory abnormal axonal transport observed with mild magnitudes of stretching below 10% (stages I and II) to irreversible axonal damage (stage III) and primary axotomy (stage IV) seen after a stretching injury above 20%.10
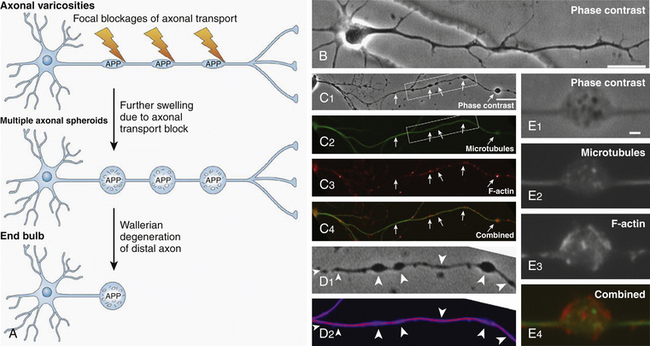
Inertial forces applied to the head at the time of TBI cause large deformations of brain tissue that may severe axons immediately or, in most cases, initiate a process of delayed axonal disruption (eFig. 133-16 A). An axonal stretching injury causes misalignment and distortion of the components of the cytoskeleton (mainly neurofilaments and microtubules) within the axon. As a consequence of the blockade of normal axonal transport, proteins and membrane-bound organelles (mainly mitochondria) are accumulated at certain areas along the traumatized axoplasm, leading to the development of varicosities (less than 10 μm) in which the progressive accumulation of vesicle-packing proteins and organelles cause the apparition of multiple focal swellings also known as axonal beads (between 10 and 50 μm).116 Finally, the progression of axonal damage causes the degeneration and phagocytosis of the axonal segments distal to the focal swellings, a process known as wallerian degeneration, with the permanence of an end bulb or retraction ball (larger than 50 μm) on the proximal axon stump (eFig. 133-16 A).116 The process of wallerian degeneration was initially described by Waller and reported to the Royal Society in 1850.117 It usually begins in injured axons 1.5 days after the head injury. The morphologic and structural changes associated with axon degeneration are poorly understood, but a new mutant mouse model, which genetically induces a delayed process of wallerian degeneration, has shown that both physical injury and blockade of axonal transport trigger a proactive axon death program (eFig. 133-16 A).116 Therefore, two methods of wallerian degeneration should be assumed after severe TBI: direct transection of axons in bundles of white matter suffering the highest degree of inertial deformation after A/D forces and most frequently delayed Wallerian degeneration observed among a large population of axons suffering more moderate levels of distortion. Wallerian degeneration should no longer be exclusively considered an atrophic process caused by failure to deliver structural components of the axon but a rather should be seen as a regulated, proactive death program similar to apoptosis. Factors that execute this program must be constantly present in the axon but maintained in an inactive form before axonal injury, just as carefully regulated effectors of apoptosis are always present in cells.
Mechanisms of Traumatic Axonal Injury: The New Concept of Mechanoporation
Nowadays, DAI is considered to be mainly the result of the process of mechanoporation, or transitory, increased axon permeability due to mechanical deformation of the axon membrane and/or stretching injury on protein-channel constituents (eFig. 133-17 A).45,118,119 The concept of mechanoporation implies the formation of temporary membrane pores large enough to allow diffusion of macromolecules without lysis of the cell and, depending on the severity of injury, the membrane’s capability for spontaneous resealing. The process of cell membrane mechanoporation has been confirmed using different in vitro models of mechanical cell injury, which induce precise and controlled tissue or cell deformation. Post-traumatic cell responses to mechanical injury observed in vivo include neuronal depolarization, dysfunction of cytoskeletal elements in astrocytes and neurons, increased intracellular calcium concentration in astrocytes, and a drop in mitochondrial membrane potential with a parallel decrease in ATP levels in astrocytes.120 More recent in vitro and in vivo models have demonstrated the formation of nonspecific membrane pores using macromolecular, fluorescently labeled tracer molecules.121 An important conclusion from these studies is that some nonaxotomized neurons undergoing increased membrane permeability after injury can reseal their mechanically damaged membranes over time without manifesting signs of necrosis.
The viability of neurons undergoing mechanical axonal injury has been demonstrated using fluorescently labeled tracers administered before and after injury.118 Using the weight drop model of diffuse TBI, Farkas et al. found that the neuronal response to TBI is heterogeneous, with three populations of injured neurons being identified: (1) one undergoing permanent mechanoporation and necrotic cell death (approximately half of injured cells), (2) a scattered population of neurons showing transitory membrane mechanoporation followed by spontaneous resealing of their membranes (approximately 25% of injured cells 8 hours after TBI), and (3) a scattered population of neurons enduring delayed membrane damage or a delayed process of mechanoporation (about 20% of neurons 8 hours after injury) (eFig. 133-17 B-D).118 Interestingly, TBI did not cause influx of the labeled tracer into glial cells. Previous in vitro studies had suggested that neuronal cell membrane resealing occurred minutes to hours after injury,121 but the mechanisms for a delayed membrane opening are unknown. It is possible that sustained high ICP after TBI could alter the neuronal cell membrane, and this factor can influence the adverse outcome observed in some patients after an apparently mild or moderate injury.5
Mechanoporation induces long-term membrane leakage that predominantly occurs at the nodes of Ranvier, leading to the uncontrolled influx of ions and other molecules at this level that is thought to be the cause of neurofilament compaction and multifocal microtubule misalignment and/or disruption. The degree of neurofilament damage varies with the severity of the A/D force, with mild TBI showing misalignment of the cytoskeleton while severe injuries cause rapid neurofilament compaction not only within the axon but also at the level of dendrites. Excess intracellular axonal calcium is considered a major cause of neurofilament compaction due to damage the neurofilament-associated sidearms observed in axons after traumatic axonal injury.23 Increased intra-axonal Ca2+ influx is driven by the large extracellular gradient and has been shown as a necessary and sufficient step to induce wallerian degeneration, probably by triggering the activation of various cysteine proteases such as calpains. Calpains are ubiquitous, calcium-dependent cysteine proteases involved in the degradation of several structural proteins such as tubulin, microtubule-associated proteins, neurofilaments, and spectrin, as well as in the activation of phospholipases that can break down the cell membrane.121 Membrane stretching may also lead to the mechanoactivated opening of channels inserted within the membrane, such as the Ca2+-permeable α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, which are present at high density at nodes of Ranvier.122
A recent in vitro model of axonal traumatic injury induced by fluid shear stress on cultured neurons was able to reproduce the development of axonal focal beading after injury and confirm the molecular hallmarks of this process, such as increase in membrane permeability, localized microtubule disruption, and organelle accumulation (eFig. 133-16 B-E).119 At the axonal beads, a transitory, brief, high increase in [Ca2+]i was observed, in addition to accumulation of F-actin and disrupted microtubules that displayed bundled on both sides of each bead. Mitochondrial dysfunction induced by increased [Ca2+]i in the axolemma is thought to be another fundamental factor contributing to the delayed cell death, through the Ca2+-activated pore transition opening in mitochondria and abolition of ATP-energy generation.123
Axonal Membrane Repair as a Therapeutic Strategy for DAI
If physical damage to the cell membrane is the precipitating event leading to immediate necrosis or delayed apoptosis in neurons, then an attractive therapeutic approach would be to minimize membrane damage with the use of chemical polymers that allow efficient blocking of the process of mechanoporation. One strategy has been the use of surfactants such as the poloxamer 188 (P188) to restore membrane integrity. The mechanism by which P188 promotes the resealing of membrane pores is not completely understood, but this molecule has been observed to insert into defects of the phospholipidic cell membrane bilayer and to lowering its surface tension.119 Indeed, the use of this polymer in several models of TBI and spinal cord injury blocks the increased axonal membrane permeability following injury, contributing to restoration of membrane integrity. The introduction of P188 into a neuronal culture was able to block the axonal bead formation in response to a stretching injury, supporting the theory that mechanoporation represents one of the major initial physiopathologic events leading to axonal dysfunction and disconnection after TBI.
Traumatic Brain Edema: The Major Gray Matter Diffuse Lesion After TBI
Brain edema is the abnormal accumulation of water within the brain tissue. This alteration represents the pathologic substrate of brain swelling, a descriptive term referring to macroscopical engorgement of the brain, frequently in association with head injury (Fig. 133-4D). Brain swelling occurs in most patients suffering from severe TBI and in 5% to 10% of those with moderate injuries. It is a long-recognized critical problem because of the direct relationship between development of brain edema and intracranial hypertension, a parameter usually associated with a poor outcome after severe TBI.59,124 Once a threshold is reached, a small increase of parenchymatous edema is associated with an exponential increase of ICP that may cause brain herniation and rapid death.39 The degree of swelling assessed in the first CT obtained after injury is highly correlated to a worse outcome.7,36
Three principal pathologic types of brain swelling are encountered in patients after TBI125: First, swelling of white matter adjacent to cerebral contusions originates from the zone of necrotic and hemorrhagic tissue, where there is a loss of arteriolar regulation, an increased permeability of blood vessels, and a leak of water to the extracellular space. Second, diffuse brain swelling is caused by a combination of transitory increased BBB permeability during the initial minutes after injury, probably due to a stretch cell injury and increased endothelial pinocytosis, and later by predominant intracellular glial swelling triggered and maintained by loss of extracellular homeostasis115,126 (Fig. 133-4D). Finally, hemispheric brain swelling, most often seen in association with an ipsilateral acute SDH, is thought to represent a cytotoxic type of edema triggered by hemispheric ischemia and acute vasomotor paralysis, given the rapid time of evolution of this type of swelling.
Types of Brain Edema
Two major types of brain edema have been differentiated according to the compartment in which excess water is accumulated and to the physiopathologic processes involved in their generation: vasogenic and cytotoxic.125,126 Vasogenic edema consists of the accumulation of water within the extracellular compartment of brain tissue, caused by dysfunction or disruption of the BBB and leading to the escape of water and plasma constituents. Because intracellular osmotic content is not affected, cell volumes remain constant while the extracellular space expands. A mechanical stretching injury may cause a primary transitory opening of the BBB (eFig. 133-14 A).115 In addition, vasogenic edema may be observed associated with hypertensive responses induced by head injuries and as a consequence of inflammatory mediators released after TBI, such as arachidonic acid, that increase the BBB permeability. Cytotoxic edema (also known as oncotic cell swelling or oncosis, from the Greek word onkos, which means “swelling”) consists of the accumulation of water within the brain cells (intracellular compartment) caused by any loss of cellular homeostasis (eFig. 133-14 B and D). Typically, this form of cellular edema is observed during hypoxic/ischemic conditions, where cells swell within seconds due to failure of the ATP-dependent Na+/K+ pump, resulting in rapid intracellular accumulation of Na+ and water driven by the electrochemical gradient.126 Cytotoxic edema can also originate from massive release of Glu to the extracellular space that leads to the opening of membrane channels, followed by Na+ influx, membrane depolarization, and secondary influx of Cl− and water. This type of cytotoxic edema due to the excitotoxic effect of Glu has been termed “neurotoxic edema” (eFig. 133-14 B).126 Cytotoxic edema causes an increase in the volume of the cell, usually followed by a compensatory increase of intracellular osmolytes, including ions and organic idiogenic osmolytes (electroneutral or charged molecules).41 The intracellular flux of water and the consequent increase in cell volume result in morphologic alterations of the membrane surface and the architecture of the cell that may signal a premorbid cellular process, leading to oncotic or necrotic cell death. In contrast, apoptosis presents with cell involution and shrinkage before death ensues.
Cellular Edema in TBI: The Role of Astroglia
Diffuse brain swelling occurs frequently after severe TBI, and its major pathologic substrate is the development of glial cell swelling involving predominantly astrocytes.126 Traditionally, the edema associated with TBI was considered to be predominantly vasogenic. Recent experimental works showed, however, that vasogenic edema usually coexists with or is the preceding cause of a cytotoxic, intracellular edema developed at later stages after TBI (eFig. 133-14 A). Indeed, TBI induces a predominant swelling of astrocytes (eFig. 133-14).41,127,128 Vasogenic edema is restricted to the early phase after the impact due to a transitory opening of BBB, lasting 15 to 30 minutes on average, although it may last longer in patients suffering from associated secondary insults such as episodes of hypotension or hypoxia (eFig. 133-14 A).39 A stretching membrane injury occurring at the moment of impact causes synchronous depolarization of neurons and massive release of K+ and Glu into the extracellular space.37 Active uptake of K+ and Glu is performed through specific channels and transporters present in the membrane of astrocyte end feet surrounding synaptic terminals. Astrocyte Glu uptake is coupled with influx of Na+ into the cytosol and followed by passive entrance of Cl− to maintain electroneutrality and then by water inflow via AQP channels to maintain isosmotic intracellular conditions (eFig. 133-14 B) (see section on functional neuron–glial coupling).95 Excess [K+]o and [Glu]o after TBI may overwhelm the buffering capability of astrocytes, leading to intracellular astrocyte swelling (eFig. 133-13 D).127,128 In addition, mechanical deformation of voltage-sensitive and agonist-gated ion channels in astrocytes can make some of these channels remain leaky for several hours, contributing to the development of cell swelling (eFig. 133-17 A). Cytotoxic edema is particularly prominent at the astrocytic end feet surrounding cerebral capillaries, because these cellular processes show the highest expression of K+ channels and AQP4 (eFig. 133-14 C). Long-term swelling of astrocyte end feet may cause an additional impairment of regional CBF due to compression of the microvasculature, worsening the edema and secondarily causing cerebral ischemia.
Recent analysis of the progression of metabolic changes experienced within edematous brain tissue in Marmarou’s experimental TBI model has been performed using high-resolution proton-MRS technology. This study has shown that post-traumatic cytotoxic cell swelling caused by TBI induces a marked increment in the content of molecules such as taurine and myoinositol within the brain tissue during the first 48 hours after the impact.41 Enhanced synthesis and accumulation of these molecules, known as organic osmolytes, is thought to reflect an adaptive mechanism to accommodate swollen cells in situations of sustained, increased cell volume, as observed in glial cells after ischemic or traumatic injury. Removal of organic osmolytes from cells after recovery of homeostasis in brain tissue is a slow process requiring the expression of specific channels in the membrane of cells and might be the reason for long-term maintenance of diffuse brain edema after TBI.128,129
Roles of AQPs in the Development of Traumatic Brain Edema
Brain function is inextricably coupled to water homeostasis. Brain metabolism of glucose yields substantial amounts of water, which needs to be transported through the lipid bilayer of neurons and glial cells through well-regulated mechanisms. In 1992, Agre et al. confirmed the existence of a family of water channels incorporated into the membrane of cells known as AQPs, which allows the rapid transport of water across cell membranes driven by osmotic and hydrostatic gradients.129a The identification of these channels meant a great advance in the comprehension of the mechanisms leading to brain edema after injury, as well as a potential promising target for the design of specific new antiedema therapies.95 AQP4 is the predominant type of AQP, expressed in the brain, and it is mainly located in the membrane of astroglial cells—specifically, at the end feet processes ensheathing cerebral capillaries and synapses, the structures showing the earliest and highest degree of edema after TBI (eFig. 133-14 C).115 Quantitative immunogold analysis has revealed a strict colocalization of AQP4 and Kir4.1 (a potassium channel involved in the clearance of extracellular K+ in glial end feet), indicating that the two channels act in concert.130 High neuronal activity is associated with rapid shrinkage of the adjacent extracellular space that is thought to be caused by siphoning of extracellular K+ coupled with water through the AQP4 and Kir4.1 channels strategically placed in pericapillary and subpial astrocyte end feet.
The first evidence linking AQP4 to brain edema was reported in the seminal study by Manley et al., who demonstrated that brain edema resulting from experimental stroke or water intoxication was reduced in AQP4 knockout mice (eFig. 133-14 D).87,88 Similar results were found in dystrophin-deficient mice, in which AQP4 loses its polarized expression in the astrocytes end feet due to the lack of molecules anchoring AQP4 to the membrane. The edema reduction is only about 50% after deleting perivascular AQP4; hence, the residual edema probably reflects water fluxes through the lipid bilayer or other type of channels. Although the mechanisms that govern the expression of AQP4 are largely unknown, the expression level of AQP4 might be subject to regulation, and up- and downregulation have been described in a number of pathologic conditions. Astrocytes lacking AQP4 have sevenfold reduced water permeability.131 A decrease in AQP4 was observed in edematous areas with BBB disruption in experimental models of TBI,132 while an increase in astrocytic end feet AQP4 occurred in conditions of elevated extracellular K+ and cytotoxic cell swelling.87,88
Regarding the role played by AQP4 in the evolution of the vasogenic type of edema, theoretically AQP4 would not affect the rate of edema formation after BBB disruption, because this edema involves fluid efflux from the vasculature directly into the extracellular space and not through a transcellular pathway. However, AQP4 can be implicated in the clearance of water from the extracellular space, an impairment that could be of equal importance in the maintenance of vasogenic edema.133 Labeled proteins injected into the extracellular space move into the cerebral ventricles by bulk flow and are eventually cleared with CSF, which is presumed to be the normal intracerebral fluid clearance pathway.134 However, a transparenchymal route for extracellular fluid resorption toward the cortical microvessels would predominate under high ICP conditions.133 This hypothesis was recently confirmed by Papadopoulos et al., who showed a greater increase in brain water content and ICP in AQP4-deficient mice after continuous infusion of artificial CSF into the cerebral extracellular space. In addition, AQP4-deficient mice subjected to BBB breakdown and vasogenic edema caused either by cortical freeze injury or by tumor/abscess implantation present with a significant increase in brain water content and high ICP.135
Treatment of Traumatic Brain Edema
The treatment of cellular edema is too complex due to the lack of knowledge about the exact mechanisms that govern the gain and loss of cell volume. Primary methods of treatment such as CSF diversion, hyperosmotic therapy, and corticoids have been the standards for more than half a century. Nevertheless, neither the use of drugs counteracting the effects of inflammatory compounds released in TBI, such as bradykinin or arachidonic acid, nor the use of hypertonic solutions, such as mannitol, have been shown to be effective in severe head injury.5 Steroids have been used in vasogenic edema on the basis of a theoretical beneficial effect in suppressing inflammatory mediator release, thereby limiting the BBB’s increased permeability and preventing the extracellular accumulation of edema fluid. Dexamethasone can improve the BBB function by increasing the tightness of the brain endothelial tight junctions and by upregulating the expression of BBB P-glycoprotein. Administration of steroids would be more effective in early stages of edema formation, but overall no significant benefits have been proved in TBI, and the last guidelines of the Brain Trauma Foundation do not recommend administrating steroids to severely head-injured patients.5 The rationale for the use of hypertonic fluids such as mannitol is based on the effectiveness of these therapies to drawing fluid from within the brain parenchyma, thereby enhancing the clearance of edema. However, mannitol is only temporarily effective, because parenchymal fluid reaccumulates if the underlying cause of edema production is not counteracted. A more rational therapy could be based on the modulation of AQP4 function. Selective suppression or enhancement of AQP4 activity could counteract the formation of edema or favor its clearance, respectively, depending on the predominant mechanism involved in edema production. However, a major problem of therapies targeting AQP4 modulation is that AQP4 facilitates bidirectional movement of water and the efflux of water from brain cells is expected to be beneficial in the resolution phase of cytotoxic brain edema. No effective drug capable of adequately modulating AQP4 expression or function has been identified to date.99
Secondary Brain Damage Caused by Cerebral Ischemia
The concept of delayed secondary neurologic damage after head injury is supported by the “lucid interval” observed in 30% to 40% of severely head-injured patients who finally die.27,46 This implies that in many cases primary impact events are not sufficiently severe to damage the brain beyond the capacity of function, emphasizing the importance of secondary damage. Hypoxic/ischemic brain damage is considered to represent the second most important factor in determining a bad outcome in patents remaining severely disabled or in a VS after DAI.14,136 This type of damage was confirmed in approximately 55% of patients included in the Glasgow database.18 The highest incidence of ischemic damage was observed in Ammon’s horn (80%), in the basal ganglia (79%), and in the cerebellum (44%). When CBF is profoundly reduced (less than 5-10 ml/100 g/min) within the distribution of one cerebral end artery for more than 60 to 90 minutes, infarction ensues.89 However, when the flow reduction is less marked (around 15-18 ml/100 g/min) and persists for more than 30 minutes, selective neuronal loss may occur, which affects the most vulnerable neuronal populations, such as the hippocampal neurons of the molecular layer, especially within the CA1 and CA3 sectors; the cerebellar granular cells; and cortical large neurons, especially in the cuneate visual cortex. Selective neuronal loss in these areas may help explain the high prevalence of memory disorders and coordination difficulties in severely head-injured survivors. Focal injuries produce zones of profoundly reduced regional CBF that induce neuronal necrosis. In adjacent zones that did not reach critical levels of ischemia, Glu neurotoxicity may play a role in producing secondary ischemia.
Molecular Cell Pathophysiology of TBI
As explained in the first section of this chapter, the classic classification of Adams et al. for post-traumatic brain lesions differentiates between primary and secondary injuries after TBI.13 This classification emphasizes the difference between unavoidable, immediate-impact lesions and avoidable, secondary lesions that occur at a variable time after injury. TBI is the result of a primary mechanical impact lasting less than 100 msec that abruptly disrupts the brain parenchyma with shearing and tearing forces acting on cerebral blood vessels and brain tissue. However, the resulting pathophysiologic events are more prolonged and are the cause of delayed cell dysfunction. Secondary injury is derived from the physiologic biomolecular alterations set in motion by the trauma. A better understanding of TBI has made it clear that traumatic brain lesions are dynamic, and new insights into the specific physiopathologic neurochemical cascades activated by mechanical injury have blurred the traditional boundaries between primary and secondary injuries.19 A recently introduced concept of great importance is that of “traumatic penumbra” to parallel the well-accepted term of “ischemic penumbra.” Traumatic penumbra can be defined as the area of especially vulnerable but still viable injured brain tissue, which can either recover or eventually undergo death, depending on the secondary pathophysiologic events triggered by the trauma. Delayed structural and functional impairment of neurons and glial cells after TBI is determined by specific biochemical and biomolecular cascades that are set in motion by mechanical cell injury and have been investigated in depth in the last decade.
Neuronal Dysfunction in TBI
Following severe TBI, a complex interplay of three major pathomechanisms at the cellular level—acting concurrently or simultaneously with a synergistic effect—are set in motion: (1) loss of ionic homeostasis with increased levels of extracellular K+ and high levels of intracellular Ca2+, (2) increased Glu release to the extracellular space causing excitotoxicity, and (3) mitochondrial dysfunction and oxidative stress.123,137 These mechanisms activate biochemical cascades within damaged brain cells that ultimately can lead to axonal transport failure and neural death, which in turn determine patient outcome.
• Loss of extracellular ionic homeostasis. The initial cellular response to trauma is widespread and massive depolarization of neurons and glial cells. The majority of voltage-sensitive and agonist-gated ion channels are sensitive to transient mechanical deformation by shearing forces that cause rapid (and sometimes persistent) Na+ and Ca2+ entry into the cells and massive K+ efflux into the extracellular space. A massive and rapid transient efflux of potassium into the extracellular fluid following the head impact has been demonstrated in experimental TBI models.37 Astrocytes uptake excess K+ from the extracellular space in an attempt to restore the ionic composition of the neuronal microenvironment, but this buffering mechanism can result in rapid astrocyte swelling.138 (See the section on glial dysfunction in TBI.)
• Post-traumatic excitotoxicity. Release of an excitatory amino acid such as Glu significantly contributes to the calcium influx into brain cells following TBI. Increased extracellular Glu concentration ([Glu]o) measured by microdialysis in severely head-injured patients correlated well with their poor outcome following the injury. The fundamental factor mediating the excitotoxic effects of Glu seems to be intracellular Ca2+ overload. Abnormally high [Glu]o activates a variety of specific Glu receptors, such as N-methyl-d-aspartic acid receptors and AMPA receptors, allowing for activation of voltage-dependent Ca2+ channels and massive influx of this ion that propagates Glu excitotoxicity by further stimulating the release of Glu in another vicious cycle.
• Mitochondrial dysfunction and oxidative stress. Post-traumatic dysfunction of mitochondria leads to the inability to generate ATP, the basic molecule that provides the energy for the vast majority of cellular functions, including the maintenance of an ionic gradient. As mitochondria produce the majority of ATP within the cell, traumatic damage of these organelles plays a crucial and central role in TBI.139 Recent experimental studies examining whole brain homogenates noted a decline in ATP levels in rats subjected to the weight-drop injury model that was maximal by 6 hours after injury.103 In addition, this dysfunction is followed by the generation of reactive oxygen species that trigger DNA fragmentation, lipid peroxidation, and disruption of cellular membranes, resulting in cell lysis and death.
It is not clear whether mitochondrial dysfunction is directly caused by the stretching injury or is secondary to a massive influx of calcium triggered by Glu excitotoxicity, and the excessive cytosolic Ca2+ influx caused in severe TBI may overwhelm the buffering capability of mitochondria and cause mitochondria swelling.139 Mitochondria play a key role in the buffering of high intracellular calcium levels. A traumatic stretching injury may cause a drop in mitochondrial membrane potential and induce mitochondrial membrane permeabilization in neurons and glial cells, an event consisting of the disruption of mitochondrial membranes that cause functional impairment of mitochondria and ultimately lead to cell death.120,140 Surprisingly, isolated neurons cultured in a silastic membrane subjected to severe stretching show a decline in mitochondrial membrane potential that is not accompanied by a change in ATP, while neurons in mixed glial–neuron cultures how a marked ATP drop even after minor–moderate stretching of the silastic membrane.120 This difference could occur because astrocytes are more rigidly attached to the silastic membrane than neurons; thus, neurons in mixed cultures experience a greater amount of stretch, a phenomenon that can similarly occur in the brain after TBI.
Mitochondrial dysfunction is a leading event in the activation of biochemical cascades, causing apoptosis and necrosis of neurons after TBI. Mitochondrial membrane permeabilization can derive from either of two distinct, but overlapping, molecular mechanisms: (1) formation of large channels in the outer membrane of mitochondria by activation of proapoptotic proteins of the BCL-2 family and (2) opening of a megachannel known as the permeability transition pore (PTP) at the inner mitochondrial membrane.139 In response to specific stimuli (Ca2+ overload and reactive oxygen species), the PTP assumes a high-conductance conformation, allowing the unregulated entry of solutes and water into the mitochondrial matrix and leading to the dissipation of mitochondrial transmembrane potential and progressive swelling of these organelles. Swelling of the mitochondrial matrix may cause the breakdown of the outer mitochondrial membrane and spillage of proteins from the intermembrane space into the cytosol, activating the caspase cascade that can cause irreversibly damage to the DNA.140 Cyclosporin A is an immunosuppressant that specifically inhibits PTP opening and preserves mitochondrial integrity. This drug has proved beneficial in experimental models of brain injury,139 as well as in recent clinical TBI trials.123
Glial Dysfunction in TBI
Traumatic injury to astrocytes can compromise the homeostasis of the extracellular microenvironment within the brain tissue and disrupt critical neural–glial interactions.141 The role played by astrocytes in the buffering of extracellular Glu may have particular importance after TBI, when large fluxes of extracellular Glu contribute to the acute excitotoxic process.37 A correlation between injured brain areas showing a loss of glial fibrillary acidic protein (GFAP) and glutamine synthetase immunoreactivity 1 hour after injury and regions showing delayed post-traumatic neuronal degeneration has been observed in experimental TBI models.141 Extracellular overload of Glu and K+ caused by massive depolarization of neurons after the impact usually overwhelms the buffering capacity of glial cells, leading to astrocyte swelling, a prominent feature of TBI. Severe astrocytic edema, dissociation of the perivascular astrocyte end feet from the capillary basement membrane, and disruption of interastrocyte gap junction are all observed in cortical biopsies of human TBI complicated by SDH.127 However, mechanical strain injury to astrocytes produces a rapid rise in [Na+]i and [Ca2+]i141 It is thought that increased intracellular sodium can augment the influx of calcium into the cells through a reversal functioning of the sodium/calcium exchanger. Mechanical strain injury to astrocytes also causes release of ATP that is a potent stimulus for gliosis.141a
Reactive Astrogliosis after TBI
Cerebral tissue damage induced by TBI is followed by the development of barriers that prevent axonal regeneration. Injuries to the CNS induce a response within the damaged tissue characterized by the development of reactive astrogliosis and a dense glial scar aimed at achieving isolation of the damaged region and restoration of its homeostasis (eFig. 133-18 A).142,143 Astrocytes respond to TBI by pronounced alterations in their morphology and functional properties, mainly by a process of astrocyte hypertrophy and proliferation, known under the global term of reactive astrogliosis (more astrocytes).142 Moderate, focal, contusive cortical injury induces robust reactive astrogliosis characterized by a marked increase of GFAP expression in most astrocytes beneath the impact site, in addition to substantial hypertrophy and actively proliferation of these cells (eFig. 133-18 A). The functional changes of reactive astrocytes in response to TBI in vivo are not well understood; specifically, it is uncertain whether reactive astrocytes upregulate or downregulate the normal functions of their nonreactive counterparts, such as the maintenance of extracellular homeostasis, clearance of extracellular K+ and Glu, water transport, and production of pro- and anti-inflammatory cytokines and growth factors. Given the range of activities carried out by astrocytes, a complex combination of protective and detrimental effects might be expected as a result of post-traumatic astrogliosis.141,144
Recent studies have demonstrated that the glial scar is more than just a physical barrier and might provide several important beneficial functions for sealing the BBB and stabilizing fragile CNS tissue after injury. In injured CNS tissue neurons spared at the primary insult are exposed to an imbalanced microenvironment that contains toxic factors such as excess excitatory amino acids and ions, reactive oxygen species, free radicals, and overwhelming inflammation. This results in further neuronal loss, a process known as secondary degeneration. It is now clear that a major role of the glial scar is to seclude the injury site from healthy tissue, preventing a cascade of uncontrolled tissue damage (eFig. 133-18 A). In the vicinity of areas with BBB disruption, reactive astrocytes are directly involved in the production of the basic components of the extracellular matrix (ECM), mainly of proteoglycans, a kind of glycoprotein destined for the basal lamina.143 One class of these molecules, the chondroitin sulfate proteoglycan (CSPG), is extremely inhibitory to axon outgrowth in culture, with axonal growth cones rapidly turning away as they contact CSPGs by selective retraction of filopodia.145
The ECM of scar tissue and its proteoglycans have a presumably beneficial key role in the repair of injured brain tissue by limiting the clotting process, sealing the lesion site, restoring homeostasis, and immobilizing the cytokines and growth factors that activate the immunologic and phagocytic response and promote regeneration.146 However, upregulation of inhibitory proteoglycans within the post-traumatic scar constitute the major obstacle to recovery in later periods after TBI, because it leads to the activation of macrophages and formation of cavities fenced by a glial scar, which inhibits axonal regeneration (eFig. 133-18 A).145 When regenerating axons encounter the environment of the glial scar, end bulbs develop and the process of axonal regeneration halts, an observation first reported by Ramón y Cajal.25 Axons that cannot regenerate through the barrier of reactive glial cells usually adopt a dystrophic appearance of stalled growth.145 On the assumption of a predominantly inhibitory, detrimental effect of the glial scar, various therapeutic scar-modulation approaches have attempted to eliminate and reorganize its chemical components to promote axonal regeneration. Enzymatic digestion of CSPGs in vivo by local or intrathecal injection of chondroitinase, the enzyme that removes the inhibitory action of this proteoglycan at the ECM, has proved to be a useful strategy for axonal regeneration in experimental models of CNS injury.143,146,147
Beneficial Roles of Glial Scar in CNS Repair
Although it is generally assumed that the participation of reactive astrocytes in the process of CNS repair after TBI is harmful overall, accumulating evidence indicates that the glial scar has beneficial effects at one phase of the recovery process and destructive effects at another phase. It is likely that growth inhibition in the early stages after the injury, rather than being an obstacle to recovery, is essential for preservation of neurons capable of regrowth. Consistent with this time-dependent view of the scar, application of xyloside, an inhibitor of CSPGs, on day 0, 2, and 7 after spinal cord injury resulted in destructive, beneficial, or no effect, respectively.146 Furthermore, the effects of astrogliosis on the early phases of post-traumatic brain damage brain tissue were recently investigated in a model of controlled cortical impact injury (CCII) using a transgenic mouse in which proliferating astrocytes expressing the herpes simplex virus–thymidine kinase enzyme can be selectively killed with the administration of ganciclovir (eFig. 133-18 B). In this model, after a moderate CCII that normally would not cause pronounced loss of neural tissue, a marked increase in the volume of damaged tissue occurred when ablation of reactive astrocytes was induced with ganciclovir administration (60% of the greater parenchymal damage 28 days after injury). In contrast, after a severe CCII causing substantial cortical destruction, the extent of injured tissue was not significantly affected by the elimination of astrogliosis (eFig. 133-18 B).148 These findings support the concept that reactive astrocytes exert a net beneficial, neuroprotective effect in the brain after injuries of moderate intensity, helping preserve the global cytoarchitecture of the contusioned tissue several weeks after the injury. Enabling axonal regrowth in the chaotic and metabolically unbalanced environment that is the injured CNS in its acute phase of recovery might cause more damage than benefit.147
Axonal Regeneration After TBI
Despite early claims that axons in the adult CNS were incapable of regeneration, experimental research has proved that CNS axons are capable of at least small amounts of regeneration through a process of sprouting.147 Two major obstacles to axonal regeneration after injury have been identified to date: the formation of the glial scar and the presence of inhibitor myelin-associated molecules.143,147,149 Before the scar matures, however, the exposure of growing axons to unstructured myelin seems to be the predominant source of growth inhibition. Ramón y Cajal was the first author to suggest that white matter could block the regeneration in the CNS.25 This fact was definitely confirmed when the use of a monoclonal antibody (IN-1) against the fraction of myelin, which inhibited neurite growth in both in vitro and in vivo situations, allowed axons to grow on myelin. In the 1990s in experiments of axonal growth performed in vitro, several research groups identified some growth-inhibitory components associated with myelin, which were thought of as major inhibitors of axonal regeneration in vivo after injury to the CNS. Among these molecules, three inhibitors of axonal growth were considered the most relevant: (1) a myelin-derived family of proteins known as Nogo, present in the oligodendrocyte surface and on the outer- and the innermost loops of the myelin membrane; (2) the myelin-associated glycoprotein located at the inner myelin loop membrane, especially in the perinodal regions; and (3) the oligodendrocyte myelin glycoprotein expressed not only by oligodendrocytes but also at high levels in neurons, specifically next to the nodes of Ranvier.149 These three myelin-associated neuron growth inhibitors induce growth cone collapse and inhibit neurite outgrowth through the interaction with the same neuronal receptor known as the Nogo receptor. The general conclusion from these studies was that myelin-associated molecules are one of the principal causes of failure of regeneration of axons in the intact adult CNS (eFig. 133-18 C1). Therapies have been designed to overcome the inhibition of axonal regrowth mediated by effects of myelin-derived molecules and encourage regeneration, specifically by blocking the Nogo receptor with IN-1, with varying degrees of success in vivo.
Axonal Regeneration through Well-Structured White Matter
The proposal that central myelin is inhibitory to axon growth did not explained why axons also fail to regenerate through gray matter, where there is no myelin. It is also counterintuitive because myelinated fiber tracts are a widely conserved anatomic structure through which axons may travel in the adult brain and spinal cord. An alternative hypothesis to the classic view of myelin as an inhibitor of axonal regeneration is that the collapse of axonal growth cones observed in vitro is actually a reflection of a physiologic repulsive contact-guidance mechanism that facilitates and accelerates regeneration of adult axons along myelinated tracts in vivo.147 If this is the case, therapies aiming to block the myelin-inhibitory mechanisms might actually prevent regeneration. It is possible that intact myelin in which few of the known inhibitors are on the surface is permissive for regeneration, and only when it is disrupted by injury are an abundance of inhibitors exposed to the growth cone (eFig. 133-18 C and D). Indeed, axonal growth from adult dorsal root ganglion neurons along adult myelinated fiber tracts has been demonstrated in vivo after microtransplantation experiments, which consisted of introducing these cells with great care into the mature spinal cord or the corpus callosum without inducing inflammation or glial scar formation and carefully avoiding myelin disruption. Under such conditions, adult neurons were able to show regeneration either within a relatively intact host glial environment or, rather unexpectedly, within areas undergoing wallerian degeneration in which abundant oligodendrocytes, degenerating myelin, and reactive astrocytes, all elements with a supposedly inhibitory effect on axon growth, were present.
Tissue cultures used to study axonal regeneration are two-dimensional structures that do not reflect the complex three-dimensional arrangements of cells and myelinated fiber tracts in vivo. Axons growing in vivo elongate their growth cones along a defined pathway until they reach the neuropil, where they branch off profusely. The purpose of the so-called inhibitory molecules that are associated with CNS myelin is to direct and facilitate long-distance growth of axons along myelinated tracts. Axonal branching is associated with the cessation of elongation, and conversely axonal elongation toward a specific target needs the action of repulsive molecules arranged along adjacent myelinated tracts that inhibit branching (eFig. 133-18 C1). The reason for the difference between the effect of myelin inhibition in vitro and the situation in vivo is one of geometric organization. In healthy tissue, in vivo inhibitory myelin–associated molecules are orderly arranged and exposed along the parallel axonal tracts supporting the growth of axons, whereas in both lesioned areas of the CNS or in vitro conditions, the unstructured myelin prevents the formation of valid pathways for axonal growth, causing the collapse of the growth cone (eFig. 133-18 C2). Although adult axons sprout vigorously in the lesion area, the sprouts remain restricted to the lesion site and cannot advance to their targets to restore functional connections.147
The narrow pathways between myelinated tracts in vivo are signaled by the arrangement of white matter astrocytes and their fine longitudinal astrocytic processes (eFig. 133-18 D). As the growing axon advances, it follows these longitudinal astrocytic processes, which guide it through the narrow channel, flanked on all sides by myelinated fibers coated with repulsive molecules. It is likely that inhibition of growth is not due to the inhibitory effects by oligodendrocytes and central myelin but is related to the proliferation of a meshwork of reactive astrocytic processes—reactive astrocytes that create an impenetrable barrier. In the absence of any myelinated fibers, the reactive astrocytes of the denervated tract change their conformational arrangement and condense to form a dense and impenetrable glial scar, thereby eliminating all channels along which axons might grow.147
1. Teasdale G., Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet. 1974;2:81-84.
2. Saatman K.E., Duhaime A.-C., Bullock R., et al. Classification of traumatic brain injury for targeted therapies. J Neurotrauma. 2008;25:719-738.
3. Prieto R., Gutiérrez-Gonzalez R., Pascual J.M., et al. Experimental models of traumatic brain injury. Neurocirugia. 2009;20:225-244.
4. Bullock M.R., Chesnut R., Ghajar J., et al. Guidelines for the surgical management of traumatic brain injury. Neurosurgery. 2006;58(S2):1-62.
5. Brain Trauma Foundation. Guidelines for the management of severe traumatic brain injury. J Neurotrauma. 2007;24:S1-S106. 3rd ed.
6. Langlois J.A., Rutland-Brown W., Wald M.M. The epidemiology and impact of traumatic brain injury. A brief overview. J Head Trauma Rehabil. 2006;21:375-378.
7. Marshall L., Marshall S., Klauber M., et al. New classification of head injury based on computerized tomography study. J Neurosurg. 1991;75:S14-S20.
8. Murray G.D., Butcher I., McHugh G.S., et al. Multivariable prognostic analysis in traumatic brain injury: results from the IMPACT study. J Neurotrauma. 2007;24:329-337.
9. Posner J.B., Saper C.B., Schiff N.D., Plum F. Plum and Posner’s Diagnosis of Stupor and Coma, 4th ed. Oxford University Press; 2007.
10. Gennarelli T.A., Thibault L.E. Biomechanics of head injury. In: Wilkins R.H., Rengachary S.S. Neurosurgery. New York: McGraw-Hill; 1985:1531-1536.
11. Lobato R.D., Cordobes F., Rivas J.J., et al. Outcome from severe head injury related to the type of intracranial lesion: a computerized tomography study. J Neurosurg. 1983;59:762-774.
12. Maas A.I., Steyerberg E.W., Butcher I., et al. Prognostic value of computerized tomography scan characteristics in traumatic brain injury: results from the IMPACT study. J Neurotrauma. 2007;24:303-314.
13. Adams J.H., Mitchel D.E., Graham D.I., et al. Diffuse brain damage of immediate impact type. Its relationship to “primary brain-stem damage” in head injury. Brain. 1977;100:489-502.
14. Graham D.I., Adams J.H. Ischemic brain damage in fatal head injuries. Lancet. 1971;1:265-266.
14a. Symonds C.P. Concussion and contusion of the brain. In: Brock S., editor. Injuries of the Skull, Brain and Spinal Cord. London: Bailliere, 1940. 1940:71-115
15. Denny-Brown D., Russell W. Experimental cerebral concussion. Brain. 1941;64:93-164.
16. Holbourn A.H.S. The mechanics of trauma with special reference to herniation of cerebral tissue. J Neurosurg. 1944;1:190-200.
17. Strich S.J. Shearing of nerve fibres as a cause of brain damage due to head injury. A pathological study of twenty cases. Lancet. 1961;2:443-448.
18. Adams J.H.. Brain damage in fatal non-missile head injury in man, Braakman R., editor, Handbook of Clinical Neurology, Head Injury, 1990;57(Vol. 13):43-63 Amsterdam: Elsevier Science Publishers BV
19. Sahuquillo J., Poca M.A. Diffuse axonal injury after head trauma. A review. Adv Tech Stand Neurosurg. 2002;27:23-86.
20. Gennarelli T.A., Thibault L.E., Adams J.H., et al. Diffuse axonal injury and traumatic coma in the primate. Ann Neurol. 1982;12:564-574.
21. Ommaya A.K., Gennarelli T.A. Cerebral concussion and traumatic unconsciousness: correlation of experimental and clinical observations of blunt head injuries. Brain. 1974;97:633-654.
22. Moruzzi G., Magoun H.W. Brain stem reticular formation and activation of EEG. Electroencephalogr Clin Neurophysiol. 1949;1:455-473.
23. Grady M.S., McLaughlin M.R., Christman C.W., et al. The use of antibodies targeted against the neurofilament subunits for the detection of diffuse axonal injury in humans. J Neuropathol Exp Neurol. 1993;52:143-152.
24. Skandsen T., Kvistad K.A., Solheim O., et al. Prevalence of diffuse axonal injury in patients with moderate and severe head injury: a cohort study of early magnetic resonance imaging findings and 1-year outcome. J Neurosurgery Oct. 23, 2009.
25. Ramón y Cajal S. Degeneration and regeneration of the nervous system. London: Oxford University Press; 1928.
26. Ramón y Cajal S. Los fenómenos precoces de la degeneración traumática de los cilindro-ejes del cerebro. Trab Lab Invest Biol Univ Madrid. 1911;9:39-95.
27. Reilly P.L., Graham D.I., Adams J.H., Jennett B. Patients with head injury who talk and die. Lancet. 1975;2:375-377.
28. Gennarelli T.A. Cerebral concussion and diffuse brain injuries. In: Cooper P.R., editor. Head injury. Baltimore: Williams & Wilkins; 1987:108-124.
29. Rodda R.A., Adams J.H., Graham D.I., Doyle D. Fatal non-missile head injury: a cohort study of patients with and without an intracranial hematoma. Clin Neuropathol. 1987;6:179-184.
30. Oprescu I. Cerebral hemispheric contusions and lacerations. In: Frowein R.A., editor. Cerebral Contusions Lacerations and Hematomas. Wien: Springer-Verlag; 1991:24-59.
31. Courville C.B. The mechanism of coup–contrecoup injuries of the brain. Bull Los Angeles Neurol. 1950;15:72-86.
32. Adams J.H., Doyle D., Graham D.I., et al. Gliding contusions in nonmissile head injury in humans. Arch Pathol Lab Med. 1986;110:485-488.
33. Scheinker I.M. Vasoparalysis of the central nervous system, a characteristic vascular syndrome. Significance in the pathology of the central nervous system. Arch Neurol Psychiat (Chicago). 1944;12:32-43.
34. Lindenberg R., Freytag E. Morphology of cortical contusions. AMA Arch Pathol. 1957;63:23-42.
35. Buki A., Povlishock J.T. All roads lead to disconnection? Traumatic axonal injury revisited. Acta Neurochir (Wien). 2006;148:181-193.
36. Marshall L.F., Toole B.M., Bowers S.A. The national Traumatic Coma Data Bank. II: Patients who talk and deteriorate: Implications for treatment. J Neurosurg. 1983;59:285-288.
37. Katayama Y., Becker D.P., Tamura T., Hovda D.A. Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J Neurosurg. 1990;73:889-900.
38. Kawamata T., Katayama Y. Cerebral contusion: a role model for lesion progression. Prog Brain Res. 2007;161:235-241.
39. Marmarou A., Signoretti S., Fatouros P.P., et al. Predominance of cellular edema in traumatic brain swelling in patients with severe head injuries. J Neurosurg. 2006;104:720-730.
40. Kawamata T., Mori T., Sato S., Katayama Y. Tissue hyperosmolality and brain edema in cerebral contusion. Neurosurg Focus E5. 2007.
41. Pascual J.M., Solivera J., Prieto R., et al. Time course of early metabolic changes following traumatic brain injury in rats as detected by 1H NMR spectroscopy. J Neurotrauma. 2007;24:944-959.
42. Alahmadi H., Vachhrajani S., Cusimano M.D. The natural history of brain contusion: radiological and clinical progression. J Neurosug. 2010;112:1139-1145.
43. Nakamura N., Yamaura A., Shigemori M., et al. Epidemiology, prevention and countermeasures against severe traumatic brain injury in Japan and abroad. Neurol Res. 2002;24:45-53.
44. Povlishock J.T., Christman C.W. The pathobiology of traumatic induced axonal injury in animals and humans: a review of current thoughts. J Neurotrauma. 1995;12:555-564.
45. Gennarelli T.A. The spectrum of traumatic axonal injury. Neuropathol Appl Neurobiol. 1996;22:509-513.
46. Adams J.H., Doyle D., Ford I., et al. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology. 1989;15:49-59.
47. Gentleman S.M., Nash M.J., Sweeting C.J., et al. β-Amyloid precursor protein (β-APP) as a marker for axonal injury after head injury. Neurosci Lett. 1993;160:139-144.
48. Mannion R.J., Cross J., Bradley P., et al. Mechanism-based MRI classification of traumatic brainstem injury and its relationship to outcome. J Neurotrauma. 2007;24:128-135.
49. McBride DQ, Patel AB, Caron M. Early repeat CT scan: Importance in detecting surgical lesions alter closed head injury. 2nd International Neurotrauma Symposium, Glasgow, July, 1993.
50. Zheng W.B., Liu G.R., Li L.P. Prediction of recovery from a post-traumatic coma state by diffusion-weighted imaging (DWI) in patients with diffuse axonal injury. Neuroradiology. 2007;49:271-279.
51. Xu J., Rasmussen Y.-A., Lagopoulos J., Haberg A. Diffuse axonal injury in severe traumatic brain injury visualized using high-resolution diffusion tensor imaging. J Neurotrauma. 2007;24:753-765.
52. Sugiyama K., Kondo T., Oouchida Y., et al. Clinical utility of diffusion tensor imaging for evaluating patients with diffuse axonal injury and cognitive disorders in the chronic stage. J Neurotrauma. 2009;26:1879-1890.
53. Shutter L., Tong K.A., Holhouser B.A. Proton MRS in acute traumatic brain injury: role for glutamate/glutamine and choline for outcome prediction. J Neurotrauma. 2004;21:1693-1705.
54. Servadei F., Compagnone C., Sahuquillo J. The role of surgery in traumatic brain injury. Curr Opin Crit Care. 2007;13:163-168.
55. van den Heever C.M., van der Merwe D.J. Management of depressed skull fractures. Selective conservative management of nonmissile injuries. J Neurosurg. 1989;71:186-190.
56. Braakman R. Depressed skull fracture: Data, treatment and follow-up in 225 consecutive cases. J Neurol Neurosurg Psychiatry. 1972;35:395-402.
57. Marshall L.F., Gautille T., Klauber M.R., et al. The outcome of severe closed head injury. Report on the Traumatic Coma Data Bank. J Neurosurg. 1991;75(Suppl)):S28-S36.
58. Boto G.R., Gómez P.A., De La Cruz J., Lobato R.D. Severe head injury and the risk of early death. J Neurol Neurosurg Psychiatry. 2006;77:1054-1059.
59. Narayan R.K., Kishore P.R.S., Becker D.P., et al. Intracranial pressure: to monitor or not to monitor? J Neurosurg. 1982;56:650-659.
60. Seelig J.M., Becker D.P., Miller J.D., et al. Traumatic acute subdural hematoma: Major mortality reduction in comatose patients treated within four hours. N Eng J Med. 1981;304:1511-1518.
61. Miller J.D., Dearden N.M., Piper I.R., et al. Control of intracranial pressure in patients with severe head injury. J Neurotrauma. 1992;9(Suppl 1):S317-S326.
62. Rivas J., Lobato R., Sarabia R., et al. Extradural hematoma: Analysis of factors influencing the courses of 161 patients. Neurosurgery. 1988;23:44-51.
63. Dunn L.T., Fitzpatrick M.O., Beard D., et al. Patients with a head injury who “talk and die” in the 1990s. J Trauma. 2003;54:497-502.
64. Lee E., Hung Y., Wang L., et al. Factors influencing the functional outcome of patients with acute epidural hematomas: Analysis of 200 patients undergoing surgery. J Trauma. 1998;45:946-952.
65. Chen T.Y., Wong C.W., Chang C.N., et al. The expectant treatment of “asymptomatic” supratentorial epidural hematomas. Neurosurgery. 1993;32:176-179.
66. Massaro F., Lanotte M., Faccani G., Triolo C. One hundred and twenty-seven cases of acute subdural haematoma operated on. Correlation between CT scan findings and outcome. Acta Neurochir (Wien). 1996;138:185-191.
67. Wilberger J.J., Harris M., Diamond D. Acute subdural hematoma: Morbidity and mortality related to timing of operative intervention. J Trauma. 1991;30:733-736.
68. Becker D.P., Miller J.D., Ward, et al. The outcome from severe head injury with early diagnosis and intensive management. J Neurosurg. 1977;47:491-502.
69. Kotwica Z., Jakubowski J. Acute head injuries in the elderly. An analysis of 136 consecutive patients. Acta Neurochir (Wien). 1992;118:98-102.
70. Servadei F., Murray G.D., Penny K., et al. The value of the “worst” computed tomographic scan in clinical studies of moderate and severe head injury. European Brain Injury Consortium. Neurosurgery. 2000;46:70-75.
71. Hirakawa K., Hashizume K., Fuchinoue T., et al. Statistical analysis of chronic subdural hematoma in 309 adult cases. Neurol Med Chir (Tokyo). 1993;12:71-83.
72. Choksey M., Crokard H.A., Sandilands M. Acute traumatic intracerebral haematomas: Determinants of outcome in a retrospective series of 202 cases. Br J Neurosurg. 1993;7:611-622.
73. Lobato R.D., Gomez P.A., Alday R., et al. Sequential computerized tomography changes and related final outcome in severe head injury patients. Acta Neurochir (Wien). 1997;139:385-391.
74. Patel N., Hoyt D., Nakaji P., et al. Traumatic brain injury: Patterns of failure of nonoperative management. J Trauma. 2000;48:367-374.
75. Baratham G., Dennyson W.G. Delayed traumatic intracerebral hemorrhage. J Neurol Neurosurg Psychiatry. 1972;35:698-706.
76. Chang E.F., Meeker M., Holland M.C. Acute traumatic intraparenchymal hemorrhage: risk factors for progression in the early postinjury period. Neurosurgery. 2006;58:647-656.
77. Roda J., Gimenez D., Perez-Higueras A., et al. Posterior fossa epidural hematomas: A review and synthesis. Surg Neurol. 1983;19:419-424.
78. d’Avella D., Servadei F., Scerrati M., et al. Traumatic intracerebrallar hemorrhages: A clinicoradiological analysis of 81 cases. Neurosurgery. 2002;50:16-25.
79. Hukkelhoven C.W., Steyergerg E.W., Rampen A.J., et al. Patient age and outcome following severe traumatic brain injury: an analysis of 5600 patients. J Neurosurg. 2003;99:666-673.
80. Signorini D.F., Andrews P.J., Jones P.A., et al. Predicting survival using simple clinical variables: a case study in traumatic brain injury. J Neurol Neurosurg Psychiatry. 1999;66:20-25.
81. Stocchetti N., Penny K.I., Dearden M., et al. Intensive care management of head injured patients in Europe: A survey from the European Brain Injury Consortium. Intensiv Care Med. 2001;27:400-406.
82. Fearnside M.R., Cook R.J., McDougall P., et al. The Westmead Head Injury Project Outcome in severe head injury. A comparative analysis of pre-hospital, clinical and CT variables. Br J Neurosurg. 1993;7:267-269.
83. Pascual-Garvi J.M., Gonzalez-Llanos F., Prieto-Arribas R., et al. La barrera hematoencefálica: desarrollo de una estructura que permite la heterogeneidad funcional del sistema nervioso central. Rev Neurol. 2004;38:565-581.
84. Abbott N.J., Ronnback L., Hansson E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat Rev Neurosci. 2006;7:41-53.
85. Jancer R.C., Raff M.C. Astrocytes induce blood–brain properties in endothelial cells. Nature. 1987;325:253-257.
86. Keogh C.L., Francis K.R., Whitaker V.R., Wei L. The blood–brain barrier. Biology, development and brain injury. In: Lajtha A., editor. Handbook of Neurochemistry and Molecular Neurobiology. 3rd ed. New York: Springer; 2009:303-320. Banik N, Swapan KR (vol eds): Brain and Spinal Cord Trauma
87. Manley G.T., Fujimura M., Ma T., et al. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159-163.
88. Manley G.T., Binder D.K., Papadopoulos M.C., Verkman A.S. New insights into water transport and edema in the central nervous system from phenotype analysis of aquaporin-4 null mice. Neuroscience. 2004;129:983-991.
89. Hossmann K.A. Viability thresholds and the penumbra of focal ischemia. Ann Neurol. 1994;36:557-565.
90. Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci. 2004;5:347-360.
91. Magistretti P.J., Pellerin L., Rothman D.L., Shulman R.G. Energy on demand. Science. 1999;283:496-497.
92. Haydon P.G. Glia: listening and talking to the synapse. Nat Rev Neurosci. 2001;2:185-193.
93. Simard M., Nedergaard M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience. 2004;129:877-896.
94. Volterra A., Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci. 2003;6:43-50.
95. Amiry-Moghaddam M., Ottersen O.P. The molecular basis of water transport in the brain. Nat Rev Neurosci. 2003;4:991-1001.
96. Kuffler S.W., Nicholls J.G., Orkand R.K. Physiological properties of glial cells in the central nervous system of amphibian. J Neurophisiol. 1966;29:768-787.
97. Nicholls J.G., Martin A.R., Wallace B.G., Fuchs P.A. Properties and functions of neuroglial cells. In: Nicholls J.G., Martin A.R., Wallace B.G., Fuchs P.A. From Neuron to Brain. 4th ed. Sunderland, Mass: Sinauer Associates Publishers; 2000:133-154.
98. Walz W. Role of glial cells in the regulation of the brain ion microenvironment. Prog Neurobiol. 1989;33:309-333.
99. Bloch O., Manley G.T. The role of aquaporin-4 in cerebral water transport and edema. Neurosurg Focus. 2007;22:E3.
100. Pascual J.M., Carceller F., Roda J.M., Cerdan S. Glutamate, glutamine and GABA as substrates for the neuronal and glial compartments after focal cerebral ischemia in rats. Stroke. 1998;29:1048-1057.
101. Nedergaard M., Ransom B., Goldman S.A. New roles for astrocytes: redefining the functional architecture of the brain. Trends Neurosci. 26, 2003. 532–530
102. Holloway R., Zhou Z., Harvey H.B., et al. Effect of lactate therapy upon cognitive deficits after traumatic brain injury in the rat. Acta Neurochir. (Wien). 2007;149:919-927.
103. Prieto R., Tavazzi B., Taya K., et al. Brain energy depletion in a rodent model of diffuse traumatic brain injury is not prevented with administration of sodium lactate. Brain Res. 2011;1404:39-49.
104. Hubel D.H., Wiesel T.N. Shape and arrangement of columns in cat’s striate cortex. J Physiol. 1963;165:559-568.
104a. Hughlings Jackson J. Remarks on evolution and dissolution of the nervous system. J Mental Sci. 1887;33:25-48.
105. Von Economo C. Sleep as a problem of localization. J Nerv Ment Dis. 1930;71:249-259.
106. Bremer F. Cerveau isolé et physiologie du sommeil CR. Soc Biol. 1935;118:1235-1241.
107. Saper C.B., Scammell T.E., Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257-1263.
108. Shaw N.A. The neurophysiology of concussion. Prog Neurobiol. 2002;67:281-344.
109. Cushing H. Some experimental and clinical observations concerning states of increased intracranial tension. Am J Med Sci. 1902;124:375-400.
110. Mokri B. The Monro-Kellie hypothesis—applications in CSF volume depletion. Neurology. 2001;56:1746-1748.
111. Carrasco R., Pascual J.M., Navas M., et al. Kernohan-Woltman notch phenomenon caused by an acute subdural hematoma. J Clin Neurosci. 2009;16:1628-1631.
112. Schwartz J.H., De Camilli P. Synthesis and trafficking of neuronal protein. In: Kandel E.R., Schwartz J.H., Jessell T.M. Principles of Neural Science. 4th ed. New York: McGraw-Hill; 2000:88-104.
113. Hirokawa N., Takemura R. Molecular motors and mechanisms of directional transport in neurons. Nat Rev Neurosci. 2005;6:201-214.
114. Conde C., Cáceres A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci. 2009;10:319-332.
115. Foda M.A.A., Marmarou A. A new model of diffuse brain injury in rats. 2. Morphological characterization. J Neurosurg. 1994;80:301-313.
116. Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6:889-898.
117. Waller A. Experiments on the section of glossopharyngeal and hypoglossal nerves of the frog, and observations of the alterations produced thereby in the structure of their primitive fibres. Phil Trans R Soc London. 1850;140:423-429.
118. Farkas O., Lifshitz J., Povlishock J. Mechanoporation induced by diffuse traumatic brain injury: an irreversible or reversible response to injury? J Neurosci. 2006;26:3130-3140.
119. Kilinc D., Gallo G., Barbee K.A. Mechanically-induced membrane poration causes axonal beading and localized cytoskeletal damage. Exp Neurol. 2008;212:422-430.
120. Ahmed S.M., Rzigalinski B.A., Willoughby K.A., et al. Stretch-induced injury alters mitochondrial membrane potential and cellular ATP in cultured astrocytes and neurons. J Neurochem. 2000;74:1951-1960.
121. Geddes D.M., Cargill R.S., LaPlaca M.C. Mechanical stretch to neurons results in a strain rate and magnitude-dependent increase in plasma membrane permeability. J Neurotrauma. 2003;20:1039-1049.
122. Spaethling J.M., Klein D.M., Singh P., Meaney D.F. Calcium-permeable AMPA receptors appear in cortical neurons after traumatic mechanical injury and contribute to neuronal fate. J Neurotrauma. 2008;25:1207-1216.
123. Mazzeo A.T., Beat A., Singh A., Bullock M.R. The role of mitochondrial transition pore, and its modulation, in traumatic brain injury and delayed neurodegeneration after TBI. Exp Neurol. 2009;218:363-370.
124. Miller J.D., Becker D.P., Ward J.D., et al. Significance of intracranial hypertension in severe head injury. J Neurosurg. 1977;47:503-516.
125. Lobato R.D. Post-traumatic brain swelling. Adv Tech Stand Neurosurg. 1993;20:3-38.
126. Marmarou A. A review of progress in understanding the pathophysiology and treatment of brain edema. Neurosurg Focus. 2007;22:E1.
127. Castejon O.J. Morphological astrocytic changes in complicated human brain trauma: a light and electron microscopic study. Brain Inj. 1998;12:402-427.
128. Mongin A.A., Kimelberg H.K. Astrocyte swelling in neuropathology. In: Kettemann H., Ransom B.R. Neuroglia. 2nd ed. New York: Oxford University Press; 2005:550-562.
129. Stover J.F., Morganti-Kosmann M.C., Stocker R., et al. Glutamate and taurine are increased in ventricular cerebrospinal fluid of severely brain-injured patients. J Neurotrauma. 1999;16:135-142.
129a. Preston G.M., Carroll T.P., Guggino WB., Agre P., et al. Appearance of water channels in xenopus oocytes expressing red cell CHIP28 protein. Science. 1992;256:385-387.
130. Nagelhus E.A., Mathiisen T.M., Ottersen O.P. Aquaporin-4 in the central nervous system: cellular and subcellular distribution and coexpression with Kir4.1. Neuroscience. 2004;129:905-913.
131. Solenov E., Watanabe H., Manley G.T., Verkman A.S. Sevenfold-reduced osmotic water permeability in primary astrocyte cultures from AQP-4–deficient mice, measured by a fluorescence quenching method. Am J Physiol Cell Physiol. 2004;286:C426-C432.
132. Kiening K.L., van Landeghem F.K., Schreiber S., et al. Decreased hemispheric aquaporin-4 is linked to evolving brain edema following controlled cortical impact injury in rats. Neurosci Lett. 2002;324:105-108.
133. Weller R.O., Djuanda E., Yow H.-Y., Carare R.O. Lymphatic drainage of the brain and the pathophysiology of neurological disease. Acta Neuropathol. 2009;117:1-14.
134. Reulen H.J., Tsuyumu M., Tack A., et al. Clearance of edema fluid into cerebrospinal fluid. A mechanism for resolution of vasogenic brain edema. J Neurosurg. 1978;48:754-764.
135. Papadopoulos M.C., Manley G.T., Krishna S., Verkman A.S. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J. 2004;18:1291-1293.
136. Gaetz M. The neurophysiology of brain injury. Clin Neurophysiol. 2004;115:4-18.
137. Maas A.I.R., Stocchetti N., Bullock R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008;7:728-741.
138. Newman E.A. High potassium conductance in astrocyte endfeet. Science. 1986;233:453-454.
139. Sullivan P.G., Rabchevsky A.G., Waldmeier P.C., Springer J.E. Mitochondrial permeability transition in CNS trauma: cause or effect of neuronal cell death? J Neurosci Res. 2005;79:321-329.
140. Galluzzi L., Blomgren K., Kroemer G. Mitochondrial membrane permeabilization in neuronal injury. Nat Rev Neurosci. 2009;10:481-494.
141. Floyd C.L., Lyeth B.G. Astroglia: Important mediators of traumatic brain injury. Prog Brain Res. 2007;161:61-79.
141a. Ahmed S.M., Rzigalinski B.A., Willoughby K.A., et al. Stretch-induced injury alters mitochondrial membrane potential and cellular ATP in cultured astrocytes and neurons. J Neurochem. 2000;74:1951-1960.
142. McGraw J., Hiebert G.W., Steeves J.D. Modulating astrogliosis after neurotrauma. J Neurosci Res. 2001;63:109-115.
143. Fitch M.T., Silver J. CNS injury, glial scars and inflammation: inhibitory extracellular matrices and regeneration failure. Exp Neurol. 2008;209:294-301.
144. Seifert G., Schilling K., Steinhauser C. Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci. 2006;7:194-206.
145. Silver J., Miller J.H. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146-156.
146. Rolls A., Shechter R., Schwartz M. The bright side of the glial scar in CNS repair. Nat Rev Neurosci. 2009;10:235-241.
147. Raisman G. Myelin inhibitors: does NO mean GO? Nat Rev Neurosci. 2004;5:157-161.
148. Myer D.J., Gurkoff G.G., Lee S.M., et al. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain. 2006;129:2761-2772.
149. Filbin M.T. Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat Rev Neurosci. 2003;4:703-713.
150. Brady S., Colman D.R., Brophy P. Subcellular organization of the nervous system: organelles and their functions. In: Squire LR, Bloom F.E., McConnell S.K., et al. Fundamental Neuroscience. 2nd ed. Amsterdam: Academic Press; 2003:79-114.