Chapter 48 Surgical Management of Neurofibromatosis Types 1 and 2
Neurofibromatosis Type 1
NF1 is an autosomal dominant genetic disorder caused by a mutation or deletion of the neurofibromin gene on the long arm of chromosome 17.1 The diagnosis of NF1 requires the presence of two or more major criteria: six or more café-au-lait spots, two cutaneous neurofibromas, one plexiform neurofibroma, certain bony abnormalities, an optic glioma, iris Lisch nodules, or a first-degree relative with NF1.2 Diagnosis has primarily been clinical, but genetic testing identifies at least 95% of patients who meet the clinical criteria. While there are no silent carriers of NF, clinical manifestations are variable, even within the same family.3 Because NF1 may affect virtually any organ system and some complications such as plexiform neurofibromas commonly involve adjacent organs, a multidisciplinary team is essential for management. Such a team should include a pediatrician, neurologist, geneticist, ophthalmologist, neurosurgeon, orthopedist, plastic surgeon, and oncologist. NF1 is a progressive disorder. Some complications worsen with age. Moreover, complications of NF1 are usually age specific. Plexiform neurofibromas can be considered congenital, although they may not require surgical intervention until later in life. Optic gliomas usually present between 18 months and 7 years of age.4 Iris Lisch nodules usually appear between 10 and 21 years of age. Cutaneous neurofibromas commonly occur in teenagers or young adults, and malignant peripheral nerve sheath tumors (MPNSTs) are a complication of young adults.5
Neurologic Complications of NF1 and Indications for Neurosurgical Intervention
The neurologic complications of NF1 include headaches, learning disabilities, seizures, peripheral nerve tumors, spinal nerve root tumors, dural ectasias, deafness, optic gliomas, areas of high-intensity signal on magnetic resonance imaging (MRI), tumors of the brain parenchyma, and aqueductal stenosis.6 Migraine headaches are a common feature.7 Learning disabilities and hyperactivity occur in at least 50% of patients.8 Deafness occurs in 10% of NF1 patients and is not caused by tumors.9 Brain tumors and optic gliomas occur in a small percentage of patients. The incidence is increased compared with the normal population.10 All patients with NF1 develop peripheral nerve tumors.
Peripheral Nerve Tumors
Five types of peripheral nerve tumors occur in NF1: schwannomas, discrete neurofibromas (sometimes called cutaneous or dermal neurofibromas), diffuse neurofibromas, plexiform neurofibromas, and MPNSTs. Schwannomas are infrequently found in patients with NF1. This tumor is more typical of NF2 and is discussed in the section on NF2. Diffuse neurofibromas most commonly present as boggy caplike lesions of the scalp that involve the subcutaneous tissue, stopping at the fascia.11 Diffuse neurofibromas of the scalp do not progress beyond the hairline and are best left alone unless there is evidence of rapid growth.
Discrete neurofibromas and plexiform neurofibromas involve a proliferation of fibroblasts, Schwann cells, perineural cells, mast cells, extracellular matrix, axons, and blood vessels.12 These two tumors differ histologically, primarily in the extent of extracellular matrix. Plexiform neurofibromas have more extracellular matrix. Both tumors cause expansion of the nerve. Nerve fibers run through the tumor. Plexiform neurofibromas may involve small peripheral nerves, large peripheral nerves, nerve trunks, plexus, or spinal roots. Motor nerves, sensory nerves, or both are affected. Plexiform neurofibromas may be associated with markedly dilated veins. Plexiform neurofibromas involve the skin with or without involvement of underlying muscle, or they may be confined to deeper tissues. Plexiform tumors are felt to be congenital or to appear within the first year of life. Growth is highly variable. Some tumors remain static, others relentlessly increase, and still others undergo spurts of growth and periods of quiescence. Plexiform neurofibromas may appear discrete and isolated, diffuse and infiltrative, or nodular with multiple grapelike clusters.13 Plexiform neurofibromas commonly infiltrate adjacent muscle and sometimes infiltrate adjacent organs, such as the bladder or esophagus. Plexiform neurofibromas occur in at least 50% of all patients.14 Large areas of hyperpigmentation with fine hair may overlie plexiform neurofibromas.
Discrete or cutaneous neurofibromas occur in all patients with NF1. These tumors usually appear in teenagers or adults.15 Early appearance of large numbers of neurofibromas is associated with complete deletion of the NF1 gene.16 Isolated neurofibromas may involve both motor and sensory nerves in the epidermis and/or dermis.
MPNSTs occur in 4% to 10% of all patients with NF1. These tumors arise within plexiform neurofibromas usually between 15 and 50 years of age.5 Earlier onset is uncommon but occurs. MPNSTs may be multifocal in some patients. MPNSTs are highly malignant, with rapid hematogenous dissemination. Outcome for patients with MPNSTs is poor. The best outcome is associated with radical resection.17
Indications for Removal of Peripheral Nerve Tumors
MPNSTs are commonly associated with pain. There are no reliable radiologic characteristics to distinguish MPNSTs from plexiform neurofibromas.12 While MPNSTs commonly enhance with contrast and lack a homogeneous appearance, the same is true of some benign plexiform tumors. A helpful distinguishing feature is that MPNSTs commonly take up gallium in radioisotope scans.18,19 Positron-emission tomography scans may also be useful in diagnosis.20 Because MPNSTs arise within plexiform neurofibromas, in which only a small portion of the tumor is malignant, biopsies can be negative. Computed tomography (CT)–directed needle biopsy is preferred when an MPNST is suspected. MPNSTs do not respond well to chemotherapy or radiation therapy.20
Spinal Nerve Root Tumors and Dural Ectasias
Dural ectasias are a weakening or expansion of the dural covering of a spinal root that is independent of a nerve root tumor. Dural ectasias commonly erode the vertebral body and may produce large dilated pockets anterior to the vertebral body (so-called anterior meningoceles). This is associated with pain, scoliosis, and sometimes vertebral instability.21 Dural ectasias increase in size or remain static. They can be seen in early childhood, suggesting that they may be congenital defects. Surgery is indicated if there is intractable pain or vertebral instability.
Brain Tumors, MRI Abnormalities, and Hydrocephalus
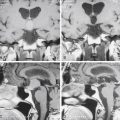
High-intensity signals are present on T2 images in MRI of the brain in roughly 50% of all patients with NF1. Common locations are the basal ganglia, cerebellum, midbrain, and pons. The lesions do not enhance and are less easily visible on T1 images. They are not visible on CT scans. These areas of increased signal are sometimes referred to as unidentified bright objects, heterotopias, or hamartomas. The latter terms are misleading, because the etiology of the lesions is unclear.22 They may be more common in children with learning disabilities but also occur in children without any cognitive difficulties. Areas of hyperintensity depend on age. They are less common after age 20.23 In younger patients, the hyperintense signals may increase or decrease over time. They are not tumors and do not require radiologic follow-up or biopsy.
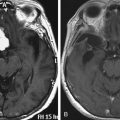
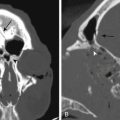
Optic gliomas or visual pathway tumors occur in 15% of patients with NF1.24 Optic gliomas are pilocytic astrocytomas (World Health Organization grade I).25 They commonly affect the chiasm, as well as one or both optic nerves. The tumors may extend into the hypothalamus or along the optic radiations.4 Impairment of vision occurs in only 20% to 30% of patients with optic gliomas.26 If treatment is required, chemotherapy is preferred.27 The tumors do not require biopsy. The age of onset is between 16 months and 8 years of age. Screening is done with regular eye exams rather than imaging. Optic gliomas are almost never symptomatic after age 8,4 but progression of tumors after treatment may occur. Not all optic gliomas respond to current chemotherapy regimes.
Tumors of the brain parenchyma (not including optic pathway tumors) occur in 2% to 3% of patients with NF1.28 The cerebellum and brain stem are the most common locations.29 Brain stem tumors involve the midbrain, pons, or medulla. They commonly have an exophytic component. Some enhancement with contrast may be seen. The natural history of brain stem tumors is usually benign.30 Almost all are grade I astrocytomas. They may be associated with recurrent coughing, intermittent difficulty swallowing, or choking, but they are not associated with any weakness or persistent cranial nerve palsies. Rarely, they produce obstructive hydrocephalus. Once a brain stem tumor has been identified, it is prudent to obtain imaging at intervals for a few years to prove that the lesion is stable.31 Brain tumors in other locations can vary from grade I to grade IV astrocytomas. In general, brain tumors in patients with NF1 are more indolent than in normal individuals. Some tumors even regress over time. Highly malignant gliomas also occur in patients with NF1. Tumors should not be biopsied unless they are clearly symptomatic or show progression over time.
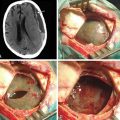
Aqueductal stenosis is a rare complication of NF1. Symptoms include headache, vomiting, progressive gait disturbance, incontinence, and cognitive difficulties.32 The onset may be insidious and recognition delayed. Surgical intervention usually results in significant improvement, even when the symptoms appear to be long-standing.
Surgical Approach to the Lesions of Neurofibromatosis Type 1
Spinal Nerve Root Tumors
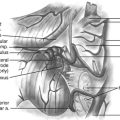
When the plexiform neurofibroma is primarily extradural, the approach is posterior with wide unilateral or bilateral bony decompression. The tumor is dissected in the epidural space. The epidural venous structures above and below are cauterized and divided. The tumor capsule is entered sharply and removed intracapsularly. We recommend the intracapsular approach to epidural tumors to preserve nerve root function. Removal of the tumor with its capsule and dural sheath interrupt both sensory and motor nerve function. In the thoracic region, radical removal can be performed, but radical removal in the cervical and lumbar region would cause significant morbidity.
Spinal Nerve Root Tumors with Large Extra-axial Components
Spinal nerve root tumors with significant extra-axial extension are particularly challenging. In the cervical spine, extension of the tumor may compress the trachea or invade the esophagus. In the lumbosacral region, extra-axial tumor commonly compresses the rectum or invades the bladder.14 An interdisciplinary surgical team is essential for resection of these tumors. The intraspinal portion of the procedure is performed identically to the procedure for purely spinal tumors, whether they be intradural or epidural. However, patient position and the incisions are dictated by the extra-axial portion of the tumor.
Lesions of the Cranial Vault and Brain
Patients with NF1 may have a variety of parenchymal abnormalities on MRI. Of the parenchymal brain lesions in NF1, only symptomatic tumors with radiologic characteristics of malignancy or progression require surgical intervention. Treatment of these lesions entails biopsy and aggressive resection, where possible, followed by adjunctive chemotherapy and radiotherapy. The treatment protocols are identical to high-grade astrocytomas in normal individuals. Treatment of the optic pathway tumors is not surgical. Diagnostic biopsy is not required. When optic pathway tumors progress, chemotherapy may be indicated.27
Neurofibromatosis Type 2
NF2 is an autosomal dominantly inherited disease due to a mutation or deletion in the long arm of chromosome 22 of the merlin gene.33 The diagnosis depends on the presence of bilateral eighth-nerve tumors or the presence of a unilateral eighth-nerve tumor before 30 years of age in an individual with a first-degree relative with NF2 or two of the following: neurofibroma, meningioma, glioma, schwannoma, or juvenile posterior cataract.9 Genetic testing is also available and may identify as many as 95% of patients with germline mutations. NF2 is characterized by the presence of multiple central nervous system (CNS) tumors. The clinical hallmark is bilateral vestibular nerve schwannomas. Patients may have multiple supratentorial meningiomas and schwannomas of the cranial nerves, in addition to vestibular tumors. Meningiomas occur along the spine, and schwannomas may develop along spinal nerve roots.34 Roughly 33% of NF2 patients have intramedullary tumors of the spinal cord or brain stem that are either ependymomas or astrocytomas.35 Juvenile posterior subcapsular cataracts and retinal hamartomas occur in 80% of patients.36 Skin tumors occur in patients with NF2 but are not particularly prominent.
The clinical presentation of NF2 in adults is usually unilateral deafness. Facial weakness, visual impairment, dizziness, or painful peripheral nerve lesions may also be presenting complaints.37 Spinal cord compression or seizures are late symptoms. Children with NF2 more commonly present because of a cataract or signs and symptoms related to cranial meningiomas, brain stem tumors, or spinal cord tumors.38 The spectrum of severity of NF2 is variable. Early studies suggested two clinical phenotypes: the Gardner phenotype with milder disease, fewer tumors, and later onset and the Wishart phenotype with more tumors, earlier onset, and rapid progression.37 Molecular studies demonstrate that more severe disease is seen in patients with frameshift or nonsense mutations. These patients are also more likely to have intramedullary spinal tumors. A milder phenotype is seen in patients with missense mutations, in-frame deletions, or large deletions.38 A mild phenotype, particularly in patients with a unilateral vestibular schwannoma with no family history of NF2, may also be due to somatic mosaicism of the NF2 gene. Somatic mosaicism is found in one third of patients with no family history of NF2 and has a lower rate of transmission of the disease than germline mutations.39
NF2 is completely distinct from NF1, although rare patients may have features of both diseases. Two additional disorders must be distinguished from NF2: Schwannomatosis is characterized by multiple schwannomas of the peripheral, spinal, or cranial nerves without evidence of a vestibular schwannoma.40 Schwannomatosis can be familial, although most cases are sporadic.41 Meningiomatosis is an autosomal dominant disorder characterized by multiple meningiomas along the spinal cord, as well as supratentorially.42
Tumors Associated with NF2
Schwannomas
Schwannomas are typically nodular masses surrounded by a fibrous capsule consisting of epineurium and some nerve fibers. The tumors consist predominantly of Schwann cells with alternating patterns of cellularity.43 Glandular or cystic areas sometimes occur. Schwannomas are virtually never malignant. Unlike neurofibromas, in which axons run through the tumor, schwannomas are usually extrinsic to the nerve and separate from the majority of the axons. However, when schwannomas involve small nerves, the tumor frequently engulfs the nerve, making separation from the nerve difficult. Schwannomas of the vestibular nerves are histologically similar to schwannomas of other nerves. Frequently, vestibular schwannomas in NF2 are multinodular and less vascular than sporadic tumors.44 Although these tumors were originally called acoustic neuromas, they arise from the vestibular nerve. They usually impair hearing, but vestibular symptoms may also be prominent at the time of presentation. Schwannomas of the other cranial nerves are found in at least 25% of patients, particularly the third and fifth cranial nerves.9 Schwannomas of the spinal nerves are present in the majority of patients.35,37
Meningiomas
Meningiomas arise from arachnoid cells of the leptomeninges. Meningiomas in patients with NF2 are predominantly fibrous, but meningothelial tumors also occur.45 Meningiomas infrequently show evidence of pleomorphism or malignancy and act more aggressively, invading bone. Orbital meningiomas may occur in childhood and must be distinguished from optic gliomas. Meningiomas of the cerebellar pontine angle are occasionally confused with vestibular schwannomas. Meningiomas of the skull base produce brain stem compression and are an important cause of mortality in NF2. An en plaque meningioma occurs in some patients with NF2 late in their disease.
Astrocytomas, Ependymomas, and Hamartomas
Astrocytomas and ependymomas occur in as many as a third of NF2 patients.35 The most common site is the brain stem or cervical cord.46,47 Syrinx formation is not uncommon. These tumors are typically indolent in NF2. A more malignant profile in spinal cord tumors is rare and usually related to prior radiation therapy. Evidence of rapid growth or symptoms related to the tumors is an indication for surgery, but radiation and chemotherapy are usually not indicated. Hamartomas of the brain are frequently found in patients with NF2. They are a mixture of Schwann cells, glia, and meningeal cells.47
Indications for Neurosurgery
NF2 is not a surgically curable disease. Lesions recur and progress. The entire CNS can be involved. The tumors of NF2 do not respond to conventional chemotherapy. However, molecular studies are beginning to suggest alternative chemotherapeutic approaches.48 Radiation therapy can be considered for lesions that are not surgically accessible. Lesions are usually addressed surgically when they are symptomatic. Special considerations involved in the surgery of the vestibular schwannomas are discussed later. In patients with the more benign phenotype, with few and slow-growing tumors, surgical intervention is reserved for prevention of impending symptoms or for symptomatic tumors. In its most aggressive form, however, the disease can progress rapidly and cause severe disability that leads to death. In that situation, surgical intervention is palliative to improve quality of life.
A variety of approaches with different goals and complications are available for vestibular schwannomas. Single or multiple fraction stereotactic radiosurgery (i.e., gamma knife, as well as other types) is advocated in some centers.49,50 Studies of NF2 patients with vestibular schwannomas treated with these techniques suggest good local disease control and a relatively low incidence of side effects that is at least comparable to results with microsurgery techniques.51,53 However, both single dose and multiple fraction radiotherapy are not without potential complications for NF2 patients.38,49,50,54 Radiotherapy may have limited usefulness and limit the options for recurrent or new vestibular tumors. We prefer a microsurgery approach to vestibular schwannomas. Regardless of the approach, all centers agree that intervention should only occur when there is documentation of tumor growth or progressive hearing loss. Because of the variety of approaches available, it is important to discuss the risks and benefits of each with the patient. For those opting for a microsurgery approach, it is important to discuss goals for surgery and define the degree of aggressiveness of the surgical approach with the patient before surgery. Aggressive removal of vestibular schwannomas may cause facial nerve injury. Facial nerve injury may be extremely distressing to patients and predisposes them to ocular injury. These complications should be discussed with patients. Consideration of cochlear implantation may also influence surgical decisions.55 Surgical removal of intramedullary spinal cord tumors is indicated only when there are signs of spinal cord compression. Because multiple tumors may develop over time along the length of the spine, the number of surgical interventions is limited. Schwannomas of the spinal nerves rarely cause problems. Surgery on these tumors should be avoided.
Surgical Approach to the Lesions of NF2
Vestibular Schwannoma (Acoustic Neuroma)
The technical approach to a vestibular schwannoma in an NF2 patient is identical to sporadic tumors. However, surgical decisions in NF2 patients are affected by the presence of bilateral disease and the knowledge that the disease is not surgically curable. Surgical approaches include radical resection, partial removal, and decompression. Suboccipital retrosigmoid, translabyrinthine, or middle cranial fossa approaches can all be appropriately used for tumor removal. The arguments for these various approaches are outlined elsewhere in this text. The translabyrinthine approach permits greater exposure of the tumor but results in deafness. The middle cranial fossa approach is used primarily for decompression. We prefer the suboccipital retrosigmoid approach, with the goal of sparing hearing.52 This operation entails drilling open the posterior aspect of the internal auditory meatus, followed by subtotal resection of the tumor, preserving facial nerve function at all costs. Electrophysiologic monitoring of the eighth cranial nerve helps to preserve hearing.
We advocate early surgery on one side if one of the vestibular tumors is less than 1.5 cm in diameter. When the tumor is small, early surgery reduces the size of the tumor and preserves hearing and facial nerve function. If the tumors are greater than 1.5 cm, we prefer to wait until there is significant motor dysfunction due to brain stem compression. When motor dysfunction is present in patients with large bilateral tumors, we recommend subtotal resection of one of the tumors. If brain stem compression is predominantly unilateral, subtotal resection of the larger tumor relieves brain stem compression and preserves facial nerve function and hearing. However, when patients have very large bilateral tumors and bilateral brain stem compression, the appropriate operation may be a radical subtotal resection on one side, sacrificing hearing and preserving facial nerve function. In the latter situation, aggressive tumor removal may compromise facial nerve function. Chemotherapy with bevacizumab may also be an option.48
Conclusions
The NFs are not surgically curable diseases. Unfortunately, the limited usefulness of adjuvant therapy leaves surgery as the primary treatment to alleviate symptoms. While surgery provides symptomatic relief, it may not alter the course of disease. Surgery should be limited to the removal of tumors that are symptomatic or that threaten to cause symptoms. The technical surgical approach to the tumors of NF1 and NF2 is generally similar to the approach of the same tumor in patients without NF. However, the decision of when to operate and to what level of aggressiveness is often affected by the disease and is of critical importance in NF. Even though the relentless progression of disease may at times be discouraging, the ability of neurosurgery to alleviate symptoms and improve longevity and quality of life is significant.
Aoki S., Barkovich A.J., Nishimura K., et al. Neurofibromatosis types 1 and 2: cranial MR findings. Radiology. 1989;172:527-534.
Balasubramniam A., Shannon P., Hodaie M., et al. Glioblastoma multiforme after stereotactic radiotherapy for acoustic neuroma: case report and review of the literature. Neuro Oncol. 2007;9:447-453.
Chan A.W., Black P., Ojemann R.G., et al. Stereotactic radiotherapy for vestibular schwannomas: favorable outcome with minimal toxicity. Neurosurgery. 2005;57:60-70.
Cohen B.H., Rothner A.D. Incidence, types, and management of cancer in patients with neurofibromatosis. Oncology. 1989;3:23-38.
Duffner P.K., Cohen M.E., Seidel F.G., et al. The significance of MRI abnormalities in children with neurofibromatosis. Neurology. 1989;39:373-378.
Evans D.G.R., Baser M.E., O’Reilly B., et al. Management of the patient and family with neurofibromatosis 2: a consensus conference statement. Br J Neurosurg. 2005;19:5-12.
Friedrich R.E., Korf B., Funsturer C., et al. Growth type of plexiform neurofibromas in NF1 determined on magnetic resonance images. Anticancer Res. 2003;23:949-952.
Huson S.M., Harper P.S., Compston D.A.S. Von Recklingausen neurofibromatosis: a clinical and population study in southeast Wales. Brain. 1988;111:1355-1381.
Ilgren E.B., Kinnier-Wilson L.M., Stiller C.A. Gliomas in neurofibromatosis: a series of 89 cases with evidence for enhanced malignancy in associated cerebellar astrocytomas. Pathol Ann. 1985;20:331-358.
Listernick R., Charrow J., Greenwald M.J., et al. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994;125:63-66.
Louis D.N., Ramesh V., Gusella J. Neuropathology and molecular genetics of neurofibromatosis 2 and related tumors. Brain Pathol. 1995;5:163-172.
Mathieu D., Kondziolka D., Flickinger J.C., et al. Stereotactic radiosurgery for vestibular schwannomas in patients with neurofibromatosis type 2: an analysis of tumor control, complications, and hearing preservation rates. Neurosurgery. 2007;60:460-470.
Mautner V.F., Tatagiba M., Lindenau M., et al. Spinal tumors in patients with neurofibromatosis type 2: MR imaging study of frequency, multiplicity, and variety. Am J Roentgenol. 1995;165:951-955.
McCormick P., Torres R., Post K., et al. Intramedullary ependymoma of the spinal cord. J Neurosurg. 1990;72:523-532.
McKenna M., Halpin C., Ojemann R., et al. Long-term hearing results in patients after surgical removal of acoustic tumors with hearing preservation. Am J Otolaryngol. 1992;13:134-136.
Mulvihill J.J., Parry D.M., Sherman J.L. NIH Conference. Neurofibromatosis 1 (Recklinghausen disease) and neurofibromatosis 2 (bilateral acoustic neurofibromatosis): an update. Ann Intern Med. 1990;113:39-52.
Neff B., Wiet R.M., Lasak J.M., et al. Cochlear implantation in the neurofibromatosis type 2 patient: long-term follow-up. Laryngoscope. 2009;117:1069-1072.
Packer R.J., Ater J., Allen J., et al. Carboplatin and vincristine chemotherapy for children with newly diagnosed progressive low-grade gliomas. J Neurosurg. 1997;86:747-754.
Plotkin S.R., Stemmer-Rachamimov A.O., Barker F.G.2nd, et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Eng J Med. 2009;361:358-367.
Pollack I.F., Shultz B., Mulvihill J.J. The management of brainstem gliomas in patients with neurofibromatosis 1. Neurology. 1996;46:1652-1660.
Samii M., Gerganov V., Samii A. Microsurgery management of vestibular schwannomas in neurofibromatosis type 2: indications and results. Prog Neurol Surg. 2008;21:169-175.
Sordillo P.P., Helson L., Hajdu S.I. Malignant schwannoma: clinical characteristics, survival, and response to therapy. Cancer. 1981;47:2503-2509.
Sorensen S.A., Mulvihill J.J., Nielsen A. Long-term follow-up of von Recklinghausen neurofibromatosis: survival and malignant neoplasms. N Engl J Med. 1986;314:1010-1015.
Tonsgard J.H., Kwak S.M., Short M.P., et al. CT imaging in adults with neurofibromatosis-1: frequent asymptomatic plexiform lesions. Neurology. 1998;50:1755-1760.
Warbey V.S., Ferner R.E., Dunn J.T., et al. [18F]FDG PET/CT in the diagnosis of malignant peripheral nerve sheath tumors in neurofibromatosis type-1. Eur J Nucl Med Mol Imaging. 2009;36:751-757.
1. Cawthon R.M., Weiss R., Xu G., et al. A major segment of the neurofibromatosis type 1 gene: CDNA sequence, genomic structure, and point mutations. Cell. 1990;62:193-201.
2. Neurofibromatosis Conference Statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45:575-578.
3. Carey J.C., Laud Hall B.D. Penetrance and variability of neurofibromatosis: a genetic study of 60 families. Birth Defects. 1979;15:271-281.
4. Listernick R., Charrow J., Greenwald M.J., et al. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994;125:63-66.
5. Ducatman B.S., Scheithauer B.W., Piepgras D.G., et al. Malignant peripheral nerve sheath tumors: a clinicopathologic study of 120 cases. Cancer. 1986;57:2006-2021.
6. Gutmann D.H., et al. Abnormalities of the nervous system. In: Friedman J.M., Gutmann D.H., MacCollin M. Neurofibromatosis: Phenotype, Natural History, and Pathogenesis. Baltimore: Johns Hopkins University Press; 1999:190-202.
7. North K. Neurofibromatosis type 1: review of the first 200 patients in an Australian clinic. J Child Neurol. 1993;8:395-402.
8. North K., Joy P., Yuille D., et al. Cognitive function and academic performance in children with Neurofibromatosis type 1. Dev Med Child Neurol. 1995;37:427-436.
9. Mulvihill J.J., Parry D.M., Sherman J.L. NIH Conference. Neurofibromatosis 1 (Recklinghausen disease) and neurofibromatosis 2 (bilateral acoustic neurofibromatosis): an update. Ann Intern Med. 1990;113:39-52.
10. Cohen B.H., Rothner A.D. Incidence, types, and management of cancer in patients with neurofibromatosis. Oncology. 1989;3:23-38.
11. Weiss S.W., Goldblum J.R., Enzinger and Weiss’s Soft Tissue Tumors, 4th ed. St. Louis, Mosby, Weiss SW, Goldblum JR. Benign tumors of peripheral nerves. In: 2001:1132-1137
12. Fisher E.R., Vuzevski V.D. Cytogenesis of schwannoma (neurilemoma), neurofibroma, dermatofibroma, and dermatofibrosarcoma as revealed by electron microscopy. Am J Clin Pathol. 1968;49:141-154.
13. Friedrich R.E., Korf B., Funsturer C., et al. Growth type of plexiform neurofibromas in NF1 determined on magnetic resonance images. Anticancer Res. 2003;23:949-952.
14. Tonsgard J.H., Kwak S.M., Short M.P., et al. CT imaging in adults with neurofibromatosis-1: frequent asymptomatic plexiform lesions. Neurology. 1998;50:1755-1760.
15. Huson S.M., Harper P.S., Compston D.A.S. Von Recklingausen neurofibromatosis: a clinical and population study in southeast Wales. Brain. 1988;111:1355-1381.
16. Tonsgard J.H., Yelavarthi K.K., Cushner S., et al. Do NF1 gene deletions result in a characteristic phenotype? Am J Med Genet. 1997;73:80-86.
17. Sordillo P.P., Helson L., Hajdu S.I. Malignant schwannoma: clinical characteristics, survival, and response to therapy. Cancer. 1981;47:2503-2509.
18. Hammond J.A., Dreidger A.A. Detection of malignant change in neurofibromatosis (von Recklinghausen’s disease) by gallium-67 scanning. Can Med Assoc J. 1978;119:352-353.
19. Kloos R.T., Rufini V., Gross M.D., et al. Bone scans in neurofibromatosis: neurofibroma, plexiform neuroma, and neurofibrosarcoma. J Nucl Med. 1996;37:1778-1783.
20. Warbey V.S., Ferner R.E., Dunn J.T., et al. [18F]FDG PET/CT in the diagnosis of malignant peripheral nerve sheath tumors in neurofibromatosis type-1. Eur J Nucl Med Mol Imaging. 2009;36:751-757.
21. Edmonson J.H., Ryan L.M., Blum R.H., et al. Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J Clin Oncol. 1993;11:1269-1275.
22. Erkulvrawatr S., El Gammai T., Hawkins J., et al. Intrathoracic meningoceles and neurofibromatosis. Arch Neurol. 1979;36:557-559.
23. Duffner P.K., Cohen M.E., Seidel F.G., et al. The significance of MRI abnormalities in children with neurofibromatosis. Neurology. 1989;39:373-378.
24. Aoki S., Barkovich A.J., Nishimura K., et al. Neurofibromatosis types 1 and 2: cranial MR findings. Radiology. 1989;172:527-534.
25. Listernick R., Charrow J., Greenwald M.J., et al. Optic gliomas in children with neurofibromatosis type 1. J Pediatr. 1989;114:788-792.
26. Kleihues P., Soylemezoglu F., Schauble B., et al. Histopathology, classification, and grading of gliomas. Glia. 1995;15:211-221.
27. Lewis R.A., Gerson L.P., Axelson K.A., et al. Von Recklinghausen neurofibromatosis: incidence of optic gliomata. Ophthalmology. 1984;91:929-935.
28. Packer R.J., Ater J., Allen J., et al. Carboplatin and vincristine chemotherapy for children with newly diagnosed progressive low-grade gliomas. J Neurosurg. 1997;86:747-754.
29. Sorensen S.A., Mulvihill J.J., Nielsen A. Long-term follow-up of von Recklinghausen neurofibromatosis: survival and malignant neoplasms. N Engl J Med. 1986;314:1010-1015.
30. Ilgren E.B., Kinnier-Wilson L.M., Stiller C.A. Gliomas in neurofibromatosis: a series of 89 cases with evidence for enhanced malignancy in associated cerebellar astrocytomas. Pathol Ann. 1985;20:331-358.
31. Bilaniuk L.T., Malloy P.T., Zimmerman R.A., et al. Neurofibromatosis type 1: brainstem tumors. Neuroradiology. 1997;39:642-653.
32. Pollack I.F., Shultz B., Mulvihill J.J. The management of brainstem gliomas in patients with neurofibromatosis 1. Neurology. 1996;46:1652-1660.
33. Horwich A., Riccardi V.M., Fracke U. Aqueductal stenosis leading to hydrocephalus—an unusual manifestation of neurofibromatosis. Am J Med Genet. 1983;14:577-581.
34. Trofatter J.A., MacCollin M.M., Rutter J.L., et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell. 1993;75:826.
35. Evans D.G.R., Huson S.M., Donnai D., et al. A clinical study of type 2 neurofibromatosis. Quart J Med. 1992;84:603-618.
36. Mautner V.F., Tatagiba M., Lindenau M., et al. Spinal tumors in patients with neurofibromatosis type 2: MR imaging study of frequency, multiplicity, and variety. Am J Roentgenol. 1995;165:951-955.
37. Bouzas E.A., Parry D.M., Eldridge R., et al. Visual impairment in patients with neurofibromatosis 2. Neurology. 1993;43:622-623.
38. Parry D.M., Eldridge R., Kaiser-Kupfer M.I., et al. Neurofibromatosis 2 (NF2): clinical characteristics of 63 affected individuals and clinical evidence of heterogeneity. Am J Med Genet. 1994;52:450-461.
39. Evans D.G.R., Baser M.E., O’Reilly B., et al. Management of the patient and family with neurofibromatosis 2: a consensus conference statement. Br J Neurosurg. 2005;19:5-12.
40. Evans D.G.R., Wallace A., Wu C., et al. Somatic mosaicism: a common cause of classic disease in tumor-prone syndromes? Lessons from type 2 neurofibromatosis. Am J Hum Genet. 1998;63:727-736.
41. MacCollin M., Woodfin W., Kronn D., et al. Schwannomatosis: a clinical and pathologic study. Neurology. 1996;46:1072-1079.
42. MacCollin M., Chiocca E.A., Evans D.G., et al. Diagnostic criteria for schwannomatosis. Neurology. 2005;64:1838-1845.
43. Pulst S.M., Rouleau G., Marineau C., et al. Familial meningioma is not allelic to neurofibromatosis 2. Neurology. 1993;43:2096-2098.
44. Sobel R.A. Vestibular (acoustic) schwannomas: Histologic features in neurofibromatosis 2 and in unilateral cases. J Neuropathol Exp Neurol. 1993;52:106-113.
45. Hamada Y., Iwaki T., Fukui M. A comparative study of embedded nerve tissue in six NF2-associated schwannomas and 17 nonassociated NF2 schwannomas. Surg Neurol. 1997;48:395-400.
46. Louis D.N., Ramesh V., Gusella J. Neuropathology and molecular genetics of neurofibromatosis 2 and related tumors. Brain Pathol. 1995;5:163-172.
47. McCormick P., Torres R., Post K., et al. Intramedullary ependymoma of the spinal cord. J Neurosurg. 1990;72:523-532.
48. Rubinstein L. The malformative central nervous system lesions in the central and peripheral forms of neurofibromatosis: a neuropathological study of 22 cases. Ann N Y Acad Sci. 1986;486:14-29.
49. Plotkin S.R., Stemmer-Rachamimov A.O., Barker F.G.2nd, et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Eng J Med. 2009;361:358-367.
50. Mathieu D., Kondziolka D., Flickinger J.C., et al. Stereotactic radiosurgery for vestibular schwannomas in patients with neurofibromatosis type 2: an analysis of tumor control, complications, and hearing preservation rates. Neurosurgery. 2007;60:460-470.
51. Chan A.W., Black P., Ojemann R.G., et al. Stereotactic radiotherapy for vestibular schwannomas: favorable outcome with minimal toxicity. Neurosurgery. 2005;57:60-70.
52. Samii M., Gerganov V., Samii A. Microsurgery management of vestibular schwannomas in neurofibromatosis type 2: indications and results. Prog Neurol Surg. 2008;21:169-175.
53. McKenna M., Halpin C., Ojemann R., et al. Long-term hearing results in patients after surgical removal of acoustic tumors with hearing preservation. Am J Otolaryngol. 1992;13:134-136.
54. Balasubramniam A., Shannon P., Hodaie M., et al. Glioblastoma multiforme after stereotactic radiotherapy for acoustic neuroma: case report and review of the literature. Neuro Oncol. 2007;9:447-453.
55. Neff B., Wiet R.M., Lasak J.M., et al. Cochlear implantation in the neurofibromatosis type 2 patient: long-term follow-up. Laryngoscope. 2009;117:1069-1072.