Surfaces and interfaces
Graham Buckton
Chapter contents
Measurement of surface tension
Solid/vapour adsorption isotherms
Interpretation of isotherm plots
Interactions between powders and water vapour
Key points
Introduction
A surface is the outer boundary of a material. In reality, each surface is the boundary between two phases: an interface, which can be solid/liquid (SL), solid/vapour (SV) or liquid/vapour (LV); or a boundary between two immiscible phases of the same state, i.e. liquid/liquid or solid/solid interfaces. There cannot be vapour/vapour interfaces, as two vapours would mix, rather than form an interface.
Pharmaceutically we often think of materials in terms of their bulk properties, such as solubility, particle size, density and melting point. However, surface material properties often bear little relationship to bulk properties, for example materials can be readily wetted by a liquid but not dissolve in it, i.e. they could have water loving surfaces but not be soluble; an example of this is glass. As contact between materials occurs at interfaces, a knowledge of surface properties is necessary if interactions between two materials are to be understood (or predicted). Indeed every process, reaction, interaction, whatever it may be, either starts, or fails to start, due to the extent of interfacial contact.
Surface tension
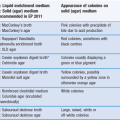
If we compare the forces acting on a molecule in the bulk of the liquid with one at the interface (see Fig. 4.1), in the bulk, the molecules are surrounded on all sides by other liquid molecules and will consequently have no net force acting on them (all attractive forces generally being balanced). At the surface, however, each liquid molecule is surrounded by other liquid molecules to the sides and below (essentially in a hemisphere below the molecule), whilst above the molecule the interactions will be with gas molecules from the vapour; these will be much weaker than those between the liquid molecules. As the molecule at the liquid surface has balanced forces pulling sideways, the imbalance is a net inward attraction in a line perpendicular to the interface. Due to the net inward force exerted on liquids, the liquid surface will tend to contract, and to form a sphere (the geometry with minimum surface area to volume ratio). The contracted liquid surface is said to exist in a state of tension – known as surface tension. The value of surface tension for a liquid will be related to the strength of the pull between the liquid molecules. The interfacial interactions are a consequence of long range forces which are electrical in nature and consist of three types: dipole, induced dipole and dispersion forces.
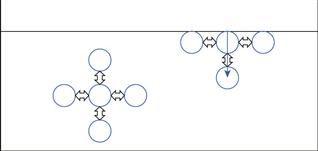
Dipole forces are due to an imbalance of charge across the structure of a molecule. This situation is quite common, certainly most drugs are ionizable, and have such an asymmetric charge distribution, as do many macromolecules and proteins. Such materials are said to have permanent dipoles, and interactive forces are due to attraction between the negative pole of one molecule when in reasonably close contact with the positive pole of another. Hydrogen bonding interactions are a specific sort of this type of bonding, resulting from the fact that hydrogen consists of only one proton and one electron, making it very strongly electronegative. When hydrogen bonds, its electron is ‘lost’, leaving an ‘exposed’ proton (i.e. one without any surrounding electrons). This unique situation causes a strong attraction between the proton and an electronegative region from another atom. The strength of the hydrogen bond results in drastically different properties of interaction, exemplified by the fact that water has such a high surface tension, melting and boiling point (in comparison with non-hydrogen bonded materials).
A bond between carbon and oxygen would be expected to be dipolar, however, if the molecule of carbon dioxide is considered (O C
C O), it can be seen that the molecule is in fact totally symmetrical, the dipole on each end of the linear molecule being in perfect balance with that on the other end. Even though these molecules do not carry a permanent dipole, if they are placed in the presence of a polarized material, a dipole will be induced on the (normally symmetrical) molecule, such that interaction can occur (dipole – induced dipole, or Debye interactions).
O), it can be seen that the molecule is in fact totally symmetrical, the dipole on each end of the linear molecule being in perfect balance with that on the other end. Even though these molecules do not carry a permanent dipole, if they are placed in the presence of a polarized material, a dipole will be induced on the (normally symmetrical) molecule, such that interaction can occur (dipole – induced dipole, or Debye interactions).
London – van der Waals forces are termed dispersion forces. These are interactions between molecules which do not have a charge imbalance, and which do not have the ability to have an induced dipole either. Essentially these are interactions between non-polar materials. These dispersion forces occur between all materials, and thus, even though the interaction forces are weak, they make a very significant contribution to the overall interaction between two molecules. Dispersion forces can be understood in a simplistic fashion by considering the fact that the electrons which spin around two neighbouring non-polarized atoms will inevitably not remain equally spaced. This will result in local imbalances in charge that lead to transient induced dipoles. These induced dipoles, and the forces which result from them will be constantly changing, and obviously the magnitude of these interactions is small compared to the permanent and induced dipole situations described above. Dispersion forces are long range, in the order of 10 nm which is significantly longer than a bond length.
Measurement of surface tension
The surface tension of a liquid is the combined strength of polar and dispersion forces that are pulling on the molecules in the surface of the liquid. There are a number of methods by which surface tension can be measured, including the rise of a liquid in a capillary, but more usually the force experienced by the surface is measured using a microbalance. To do this, an object either in the form of a thin plate (Wilhelmy plate) or ring (Du Nouy ring), is introduced to the surface and then pulled free, with the force at detachment being measured. For the Wilhelmy plate method, a plate (usually very clean glass or platinum) is positioned edge on in the surface whilst suspended from a microbalance arm, the force is then measured as the plate is pulled out of the liquid. The surface tension is obtained by dividing the measured force at the point of detachment by the perimeter of the plate.
Water is the liquid with the highest value for its surface tension of all commonly used liquids in the pharmaceutical field (although metals have much higher surface tensions than water, e.g. mercury with 380 mN m−1). Water is also of great pharmaceutical interest, being the vehicle used for the large majority of liquid formulations, and being the essential component of all biological fluids. At the standard reporting temperature, the surface tension of water is 72.6 mN m−1.
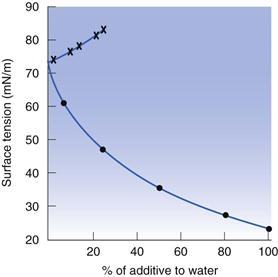
The addition of small quantities of impurities will alter the surface tension. In general, organic impurities are found to lower the surface tension of water significantly. Take for example the addition of methanol to water. The surface tension of methanol is 22.7 mN m−1, but the surface tension of a 7.5% solution of methanol in water is 60.9 mN m−1 (Fig. 4.2). On the basis of a linear reduction in surface tension in proportion to the concentration of methanol added, the surface tension of this mixture would be expected to be about 68.9 mN m−1, thus the initial reduction in surface tension upon addition of an organic impurity is dramatic, and cannot be explained by the weighted mean of the surface tensions of the two liquids. Methanol has been used as the example here, as it is one of the more polar organic liquids, containing just 1 carbon, attached to a polar hydroxyl group. However, it is its hydrophobicity that causes the significant reduction in surface tension. The reason for the large effect on surface tension is that the water molecules have a greater attraction to each other than to methanol, consequently the methanol is concentrated at the water/air interface, rather than in the bulk of the water. The methanol here is said to be surface active (surface active agents are discussed elsewhere in this book; in particular in Chapters 5 and 27). Water obtained directly from a tap can have a surface tension greater than 72.6 mN m−1, due to the presence of ionic impurities, such as sodium chloride, which are concentrated preferentially in the bulk of water rather than at the surface. Inorganic additives also strengthen the bonding within water, so the surface tension is increased in their presence.
Solid wettability
The vast majority of pharmaceutically active compounds exist in the solid state at standard temperatures and pressures. Inevitably, the solid drug will come into contact with a liquid phase, either during processing, and/or in the formulation, and also ultimately during use in the body. Consequently, the solid/liquid interface is of great importance. Here the term wettability is used to assess the extent to which a solid will come into contact with a liquid. Obviously a material which is potentially soluble, but which is not wetted by the liquid (i.e. the liquid does not spread over the solid) will have limited contact with the liquid and this will certainly reduce the rate at, and potentially the extent to, which the solid will dissolve. When formulating an active pharmaceutical ingredient, it is important that the powder ultimately becomes wetted by body fluids in order that it will dissolve.
As with liquid surfaces, there is a net imbalance of forces in the surface of a solid, and so solids will have a surface energy. The surface energy of a solid is a reflection of the ease of making new surface, and in simple terms can be considered to be the same as surface tension for a liquid. With liquids, the surface molecules are free to move, and consequently surface levelling is seen, resulting in a consistent surface tension/energy over the entire surface. However, with solids the surface molecules are held much more rigidly, and are consequently less able to move. The shape of solids is dependent upon previous history (perhaps crystallisation or milling techniques). These processes may yield rough surfaces with different regions of the same solid’s surface having different surface energies. Certainly different crystal faces and edges can all be expected to have a different surface nature due to the local orientation of the molecules presenting different functional groups at the surface of different faces of the crystal – some more and some less polar, and therefore some regions more water loving and other regions less so.
Contact angle
The properties of solids raise many problems with respect to surface energy determination, not least the fact that it is not possible to measure directly the forces exerted on the surface. The methods that are used for liquid surface tension measurement, such as immersing a Wilhelmy plate and measuring the force as it is pulled from the liquid, cannot be used as the plate cannot gain access into the solid. This means that surface properties of solids must be derived from techniques such as contact angle measurement. The tendency for a liquid to spread is estimated from the magnitude of the contact angle (θ), which is defined as the angle formed between the tangent drawn to the liquid drop at the three phase interface and the solid surface, measured through the liquid (Fig. 4.3). The contact angle is a consequence of a balance of the three interfacial forces; γSV acting to aid spreading; γSL acting to prevent spreading and γLV which acts along the tangent to the drop. The interfacial forces are related to contact angle by Young’s equation:
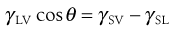 (4.1)
(4.1)
A low value for the contact angle indicates good wettability, with total spreading being described by an angle of 0 degrees. Conversely, a high contact angle indicates poor wettability, with an extreme being total non-wetting with a contact angle of 180 degrees. The contact angle provides a numerical assessment of the tendency of a liquid to spread over a solid, and as such is a measure of wettability.
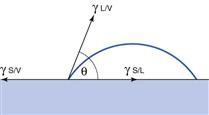
If a contact angle were measured on an ideal (perfectly smooth, homogeneous and flat) surface, with a pure liquid, then there would be only one value for the contact angle. In reality there are many contact angles that can be formed on a solid surface. The simplest analogy is water on glass. The contact angle of pure water on clean glass is zero, which provides the basis of surface tension experiments (as a finite contact angle would prevent such measurements). However, whenever raindrops are seen to form on a glass window, they do not spread, but rather form drops. The reason for this is that the window will not be clean and the liquid not pure. If raindrops fall on to a plate of glass which is horizontal, each drop will have the same contact angle all around its circumference. This value is termed the equilibrium contact angle, θE. If the glass plate is displaced from the horizontal, the drops will run down the surface, forming a tear shape. The leading edge of this drop will always have a larger contact angle than the trailing edge. The angle formed at the leading edge is termed the advancing contact angle (θA) and the other angle is termed the receding contact angle (θR). The difference between θA and θR defines the contact angle hysteresis. There are two possible reasons for contact angle hysteresis, surface roughness and contamination or variability of the composition of the surface, i.e. surface heterogeneity.
There are many different methods by which it is possible to measure a contact angle formed by a liquid on a solid. The vast majority of studies deal with smooth flat surfaces, such as polymer films, onto which it is comparatively simple to position a drop of liquid. The approaches to determination of the angle for such systems include direct measurement of the angle on a video image.
The Wilhelmy plate apparatus is described above as a method by which it is possible to measure surface tension. To do so it is necessary for the liquid to have a zero contact angle on the plate. Conversely, it is possible to assess the contact angle (θ) between the solid plate and the liquid if the surface tension of the liquid (γLV) is known. The force detected by the balance (F) is:
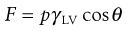 (4.2)
(4.2)
where p is the perimeter of the plate, and from this the value of the contact angle can be determined.
As mentioned above, certain polymeric systems are readily formed into smooth flat plates for contact angle studies, however, most pharmaceutical materials exist as powders, for which such a physical state is not readily achievable. A full understanding of powder surface energetics, and an ability to alter and control powder surface properties, would be a major advantage to the pharmaceutical scientist.
A drug crystal will consist of a number of different faces which may each consist of different proportions of the functional groups of the drug molecule; thus a contact angle for a powder will in fact be, at best, an average of the contact angles of the different faces, with contributions from crystal edges and defects. Also, impurities in the crystallising solvent can cause an adjustment of habit, and crystals of the same drug can exist in different polymorphic forms; such changes in molecular packing will potentially alter the surface properties. A final complication is that despite the fact that most pharmaceutical powders have a very high degree of crystallinity (and are called crystals), in reality sometimes they will have a small degree of amorphous content which is likely to be present at the surface. Thus, drug powders have heterogeneous surfaces of different shapes and sizes, which can readily change their surface properties. It is clear that all contact angle data for powders and the appropriate choice of methodology must be viewed in full knowledge of the inherent difficulties of the solid sample.
The most cited method of obtaining a contact angle for powders is to prepare a compact in order to produce a smooth surface, and then to place a drop on the surface in order to measure the contact angle that is formed. The first major problem with compacted samples is that the very process of compaction will potentially change the surface energy of the sample. Compacts form by processes of brittle fracture and plastic deformation, thus new surfaces will be formed during compaction, which can mask subtle differences in the original surface nature. The alternative is to not compact the powder, for example sticking fine powder to a piece of doubled sided adhesive tape. This presents a rough surface which gives rise to hysteresis and potentially also has a contribution from the surface property of the adhesive. There is no solution to these sample preparation difficulties, so a compromise decision has to be made in order to proceed with measurements.
Alternatives to placing a drop on the surface of a material exist for powder contact angle measurement, including making the powder into a plate and adapting the Wilhelmy plate method, and also measuring the rate at which liquid penetrates into a packed bed of the powder. These methods and their limitations have been reviewed elsewhere (Buckton, 1995). The different methods by which contact angle is measured for powders gives rise to different results, so comparison of data should take this into account.
An alternative to contact angle measurement is to use inverse gas chromatography (IGC). Whilst IGC has been used for many years in other sectors, it has only been used pharmaceutically to any great extent in recent years. Further discussion of IGC is presented below.
Adsorption at interfaces
Adsorption is the presence of a greater concentration of a material at the surface than is present in the bulk. The material which is adsorbed is called the adsorbate, and that which does the adsorbing is the adsorbent. Adsorption can be due to physical bonding between adsorbent and adsorbate (physisorption) or chemical bonding (chemisorption). The differences between physisorption and chemisorption are that physisorption is by weak bonds (such as hydrogen bonding, with energies up to 40 kJ mol−1) whilst chemisorption is due to strong bonding (greater than 80 kJ mol−1); physisorption is reversible, whilst chemisorption seldom is; physisorption may progress beyond a single layer coverage of molecules on the surface (monolayer formation to multilayer formation), whilst chemisorption can only proceed to monolayer coverage.
Solid/liquid interfaces
The usual pharmaceutical situation is to have a liquid (solvent), particles of a solid dispersed in that liquid and another component dissolved into the liquid (solute). This forms the basis of stabilising suspension formulations, where there may be water with suspended active pharmaceutical ingredient and in order to help stabilize the suspension (keep the solid particles from joining together) there may be a surface active agent dissolved in the water. The surface active agent will adsorb on the surface of the powder particles and help to keep them separated from each other (steric stabilisation). It is also possible to use this surface interaction in the treatment of drug overdose, where charcoal of high surface area can be administered and the excess of drug in the patient’s gastrointestinal tract can be adsorbed from solution onto the surface of the charcoal, which is then cleared from the patient. Kaolin is administered as a therapy in order to adsorb toxins in the stomach and so reduce gastrointestinal disturbances. A further example is analysis by HPLC – where molecules in solution are adsorbed onto a column to achieve separation. As a final example, the loss of active pharmaceutical ingredient, or preservative, from a solution product to a container can be a damaging effect of adsorption from solution to a solid.
The quantity of solute which adsorbs will be related to its concentration in the liquid. The adsorption will proceed until equilibrium is reached between that which has been adsorbed at the interface and that which is in the bulk.
Many factors will affect adsorption from solution onto a solid, these include temperature, concentration, and the nature of the solute, solvent and solid. The effect of temperature is almost always that an increase in temperature will result in a decrease in adsorption. This can be viewed as a consequence of giving the solute molecules more energy, and thus allowing them to escape the forces of adsorption, or simply viewed as the fact that adsorption is almost always exothermic, and thus an increase in temperature will cause it to decrease.
pH is important as many materials are ionizable, and the tendency to interact will vary greatly if they exist as polar ions, rather than a non-polar unionized material. In most pharmaceutical examples (chromatographic separation being an obvious exception), adsorption will be from aqueous fluids, and for these adsorption will tend to be greatest when the solute is in its unionized form, i.e. at low pH for weak acids, at high pH for weak bases, and at the isoelectric points for amphoteric compounds (those which exhibit acid and basic regions), although at other pH values the solubility in water will be higher (due to greater ionization favouring the interaction with water) and there will still be some unionized molecules present, which will usually adsorb on surfaces in preference to maintaining a disfavoured interaction with water.
The effect of solute solubility will influence adsorption as the greater the affinity of the solute for the liquid, the lower the tendency to adsorb to a solid. Thus, adsorption from solution is approximately inversely related to solubility.
The nature of the solid (the adsorbent) will be very important, both in terms of its chemical composition and its physical form. The physical form is the easiest to deal with, as it relates largely to available surface area. Materials such as carbon black (a very finely divided form of carbon) have extremely large surface areas, and as such are excellent adsorbents, both from solution (e.g. as an antidote as mentioned above) and from the vapour state where it has been used for gas masks. The chemical nature of the adsorbent solid is important, as it can be a nonpolar hydrophobic surface, or a polar (charged) surface. Obviously, adsorption to a non-polar surface will be predominantly by dispersion force interactions, whilst charged materials can also interact by ionic or hydrogen bonding processes.
Solid/vapour interfaces
When considering the solid/vapour interface, it is necessary to understand processes of adsorption and absorption. Adsorption has already been defined as the presence of greater concentrations of a material at the surface than is present in the bulk. Pharmaceutically, absorption is usually considered as the passage of a molecule across a barrier membrane, and is the essential requirement for enteral drug delivery routes to the systemic circulation. However, absorption should be considered as the movement into something, for example a gas or vapour can pass into the structure of an amorphous material, such that the uptake onto/into the solid is a sum of adsorption (to the surface) and absorption (into the bulk). If the uptake is thought to consist of both adsorption and absorption processes, it is often referred to by the general term, sorption.
There are many processes at the solid/vapour interface which are of pharmaceutical interest, but two of the most important are water vapour/solid interactions, and surface area determination using nitrogen (or similar inert gas)/solid interactions.
Solid/vapour adsorption isotherms
As with adsorption at the solid/liquid interface, the process can be due to chemisorption or physisorption. Most usually we will be concerned with physisorption.
Adsorption isotherms of vapours onto solids are representations of experimental data, usually plotted as amount adsorbed as a function of pressure of the gas, at a constant temperature. For such a plot, the pressure of the gas can be varied from zero to the saturated vapour pressure of the gas at that temperature (Po), and in each case the amount adsorbed can be determined (often by monitoring the change of weight of the sample). The concept of named adsorption isotherms (e.g. the Langmuir isotherm) is simply one of observing whether the experimental data fit to one of the existing mathematical models. If the data can be fitted, then there are several advantages: firstly, it becomes possible to define the adsorption process numerically, and thus exact comparisons can be made with similar data for other materials, and secondly, the models provide clues as to the nature of the adsorption process that has occurred (e.g. indicating whether the process is monolayer or multilayer, etc.).
Langmuir (Type I) isotherm
The Langmuir isotherm (one which fits the equation developed by Langmuir) is shown schematically in Figure 4.4. It has a characteristic shape of fairly rapid adsorption at low pressures of gas/vapour, which reaches a plateau well below Po, after which any further increases in pressure do not cause an increase in adsorption. This is the idealized model for monolayer adsorption, in that initially the surface is ‘clean’ and consists entirely of adsorption sites. Thus, a small amount of vapour allows rapid and extensive adsorption. Subsequently, more and more of the available adsorption sites become occupied, and thus further increases in pressure result in comparatively little increase in the amount adsorbed. At a certain pressure, all the adsorption sites will be occupied, i.e. monolayer coverage has been achieved, after which adsorption stops, giving a plateau region in which further increases in pressure have no effect on the amount adsorbed.
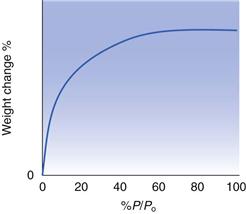
The Langmuir isotherm can only occur in situations where the entire surface is covered with equally accessible, identical adsorption sites, and the presence of an adsorbed molecule on one site does not hinder (or encourage) adsorption to a neighbouring site. For a system to follow a Langmuir isotherm, there must be a strong non-specific interaction between the adsorbate and the adsorbent (such that adsorption is desirable over the entire surface), and there must be little adsorbate – adsorbate interaction (in terms of attraction or repulsion).
Type II isotherms
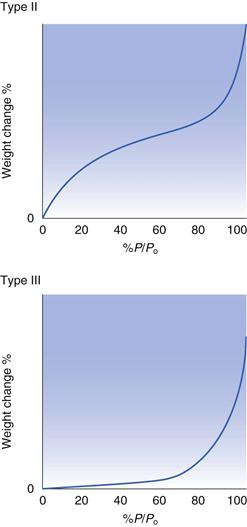
The Langmuir isotherm (Fig. 4.4) which describes adsorption of a monolayer only is often referred to as a Type I physical adsorption isotherm. There are other common shapes for adsorption isotherms, each of which can be taken to give an indication of the nature of the adsorption process. The schematic shapes of some other isotherms are shown in Figure 4.5. Type II isotherms are thought to correspond to a process which initially follows the Langmuir type of isotherm, in that there is a build-up of a monolayer, after this monolayer region, however, further increases in the vapour content result in further, and extensive adsorption. This subsequent adsorption is multilayer coverage, and is a consequence of strong interactions between the molecules of the adsorbate. These post-monolayer regions can be regarded as being analogous to condensation, and the isotherm rises as the pressure approaches Po.
Type III isotherms
Type III isotherms are typical of the situation where the interaction between adsorbate molecules is greater than that between the adsorbate and adsorbent molecules, i.e. the solid and the vapour have no great affinity for each other. This results in an isotherm shape for which it is necessary to have a significant presence of vapour before the adsorption process becomes significant, but once the surface starts to be covered with adsorbate, then the favourable adsorbate-adsorbate interaction results in a dramatic increase in adsorption for limited further increases in vapour concentration.
Brunauer, Emmett and Teller isotherm
The isotherm derived by Brunauer, Emmett and Teller is eponymously known as the BET isotherm. It is widely used as a standard method of determining surface area for solids. Just as the Langmuir isotherm fits to the Type I physical isotherm, the BET isotherm fits those situations which follow the Type II isotherm. The Type II isotherm is perhaps the most commonly encountered practically determined isotherm.
Interpretation of isotherm plots
With the Langmuir isotherm it can be assumed that the plateau region corresponds to monolayer formation, thus the quantity of gas adsorbed at monolayer is known, and consequently, as the area of each molecule of gas is known, the surface area of the solid can be determined. With a Type II isotherm, the system passes through monolayer coverage, at a region on the isotherm, this is rather difficult to define with any certainty from the graphical isotherm, but can easily be obtained from the BET equation.
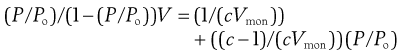 (4.3)
(4.3)
where P is vapour pressure; Po is saturated vapour pressure (note P/Po for water is the relative humidity), V is the volume of gas adsorbed, Vmon is the volume of gas adsorbed at monolayer coverage and c is a constant.
If (P/Po)/(1 – (P/Po)) V is plotted as a function of P/Po, the slope will be (c-1)/(cVmon) and intercept 1/(cVmon). From this it is possible to calculate Vmon, is the volume of the adsorbed gas which covers a monolayer. If the volume of gas is known, the number of gas molecules can be calculated, and then if the area occupied by each gas molecule is known, then the surface area of the solid is obtained.
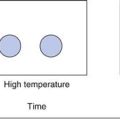
The measured surface area can vary depending upon the gas/vapour used to determine the isotherm. The most commonly used gas for surface area determination is nitrogen. The concept of fractal geometries brings into question all definitions of length, and consequently surface area. The standard typical illustration of fractals is shown in Figure 4.6, in which it can be seen that the length of an irregular, rough object can be altered enormously depending upon the resolution used in its measurement. For example, it is easy to consider the length of coastline on low magnification, but it becomes hard to know at what magnification one should reasonably stop, as with each magnification the length will increase by a factor proportional to that magnification. This caution is included, as the surface area of most solids is determined in relation to a nitrogen molecule as a probe. There will be many indentations in solids which may not be readily accessed by nitrogen gas, so a different probe gas (different size of molecule, e.g. krypton gas) can access different regions of the solid surface and the calculated surface area will change.
Isotherm models, other than Langmuir and BET, exist which can also be used to understand powder – vapour interactions, but these will not be discussed here.
Interactions between powders and water vapour
The interaction between water and a product is a consideration for almost every pharmaceutical product. Water may be important during formulation/preparation (for example, affecting powder flow, in wet granulation process, drying processes, ease of compaction, as a film coating solvent, aqueous liquid formulations, etc.), during storage (where it may influence chemical stability, physical transitions such as crystallization, or microbial spoilage), and during use (where there is a need to contact aqueous body fluids).
It is clear from the above paragraph that interaction with water is essential at certain stages, but undesirable in other situations. Consequently, an understanding of how, why, where, when and how much water will associate with a solid is an important issue in the development of pharmaceutical products. Water may interact with surfaces by adsorption and condensation, with some solids by absorption, as well as by inclusion into crystal structures as hydrates.
Water adsorption
Water is able to adsorb to a wide range of different materials over a wide range of temperature and humidity. Most gasses that have been mentioned so far, such as nitrogen, are thought to adsorb uniformly across surfaces, whilst water is thought to selectively bind to polar regions of a solid surface. Thus, the extent of adsorption of water to a solid surface is related to the degree of polarity of the solid itself.
It has been reported (van Campen et al., 1983; Kontny et al., 1987) that the adsorption of water onto most crystalline solids is not able to cause the solids to dissolve. This is because only a few layers of water molecules form as a ‘multilayer’ on solids, and this is a very small volume for dissolution. Furthermore, the structure of water adsorbed to the surface of a solid is different to the tetrahedral structure of bulk water, so the adsorbed material cannot be expected to have the same properties as a solvent as would be expected of bulk water. Given the observation of Kontny et al. (1987), that water which is adsorbed to the surface is only a few molecules thick and is not acting as bulk liquid, the question must be asked as to why water can have such a huge influence on the properties of materials, and on their physical and chemical stability. It can easily be calculated that the quantities of water which are said to be associated with solids are greatly in excess of that which can be accommodated in a few layers around their surface. Water can also interact with powders by being condensed in capillaries (or at other regions), or can be absorbed in amorphous regions, which is water that has the properties of bulk water and the ability to cause instability and spoilage.
The water content can be divided into different regions by considering the shape of the isotherm. The standard Type II isotherm (Fig. 4.7) has two inflection points, the first of which is termed Wm (the water content at the point which is thought to be the onset of monolayer coverage) and the second Wf (the water content regarded as free). At all humidities below that which corresponds to Wm the water can be regarded as tightly bound. At all points above Wf the water is considered to be liquid at room temperature and freezable.
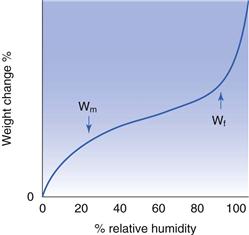
The condensation of water into capillaries is a consequence of the small pore sizes reducing the relative pressure at which condensation is possible. It can be estimated that the relative humidity at which water would condense would be 99% for pores of 100 nm, but only 50% for pores of 1.5 nm. It follows that materials which have surfaces which consist of many thousands of large volume micropores will adsorb huge quantities of water by capillary condensation. Materials such as silica gel have this type of structure, but it is comparatively rare to find pharmaceuticals which have microporous surfaces.
Water absorption
It is incorrect to assume that most pharmaceuticals are fully crystalline, or that most water association with pharmaceuticals is by adsorption. It has already been stated that pharmaceuticals can have amorphous regions, and that even those which are regarded as crystalline can have amorphous surfaces. Amorphous surfaces result from physical treatment moving surface molecules, and there being no mechanism by which they can recrystallize rapidly. The amorphous regions can result in chemical instability, and altered interactions between surfaces.
For amorphous materials, experimental evidence points to water uptake being due to absorption of water, as the quantity of water sorbed is related to the weight of materials present, and not the surface area (as would be the case for adsorption). It is also common for sorption and desorption isotherms for amorphous materials to show considerable hysteresis, despite the absence of microporous structure (the other main cause of such effects).
The interpretation of isotherms for systems which are suspected to have undergone absorption must be undertaken with care. The value Wm, for example, will still exist as a Type II isotherm will be a common occurrence; however, it can no longer be expected to represent monolayer coverage. For amorphous materials the value of Wm reflects the polarity of the solid, the higher the value, the more polar the solid. The second inflection point (Wf) for amorphous materials is believed to be the point at which the water has so plasticized the solid that the glass transition temperature (Tg) of the amorphous mass has fallen, such that it now equals the temperature of the experiment.
The glass transition temperature of an amorphous material is the point at which it shows a change in properties. Below the Tg materials are brittle and are said to be in the glass (or glassy) state. For example, window glass has a Tg of about 1000 °C, and as such is brittle at ambient conditions. Above the Tg a material becomes more rubbery. It is often desirable to have materials of a rubbery nature at room temperature, for example for the production of bottles which are less prone to shatter than glass. It is possible to mix another material with the main component, the minor component will fit between the molecules of the first, and will allow greater molecular movement, thus lowering Tg. The additive is called a plasticizer. It is possible to estimate the effect of a plasticizer by use of the following simple equation:
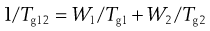 (4.4)
(4.4)
where W1 is the weight fraction of material 1 (with Tg= Tg1), and W2 is the weight fraction of material 2 (with Tg= Tg2) and Tg12 is the Tg of the mixture. Thus, a plasticizer is a material which has a lower Tg than the material, and which can gain access to regions within the molecules of the material. Water has a Tg of about −138 °C, and as such can efficiently plasticize many amorphous materials.
The process of amplification has been explained by Ahlneck and Zografi (1990) who regard absorption into amorphous regions as being the preferred form of interaction between powders and water vapour. It is argued that the amorphous regions are energetic ‘hot spots’, such that water would rather absorb than adsorb to the general surface. If we accept this hypothesis, which does seem entirely reasonable, then there must be great concern about materials which have a very small amorphous content and a small amount of associated water. It is quite usual for materials to contain 0.5% moisture, which sounds insignificant, however, if the material is 0.5% amorphous then it is likely that 0.5 % moisture is in 0.5% of the solid, and is thus present in a 50 : 50 ratio of water to solid. This would provide a region of enormous potential for physical transition, chemical reaction or microbial spoilage. The example does not have to be as extreme as this; it has been calculated (Ahlneck and Zografi, 1990) that only 0.1 % moisture content is needed in a sucrose sample which is 1 % amorphous in order to plasticize the Tg of the amorphous sucrose to below room temperature.
It is clear then that the critical drastic consequences of water/solid interaction are much more likely to result as a consequence of amplification of water into the minor regions of amorphous surface material, than by surface adsorption. It follows that materials can be expected to change their properties as a consequence of any process which can re-order surface molecules, such as milling, spray drying, etc.
It is worth restating that the great increases in molecular mobility that accompany the transition from glass to rubber state will be sufficient to trigger physical changes and to initiate, or speed up, chemical degradation processes. This can occur in any amorphous material, which includes surface regions of ‘crystalline’ drugs and excipients.
The presence of high proportions of water in amorphous regions of solids is often enough to promote surface recrystallisation. The surface need not have dissolved in the true dissolution sense of the word, but may simply have been plasticized to give sufficient reduction in viscosity to allow molecular realignment. It is now a matter of some commercial interest that surfaces will behave in totally different manners depending upon whether they are partially amorphous, or crystalline, this will relate to ease of use, stability on storage and ease of manufacture (see examples in Chapter 8).
Deliquescence
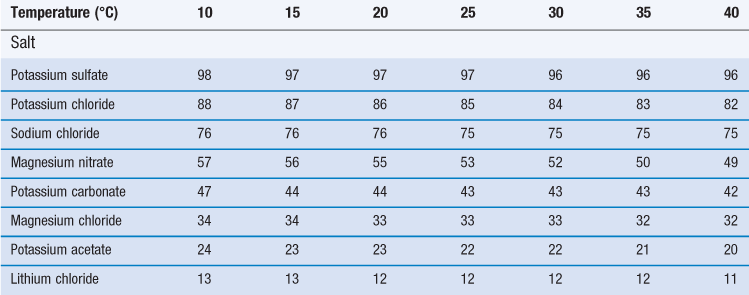
Certain saturated solutions of salts are known to produce an atmosphere of a certain relative humidity above their surface. If any of these salts are stored in solid form at any humidity above the values that would be produced above their saturated solutions, then they will dissolve in the vapour. If they are stored below that critical humidity value then they will adsorb water vapour, but will not dissolve. Such materials which dissolve in water vapour are known as deliquescent.
A major characteristic of deliquescent materials is that they are very soluble, and have a large colligative effect on the solution formed, such that the vapour pressure of water is drastically reduced by the presence of the dissolved solute. The stage of events in deliquescence is that some water is adsorbed/absorbed. At a critical humidity, a small amount of the highly soluble solid dissolves, this lowers the vapour pressure of water, leading to extensive condensation, and an auto-catalytic process develops (i.e. as more solid dissolves the vapour pressure lowers, which causes more condensation to occur, which causes more solid to dissolve). The process will continue until all the material has dissolved, or until the relative humidity falls below that which is exhibited above the saturated solution of the salt. The reason that different salts produce such a range of relative humidities above their saturated solution is due to the colligative action of their respective molecules reducing the activity of water. The relative humidity produced in the vapour space above saturated solutions of certain salts is reported in Table 4.1.
Inverse phase gas chromatography (IGC)
As mentioned above, there are practical issues with measuring the contact angle for powdered systems. An alternative is to study the interaction between the powder and a vapour. Gas chromatography is a well-established analytical method. A column is packed with a powder and a test sample is injected into a constant flow of gas that is passing through the column, which is held at constant temperature. A detector is positioned at the end of the column. The test sample will be carried through the column by the carrier gas, however as it interacts with the powder in the column, components of the test sample will be slowed to different extents based on the extent of interaction between them and the powder in the column. This achieves separation and good analysis. Inverse phase gas chromatography is where a known substance is injected and the test material is the powder packed into the column. For example, the known gas could be hexane vapour and the powder packed in the column is the material for which we wish to know the nature of its surface. It would be usual to inject vapours of a series of alkanes, say hexane, heptane, octane, nonane, and also to inject a number of polar vapours. From the retention times of the injected vapour it is possible to understand the dispersive surface energy (from the retention of the alkanes) and the polar surface energy (form the retention of polar probes) of the test solid. This allows the surface nature of different solids to be compared without the need to compact the sample and measure a contact angle.
References
1. Ahlneck C, Zografi G. The molecular basis of moisture effects on the physical and chemical stability of drugs in the solid state. International Journal of Pharmaceutics. 1990;62:87–95.
2. Kontny MJ, Grandolfi GP, Zografi G. Water vapour sorption in water soluble substances: Studies of crystalline solids below their critical relative humidity. Pharmaceutical Research. 1987;4:247–254.
3. Van Campen L, Amidon GL, Zografi G. Moisture sorption kinetics for water-soluble substances 1) Theoretical considerations of heat transport control. Journal of Pharmaceutical Science. 1983;72:1381–1388.
4. Wade A, ed. Pharmaceutical Handbook. London: Pharmaceutical Press; 1980.
5. Weast RC, ed. Handbook of Chemistry and Physics. CRC Press 1988; Boca Raton.
Bibliography
1. Buckton G. Interfacial Phenomena in Drug Delivery and Targetting. Amsterdam: Harwood Academic Press; 1995.