[level-membership-for-basic-science-category]
Sterilization in practice
Jean-Yves Maillard and Susannah E. Walsh
Chapter contents
Determination of sterilization protocols
Recommended pharmacopoeial sterilization processes
Steam (under pressure) sterilization
Integrated lethality in sterilization practice
Statistical considerations of sterility testing and sterility assurance level
Test for sterility of the product
Validation of a sterilization process
Key points
Sterile products
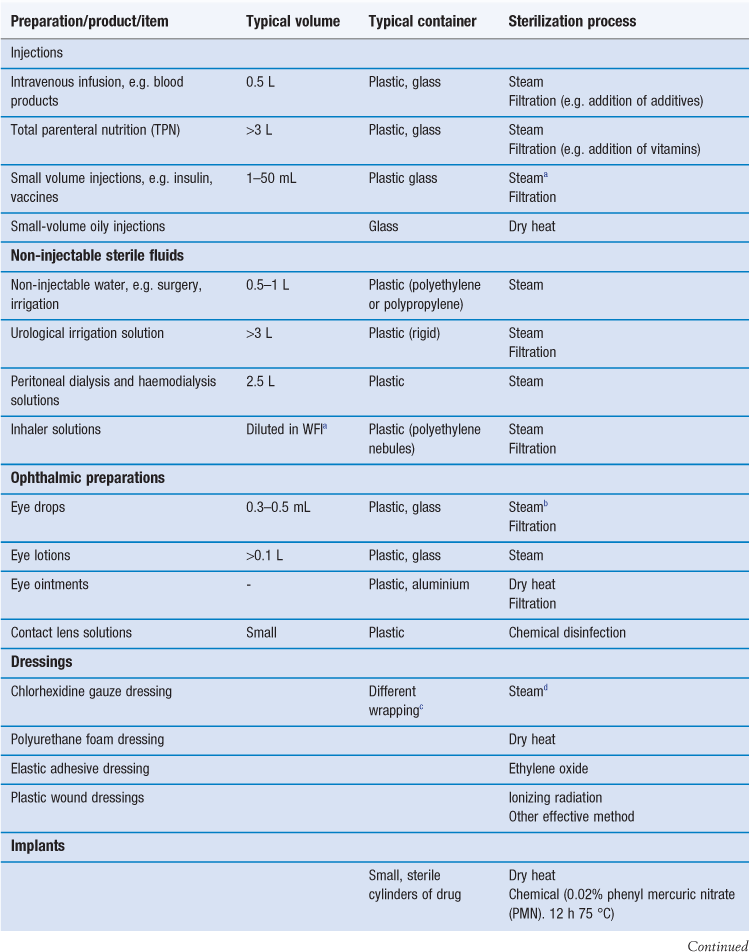
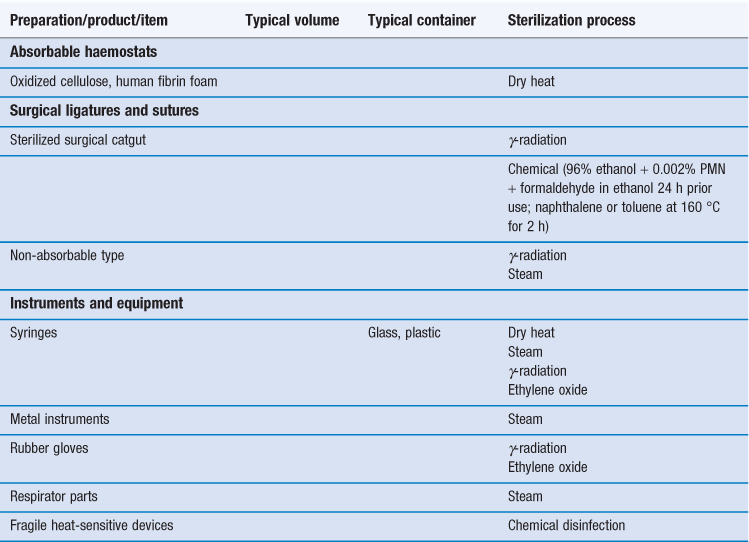
Sterilization is an essential part of the processing of pharmaceutical dosage forms that are required to be sterile. By definition, a sterile product is completely free of viable microorganisms. In addition to the pharmaceutical products that require to be sterile, a number of medical devices that come into contact with sterile parts of the body or are reused in patients also need to be free of microorganisms (Table 17.1). The diversity of the items to be sterilized in terms of properties (e.g. heat sensitive or not), bulk, content, and the number of items to be sterilized per load requires the use of distinct sterilization processes. The British Pharmacopoeia (2013a), as an example, describes the use of five main sterilization processes to accommodate the range of products to be sterilized: steam, dry heat, gaseous, ionizing radiation and filtration sterilization. The first four methods are usually used to process products in their final containers (terminal sterilization). Regardless of the sterilization methodology used, it is important that the process itself is fully validated. A number of guidelines and European/international standard documents for specific product-sterilization method combinations exist and are followed by manufacturers and end users. Failure to control and/or document adequately a sterilization process can lead to serious incidents. This chapter aims to provide a brief overview of the recommended sterilization processes, their control and validation.
Determination of sterilization protocols
There are various technologies available to achieve sterility of pharmaceutical preparations and medical devices (Table 17.2). Generally, sterilization of the product in its final container (terminal sterilization) is preferred. This infers that the container must not impinge on the optimum sterilization to be delivered and that the container and closure maintain the sterility of the product throughout its shelf-life. The selected sterilization process must be suitable for its purpose, i.e. the sterilization of a given product, device and preparation, which means that the product and its container have to be rendered sterile and must not be damaged by the process or post process.

The choice of an appropriate sterilization process depends on a number of factors (Table 17.3) related to the product to be sterilized, such as type and composition of product and also the quantity to be sterilized. Additionally, the composition and the packaging of the product are significant factors that rule out some sterilization processes. For example, a heat-labile preparation would not be sterilized by heat and a small oily injection would not be sterilized by steam sterilization (further examples are given in Table 17.1). For specific types of products such as dressings, although moist heat sterilization is generally the method of choice, only certain types of autoclave, such as vacuum and pressure-pulsing autoclaves are appropriate.
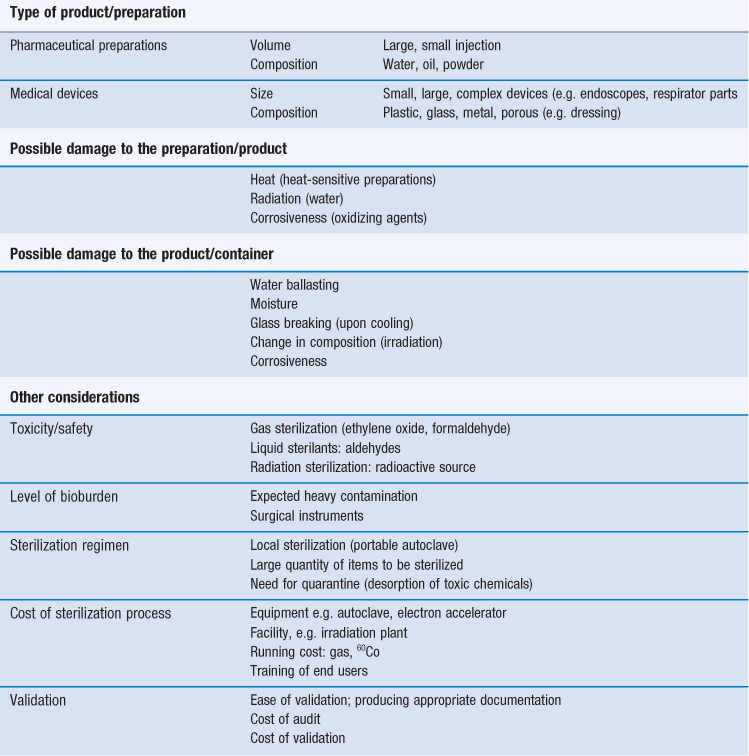
For any given preparation or product, it is difficult to predict the microbial bioburden prior to sterilization. It is assumed that the bioburden of pharmaceutical preparations will be minimal as the manufacturing process should adhere to good manufacturing practice (GMP) (Box 17.1). However, a sterilization process should be able to deal with a worst case scenario. This is usually exemplified by the use of biological indicators (see the ‘Process indicators’ section later in this chapter) such as bacterial spores, which are considered as the most resistant infectious agents (with the exception of prions, the agents responsible for spongiform encephalopathies). This is usually the situation for official sterilization methods. Pharmacopoeial recommendations as well as guideline documents are derived from data generated from the use of bacterial spores (biological indicators) for a given sterilization process.
When a fully validated sterilization process has been conducted, the release of a batch of product can be based on process data during sterilization rather than the results from sterility testing. Any change in the sterilization procedure (e.g. product load, type of containers) requires re-validation to take place.
Re-sterilization of products/devices can cause their degradation (e.g. repeated irradiation or autoclaving) or may even cause them to become toxic (e.g. with ethylene oxide; Richards 2004). Therefore any proposed re-sterilization must be carefully investigated.
Recommended pharmacopoeial sterilization processes
Five main sterilization processes which possess different characteristics are usually recommended by pharmacopoeias:
• steam (under pressure) sterilization (terminal)
• dry heat sterilization (terminal)
• ionizing radiation sterilization (terminal)
Although the use of other sterilization methodologies is not necessarily precluded, appropriate validation documentations for each product need to be provided. More information can be found in Chapters 15 and 16, or by consulting the relevant pharmacopoeia. At the time of writing, examples of these include the European Pharmacopoeia (2011), the United States Pharmacopeia (2012) and the British Pharmacopoeia (2013a) but it is always important to consult the most up-to-date texts and guidelines.
Steam (under pressure) sterilization
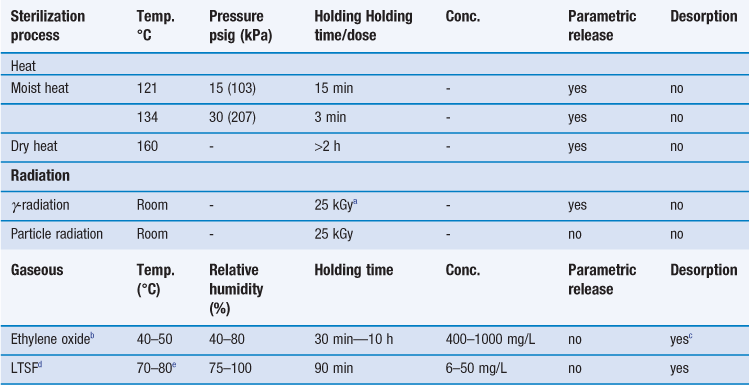
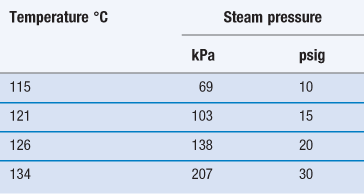
Steam sterilization is the most reliable, versatile and universally used form of sterilization and relies on the combination of steam, temperature and pressure. The typical cycle consists of a holding time of 15 minutes at a temperature of 121 °C under 15 psi (103 kPa) gauge pressure (Table 17.4). The aim is to deliver steam at the phase boundary (dry saturated steam; see Chapter 16, Fig. 16.2) to all areas of the load. This is achieved using steam and pressure (Table 17.5).
Table 17.4
Typical terminal sterilization cycles
cDesorption could take up to 15 days; maximum threshold of ethylene oxide residues and evaluation documented in ISO 10993–7 (1996).
dLow temperature steam formaldehyde; values can differ slightly depending upon the literature.
eLower temperature of 55–56 °C can be used depending upon the thermotolerance of the preparation.
Steam under pressure is commonly used unless prohibited by lack of load penetration or heat and/or moisture damage. Steam can only kill microorganisms if it makes direct contact, so it is very important to avoid air pockets in the sterilizer during a sterilization process. In addition, air can reduce the partial pressure of the steam so that the temperature reached on surfaces will be less than that expected with the pressure used. Hence, removal of air is an essential part of the process to ensure effective sterilization. To remove the air present when an autoclave is loaded, autoclaves are equipped with air removal/displacement systems (e.g. vacuum and displacement autoclaves, etc.). For porous loads, gravity displacement systems (downward-displacement autoclaves) are not adequate and vacuum and pressure-pulsing autoclaves are the method of choice (McDonnell 2007). Non-condensable gases must also be removed and monitored; these are atmospheric gases like nitrogen and oxygen that form part of the initial atmosphere of the sterilizer. Other factors that affect the efficacy of steam sterilization are water content and steam purity. The optimal sterilization is obtained with saturated steam (as discussed in Chapter 16). Supersaturated steam (i.e. wetter steam) is associated with condensation and poor penetration. Superheated steam (i.e. drier steam) behaves like dry heat and is less efficient. Steam purity is determined by the quality of the water, which can be affected by a number of contaminants (e.g. pyrogens, amines, toxic metals, iron, chlorides, etc.) that can render the sterile product unsafe (e.g. toxicity caused by pyrogenic reactions, metallic poisoning) or damaged (e.g. discoloration of packaging, corrosion caused by iron and chlorides).
Steam under pressure is generated in autoclaves which can vary greatly in size and shape from portable bench-top units to industrial production facilities (Fig. 17.1). A cross-section through an autoclave is shown in Figure 17.2.



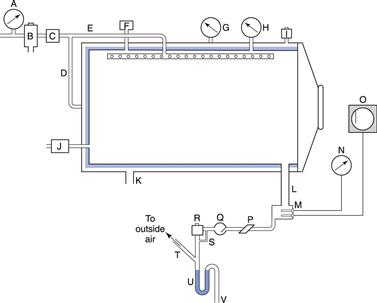
Steam sterilization applications are informed/regulated by a number of European and international guidelines and standards providing information on sterilizer design and installation, quality of steam, requirement for pressure, development and validation and routine control, etc.
Dry heat sterilization
The most common dry heat sterilization method uses hot air ovens (Fig. 17.3). Other procedures, such as sterilizing tunnels utilizing high-temperature filtered laminar air flow or infrared irradiation to achieve rapid heat transfer, are also available. Hot air ovens are usually heated electrically and often have heaters under a perforated bottom plate to provide convection currents (gravity convection type). Mechanical convection hot air ovens are equipped with a fan to assist air circulation and increase heat transfer by convection (Joslyn 2001). Dry heat sterilization is less expensive than steam sterilization and is effective for the depyrogenation of containers/packaging (e.g. glassware). Overloading should be avoided, wrappings and other barriers minimized and the load positioned to allow optimal air circulation. Other problems include long heating up times (e.g. with large loads of instruments) and the charring or baking of organic matter onto items. Dry heat sterilization cycles are generally longer than for moist heat sterilization, typically 2 hours at 160 °C (see Table 17.4). The process is thermostatically controlled and monitored using thermocouples.
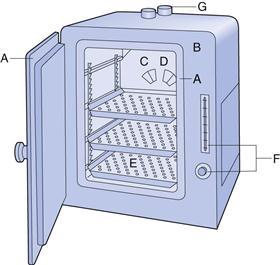
Dry heat sterilization cannot be used for a number of products such as rubber, plastics and other thermolabile items, or for aqueous solutions.
Integrated lethality in sterilization practice
All heat sterilization processes must include heating up and cooling down time periods. These prolonged time periods at a raised temperature may increase the degradation of the product. Integrated lethality attempts to examine the effects of heat on the inactivation process during these time periods.
For steam sterilization, the Fo concept (‘reference unit of lethality’) is used. This takes into account the heating up and cooling down stages of the cycle and is expressed as the equivalent time in minutes at a temperature of 121 °C delivered by the process to the product in its final container with reference to microorganisms possessing a Z value of 10. Its calculation is complex and further information can be found in the relevant pharmacopoeias. In practice, computer programs can be used to calculate the combined effect of whole processes, allowing a reduction in the total process time. It is important that the appropriate sterility assurance level is consistently achieved and the routine use of biological indicators is recommended, although following process validation, parametric release might be preferred.
Gaseous sterilization
The gaseous sterilization method recommended by pharmacopoeias mainly employs ethylene oxide. It is usually used on a commercial scale for the sterilization of catheters, infusion giving sets, syringes, prostheses and some plastic containers and thermolabile powders (if humidity is not a problem; Sharp 2000). The ethylene oxide sterilization cycle is complex since many factors need to be controlled over a long period of time (see Table 17.4). The control of the temperature, concentration and relative humidity is critical. In addition, ethylene oxide is very flammable and can form explosive mixtures in air. It is therefore combined with an inert gas carrier (e.g. carbon dioxide, nitrogen or chlorofluorocarbon). Ethylene oxide is also toxic, mutagenic and a possible human carcinogen. It is nevertheless a popular sterilization process, mainly because of the low temperature used during sterilization, but also because of the amount of information acquired on ethylene oxide sterilization processes over the years. The sterilization procedure is usually carried out in a purpose-built, gas-tight stainless steel chamber which can withstand high pressures and vacuum (Sharp 2000). However, systems utilizing a slight negative pressure rather than drawing a full vacuum are available (Fig. 17.4) and these are suitable for smaller, vacuum-sensitive loads.

Packaging should be permeable to air, water vapour and ethylene oxide. The sterilized products need to be quarantined post process to allow the removal of gas. The European Pharmacopoeia and other international standards set limits for ethylene oxide residue levels (e.g. a maximum of 10 ppm for plastic syringes).
Low-temperature steam formaldehyde (LTSF, discussed in Chapter 16), although not included in this chapter’s list of recommended methods, is used for the sterilization of certain preparations. As with ethylene oxide, its sterilization cycle is rather complex, since several parameters have to be controlled (see Table 17.4).
Radiation sterilization
There are two types of radiation unit. The becquerel (Bq) measures the activity of a source of radiation (physical radiation). One Bq equates to a source that has one nuclear disintegration per second. The gray (Gy) measures the effect of radiation on living tissue. One Gy is equal to the transfer of 1 J of energy to 1 kg of living tissue. The gray has replaced the rad that quantified radiation absorbed dose. Electron volt measures the energy of radiation and is usually expressed as millions of electron volts (MeV).
The source of γ-rays for sterilization is usually cobalt 60. Caesium 137 can also be used but has less penetrating power. Cobalt 60 decays with the emission of two high-energy γ-rays (1.17 and 1.33 MeV) and a lower energy (0.318 MeV) β particle. Gamma radiation is highly penetrative, causes negligible heating of the sterilized product at normal doses and induces no radioactivity in the final product.
Irradiation of product can be carried out in batches but is more commonly a continuous process using a conveyor system. The products pass through the irradiation chamber and are irradiated from one or two sides. The source is shielded with concrete to protect the operators and the environment. The intensity of radiation decreases as it penetrates. For example, 100 mm of a product with a density of 1 g/cm3 would reduce the cobalt 60 intensity by 50%. A cobalt 60 source of 1–4 × 1016 Bq is used for industrial irradiation and this provides a radiation dose in excess of 25 kGy. In most of Europe, 25 kGy is the standard dose (e.g. European Pharmacopoeia 2011) but in Scandinavia doses of up to 45 kGy are recommended (Lambert 2004). When not in use, the radioactive source is submerged in water for shielding and cooling.
Particle radiation sterilization utilizes β particles that are accelerated to a high energy level by application of high-voltage potentials (no radioactivity required). Their low energy means that beams from particle accelerators are less penetrating than γ-rays, with only 10 mm of a 1 g/cm3 material being penetrated per million electron volts (MeV). However, an important advantage of particle radiation is that the source can be turned off and is directional (Lambert 2004). The design of an accelerator can be customized to applications by including different energy and power requirements. The beam source is shielded with concrete and products are conveyed through the exposure area and irradiated. Another advantage is that shorter exposure times are required than those needed for γ irradiation. High-energy beams with energies of 5–10 MeV are used for sterilization, the accelerating field being generated using radiofrequency or microwave energy. Once it has been accelerated to the required energy, the beam of electrons is managed by magnetic fields which can alter its size, shape or direction (McDonnell 2007).
Radiation can affect a number of materials (e.g. polyethylene, silicone rubber, polypropylene, Teflon) and aqueous solutions (e.g. through the process of water radiolysis), and its packaging (discussed further in the ‘Limitation of sterilization methods’ section later in this chapter). Although radiation is considered as a ‘cold’ process, intense radiation can cause an increase in temperature and as such possible overheating needs to be considered for a specific load.
Validation of radiation sterilization involves the use of Bacillus pumilus as a biological indicator and dosimetric analysis (discussed later in this chapter). The routine monitoring involves measurements to ensure that all products are receiving the required dose. The radiation sterilization procedure is highly regulated and there are a number of European and international standards and guidelines available with information on requirements for the development, validation and routine control of the process (e.g. EN552, ISO 11137-1) and the dose required for sterilization (e.g. ISO 11137-2).
Filtration
Filtration is employed for non-terminal sterilization and has to be used under strict aseptic conditions. It is used for those preparations that cannot be sterilized by a terminal process or to which an agent (e.g. additive, heparin, vitamin, etc.) is added post-sterilization. Filtration is used to sterilize aqueous liquid, oils and organic solutions, and also air and other gases. Membrane filtration is an absolute process which ensures the exclusion of all particles above a defined size. Although many materials have been used to make filters, only a few are suitable for sterilization of pharmaceutical products.
Depth and surface filters are suitable for prefiltration of pharmaceutical products as they can retain large amounts of particles. Depth filters can be made of fibrous, granular or sintered material that is bonded into a maze of channels that trap particles throughout their depth. Surface filters are made of multiple layers of a substance such as glass or polymeric microfibres. Any particles that are larger than the spaces between the fibres are retained and smaller particles may be trapped in the matrix (McDonnell 2007). A membrane filter downstream is needed to retain any fibres shed from these filters as well as small particles and microorganisms.
To sterilize a product, it is often necessary to combine several types of filtration (e.g. depth, surface and membrane filters) to achieve the removal of microorganisms. Depth and surface filtration are used to remove the majority of particles by acting as prefilters. The final filtration step is accomplished using a membrane filter. This combined approach removes particles and microorganisms without the membrane filter blocking up rapidly with large particles.
High-level disinfection
In addition to the processes described above, high-level disinfectants (chemical biocides) have to be mentioned, since they are used for the chemosterilization of medical devices, particularly high-risk items that come into contact with sterile parts of the body, such as surgical instruments, intrauterine devices, endoscopes (which are used for a wide range of diagnostic and therapeutic procedures) (see Table 17.1).
Like the gaseous biocides, the activity of high-level liquid disinfectants depends upon a number of factors (Maillard 2005). Consequently, the training of the end user is of prime importance. Guidelines are often available from professional societies regarding the use of chemical biocides and specific devices; for example, the sterilization procedure and risk assessment for gastroscopes is published by the British Society of Gastroenterology (2008).
To ensure the efficacy of high-level disinfection, knowledge of the factors affecting efficacy, education of end users and compliance with manufacturer’s instructions is essential (Maillard 2005). The main advantage of using high-level disinfection is the low temperature used in processing medical devices. However, high-level disinfection might not give the same level of sterility assurance, and where possible, physical processing (e.g. steam sterilization) should be the method of choice.
The main disadvantages of high-level disinfection are exposure toxicity to the end users, damage to materials and potential emerging microbial resistance; all high-level disinfectants are toxic at the concentration used. For example there have been many reports of exposure toxicity to glutaraldehyde following endoscope reprocessing and this has resulted in abandoning the use of the dialdehyde in many countries. Damage to the materials following reprocessing can take the form of corrosion to metallic surfaces and increased rigidity of plastics. Problems associated with inappropriate high-level disinfection regimens, which resulted in microbial contamination have been described since the 1990s. It has been suggested that as many as 270,000 infections are transmitted by endoscopes per year (Lewis 1999). These events are quite distinct from the recent reports that microorganisms are becoming resistant to the in use concentration of these high-level disinfectants (Maillard 2010).
Statistical considerations of sterility testing and sterility assurance level
The strict definition of sterility is the complete absence of viable microorganisms. In other words, after a successful sterilization process, the number of microbial survivors should be zero. This is an absolute definition which cannot be guaranteed especially from a microbial point of view. To ensure the absence of viable microorganisms, one has to ensure all viable microorganisms can be detected and cultured. When one looks at microbial inactivation following, for example, exposure to heat or radiation, the inactivation usually follows first-order kinetics (see Chapters 15 and 16), although in practice microorganisms are inactivated at different rates producing a deviation from linear inactivation. Thus, assuring the complete elimination of microbial contaminants and thus sterility of the product cannot be guaranteed mathematically or practically.
Instead of defining sterility in a strict microbiological sense, it is more appropriate to consider the likelihood of a preparation being free of microorganisms. This is best expressed as the probability of a product containing a surviving microorganism after a given sterilization process. Survival depends upon the number and the type of microorganisms, soiling and the environmental conditions within sterilizing equipment. The concept of a sterility assurance level (SAL) or microbial safety index provides a numerical value to the probability of survival of a single microorganism. The SAL is therefore the degree of assurance for a sterilizing process to render a population of products sterile. For pharmaceutical preparations a SAL of < 10–6 is required. This equates to not more than one viable microorganism per million items/units processed. Practically, the lethality of a sterilization process and in particular the number of log cycles required need to be calculated.
The inactivation factor (IF) which measures the reduction in the number of microorganisms (of a known D value; see Chapters 15 and 16) brought about by a defined sterilization process can be calculated as follows:
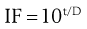 (17.1)
(17.1)
where t is the contact time (for heat or gaseous process) or radiation dose (for ionizing radiation) and D is the D value appropriate to the process employed. For example, if we consider steam sterilization, for an initial bioburden of 104 spores of Geobacillus stearothermophilus, an IF of 1010 will be required to achieve a SAL of 10–6. G. stearothermophilus has a D value of 1.5 for steam sterilization. Thus according to Equation 17.1, a 15-minute sterilization process (i.e. holding time) at 121 °C will be required to achieve an IF of 1010 (i.e. 1015/1.5). The process will therefore reduce the level of microorganisms by 10 log cycles.
Calculation of IF is based on obtaining an inactivation kinetic that follows a first-order kinetic. In reality, this is not always the case. In the food industry, the calculation of the most probable effective dose (MPED) is preferred as it is independent of the slope of the survivor curve for the process. However, to establish a MPED that will achieve the required reduction in a number of microorganisms, complex calculations are required.
Test for sterility of the product
Sterility testing assesses whether a sterilized pharmaceutical or medical product is free from microorganisms by incubating all or part of the product with a nutrient medium. Testing for sterility is a destructive process. For an item to be shown not to contain organisms, unfortunately it has to be destroyed. Due to the destructive nature of the test and the probabilities involved in sampling only a portion of a batch, it is only possible to say that no contaminating microorganisms have been found in the sample examined in the conditions of the test (British Pharmacopoeia 2013b). Thus the measurement of sterility relies on statistical probability. In other words, it is impossible to prove sterility since sampling may fail to select non-sterile containers and culture techniques have limited sensitivity. In addition, not all types of microorganisms that might be present can be detected by conventional methods as not all microorganisms are affected by a sterilization process in the same way. It is possible that some may not be killed or removed. For example, a filter pore size of 0.22 µm is usually used for filtration sterilization which means that smaller microorganisms such as viruses are allowed through.
Detailed sampling and testing procedures are given in pharmacopoeias and further details can be found in Chapter 14. For terminally sterilized products, biologically based and automatically documented physical proofs that show correct treatment during sterilization are of greater assurance than the sterility test. This method of assuring sterility is termed parametric release and is defined as the release of a sterile product based on process compliance to physical specifications. Parametric release is acceptable for all terminal sterilization processes recommended by the European Pharmacopoeia.
Validation of a sterilization process
The British Pharmacopoeia (2013a) states:
The sterility of a product cannot be guaranteed by testing; it has to be assured by the application of a suitably validated production process. It is essential that the effect of the chosen sterilization procedure on the product (including its final container or package) is investigated to ensure effectiveness and the integrity of the product and that the procedure is validated before being applied in practice.
Clearly this statement points out that testing for sterility is not enough and a suitable production process should be appropriately validated. Any changes in the sterilization procedure (i.e. change in sterilization process, product packaging or load) require revalidation. For pharmaceutical preparations, good manufacturing practices (GMP) have to be observed for the entire manufacturing process, not just the sterilization procedure.
The process of validation requires that the appropriate documentation is obtained to show that a process is consistently complying with predetermined specifications. International organizations such as the International Standards Organization (www.iso.org) and the Food and Drug Administration in the USA (www.fda.gov) provide detailed documentation for the validation of sterilization of healthcare products or medical devices with various processes (e.g. steam, radiation and gaseous). For the validation of sterilization processes, two types of data are required: commissioning data and performance qualification data (Box 17.2). Commissioning data refer mainly to the installation and characteristics of the equipment and the performance data ensure that the equipment will produce the required sterility assurance level. The performance qualification data can be divided into physical and biological performance data (Box 17.2).
Obtaining biological performance data is required for the validation and revalidation of the sterilization process of new preparations, new loads and new sterilization regimens and is usually not used routinely except when sterilization conditions are not well defined (e.g. gaseous sterilization) or with non-standard methods. The use of biological indicators (discussed below) requires a good knowledge of the inactivation kinetics (e.g. D value) for a given process. Performance qualification data must be reevaluated following a change to the preparation or product and its packaging, the loading pattern or the sterilization cycle.
Process indicators
For all methods of sterilization it is essential that the equipment used works correctly. Routine tests are carried out to demonstrate that all parts of the sterilizer have been correctly installed (installation qualification) and that they operate properly, with sterilizing conditions reaching every part of the load (operation qualification; McDonnell 2007). The test methods used vary according to the sterilization method and may involve the use of physical indicators, chemical indicators and biological indicators.
Physical indicators measure parameters such as heat distribution (i.e. temperature) by thermocouples, pressure variation by gauges or transducers, gas concentration, steam purity, relative humidity by hygrometers or direct calorimetry, delivered dose and time exposure. Sensors must be maintained and calibrated regularly. They are usually the first indicator of a problem with a sterilization process and if documented correctly can be sufficient to meet the requirements for parametric release (Berube et al 2001).
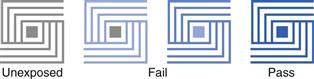
Chemical indicators vary depending on the sterilization method but essentially they all change in physical or chemical nature in response to one or more parameters. There are several types of chemical indicators (Fig. 17.5); temperature-specific indicators just show whether a specific temperature has been reached (single variable indicators) whereas multiparameter/multivariable indicators can measure more than one variable at a time, for example heat and time or gas concentration and time or time, steam and temperature.


Process indicators demonstrate that an indicator has gone through a process but they do not guarantee that sterilization was satisfactory. A common example is autoclave tape (single end-point indicator), which reflects the conditions inside the chamber environment but is not able to demonstrate that an item has been sterilized. Another example is Temptubes®, which are glass tubes containing chemical with a specific melting point indicated by a colour change. More specific indicators, such as the ‘Bowie Dick tests’, are used to monitor air removal from autoclaves. They must be used in the first cycle of the day as an equipment function test (McDonnell 2007). The standardized test pack is placed in the centre of porous load sterilizers and if the process is correct (i.e. air removal is appropriate), uniform colour change occurs across the test package (Fig. 17.6).
A common example of multivariable indicators is sterilization control tubes (e.g. Browne’s tubes), which produce a colour change when the appropriate temperature and exposure time have been achieved. Other chemical indicators are quantitative and indicate a combination of critical variables within a process. This is the case with dosimeters (e.g. Perspex®, which gradually change colour upon exposure to radiation sterilization. It should be noted that the performance of chemical indicators can be altered by storage conditions before and after use and by the method of use.
Biological indicators consist of a carrier or package containing a standardized preparation of defined microorganisms of a known resistance to a specific mode of sterilization (Berube et al 2001; see Fig. 17.5). The carriers used are usually made of filter paper, a glass slide, stainless steel or a plastic tube. Some new versions incorporate ampoules containing a growth medium. The carrier is covered to prevent deterioration or contamination while still allowing entry of the sterilizing agent (British Pharmacopoeia 2011b). Different organisms are used for different processes (Table 17.6) but biological indicators usually consist of bacterial spores (<106). After exposure to the sterilization process, the indicators are removed aseptically and incubated in suitable media to detect the presence of surviving microorganisms. If no growth occurs, the sterilization process is said to have had sufficient lethality (Berube et al 2001).
Table 17.6
| Sterilization process | Spores used as a biological indicator |
| Dry heat | Bacillus subtilis var. niger ATCC 9372. |
| NCIMB 8058 or CIP 77.18 | |
| Moist heat | Geobacillus stearothermophilius ATCC 7953. |
| NCTC 10007. NCIMB 8157 or CIP 52.81 | |
| Ethylene oxide | Bacillus subtilis var. niger ATCC 9372. |
| NCIMB 8058 or CIP 77.18 | |
| Radiation | Bacillus pumilus ATCC 27.142. NCTC 10327. |
| NCIMB 10692 or CIP 77.25 |
Testing filtration efficacy
Compared to other sterilization methods, the potential risk of failure is higher for filtration sterilization. This means that it may be advisable to add an extra prefiltration stage using a bacteria-retentive filter. Confidence in the filters used is of prime importance during filtration sterilization. Each batch of filters is tested to ensure that they meet the specifications for release of particulate materials, mechanical strength, chemical characteristics (e.g. oxidizable materials and leaching of materials) and filtration performance. The methods for testing filtration performance involve either a challenge test (which is destructive so cannot be carried out on every filter in a batch) or an integrity test (Denyer & Hodges 2004).
The microbial challenge test is used to demonstrate that a filter is capable of retaining microorganisms. This is normally carried out using a suspension of at least 107 cfu (colony-forming units; see Chapter 14) of Brevundimonas diminuta per cm2 of active filter surface. Brevundimonas diminuta is a small (0.2–0.9 µm) Gram-negative short rod that is a natural choice for this test due to its size and because it was originally isolated from contaminated filtered solutions (Levy 2001). After filtration of a bacterial suspension prepared in tryptone soya broth, the filtrate is collected and incubated at 32 °C.
Integrity tests are used to verify the integrity of an assembled sterilizing filter before use and confirmed integrity after use. The tests used must be appropriate to the filter type and the stage of testing and may include bubble point tests, pressure hold tests and diffusion rate tests. The bubble point test is the oldest and one of the most widely used non-destructive tests. It measures the pressure (bubble point pressure) needed to pass gas through the largest pore of a wetted filter. In practice, the pressure required to produce a steady stream of gas bubbles through a wetted filter is often used as the bubble point. The basis of the test relies on the holes through the filter resembling uniform capillaries passing from one side to another. If these capillaries become wet then they will retain liquid via surface tension and the force needed to expel the liquid using a gas is proportional to the diameter of the capillaries (pore diameter). The main limitations of this technique are that it is reliant on operator judgement and on the holes in the filter being perfect uniform capillaries (Denyer & Hodges 2004).
Diffusion rate tests are especially useful for large area filters. They measure the rate of flow of a gas as it diffuses through the water in a wetted filter. The pressure required to cause migration of the gas through the liquid in the pores can be compared to data specified by the filter’s manufacturer to establish if the filter has defects (Levy 2001).
Limitations of sterilization methods
Sterilization processes can involve some extreme conditions such as high temperatures, pressure, vacuum and pressure pulsing, or the use of toxic substances, that can damage the products and/or its packaging. The alteration of a pharmaceutical preparation might lead to a reduced therapeutic efficacy or patient acceptability, and damage to the container might lead to the post-sterilization contamination of the product. There needs to be a balance between acceptable sterility assurance and acceptable damage to the product and container. Knowledge of the preparations and packaging design and the choice and understanding of the sterilization technologies help in making the appropriate selection to achieve maximum kill while decreasing the risk of product and packaging deterioration.
Nevertheless, each sterilization technology is associated with its own limitations (Table 17.7). Limitations associated with established and recommended procedures are usually linked to the nature of the process (e.g. heat, irradiation), whereas newer technologies tend to suffer from a lack of reproducibility.
Table 17.7
| Sterilization processes | Limitations |
| Heat sterilization | |
| Steam | Heat; damage to preparation |
| Vapour; damage to the container (wetting of final product, risk of contamination post sterilization) | |
| Pressure: air ballasting: damage to the container | |
| Dry heat | Heat: damage to preparation |
| Potentially longer exposure time needed | |
| Gaseous sterilization | |
| Ethylene oxide | High toxicity: risk to the operator |
| Decontamination required post process | |
| Explosive: risk to the operator | |
| Slow processa | |
| Many factors to control | |
| Formaldehyde | High toxicity: risk to the operator |
| Damage to some materials (e.g. cellulose-made materials) | |
| Decontamination required post process | |
| Slow processa | |
| Many factors to control | |
| Radiation sterilization | |
| γ-radiation | Risk to the operator |
| Water radiolysis: damage to the product | |
| Discolouration of some glasses and plastics (including PVC), destructive process may continue after sterilization finished | |
| Liberation of gases (e.g. hydrogen chloride from PVC) | |
| Hardness and brittleness properties of metals may change | |
| Butyl and chlorinated rubber are degraded | |
| Changes in potency can occur | |
| High costs | |
| Particle radiation | β-radiation: risk to the operator |
| Water radiolysis: damage to the product | |
| Poor penetration of electrons exacerbated by density of product | |
| Significant product heating may take place at high doses | |
| High costs | |
| Chemosterilants | |
| Glutaraldehyde and ortho-phthaladehyde | Toxicity: risk to the operator |
| Activity: reports of microbial resistance | |
| Peracetic acid | Corrosiveness: damage to the product/device |
| Activity: reports of microbial resistance | |
| Filtration sterilization | |
| Not efficient for small particles (viruses, prions) | |
| Requires strict aseptic techniques | |
| Integrity of membrane filter | |
| Growth of microbial contaminants in depth filter | |
| Shedding of materials from depth filter |
Summary
The achievement of sterility is a complex process that requires proper documentation. Sterility in the microbiological sense cannot be guaranteed. Therefore the sterility of a product has to be assured by the application of an appropriate validation process. It is important that the sterilization methodology is compatible with the preparation or product, including its final container or packaging, and combines effectiveness and the absence of detrimental effects. Although not described in detail in this chapter, the choice of the container/packaging must allow the optimum sterilization to be applied and assure that sterility is maintained post process. Sterilization occurs at the end of manufacturing but it does not replace or permit a relaxation of the principles of good manufacturing practice. In particular, the microbiological quality of ingredients for pharmaceutical preparations and the removal of bioburden must be monitored. Monitoring the critical parameters of the sterilization process will ensure that the predetermined conditions (during validation) are met. The lack of validation, or failure to follow a validated process, carries the risk of a non-sterile product, deterioration and possible infection.
Where possible, terminal sterilization is the method of choice. Processes that are fully validated allow the parametric release of the preparation/product and hence their rapid commercialization, since sterility testing, and the delay it incurs, might not be necessary.
A clear understanding of the methodology, the product to be sterilized (including its packaging), the validation process and the overall documentation required is therefore necessary to carry out a successful sterilization.
References
1. Berube R, Oxborrow GS, Gaustad JW. Sterility testing: validation of sterilization processes and sporicide testing. In: Block SS, ed. Disinfection, Sterilization, and Preservation. 5th edn and Wilkins, Philadelphia: Lippincott Williams; 2001.
2. British Pharmacopoeia, (2013a). Appendix XVIII Methods of sterilization (methods of preparation of sterile products). Pharmaceutical Press, London.
3. British Pharmacopoeia, (2013b). Appendix XVI A. Sterility. Pharmaceutical Press, London.
4. British Society for Gastroenterology. BSG Guidelines for Decontamination of Equipment for Gastrointestinal Endoscopy. The Report of a Working Party of the British Society of Gastroenterology Endoscopy Committee 2008.
5. Denyer SP, Hodges NA. Sterilization: filtration sterilization. In: Fraise AP, Lambert PA, Maillard J-Y, eds. Principles and Practice of Disinfection, Preservation and Sterilization. 4th edn Oxford: Blackwell Science; 2004.
6. European Pharmacopoeia 2011 Methods of Preparation of Sterile Products. 5.1.1, 5th Council of Europe, Strasbourg.
7. International Standards Organization (ISO). Biological evaluation of medical devices – part 7: ethylene oxide sterilization residuals. ISO 10993–7 Geneva: ISO; 1996.
8. Joslyn LJ. Sterilization by heat. In: Block SS, ed. Disinfection, Sterilization, and Preservation. 5th edn Philadelphia: Lippincott Williams and Wilkins; 2001.
9. Lambert PA. Sterilization: radiation sterilization. In: Fraise AP, Lambert PA, Maillard J-Y, eds. Principles and Practice of Disinfection, Preservation and Sterilization. 4th edn Oxford: Blackwell Science; 2004.
10. Levy RV. Sterile filtration of liquids and gases. In: Block SS, ed. Disinfection, Sterilization, and Preservation. 5th edn Philadelphia: Lippincott Williams and Wilkins; 2001.
11. Lewis DL. A sterilization standard for endoscopes and other difficult to clean medical devices. Practice in Gastroenterology. 1999;23:28–56.
12. Maillard J-Y. Usage of antimicrobial biocides and products in the healthcare environment: efficacy, policies, management and perceived problems. Therapeutics and Clinical Risk Management. 2005;1:340–370.
13. Maillard J-Y. Emergence of bacterial resistance to microbicides and antibiotics. Microbiology Australia. 2010;Nov:159–165.
14. Richards RME. Principles and methods of sterilization. In: Winfield AJ, Richards RME, eds. Pharmaceutical Practice. London: Churchill Livingstone; 2004.
15. Sharp J. Sterile products: basic concepts and principles. In: Quality in the Manufacture of Medicines and Other Healthcare Products. London: Pharmaceutical Press; 2000.
16. United States Pharmacopeia (2012) USP 36, NF 31. United States Pharmacopeial Convention, Maryland.
Bibliography
1. Block SS, ed. Disinfection, Sterilization, and Preservation. 5th edn Philadelphia: Lippincott Williams and Wilkins; 2001.
2. Fraise AP, Lambert PA, Maillard J-Y, eds. Principles and Practice of Disinfection, Preservation and Sterilization. 4th edn Oxford: Blackwell Science; 2004.
3. McDonnell G, ed. Antisepsis, Disinfection and Sterilization: Types, Action and Resistance. Washington DC: ASM Press; 2007.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Sterilization in practice
Jean-Yves Maillard and Susannah E. Walsh
Chapter contents
Determination of sterilization protocols
Recommended pharmacopoeial sterilization processes
Steam (under pressure) sterilization
Integrated lethality in sterilization practice
Statistical considerations of sterility testing and sterility assurance level
Test for sterility of the product
Validation of a sterilization process
Key points
Sterile products
Sterilization is an essential part of the processing of pharmaceutical dosage forms that are required to be sterile. By definition, a sterile product is completely free of viable microorganisms. In addition to the pharmaceutical products that require to be sterile, a number of medical devices that come into contact with sterile parts of the body or are reused in patients also need to be free of microorganisms (Table 17.1). The diversity of the items to be sterilized in terms of properties (e.g. heat sensitive or not), bulk, content, and the number of items to be sterilized per load requires the use of distinct sterilization processes. The British Pharmacopoeia (2013a), as an example, describes the use of five main sterilization processes to accommodate the range of products to be sterilized: steam, dry heat, gaseous, ionizing radiation and filtration sterilization. The first four methods are usually used to process products in their final containers (terminal sterilization). Regardless of the sterilization methodology used, it is important that the process itself is fully validated. A number of guidelines and European/international standard documents for specific product-sterilization method combinations exist and are followed by manufacturers and end users. Failure to control and/or document adequately a sterilization process can lead to serious incidents. This chapter aims to provide a brief overview of the recommended sterilization processes, their control and validation.
Determination of sterilization protocols
There are various technologies available to achieve sterility of pharmaceutical preparations and medical devices (Table 17.2). Generally, sterilization of the product in its final container (terminal sterilization) is preferred. This infers that the container must not impinge on the optimum sterilization to be delivered and that the container and closure maintain the sterility of the product throughout its shelf-life. The selected sterilization process must be suitable for its purpose, i.e. the sterilization of a given product, device and preparation, which means that the product and its container have to be rendered sterile and must not be damaged by the process or post process.
The choice of an appropriate sterilization process depends on a number of factors (Table 17.3) related to the product to be sterilized, such as type and composition of product and also the quantity to be sterilized. Additionally, the composition and the packaging of the product are significant factors that rule out some sterilization processes. For example, a heat-labile preparation would not be sterilized by heat and a small oily injection would not be sterilized by steam sterilization (further examples are given in Table 17.1). For specific types of products such as dressings, although moist heat sterilization is generally the method of choice, only certain types of autoclave, such as vacuum and pressure-pulsing autoclaves are appropriate.
For any given preparation or product, it is difficult to predict the microbial bioburden prior to sterilization. It is assumed that the bioburden of pharmaceutical preparations will be minimal as the manufacturing process should adhere to good manufacturing practice (GMP) (Box 17.1). However, a sterilization process should be able to deal with a worst case scenario. This is usually exemplified by the use of biological indicators (see the ‘Process indicators’ section later in this chapter) such as bacterial spores, which are considered as the most resistant infectious agents (with the exception of prions, the agents responsible for spongiform encephalopathies). This is usually the situation for official sterilization methods. Pharmacopoeial recommendations as well as guideline documents are derived from data generated from the use of bacterial spores (biological indicators) for a given sterilization process.
When a fully validated sterilization process has been conducted, the release of a batch of product can be based on process data during sterilization rather than the results from sterility testing. Any change in the sterilization procedure (e.g. product load, type of containers) requires re-validation to take place.
Re-sterilization of products/devices can cause their degradation (e.g. repeated irradiation or autoclaving) or may even cause them to become toxic (e.g. with ethylene oxide; Richards 2004). Therefore any proposed re-sterilization must be carefully investigated.
Recommended pharmacopoeial sterilization processes
Five main sterilization processes which possess different characteristics are usually recommended by pharmacopoeias:
• steam (under pressure) sterilization (terminal)
• dry heat sterilization (terminal)
• ionizing radiation sterilization (terminal)
Although the use of other sterilization methodologies is not necessarily precluded, appropriate validation documentations for each product need to be provided. More information can be found in Chapters 15 and 16, or by consulting the relevant pharmacopoeia. At the time of writing, examples of these include the European Pharmacopoeia (2011), the United States Pharmacopeia (2012) and the British Pharmacopoeia (2013a) but it is always important to consult the most up-to-date texts and guidelines.
Steam (under pressure) sterilization
Steam sterilization is the most reliable, versatile and universally used form of sterilization and relies on the combination of steam, temperature and pressure. The typical cycle consists of a holding time of 15 minutes at a temperature of 121 °C under 15 psi (103 kPa) gauge pressure (Table 17.4). The aim is to deliver steam at the phase boundary (dry saturated steam; see Chapter 16, Fig. 16.2) to all areas of the load. This is achieved using steam and pressure (Table 17.5).
Table 17.4
Typical terminal sterilization cycles
cDesorption could take up to 15 days; maximum threshold of ethylene oxide residues and evaluation documented in ISO 10993–7 (1996).
dLow temperature steam formaldehyde; values can differ slightly depending upon the literature.
eLower temperature of 55–56 °C can be used depending upon the thermotolerance of the preparation.
Steam under pressure is commonly used unless prohibited by lack of load penetration or heat and/or moisture damage. Steam can only kill microorganisms if it makes direct contact, so it is very important to avoid air pockets in the sterilizer during a sterilization process. In addition, air can reduce the partial pressure of the steam so that the temperature reached on surfaces will be less than that expected with the pressure used. Hence, removal of air is an essential part of the process to ensure effective sterilization. To remove the air present when an autoclave is loaded, autoclaves are equipped with air removal/displacement systems (e.g. vacuum and displacement autoclaves, etc.). For porous loads, gravity displacement systems (downward-displacement autoclaves) are not adequate and vacuum and pressure-pulsing autoclaves are the method of choice (McDonnell 2007). Non-condensable gases must also be removed and monitored; these are atmospheric gases like nitrogen and oxygen that form part of the initial atmosphere of the sterilizer. Other factors that affect the efficacy of steam sterilization are water content and steam purity. The optimal sterilization is obtained with saturated steam (as discussed in Chapter 16). Supersaturated steam (i.e. wetter steam) is associated with condensation and poor penetration. Superheated steam (i.e. drier steam) behaves like dry heat and is less efficient. Steam purity is determined by the quality of the water, which can be affected by a number of contaminants (e.g. pyrogens, amines, toxic metals, iron, chlorides, etc.) that can render the sterile product unsafe (e.g. toxicity caused by pyrogenic reactions, metallic poisoning) or damaged (e.g. discoloration of packaging, corrosion caused by iron and chlorides).
Steam under pressure is generated in autoclaves which can vary greatly in size and shape from portable bench-top units to industrial production facilities (Fig. 17.1). A cross-section through an autoclave is shown in Figure 17.2.
Steam sterilization applications are informed/regulated by a number of European and international guidelines and standards providing information on sterilizer design and installation, quality of steam, requirement for pressure, development and validation and routine control, etc.
Dry heat sterilization
The most common dry heat sterilization method uses hot air ovens (Fig. 17.3). Other procedures, such as sterilizing tunnels utilizing high-temperature filtered laminar air flow or infrared irradiation to achieve rapid heat transfer, are also available. Hot air ovens are usually heated electrically and often have heaters under a perforated bottom plate to provide convection currents (gravity convection type). Mechanical convection hot air ovens are equipped with a fan to assist air circulation and increase heat transfer by convection (Joslyn 2001). Dry heat sterilization is less expensive than steam sterilization and is effective for the depyrogenation of containers/packaging (e.g. glassware). Overloading should be avoided, wrappings and other barriers minimized and the load positioned to allow optimal air circulation. Other problems include long heating up times (e.g. with large loads of instruments) and the charring or baking of organic matter onto items. Dry heat sterilization cycles are generally longer than for moist heat sterilization, typically 2 hours at 160 °C (see Table 17.4). The process is thermostatically controlled and monitored using thermocouples.
Dry heat sterilization cannot be used for a number of products such as rubber, plastics and other thermolabile items, or for aqueous solutions.
Integrated lethality in sterilization practice
All heat sterilization processes must include heating up and cooling down time periods. These prolonged time periods at a raised temperature may increase the degradation of the product. Integrated lethality attempts to examine the effects of heat on the inactivation process during these time periods.
For steam sterilization, the Fo concept (‘reference unit of lethality’) is used. This takes into account the heating up and cooling down stages of the cycle and is expressed as the equivalent time in minutes at a temperature of 121 °C delivered by the process to the product in its final container with reference to microorganisms possessing a Z value of 10. Its calculation is complex and further information can be found in the relevant pharmacopoeias. In practice, computer programs can be used to calculate the combined effect of whole processes, allowing a reduction in the total process time. It is important that the appropriate sterility assurance level is consistently achieved and the routine use of biological indicators is recommended, although following process validation, parametric release might be preferred.
Gaseous sterilization
The gaseous sterilization method recommended by pharmacopoeias mainly employs ethylene oxide. It is usually used on a commercial scale for the sterilization of catheters, infusion giving sets, syringes, prostheses and some plastic containers and thermolabile powders (if humidity is not a problem; Sharp 2000). The ethylene oxide sterilization cycle is complex since many factors need to be controlled over a long period of time (see Table 17.4
[/not-level-membership-for-basic-science-category]
