[level-membership-for-basic-science-category]
Pharmaceutical applications of microbiological techniques
Norman A. Hodges
Chapter contents
Measurement of antimicrobial activity
Factors to be controlled in the measurement of antimicrobial activity
Minimum inhibitory concentration determinations (MICs)
Preservative efficacy tests (PETs or challenge tests)
Key points
Introduction
The purpose of this chapter is to bring together those microbiological methods and procedures that are relevant to the design and production of medicines and medical devices. These are methods used (a) to determine the potency or activity of antimicrobial chemicals, e.g. antibiotics, preservatives and disinfectants, and (b) as part of the microbiological quality control of manufactured sterile and non-sterile products. The chapter describes the experimental procedures that are unique or particularly relevant to pharmacy, rather than those that are common to microbiology as a whole. In the latter category, for example, are procedures used to identify and enumerate microorganisms. These, together with staining and microscopical techniques, are described in Chapter 13.
Several of the methods and tests discussed here are the subject of monographs or appendices in pharmacopoeias or they are described in national and international standards or other recognized reference works. It is not the intention to reproduce these official testing procedures in detail but rather to explain the principles of the tests, to draw attention to difficult or important aspects, and to indicate the advantages, problems or shortcomings of the various methods.
Measurement of antimicrobial activity
In most of the methods used to assess the activity of antimicrobial chemicals, an inoculum of the test organism is added to a solution of the chemical under test, samples are removed over a period of time, the chemical is inactivated and the proportion of surviving cells determined. Alternatively, culture medium is present together with the chemical and the degree of inhibition of growth of the test organism is measured. In each case it is necessary to standardize and control such factors as the concentration of the test organism, its origin, i.e. the species and strain employed, together with the culture medium in which it was grown, the phase of growth from which the cells were taken, and the temperature and time of incubation of the cells after exposure to the chemical. Because such considerations are common to several of the procedures described here, e.g. antibiotic assays, preservative efficacy (challenge) tests and determinations of minimum inhibitory concentration (MIC), it is appropriate that they should be considered first, both to emphasize their importance and to avoid repetition.
Factors to be controlled in the measurement of antimicrobial activity
Origin of the test organism
Although two cultures may bear the same generic and specific name, i.e. they may both be called Escherichia coli, this does not mean that they are identical. Certainly, they would normally be similar in many respects, e.g. morphology (appearance), cultural requirements and biochemical characteristics, but they may exhibit slight variations in some of these properties; such variants are described as strains of E. coli. A variety of strains of a single species may normally be obtained from a culture collection, e.g. the National Collection of Industrial and Marine Bacteria or the National Collection of Type Cultures. Different strains may also occur in hospital pathology laboratories by isolation from swabs taken from infected patients or by isolation from contaminated food, cosmetic or pharmaceutical products, and from many other sources. Strains obtained in these ways are likely to exhibit variations in resistance to antimicrobial chemicals. Strains from human or animal infections are frequently more resistant to antimicrobial chemicals, particularly antibiotics, than those from other sources. Similarly, strains derived from contaminated medicines may be more resistant to preservative chemicals than those obtained from culture collections. Therefore, in order to achieve results that are reproducible by a variety of laboratories, it is necessary to specify the strain of the organism used for the determination.
It is becoming increasingly common, too, for official testing methods to limit the number of times the culture collection specimen may be re-grown in fresh medium (called the number of subcultures or passages) before it must be replaced. This is because the characteristics of the organism (including its resistance to antimicrobial chemicals) may progressively change as a result of mutation and natural selection through the many generations that might arise during months or years of laboratory cultivation.
Composition and pH of the culture medium
There are several methods of assessing antimicrobial activity which all have in common the measurement of inhibition of growth of a test organism when the antimicrobial chemical is added to the culture medium. In such cases the composition and pH of the medium may influence the result. The medium may contain substances that antagonize the action of the test compound, e.g. high concentrations of thymidine or para-aminobenzoic acid will interfere with sulfonamide activity.
The antimicrobial activities of several groups of chemical are influenced by the ease with which they cross the cell membrane and interfere with the metabolism of the cell. This, in turn, is influenced by the lipid solubility of the substance, because the membrane contains a high proportion of lipid and tends to permit the passage of lipid-soluble substances. Many antimicrobial chemicals are weak acids or weak bases, which are more lipid soluble in the unionized form. The pH of the environment therefore affects their degree of ionization, hence their lipid solubility and so, ultimately, their antimicrobial effect. Benzoic acid, for example, is a preservative used in several oral mixtures which has a much greater activity in liquids buffered to an acid pH value than those which are neutral or alkaline. Conversely, the aminoglycoside antibiotics, e.g. streptomycin, neomycin and gentamicin, which are weak bases, are more active at slightly alkaline pH values, although this is more a consequence of the transport systems by which the molecules enter the bacterial cell working better at alkaline pH than of enhanced lipid solubility. The presence of organic matter, e.g. blood, pus or serum, is likely to have a marked protective effect on the test organism and so antimicrobial chemicals may appear less active in the presence of such material. The activity of several antibiotics, notably tetracyclines and aminoglycosides, is reduced by the presence of high concentrations of di- or trivalent cations in the medium.
Exposure and incubation conditions
The temperature, duration and redox conditions of exposure to the antimicrobial chemical (or incubation of survivors after exposure) may all have a significant effect on its measured activity. Increasing the temperature of exposure of the test organism to the chemical increases the antimicrobial activity by a factor which is quantified by the temperature coefficient (Q10 value: the number of times increase in activity for a 10 °C rise in temperature). Phenols and alcohols, for example, may respectively exhibit Q10 values of 3–5 and > 10, and so a variation of 5 °C in the temperature of exposure (which is permitted by pharmacopoeial preservative efficacy tests) may lead to a markedly different rate of kill of the organism in question.
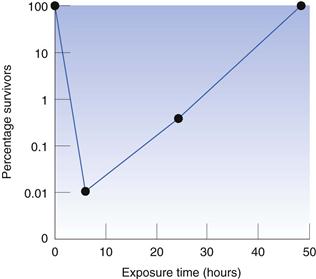
The period of time for which the test organism is exposed to the antimicrobial chemical may influence the recorded result because it is possible for the organism to adapt and become resistant to the presence of the chemical. In preservative efficacy tests, the exposure period is normally 28 days, which is sufficient time for any cells that are not killed during the first 24–48 hours to recover and start to reproduce, so that the final bacterial concentration may be much higher than that at the start. This is illustrated in Figure 14.1, which shows the effect of the quaternary ammonium preservative benzethonium chloride on Pseudomonas aeruginosa. The concentration of bacteria was reduced to approximately 0.01% of the initial value during the first 6 hours, but the bacteria that survived this early period recovered to the original level within 2 days. There is the potential for a similar phenomenon to arise in other situations, e.g. in minimum inhibitory concentration (MIC) determinations of bacteriostatic agents (those that do not kill but merely inhibit the growth of the test organism), although it is not common in MICs because the exposure (incubation) time is much shorter than that in preservative testing.
The effect of some antibiotics may be influenced by the redox conditions during their period of contact with the test organism. Aminoglycosides, for example, are far less active, and metronidazole is far more active, under conditions of low oxygen availability. Such effects may even be seen during agar diffusion antibiotic assays, in which the antibiotic diffuses from a well into an agar gel inoculated with the test organism; the diameter of the zone of growth inhibition that surrounds a well filled with neomycin solution, for example, may be significantly greater at the surface of the agar (where there is abundant oxygen) than at its base, where the oxygen concentration is limited by its poor diffusion through the gel.
Inoculum concentration and physiological state
It is perhaps not surprising that the concentration of the inoculum can markedly affect antimicrobial action, with high inoculum levels tending to result in reduced activity. There are two main reasons for this. First, there is the phenomenon of drug adsorption on to the cell surface or absorption into the interior of the cell. If the number of drug molecules in the test tube is fixed yet the number of cells present is increased, this obviously results in fewer molecules available per cell and consequently the possibility of a diminished effect. In addition to this there is the second, more specialized case, again concerning antibiotics, where it is frequently observed that certain species of bacteria can synthesize antibiotic-inactivating enzymes, the most common of which are the various types of β-lactamases (those destroying penicillin, cephalosporin and related antibiotics). Thus a high inoculum means a high carryover of enzyme with the inoculum cells, or at least a greater potential synthetic capacity.
Perhaps less predictable than the inoculum concentration effect is the possibility of the inoculum history influencing the result. There is a substantial amount of evidence to show that the manner in which the inoculum of the test organism has been grown and prepared can significantly influence its susceptibility to toxic chemicals. Features such as the nature of the culture medium, e.g. nutrient broth or a defined glucose-salts medium, the metal ion composition of the medium and hence of the cells themselves, and the physiological state of the cells, i.e. ‘young’ actively growing cells from the logarithmic growth phase or ‘old’ non-dividing cells from the stationary phase, all have the potential to influence the observed experimental values. Generally, antimicrobial chemicals are more effective against actively growing cells than slowly growing or dormant ones, e.g. bacterial spores.
Antibiotic assays
Methods of assaying antibiotics may be broadly divided into three groups:
Biological methods offer the advantage that the parameter being measured in the assay (growth inhibition) is the property for which the drug is used, and so inactive impurities or degradation products will not interfere and lead to an inaccurate result. Biological methods also offer other advantages (Table 14.1) but they have several significant limitations and non-biological methods are now generally preferred.
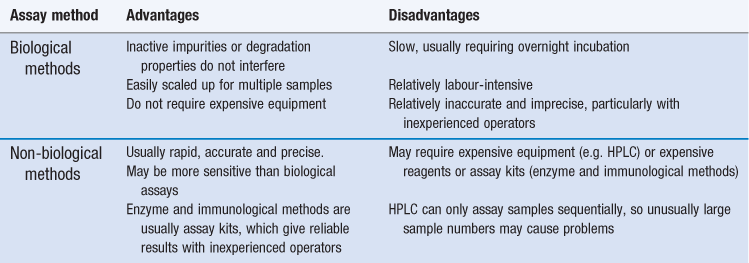
Enzyme-based and immunoassay kits are used in hospitals, notably for therapeutic monitoring of toxic antibiotics (e.g. aminoglycosides and vancomycin), whereas HPLC tends to be preferred in the pharmaceutical industry, particularly for quality assurance applications. Biological assays are most likely to be used when the alternatives are inappropriate, especially when the active antibiotic cannot readily be separated from inactive impurities, degradation products or interfering substances, or it cannot easily be assayed by HPLC without derivatization to enhance ultraviolet absorption (e.g. aminoglycosides). These situations may arise:
Biological antibiotic assays, or bioassays as they are frequently known, may be of two main types: agar diffusion and turbidimetric. The European Pharmacopoeia (PhEur) (2011) describes experimental details for both methods, e.g. test microorganisms, solvents, buffers, culture media and incubation conditions. In each case, a reference material of known activity must be available. When antibiotics were in their infancy, few could be produced in the pure state free from contaminating material, and specific chemical assays were rarely available. Thus the potency or activity of reference standards was expressed in terms of (international) units of activity. There are few antibiotics for which dosage is still normally expressed in units: nystatin and polymyxin are two of the remaining examples. More commonly, potencies are recorded in terms of µg mL−1 of solution or µg antibiotic mg−1 of salt, with dosages expressed in mg. Antibiotic assay results are usually in the form of a potency ratio of the activity of the unknown or test solution divided by that of the standard.
Agar diffusion assays
In this technique, the agar medium in a Petri dish or a larger assay plate is inoculated with the test organism, wells are created by removing circular plugs of agar, and these wells are filled with a solution of the chemical under test (Fig. 14.2).
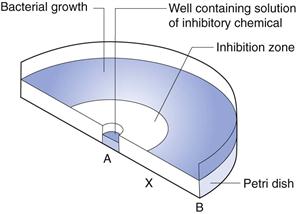
The chemical diffuses through the gel from A towards B and the concentration falls steadily in that direction. The concentration in the region A to X is sufficiently high to prevent growth, i.e. it is an inhibitory concentration. Between X and B the concentration is sub-inhibitory and growth occurs. The concentration at X at the time the zone edge is formed is known as the critical inhibitory concentration (CIC). After incubation, the gel between A and X is clear and that between X and B is opaque as a result of microbial growth which, with the common test organisms, is usually profuse. A zone of inhibition is therefore created, the diameter of which will increase as the concentration of chemical in the well increases.
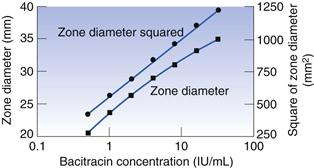
A graph may be constructed which relates zone diameter to the logarithm of the concentration of the solution in the well (Fig. 14.3). It is normally found to be linear over a small concentration range but the square of the diameter must be plotted to achieve linearity over a wide range. A plot such as that in Figure 14.3 may, quite correctly, be used to calculate the concentration of a test solution of antibiotic. In practice, however, it is found to be more convenient to obtain reliable mean zone diameters for the standard at just two or three concentrations, rather than somewhat less reliable values for six or seven concentrations. There is no reason why an assay should not be based upon a two- or three-point line, provided that those points are reliable and that preliminary experiments have shown that the plotted relationship over the concentration range in question is linear.
It is not common to conduct antibiotic assays in Petri dishes because too few zones may be accommodated on a standard-sized dish to permit the replication necessary to obtain the required accuracy and precision. Antibiotic assays, when performed on a large scale, are more often conducted using large assay plates 300 mm or more square (Fig. 14.4). The wells are created in a square design and the number that may be accommodated will depend upon the anticipated zone diameters: 36 or 64 wells are common (6 × 6 or 8 × 8, respectively). The antibiotic standard material may be used in solution at three known concentrations (frequently referred to as ‘doses’) and the antibiotic solution of unknown concentration treated likewise; alternatively, each may be employed at two concentrations. A randomization pattern known as a Latin square is used to ensure that there is a suitable distribution of the solutions over the plate, thereby minimizing any errors due to uneven agar thickness.

Fig. 14.4 Antibiotic agar diffusion assay conducted using a 6 x 6 assay design in a 300 mm square assay plate.
In the case of an assay based upon standard solutions used at two concentrations, the potency ratio may be calculated directly from the graph (as shown in Fig. 14.5) or by using the formula below:
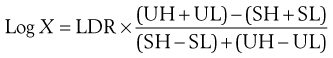 (14.1)
(14.1)
where X is the potency ratio, LDR is the logarithm of the dose ratio (i.e. ratio of concentrations of standard solutions), UH, UL, SH and SL are the mean zone diameters for the unknown and standard high and low doses. The derivation of this is described in detail by Wardlaw (1999), who deals extensively with the subject of antibiotic assays. The tests for acceptable limits of parallelism between the line joining the standards and that joining the test points, together with confidence limits applicable to the calculated potency ratios, are described in the current PhEur.
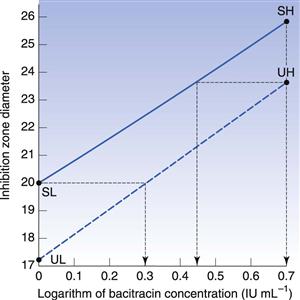
In calculating the potency ratio directly from Figure 14.5, the zone diameters for the standard and unknown high concentrations are plotted at the same abscissa values, and those for the low concentrations similarly. Two zone diameters are considered which are as widely separated on the ordinate as possible while still being covered by the standard and the test lines. The ratio of the concentrations required to achieve the selected diameter is thus an estimate of the potency ratio. The mean of the two estimates taken at the extremes of the range of common zone diameters should be identical to the value by calculation from the formula. Thus, in Figure 14.5, at a zone diameter of 23.75 mm the first estimate of potency ratio is 0.557 (antilog of 0.445 divided by antilog of 0.699); the second is 0.507 (antilog of zero divided by the antilog of 0.295). The mean value of 0.53 indicates the unknown solution to have approximately half the activity of the standard.
Practical aspects of the conduct of agar diffusion assays.
The agar may be surface inoculated or inoculated throughout while in the molten state prior to pouring. In the latter case zones may arise which are different in diameter at the agar surface than at the base of the Petri dish; this may complicate the recording of zone diameters. Zones which are not perfectly circular may be disregarded, although it may be appropriate to record the mean of the long and short axes. Such zones may result from non-circular wells, careless filling or uneven drying of the agar gel owing to a poorly fitting plate cover. The zones may be read directly with callipers or, more conveniently, after enlargement by projection onto a screen. Automatic zone readers incorporating a series of photocells that detect opacity changes at the zone edge are available, and may be linked to a personal computer which rapidly calculates the result together with the appropriate statistical analyses. The size of the zone is determined by the relative rates of diffusion of the drug molecule and growth of the test organism. If the assay plates are left at room temperature for 1–4 hours prior to incubation, growth is retarded whereas diffusion proceeds. This may result in larger zones and improved precision.
The zone diameter is affected by most of the factors previously stated to influence antimicrobial activity and, in addition, gel strength and the presence of other solutes in the antibiotic solution, e.g. buffer salts. If the antibiotic has been extracted from a formulated medicine, e.g. cream, lotion or mixture, excipients may be simultaneously removed and influence the diffusion of the antibiotic in the gel; sugars are known to have this effect. Because antibiotic assays involve a comparison of two solutions that are similarly affected by changes in experimental conditions, day-to-day variations in, for example, inoculum concentration will not have a great effect on the accuracy of the potency ratio obtained. However, the precision may be affected. The volume of liquid in the well is of minimal importance; it is usually of the order of 0.1 mL and is delivered by semi-automatic pipette. As an alternative to wells, the antibiotic may be introduced on to the agar using absorbent paper discs, metal cylinders or ‘fish spine’ beads (beads having a hole drilled in them which contains the liquid).
For many antibiotics, the test organism is a Bacillus species and the inoculum is in the form of a spore suspension, which is easy to prepare, standardize and store. Alternatively, frozen inocula from liquid nitrogen may be used as a means of improving reproducibility.
Careful storage and preparation of the reference standards are essential. The reference antibiotic is usually stored at low temperature in a freeze-dried condition.
Turbidimetric assays
In this case, antibiotic standards at several concentrations are incorporated into liquid media and the extent of growth inhibition of the test organism is measured turbidimetrically using a nephelometer or spectrophotometer. The unknown or test antibiotic preparation is run simultaneously, again at several concentrations, and the degree of growth inhibition compared. Such assays are less commonly used than agar diffusion methods because their precision is rather inferior but they do have the advantage of speed: the result may be available after an incubation period as short as 3–4 hours. They are also more sensitive than diffusion assays and consequently may be applied to low-activity preparations.
The shape and slope of the dose–response plot for a turbidimetric assay may be more variable than that for agar diffusion, and non-linear plots are common. Typical dose–response plots are shown in Hewitt & Vincent (1989). The plotted points are usually the mean turbidity values obtained from replicate tubes and the assay may be conducted using a Latin square arrangement of tubes incubated in a shaker, which is necessary to ensure adequate aeration and uniform growth throughout the tube.
Practical aspects of the conduct of turbidimetric assays.
Incubation time is critical in two respects. First, it is necessary to ensure that the culture in each of the many tubes in the incubator has exactly the same incubation period, because errors of a few minutes become significant in a total of only 3–4 hours’ incubation. Care must therefore be taken to ensure that the tubes are inoculated in a precise order, and that growth is stopped in the same order by the addition of formalin, heating or other means.
The incubation period must be appropriate to the inoculum level so that the cultures do not achieve maximal growth. At the concentrations used for such assays, the antibiotics usually reduce growth rate but do not limit total growth. Therefore, if the incubation period is sufficiently long, all the cultures may achieve the same cell density regardless of the antibiotic concentration.
There are certain other limitations to the use of turbidimetric assays. Because it is the ‘cloudiness’ of the culture that is measured, standard and test solutions in which the organisms are suspended should, ideally, be clear before inoculation. Cloudy or hazy solutions which may result from the extraction of the antibiotic from a cream, for example, can only be determined after similarly compensating the standards or otherwise eliminating the error. Test organisms that produce pigments during the course of the incubation period should be avoided; so too should those that normally clump in suspension.
The rate of growth of the test organism may vary significantly from one batch of medium to another. Thus it is important to ensure that all the tubes in the assay contain medium from the same batch, and were prepared and sterilized at the same time. Many liquid media become darker brown on prolonged heating, and so samples from the same batch may differ in colour if the sterilizing time is not strictly controlled.
Minimum inhibitory concentration determinations (MICs)
The MIC is the lowest concentration of an antimicrobial chemical found to inhibit the growth of a particular test organism. It is therefore a fundamental measure of the intrinsic antimicrobial activity (potency) of a chemical, which may be an antiseptic, disinfectant, preservative or antibiotic. MIC determinations are applied to chemicals in the pure state, i.e. they are particularly relevant to raw materials rather than to the final formulated medicines; the latter are usually subject to preservative efficacy (challenge) tests to assess their antimicrobial activity. MIC values are usually expressed in terms of µg mL−1 or, less commonly, as in the case of some antibiotics, units mL−1. It is important to recognize that the test organism is not necessarily killed at the MIC. Whether or not the cells die or merely cease growing depends upon the mode of action of the antimicrobial agent in question. MIC values are commonly used to indicate the sensitivity of a particular organism to an antibiotic, so for the antibiotic to be effective in treating an infection its concentration at the infection site must comfortably exceed the MIC for the organism in question.
An MIC is an absolute value which is not based upon a comparison with a standard/reference preparation, as in the case of antibiotic assays and certain disinfectant tests. For this reason, inadequate control of experimental conditions is particularly likely to have an adverse effect on results. Discrepancies in MIC values measured in different laboratories are often attributable to slight variations in such conditions, and care must be taken to standardize all the factors previously stated to influence the result. It is important also to state the experimental details concerning an MIC determination. A statement such as ‘the MIC for phenol against E. coli is 0.1% w/v’ is not, by itself, very useful. It has far more value if the strain of E. coli, the inoculum concentration, the culture medium, etc. are also stated.
MIC test methods
The most common way to conduct MIC determinations is to incorporate the antimicrobial chemical at a range of concentrations into a liquid medium, the containers of which are then inoculated, incubated and examined for growth.
Test tubes may be used, but microtitre plates (small rectangular plastic trays with, usually, 96 wells each holding approximately 0.1 mL liquid) and other miniaturized systems are common. It is also possible to incorporate the chemical into molten agar, which is then poured into Petri dishes and allowed to set. An advantage of using a microtitre plate or series of Petri dishes is that several organisms can be tested at the same time using a multipoint inoculator; there is also a greater chance of detecting contaminating organisms (as uncharacteristic colonies) on the agar surface than in liquid media. Usually the presence or absence of growth is easier to distinguish on the surface of agar than in liquid media. In tubes showing only faint turbidity, it is often difficult to decide whether growth has occurred or not. Regardless of the method used, the principle is the same and the MIC is the lowest concentration at which growth is inhibited.
In addition to the other experimental details that should be described in order to make the measured result meaningful, it is necessary to specify the increment by which the concentration of test chemical changes from one container to the next. The operator could, for example, change the concentration 10-fold from one tube to the next in the rare circumstance where even the likely order of magnitude of the MIC is not known. Far more commonly, however, the concentration changes by a factor of 2, and this is almost invariably the case when antibiotic MIC values are determined; thus, reference is made to ‘doubling dilutions’ of the antibiotic. If, for example, an MIC were to be measured using test tubes, an aqueous solution of the chemical would normally be mixed with an equal volume of double-strength growth medium in the first tube in the series, then half the contents of the first tube added to an equal volume of single-strength medium in the second, and so on. In this case half the contents of the last tube in the series would have to be discarded prior to inoculation in order to maintain the same volume in each tube. Control tubes may be included to demonstrate (a) that the inoculum culture was viable and that the medium was suitable for its growth (a tube containing medium and inoculum but no test chemical) and (b) that the operator was not contaminating the tubes with other organisms during preparation (a tube with no test chemical or added inoculum). It is possible to use an arithmetic series of concentrations of test chemical, e.g. 0.1, 0.2, 0.3, 0.4, … rather than 0.1, 0.2, 0.4, 0.8, … µg mL−1. The potential problem with this approach is that there may be merely a gradation in growth inhibition, rather than a sharp point of demarcation with obvious growth in one tube in the series and no growth in the next.
All the solutions used must be sterilized; it must not be assumed that the test chemical is self-sterilizing. Most disinfectants, antiseptic and preservative chemicals are bactericidal but they are unlikely to kill bacterial spores. Also, several antibiotics act by inhibiting growth and so would not necessarily kill vegetative cells with which they might be contaminated. If the experiment is conducted in tubes, all the tube contents must be mixed before inoculation as well as after, otherwise there is the possibility of the inoculum cells being killed by an artificially high concentration of the test chemical towards the top of the tube. If there is any risk of precipitation of the test chemical or the medium components during incubation, a turbidity comparison must be available for each concentration (same tube contents without inoculum); alternatively, in the case of bactericidal chemicals, the liquid in each tube may be subcultured into pure medium to see whether the inoculum has survived. Each of the tubes in the series may be prepared in duplicate or triplicate if it is considered desirable. This is the case where the incremental change in concentration is small.
Distinction between MICs conducted in agar and the assessment of sensitivity using agar diffusion methods
It is important to understand that when MICs are determined in Petri dishes the antimicrobial chemical is dissolved in the agar and is uniformly distributed through the gel when the test organism is inoculated into the surface. This is a fundamental difference from the test procedure used for antibiotic bioassays where the antibiotic diffuses through the agar to create a growth inhibition zone. When MICs are conducted in agar there is no diffusion and no zones of growth inhibition; the result merely depends on the presence or absence of growth of the test organism.
If the agar diffusion method were used to measure the size of the inhibition zones from a series of solutions of progressively decreasing concentration, it would obviously be possible to identify the concentration that just fails to produce an inhibition zone. This is sometimes incorrectly described as the MIC value for the chemical in question; such a procedure, however, gives the critical inhibitory concentration (CIC), not the MIC. CIC values usually exceed MIC values by a factor of 2–4. Not only is this misconception about agar diffusion methods giving MIC values commonly found in the pharmaceutical and chemical literature but misinterpretations of agar diffusion data are, unfortunately, also common. The diameter of a growth inhibition zone depends upon several factors. Whilst the sensitivity of the test organism, its concentration and that of the chemical are paramount, the incubation conditions, the physicochemical composition of the gelled culture medium and the properties of the diffusing molecule are also important. It is tempting to take the simplistic view that if two chemicals are used at the same concentration and one produces a larger zone of growth inhibition than the other, that is a direct reflection of their intrinsic antimicrobial activities. Unfortunately, that is often not the case because it fails to take into account both the diffusion coefficients of the different molecules and their concentration exponents (Chapter 15). To diffuse well in agar, a molecule should be small, water soluble and of a charge that does not interact with the components of the gel. There are several very effective antimicrobial chemicals that either do not diffuse well in agar or possess a high concentration exponent, both of which properties would predispose to small zones. If these agents were to be assessed purely on the basis of inhibition zone diameter they would be incorrectly dismissed as virtually inactive. Parabens and phenols are prime examples. Even saturated solutions of parabens in water can fail to give inhibition zones by agar diffusion (Fig. 14.6) but they are, nevertheless, amongst the most effective and widely used antimicrobial preservatives. Likewise phenols, with their high concentration exponents, only give small inhibition zones and this has led to misleading comparisons; manuka honey, for example, has been claimed to possess antibacterial activity equivalent to 10% phenol on the basis that the inhibition zone diameters are similar. This fundamental limitation of agar diffusion as a method of assessing antimicrobial potency is all too frequently overlooked.

Preservative efficacy tests (PETs or challenge tests)
These are tests applied to the formulated medicine in its final container to determine whether it is adequately protected against microbial spoilage; they are normally used only during product development and are not part of the routine quality control applied to batches of manufactured medicines. Preservative efficacy tests (rather than chemical assays of preservatives) are used to assess vulnerability to spoilage because it is not normally possible to predict how the activity of a preservative chemical will be influenced by the active ingredients, the excipients and the container itself.
Certain products may contain no added preservative, either because the active ingredients have sufficient antimicrobial activity themselves or because they already contain high concentrations of sugar or salts which restrict the growth of microorganisms. However, such products are rare; multidose injections or eye drops, the majority of oral mixtures, linctuses and similar preparations, together with creams and lotions, all contain preservatives. They are not normally required in anhydrous products, e.g. ointments, or in single-dose injections.
Again, it must not be assumed that products containing antimicrobial agents as the active ingredients are self-sterilizing, It is quite possible for an antibiotic cream, for example, to be active against certain bacteria yet fail to restrict the growth of contaminating yeasts or moulds.
The basic principle of a preservative test is to inoculate separate containers of the product with known concentrations of a variety of test organisms, then remove samples from each container over a period of time and determine the proportion of the inoculum that has survived. When first introduced into national pharmacopoeias, preservative efficacy tests differed to some extent in experimental detail and differed markedly in the required performance criteria for preservatives to be used in different product categories. In the late 1990s moves towards international harmonization of preservative testing procedures in the European, United States and Japanese pharmacopoeias (PhEur, USP and JP, respectively) meant that many (but not all) of the discrepancies in experimental detail were eliminated. The differences in performance criteria remain, however, with the PhEur generally requiring a greater degree of microbial inactivation for the preservative to be considered satisfactory than the USP and JP which, in this respect, are very similar.
The PhEur (2011) recommends the routine use of four test organisms, each at a final concentration of 105–106 cells mL−1 or g−1 in the product. Counts are performed on samples removed at Oh, 6h, 24h, 48h, 7 days, 14 days and 28 days. Various aspects of the test are considered in more detail below.
Choice of test organisms and inoculum concentration
The test organisms used are the bacteria Staphylococcus aureus, Pseudomonas aeruginosa and E. coli (which is used for testing all product types in the USP test but for oral products only in the PhEur test), together with the yeasts/moulds Candida albicans and Aspergillus brasiliensis (plus the osmophilic Zygosaccharomyces rouxii in the PhEur test for oral syrups). The current PhEur recommends that the designated organisms be supplemented, where appropriate, by other strains or species that may represent likely contaminants to the preparation. A similar recommendation was contained in earlier versions of the USP preservative test but this has been deleted from the current test (2010).
One problem with adding other organisms (such as those isolated from the manufacturing environment) is that they are not universally available and so a particular product could be tested at different manufacturing sites of the same company and pass in one location yet fail in another simply because the organisms used locally were not the same. The possibility of using resistant strains isolated from previous batches of spoilt product has been advocated, but this too may pose problems because organisms may rapidly lose their preservative resistance unless routinely grown on media supplemented with the preservative in question.
The inoculum concentration of 105–106 microorganisms mL−1 or g−1 of the preparation under test has been criticized as being unrealistic because it is much higher than that which would be acceptable in a freshly manufactured product. It is adopted, however, in order for the 1000-fold fall in microbial concentration that would be required from an effective parenteral or ophthalmic preservative to be easily measured. The test organisms are added separately to different containers rather than as a mixed inoculum.
Inactivation of preservative
It is quite possible for sufficient of the preservative to be contained in, and carried over with, the sample removed from the container to prevent or retard growth of colonies on the Petri dishes. If the inoculum level of the test organism initially is about 106 cells mL−1 or g−1 of product, the problem of carryover may not arise because a dilution factor of 103 or 104 would be required to achieve a countable number of colonies on a plate; at this dilution most preservatives would no longer be active. When a high proportion of the cells in the product has died, however, little or no such dilution is required, so preservative carryover is a real problem which may artificially depress the count even more. To avoid this, preservative inhibitors or antagonists may be used. There are several of these; common examples being glycine for aldehydes, thioglycollate or cysteine for heavy metals, and mixtures of lecithin and polysorbate-80 with or without Lubrol W for quaternary ammonium compounds, chlorhexidine and parabens. The use of these and other inactivators has been tabulated by Gilmore et al (2011).
An alternative method of removing residual preservative is to pass the sample of inoculated product through a bacteria-proof membrane so that surviving organisms are retained and washed on the surface of the membrane and the preservative is thus physically separated from them. After washing, the membrane is transferred to the surface of a suitable agar medium and colonies of microorganisms develop on it in the normal way. It is necessary to incorporate controls (validate the method) to demonstrate both that the inactivator really works and that it is not, itself, toxic. The former usually involves mixing the inactivator with the concentrations of preservative likely to be carried over, then inoculating and demonstrating no viability loss. Details of these validation procedures are described more fully in chapter <1227> of the USP (2010).
One further control is a viable count of the inoculum performed by dilution in peptone water to check the actual number of cells introduced into the product. This is necessary because even a ‘zero time sample’ of the product will contain cells that have been exposed to the preservative for a short period as it usually takes 15–45 seconds or more to mix the inoculum with the product and then remove the sample. Some of the cells may be killed even in such a short time and so a viable count of the inoculum culture will reflect this.
Interpretation of results
The extent of microbial killing required at the various sampling times for a preservative to be considered acceptable for use in parenteral or ophthalmic products is greater than that required for a preservative to be used in topical products, which in turn exceeds that for an oral product preservative (Table 14.2).
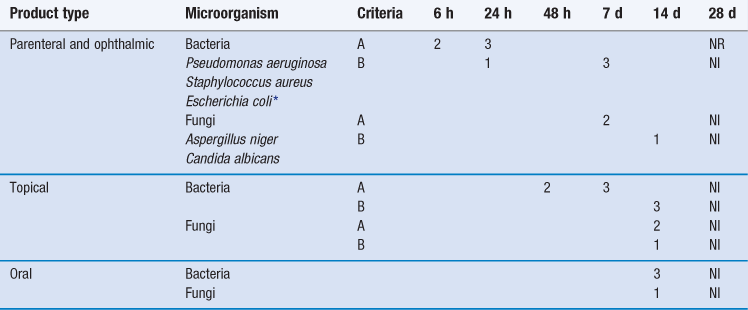
In the case of the first two product categories, the PhEur specifies two alternative performance criteria, designated A and B. The A criteria express the recommended efficacy to be achieved, whereas the B criteria must be satisfied in justified cases where the A criteria cannot be attained, for example because of an increased risk of adverse reactions. The baseline used as the reference point to assess the extent of killing is the concentration of microorganisms expected to arise in the product after addition and mixing of the inoculum, as calculated from a viable count performed on the concentrated inoculum suspension prior to its addition to the product. The viable count of the time-zero samples removed from the inoculated product is not the baseline.
Disinfectant evaluation
A variety of tests have been described over many years for the assessment of disinfectant activity. Those developed during the early part of the 20th century, e.g. the Rideal–Walker and Chick–Martin tests, were primarily intended for testing phenolic disinfectants against pathogenic organisms such as Salmonella typhi. Such phenol coefficient tests are now outmoded because S. typhi is no longer endemic in Britain and phenolics are no longer preeminent; indeed, they now represent a minor fraction of the total biocides used for floor disinfection in aseptic dispensing areas in British hospital pharmacies (Murtough et al 2000).
In the second half of the 20th century several other testing procedures were described for use in the UK which reduced the sampling or other problems associated with the early phenol coefficient tests; these included the Berry and Bean method, the British Standard 3286 test for quaternary ammonium compounds and the Kelsey–Sykes test. Other countries adopted procedures that were similar in concept but which differed in experimental detail; these and other tests used in the UK, Europe and the USA are described by Reybrouck (2004). At present there is no internationally applicable and officially recommended disinfectant testing procedure; although good uniformity exists in Europe as a result of the establishment by the European Committee for Standardization in 1990 of Technical Committee (TC) 216 that has a responsibility for chemical disinfectants and antiseptics. The European Standard BS EN 1276 (1997) was the first result of the work of TC 216; this deals with assessment of bactericidal activity of disinfectants on bacteria in aqueous suspension. Other procedures applicable to more specialized situations, e.g. disinfection of solid surfaces, are described in various European Standards and have been reviewed by Hanlon (2010).
A confusing variety of methods for describing and categorizing test procedures is in use. Thus, some schemes classify tests according to the organisms to be killed (bactericidal, fungicidal, virucidal, etc.) but classification based upon test design is more common, for example:
• carrier tests, where the organism is loaded or dried on to a carrier
• in-use tests, which are intended to simulate actual conditions of use as closely as possible.
Most suspension tests of disinfectants have in common the addition of a defined concentration of test organism to the disinfectant solution at a specified temperature, followed by assessment of viability in samples removed after suitable time periods. However, there are four aspects of disinfectant testing that merit special note.
Microbiological quality of pharmaceutical materials
Non-sterile products
Non-sterile pharmaceutical products obviously differ from sterile products in that they are permitted to contain some viable microorganisms, but the PhEur (2011) specifies the maximum concentrations acceptable in different types of product and the species of organism that are not permitted at all (Table 14.3). Similar specifications arise in the US and other pharmacopoeias.
Table 14.3
European Pharmacopoeia (2011) specifications for the microbiological quality of major categories of pharmaceutical products*

*Specifications also exist for transdermal patches, inhalations and certain oral products of animal, vegetable or mineral origin.
Note cfu is a colony-forming unit (defined in Chapter 13).
The required microbiological quality of the manufactured medicine cannot be achieved by the application of an antimicrobial process (heating, radiation, etc.) as the final production step for two reasons: first, an approach that uses poor-quality raw materials and manufacturing procedures and then attempts to ‘clean up’ the product at the end is not acceptable to the licensing authorities; and second, some products would not withstand such antimicrobial treatment, e.g. heating an emulsion may cause cracking or creaming. Thus, the most reliable approach to ensure that the manufactured medicine complies with the pharmacopoeial specification is to ensure that the raw materials are of good quality and that the manufacturing procedures conform to the standards laid down in the latest edition of Rules and Guidance for Pharmaceutical Manufacturers and Distributors.
Implicit in these standards is the principle that the extent of product contamination originating from the manufacturing environment and production personnel should be subject to regular monitoring and control.
Environmental monitoring
Environmental monitoring is normally taken to mean regular monitoring of the levels of microbial contamination of the atmosphere, of solid surfaces and, less frequently, of the personnel in the production areas. Water used to clean floors, benches and equipment (as distinct from water incorporated in the product) may be considered as part of environmental monitoring but will not be considered here as the procedures for counting microorganisms in water are described below.
Atmospheric monitoring is most commonly undertaken by means of settle plates, which are simply Petri dishes containing media suitable for the growth of bacteria and/or yeasts and moulds, e.g. tryptone soya agar, which are exposed to the atmosphere for periods of, typically, 1–4 hours. Microorganisms in the air may exist as single cells, e.g. mould spores, but more commonly they are attached to dust particles, so that any organisms in the latter category (for which the culture medium is suitable) will grow into visible colonies during incubation after dust particles have settled on the agar surface. The colony counts recorded on the plates are obviously influenced by:
The disadvantage of settle plates is that it is not possible to relate colony counts directly to air volume. This limitation is overcome in active sampling methods, whereby a known volume of air is drawn over, or caused to impact upon, the agar surface. These methods and the equipment available for active sampling have been reviewed by Johnson (2003).
Surface and equipment sampling is most frequently undertaken by swabbing or the use of contact plates (also known as RODAC – replicate organism detection and counting – plates). Swabbing a known area of bench, floor or equipment with a culture medium-soaked swab is convenient for irregular surfaces. The organisms on the swab may be counted after they have been dispersed by agitation into a fixed volume of suspending medium but it is not easy to quantify either the proportion of total organisms removed from the swabbed surface or the proportion dispersed in the diluent. This second limitation is overcome using contact plates, which are simply specially designed Petri dishes slightly overfilled with molten agar which, on setting, present a convex surface that projects above the rim of the plate. When the plate is inverted on to the surface to be sampled, microorganisms are transferred directly on to the agar (Fig. 14.7).
Sampling of manufacturing personnel usually consists of sampling clothing, face masks or, more commonly, gloves. ‘Finger dabs’ is the phrase used to describe the process whereby an operator rolls the gloved surface of each finger over a suitable solid medium in a manner similar to that in which fingerprints are taken. Operator sampling by any means other than finger dabs is rare, particularly outside aseptic manufacturing areas.
Counting of microorganisms in pharmaceutical products
Most pharmaceutical raw materials are contaminated with microorganisms. The levels of contamination are often a reflection of the source of the raw material in question, with ‘natural’ products derived from vegetable or animal sources, or mined minerals such as kaolin and talc, being more heavily contaminated than synthetic materials whose microbial burden has been reduced by heat, extremes of pH or organic solvents during the course of manufacture. Determining the bioburden in these materials is often straightforward, utilizing without modification the viable counting procedures described in Chapter 13. Occasionally the physical nature of the raw material makes this difficult or impossible, and this is often found to be the case with the finished manufactured medicine, where problems of dispersibility, sedimentation or viscosity cause complications. As a consequence, modifications to the standard viable counting procedures are necessary to reduce errors. Some of modifications and the circumstances that necessitate them are considered below.
Very low concentrations of microorganisms in aqueous solutions.
The reliability of calculated viable cell concentrations becomes much reduced when they are based upon colony counts much lower than about 10–15 per Petri dish. Using a surface-spread method, it is rarely possible to place more than about 0.5 mL of liquid on to the agar surface in a standard Petri dish because it will not easily soak in. By a pour-plate method, 1 mL or more may be used but a point is reached where the volume of sample significantly dilutes the agar and nutrients. Thus, using a conventional plating technique, the lowest concentration conveniently detectable is of the order of 10–50 cells mL−1. When the cell concentration is below this value it is necessary to pass a known quantity of the liquid – 10–100 mL or more – through a filter membrane having a pore size sufficiently small to retain bacteria. The membrane is then placed with the organisms uppermost on to the agar surface in a Petri dish, which is incubated without inversion. As a result of diffusion of nutrients through the membrane, colonies grow on the surface in the normal way (Fig. 14.8). Diffusion may be assisted by the inclusion of a medium-soaked pad between the membrane and the agar. It is important to ensure that all the membrane is in contact with the pad or agar, otherwise elevated areas may become dry and no colonies will appear upon them.
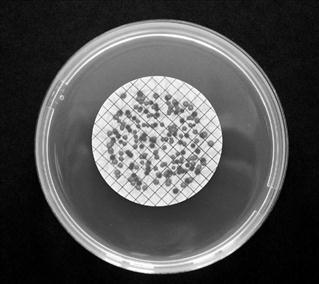
Insoluble solids.
It is necessary to suspend an insoluble solid in a medium that will permit uniform dispersion and adequate wetting of the suspended material. Nutrient broth, peptone water or a buffered salt solution is frequently used and a low concentration of a surfactant may be incorporated to promote wetting, e.g. polysorbate 80 (0.01–0.05%). Suspension in distilled water alone carries the risk of osmotic damage to sensitive cells, with a consequently low count; for this reason it is best avoided. Having obtained the suspension, there are two options available depending upon the nature and concentration of the suspended material.
The first is to remove a sample of the continuously mixed suspension, dilute if necessary, and plate in or on a suitable medium using a pour- or spread-plate method. If the concentration of suspended material is low it may still be possible to see clearly the developing colonies. High concentrations may obscure the colonies and make counting impossible. The alternative is to dislodge the microbial cells from the solid to which they are attached, allow the solid to sediment out and then sample the supernatant. Methods of removal include vigorous manual shaking, use of a vortex mixer or equipment designed for the purpose, e.g. the Colworth ‘stomacher’ in which the aqueous suspension is placed in a sealed sterile bag which is repeatedly agitated by reciprocating paddles. The use of ultrasonics to dislodge the cells carries the risk of damage to or lysis of the cells themselves.
Assuming the suspended material has no antimicrobial activity, plating the ‘whole suspension’ is probably the easiest and most reliable method. The alternative strategy of sampling the supernatant involves the assumption that all the cells have been removed from the solid but this would have to be confirmed by control (validation) experiments in which a known quantity of similar organisms was artificially dried on to sterile samples of the material. The second method also relies upon the solid sedimenting sufficiently rapidly for it to be separated from the bacteria in aqueous suspension above. If all or part of the sample has a particle size similar to that of bacteria, yeasts or mould spores, i.e. approximately 1–5 µm, then a separation cannot easily be achieved.
Oils and hydrophobic ointments.
These materials are usually not heavily contaminated because they are anhydrous and microorganisms will not multiply without water. Thus the microorganisms contained in oily products have usually arisen by contamination from the atmosphere, equipment used for manufacture and from storage vessels. To perform a viable count the oil sample must be emulsified or solubilized without the aid of excessive heat or any other agent that might kill the cells.
An oil-in-water emulsion must be produced using a suitable surfactant; non-ionic emulsifiers generally have little antimicrobial activity. The proportion of surfactant to use must be determined experimentally and validation experiments conducted to confirm that the surfactant is not, itself, toxic to the species that typically arise as contaminants of the sample in question; Millar (2000) has described the use of up to 5 g of polysorbate 80 added to a 10 g sample. Such an emulsion may be diluted in water or buffered salts solution if necessary, and aliquots placed on or in the agar medium in the usual way. Alternatively, the oil may be dissolved in a sterile, non-toxic solvent and passed through a membrane filter. Isopropyl myristate, for example, is recommended in pharmacopoeial sterility testing procedures as a solvent for anhydrous materials but it may kill a significant fraction of the cells of some sensitive species, even during an exposure period of only a few minutes.
Creams and lotions.
Oil-in-water emulsions do not usually represent a problem because they are miscible with water and thus are easily diluted. Water-in-oil creams, however, are not miscible and cannot be plated directly because bacteria may remain trapped in a water droplet suspended in a layer of oil on the agar surface. Such bacteria may not form colonies because the diffusion of nutrients through the oil would be inadequate. These creams are best diluted, dispersed in an aqueous medium and membrane filtered or converted to an oil-in-water type, and then counted by normal plating methods.
Dilution and emulsification of the cream in broth containing Lubrol W, polysorbate 80 or Triton X 100 is probably the best procedure, although the addition of approximately 0.1 g of the w/o emulsion sample to 25 g of isopropyl myristate followed by membrane filtration may be satisfactory.
Detection of specific hazardous organisms
In addition to placing limits on the maximum concentration of microorganisms that is acceptable in different materials, pharmacopoeias usually specify certain organisms that must not be present at all. In practice, this means that detection methods which are described in the pharmacopoeia must be applied to a known weight of material (typically 1–10 g), and the sample passes the test if, on the culture plates, no organisms arise that conform to the standard textbook descriptions of those to be excluded. Typically, the pharmacopoeial methods involve preliminary stages using selective liquid culture media; these are designed to increase the concentration of the organism that is the subject of the test (‘target’ organism) and so render it more readily detectable. Commercially available identification kits or specific supplementary biochemical tests may also be used to confirm the identity of any isolates having the typical appearance of the target organisms. The PhEur used to recommend appropriate supplementary tests but these have been removed from the current edition, not because of a lack of reliability but because identification kits have become more common.
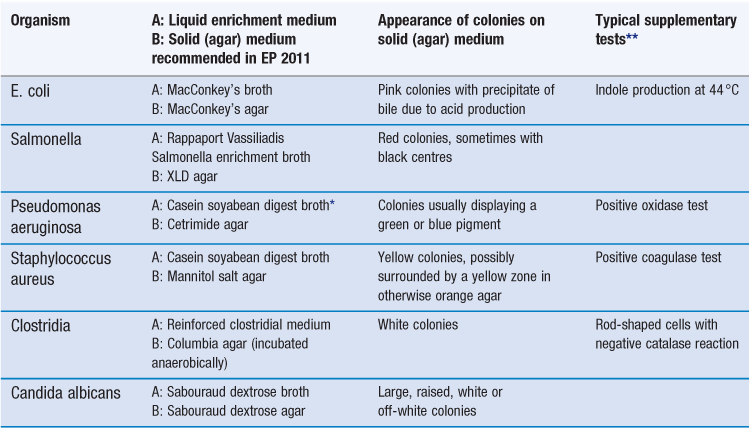
Both the PhEur (2011) and the USP (2010) describe detection tests for Staphylococcus aureus, Pseudomonas aeruginosa, Escherichia coli, salmonellae and Candida albicans. In addition, the PhEur describes a test for clostridia, but this is unlikely to be applied to any material other than mined minerals, e.g. talc and bentonite. The five organisms common to both pharmacopoeias are the subject of these tests primarily because of their potential to cause infections. However, they may also represent common contaminants of the products to which the tests are applied, or their presence may be indicative of the quality of the raw material or finished manufactured product. E. coli, for example, is a natural inhabitant of mammalian intestines and so its presence in a material such as gelatin (which originates in the slaughterhouse) would indicate unacceptable quality. The most likely source of Staphylococcus aureus in a manufactured medicine is the production personnel, so that if this origin were confirmed it would indicate the need for higher manufacturing standards. In general, the tests are applied to pharmaceutical raw materials of ‘natural’ origin, e.g. carbohydrates, cellulose derivatives, gums and vegetable drugs. In addition, there is a requirement that products for use in the mouth, nose, ears or on the skin should be free of both Pseudomonas aeruginosa and Staphylococcus aureus and vaginal products should also be free from Candida albicans. Table 14.4 summarizes the PhEur (2011) testing schemes for the five principal organisms of interest. These schemes are described in more detail elsewhere, together with photographs of the typical appearance of the organisms in question (Hodges 2000).
Microbiological assays of B-group vitamins
Just as HPLC has become the favoured method of antibiotic assay, so too has it become the method of choice for assaying B-group vitamins. Turbidimetric assays are still occasionally used, however, for example when insurmountable problems arise in resolving the many peaks that might arise on an HPLC chromatogram from a multivitamin product (which may contain 10 or more active ingredients plus excipients, all of which may cause assay interference).
Microbiological assays of B-group vitamins employ similar techniques to turbidimetric assays of antibiotics (see earlier in this chapter). A culture medium is used which is suitable for the assay organism, except for the omission of the vitamin in question. The extent of bacterial growth in the medium is thus directly proportional to the amount of reference standard or test vitamin added. It is important to select an assay organism that has an absolute requirement for the substance in question and is unable to obtain it by metabolism of other medium components; species of Lactobacillus are often used for this purpose. ‘Carryover’ of the vitamin with the inoculum culture must be avoided because this results in some growth even when none of the test material has been added. Growth may be determined turbidimetrically or by acid production from sugars.
Sterile products
Sterile products must, by definition, be free of viable microorganisms and it is important to understand that this is an absolute requirement. Thus, the presence of one single surviving microbial cell is sufficient to render the product non-sterile. There is not a level of survivors which is so small as to be regarded as negligible and therefore acceptable.
The principal component of microbiological quality assurance which has traditionally been applied to sterile products is, of course, the test for sterility itself. In essence, this is quite simple: a sample of the material to be tested is added to culture medium which is incubated and then examined for signs of microbial growth. If growth occurs, the assumption is made that the contamination arose from the sample, which consequently fails the test. However, the limitations of this simplistic approach became more widely recognized in the second half of the 20th century, and there was an increasing awareness of the fact that contaminated products could pass the test and sterile ones apparently fail it (because of contamination introduced during the testing procedure itself). For these reasons the sterility test alone could no longer be relied upon to provide an assurance of sterility, and that assurance is now derived from a strict adherence to high quality-standards throughout the manufacturing process. These encompass:
• the adoption of the highest possible specifications for the microbiological quality of the raw materials. The rationale here is that sterilization processes are more likely to be effective when the levels of microorganisms to be killed or removed (bioburdens) are as low as possible to begin with. Procedures used to determine bioburdens are described in Chapter 13 and earlier in this chapter
The pharmacopoeias and regulatory authorities require a sterility assurance level for terminally sterilized products of 10−6 or better. This means that the probability of non-sterility in an item selected at random from a batch should be no more than 1 in 1 million. This sterility assurance level (SAL) may be demonstrated in the case of some terminally sterilized products simply by reference to data derived from bioburdens, environmental monitoring and in-process monitoring of the sterilization procedure itself. In this case the sterility test may be unnecessary and omitted; the term ‘parametric release’ is used to describe the release of products for sale or use under these circumstances, although it should be emphasized that manufacturers must seek approval for parametric release from regulatory authorities; the decision is not made by the manufacturers themselves.
Sterilization monitoring
Sterilization processes may be monitored physically, chemically or biologically (Denyer et al 2011). Physical methods are exemplified by thermocouples, which are routinely incorporated at different locations within an autoclave load, whereas chemical indicators usually exhibit a colour change after exposure to a heat sterilization process. Biological indicators consist of preparations of spores of the Bacillus species that exhibits the greatest degree of resistance to the sterilizing agent in question. The principle of their use is simply that if such spores are exposed to the sterilization process and fail to survive, it can be assumed that all other common organisms will also have been killed and the process is safe. Spores of Bacillus stearothermophilus (strictly speaking now Geobacillus stearothermophilus, although this name is still not in common use in the pharmaceutical literature) are used to monitor autoclaves and gaseous hydrogen peroxide or peracetic acid sterilization processes, whereas Bacillus atrophaeus is the organism normally employed for dry heat, ethylene oxide and low-temperature steam-formaldehyde methods; Bacillus pumilus is used in radiation sterilization procedures.
Such biological indicators are regularly employed for validation of a sterilization process which is under development for a new product, or when a new autoclave is being commissioned; they are not normally used for routine monitoring during product manufacture. Spores possess the advantage that they are relatively easy to produce, purify and dry on to an inert carrier, which is frequently an absorbent paper strip or disc, or a plastic or metal support. Spore resistance to the sterilizing agent must be carefully controlled and so rigorous standardization of production processes followed by observance of correct storage conditions and expiry dates are essential.
Tests for sterility
It is sufficient here to repeat that the test is really one for demonstrating the absence of gross contamination with readily grown microorganisms, and is not capable of affording a guarantee of sterility in any sample that passes.
The experimental details of these procedures are described in the PhEur (2011). This section is therefore restricted to an account of the major features of the test and a more detailed consideration of those practical aspects that are important or problematical.
It is obviously important that materials to be tested for sterility are not subject to contamination from the operator or the environment during the course of the test. For this reason it is essential that sterility tests are conducted in adequate laboratory facilities by competent and experienced personnel. Clearly, the consequences of recording an incorrect sterility result may be very severe. If a material which was really sterile were to fail the test it would need to be resterilized or, more probably, discarded. This would have significant cost implications. If, on the other hand, a contaminated batch were to pass a test for sterility and be released for use this would obviously represent a significant health hazard. For these reasons sterility testing procedures have improved significantly in recent years and failures are now viewed very seriously by the regulatory authorities. If a product does fail, it means either that the item in question is really contaminated, in which case the manufacturing procedures are seriously inadequate, or that the item is in fact sterile but the testing procedure is at fault. Either way, it is not possible to dismiss a failure lightly.
Sterility tests may be conducted in clean rooms or laminar flow cabinets which provide a grade A atmosphere as defined by the Rules and Guidance for Pharmaceutical Manufacturers and Distributors. However, it is becoming increasingly common for testing to be undertaken in an isolator that physically separates the operator from the test materials and so reduces the incidence of false-positive test results due to extraneous contamination introduced during the test itself. Such isolators are similar in principle to a glove box, and typically consist of a cabinet (supported on legs or a frame) that is sufficiently large for the operator, who is covered by a transparent hood of moulded flexible plastic forming the cabinet base, to sit or stand within it.
A sterility test may be conducted in two ways. The direct inoculation method involves the removal of samples from the product under test and their transfer to a range of culture media that might be expected to support the growth of contaminating organisms. After incubation the media are examined for evidence of growth which, if present, is taken to indicate that the product may not be sterile. It is not certain that the product is contaminated because the organisms responsible for the growth may have arisen from the operator or have been already present in the media to which the samples were transferred, i.e. the media used for the test were not themselves sterile. Thus, in conducting a sterility test it is necessary to include controls that indicate the likelihood of the contaminants arising from these sources; these are discussed below. The size and number of the samples to be taken are described in the PhEur (2011).
Again it is necessary to inactivate any antimicrobial substances contained in the sample. These may be the active drug, e.g. an antibiotic, or a preservative in an eye drop or multidose injection. Suitable inactivators may be added to the liquid test media to neutralize any antimicrobial substances, but in the case of antibiotics particularly, no such specific inactivators are available (with the exception of β-lactamases which hydrolyze penicillins and cephalosporins). This problem may be overcome using a membrane filtration technique. This alternative method of conducting sterility tests is obviously only applicable to aqueous or oily solutions that will pass through a membrane having a pore size sufficiently small to retain bacteria. The membrane, and hence the bacteria retained on it, is washed with isotonic salts solution, which should remove any last traces of antimicrobial substances. It is then placed in a suitable liquid culture medium. This method is certainly to be preferred to direct inoculation because there is a greater chance of effective neutralization of antimicrobial substances.
Solids may be dissolved in an appropriate solvent. This is almost invariably water because most other common solvents have antimicrobial activity. If no suitable solvent can be found the broth dilution method is the only one available. If there is no specific inactivator available for antimicrobial substances that may be present in the solid then their dilution to an ineffective concentration by use of a large volume of medium is the only course remaining.
The controls associated with a sterility test are particularly important because incomplete control of the test may lead to erroneous results. Failure to neutralize a preservative completely may lead to contaminants in the batch going undetected and subsequently initiating an infection when the product is introduced into the body.
The PhEur (2011) recommends that four controls are incorporated. The so-called growth promotion test simply involves the addition of low inocula (not more than 100 cells or spores per container) of suitable test organisms into the media used in the test to show that they do support the growth of the common contaminants for which they are intended. Staphylococcus aureus, Bacillus subtilis and Pseudomonas aeruginosa are the three aerobic bacteria used, Clostridium sporogenes the anaerobic bacterium and Candida albicans and Aspergillus brasiliensis the fungi. Organisms having particular nutritional requirements, such as blood, milk or serum, are not included, so they, in addition to the more obvious omissions such as viruses, cannot be detected in a routine sterility test because suitable cultural conditions are not provided. On the other hand, it is impossible to design an all-purpose medium, and sterilization processes that kill the spore-forming bacteria and other common contaminants are likely also to eradicate the more fastidious pathogens such as streptococci and Haemophilus species, which would be more readily detected on blood-containing media. This argument does not, however, cover the possibility of such pathogens entering the product, perhaps via defective seals or packaging, after the sterilization process itself and then going undetected in the sterility test.
The second control, termed the method suitability test, is intended to demonstrate that any preservative or antimicrobial substance has been effectively neutralized. This requires the addition of test organisms to containers of the various media as before but, in addition, samples of the material under test must also be added to give the same concentrations as those arising in the test itself. For the sterility test as a whole to be valid, growth must occur in each of the containers in these controls.
It is necessary also to incubate several tubes of the various media just as they are received by the operator. If the tubes are not opened but show signs of growth after incubation this is a clear indication that the medium is itself contaminated. This should be an extremely rare occurrence but, in view of the small additional cost or effort, the inclusion of such a control is worthwhile.
A control to check the likelihood of contamination being introduced during the test should be included in the programme of regular monitoring of test facilities. The PhEur 2011 recommends the use of ‘negative controls’, which may be employed to check the adequacy of facilities and operator technique. These items, identical to the sample to be tested, are manipulated in exactly the same way as the test samples. If, after incubation, there are signs of microbial growth in the media containing these negative controls, the conclusion is drawn that the contamination arose during the testing process itself.
Some items present particular difficulties in sterility testing because of their shape or size, e.g. surgical dressings and medical devices. These problems are most conveniently overcome simply by testing the whole sample rather than attempting to withdraw a portion of it. So, for example, large clear plastic bags which have been radiation sterilized may be used to hold the entire medical device or complete roll or pack of dressings, which would then be totally immersed in culture medium. This method would only be valid if the culture medium gained access to the entire sample; otherwise the possibility exists, for example, of aerobic bacterial spores trapped within it failing to grow owing to insufficient diffusion of oxygen. This approach has the advantage of imposing a more rigorous test because a much larger sample is used. In the case of dressings, it may also reduce the risk of operator-induced contamination compared to the alternative approach, which would require the withdrawal of representative samples for testing from different areas of the roll or pack.
The final aspect of the test which is worthy of comment is the interpretation of results. If there is evidence that any of the test samples are contaminated, the batch fails the test. If, however, there is convincing evidence that the test was invalid because the testing facility, procedure or media were inadequate, a single retest is permitted; this contrasts with earlier pharmacopoeial protocols, which under certain circumstances permitted two retests.
Endotoxin and pyrogen testing
This is an aspect of microbial contamination of medicines which is not normally considered part of microbiology but is discussed here because pyrogens are normally the products of microbial growth. A pyrogen is a material which when injected into a patient will cause a rise in body temperature (pyrexia). The lipopolysaccharides that comprise a major part of the cell wall of Gram-negative bacteria are called endotoxins, and it is these that are the most commonly encountered pyrogens (although any other substance that causes a rise in body temperature may be classified under the same heading). Bacterial cells may be pyrogenic even when they are dead and when they are fragmented, and so a solution or material that passes a test for sterility will not necessarily pass a pyrogen test. It follows from this that the more heavily contaminated with bacteria an aqueous injection becomes during manufacture, the more pyrogenic it is likely to be at the end of the process.
Two main procedures are used for the detection of pyrogens. The traditional method requires the administration of the sample to laboratory rabbits, whose body temperature is monitored for a period of time thereafter. The alternative procedure, which is now by far the most common, is to use the Limulus Amoebocyte Lysate Test (LAL), in which the pyrogen-containing sample causes gel formation in the lysis product of amoebocyte cells of the giant horseshoe crab Limulus polyphemus. A detailed account of endotoxin testing is outside the scope of this chapter but the review by Baines (2000) provides a comprehensive account of the practicalities of the method.
References
1. Baines A. Endotoxin testing. In: Baird RM, Hodges NA, Denyer SP, eds. Handbook of Microbiological Quality Assurance. London: Taylor and Francis; 2000.
2. BS EN 1276. Quantitative suspension test for the evaluation of bactericidal activity of chemical disinfectants and antiseptics used in food, industrial, domestic and institutional areas. London: British Standards Institute; 1997.
3. Denyer SP, Hodges NA, Talbot C. Sterilization procedures and sterility assurance. In: Denyer SP, Hodges N, Gorman SP, Gilmore BF, eds. Hugo and Russell’s Pharmaceutical Microbiology. 8th edn Oxford: Wiley-Blackwell; 2011.
2011. European Pharmacopoeia. 7th edn Strasbourg: Council of Europe; 2011.
5. Gilmore BF, Ceri H, Gorman SP. Laboratory evaluation of antimicrobial agents. In: Denyer SP, Hodges N, Gorman SP, Gilmore BF, eds. Hugo and Russell’s Pharmaceutical Microbiology. 8th edn Oxford: Wiley-Blackwell; 2011.
6. Hanlon G. Disinfectant testing and the measurement of biocide effectiveness. In: Hodges NA, Hanlon GW, eds. Industrial Pharmaceutical Microbiology: Standards and Controls. Haslemere, UK: Euromed Communications; 2010.
7. Hewitt W, Vincent S. Theory and Application of Microbiological Assay. London: Academic Press; 1989.
8. Hodges NA. Pharmacopoeial methods for the detection of specified microorganisms. In: Baird RM, Hodges NA, Denyer SP, eds. Handbook of Microbiological Quality Assurance. London: Taylor and Francis; 2000.
9. Johnson SM. Microbiological environmental monitoring. In: Hodges NA, Hanlon GW, eds. Industrial Pharmaceutical Microbiology: Standards and Controls. Haslemere, UK: Euromed Communications; 2003.
10. Millar R. Enumeration. In: Baird RM, Hodges NA, Denyer SP, eds. Handbook of Microbiological Quality Assurance. London: Taylor and Francis; 2000.
11. Murtough SM, Hiom SJ, Palmer M, Russell AD. A survey of disinfectant use in hospital pharmacy aseptic preparation areas. Pharmaceutical Journal. 2000;264:446–448.
12. Reybrouck G. Evaluation of the antibacterial and antifungal activity of disinfectants. In: Fraise AP, Lambert PA, Maillard JY, eds. Principles and Practice of Disinfection Preservation and Sterilization. 4th edn Oxford: Blackwell Science; 2004.
2007. Rules and Guidance for Pharmaceutical Manufacturers and Distributors. London: Pharmaceutical Press; 2007.
2010. United States Pharmacopoeia. 33rd edn Rockville, Maryland: US Pharmacopoeial Convention Inc.; 2010.
15. Wardlaw AC. Practical Statistics for Experimental Biologists. 2nd edn Chichester: Wiley; 1999.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Pharmaceutical applications of microbiological techniques
Norman A. Hodges
Chapter contents
Measurement of antimicrobial activity
Factors to be controlled in the measurement of antimicrobial activity
Minimum inhibitory concentration determinations (MICs)
Preservative efficacy tests (PETs or challenge tests)
Key points
Introduction
The purpose of this chapter is to bring together those microbiological methods and procedures that are relevant to the design and production of medicines and medical devices. These are methods used (a) to determine the potency or activity of antimicrobial chemicals, e.g. antibiotics, preservatives and disinfectants, and (b) as part of the microbiological quality control of manufactured sterile and non-sterile products. The chapter describes the experimental procedures that are unique or particularly relevant to pharmacy, rather than those that are common to microbiology as a whole. In the latter category, for example, are procedures used to identify and enumerate microorganisms. These, together with staining and microscopical techniques, are described in Chapter 13.
Several of the methods and tests discussed here are the subject of monographs or appendices in pharmacopoeias or they are described in national and international standards or other recognized reference works. It is not the intention to reproduce these official testing procedures in detail but rather to explain the principles of the tests, to draw attention to difficult or important aspects, and to indicate the advantages, problems or shortcomings of the various methods.
Measurement of antimicrobial activity
In most of the methods used to assess the activity of antimicrobial chemicals, an inoculum of the test organism is added to a solution of the chemical under test, samples are removed over a period of time, the chemical is inactivated and the proportion of surviving cells determined. Alternatively, culture medium is present together with the chemical and the degree of inhibition of growth of the test organism is measured. In each case it is necessary to standardize and control such factors as the concentration of the test organism, its origin, i.e. the species and strain employed, together with the culture medium in which it was grown, the phase of growth from which the cells were taken, and the temperature and time of incubation of the cells after exposure to the chemical. Because such considerations are common to several of the procedures described here, e.g. antibiotic assays, preservative efficacy (challenge) tests and determinations of minimum inhibitory concentration (MIC), it is appropriate that they should be considered first, both to emphasize their importance and to avoid repetition.
Factors to be controlled in the measurement of antimicrobial activity
Origin of the test organism
Although two cultures may bear the same generic and specific name, i.e. they may both be called Escherichia coli, this does not mean that they are identical. Certainly, they would normally be similar in many respects, e.g. morphology (appearance), cultural requirements and biochemical characteristics, but they may exhibit slight variations in some of these properties; such variants are described as strains of E. coli. A variety of strains of a single species may normally be obtained from a culture collection, e.g. the National Collection of Industrial and Marine Bacteria or the National Collection of Type Cultures. Different strains may also occur in hospital pathology laboratories by isolation from swabs taken from infected patients or by isolation from contaminated food, cosmetic or pharmaceutical products, and from many other sources. Strains obtained in these ways are likely to exhibit variations in resistance to antimicrobial chemicals. Strains from human or animal infections are frequently more resistant to antimicrobial chemicals, particularly antibiotics, than those from other sources. Similarly, strains derived from contaminated medicines may be more resistant to preservative chemicals than those obtained from culture collections. Therefore, in order to achieve results that are reproducible by a variety of laboratories, it is necessary to specify the strain of the organism used for the determination.
It is becoming increasingly common, too, for official testing methods to limit the number of times the culture collection specimen may be re-grown in fresh medium (called the number of subcultures or passages) before it must be replaced. This is because the characteristics of the organism (including its resistance to antimicrobial chemicals) may progressively change as a result of mutation and natural selection through the many generations that might arise during months or years of laboratory cultivation.
Composition and pH of the culture medium
There are several methods of assessing antimicrobial activity which all have in common the measurement of inhibition of growth of a test organism when the antimicrobial chemical is added to the culture medium. In such cases the composition and pH of the medium may influence the result. The medium may contain substances that antagonize the action of the test compound, e.g. high concentrations of thymidine or para-aminobenzoic acid will interfere with sulfonamide activity.
The antimicrobial activities of several groups of chemical are influenced by the ease with which they cross the cell membrane and interfere with the metabolism of the cell. This, in turn, is influenced by the lipid solubility of the substance, because the membrane contains a high proportion of lipid and tends to permit the passage of lipid-soluble substances. Many antimicrobial chemicals are weak acids or weak bases, which are more lipid soluble in the unionized form. The pH of the environment therefore affects their degree of ionization, hence their lipid solubility and so, ultimately, their antimicrobial effect. Benzoic acid, for example, is a preservative used in several oral mixtures which has a much greater activity in liquids buffered to an acid pH value than those which are neutral or alkaline. Conversely, the aminoglycoside antibiotics, e.g. streptomycin, neomycin and gentamicin, which are weak bases, are more active at slightly alkaline pH values, although this is more a consequence of the transport systems by which the molecules enter the bacterial cell working better at alkaline pH than of enhanced lipid solubility. The presence of organic matter, e.g. blood, pus or serum, is likely to have a marked protective effect on the test organism and so antimicrobial chemicals may appear less active in the presence of such material. The activity of several antibiotics, notably tetracyclines and aminoglycosides, is reduced by the presence of high concentrations of di- or trivalent cations in the medium.
Exposure and incubation conditions
The temperature, duration and redox conditions of exposure to the antimicrobial chemical (or incubation of survivors after exposure) may all have a significant effect on its measured activity. Increasing the temperature of exposure of the test organism to the chemical increases the antimicrobial activity by a factor which is quantified by the temperature coefficient (Q10 value: the number of times increase in activity for a 10 °C rise in temperature). Phenols and alcohols, for example, may respectively exhibit Q10 values of 3–5 and > 10, and so a variation of 5 °C in the temperature of exposure (which is permitted by pharmacopoeial preservative efficacy tests) may lead to a markedly different rate of kill of the organism in question.
The period of time for which the test organism is exposed to the antimicrobial chemical may influence the recorded result because it is possible for the organism to adapt and become resistant to the presence of the chemical. In preservative efficacy tests, the exposure period is normally 28 days, which is sufficient time for any cells that are not killed during the first 24–48 hours to recover and start to reproduce, so that the final bacterial concentration may be much higher than that at the start. This is illustrated in Figure 14.1, which shows the effect of the quaternary ammonium preservative benzethonium chloride on Pseudomonas aeruginosa. The concentration of bacteria was reduced to approximately 0.01% of the initial value during the first 6 hours, but the bacteria that survived this early period recovered to the original level within 2 days. There is the potential for a similar phenomenon to arise in other situations, e.g. in minimum inhibitory concentration (MIC) determinations of bacteriostatic agents (those that do not kill but merely inhibit the growth of the test organism), although it is not common in MICs because the exposure (incubation) time is much shorter than that in preservative testing.
The effect of some antibiotics may be influenced by the redox conditions during their period of contact with the test organism. Aminoglycosides, for example, are far less active, and metronidazole is far more active, under conditions of low oxygen availability. Such effects may even be seen during agar diffusion antibiotic assays, in which the antibiotic diffuses from a well into an agar gel inoculated with the test organism; the diameter of the zone of growth inhibition that surrounds a well filled with neomycin solution, for example, may be significantly greater at the surface of the agar (where there is abundant oxygen) than at its base, where the oxygen concentration is limited by its poor diffusion through the gel.
Inoculum concentration and physiological state
It is perhaps not surprising that the concentration of the inoculum can markedly affect antimicrobial action, with high inoculum levels tending to result in reduced activity. There are two main reasons for this. First, there is the phenomenon of drug adsorption on to the cell surface or absorption into the interior of the cell. If the number of drug molecules in the test tube is fixed yet the number of cells present is increased, this obviously results in fewer molecules available per cell and consequently the possibility of a diminished effect. In addition to this there is the second, more specialized case, again concerning antibiotics, where it is frequently observed that certain species of bacteria can synthesize antibiotic-inactivating enzymes, the most common of which are the various types of β-lactamases (those destroying penicillin, cephalosporin and related antibiotics). Thus a high inoculum means a high carryover of enzyme with the inoculum cells, or at least a greater potential synthetic capacity.
Perhaps less predictable than the inoculum concentration effect is the possibility of the inoculum history influencing the result. There is a substantial amount of evidence to show that the manner in which the inoculum of the test organism has been grown and prepared can significantly influence its susceptibility to toxic chemicals. Features such as the nature of the culture medium, e.g. nutrient broth or a defined glucose-salts medium, the metal ion composition of the medium and hence of the cells themselves, and the physiological state of the cells, i.e. ‘young’ actively growing cells from the logarithmic growth phase or ‘old’ non-dividing cells from the stationary phase, all have the potential to influence the observed experimental values. Generally, antimicrobial chemicals are more effective against actively growing cells than slowly growing or dormant ones, e.g. bacterial spores.
Antibiotic assays
Methods of assaying antibiotics may be broadly divided into three groups:
Biological methods offer the advantage that the parameter being measured in the assay (growth inhibition) is the property for which the drug is used, and so inactive impurities or degradation products will not interfere and lead to an inaccurate result. Biological methods also offer other advantages (Table 14.1) but they have several significant limitations and non-biological methods are now generally preferred.
Enzyme-based and immunoassay kits are used in hospitals, notably for therapeutic monitoring of toxic antibiotics (e.g. aminoglycosides and vancomycin), whereas HPLC tends to be preferred in the pharmaceutical industry, particularly for quality assurance applications. Biological assays are most likely to be used when the alternatives are inappropriate, especially when the active antibiotic cannot readily be separated from inactive impurities, degradation products or interfering substances, or it cannot easily be assayed by HPLC without derivatization to enhance ultraviolet absorption (e.g. aminoglycosides). These situations may arise:
Biological antibiotic assays, or bioassays as they are frequently known, may be of two main types: agar diffusion and turbidimetric. The European Pharmacopoeia (PhEur) (2011) describes experimental details for both methods, e.g. test microorganisms, solvents, buffers, culture media and incubation conditions. In each case, a reference material of known activity must be available. When antibiotics were in their infancy, few could be produced in the pure state free from contaminating material, and specific chemical assays were rarely available. Thus the potency or activity of reference standards was expressed in terms of (international) units of activity. There are few antibiotics for which dosage is still normally expressed in units: nystatin and polymyxin are two of the remaining examples. More commonly, potencies are recorded in terms of µg mL−1 of solution or µg antibiotic mg−1 of salt, with dosages expressed in mg. Antibiotic assay results are usually in the form of a potency ratio of the activity of the unknown or test solution divided by that of the standard.
Agar diffusion assays
In this technique, the agar medium in a Petri dish or a larger assay plate is inoculated with the test organism, wells are created by removing circular plugs of agar, and these wells are filled with a solution of the chemical under test (Fig. 14.2).
The chemical diffuses through the gel from A towards B and the concentration falls steadily in that direction. The concentration in the region A to X is sufficiently high to prevent growth, i.e. it is an inhibitory concentration. Between X and B the concentration is sub-inhibitory and growth occurs. The concentration at X at the time the zone edge is formed is known as the critical inhibitory concentration (CIC). After incubation, the gel between A and X is clear and that between X and B is opaque as a result of microbial growth which, with the common test organisms, is usually profuse. A zone of inhibition is therefore created, the diameter of which will increase as the concentration of chemical in the well increases.
A graph may be constructed which relates zone diameter to the logarithm of the concentration of the solution in the well (Fig. 14.3). It is normally found to be linear over a small concentration range but the square of the diameter must be plotted to achieve linearity over a wide range. A plot such as that in Figure 14.3 may, quite correctly, be used to calculate the concentration of a test solution of antibiotic. In practice, however, it is found to be more convenient to obtain reliable mean zone diameters for the standard at just two or three concentrations, rather than somewhat less reliable values for six or seven concentrations. There is no reason why an assay should not be based upon a two- or three-point line, provided that those points are reliable and that preliminary experiments have shown that the plotted relationship over the concentration range in question is linear.
It is not common to conduct antibiotic assays in Petri dishes because too few zones may be accommodated on a standard-sized dish to permit the replication necessary to obtain the required accuracy and precision. Antibiotic assays, when performed on a large scale, are more often conducted using large assay plates 300 mm or more square (Fig. 14.4). The wells are created in a square design and the number that may be accommodated will depend upon the anticipated zone diameters: 36 or 64 wells are common (6 × 6 or 8 × 8, respectively). The antibiotic standard material may be used in solution at three known concentrations (frequently referred to as ‘doses’) and the antibiotic solution of unknown concentration treated likewise; alternatively, each may be employed at two concentrations. A randomization pattern known as a Latin square is used to ensure that there is a suitable distribution of the solutions over the plate, thereby minimizing any errors due to uneven agar thickness.
Fig. 14.4 Antibiotic agar diffusion assay conducted using a 6 x 6 assay design in a 300 mm square assay plate.
In the case of an assay based upon standard solutions used at two concentrations, the potency ratio may be calculated directly from the graph (as shown in Fig. 14.5) or by using the formula below:
(14.1)
where X is the potency ratio, LDR is the logarithm of the dose ratio (i.e. ratio of concentrations of standard solutions), UH, UL, SH and SL are the mean zone diameters for the unknown and standard high and low doses. The derivation of this is described in detail by Wardlaw (1999), who deals extensively with the subject of antibiotic assays. The tests for acceptable limits of parallelism between the line joining the standards and that joining the test points, together with confidence limits applicable to the calculated potency ratios, are described in the current PhEur.
In calculating the potency ratio directly from Figure 14.5, the zone diameters for the standard and unknown high concentrations are plotted at the same abscissa values, and those for the low concentrations similarly. Two zone diameters are considered which are as widely separated on the ordinate as possible while still being covered by the standard and the test lines. The ratio of the concentrations required to achieve the selected diameter is thus an estimate of the potency ratio. The mean of the two estimates taken at the extremes of the range of common zone diameters should be identical to the value by calculation from the formula. Thus, in Figure 14.5, at a zone diameter of 23.75 mm the first estimate of potency ratio is 0.557 (antilog of 0.445 divided by antilog of 0.699); the second is 0.507 (antilog of zero divided by the antilog of 0.295). The mean value of 0.53 indicates the unknown solution to have approximately half the activity of the standard.
Practical aspects of the conduct of agar diffusion assays.
The agar may be surface inoculated or inoculated throughout while in the molten state prior to pouring. In the latter case zones may arise which are different in diameter at the agar surface than at the base of the Petri dish; this may complicate the recording of zone diameters. Zones which are not perfectly circular may be disregarded, although it may be appropriate to record the mean of the long and short axes. Such zones may result from non-circular wells, careless filling or uneven drying of the agar gel owing to a poorly fitting plate cover. The zones may be read directly with callipers or, more conveniently, after enlargement by projection onto a screen. Automatic zone readers incorporating a series of photocells that detect opacity changes at the zone edge are available, and may be linked to a personal computer which rapidly calculates the result together with the appropriate statistical analyses. The size of the zone is determined by the relative rates of diffusion of the drug molecule and growth of the test organism. If the assay plates are left at room temperature for 1–4 hours prior to incubation, growth is retarded whereas diffusion proceeds. This may result in larger zones and improved precision.
The zone diameter is affected by most of the factors previously stated to influence antimicrobial activity and, in addition, gel strength and the presence of other solutes in the antibiotic solution, e.g. buffer salts. If the antibiotic has been extracted from a formulated medicine, e.g. cream, lotion or mixture, excipients may be simultaneously removed and influence the diffusion of the antibiotic in the gel; sugars are known to have this effect. Because antibiotic assays involve a comparison of two solutions that are similarly affected by changes in experimental conditions, day-to-day variations in, for example, inoculum concentration will not have a great effect on the accuracy of the potency ratio obtained. However, the precision may be affected. The volume of liquid in the well is of minimal importance; it is usually of the order of 0.1 mL and is delivered by semi-automatic pipette. As an alternative to wells, the antibiotic may be introduced on to the agar using absorbent paper discs, metal cylinders or ‘fish spine’ beads (beads having a hole drilled in them which contains the liquid).
For many antibiotics, the test organism is a Bacillus species and the inoculum is in the form of a spore suspension, which is easy to prepare, standardize and store. Alternatively, frozen inocula from liquid nitrogen may be used as a means of improving reproducibility.
Careful storage and preparation of the reference standards are essential. The reference antibiotic is usually stored at low temperature in a freeze-dried condition.
Turbidimetric assays
In this case, antibiotic standards at several concentrations are incorporated into liquid media and the extent of growth inhibition of the test organism is measured turbidimetrically using a nephelometer or spectrophotometer. The unknown or test antibiotic preparation is run simultaneously, again at several concentrations, and the degree of growth inhibition compared. Such assays are less commonly used than agar diffusion methods because their precision is rather inferior but they do have the advantage of speed: the result may be available after an incubation period as short as 3–4 hours. They are also more sensitive than diffusion assays and consequently may be applied to low-activity preparations.
The shape and slope of the dose–response plot for a turbidimetric assay may be more variable than that for agar diffusion, and non-linear plots are common. Typical dose–response plots are shown in Hewitt & Vincent (1989). The plotted points are usually the mean turbidity values obtained from replicate tubes and the assay may be conducted using a Latin square arrangement of tubes incubated in a shaker, which is necessary to ensure adequate aeration and uniform growth throughout the tube.
Practical aspects of the conduct of turbidimetric assays.
Incubation time is critical in two respects. First, it is necessary to ensure that the culture in each of the many tubes in the incubator has exactly the same incubation period, because errors of a few minutes become significant in a total of only 3–4 hours’ incubation. Care must therefore be taken to ensure that the tubes are inoculated in a precise order, and that growth is stopped in the same order by the addition of formalin, heating or other means.
The incubation period must be appropriate to the inoculum level so that the cultures do not achieve maximal growth. At the concentrations used for such assays, the antibiotics usually reduce growth rate but do not limit total growth. Therefore, if the incubation period is sufficiently long, all the cultures may achieve the same cell density regardless of the antibiotic concentration.
There are certain other limitations to the use of turbidimetric assays. Because it is the ‘cloudiness’ of the culture that is measured, standard and test solutions in which the organisms are suspended should, ideally, be clear before inoculation. Cloudy or hazy solutions which may result from the extraction of the antibiotic from a cream, for example, can only be determined after similarly compensating the standards or otherwise eliminating the error. Test organisms that produce pigments during the course of the incubation period should be avoided; so too should those that normally clump in suspension.
The rate of growth of the test organism may vary significantly from one batch of medium to another. Thus it is important to ensure that all the tubes in the assay contain medium from the same batch, and were prepared and sterilized at the same time. Many liquid media become darker brown on prolonged heating, and so samples from the same batch may differ in colour if the sterilizing time is not strictly controlled.
Minimum inhibitory concentration determinations (MICs)
[/not-level-membership-for-basic-science-category]
