Chapter 5 Stupor and Coma
Definitions
Consciousness may be defined as a state of awareness of self and surroundings. Alterations in consciousness are conceptualized into two types. The first type affects arousal and is the subject of this chapter. The second type involves cognitive and affective mental function, sometimes referred to as the “content” of mental function. Examples of the latter type of alteration in consciousness are dementia (see Chapter 6), delusions, confusion, and inattention (see Chapter 9). These altered states of consciousness, with the exception of advanced dementia, do not affect the level of arousal. Sleep, the only normal form of altered consciousness, is discussed in Chapter 68.
Conditions That May Mimic Coma
Several different states of impaired cognition or consciousness may appear similar to coma or be confused with it (Table 5.1). Moreover, patients who survive the initial coma may progress to certain of these syndromes after varying lengths of time. Once sleep/wake cycles become established, true coma is no longer present. Differentiation of these states from true coma is important to allow administration of appropriate therapy and help determine prognosis.
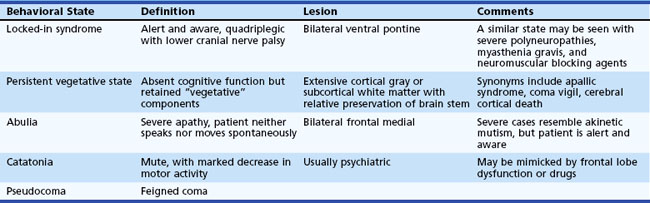
In the persistent vegetative state (PVS), patients have lost cognitive neurological function but retain vegetative or noncognitive neurological function such as cardiac action, respiration, and maintenance of blood pressure. This state follows coma and is characterized by absence of cognitive function or awareness of the environment despite a preserved sleep/wake cycle. Spontaneous movements may occur, and the eyes may open in response to external stimuli, but the patient does not speak or obey commands. Diagnostic criteria for PVS are provided in Box 5.1. Diagnosis of this condition should be made cautiously and only after extended periods of observation. A number of poorly defined syndromes have been used synonymously with PVS, including apallic syndrome or state, akinetic mutism, coma vigil, alpha coma, neocortical death, and permanent unconsciousness. These terms, used variously by different authors, probably are best avoided because of their lack of precision.
Box 5.1
Criteria for Diagnosis of Persistent Vegetative State
1. No evidence of awareness of themselves or their environment; they are incapable of interacting with others
2. No evidence of sustained, reproducible, purposeful, or voluntary behavioral responses to visual, auditory, tactile, or noxious stimuli
3. No evidence of language comprehension or expression
4. Intermittent wakefulness manifested by the presence of sleep/wake cycles
5. Sufficiently preserved hypothalamic and brainstem autonomic functions to survive if given medical and nursing care
6. Bowel and bladder incontinence
7. Variably preserved cranial nerve (pupillary, oculocephalic, corneal, vestibulo-ocular, and gag) and spinal reflexes
Data from The Multi-Society Task Force on PVS, 1994. Medical aspects of the persistent vegetative state. N Engl J Med 330, 1499-1508, 1572-1579.
A condition that has been estimated to be 10 times more common than PVS is the minimally conscious state, in which severe disability accompanies minimal awareness. A set of diagnostic criteria for the minimally conscious state has been proposed (Box 5.2). Abulia is a severe apathy in which patients have blunting of feeling, drive, mentation, and behavior such that they neither speak nor move spontaneously.
Box 5.2
Criteria for the Minimally Conscious State
2. Gestural or verbal yes/no responses (regardless of accuracy)
4. Purposeful behavior, including movements or affective behaviors that occur in contingent relationship to relevant environmental stimuli and are not due to reflexive activity
Data from Giacino, J.T., Ashwal, S., Childs, N., et al., 2002. The minimally conscious state: definition and diagnostic criteria. Neurology 58, 349-353.
Approach to the Patient in Coma
The initial clinical approach to the patient in a state of stupor or coma is based on the principle that all alterations in arousal constitute acute life-threatening emergencies until vital functions such as blood pressure and oxygenation are stabilized, potentially reversible causes of coma are treated, and the underlying cause of the alteration in arousal is understood. Urgent steps may be necessary to avoid or minimize permanent brain damage from reversible causes. In view of the urgency of this situation, every physician should develop a diagnostic and therapeutic routine to use with a patient with an alteration in consciousness. A basic understanding of the mechanisms that lead to impairment in arousal is necessary to develop this routine. The anatomical and physiological bases for alterations in arousal are discussed in Chapter 68.
Although it is essential to keep in mind the concept of a spectrum of arousal, for the sake of simplicity and brevity only the term coma is used in the rest of this chapter. Table 5.2 lists many of the common causes of coma. More than half of all cases of coma are due to diffuse and metabolic brain dysfunction. In Plum and Posner’s landmark study (1980, see 2007 revision) of 500 patients initially diagnosed as having coma of unknown cause (in whom the diagnosis was ultimately established), 326 patients had diffuse and metabolic brain dysfunction. Almost half of these had drug poisonings. Of the remaining patients, 101 had supratentorial mass lesions, including 77 hemorrhagic lesions and 9 infarctions; 65 had subtentorial lesions, mainly brainstem infarctions; and 8 had psychiatric coma.
| I. SYMMETRICAL-NONSTRUCTURAL |
| Toxins |
* Relatively common asymmetrical presentation.
Data from Plum, F., Posner, J.B., 1980. The Diagnosis of Stupor and Coma, third ed. F.A. Davis, Philadelphia; and from Fisher, C.M., 1969. The neurological examination of the comatose patient. Acta Neurol Scand 45, 1-56.
Rapid Initial Examination and Emergency Therapy
The risk of herniation from an LP in patients with evidence of increased intracerebral pressure is difficult to ascertain from the literature; estimates range from 1% to 12%, depending on the series (Posner et al., 2007). It is important to recognize that both central and tonsillar herniation may increase neck tone.
History
The neurologist often is called when patients do not awaken after surgery or when coma supervenes following a surgical procedure. Postoperative causes of coma include many of those listed in Table 5.4. In addition, the physician also must have a high index of suspicion for certain neurological conditions that occur in this setting, including fat embolism, addisonian crisis, hypothyroid coma (precipitated by acute illness or surgical stress), Wernicke encephalopathy from carbohydrate loading without adequate thiamine stores, and iatrogenic overdose of a narcotic analgesic.
Finally, family members or friends may be invaluable in identifying psychiatric causes of unresponsiveness. The family may describe a long history of psychiatric disease, previous similar episodes from which the patient recovered, current social stresses on the patient, or the patient’s unusual idiosyncratic response to stress. Special care must be taken with psychiatric patients because of the often biased approach to these patients, which may lead to incomplete evaluation. Psychiatric patients are subject to all the causes of coma listed in Table 5.4.
General Examination
Integument Examination
Systematic examination of the integument includes inspection of the skin, nails, and mucous membranes. A great deal of information can be gained by a brief examination of the skin (Table 5.3). Hot, dry skin is a feature of heatstroke. Sweaty skin is seen with hypotension or hypoglycemia. Drugs may cause macular-papular, vesicular, or petechial-purpuric rashes or bullous skin lesions. Bullous skin lesions most often are a result of barbiturates but also may be caused by imipramine, meprobamate, glutethimide, phenothiazine, and carbon monoxide. Kaposi sarcoma, anogenital herpetic lesions, or oral candidiasis should suggest the acquired immunodeficiency syndrome (AIDS), with its plethora of CNS abnormalities.
| Lesion or Rash | Possible Cause |
|---|---|
| Antecubital needle marks | Opiate drug abuse |
| Pale skin | Anemia or hemorrhage |
| Sallow, puffy appearance | Hypopituitarism |
| Hypermelanosis (increased pigment) | Porphyria, Addison disease, chronic nutritional deficiency, disseminated malignant melanoma, chemotherapy |
| Generalized cyanosis | Hypoxemia or carbon dioxide poisoning |
| Grayish-blue cyanosis | Methemoglobin (aniline or nitrobenzene) intoxication |
| Localized cyanosis | Arterial emboli or vasculitis |
| Cherry-red skin | Carbon monoxide poisoning |
| Icterus | Hepatic dysfunction or hemolytic anemia |
| Petechiae | Disseminated intravascular coagulation, thrombotic thrombocytopenic purpura, drugs |
| Ecchymosis | Trauma, corticosteroid use, abnormal coagulation from liver disease or anticoagulants |
| Telangiectasia | Chronic alcoholism, occasionally vascular malformations of the brain |
| Vesicular rash | Herpes simplex, varicella, behçet disease, drugs |
| Petechial-purpuric rash | Meningococcemia, other bacterial sepsis (rarely), gonococcemia, staphylococcemia, pseudomonas, subacute bacterial endocarditis, allergic vasculitis, purpura fulminans, Rocky Mountain spotted fever, typhus, fat emboli |
| Macular-papular rash | Typhus, candida, cryptococcus, toxoplasmosis, subacute bacterial endocarditis, staphylococcal toxic shock, typhoid, leptospirosis, pseudomonas sepsis, immunological disorders: Systemic lupus erythematosus Dermatomyositis Serum sickness |
| Other skin lesions: | |
| Ecthyma gangrenosum | Necrotic eschar often seen in the anogenital or axillary area in Pseudomonas sepsis |
| Splinter hemorrhages | Linear hemorrhages under the nail, seen in subacute bacterial endocarditis, anemia, leukemia, and sepsis |
| Osler nodes | Purplish or erythematous painful, tender nodules on palms and soles, seen in subacute bacterial endocarditis |
| Gangrene of digits, extremities | Emboli to larger peripheral veins or arteries |
Data on diseases associated with rashes from Corey, L., Kirby, P., 1987. Rash and fever. In: Braunwald, E., Isselbacher, K.J., Petersdorf, R.G. (Eds.), Harrison’s Principles of Internal Medicine, eleventh ed. McGraw-Hill, New York, pp. 240-244.
Neurological Examination
State of Consciousness
The importance of a detailed description of the state of consciousness is worth reemphasizing. It is imperative that the exact stimulus and the patient’s specific response be recorded. Several modes of stimulation should be used, including auditory, visual, and noxious. Stimuli of progressively increasing intensity should be applied, with the maximal state of arousal noted and the stimuli, the site of stimulation, and the patient’s exact response described. The examiner should start with verbal stimuli, softly and then more loudly calling the patient’s name or giving simple instructions to open the eyes. If there is no significant response, more threatening stimuli such as taking the patient’s hand and advancing it toward the patient’s face are applied. However, a blink response to visual threat need not indicate consciousness (Vanhaudenhuyse et al., 2008). Finally, painful stimuli may be needed to arouse the patient. All patients in apparent coma should be asked to open or close the eyes and to look up and down; these voluntary movements are preserved in the locked-in syndrome but cannot be elicited in coma—an important distinction.
The Glasgow Coma Scale (GCS; Table 5.4) is used widely to assess the initial severity of traumatic brain injury. This battery assesses three separate aspects of a patient’s behavior: the stimulus required to induce eye opening, the best motor response, and the best verbal response. Degrees of increasing dysfunction are scored. Its reproducibility and simplicity make the GCS an ideal method of assessment for non-neurologists involved in the care of comatose patients, such as neurological intensive care nurses. Its failure to assess other essential neurological parameters, however, limits its utility. Additionally, in patients who are intubated or who have suffered facial trauma, assessment of certain components of the GCS, such as eye opening and speech, may be difficult or impossible. Wijdicks and colleagues (1998) have suggested two new tools—the continuous performance test and the hand position test—that may serve as substitutes for the GCS in such patients, as well as in those with fluctuating levels of consciousness. The continuous performance test monitors level of alertness and requires the patient to raise a hand every time he or she hears a certain letter sound in a standardized sentence spoken by the examiner. The hand position test is a test of praxis in which the patient must mimic three different hand positions demonstrated by the examiner.
| BEST MOTOR RESPONSE | M |
| Obeys | 6 |
| Localizes | 5 |
| Withdraws | 4 |
| Abnormal flexion | 3 |
| Extensor response | 2 |
| Nil | 1 |
| VERBAL RESPONSE | V |
| Oriented | 5 |
| Confused conversation | 4 |
| Inappropriate words | 3 |
| Incomprehensible sounds | 2 |
| Nil | 1 |
| EYE OPENING | E |
| Spontaneous | 4 |
| To speech | 3 |
| To pain | 2 |
| Nil | 1 |
Respiration
Normal breathing is quiet and unlabored. The presence of any respiratory noise implies airway obstruction, which must be dealt with immediately to prevent hypoxia. Normal respiration depends on (1) a brainstem mechanism, located between the midpons and cervical medullary junction, that regulates metabolic needs; and (2) forebrain influences that subserve behavioral needs such as speech production. The organization and function of brainstem mechanisms responsible for respiratory rhythm generation, as well as forebrain influences, are complex and beyond the scope of this chapter. Neuropathological correlates of respiration are presented in Fig. 5.1.
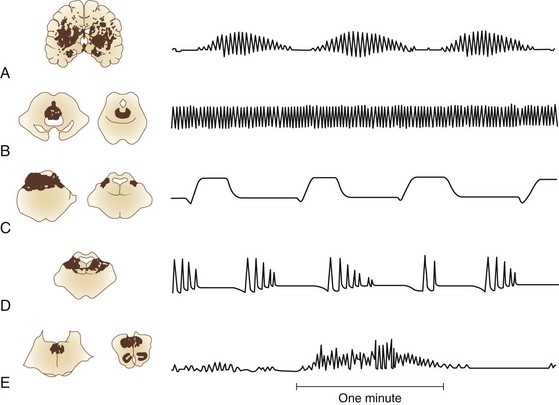
Cheyne-Stokes respiration is associated with bilateral hemispheric or diencephalic insults, but it may occur as a result of bilateral damage anywhere along the descending pathway between the forebrain and upper pons. It also is seen with cardiac disorders that prolong circulation time. Alertness, pupillary size, and heart rhythm may vary during Cheyne-Stokes respiration (Posner et al., 2007). Patients are more alert during the waxing portion of breathing.
Two breathing patterns similar to Cheyne-Stokes respiration should not be confused with it. Short-cycle periodic breathing is a respiratory pattern with a shorter cycle (faster rhythm) than Cheyne-Stokes respiration, with one or two waxing breaths followed by two to four rapid breaths, then one or two waning breaths. It is seen with increased ICP, lower pontine lesions, or expanding lesions in the posterior fossa (Posner et al., 2007). A similar type of respiration, in which there are short bursts of seven to ten rapid breaths, then apnea without a waning and waxing prodrome, has been erroneously referred to as Biot’s breathing. Biot, in fact, described an ataxic respiratory pattern, which is described later.
Central neurogenic hyperventilation refers to rapid breathing, from 40 to 70 breaths per minute, usually due to central tegmental pontine lesions just ventral to the aqueduct or fourth ventricle (Posner et al., 2007). This type of breathing is rare and must be differentiated from reactive hyperventilation due to metabolic abnormalities of hypoxemia secondary to pulmonary involvement. Large CNS lesions may cause neurogenic pulmonary edema with associated hypoxemia and increased respiratory rate. Increased intracerebral pressure causes spontaneous hyperpnea. Hyperpnea cannot be ascribed to a CNS lesion when arterial oxygen partial pressure is less than 70 to 80 mm Hg or carbon dioxide partial pressure is greater than 40 mm Hg.
Kussmaul breathing is a deep, regular respiration observed with metabolic acidosis. Apneustic breathing is a prolonged inspiratory gasp with a pause at full inspiration. It is caused by lesions of the dorsolateral lower half of the pons (Posner et al., 2007). Cluster breathing, which results from high medullary damage, involves periodic respirations that are irregular in frequency and amplitude, with variable pauses between clusters of breaths.
Pupil Size and Reactivity
Normal pupil size in the comatose patient depends on the level of illumination and the state of autonomic innervation. The sympathetic efferent innervation consists of a three-neuron arc. The first-order neuron arises in the hypothalamus and travels ipsilaterally through the posterolateral tegmentum to the ciliospinal center of Budge at the T1 level of the spinal cord. The second-order neuron leaves this center and synapses in the superior cervical sympathetic ganglion. The third-order neuron travels along the internal carotid artery and then through the ciliary ganglion to the pupillodilator muscles. Parasympathetic efferent innervation of the pupil arises in the Edinger-Westphal nucleus and travels in the oculomotor nerve to the ciliary ganglion, from which it innervates the pupillosphincter muscle (see Figs. 16.1 and 16.2 in Chapter 16).
Afferent input to the papillary reflex depends on the integrity of the optic nerve, optic chiasm, optic tract, and projections into the midbrain tectum and efferent fibers through the Edinger-Westphal nucleus and oculomotor nerve. Abnormalities in pupil size and reactivity help delineate structural damage between the thalamus and pons (Fig. 5.2), act as a warning sign heralding brainstem herniation, and help differentiate structural causes of coma from metabolic causes.
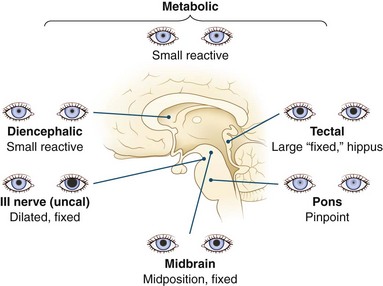
Fig. 5.2 Pupils in comatose patients.
(Reprinted from Plum, F., Posner, J.B., 1995. The Diagnosis of Stupor and Coma, third ed. Oxford University Press, New York. Copyright 1966, 1972, 1980, 1996, Oxford University Press, Inc. Used by permission of Oxford University Press, Inc.)
1. Dorsal tectal lesions interrupt the pupillary light reflex, resulting in midposition pupils, which are fixed to light but react to near vision; the latter is impossible to test in the comatose patient. Spontaneous fluctuations in size occur, and the ciliospinal reflex is preserved.
2. Nuclear midbrain lesions usually affect both sympathetic and parasympathetic pathways, resulting in fixed, irregular midposition pupils, which may be unequal.
3. Lesions of the third nerve fascicle in the brainstem, or after the nerve has exited the brainstem, cause wide pupillary dilation unresponsive to light. Pontine lesions interrupt sympathetic pathways and cause small, so-called pinpoint pupils which remain reactive, although magnification may be needed to observe this feature. Lesions above the thalamus and below the pons should leave pupillary function intact, except for Horner syndrome in medullary or cervical spinal cord lesions. The pathophysiology of pupillary response is discussed further in Chapters 16 and 36.
Ocular Motility
Normal ocular motility (see Chapters 16 and 35) depends on the integrity of a large portion of the cerebrum, cerebellum, and brainstem. Preservation of normal ocular motility implies that a large portion of the brainstem from the vestibular nuclei at the pontomedullary junction to the oculomotor nucleus in the midbrain is intact. Voluntary ocular motility cannot be judged in the comatose patient, so the examiner must rely on reflex eye movements that allow for assessment of the ocular motor system. The eye movements normally are conjugate, and eyes are in the midposition in the alert person. Sleep or obtundation alone may unmask a latent vertical or horizontal strabismus, resulting in dysconjugacy; therefore, patients must be examined when maximally aroused. The eyes return to the midposition in brain-dead patients.
Abnormalities in Resting Position
Pontaneous eye deviation may be conjugate or dysconjugate. Conjugate lateral eye deviation usually is due to an ipsilateral lesion in the frontal eye fields but may be due to a lesion anywhere in the pathway from the ipsilateral eye fields to the contralateral parapontine reticular formation (see Chapter 35). Dysconjugate lateral eye movement may result from a sixth nerve palsy in the abducting eye, a third nerve palsy in the adducting eye, or an internuclear ophthalmoplegia. An internuclear ophthalmoplegia may be differentiated from a third nerve palsy by the preservation of vertical eye movements.
Spontaneous Eye Movements
Spontaneous eye movements (see Chapter 16) are of many types. Purposeful-appearing eye movements in a patient who otherwise seems unresponsive should lead to consideration of the locked-in syndrome, catatonia, pseudocoma, or PVS. Roving eye movements are slow, conjugate, lateral to-and-fro movements. For roving eye movements to be present, the ocular motor nuclei and their connections must be intact. Generally when roving eye movements are present, the brainstem is relatively intact and coma is due to a metabolic or toxic cause or bilateral lesions above the brainstem. Detection of roving eye movements may be complicated by ocular palsies or internuclear ophthalmoplegia. These superimposed lesions produce relatively predictable patterns but often obscure the essential roving nature of the movement for the inexperienced observer.
Spontaneous conjugate vertical eye movements are separated into different types according to the relative velocities of their downward and upward phases. In ocular bobbing, rapid downward jerks of both eyes are observed, followed by a slow return to the midposition (Leigh and Zee, 2006). In the typical form, there is associated paralysis of both reflex and spontaneous horizontal eye movements. Monocular or paretic bobbing occurs when a coexisting ocular motor palsy alters the appearance of typical bobbing. The term atypical bobbing refers to all other variations of bobbing that cannot be explained by an ocular palsy superimposed on typical bobbing. Most commonly, this term is used to describe ocular bobbing when lateral eye movements are preserved. Typical ocular bobbing is specific but not pathognomonic for acute pontine lesions. Atypical ocular bobbing occurs with anoxia and is nonlocalizing. Ocular dipping, also known as inverse ocular bobbing, refers to spontaneous eye movements in which an initial slow downward phase is followed by a relatively rapid return. Reflex horizontal eye movements are preserved. It usually is associated with diffuse cerebral damage. In reverse ocular bobbing, there is a slow initial downward phase followed by a rapid return that carries the eyes past the midposition into full upward gaze. Then the eyes slowly return to the midposition. Reverse ocular bobbing is nonlocalizing.
Reflex Ocular Movements
Examination of ocular movement is not complete in the comatose patient without assessment of reflex ocular movements, including the oculocephalic reflex (“doll’s eye phenomenon”) and, if necessary, caloric (thermal) testing. In practice, the terms doll’s eye phenomenon and doll’s eye maneuver are used synonymously to refer to the oculocephalic reflex, but these terms are often confusing to the neophyte neurologist. It is better to use the term oculocephalic reflex followed by a description of the response. To test for this reflex, the examiner briskly rotates the patient’s head in both directions laterally, then flexes and extends the neck, continually observing the motion of the eyes. When supranuclear influences on the ocular motor nerves are removed, the eyes move in the orbit opposite to the direction of the head turn and maintain their position in space. This maneuver should not be performed on any patient until the stability of the neck has been adequately assessed. If there is any question of neck stability, a neck brace should be applied and caloric testing substituted. In the normal oculocephalic reflex (normal or positive doll’s eye phenomenon), the eyes move conjugately in a direction opposite to the direction of movement of the head. Cranial nerve palsies predictably alter the response to this maneuver (Table 5.5).
| Method | Response | Interpretation |
|---|---|---|
| Lateral head rotation | Eyes remain conjugate, move in direction opposite to head movement and maintain position in space | Normal |
| No movement in either eye on rotating head to left or right | Bilateral pontine gaze palsy, bilateral labyrinthine dysfunction, drug intoxication, anesthesia | |
| Eyes move appropriately when head is rotated in one direction but do not move when head is rotated in opposite direction | Unilateral pontine gaze palsy | |
| One eye abducts, the other eye does not adduct | Third nerve palsy | |
| Internuclear ophthalmoplegia | ||
| Vertical head flexion and extension | Eyes remain conjugate, move in direction opposite to head movement and maintain position in space | Normal |
| No movement in either eye | Bilateral midbrain lesions | |
| Only one eye moves | Third nerve palsy | |
| Bilateral symmetrical limitation of upgaze | Aging |
* To be performed only after neck stability has been ascertained.
Clinical caloric testing (as distinct from quantitative calorics, used to assess vestibular end-organ disorders; see Chapter 37) is commonly done by applying cold water to the tympanic membrane. With the patient supine, the head should be tilted forward 30 degrees to allow maximal stimulation of the lateral semicircular canal, which is most responsible for reflex lateral eye movements. After the ear canal is carefully checked to ensure that it is patent and the tympanic membrane is free of defect, 10 mL of ice-cold water is slowly instilled into one ear canal. For purposes of the neurological examination, irrigation of each ear with 10 mL of ice water generally is sufficient.
False-negative or misleading responses on caloric testing occur with preexisting inner ear disease, vestibulopathy such as that due to ototoxic drugs like streptomycin, vestibular paresis caused by illnesses such as Wernicke encephalopathy, and drug effects. Subtotal labyrinthine lesions decrease the response; there is no response when the labyrinth is destroyed. Lesions of the vestibular nerve cause a decreased or absent response. Drugs that suppress either vestibular or ocular motor function (or both) include sedatives, anticholinergics, anticonvulsants, tricyclic antidepressants, and neuromuscular blocking agents. If the response from one ear is indeterminate, both cold- and warm-water stimuli should be applied to the other ear. If the test remains equivocal, superimposition of the doll’s eye maneuver is recommended. Interpretation of abnormal cold caloric responses is summarized in Table 5.6. An unusual ocular reflex that has been observed in the setting of PVS is reflex opening of both eyes triggered by flexion of an arm at the elbow. This reflex is distinct from reflex eye opening in the comatose patient induced by raising the head or turning it from side to side.
| Method | Response | Interpretation |
|---|---|---|
| Cold water instilled in right ear | Slow phase to right, fast (corrective) phase to the left | Normal |
| No response (make sure canal is patent, apply warm-water stimulus to opposite ear) | Obstructed ear canal, “dead” labyrinth, eighth nerve or nuclear dysfunction, false-negative result (see text) | |
| Slow phase to right, no fast phase | Toxic-metabolic disorder, drugs, structural lesion above brainstem | |
| Downbeating nystagmus | Horizontal gaze palsy | |
| Cold water instilled in left ear | Responses should be opposite those for right ear | Peripheral eighth nerve or labyrinth disorder on right (provided that right canal is patent) |
| Warm water instilled in left ear after no response from cold water in right ear | Slow phase to right, fast phase to left |
Coma and Brain Herniation
Herniation syndromes are explained in Chapter 50. Knowledge of some of the clinical signs of herniation is especially important in the clinical approach to coma. Traditional signs of herniation due to supratentorial masses usually are variations of either an uncal or a central pattern. Classically, the uncal pattern includes early signs of third nerve and midbrain compression. The pupil initially dilates as a result of third nerve compression but later returns to the midposition with midbrain compression that involves the sympathetic as well as the parasympathetic tracts. In the central pattern, the earliest signs are mild impairment of consciousness, with poor concentration, drowsiness, or unexpected agitation; small but reactive pupils; loss of the fast component of cold caloric testing; poor or absent reflex vertical gaze; and bilateral corticospinal tract signs, including increased tone of the body ipsilateral to the hemispheric mass lesion responsible for herniation (Posner et al., 2007).
Signs of herniation tend to progress generally in a rostrocaudal manner. An exception occurs when intraventricular bleeding extends to the fourth ventricle and produces a pressure wave, compressing the area around the fourth ventricle. Also, when an LP reduces CSF pressure suddenly in the face of a mass lesion that produced increased ICP, sudden herniation of the cerebellar tonsils through the foramen magnum may result (Posner et al., 2007). Both of these clinical scenarios may be associated with sudden unexpected failure of medullary functions that support respiration or blood pressure. In patients with herniation syndromes, the clinical picture may be confusing because of changing signs or the expression of scattered, isolated signs of dysfunction in separate parts of the brain. In addition, certain signs may be more prominent than others.
Differential Diagnosis
Differentiating Toxic-Metabolic Coma from Structural Coma
• State of consciousness. Patients with metabolic problems often have milder alterations in arousal, typically with waxing and waning of the behavioral state. Patients with acute structural lesions tend to stay at the same level of arousal or progressively deteriorate. Toxins may also cause progressive decline in level of arousal.
• Respiration. Deep, frequent respiration most commonly is due to metabolic abnormalities, though rarely it is caused by pontine lesions or by neurogenic pulmonary edema secondary to acute structural lesions.
• Funduscopic examination. Subhyaloid hemorrhage or papilledema are almost pathognomonic of structural lesions. Papilledema due to increased ICP may be indicative of an intracranial mass lesion or hypertensive encephalopathy. Papilledema does not occur in metabolic diseases except hypoparathyroidism, lead intoxication, and malignant hypertension.
• Pupil size. The pupils usually are symmetrical in coma from toxic-metabolic causes. Patients with metabolic or toxic encephalopathies often have small pupils with preserved reactivity. Exceptions occur with methyl alcohol poisoning, which may produce dilated and unreactive pupils, or late in the course of toxic or metabolic coma if hypoxia or other permanent brain damage has occurred. In terminal asphyxia, the pupils dilate initially and then become fixed at midposition within 30 minutes. The initial dilation is attributed to massive sympathetic discharge.
• Pupil reactivity. Assessment of the pupillary reflex is one of the most useful means of differentiating metabolic from structural causes of coma. Pupillary reactivity is relatively resistant to metabolic insult and usually is spared in coma from drug intoxication or metabolic causes, even when other brainstem reflexes are absent. Hypothermia may fix pupils, as does severe barbiturate intoxication. Neuromuscular blocking agents produce midposition or small pupils, and glutethimide and atropine dilate them.
• Ocular motility. Asymmetry in oculomotor function typically is a feature of structural lesions.
• Spontaneous eye movements. Roving eye movements with full excursion are most often indicative of metabolic or toxic abnormalities.
• Reflex eye movements. Reflex eye movements normally are intact in toxic-metabolic coma, except rarely in phenobarbital or phenytoin intoxication or deep metabolic coma from other causes.
• Adventitious movement. Periods of motor restlessness, tremors, or spasm punctuating coma often are due to drugs or toxins such as chlorpromazine or lithium. Brainstem herniation or intermittent CNS ischemia also may produce unusual posturing movements. Myoclonic jerking generally is metabolic and often anoxic in origin.
• Muscle tone. Muscle tone usually is symmetrical and normal or decreased in metabolic coma. Structural lesions cause asymmetrical muscle tone. Tone may be increased, normal, or decreased by structural lesions.
The examiner should be aware of common structural lesions that mimic toxic-metabolic causes and, conversely, toxic or metabolic causes of coma that may be associated with focal abnormalities on examination. Structural lesions that may mimic toxic-metabolic causes include subarachnoid hemorrhage, sinus vein thrombosis, chronic or bilateral subdural hemorrhage, and other diffuse or multifocal disorders such as vasculitis, demyelinating diseases, or meningitis. Any toxic-metabolic cause of coma may be associated with focal features; however, such features most often are observed with barbiturate or lead poisoning, hypoglycemia, hepatic encephalopathy, and hyponatremia. Old structural lesions such as prior stroke may be the origin of residual abnormalities found on neurological examination in a patient who is comatose from toxic or metabolic causes. Moreover, metabolic abnormalities such as hypoglycemia may unmask relatively silent structural abnormalities. Detailed descriptions of the toxic and metabolic encephalopathies are provided in Chapter 56.
Helpful Laboratory Studies
Laboratory tests that are extremely helpful in evaluating the comatose patient are listed in Table 5.7. Arterial blood gas determinations rule out hypoxemia and carbon dioxide narcosis and help differentiate primary CNS problems from secondary respiratory problems. Liver disease, myopathy, and rhabdomyolysis all elevate alanine aminotransferase and aspartate aminotransferase levels. Liver function test results may be misleading in end-stage liver disease, as values may be normal or only mildly elevated with markedly abnormal liver function. Although the blood ammonia level does not correlate well with the level of hepatic encephalopathy, it often may be markedly elevated and thus helpful in cases of suspected liver disease with relatively normal liver function studies. Hepatic encephalopathy may continue for up to 3 weeks after liver function values return to normal.
Table 5.7 Laboratory Tests Helpful in Differential Diagnosis for Coma
| Laboratory Study | Result | Associated Disorders |
|---|---|---|
| Electrolytes (Na, K, Cl, CO2) | See Chapters 49A and 56 for discussion of disorders associated with abnormalities of electrolytes, glucose, BUN, calcium, and magnesium | |
| Glucose | ||
| BUN | ||
| Creatinine | ||
| Calcium | ||
| Magnesium | ||
| Complete blood count with differential | Hematocrit: | |
| Increased | Volume depletion, underlying lung disorder, myeloproliferative disorder, cerebellar hemangioblastoma; may be associated with vascular sludging (hypoperfusion) | |
| Decreased | Anemia, hemorrhage | |
| White blood cell count: | ||
| Increased | Infection, acute stress reaction, steroid therapy, after epileptic fit, myeloproliferative disorder | |
| Decreased | Chemotherapy, immunotherapy, viral infection, sepsis | |
| Lymphocyte count: | ||
| Decreased | Viral infection, malnutrition, AIDS | |
| Platelet count | Decreased | Sepsis, disseminated intravascular coagulation, thrombotic thrombo-cytopenic purpura, idiopathic thrombocytopenic purpura, drugs; may be associated with intracranial hemorrhage |
| PT | Increased | Coagulation factor deficiency, liver disease, anticoagulants, disseminated intravascular coagulation |
| PTT | Increased | Heparin therapy, lupus anticoagulant |
| Arterial blood gases | See text | |
| Creatine phosphokinase | See text | |
| Liver function studies | See text | |
| Thyroid function studies | See text | |
| Plasma cortisol level | See text | |
| Drug and toxin screen | See text | |
| Serum osmolality | See text |
AIDS, Acquired immunodeficiency syndrome; BUN, blood urea nitrogen; PT, prothrombin time; PTT, partial thromboplastin time.
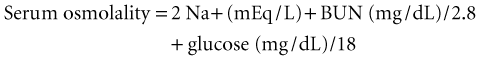
Other Useful Studies
Neuroradiological Imaging
The value of the CT scan in demonstrating mass lesions and hemorrhage is undeniable. Furthermore, it may demonstrate features of brain herniation. Uncal herniation is characterized on CT scan by (1) displacement of the brainstem toward the contralateral side, with increase in width of subarachnoid space between the mass and ipsilateral free edge, (2) medial stretching of the posterior cerebral and posterior communicating arteries, (3) obliteration of the interpeduncular cistern, (4) occipital lobe infarction, and (5) distortion and elongation of the U-shaped tentorial incisura. The clinician should be aware that the CT scan may miss early infarction, encephalitis, and isodense subdural hemorrhage. Special caution must be taken in evaluating CT scans in comatose patients, especially before LP, to rule out isodense subdural or bilateral subdural hemorrhage. Interpretation of CT scans is discussed in Chapter 33A.
In severe head injury, studies of cerebral metabolism employing single photon emission computed tomography (SPECT) may be of prognostic value (Della Corte et al., 1997). Although cerebral blood flow in the first 48 hours after trauma does not appear to correlate with severity or prognosis, the cerebral metabolic rate of oxygen (CMRo2), like the GCS, may be useful in predicting prognosis.
Electroencephalography
The EEG is helpful in many situations and disorders: confirming underlying cortical structural damage in patients too unstable to travel to the CT scanner; postictal states in patients slow to wake after a presumed seizure; partial complex seizures; electroencephalographic or nonconvulsive status epilepticus, such as is seen in comatose patients after anoxic ischemic damage; and toxic-metabolic disturbances. With metabolic disorders, the earliest EEG changes typically are a decrease in the frequency of background rhythms and the appearance of diffuse theta activity that progresses to more advanced slowing in association with a decrease in the level of consciousness. In hepatic encephalopathy, bilaterally synchronous and symmetrical, medium- to high-amplitude, broad triphasic waves, often with a frontal predominance, may be observed. Herpes simplex encephalitis may be suggested by the presence of unilateral or bilateral periodic sharp waves with a temporal preponderance. The EEG also can help confirm a clinical impression of catatonia, pseudocoma, the locked-in syndrome, PVS, and brain death (Brenner, 2005). EEGs are discussed further in Chapter 32A.
Evoked Potentials
Evoked potentials may help in evaluating brainstem integrity and assessing prognosis for comatose patients. A study of 50 hemodynamically stable patients remaining in coma 4 hours after resuscitation from cardiopulmonary arrest with short-latency somatosensory evoked potentials within 8 hours after arrest found that none of the 30 patients without cortical potentials recovered cognition. Five of the 20 patients with cortical potentials recovered. Forty percent of the patients who did not recover had preserved brainstem reflexes, allowing some evaluation of prognosis in a group of patients in whom prognosis is difficult to assess by other means. Event-related potentials may prove particularly useful as an objective assessment of cognitive function in patients with the locked-in syndrome (Onofrj et al., 1997). The N100 component of the auditory evoked potential and cognitive evoked potentials (mismatch negativity obtained after novel stimuli) appear to have predictive value for awakening from coma, but the pupillary reflex remains the strongest prognostic variable (Fischer et al., 2004). Absence of evoked potentials in response to somatosensory stimuli also is highly predictive of nonawakening from coma (Robinson et al., 2003).
Prognosis
Nontraumatic Coma
Only about 15% of patients in nontraumatic coma make a satisfactory recovery. Functional recovery is related to the cause of coma. Diseases causing structural damage, such as cerebrovascular disease including subarachnoid hemorrhage, carry the worst prognosis; coma from hypoxia-ischemia due to such causes as cardiac arrest has an intermediate prognosis; coma due to hepatic encephalopathy and other metabolic causes has the best ultimate outcome. Age does not appear to be predictive of recovery. The longer a coma lasts, the less likely the patient is to regain independent functioning. Factors that adversely impact brain injury following cardiac arrest include cerebral edema, pyrexia, hyperglycemia, and seizures (Neumar et al., 2008).
In the early days after the onset of nontraumatic coma, it is not possible to predict with certainty which patients will ultimately enter or remain in a vegetative state. Although rare cases have been reported of patients awakening after prolonged vegetative states, patients with nontraumatic coma who have not regained awareness by the end of 1 month are unlikely to do so. Even if they do regain consciousness, they have practically no chance of achieving an independent existence. A large multi-institutional study determined that within 3 days of cardiac arrest, evaluation in the intensive care unit is sufficiently predictive of neurological outcome to allow for informed decisions regarding life support. Absence of pupillary light or corneal reflexes, and motor response to noxious stimuli no greater than extension, suggest a poor prognosis for recovery. Other poor prognostic signs are myoclonic status epilepticus, bilateral absence of the N20 response from the somatosensory cortex, and several neuroimaging signs (Young, 2009).
Persistent Vegetative State
As a rule, PVS can be reliably diagnosed 12 months after a traumatic brain injury and 6 months after other cerebral insults. Recovery after 3 years for patients in PVS has not been reported (Wijdicks and Cranford, 2005). Those patients who have been reported to “improve” remain severely disabled, bed- or wheelchair-bound, and fully dependent on care. At 5 years, the mortality rate for PVS is in excess of 80%. Prolonged survival is rare and requires exquisite medical attention. Death typically results from untreated infection or overwhelming sepsis. “Miracle awakenings,” such as with the sudden appearance of communicative speech, have been observed rarely in patients in the minimally conscious state but not in those in PVS (Wijdicks and Cranford, 2005).
Brain Death
Clinical Approach to Brain Death
• Coma. The patient should exhibit an unarousable unresponsiveness. There should be no meaningful response to noxious, externally applied stimuli. The patient should not obey commands or demonstrate any verbal response, either reflexively or spontaneously. Spinal reflexes, however, may be retained.
• No spontaneous respirations. The patient should be removed from ventilatory assistance, and carbon dioxide should be allowed to build up because of the respiratory drive that hypercapnia produces. The diagnosis of absolute apnea requires the absence of spontaneous respiration at a carbon dioxide tension of at least 60 mm Hg. A safe means of obtaining this degree of carbon dioxide retention involves the technique of apneic oxygenation, in which 100% oxygen is delivered endotracheally through a thin sterile catheter for 10 minutes. Arterial blood gas levels should be obtained to confirm the arterial carbon dioxide pressure.
• Absence of brainstem reflexes. Pupillary, oculocephalic, corneal, and gag reflexes all must be absent, and there should not be any vestibulo-ocular responses to cold calorics.
• Electrocerebral silence. An isoelectric EEG should denote the absence of cerebrocortical function. Some authorities do not regard the performance of an EEG as mandatory in assessing brain death, and instances of preserved cortical function despite irreversible and complete brainstem disruption have been reported.
• Absence of cerebral blood flow. Cerebral contrast angiography or radionuclide angiography can substantiate the absence of cerebral blood flow, which is expected in brain death. These tests are considered confirmatory rather than mandatory. On rare occasions, in the presence of supratentorial lesions with preserved blood flow to the brainstem and cerebellum, findings on cerebral radionuclide angiography may be misleading.
• Absence of any potentially reversible causes of marked CNS depression. Such causes include hypothermia (temperature 32°C [89.6°F] or less), drug intoxication (particularly barbiturate overdose), and severe metabolic disturbance.
Brain Death Survival
Despite aggressive therapeutic measures, survival of “brain-dead” persons for more than 1 week has been considered unlikely. Shewmon (1998) reviewed a series of 175 cases surviving longer than 1 week after diagnosis of “brain death.” Survival potential decreased exponentially, with an initial half-life of 2 to 3 months, followed at 1 year by a slow decline. One patient survived for more than 14 years. Survival was found to correlate inversely with age, and prolonged survival was more common with primary brain pathology. The tendency to cardiovascular collapse in brain death may be transient and is more likely to be attributable to systemic than to brain pathology.
Brenner R.P. The interpretation of the EEG in stupor and coma. Neurologist. 2005;11:271-284.
Della Corte F., Galli G., Campioni P., et al. Quantitative cerebral blood flow and metabolism determination in the first 48 hours after severe head injury with a new dynamic SPECT device. Acta Neurochir (Wien). 1997;139:636-641.
Fischer C., Luauté J., Adeleine P., et al. Predictive value of sensory and cognitive evoked potentials for awakening from coma. Neurology. 2004;63:669-673.
Leigh R.J., Zee D.S. The Neurology of Eye Movements, fourth ed. New York: Oxford University Press; 2006.
Neumar R.W., Nolan J.P., Adrie C., et al. Post cardiac arrest syndrome. Epidemiology, pathophysiology, treatment and prognostication. A consensus statement from the International Liaison Committee on Resuscitation. Circulation. 2008;108:1-32.
Onofrj M., Thomas A., Paci C., et al. Event-related potentials recorded in patients with locked-in syndrome. J Neurol Neurosurg Psychiatry. 1997;63:759-764.
Posner J.B., Saper C.B., Schiff N.D., et al. Plum and Posner’s Diagnosis of Stupor, fourth ed. New York: Oxford University Press; 2007.
Robinson L.R., Micklesen P.J., Tirschwell D.L., et al. Predictive value of somatosensory evoked potentials for awakening from coma. Crit Care Med. 2003;31:960-967.
Shewmon D.A. Chronic “brain death”: meta-analysis and conceptual consequences. Neurology. 1998;51:1538-1545.
Wijdicks E.F.M., Cranford R.E. Clinical diagnosis of prolonged states of impaired consciousness in adults. Mayo Clin Proc. 2005;80:1037-1104.
Wijdicks E.F.M., Kokmen E., O’Brien P.C. Measurement of impaired consciousness in the neurological intensive care unit: a new test. J Neurol Neurosurg Psychiatry. 1998;64:117-119.
Vanhaudenhuyse A., Giacinio J., Schnakers C., et al. Blink to visual threat does not herald consciousness in the vegetative state. Neurology. 2008;71:1374-1375.
Young G.B. Neurologic prognosis after cardiac arrest. N Engl J Med. 2009;361:605-611.