[level-membership-for-opthalmology-category]
Chapter 14 Structure and Function of Rod and Cone Photoreceptors
Introduction
Our visual experience is initiated by rod and cone photoreceptors in the retina. The human eye contains over 100 million rods and about 6 million cones, which are located within the outer nuclear layer of the retina and allow our visual experience to extend over 12 orders of magnitude in light intensity by splitting this range. Rod photoreceptors mediate vision under conditions of dim illumination, and allow our visual system to reach the limit imposed by the absorption of single photons.1,2 Cones are less sensitive by ~100-fold, but their tremendous capacity for adaptation allows them to encode light intensities in the brightest of days.3,4 Over the last decade, our understanding of structure and function of these cells has increased dramatically. Over 150 genes have been cloned or linked to retinal diseases, and, surprisingly, as many as half of these genes are specifically expressed or highly enriched in the photoreceptor cells. Discovery of the molecular constituents of the rods and cones is progressing at an increasing rate, particularly enhanced by the availability of “complete” genomic sequences for both human and mouse. While photoreceptor genes can be identified through association with a retinal disease (linkage), information about their function does not accompany their identification.5 Basic science research must then be undertaken to explain their role in both the normal, healthy photoreceptor as well as in photoreceptor diseases.
Though much of what we know about the structure and function of photoreceptor cells has come from studying animal models of inherited blindness,6,7 more recently, transgenic and knockout animal technologies have established themselves as powerful tools for understanding function and studying disease. After new photoreceptor genes are identified from patients, mutant animals can be engineered to emulate human photoreceptor pathologies. Before these molecular technologies were available, most data about retinal disease were gleaned from the rare “informative” patient or donor retina that had surviving photoreceptors to examine.8 Other major sources of information were studies of animal models that occurred through inbreeding or random inheritance of mutations in photoreceptor genes. Examples of these are the Irish Setter dog,9 the Briard dog,10 the Abyssinian cat,11 the Royal College of Surgeons (RCS) rat,12 and the rd13,14 and rds15,16 mouse models. Now, the ability to engineer transgenic animals has made the search for “informative” patients and naturally occurring animal models less acute, since any single gene of interest can be introduced17–19 or removed20–24 from the photoreceptor. However, the most instructive examples of structure–function relationships occur when there are patients and animal models with analogous mutation(s) and/or disease phenotype.
Photoreceptor fundamentals
The rod and cone photoreceptors are specialized sensory neurons that contain the protein machinery necessary to convert incident light into a signal that can be interpreted by the nervous system. Rod photoreceptors are more numerous than cones in most mammalian retina, and are highly sensitive. In the fully dark-adapted state, rods can reliably report the absorption of single photons to the retinal output, and they permit our scotopic, or night, vision. Cone photoreceptors are morphologically and functionally distinct from rods and express several types of visual pigments, or opsins, whose spectral sensitivity varies based on the cone’s subtype. In humans, three classes of cones confer robust color vision: S, M, and L cones (see Chapter 10, Color vision and night vision). Cones are also less sensitive than rods and generate light responses that are temporally briefer. This allows cones to mediate our photopic, or day, vision with improved temporal resolution. The concerted action of these two types of photoreceptors, and the retinal circuitry that carries their signals to the retinal output, ultimately underlie our rich visual experience.
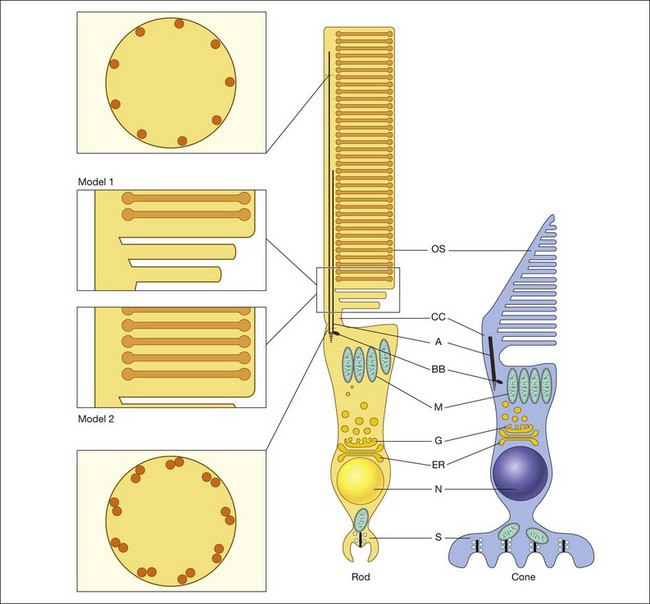
Both rod and cone photoreceptors are highly polarized elongated cells that can be described as having four subcellular compartments: the outer segment (OS), the inner segment (IS), the nucleus, and the synaptic terminal (Figs 14.1 and 14.2). The OS is where photons are captured and activation of the phototransduction cascade begins. The IS lies immediately proximal to the OS, and contains the cell’s protein synthesis (Golgi apparatus and endoplasmic reticulum) and metabolic (mitochondria) machinery. Light-evoked signals are relayed passively down the photoreceptor axon (up to 75 µm long) to the synaptic terminals in the outer plexiform layer. The structure of photoreceptor terminals is unique in the nervous system, as they contain a specialized structure called a ribbon that facilitates the release of the excitatory neurotransmitter glutamate on to second-order retinal neurons (bipolar and horizontal cells). Thus, the photoreceptor cells transduce the sensory stimulus, light, and pass on a signal to retinal circuits that carry this information to higher visual centers.
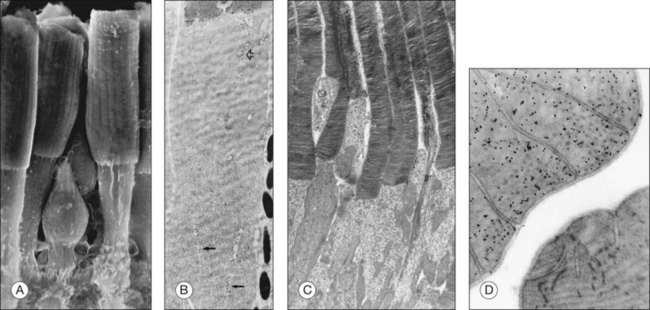
Photoreceptor outer-segment structure
The OS compartment contains all components necessary for phototransduction, which is a set of biochemical reactions that convert photon capture to a change in a cationic current at the plasma membrane. Rods have cylindrical OS about 1.3 µm in diameter and length that ranges from 25 to 45 µm,25–28 that depends on numerous factors, including the time of day, the light intensity, their location in the retina, and the animal species. The cone OS is shorter, typically half the length of rods, with a larger diameter at the base that gradually tapers towards the tip (Fig. 14.2). Mouse rod OS contains an average of 810 membranous discs, which occupy two-thirds of the volume of the OS.29 The majority of the protein on these discs is rod opsin (or rhodopsin when the opsin is bound to its chromophore, 11-cis retinal). The density of rhodopsin on the disc membrane has been measured to be about 24 000 molecules/µm2.30 Despite this dense packing, rhodopsin freely diffuses in the membrane,31 which facilitates its encounter with, and activation of, transducin molecules to amplify the light signal. Many G-protein-coupled receptors are known to form dimers and higher-order oligomers. Rhodopsin has been observed to form a paracrystalline array of dimers when native disc membrane is viewed under atomic force microscopy.32 Whether it exists as dimers in vivo is still an area of controversy. However, it is known that rhodopsin is functional as monomers.33
Proteins that stabilize the structure of outer-segment discs
Rhodopsin is not only integral to the phototransduction cascade, it is also required for the formation and maintenance of OS discs; in the absence of rhodopsin, the OS structure is not formed.22 Peripherin and Rom-115,34,35 are two other disc membrane components that contribute to maintaining the flattened structure of the disc. Both of these proteins belong to the tetraspanin family of integral membrane proteins that form large multiprotein complexes known as the tetraspanin web or tetraspanin-enriched microdomains.36 Peripherin/rds is normally found within the edge or “rim” of the disc. It self-associates to form higher-order complexes and also interacts with Rom-1.37 Peripherin/rds and Rom-1 likely function as adhesion molecules that assist in keeping the discs vertically aligned, and may stabilize the disc stack with bridges to the overlying plasma membrane. Peripherin/rds is also thought to function in forming the curvature of the disc rim.38
Deletion or disruption of peripherin/rds results in malformation of discs at the OS base. The naturally occurring retinal degeneration in the mouse named “retinal degeneration, slow” (rds) has been shown to be due to a defect in peripherin/rds. The absence of peripherin/rds in the rds/rds mouse prevents normal development of the photoreceptor OS and leads to photoreceptor cell death.15,39 Transgenic mice that lack peripherin/rds fail to form an OS. When the level is reduced, as in the heterozygous mice, large whorls of disc membrane are formed instead of an organized stack of uniform discs.15,39 Mutations that affect the quaternary structure of peripherin/rds also lead to malformed discs. These observations underscore the importance of peripherin/rds in the formation and maintenance of disc structure in the OS.
Although Rom-1 is analogous to peripherin in structure and function, its absence in rods shows a less severe phenotype than peripherin/rds knockout mice, suggesting that peripherin/rds is more critical for disc formation. Although the discs grow larger than normal sizes, photoreceptor function appears to be retained to a much higher level than when peripherin/rds is absent. Nevertheless, abnormal Rom-1 does lead to slow and progressive photoreceptor degeneration both in mice and in humans.40
Functional differences of peripherin/rds and Rom-1 have been noted in rods and cones. Cones appear to have a lower ratio of Rom-1/peripherin,36 and different peripherin/rds mutations appear to affect rods and cones differentially. For example, the C214S and N244K mutations have a greater effect on rods whereas R172W and N244H tend to cause cone-dominant diseases such as macular dystrophy.36
Transmission electron microscopy has revealed structural elements between the lamellar discs as well as between the discs and the plasma membrane that are mainly localized to the rim region and incisures of discs.41–44 A study of OS structure using cryoelectron tomography of vitrified OS showed previously unobserved spacers distributed throughout the discs.30 These structural elements likely maintain the proper distance between adjacent discs and between discs and the plasma membrane. The protein identities of these spacers are not known, but peripherin/rds and Rom-1 may be candidates.
Another component of the rod disc is ABCR (photoreceptor cell-specific ATP-binding cassette transporter).45–47 Mutations in ABCR are responsible for a large variety of retinal degenerations, including Stargardt macular dystrophy, fundus flavimaculatus, some forms of cone–rod degeneration, and retinitis pigmentosa (RP). Other ABCR mutations are thought to increase the risk of developing age-related macular degeneration (AMD). ABCR is a member of the ABC superfamily. This is a large family of transmembrane proteins involved in energy-dependent transport of many different substrates across membrane “barriers.” The localization of ABCR at the disc rim suggests that it is involved in the movement of molecules from the disc lumen into the cytosol. This hypothesis was supported by studies in ABCR knockout mice, which show delayed dark adaptation, increased all-trans retinal following light exposure, and elevated phosphatidylethanolamine (PE) in the rod OS.48 Biochemical analysis of retinas from ABCR knockout mice revealed accumulation of a novel complex of all-trans retinal and PE, termed N-retinylidene-PE, which is not found in normal retina. Once in the cytosol, the all-trans retinol moves to the retinal pigment epithelium (RPE) for recycling and chromophore (11-cis retinal) regeneration (see Chapter 16, Cell biology of the retinal pigment epithelium). The primary pathologic defect in Stargardt disease, and also in ABCR knockout mice, is accumulation of toxic lipofuscin pigments, such as A2E, in RPE cells. Thus the phenotype is remarkably similar between mice and that observed in the fundus of Stargardt patients.
The animal model and further biochemical investigations49 suggest that ABCR functions as an outwardly directed flippase for N-retinylidene-PE. Delayed dark adaptation is likely due to accumulation (in discs) of the noncovalent complex between opsin and all-trans retinal. ABCR-mediated retinal degeneration in patients may result from “poisoning” of the RPE due to A2E accumulation, with secondary photoreceptor degeneration due to loss of the ABCR support role.
Though ABCR most definitely plays a role in the structure of discs, no mutations have been conclusively associated with abnormal structural features per se. In other words, it appears that mutations do not directly affect folding at the disc rim, and more likely affect the biochemical function of the transporter. In corroboration of these data, Radu and colleagues tested the effects of isotretinoin on lipofuscin accumulation in ABCR knockout mice,50 a model of recessive Stargardt disease. They observed by electron microscopy that isotretinoin blocked both the formation of A2E biochemically, and the accumulation of lipofuscin pigments. Further, no significant visual loss was observed in ABCR null mice by electroretinography (when treated), and isotretinoin also blocked the slower, age-dependent accumulation of lipofuscin in wild-type mice. The results suggest that treatment with isotretinoin may inhibit lipofuscin accumulation and delay the onset of visual loss in patients with Stargardt disease and may be an effective treatment for other forms of retinal or macular degeneration associated with lipofuscin accumulation, though “normal” visual function may be somewhat compromised by such treatment.
The presence of an OS is clearly not optional, as there are many examples of retinal disease in which the photoreceptor cell dies shortly after loss of the OS.51–53 Why loss of the OS triggers rod cell death is not obvious, since all of the required cellular organelles inhabit the IS and cell body; the OS appears devoid of organelles. The necessity to maintain an OS is further surprising since photoreceptors continually replace all the OS components. In fact, the renewal of rod cell OS components is perhaps more readily seen in the photoreceptor than in any other cell in the body. The disc and plasma membrane components of the OS are completely replaced within 2 weeks.54,55 To support this turnover there is an influx of newly synthesized proteins and lipids from the IS. These components are transported to the base of the OS, and from there discs gradually migrate towards the RPE for phagocytosis and recycling. One idea is that the high oxygen tension coming from the choroidal vasculature may interfere with the cellular machinery in the IS and nucleus of the photoreceptor, and the presence of the OS puts a protective distance between the choroid and the photoreceptor nuclei.56
Disc morphogenesis
Although it is known that new discs are formed at the base of the OS, the process by which these discs are formed is not fully resolved (Fig. 14.2). One model was described in the work of Anderson et al. (model 1).57,58 Based on morphologic studies of adult monkey rods, it was observed that the folded membrane stacks at the base of the OS are continuous with the plasma membrane of the connecting cilium, whereas the older discs are closed off and separated from the plasma membrane. From this appearance a model was proposed that the newly arrived rhodopsin-bearing vesicles fuse with the plasma membrane, and the growing membrane evaginates to form open discs. Eventually these discs pinch off and form the mature closed discs. Another model (model 2), the vesicular targeting model, was recently proposed by Chuang and colleagues.59 They provide molecular evidence that rhodopsin’s carboxyl terminus interacts with a protein called SARA (smad anchor for receptor activation), as well as phosphatidylinositol 3-phosphate (PI3P), and syntaxin 3. In their model, the new discs are already closed and enveloped by the OS plasma membrane. Their evidence suggests that the protein–protein and protein–lipid interactions organized by SARA regulate the vesicular targeting of axonemal rhodopsin-bearing vesicles to the newly formed discs. Thus the discs grow by fusing with the rhodopsin-bearing vesicles and not from the evaginated plasma membrane. This model is more consistent with the morphology of their experimental model system, the mouse rods, which do not display open discs at the base of the OS.
Outer-segment plasma membrane
It is clear that, in addition to its obvious role in phototransduction, the OS plasma membrane contains a rich array of specialized proteins, many of which regulate the movement of ions into and out of the OS. The best-characterized proteins of this type are the retinal cGMP-gated (CNG) cation channels. In the dark-adapted rods and cones, Na+ and Ca2+ flow into the OS through these channels in the plasma membrane. Calcium comprises about 10% of the dark current carried by these channels in rods,60 and perhaps 20% or more in cones.61,62 In both rods and cones Na+ is extruded from the IS through the Na+/K+ pump. This flow of ions sets up the circulating dark current, of which the vast majority is carried by the Na+. The probability of the opening of the CNG channel, which in turn determines the size of the circulating current, depends on the amount of free [cGMP], which in the dark is estimated to be 3–4 µM.63 At this concentration, the probability of channel opening is estimated to be only 0.1–0.2.64,65 This underscores the impact that elevated [cGMP] can have on the number of open channels in the diseased state. The influx of Ca2+ through the channel is balanced by an efflux of Ca2+ by the Na+/Ca2+-K+ exchanger (NCKX) in the rod OS plasma membrane, thereby maintaining the intracellular level of Ca2+ at a relatively constant level.66 Kaupp and colleagues67 cloned the cGMP channel from bovine retina, while Pittler and colleagues68 determined the primary structures of the human and mouse retinal rod cGMP-gated cation channel. These studies found that the sequence of the cGMP channel has significant similarity (59%) to the olfactory cAMP-gated channel. The retinal rod CNG channel is a hetero-oligomer composed of three alpha (CNGA1) and one beta (CNGB1) subunits,69,70 each with cytoplasmic amino (N)- and carboxyl (C)-termini, six putative transmembrane domains, and a pore region.71,72 Mutations in CNGA1 and CNGB1 have been linked to disease. A point mutation in CNGB173 and several mutations in CNGA174 have been identified and linked to recessive RP in humans. The CNG channel in cones consists of two CNGA3 and two CNGB3 subunits.75 Together, mutations in either of these genes account for ~75% of complete acromatopsia.76,77
In addition to the CNG channel, there are several other channels and transporters in the plasma membrane that serve to regulate the intracellular contents of the OS. The best studied of these is the NCKX. Rods express NCKX1 whereas cones express NCKX2.78 The exchanger moves with every cycle 4 Na+ into the OS, and in exchange, moves one Ca2+ and one K+ into the subretinal space; exchange is thus electrogenic. Other transmembrane proteins, such as the GLUT-1 glucose transporter, have been shown to be present on both rod and cone OS.79
Outer-segment lipids
Docosahexaenoic acid (DHA, 22:6n-6) is the major fatty acid found in the retinal rod OS, and rod photoreceptors have higher levels of DHA than in any other membrane system examined.80 Numerous studies have established that the high level of DHA in rod OS membranes provides an optimal microenvironment for rhodopsin. DHA belongs to the n-3 family of essential polyunsaturated fatty acids. These fatty acids cannot be synthesized by vertebrates and they, or their shorter-chain precursors, must therefore be obtained in the diet.81
Humans with RP and dogs with progressive rod–cone degeneration (prcd) have lower than normal blood levels of long-chain polyunsaturated fatty acids, including DHA. In addition, prcd-affected dogs have lower levels of DHA in their rod OS than control animals.82 The reason for the reduced level of DHA in rod OS of animals with inherited retinal degeneration is not known. However, the fatty acid composition of the rod OS of these animals suggests that the synthesis of DHA-containing glycerolipids is downregulated in retinas of animals with inherited retinal degenerations.
Phototransduction
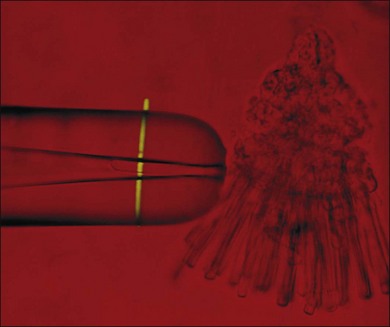
Visual transduction is initiated by the phototransduction cascade, which is perhaps the best characterized of all G-protein receptor-coupled signaling pathways. Our knowledge about phototransduction is relatively advanced because the components of this pathway are located selectively in the photoreceptor OS and can be isolated in quantities suitable for biochemical analysis. Photoreceptors can also be isolated easily for electrophysiological measurements, either as single cells using suction electrodes (Fig. 14.3), or en masse using field potentials called electoretinograms (ERG: see Chapter 7, Electrogenesis of the electroretinogram). The phototransduction cascade within rods is so robust that single-photon absorption by rhodopsin gives rise to a change in current that can be monitored by suction electrode recordings. Similarly, the remarkable ability of cone phototransduction to adapt to increases in background light intensity can also be monitored with suction electrodes. The combination of transgenic technology and electrophysiological analysis has greatly enhanced our understanding of signal transduction within these photoreceptor cells. In particular, transgenic technologies have allowed the introduction of targeted changes into specific components of the signaling pathway. These genetically altered intact photoreceptor cells can then be subjected to electrophysiological measurements, and their tissues can subsequently be used in biochemical or immunocytochemical assays to pinpoint the molecular mechanism behind the physiological phenotype. In this section we summarize the current state of our understanding of phototransduction in retinal photoreceptors.
Signal activation and amplification
A hallmark of rod phototransduction is its high sensitivity. Fully dark-adapted rods achieve sensitivity that reaches the theoretical maximum, the ability to detect individual quanta of light.1,2 This high sensitivity is generated through several stages of amplification, resulting in a tremendous increase in the signal gain (Fig. 14.4). This cascade of amplification begins at the light-activated G-protein-coupled receptor rhodopsin (R). Absorption of a photon by R leads to a conformation change (R*), allowing it to interact with the heterotrimeric G-protein transducin (Tα, β, and γ), thereby promoting the exchange of bound GDP for GTP. The complex then dissociates into Tα-GTP and Tβγ, and R* is free to activate many other transducin molecules during its catalytic lifetime.83 In the subsequent step, Tα-GTP binds and removes the inhibitory γ-subunit of the cGMP-PDE so that it can now hydrolyze cGMP to 5’-GMP; the enzymatic activity of PDE (PDE*) in turn degrades thousands of cGMP molecules. The decrease in cytoplasmic cGMP concentration then leads to closure of the cGMP-gated cation conductance channel in the rod plasma membrane, resulting in a reduction in the influx of ~ 1 000 000 Na+ ions.84 This is a graded effect: a slight lowering in the cGMP concentration leads to closure of some channels, while a large decrease will eventually lead to closure of all channels. In this instance, the rod cell is said to be saturated.
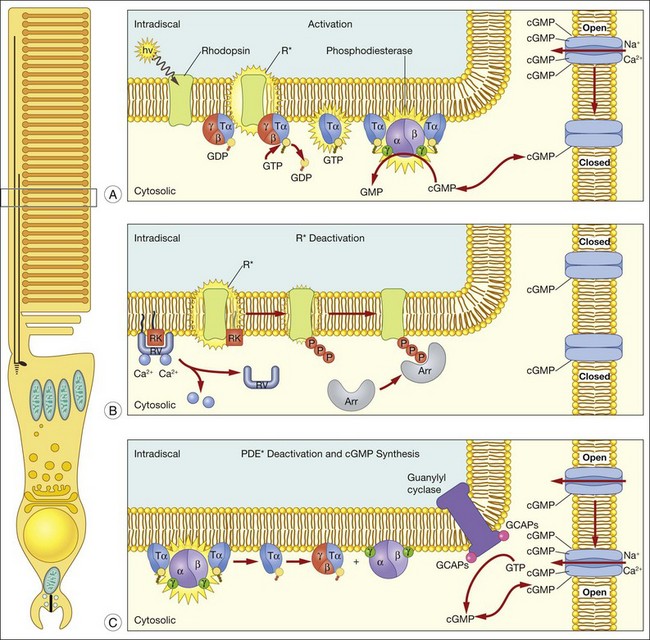
In darkness, glutamate, the transmitter used by both rods and cones, is released at a steady rate because the photoreceptor’s membrane potential is in a relatively depolarized state. The closure of CNG channels results in a graded hyperpolarization of the cell, leading to a decrease in glutamate release at the synapse. In this manner, the first sensation of light perception is transmitted from the photoreceptor cell to second-order cells of the retina, where signals are further processed and ultimately conveyed to the retinal ganglion cells (see Chapter 15, Functional anatomy of the mammalian retina).
Signal deactivation
A reversal of the activation steps is required ultimately for the photoreceptor to return to its resting state. First, the catalytically active components of the phototransduction cascade, R* and PDE*, must be quenched, then cGMP needs to be resynthesized (Fig. 14.4). These events must be rapid and reproducible for the photoreceptor to maintain its high sensitivity and respond to subsequent stimuli. Our current understanding of these processes is detailed below.
Quenching R*: phosphorylation and arrestin binding
Since the early 1980s, phosphorylation was recognized to play an important role in deactivation of R*.85,86 To determine how receptor deactivation occurs in vivo, transgenic mouse models were developed to assess the contribution of receptor phosphorylation to R* shutoff. In early experiments, a rhodopsin truncation mutation, S334ter, was expressed in the photoreceptors of transgenic mice17 that removed the terminal 15 amino acid residues and thus all putative Ser and Thr phosphorylation sites. Single-photon responses produced by S334ter R* failed to shut off in a timely and stereotyped manner, indicating that this domain is important for R* quenching. In the 1990s, numerous biochemical experiments suggested that specific Ser residues on the C-terminus are crucial in R* shutoff.87–89 However, single-cell recordings from transgenic mice rods expressing rhodopsin where these phosphorylation sites removed in different combinations showed that all of the Ser and Thr sites at the C-terminus are required for rapid and reproducible R* deactivation; the cluster of Ser and Thr sites at rhodopsin’s C-terminus functions to ensure fast and reproducible R* deactivation.90 It is clear that rhodopsin kinase is solely responsible for phosphorylating R*, since phosphorylation of R* does not occur in mice lacking rhodopsin kinase.91
Subsequent to C-terminus phosphorylation, the catalytic activity of R* is quenched fully by binding of visual arrestin. Arrestins are soluble cytoplasmic proteins that bind to G-protein-coupled receptors, thus switching off activation of the G-protein and terminating the signaling pathway that triggers the cellular response; the most commonly studied arrestin is β-arrestin. Visual arrestin exhibits exquisite specificity to phosphorylated R*. Our understanding of how this specificity is conferred emerges from extensive mutagenesis studies, and the crystal structure of visual arrestin.92–95 These studies reveal that arrestin is constrained into a latent, inactive structure by a network of intramolecular interactions.92 According to the current model of arrestin activation, these intramolecular constraints are released by: (1) interaction with multiple phosphates on rhodopsin’s C-terminus; and (2) interaction with the cytoplasmic loops of R*. The physiological role of visual arrestin in controlling the rod single-photon response was studied by Xu and colleagues,24 who created transgenic mice lacking visual arrestin. Suction electrode recordings of photoresponses from the OS of these transgenic rods displayed a rapid, partial recovery, followed by a prolonged final recovery phase. The timing of the slow phase of shutoff is consistent with the spontaneous decay of the R*. Thus, while R* phosphorylation is required to terminate rapidly its catalytic activity, the full quenching of R* activity requires visual arrestin binding.
Deactivating PDE: control of transducin’s GTPase activity
Quenching of the phototransduction cascade also requires the shutoff of PDE*, which occurs through the deactivation of Tα. The GTPase activity of Tα allows the reassociation of the inhibitory γ-subunit to the catalytic PDE subunits,96 a process which is speeded through the participation of a photoreceptor-specific RGS protein, RGS-9, and its binding partner, Gβ5.97 These proteins act sequentially on Tα to facilitate the hydrolysis of bound GTP. The physiological role of all these proteins in the shutoff of the rod photoresponse was evaluated with transgenic mice that had alterations in each of these components. Rod photoresponses from transgenic mice expressing a PDE-γ with an impaired ability to stimulate GTP hydrolysis98 showed abnormally slowed recovery, demonstrating the necessary role of PDE-γ in Tα deactivation.99 Similarly, mice lacking RGS-9 showed profoundly slowed response recovery,100 as do mice lacking Gβ5.101 RGS-9 is anchored to photoreceptor membranes by the adaptor protein, R9AP.102–104 Interestingly, human patients with mutations in R9AP have difficulties adapting to sudden changes in light levels that are mediated by cones,105 consistent with the higher concentration of RGS-9 complex in cones compared to rods.106 Together, the current data support an important role of PDE-γ, RGS-9/Gβ5, and R9AP in Tα deactivation, and consequently, response recovery, in both rods and cones.
Resynthesis of cGMP: Ca2+ dependence of guanylyl cyclase
Recovery of the dark current also requires that cGMP is resynthesized to allow CNG channels to open. cGMP is synthesized in photoreceptor OS by a retinal guanylyl cyclase (RetGC), which is regulated by the protein GCAPs (GCAP 1 and 2).107 GCAPs is an EF hand Ca2+-binding protein that confers on RetGC a highly cooperative Ca2+ dependence for cGMP synthesis.108 Near the dark resting Ca2+ concentration RetGC’s activity is inhibited, but as OS Ca2+ falls in response to illumination as CNG channels close, RetGC’s activity accelerates to restore the resting cGMP and reopen CNG channels. The physiological actions of GCAPs have been evaluated in physiological recordings. For instance, when GCAP1 is dialyzed into gecko rods, the recovery phase of the light response is accelerated.109 In addition, suction electrode recordings from rods of GCAPs knockout mice show that the Ca2+ dependence of cGMP synthesis is required to set the time course of the rod’s photoresponse, and to suppress noise within the phototransduction cascade.110
Light adaptation
In a normal cycle of day and night, the illumination at the Earth’s surface varies over 11 orders of magnitude, making photoreceptor adaptation fundamentally important to the normal functioning of the vertebrate visual system. Rods cells are able to adjust their sensitivity over 2–3 log units of light intensities, while cone cells exhibit no response saturation over 6–7 log units.3,4 The switch between rod and cone function from night to bright daylight covers a large portion of vision’s functional range, and downstream circuitry accounts for the remainder of the range extension. While the molecular events underlying the amplification cascade following photon capture by rhodopsin are well delineated, the mechanisms underlying photoreceptor adaptation are less well understood; however, Ca2+ is known to play a feedback role in the adaptation process.111,112
The role of Ca2+ feedback
In darkness, Na+ ions enter the OS through the CNG channels and are extruded in the IS through Na+/K+ ATPase pumps. This steady-state influx and efflux of Na+ is largely the basis for the circulating dark current in photoreceptor cells. Similarly, Ca2+ enters through CNG channels in the OS, but is extruded locally by NCKX exchangers. In mouse rods the steady-state influx and efflux of Ca2+ hold concentration near 250 nM,113 but as light closes CNG channels, Ca2+ entry is blocked, Ca2+ efflux continues, and [Ca2+]i falls. This change in [Ca2+]i is graded just like the dark current: the degree of lowered [Ca2+]i is proportional to the intensity of background light.
This decrease in [Ca2+]i triggers a feedback signal that is necessary for light adaptation, since, when the [Ca2+]i decrease is blocked, adaptation to background illumination is greatly compromised.114,115 Existing evidence suggests that this feedback is rather complex, as it orchestrates several pathways directed toward different components of the transduction machinery.116–118 Here we review the role of Ca2+ on each of these components.
Adaptation mediated by Ca2+ feedback to retinal guanylyl cyclase
It has long been recognized that feedback controlling accelerated recovery contributes to light adaptation.108 Recovery of the light response requires the resynthesis of cGMP, which is needed to reopen the CNG channels and re-establish the circulating current. The control of cGMP synthesis by RetGC/GCAPs is central to this process. The contribution of accelerated cGMP synthesis to adaptation in rod photoreceptors was investigated in GCAP knockout mice.23,110 This work showed that the presence of GCAPs extends the sensitivity to background light in rod photoreceptors ~10-fold (Fig. 14.5), and can account for about half of the rod’s capacity to adapt to background light. Recordings from cones in GCAPs knockout mice also show that GCAP contributes to background adaptation, but to a lesser extent than it does in mouse rods.119
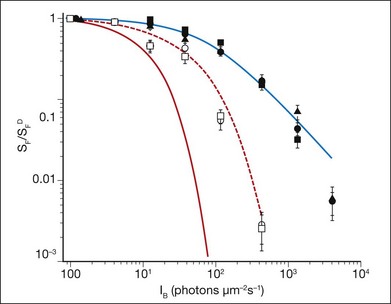
Fig. 14.5 Sensitivity as a function of background light was measured from mouse rod photoreceptors using suction electrodes (Fig. 14.3). Sensitivity was calculated from dim flash responses as the peak response amplitude divided by the flash strength. Flash sensitivity (SF) in the presence of increasing background light, background light (IB) was normalized against the flash sensitivity in darkness (SFD) and is plotted for the rods of several transgenic mice with particular elements of the phototransduction cascade altered. These mice include wild-type (•), mice lacking recoverin ( ), mice where the calmodulin-binding site on the cyclic nucleotide gated (CNG) channels has been eliminated (
), mice where the calmodulin-binding site on the cyclic nucleotide gated (CNG) channels has been eliminated ( ), mice lacking guanylyl cyclase-activating proteins 1 and 2 (
), mice lacking guanylyl cyclase-activating proteins 1 and 2 ( ), and mice where both the calmodulin-binding site on the CNG channel and guanylyl cyclase-activating proteins 1 and 2 have been eliminated (
), and mice where both the calmodulin-binding site on the CNG channel and guanylyl cyclase-activating proteins 1 and 2 have been eliminated ( ). The red line shows the predicted decline in sensitivity as a function of background light intensity if rods displayed no light adaptation. The blue line is best-fitting Weber–Fechner function for wild-type rods, and shows that light adaptation extends the sensitivity of rod photoreceptors to 100-fold higher background light levels. Rods from mice lacking recoverin, or the calmodulin-binding site on the CNG channels, show little deviation from wild-type rods, indicating that these proteins do not play a significant role in light adaptation. However, in mice lacking guanylyl cyclase-activating protein 1 and 2, light adaptation is impaired, but not absent, even when the calmodulin-binding site on the CNG channel is also eliminated (red dashed line). The remaining mechanisms that control light adaptation in rod photoreceptors remain unidentified.
). The red line shows the predicted decline in sensitivity as a function of background light intensity if rods displayed no light adaptation. The blue line is best-fitting Weber–Fechner function for wild-type rods, and shows that light adaptation extends the sensitivity of rod photoreceptors to 100-fold higher background light levels. Rods from mice lacking recoverin, or the calmodulin-binding site on the CNG channels, show little deviation from wild-type rods, indicating that these proteins do not play a significant role in light adaptation. However, in mice lacking guanylyl cyclase-activating protein 1 and 2, light adaptation is impaired, but not absent, even when the calmodulin-binding site on the CNG channel is also eliminated (red dashed line). The remaining mechanisms that control light adaptation in rod photoreceptors remain unidentified.
(Data replotted from Chen J, Woodruff ML, Wang T, et al. Channel modulation and the mechanism of light adaptation in mouse rods. J Neurosci 2010;30:16232–40.)
Recoverin and control of rhodopsin kinase
The accelerated response recovery that mediates light adaptation is also achieved through the speeded deactivation of R*. The first step in the quenching of R* activity is the phosphorylation of its C-terminus by rhodopsin kinase, a process that is believed to be regulated in a Ca2+-dependent manner by the protein recoverin. Recoverin is a highly conserved Ca2+-binding protein found in both rod and cone photoreceptors. In vitro, recoverin binds and inhibits rhodopsin kinase when it is Ca2+-bound, and accelerates rhodopsin kinase activity as Ca2+ is removed.120,121 Since [Ca2+]i is high in darkness, it has been suggested that, under dark-adapted conditions, recoverin prolongs the catalytic activity of R* by inhibiting rhodopsin phosphorylation. Early evidence in experiments that introduce recombinant recoverin into gecko and salamander rods indicated that recoverin prolongs the light response.122,123 However, recoverin’s effect was most prominent at Ca2+ concentrations outside the rod’s physiological range (>1 µM). The Ca2+ requirement for rhodopsin kinase inhibition by recoverin in reconstituted biochemical assays was similarly high.120 These results placed into doubt the role of recoverin in mediating light adaptation. Suction electrode recordings from the rods of transgenic mice lacking recoverin, however, reveal a response speeding.124 This speeding effect is exacerbated in rods that lack recoverin, but also have a reduced expression of rhodopsin kinase.125 Thus, in the physiological range of Ca2+ concentrations, recoverin can influence R* lifetime. Despite this effect on R* lifetime, recoverin appears to play a relatively minor role in rod light adaptation (Fig. 14.5).124 It remains to be seen in cone photoreceptors what is the efficacy of recoverin’s action. However, one might expect a greater contribution to adaptation as experiments from salamander L cones show that R* lifetime dominates the light response.126
Feedback regulation of the cGMP-gated channel
Another target of the Ca2+ feedback is the CNG channel itself. In vitro data have shown that the presence of Ca2+ calmodulin causes a decrease in the apparent affinity of the CNG channels for cGMP.127 This effect is reversed in light when [Ca2+]i is low, such that more channels tend to open despite the lowering of cGMP levels. However, the role of CNG channel modulation in intact rods remains controversial, because, based upon the measured effect of Ca2+ on the apparent affinity of the channel for cGMP and the intracellular range of cGMP concentration, it has been predicted theoretically that the channel contributes little to the overall adaptive behavior of the rod photoreceptor.117 The site that confers Ca2+ calmodulin sensitivity has been identified in the β-subunit of the CNG channel.128,129 Transgenic mice with this Ca2+-binding site disrupted show little influence on the light response or in the adaptive features of rod phototransduction,130 consistent with previous conjecture (Fig. 14.5). The role of CNG channel modulation in cones, however, may be more profound than for rods. For instance, the cones of the ground squirrel demonstrate strong Ca2+-dependent modulation of the CNG-gated current in the normal range of Ca2+ concentrations, while the rods of the same species do not.131 In addition, Ca2+ concentration varies over a larger dynamic range in cones than rods.132 Modulation of cone CNG channels may also involve an unidentified soluble factor instead of calmodulin.133 Ultimately how this modulation influences light adaptation in cones remains to be seen.
Other (Ca2+-independent) adaptation mechanisms: protein translocation
A mechanism operating at a longer timescale (minutes rather than seconds) that may contribute to regulating photoreceptor cell performance is the light-driven redistribution of specific signal transduction proteins between the compartments of the photoreceptor cell. The immunoreactivity of both Tα and visual arrestin is known to redistribute in rods in response to light.134–137 Visual arrestin immunostaining predominates at rod IS, as well as at the outer nuclear layer and outer plexiform layers of dark-adapted retinas, and shifts to rod OS upon light exposure. Tα immunoreactivity shifts in the opposite direction in response to light.136–138 Quantitative measurements show that bright light exposure is required to trigger translocation: for visual arrestin the threshold is 1000 rhodopsin excitations per rod per second, a light intensity at the upper limits of rod vision; for Tα at least 10 000 rhodopsin excitations are required.139 Sokolov and colleagues confirmed the physiological movement of Tα by combining serial tangential cryosectioning of the retina with Western blot analysis, and demonstrated that such movement may help to extend the range of light intensities in which the rods are able to operate.140 Similarly, visual arrestin movement may regulate rod performance under varying light environments.
Our current understanding of protein translocation is that it occurs by diffusion. In the case of Tα, the lipid modifications on the α- and γ-subunits act synergistically to anchor Tα GDP on the membrane. Upon activation, the subunits dissociate and become solubilized.141 In an elegant set of experiments, Lobanova and colleagues showed that light threshold can be shifted to either a lower or higher light intensity using mutant mice whose GTPase-activating (GAP) complex is enhanced or decreased.142 Their data suggest that, when the GAP complex is saturated, Tα GTP can escape the OS compartment and diffuse to the IS. Whether such a mechanism also governs cone translocation is uncertain. It has been observed that cones contain a higher concentration of GAP proteins,106,143 a reason that could explain a much higher threshold for cone Tα translocation.144 However, others have observed that cone Tα readily translocates.145 In the case of rod arrestin, its interaction with microtubules restricts its location to the IS in the dark. Light exposure creates its high-affinity binding partner, R*, which drives its movement towards the OS.146
It has become increasingly clear that Ca2+-dependent light adaptation results from the sum of multiple mechanisms that act at different sites in the phototransduction cascade. While biochemical experiments have identified the Ca2+-sensitive steps in the phototransduction cascade, a full understanding of how these steps contribute to visual adaptation requires a method that critically evaluates the timing, as well as the strength, of feedback modulation under physiological conditions. Given the complexity of Ca2+ feedback, coupled with the possibility that other adaptation mechanisms may overlay on this feedback regulation, visual adaptation, particularly in cones, remains one of the least understood areas in phototransduction. This complexity is being unraveled by the systematic removal of Ca2+ feedback pathways in vivo using mouse genetics, and by measuring the resultant change in adaptation with electrophysiological measurements. The molecular mechanism behind the observed phenotype can then be confirmed by biochemical experiments. Using this approach, the Ca2+ feedback loops can be evaluated independently, as well as in sum, since the mice can be bred to combine the individual components (Fig. 14.5). Eventually, as each and every Ca2+-dependent pathway is peeled away, the remaining (perhaps Ca2+-independent) pathways will be revealed.
Differences between rod and cone phototransduction
What factors underlie the specialized function of rods and cones? This remains a very important problem in phototransduction. Investigation of rod phototransduction has benefited from rod-dominated species where biochemical and physiological assays are robust. The relatively low number of cones in most mammalian species has made a biochemical analysis of cone phototransduction less fruitful, although the Nrl knockout mouse has proven useful for generating cone-dominant retinas.147 As described above, some quantitative differences may arise for differential modulation of components of the phototransduction cascade. In addition, differences in their characteristic morphology may play a role in setting the response properties (i.e., the continuously invaginating OS plasma membrane in cones provides greater surface area for ion exchange and chromophore recycling). Also important may be their signal transduction cascades: molecular cloning has revealed that phototransduction in the vertebrate rods and cones is regulated by structurally homologous but distinct groups of signaling proteins. Therefore, it is reasonable to assume that the phototransduction in rods and cones is qualitatively similar, and that quantitative differences in the transduction steps underlie the characteristic rod and cone behavior. For example, R* may activate more rod Tα molecules than cone opsin R* in activating cone Tα, resulting in a higher gain and a larger response amplitude.
To determine the roles of rod and cone phototransduction proteins in setting the response properties of the photoreceptor cells, transgenic technology has recently been used to express rod components in cones, and vice versa. For example, Kefalov and colleagues148 expressed an L-cone opsin in Xenopus rods, and rhodopsin in Xenopus cones. They surprisingly found that the activation of rhodopsin and L-cone opsin within the same cell yielded nearly identical photoresponses. In addition, similar photoresponses were observed in mouse rods transgenically expressing an S-cone pigment.149 These results collectively rule out the visual pigment from being a major contributor to rod–cone functional differences. Similar experiments have also addressed the role of Tα. Deng and colleagues150 showed no functional differences in mouse rods expressing cone Tα, or mouse cones expressing rod Tα. However, Chen and colleagues151 found to the contrary that complete exchange of rod Tα with cone Tα produced rod photoresponses that were 10-fold desensitized, and thus transducin might contribute partially to observed rod–cone functional differences. These results, as well as homolog substitution experiments with other components of phototransduction, require further evaluation to determine how differences in rod and cone photoresponses are established.
Inner segment and connecting cilia
Inner segment
The functional role of the IS can be appreciated by examination of its unique anatomy, which is highly specialized to fuel the high energy and protein synthesis requirements of the photoreceptor. The metabolic functions of the IS are constantly and highly active, generating most of the rod cell’s energy as well as being the primary site for synthesizing new proteins. The proximal portion of the IS, termed the myoid, contains the endoplasmic reticulum and Golgi apparatus, while the distal portion, termed the ellipsoid, is densely packed with elongated mitochondria that are aligned circumferentially along the axis of the rod cell (Fig. 14.2). Cones have a larger IS, perhaps reflecting their higher metabolic needs. This photoreceptor cell compartment is structured for high O2 consumption, as well as for glycolysis at very high rates. One major metabolic demand comes from the extrusion of Na+ from the cytoplasm by the Na+/K+ pump at the IS plasma membrane, which balances the influx of Na+ through the cGMP-gated channels at the OS. In darkness, these pumps operate at a very high rate to accommodate the high fluxes generated by many open channels in the OS, setting up the “dark” circulating current.152 The rate that these pumps can operate is related to the supply of ATP153 generated by the mitochondria in the ellipsoid. This ATP synthesis rate is coupled with the O2 delivery to the IS, which is completely supplied by the choroidal circulation.
Another major metabolic need is a high rate of protein synthesis that occurs in the myoid region of the IS to meet the demand of high levels of phototransduction proteins at the OS. In particular, each rod contains ~5 × 107 rhodopsin molecules, of which 10% at the tip of the OS are phagocytosed by the RPE, beginning at the onset of light each day. To maintain homeostasis, ~5 × 106 molecules are synthesized daily and added to the base of the OS. Similar events occur in cones, although they express about half the amount of visual pigment as rods. As with other cells, the soluble and peripheral membrane proteins are made in the cytosol, while the transmembrane proteins are made in the endoplasmic reticulum. To facilitate the speed of molecular collisions, many of the phototransduction proteins are associated with the membrane through lipid modifications and protein–protein interactions. These interactions are dynamic, with functional complexes forming and dissociating on the membrane surface on a rapid timescale. For example, the rod Tα is heterogeneously acylated at the amino-terminal glycine residue while the γ-subunit is carboxyl-methylated and prenylated at the cysteine residue. PDE6α and rhodopsin kinase are farnesylated, while PDE6β is geranylgeranylated.154,155 In some instances, the lipid modification is exposed as a consequence of a change in protein structure. An example is recoverin that, upon binding calcium, exposes its amino-terminal myristoyl group.156 GCAP1 and GCAP2 are also calcium-binding proteins that are myristoylated, but exposure of their lipid group is not regulated by calcium binding. These proteins are complexed with RetGCs and regulate their activity in a calcium-dependent manner.
Targeting of phototransduction proteins from the inner segment to the outer segment
How phototransduction proteins traffic from the IS, the site of synthesis, to the OS, the site of phototransduction, is an area of active research. This question is made more important by the fact that defective trafficking can cause photoreceptor cell death. For example, a cluster of naturally occurring mutations on rhodopsin’s carboxyl-terminus causes autosomal dominant RP in human patients.157 These mutations do not affect the biochemical properties of rhodopsin with respect to its ability to bind 11-cis retinal and form a visual pigment, nor its ability to activate Tα following photon absorption in reconstituted systems.158–160 The underlying defect remained a mystery until these mutants were expressed in transgenic mice, whereupon it was observed that they are mislocalized throughout the cell and caused retinal degeneration.161–163 It is now known that the VxPx motif at the rhodopsin’s carboxyl terminus is important for sorting of rhodopsin into transport carriers from the trans-Golgi network towards the connecting cilium en route to the OS.164 This is a recognition site for transport proteins that include the small GTPase Arf4, ASAP1, Rab11, and FIP3.165 The VxPx motif is also present in cone opsins. Other integral membrane proteins contain their own trafficking motif. These motifs have been identified in peripherin/rds166 and the cGMP-gated channel in rods.167 Often defects in transport of one transmembrane protein do not affect transport of others, suggesting that transport occurs independently.
Trafficking of membrane-associated proteins to the OS may occur through their interaction with transmembrane proteins as they are delivered out of the trans-Golgi network. It was also recently discovered that membrane-associated phototransduction proteins are transported by prenyl- or acyl-chain-binding proteins. For example, rhodopsin kinase and cone PDE6α’ require PrBP/δ, which contains a binding site for prenyl group, for transport to the OS,168 while the α-subunit of rod transducin requires UNC119, which binds acyl chains.169 Thus diverse mechanisms are responsible for correct targeting of phototransduction proteins from the IS to the OS compartment.
The connecting cilium
A slender nonmotile connecting cilium that is 0.3 µm in diameter connects the IS to the OS. The microtubule-based axoneme of the connecting cilium lacks the central microtubule pair of the motile cilium, and therefore has a 9 × 2 + 0 microtubule structure towards the base of the cilia. The doublet microtubules transition to a 9 × 1 + 0 singlet microtubule arrangement more distally from the basal body and continue through much of the OS (Fig. 14.2). The axoneme arises from basal bodies which, together with the centrioles, act as microtubule organizing centers of the photoreceptor cell. It is the site of tremendous vectorial flow of lipids and proteins from their site of synthesis in the IS to the OS. It has been estimated that 2000 opsin molecules are transported every minute from the IS to the OS.170 As post-Golgi carrier vesicles are delivered to the connecting cilium, proteins destined for the OS need to be sorted between the disc lamellar region, the disc rim region, and the plasma membrane in the OS. How the protein cargo is sorted at this point is not understood fully, but is thought to involve recognition of the specific trafficking motif displayed on the protein cargo by their cognate transport complex that also includes the intraflagellar transport (IFT) particles.170 The IFT particles associate with the post-Golgi carrier vesicles at the base of the cilium and transport them along the connecting cilium to the OS. IFTs are large protein complexes that move along the axoneme by motor proteins and may be viewed as conveyer belts that deliver different cargos to their respective destination.
Actin and myosin, two proteins common to many cells in the body, are also present in photoreceptors. Both myosin and actin filaments are localized to the photoreceptor connecting cilium and IS/OS. Disruption of either of these proteins causes retinal degeneration.171 Actin depolymerization promotes disc overgrowth and photoreceptor death,172 and myosin mutants appear to have defects in the transport of molecules from the IS to the OS. Myosin defects cause numerous diseases, including Usher syndrome, that affect both hearing and vision.173–175
Other proteins are receiving increased attention because of their association with the connecting cilium and human retinal disease. First described nearly two decades ago, one such protein, RP1, is responsible for 5–10% of all RP cases.176 RP1 is a large protein localized to the connecting cilium in both rods and cones and is especially concentrated at the site of nascent OS discs. RP1 has recently been shown to bind microtubules, suggesting that this interaction may stabilize microtubules in the axoneme.177–179 An RP1 knockout mouse model shows that discs are misshapen, overgrown, and fail to align properly.177 This phenotype correlates well with ERG abnormalities in RP1 patients.
The RP GTPase regulator (RPGR) is a GTPase regulator found in rods. Mutations in RPGR180 cause X-linked RP3, a severe and progressive retinal dystrophy. Antibodies localize RPGR and a related protein, RPGRIP (RPGR interacting protein) throughout the human rod and cone OS,181 as well as in amacrine cells. RPGR is found in connecting cilia of rods and cones and in the cilia of airway epithelia.182 An RPGR gene knockout mouse (an animal model of RP3)183 demonstrated mislocalization of opsin in cones, and reduced quantities of rhodopsin in rods prior to photoreceptor degeneration. These data suggest a role for RPGR in maintaining the polarized distribution of proteins between the IS and the OS through the connecting cilium.
Centrins are calcium-binding proteins associated with centrosome-related structures. The mammalian rods and cones express four centrin isoforms; three of these (Cen1p–3p) are in the connecting cilium and three (Cen2p-4p) are in the basal body.184 These centrins may participate in the alignment of the photoreceptor OS. Inasmuch as centrins were shown to interact with transducin in a calcium-dependent manner, they have been proposed to regulate the light-dependent translocation of Tα from the OS to the IS.184
The ciliary rootlet, first recognized over a century ago, is a prominent structure originating from the basal body at the proximal end of a cilium. Rootlets appear to be particularly robust in retinal photoreceptors, extending from the basal bodies to the synaptic terminals, anchoring endoplasmic reticulum membranes along their length. Recent studies indicate that rootlets are composed of a structural protein, aptly named rootletin. Rootletin protofilaments are bundled into variably shaped thick filaments within the cilium.185
Additional proteins, such as TULP1,186,187 cadherin,188 and myocilin,189 are associated with the connecting cilium: several are known to be associated with rod photoreceptor disease. It suggests, not surprisingly, that the highly specialized connecting cilium is a complicated structure connecting the IS to OS, and that there will undoubtedly be additional proteins associated with this unique subcellular structure.
Photoreceptor synaptic terminal
The light response generated by the phototransduction cascade in the OS is passively relayed to the synaptic terminal, called a spherule for rods and a pedicle for cones, where it modulates the rate of glutamate release on to second-order retinal neurons (Fig. 14.6). Since open CNG channels leave the membrane potential of photoreceptors relatively depolarized in darkness, these synapses support a high level of Ca2+ influx and thus a high level of glutamate release. Light activates the phototransduction cascade, leading to the closure of CNG channels, the hyperpolarization of the membrane potential, and a reduction in Ca2+ influx, which in turn reduces glutamate release. This reduction in glutamate release constitutes the “light response” that is passed to the retinal circuitry.
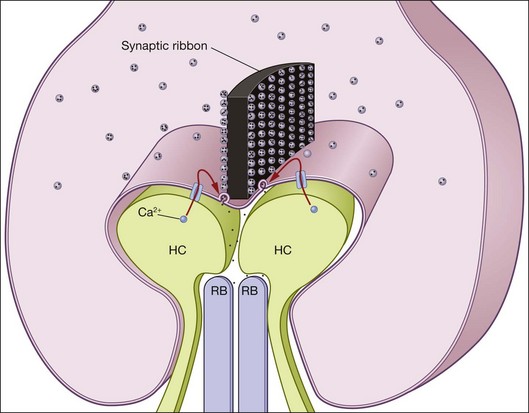
To allow high levels of glutamate release, the photoreceptor synaptic terminals house a specialized synaptic structure, called a ribbon, which is composed of the protein ribeye.190 The ribbon tethers glutamatergic vesicles and is believed to act like a conveyor belt to bring these vesicles to the active zone, where they promote vesicle priming and fusion with the plasma membrane to release glutamate into the synaptic cleft191,192 (Fig. 14.6). While the spherules of rods contain only one ribbon (i.e., one release site) in mammalian species,193 the cone pedicles contain many ribbons whose numbers vary by species. In mice, for instance, cone pedicles contain ~10 ribbons,194 while pedicles in other species like ground squirrels may contain ~15 or more ribbons.195 Primate cone pedicles may also contain in excess of 25 ribbons.196,197 Multiple ribbons in each cone pedicle allow the output of the cone to be relayed to many classes of cone bipolar cells (see Chapter 15, Functional anatomy of the mammalian retina).
Compared to conventional central synapses, photoreceptor cells release glutamate in the absence of the stimulus. Such a configuration places fundamental constraints on the mechanisms that regulate glutamate release. For instance, the persistent depolarization of photoreceptors in darkness would cause many types of ion channels to desensitize. To allow robust control of glutamate release with changes in membrane potential, a Cav1.4 channel located close to the active zone in the rod spherule and cone pedicle allows the Ca2+ influx that regulates release. These L-type Ca2+ channels display little voltage or Ca2+-dependent desensitization,198,199 and thus are ideal to support continuous glutamate release in darkness. In addition, in rod spherules these channels associate with the modulatory Ca2+-binding protein, CABP4.200 The association of Cav1.4 with CABP4 shifts the voltage sensitivity of Cav1.4 to more hyperpolarized membrane potentials.200,201 This allows the gating of Cav1.4 channels to be modulated robustly over the rod photoreceptor’s physiological voltage range.
Additional control of glutamate release may also involve the interaction of other proteins with the synaptic machinery. For instance, visual arrestin serves to quench fully phosphorylated rhodopsin in the rod OS, but has been shown to interact with N-ethylmaleimide-sensitive factor at the rod spherule to increase the turnover rate of SNARE complexes.202 Similarly, the protein recoverin, which serves as a modulator of rhodopsin phosphorylation in the rod OS, has a separate action in the rod spherule that increases the magnitude of the reduction in glutamate release on to rod bipolar cells.203 Such modulation may also be controlled by the light-dependent translocation of these components,135,204 or in the activity of signaling cascades initiated by metabotropic glutamate receptors in the photoreceptor terminal.205,206 Finally, the control of Ca2+ homeostasis in photoreceptor terminals can also modulate glutamate release.207–209
It should be noted that most of our understanding of the structure and function of ribbon synapses has emerged from the study of rod photoreceptors in the amphibian retina, particularly the salamander. However, the characteristics of glutamate release in cones can also be studied in this same species and reveal characteristics that are tuned for the faster kinetics of the light response in cones (reviewed by Thoreson210). Furthermore, since the cone photocurrent is difficult to saturate, increased vesicle turnover and recruitment are required to maintain the high level of glutamate release.211,212 Ultimately, the mechanisms that produce these functional distinctions between rod spherules and cone pedicles remain largely unidentified and are currently under investigation.
Photoreceptor dysfunction and disease
Rhodopsin mutations
More than 100 different mutations of rhodopsin have been associated with autosomal dominant and recessive RP (adRP and arRP) and autosomal dominant congenital stationary night blindness.213 This phenotypic heterogeneity points to diverse disease mechanisms arising from the same gene product. In the case of adRP, it has been proposed that some mutations lead to misfolding of the opsin protein, which then accumulates in the endoplasmic reticulum and elicits the unfolded protein response.214 The accumulation of misfolded opsin may also interfere with the sorting and trafficking of normal proteins destined for the OS or other parts of the cell.215 This may result in a pathologic mechanism where the cell cannot replenish other required proteins that do not harbor mutations.
Other rhodopsin mutations appear to affect trafficking of rhodopsin from the IS to the OS. As mentioned previously, mutations involving the last five carboxyl amino acids, such as P347A, P347S or S344ter, allow rhodopsin to appear to fold normally and to function normally in terms of Tα activation and deactivation by phosphorylation and visual arrestin binding. Yet, mutations in these residues cause adRP. Several lines of evidence suggest that the C-terminus of opsin contains a signal for vectorial transport of newly synthesized opsin toward the OS. In a retinal cell-free system, it was demonstrated that a monoclonal antibody to the C-terminal domain of rhodopsin inhibited post-Golgi vesicle formation and arrested newly synthesized rhodopsin in the trans-Golgi network.216 A wild-type C-terminal peptide, but not a mutant peptide bearing the mutation at the P347 position, can also block this process.217 Another line of evidence comes from an in vivo model where a truncation mutant, S344ter, was expressed in the photoreceptors of transgenic mice. Antibodies that specifically recognize the truncated opsin showed strong immunoreactivity in the IS, indicating a block in the transport to the OS.163 However, some truncated opsins are also cotransported with the full-length protein to the OS. The role of the rhodopsin C-terminal domain in vectorial transport was also investigated in MDCK cells.218 This cell line exhibits polarized structure when it is grown on a porous support. When transfected into this cell line, wild-type opsin was transported to the apical membrane surface, whereas an opsin mutant lacking the terminal 32 amino acids was not.
Constitutive phototransduction and retinal disease
It is thought that constitutive phototransduction causes some forms of retinal disorders. Unabated signal flow can arise from different steps in the visual cascade. For example, certain mutations in rhodopsin, particularly those that affect the salt bridge between Lys-296 and Glu-113 (such as A292E and G90D), can lead to constitutive activity.219 The interaction between Lys-296 and Glu-113 constrains the chromophore-free opsin to an inactive conformation.220 Disruption of this bond leads to an opsin conformation that can support Tα activation. This activity is suppressed when the mutant opsin is regenerated with the chromophore 11-cis retinal. In the case of G90D and A292E mutations, light exposure leads to formation of opsin that can still activate the visual cascade. This can lead to impairment of visual function because the transduction machinery is overwhelmed by this constitutive activity. Patients carrying these mutations suffer from night blindness because phototransduction in rods cannot be fully quenched.221,222 However, in the instance of the rhodopsin K296E mutation, no evidence of constitutive activity was observed when it was expressed in rod photoreceptors of transgenic mice; instead, it was found bound to arrestin.223 It was found subsequently that stable rhodopsin–arrestin complex is toxic, and rod photoreceptor survival can be prolonged if such complex is prevented in transgenic mice that lack visual arrestin and Tα.224
Defective shutoff can also be caused by the lack of visual arrestin or rhodopsin kinase. These genes are mutated in patients diagnosed with the recessive genetic disorder Oguchi disease,225,226 a nonprogressive disorder characterized by early onset and stationary night blindness. These mutations are thought to cause a loss of function in the visual arrestin or rhodopsin kinase gene. Because of the inability to shut off signal flow, the rod photoreceptor becomes saturated even at very low light levels, leading to night blindness. Interestingly, some patients with the visual arrestin gene mutation also suffer from RP, pointing to a potential causal role between constitutive signal flow and photoreceptor cell death. Using the visual arrestin knockout mouse model, Chen et al. demonstrated that the absence of visual arrestin in the rod photoreceptor does lower the threshold for light damage.227 Therefore, protection from light exposure is an important preventive measure to prolong photoreceptor cell survival in those instances where constitutive signal flow is the disease mechanism.
Transducin defects and retinal disease
Transducin is composed of three subunits: α, β, and γ. Extensive genetic screening has revealed mutations only in the rod Tα.228 The associated disease is autosomal dominant congenital stationary night blindness. The mutation is in the position homologous to that affected by the oncogenic mutation in p21ras, a small G-protein. It is therefore hypothesized that this mutation may affect Tα deactivation, leading to the night blindness phenotype. Interestingly, mice that lack rod Tα have normal retinal structure, indicating that this abundant visual G-protein has no structural function in the photoreceptor, and that signal flow is not a prerequisite for rod photoreceptor cell survival.20
cGMP and photoreceptor cell physiology
One of the first characterized mutations affecting components in the phototransduction cascade is the null mutation in the β-subunit of PDE6, leading to the rapid retinal degeneration phenotype in the rd mouse.13,14 Subsequently, null mutations in PDE6β have been found in patients diagnosed with arRP.229 The complete PDE6 enzyme is made up of two catalytic chains (α and β), each associated with an inhibitory chain (γ). Since both α- and β-subunits are necessary for the formation of a functional PDE holoenzyme, the mutation in the γ-subunit renders the holoenzyme inactive. Farber and Lolley230 showed that photoreceptor cell death is preceded by an accumulation of cGMP in the retina, which is consistent with the role of this enzyme in degrading cGMP. Another study investigating the relationship between cGMP and photoreceptor cell death showed that external application of cGMP on to cultured eyecups resulted in rapid photoreceptor cell death.231 However, the relevance of this finding is uncertain because the site of cGMP accumulation and action is intracellular. For example, the CNG channels open only when cGMP is dialyzed inside the truncated OS, not when it is applied externally.232
Although PDE6 is responsible for hydrolyzing cGMP, RetGCs are responsible for synthesizing the cGMP that is necessary for reopening of the CNG channel leading to recovery of the dark current. Two membrane forms of RetGC are expressed specifically in the retina of several mammalian species, including rat,233 human,234,235 and bovine.236 Whereas other membrane cyclases are activated by the binding of peptide ligands to their extracellular domains, the activity of RetGCs is controlled by interaction with GCAPs through their intracellular domain.237,238 Because the extracellular domain of RetGCs is situated in the enclosed and seemingly inaccessible intradiscal space, it is questionable whether the extracellular domain serves as a receptor. The retinal cyclases are therefore termed “orphan” receptors because no ligands have yet been identified. Although two forms of RetGC are expressed in the photoreceptor layer, there is evidence that they are not functionally redundant. First, they appear to be expressed at different levels in different photoreceptors. For example, it was observed that RetGC1 immunoreactivity is stronger in cone OS than in rod OS239; RetGC2, on the other hand, is expressed in the rod OS,240 but evidence for cone expression is lacking. Second, RetGC1 and RetGC2 behave differently biochemically in terms of their regulation by GCAPs: RetGC1 is regulated by both GCAP1 and GCAP2, whereas RetGC2 is regulated by GCAP2 but not by GCAP1.234,237–239,241 Third, loss of function of RetGC1 appears to affect cones more than rods; a mouse knockout of RetGC1 lacked cone function altogether, but rod function was only reduced.242 The importance of RetGC1 in cones is also reflected in the rd chicken model where a loss of RetGC1 function is thought to cause retinal degeneration in this cone-dominant animal.243 These animal models will be valuable for evaluating the pathophysiology for Leber’s congenital amaurosis, an early-onset blindness caused by a naturally occurring mutation in RetGC1.
Another naturally occurring mutation that may influence intracellular cGMP concentration is the Y99C mutation in GCAP1, a mutation that is thought to cause cone dystrophy in an autosomal dominant manner.244 This mutation renders GCAP1 capable of stimulating RetGC, independently of Ca2+ concentration.245,246 Constitutive activity of RetGC may therefore lead to an accumulation of intracellular cGMP.
It is known that a small increase in the level of free cGMP can profoundly affect the open probability of CNG channels, such that a greater number of channels will be open. This will in turn influence the ion influx, including Ca2+, which would then directly and/or indirectly influence the physiology of the photoreceptor cell, leading to cell death. In support of this idea, survival of rd1 photoreceptors, whose mutation in PDE6β leads to buildup of cGMP, was enhanced when the mutation was put into a CNG channel knockout mouse background.247 This observation motivates effort to synthesize small molecules that specifically block the CNG channel as a means of prolonging photoreceptor cell survival.248 A loss of RetGC1, on the other hand, is expected to lead to lowered cGMP levels. This might be functionally equivalent to constant light and may lead to cell death. However, further experiments will be needed to evaluate rigorously the relationship between cGMP concentration and retinal degeneration.
1 Baylor DA, Lamb TD, Yau KW. Responses of retinal rods to single photons. J Physiol. 1979;288:613–634.
2 Hecht S, Schlaer S, Pirenne MH. Energy, quanta, and vision. J Gen Physiol. 1942;25:819–840.
3 Burkhardt DA. Light adaptation and photopigment bleaching in cone photoreceptors in situ in the retina of the turtle. J Neurosci. 1994;14:1091–1105.
4 Normann RA, Perlman I. The effects of background illumination on the photoresponses of red and green cones. J Physiol. 1979;286:491–507.
5 Morimura H, Fishman GA, Grover SA, et al. Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or Leber congenital amaurosis. Proc Natl Acad Sci U S A. 1998;95:3088–3093.
6 Daiger SP. Identifying retinal disease genes: how far have we come, how far do we have to go? Novartis Found Symp. 2004;255:17–27. discussion 36, 177–8
7 Sheffield VC, Fishman GA, Beck JS, et al. Identification of novel rhodopsin mutations associated with retinitis pigmentosa by GC-clamped denaturing gradient gel electrophoresis. Am J Hum Genet. 1991;49:699–706.
8 Flannery JG, Farber DB, Bird AC, et al. Degenerative changes in a retina affected with autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1989;30:191–211.
9 Suber ML, Pittler SJ, Qin N, et al. Irish setter dogs affected with rod/cone dysplasia contain a nonsense mutation in the rod cGMP phosphodiesterase beta-subunit gene. Proc Natl Acad Sci U S A. 1993;90:3968–3972.
10 Wrigstad A, Narfstrom K, Nilsson SE. Slowly progressive changes of the retina and retinal pigment epithelium in Briard dogs with hereditary retinal dystrophy. A morphological study. Doc Ophthalmol. 1994;87:337–354.
11 Narfstrom K. Hereditary progressive retinal atrophy in the Abyssinian cat. J Hered. 1983;74:273–276.
12 Edwards RB, Szamier RB. Defective phagocytosis of isolated rod outer segments by RCS rat retinal pigment epithelium in culture. Science. 1977;197:1001–1003.
13 Bowes C, Li T, Danciger M, et al. Retinal degeneration in the rd mouse is caused by a defect in the beta subunit of rod cGMP-phosphodiesterase. Nature. 1990;347:677–680.
14 Pittler SJ, Baehr W. Identification of a nonsense mutation in the rod photoreceptor cGMP phosphodiesterase beta-subunit gene of the rd mouse. Proc Natl Acad Sci U S A. 1991;88:8322–8326.
15 Connell G, Bascom R, Molday L, et al. Photoreceptor peripherin is the normal product of the gene responsible for retinal degeneration in the rds mouse. Proc Natl Acad Sci U S A. 1991;88:723–726.
16 Travis GH, Brennan MB, Danielson PE, et al. Identification of a photoreceptor-specific mRNA encoded by the gene responsible for retinal degeneration slow (rds). Nature. 1989;338:70–73.
17 Chen J, Makino CL, Peachey NS, et al. Mechanisms of rhodopsin inactivation in vivo as revealed by a COOH-terminal truncation mutant. Science. 1995;267:374–377.
18 Lem J, Flannery JG, Li T, et al. Retinal degeneration is rescued in transgenic rd mice by expression of the cGMP phosphodiesterase beta subunit. Proc Natl Acad Sci U S A. 1992;89:4422–4426.
19 Travis GH, Groshan KR, Lloyd M, et al. Complete rescue of photoreceptor dysplasia and degeneration in transgenic retinal degeneration slow (rds) mice. Neuron. 1992;9:113–119.
20 Calvert PD, Krasnoperova NV, Lyubarsky AL, et al. Phototransduction in transgenic mice after targeted deletion of the rod transducin alpha-subunit. Proc Natl Acad Sci U S A. 2000;97:13913–13918.
21 Humphries MM, Rancourt D, Farrar GJ, et al. Retinopathy induced in mice by targeted disruption of the rhodopsin gene. Nat Genet. 1997;15:216–219.
22 Lem J, Krasnoperova NV, Calvert PD, et al. Morphological, physiological, and biochemical changes in rhodopsin knockout mice. Proc Natl Acad Sci U S A. 1999;96:736–741.
23 Mendez A, Burns ME, Sokal I, et al. Role of guanylate cyclase-activating proteins (GCAPs) in setting the flash sensitivity of rod photoreceptors. Proc Natl Acad Sci U S A. 2001;98:9948–9953.
24 Xu J, Dodd RL, Makino CL, et al. Prolonged photoresponses in transgenic mouse rods lacking arrestin. Nature. 1997;389:505–509.
25 von Greeff R. Mikroskopische Anatomie des Sehnerven und der Netzhaut. In: Graefe u. Saemisch Handbuch d. Ges. Augenheilkunde, 2. Aufl., 1, Teil, bd 1, Kap. V. Leipzig: 1900.
26 von Greeff R. A guide to the microscopic examination of the eye. Philadelphia: Blackiston; 1902.
27 Hendrickson A, Drucker D. The development of parafoveal and mid-peripheral human retina. Behav Brain Res. 1992;49:21–31.
28 Detwiler SR. Vertebrate photoreceptors. New York: Macmillan; 1943.
29 Liang Y, Fotiadis D, Maeda T, et al. Rhodopsin signaling and organization in heterozygote rhodopsin knockout mice. J Biol Chem. 2004;279:48189–48196.
30 Nickell S, Park PS, Baumeister W, et al. Three-dimensional architecture of murine rod outer segments determined by cryoelectron tomography. J Cell Biol. 2007;177:917–925.
31 Poo M, Cone RA. Lateral diffusion of rhodopsin in the photoreceptor membrane. Nature. 1974;247:438–441.
32 Liang Y, Fotiadis D, Filipek S, et al. Organization of the G protein-coupled receptors rhodopsin and opsin in native membranes. J Biol Chem. 2003;278:21655–21662.
33 Bayburt TH, Vishnivetskiy SA, McLean MA, et al. Monomeric rhodopsin is sufficient for normal rhodopsin kinase (GRK1) phosphorylation and arrestin-1 binding. J Biol Chem. 2011;286:1420–1428.
34 Arikawa K, Molday LL, Molday RS, et al. Localization of peripherin/rds in the disk membranes of cone and rod photoreceptors: relationship to disk membrane morphogenesis and retinal degeneration. J Cell Biol. 1992;116:659–667.
35 Travis GH, Sutcliffe JG, Bok D. The retinal degeneration slow (rds) gene product is a photoreceptor disc membrane-associated glycoprotein. Neuron. 1991;6:61–70.
36 Conley SM, Stuck MW, Naash MI. Structural and functional relationships between photoreceptor tetraspanins and other superfamily members. Cell Mol Life Sci. 2012;69:1035–1047.
37 Bascom RA, Manara S, Collins L, et al. Cloning of the cDNA for a novel photoreceptor membrane protein (rom-1) identifies a disk rim protein family implicated in human retinopathies. Neuron. 1992;8:1171–1184.
38 Molday RS, Warren R, Loewen C, et al. Cyclic GMP-gated channel and peripherin/rds-rom-1 complex of rod cells. Novartis Found Symp. 1999;224:249–261. discussion 61–4
39 McNally N, Kenna PF, Rancourt D, et al. Murine model of autosomal dominant retinitis pigmentosa generated by targeted deletion at codon 307 of the rds-peripherin gene. Hum Mol Genet. 2002;11:1005–1016.
40 Clarke G, Goldberg AF, Vidgen D, et al. Rom-1 is required for rod photoreceptor viability and the regulation of disk morphogenesis. Nat Genet. 2000;25:67–73.
41 Corless JM, Schneider TG. Patterns of interdisk connections within the lamellar domains of retinal rod outer segment disks: observations relevant to the axial propagation of incisures. Exp Eye Res. 1987;45:883–905.
42 Kajimura N, Harada Y, Usukura J. High-resolution freeze-etching replica images of the disk and the plasma membrane surfaces in purified bovine rod outer segments. J Electron Microsc (Tokyo). 2000;49:691–697.
43 Miyaguchi K, Kuo CH, Miki N, et al. Topography of opsin within disk and plasma membranes revealed by a rapid-freeze deep-etch technique. J Neurocytol. 1992;21:807–819.
44 Roof DJ, Korenbrot JI, Heuser JE. Surfaces of rod photoreceptor disk membranes: light-activated enzymes. J Cell Biol. 1982;95:501–509.
45 Allikmets R, Gerrard B, Hutchinson A, et al. Characterization of the human ABC superfamily: isolation and mapping of 21 new genes using the expressed sequence tags database. Hum Mol Genet. 1996;5:1649–1655.
46 Allikmets R, Shroyer NF, Singh N, et al. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science. 1997;277:1805–1807.
47 Allikmets R, Singh N, Sun H, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15:236–246.
48 Weng J, Mata NL, Azarian SM, et al. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt’s disease from the phenotype in abcr knockout mice. Cell. 1999;98:13–23.
49 Molday LL, Rabin AR, Molday RS. ABCR expression in foveal cone photoreceptors and its role in Stargardt macular dystrophy. Nat Genet. 2000;25:257–258.
50 Radu RA, Mata NL, Nusinowitz S, et al. Treatment with isotretinoin inhibits lipofuscin accumulation in a mouse model of recessive Stargardt’s macular degeneration. Proc Natl Acad Sci U S A. 2003;100:4742–4747.
51 Sanyal S, De Ruiter A, Hawkins RK. Development and degeneration of retina in rds mutant mice: light microscopy. J Comp Neurol. 1980;194:193–207.
52 Sanyal S, Jansen HG. Absence of receptor outer segments in the retina of rds mutant mice. Neurosci Lett. 1981;21:23–26.
53 van Nie R, Ivanyi D, Demant P. A new H-2-linked mutation, rds, causing retinal degeneration in the mouse. Tissue Antigens. 1978;12:106–108.
54 Young RW. The renewal of rod and cone outer segments in the rhesus monkey. J Cell Biol. 1971;49:303–318.
55 Young RW. Visual cells and the concept of renewal. Invest Ophthalmol Vis Sci. 1976;15:700–725.
56 Travis GH. Mechanisms of cell death in the inherited retinal degenerations. Am J Hum Genet. 1998;62:503–508.
57 Anderson DH, Fisher SK, Steinberg RH. Mammalian cones: disc shedding, phagocytosis, and renewal. Invest Ophthalmol Vis Sci. 1978;17:117–133.
58 Steinberg RH, Fisher SK, Anderson DH. Disc morphogenesis in vertebrate photoreceptors. J Comp Neurol. 1980;190:501–508.
59 Chuang JZ, Zhao Y, Sung CH. SARA-regulated vesicular targeting underlies formation of the light-sensing organelle in mammalian rods. Cell. 2007;130:535–547.
60 Nakatani K, Yau KW. Calcium and magnesium fluxes across the plasma membrane of the toad rod outer segment. J Physiol. 1988;395:695–729.
61 Perry RJ, McNaughton PA. Response properties of cones from the retina of the tiger salamander. J Physiol. 1991;433:561–587.
62 Picones A, Korenbrot JI. Permeability and interaction of Ca2+ with cGMP-gated ion channels differ in retinal rod and cone photoreceptors. Biophys J. 1995;69:120–127.
63 Pugh EN, Jr., Lamb TD. Amplification and kinetics of the activation steps in phototransduction. Biochim Biophys Acta. 1993;1141:111–149.
64 Nakatani K, Yau KW. Guanosine 3’,5’-cyclic monophosphate-activated conductance studied in a truncated rod outer segment of the toad. J Physiol. 1988;395:731–753.
65 Ruiz ML, Karpen JW. Single cyclic nucleotide-gated channels locked in different ligand-bound states. Nature. 1997;389:389–392.
66 Yau KW, Nakatani K. Electrogenic Na-Ca exchange in retinal rod outer segment. Nature. 1984;311:661–663.
67 Kaupp UB, Niidome T, Tanabe T, et al. Primary structure and functional expression from complementary DNA of the rod photoreceptor cyclic GMP-gated channel. Nature. 1989;342:762–766.
68 Pittler SJ, Lee AK, Altherr MR, et al. Primary structure and chromosomal localization of human and mouse rod photoreceptor cGMP-gated cation channel. J Biol Chem. 1992;267:6257–6262.
69 Zheng J, Trudeau MC, Zagotta WN. Rod cyclic nucleotide-gated channels have a stoichiometry of three CNGA1 subunits and one CNGB1 subunit. Neuron. 2002;36:891–896.
70 Zhong H, Molday LL, Molday RS, et al. The heteromeric cyclic nucleotide-gated channel adopts a 3A:1B stoichiometry. Nature. 2002;420:193–198.
71 Molday LL, Cook NJ, Kaupp UB, et al. The cGMP-gated cation channel of bovine rod photoreceptor cells is associated with a 240-kDa protein exhibiting immunochemical cross-reactivity with spectrin. J Biol Chem. 1990;265:18690–18695.
72 Molday RS, Molday LL, Dose A, et al. The cGMP-gated channel of the rod photoreceptor cell characterization and orientation of the amino terminus. J Biol Chem. 1991;266:21917–21922.
73 Bareil C, Hamel CP, Delague V, et al. Segregation of a mutation in CNGB1 encoding the beta-subunit of the rod cGMP-gated channel in a family with autosomal recessive retinitis pigmentosa. Hum Genet. 2001;108:328–334.
74 Dryja TP, Finn JT, Peng YW, et al. Mutations in the gene encoding the alpha subunit of the rod cGMP-gated channel in autosomal recessive retinitis pigmentosa. Proc Natl Acad Sci U S A. 1995;92:10177–10181.
75 Peng C, Rich ED, Varnum MD. Subunit configuration of heteromeric cone cyclic nucleotide-gated channels. Neuron. 2004;42:401–410.
76 Kohl S, Marx T, Giddings I, et al. Total colourblindness is caused by mutations in the gene encoding the alpha-subunit of the cone photoreceptor cGMP-gated cation channel. Nat Genet. 1998;19:257–259.
77 Kohl S, Varsanyi B, Antunes GA, et al. CNGB3 mutations account for 50% of all cases with autosomal recessive achromatopsia. Eur J Hum Genet. 2005;13:302–308.
78 Lytton J. Na+/Ca2+ exchangers: three mammalian gene families control Ca2+ transport. Biochem J. 2007;406:365–382.
79 Hsu SC, Molday RS. Glycolytic enzymes and a GLUT-1 glucose transporter in the outer segments of rod and cone photoreceptor cells. J Biol Chem. 1991;266:21745–21752.
80 Fliesler SJ, Anderson RE. Chemistry and metabolism of lipids in the vertebrate retina. Prog Lipid Res. 1983;22:79–131.
81 Tinoco J. Dietary requirements and functions of alpha-linolenic acid in animals. Prog Lipid Res. 1982;21:1–45.
82 Anderson RE, Maude MB, Bok D. Low docosahexaenoic acid levels in rod outer segment membranes of mice with rds/peripherin and P216L peripherin mutations. Invest Ophthalmol Vis Sci. 2001;42:1715–1720.
83 Fung BK, Hurley JB, Stryer L. Flow of information in the light-triggered cyclic nucleotide cascade of vision. Proc Natl Acad Sci U S A. 1981;78:152–156.
84 Yee R, Liebman PA. Light-activated phosphodiesterase of the rod outer segment. Kinetics and parameters of activation and deactivation. J Biol Chem. 1978;253:8902–8909.
85 Wilden U, Hall SW, Kühn H. Phosphodiesterase activation by photoexcited rhodopsin is quenched when rhodopsin is phosphorylated and binds the intrinsic 48-kDa protein of rod outer segments. Proc Natl Acad Sci USA. 1986;83:1174–1178.
86 Wilden U, Kuhn H. Light-dependent phosphorylation of rhodopson: number of phosphorylation sites. Biochemistry. 1982;21:3014–3022.
87 Ohguro H, Johnson RS, Ericsson LH, et al. Control of rhodopsin multiple phosphorylation. Biochemistry. 1994;33:1023–1028.
88 Ohguro H, Palczewski K, Ericsson LH, et al. Sequential phosphorylation of rhodopsin at multiple sites. Biochemistry. 1993;32:5718–5724.
89 Ohguro H, Van Hooser JP, Milam AH, et al. Rhodopsin phosphorylation and dephosphorylation in vivo. J Biol Chem. 1995;270:14259–14262.
90 Mendez A, Burns ME, Roca A, et al. Rapid and reproducible deactivation of rhodopsin requires multiple phosphorylation sites. Neuron. 2000;28:153–164.
91 Chen CK, Burns ME, Spencer M, et al. Abnormal photoresponses and light-induced apoptosis in rods lacking rhodopsin kinase. Proc Natl Acad Sci U S A. 1999;96:3718–3722.
92 Gurevich VV, Gurevich EV. The molecular acrobatics of arrestin activation. Trends Pharmacol Sci. 2004;25:105–111.
93 Hirsch JA, Schubert C, Gurevich VV, et al. The 2.8 A crystal structure of visual arrestin: a model for arrestin’s regulation. Cell. 1999;97:257–269.
94 Vishnivetskiy SA, Hosey MM, Benovic JL, et al. Mapping the arrestin–receptor interface. Structural elements responsible for receptor specificity of arrestin proteins. J Biol Chem. 2004;279:1262–1268.
95 Vishnivetskiy SA, Paz CL, Schubert C, et al. How does arrestin respond to the phosphorylated state of rhodopsin? J Biol Chem. 1999;274:11451–11454.
96 Heck M, Hofmann K. G-protein effector coupling – a real time light-scattering assay for transducin phosphodiesterase interaction. Biochemistry. 1993;32:8220–8227.
97 He W, Cowan CW, Wensel TG. RGS9, a GTPase accelerator for phototransduction. Neuron. 1998;20:95–102.
98 Slepak VZ, Artemyev NO, Zhu Y, et al. An effector site that stimulates G-protein GTPase in photoreceptors. J Biol Chem. 1995;270:14319–14324.
99 Tsang SH, Burns ME, Calvert PD, et al. Role for the target enzyme in deactivation of photoreceptor G protein in vivo. Science. 1998;282:117–121.
100 Chen CK, Burns ME, He W, et al. Slowed recovery of rod photoresponse in mice lacking the GTPase accelerating protein RGS9–1. Nature. 2000;403:557–560.
101 Krispel CM, Chen CK, Simon MI, et al. Prolonged photoresponses and defective adaptation in rods of Gbeta5-/- mice. J Neurosci. 2003;23:6965–6971.
102 Hu G, Wensel TG. R9AP, a membrane anchor for the photoreceptor GTPase accelerating protein, RGS9–1. Proc Natl Acad Sci U S A. 2002;99:9755–9760.
103 Hu G, Zhang Z, Wensel TG. Activation of RGS9–1GTPase acceleration by its membrane anchor, R9AP. J Biol Chem. 2003;278:14550–14554.
104 Lishko PV, Martemyanov KA, Hopp JA, et al. Specific binding of RGS9-Gbeta 5L to protein anchor in photoreceptor membranes greatly enhances its catalytic activity. J Biol Chem. 2002;277:24376–24381.
105 Nishiguchi KM, Sandberg MA, Kooijman AC, et al. Defects in RGS9 or its anchor protein R9AP in patients with slow photoreceptor deactivation. Nature. 2004;427:75–78.
106 Cowan CW, Fariss RN, Sokal I, et al. High expression levels in cones of RGS9, the predominant GTPase accelerating protein of rods. Proc Natl Acad Sci U S A. 1998;95:5351–5356.
107 Gorczyca WA, Gray-Keller MP, Detwiler PB, et al. Purification and physiological evaluation of a guanylate-cyclase activating protein from retinal rods. Proc Natl Acad Sci U S A. 1994;91:4014–4018.
108 Koch KW, Stryer L. Highly cooperative feedback-control of retinal rod guanylate-cyclase by calcium-ions. Nature. 1988;334:64–66.
109 Gorczyca WA, Polans AS, Surgucheva IG, et al. Guanylyl cyclase-activating protein – a calcium-sensitive regulator of phototransduction. J Biol Chem. 1995;270:22029–22036.
110 Burns ME, Mendez A, Chen J, et al. Dynamics of cyclic GMP synthesis in retinal rods. Neuron. 2002;36:81–91.
111 Detwiler PB, Gray-Keller MP. The mechanisms of vertebrate light adaptation: speeded recovery versus slowed activation. Curr Opin Neurobiol. 1996;6:440–444.
112 Krispel CM, Chen CK, Simon MI, et al. Novel form of adaptation in mouse retinal rods speeds recovery of phototransduction. J Gen Physiol. 2003;122:703–712.
113 Woodruff ML, Sampath AP, Matthews HR, et al. Measurement of cytoplasmic calcium concentration in the rods of wild-type and transducin knock-out mice. J Physiol. 2002;542:843–854.
114 Matthews HR, Murphy RLW, Fain GL, et al. Photoreceptor light adaptation is mediated by cytoplasmic calcium concentration. Nature. 1988;334:67–69.
115 Nakatani K, Yau K-W. Calcium and light adaptation in retinal rods and cones. Nature. 1988;334:69–71.
116 Fain GL, Matthews HR, Cornwall MC, et al. Adaptation in vertebrate photoreceptors. Physiol Rev. 2001;81:117–151.
117 Koutalos Y, Yau K-W. Regulation of sensitivity in vertebrate rod photoreceptors by calcium. TINS. 1996;19:73–81.
118 Pugh EN, Jr., Nikonov S, Lamb TD. Molecular mechanisms of vertebrate photoreceptor light adaptation. Curr Opin Neurobiol. 1999;9:410–418.
119 Sakurai K, Chen J, Kefalov VJ. Role of guanylyl cyclase modulation in mouse cone phototransduction. J Neurosci. 2011;31:7991–8000.
120 Chen CK, Inglese J, Lefkowitz RJ, et al. Ca(2+)-dependent interaction of recoverin with rhodopsin kinase. J Biol Chem. 1995;270:18060–18066.
121 Kawamura S. Rhodopsin phosphorylation as a mechanism of cyclic GMP phosphodiesterase regulation by S-modulin. Nature. 1993;362:855–857.
122 Erickson MA, Lagnado L, Zozulya S, et al. The effect of recombinant recoverin on the photoresponse of truncated rod photoreceptors. Proc Natl Acad Sci U S A. 1998;95:6474–6479.
123 Gray-Keller MP, Polans AS, Palczewski K, et al. The effect of recoverin-like calcium-binding proteins on the photoresponse of retinal rods. Neuron. 1993;10:523–531.
124 Makino CL, Dodd RL, Chen J, et al. Recoverin regulates light-dependent phosphodiesterase activity in retinal rods. J Gen Physiol. 2004;123:729–741.
125 Chen CK, Woodruff ML, Chen FS, et al. Background light produces a recoverin-dependent modulation of activated-rhodopsin lifetime in mouse rods. J Neurosci. 2010;30:1213–1220.
126 Matthews HR, Sampath AP. Photopigment quenching is Ca2+ dependent and controls response duration in salamander L-cone photoreceptors. J Gen Physiol. 2010;135:355–366.
127 Hsu Y-T, Molday RS. Modulation of the cGMP-gated channel of rod photoreceptor cells by calmodulin. Nature. 1993;361:76–79.
128 Grunwald ME, Yu W-P, Yu H-H, et al. Identification of a domain on the ß-subunit of the rod cGMP-gated cation channel that mediates inhibition by calcium-calmodulin. J Biol Chem. 1998;273:9148–9157.
129 Weitz D, Zoche M, Müller F, et al. Calmodulin controls the rod photoreceptor CNG channel through an unconventional binding site in the N-terminus of the ß-subunit. EMBO J. 1998;17:2273–2284.
130 Chen J, Woodruff ML, Wang T, et al. Channel modulation and the mechanism of light adaptation in mouse rods. J Neurosci. 2010;30:16232–16240.
131 Rebrik TI, Korenbrot JI. In intact mammalian photoreceptors, Ca2+-dependent modulation of cGMP-gated ion channels is detectable in cones but not in rods. J Gen Physiol. 2004;123:63–75.
132 Sampath AP, Matthews HR, Cornwall MC, et al. Light-dependent changes in outer segment free-Ca2+ concentration in salamander cone photoreceptors. J Gen Physiol. 1999;113:267–277.
133 Rebrik TI, Korenbrot JI. In intact cone photoreceptors, a Ca2+-dependent, diffusible factor modulates the cGMP-gated ion channels differently than in rods. J Gen Physiol. 1998;112:537–548.
134 Broekhuyse RM, Janssen AP, Tolhuizen EF. Effect of light-adaptation on the binding of 48-kDa protein (S-antigen) to photoreceptor cell membranes. Curr Eye Res. 1987;6:607–610.
135 Broekhuyse RM, Tolhuizen EF, Janssen AP, et al. Light induced shift and binding of S-antigen in retinal rods. Curr Eye Res. 1985;4:613–618.
136 Philp NJ, Chang W, Long K. Light-stimulated protein movement in rod photoreceptor cells of the rat retina. FEBS Lett. 1987;225:127–132.
137 Whelan JP, McGinnis JF. Light-dependent subcellular movement of photoreceptor proteins. J Neurosci Res. 1988;20:263–270.
138 McGinnis JF, Whelan JP, Donoso LA. Transient, cyclic changes in mouse visual cell gene products during the light-dark cycle. J Neurosci Res. 1992;31:584–590.
139 Calvert PD, Strissel KJ, Schiesser WE, et al. Light-driven translocation of signaling proteins in vertebrate photoreceptors. Trends Cell Biol. 2006;16:560–568.
140 Sokolov M, Lyubarsky AL, Strissel KJ, et al. Massive light-driven translocation of transducin between the two major compartments of rod cells: a novel mechanism of light adaptation. Neuron. 2002;34:95–106.
141 Rosenzweig DH, Nair KS, Wei J, et al. Subunit dissociation and diffusion determine the subcellular localization of rod and cone transducins. J Neurosci. 2007;27:5484–5494.
142 Lobanova ES, Finkelstein S, Song H, et al. Transducin translocation in rods is triggered by saturation of the GTPase-activating complex. J Neurosci. 2007;27:1151–1160.
143 Zhang X, Wensel TG, Kraft TW. GTPase regulators and photoresponses in cones of the eastern chipmunk. J Neurosci. 2003;23:1287–1297.
144 Lobanova ES, Herrmann R, Finkelstein S, et al. Mechanistic basis for the failure of cone transducin to translocate: why cones are never blinded by light. J Neurosci. 2010;30:6815–6824.
145 Chen J, Wu M, Sezate SA, et al. Light threshold-controlled cone alpha-transducin translocation. Invest Ophthalmol Vis Sci. 2007;48:3350–3355.
146 Nair KS, Hanson SM, Mendez A, et al. Light-dependent redistribution of arrestin in vertebrate rods is an energy-independent process governed by protein-protein interactions. Neuron. 2005;46:555–567.
147 Mears AJ, Kondo M, Swain PK, et al. Nrl is required for rod photoreceptor development. Nat Genet. 2001;29:447–452.
148 Kefalov V, Fu Y, Marsh-Armstrong N, et al. Role of visual pigment properties in rod and cone phototransduction. Nature. 2003;425:526–531.
149 Shi G, Yau KW, Chen J, et al. Signaling properties of a short-wave cone visual pigment and its role in phototransduction. J Neurosci. 2007;27:10084–10093.
150 Deng WT, Sakurai K, Liu J, et al. Functional interchangeability of rod and cone transducin alpha-subunits. Proc Natl Acad Sci U S A. 2009;106:17681–17686.
151 Chen CK, Woodruff ML, Chen FS, et al. Replacing the rod with the cone transducin subunit decreases sensitivity and accelerates response decay. J Physiol. 2010;588:3231–3241.
152 Okawa H, Sampath AP, Laughlin SB, et al. ATP consumption by mammalian rod photoreceptors in darkness and in light. Curr Biol. 2008;18:1917–1921.
153 Trachtenberg MC, Packey DJ, Sweeney T. In vivo functioning of the Na+, K+-activated ATPase. Curr Top Cell Regul. 1981;19:159–217.
154 Karan S, Zhang H, Li S, et al. A model for transport of membrane-associated phototransduction polypeptides in rod and cone photoreceptor inner segments. Vision Res. 2008;48:442–452.
155 Wensel TG. Signal transducing membrane complexes of photoreceptor outer segments. Vision Res. 2008;48:2052–2061.
156 Ames JB, Ishima R, Tanaka T, et al. Molecular mechanics of calcium-myristoyl switches. Nature. 1997;389:198–202.
157 Malanson KM, Lem J. Rhodopsin-mediated retinitis pigmentosa. Prog Mol Biol Transl Sci. 2009;88:1–31.
158 Kaushal S, Khorana HG. Structure and function in rhodopsin. 7. Point mutations associated with autosomal dominant retinitis pigmentosa. Biochemistry. 1994;33:6121–6128.
159 Mendes HF, van der Spuy J, Chapple JP, et al. Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol Med. 2005;11:177–185.
160 Sung CH, Davenport CM, Nathans J. Rhodopsin mutations responsible for autosomal dominant retinitis pigmentosa. Clustering of functional classes along the polypeptide chain. J Biol Chem. 1993;268:26645–26649.
161 Concepcion F, Mendez A, Chen J. The carboxyl-terminal domain is essential for rhodopsin transport in rod photoreceptors. Vision Res. 2002;42:417–426.
162 Li T, Snyder WK, Olsson JE, et al. Transgenic mice carrying the dominant rhodopsin mutation P347S: evidence for defective vectorial transport of rhodopsin to the outer segments. Proc Natl Acad Sci U S A. 1996;93:14176–14181.
163 Sung CH, Makino C, Baylor D, et al. A rhodopsin gene mutation responsible for autosomal dominant retinitis pigmentosa results in a protein that is defective in localization to the photoreceptor outer segment. J Neurosci. 1994;14:5818–5833.
164 Deretic D. A role for rhodopsin in a signal transduction cascade that regulates membrane trafficking and photoreceptor polarity. Vision Res. 2006;46:4427–4433.
165 Deretic D, Williams AH, Ransom N, et al. Rhodopsin C terminus, the site of mutations causing retinal disease, regulates trafficking by binding to ADP-ribosylation factor 4 (ARF4). Proc Natl Acad Sci U S A. 2005;102:3301–3306.
166 Tam BM, Moritz OL, Papermaster DS. The C terminus of peripherin/rds participates in rod outer segment targeting and alignment of disk incisures. Mol Biol Cell. 2004;15:2027–2037.
167 Huttl S, Michalakis S, Seeliger M, et al. Impaired channel targeting and retinal degeneration in mice lacking the cyclic nucleotide-gated channel subunit CNGB1. J Neurosci. 2005;25:130–138.
168 Zhang H, Li S, Doan T, et al. Deletion of PrBP/delta impedes transport of GRK1 and PDE6 catalytic subunits to photoreceptor outer segments. Proc Natl Acad Sci U S A. 2007;104:8857–8862.
169 Zhang H, Constantine R, Vorobiev S, et al. UNC119 is required for G protein trafficking in sensory neurons. Nat Neurosci. 2011;14:874–880.
170 Insinna C, Besharse JC. Intraflagellar transport and the sensory outer segment of vertebrate photoreceptors. Dev Dyn. 2008;237:1982–1992.
171 Williams DS. Transport to the photoreceptor outer segment by myosin VIIa and kinesin II. Vision Res. 2002;42:455–462.
172 Williams DS, Linberg KA, Vaughan DK, et al. Disruption of microfilament organization and deregulation of disk membrane morphogenesis by cytochalasin D in rod and cone photoreceptors. J Comp Neurol. 1988;272:161–176.
173 Adato A, Weil D, Kalinski H, et al. Mutation profile of all 49 exons of the human myosin VIIA gene, and haplotype analysis, in Usher 1B families from diverse origins. Am J Hum Genet. 1997;61:813–821.
174 Weil D, Blanchard S, Kaplan J, et al. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature. 1995;374:60–61.
175 Weil D, Kussel P, Blanchard S, et al. The autosomal recessive isolated deafness, DFNB2, and the Usher 1B syndrome are allelic defects of the myosin-VIIA gene. Nat Genet. 1997;16:191–193.
176 Pierce EA, Quinn T, Meehan T, et al. Mutations in a gene encoding a new oxygen-regulated photoreceptor protein cause dominant retinitis pigmentosa. Nat Genet. 1999;22:248–254.
177 Gao J, Cheon K, Nusinowitz S, et al. Progressive photoreceptor degeneration, outer segment dysplasia, and rhodopsin mislocalization in mice with targeted disruption of the retinitis pigmentosa-1 (Rp1) gene. Proc Natl Acad Sci U S A. 2002;99:5698–5703.
178 Liu Q, Lyubarsky A, Skalet JH, et al. RP1 is required for the correct stacking of outer segment discs. Invest Ophthalmol Vis Sci. 2003;44:4171–4183.
179 Liu Q, Zuo J, Pierce EA. The retinitis pigmentosa 1 protein is a photoreceptor microtubule-associated protein. J Neurosci. 2004;24:6427–6436.
180 Meindl A, Dry K, Herrmann K, et al. A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3). Nat Genet. 1996;13:35–42.
181 Mavlyutov TA, Zhao H, Ferreira PA. Species-specific subcellular localization of RPGR and RPGRIP isoforms: implications for the phenotypic variability of congenital retinopathies among species. Hum Mol Genet. 2002;11:1899–1907.
182 Hong DH, Pawlyk B, Sokolov M, et al. RPGR isoforms in photoreceptor connecting cilia and the transitional zone of motile cilia. Invest Ophthalmol Vis Sci. 2003;44:2413–2421.
183 Hong DH, Pawlyk BS, Shang J, et al. A retinitis pigmentosa GTPase regulator (RPGR)-deficient mouse model for X-linked retinitis pigmentosa (RP3). Proc Natl Acad Sci U S A. 2000;97:3649–3654.
184 Trojan P, Krauss N, Choe HW, et al. Centrins in retinal photoreceptor cells: regulators in the connecting cilium. Prog Retin Eye Res. 2008;27:237–259.
185 Yang J, Liu X, Yue G, et al. Rootletin, a novel coiled-coil protein, is a structural component of the ciliary rootlet. J Cell Biol. 2002;159:431–440.
186 Hagstrom SA, Adamian M, Scimeca M, et al. A role for the Tubby-like protein 1 in rhodopsin transport. Invest Ophthalmol Vis Sci. 2001;42:1955–1962.
187 Lewis CA, Batlle IR, Batlle KG, et al. Tubby-like protein 1 homozygous splice-site mutation causes early-onset severe retinal degeneration. Invest Ophthalmol Vis Sci. 1999;40:2106–2114.
188 Rattner A, Chen J, Nathans J. Proteolytic shedding of the extracellular domain of photoreceptor cadherin. Implications for outer segment assembly. J Biol Chem. 2004;279:42202–42210.
189 Kubota R, Noda S, Wang Y, et al. A novel myosin-like protein (myocilin) expressed in the connecting cilium of the photoreceptor: molecular cloning, tissue expression, and chromosomal mapping. Genomics. 1997;41:360–369.
190 Schmitz F, Konigstorfer A, Sudhof TC. RIBEYE, a component of synaptic ribbons: a protein’s journey through evolution provides insight into synaptic ribbon function. Neuron. 2000;28:857–872.
191 Jackman SL, Choi SY, Thoreson WB, et al. Role of the synaptic ribbon in transmitting the cone light response. Nat Neurosci. 2009;12:303–310.
192 Snellman J, Mehta B, Babai N, et al. Acute destruction of the synaptic ribbon reveals a role for the ribbon in vesicle priming. Nat Neurosci.. 2011;14:1135–1141.
193 Carter-Dawson LD, LaVail MM. Rods and cones in the mouse retina. I. Structural analysis using light and electron microscopy. J Comp Neurol. 1979;188:245–262.
194 Tsukamoto Y, Morigiwa K, Ueda M, et al. Microcircuits for night vision in mouse retina. J Neurosci. 2001;21:8616–8623.
195 West RW, Dowling JE. Anatomical evidence for cone and rod-like receptors in the gray squirrel, ground squirrel, and prairie dog retinas. J Comp Neurol. 1975;159:439–460.
196 Chun MH, Grunert U, Martin PR, et al. The synaptic complex of cones in the fovea and in the periphery of the macaque monkey retina. Vision Res. 1996;36:3383–3395.
197 Hopkins JM, Boycott BB. The cone synapses of cone bipolar cells of primate retina. J Neurocytol. 1997;26:313–325.
198 Koschak A, Reimer D, Walter D, et al. Cav1.4alpha1 subunits can form slowly inactivating dihydropyridine-sensitive L-type Ca2+ channels lacking Ca2+-dependent inactivation. J Neurosci. 2003;23:6041–6049.
199 Wahl-Schott C, Baumann L, Cuny H, et al. Switching off calcium-dependent inactivation in L-type calcium channels by an autoinhibitory domain. Proc Natl Acad Sci U S A. 2006;103:15657–15662.
200 Haeseleer F, Imanishi Y, Maeda T, et al. Essential role of Ca2+-binding protein 4, a Cav1.4 channel regulator, in photoreceptor synaptic function. Nat Neurosci. 2004;7:1079–1087.
201 Morgans CW, Bayley PR, Oesch NW, et al. Photoreceptor calcium channels: insight from night blindness. Vis Neurosci. 2005;22:561–568.
202 Huang SP, Brown BM, Craft CM. Visual Arrestin 1 acts as a modulator for N-ethylmaleimide-sensitive factor in the photoreceptor synapse. J Neurosci. 2010;30:9381–9391.
203 Sampath AP, Strissel KJ, Elias R, et al. Recoverin improves rod-mediated vision by enhancing signal transmission in the mouse retina. Neuron. 2005;46:413–420.
204 Strissel KJ, Lishko PV, Trieu LH, et al. Recoverin undergoes light-dependent intracellular translocation in rod photoreceptors. J Biol Chem. 2005;280:29250–29255.
205 Higgs MH, Lukasiewicz PD. Activation of group II metabotropic glutamate receptors inhibits glutamate release from salamander retinal photoreceptors. Vis Neurosci. 2002;19:275–281.
206 Koulen P, Kuhn R, Wassle H, et al. Modulation of the intracellular calcium concentration in photoreceptor terminals by a presynaptic metabotropic glutamate receptor. Proc Natl Acad Sci U S A. 1999;96:9909–9914.
207 Duncan JL, Yang H, Doan T, et al. Scotopic visual signaling in the mouse retina is modulated by high-affinity plasma membrane calcium extrusion. J Neurosci. 2006;26:7201–7211.
208 Szikra T, Barabas P, Bartoletti TM, et al. Calcium homeostasis and cone signaling are regulated by interactions between calcium stores and plasma membrane ion channels. PLoS One. 2009;4:e6723.
209 Szikra T, Cusato K, Thoreson WB, et al. Depletion of calcium stores regulates calcium influx and signal transmission in rod photoreceptors. J Physiol. 2008;586:4859–4875.
210 Thoreson WB. Kinetics of synaptic transmission at ribbon synapses of rods and cones. Mol Neurobiol. 2007;36:205–223.
211 Choi SY, Jackman S, Thoreson WB, et al. Light regulation of Ca2+ in the cone photoreceptor synaptic terminal. Vis Neurosci. 2008;25:693–700.
212 Sheng Z, Choi SY, Dharia A, et al. Synaptic Ca2+ in darkness is lower in rods than cones, causing slower tonic release of vesicles. J Neurosci. 2007;27:5033–5042.
213 Gal A, Apfelstedt-Sylla E, Janecke AR, et al. Rhodopsin mutations in inherited retinal dystrophies and dysfunctions. Prog Ret Eye Res. 1997;16:51–79.
214 Lin JH, Li H, Yasumura D, et al. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949.
215 Colley NJ, Cassill JA, Baker EK, et al. Defective intracellular transport is the molecular basis of rhodopsin-dependent dominant retinal degeneration. Proc Natl Acad Sci U S A. 1995;92:3070–3074.
216 Deretic D, Puleo-Scheppke B, Trippe C. Cytoplasmic domain of rhodopsin is essential for post-Golgi vesicle formation in a retinal cell-free system. J Biol Chem. 1996;271:2279–2286.
217 Deretic D, Schmerl S, Hargrave PA, et al. Regulation of sorting and post-Golgi trafficking of rhodopsin by its C-terminal sequence QVS(A)PA. Proc Natl Acad Sci U S A. 1998;95:10620–10625.
218 Chuang JZ, Sung CH. The cytoplasmic tail of rhodopsin acts as a novel apical sorting signal in polarized MDCK cells. J Cell Biol. 1998;142:1245–1256.
219 Rao VR, Oprian DD. Activating mutations of rhodopsin and other G protein-coupled receptors. Annu Rev Biophys Biomol Struct. 1996;25:287–314.
220 Robinson PR, Cohen GB, Zhukovsky EA, et al. Constitutively active mutants of rhodopsin. Neuron. 1992;9:719–725.
221 Rao VR, Cohen GB, Oprian DD. Rhodopsin mutation G90D and a molecular mechanism for congenital night blindness. Nature. 1994;367:639–642.
222 Sieving PA, Richards JE, Naarendorp F, et al. Dark-light: model for nightblindness from the human rhodopsin Gly-90–>Asp mutation. Proc Natl Acad Sci U S A. 1995;92:880–884.
223 Li T, Franson WK, Gordon JW, et al. Constitutive activation of phototransduction by K296E opsin is not a cause of photoreceptor degeneration. Proc Natl Acad Sci U S A. 1995;92:3551–3555.
224 Chen J, Shi G, Concepcion FA, et al. Stable rhodopsin/arrestin complex leads to retinal degeneration in a transgenic mouse model of autosomal dominant retinitis pigmentosa. J Neurosci. 2006;26:11929–11937.
225 Fuchs S, Nakazawa M, Maw M, et al. A homozygous 1-base pair deletion in the arrestin gene is a frequent cause of Oguchi disease in Japanese. Nat Genet. 1995;10:360–362.
226 Yamamoto S, Sippel KC, Berson EL, et al. Defects in the rhodopsin kinase gene in the Oguchi form of stationary night blindness. Nat Genet. 1997;15:175–178.
227 Chen J, Simon MI, Matthes MT, et al. Increased susceptibility to light damage in an arrestin knockout mouse model of Oguchi disease (stationary night blindness). Invest Ophthalmol Vis Sci. 1999;40:2978–2982.
228 Dryja TP, Hahn LB, Reboul T, et al. Missense mutation in the gene encoding the alpha subunit of rod transducin in the Nougaret form of congenital stationary night blindness. Nat Genet. 1996;13:358–360.
229 McLaughlin ME, Sandberg MA, Berson EL, et al. Recessive mutations in the gene encoding the beta-subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nat Genet. 1993;4:130–134.
230 Farber DB, Lolley RN. Cyclic guanosine monophosphate: elevation in degenerating photoreceptor cells of the C3H mouse retina. Science. 1974;186:449–451.
231 Lolley RN, Farber DB, Rayborn ME, et al. Cyclic GMP accumulation causes degeneration of photoreceptor cells: simulation of an inherited disease. Science. 1977;196:664–666.
232 Fesenko EE, Kolesnikov SS, Lyubarsky AL. Induction by cyclic GMP of cationic conductance in plasma membrane of retinal rod outer segment. Nature. 1985;313:310–313.
233 Yang RB, Foster DC, Garbers DL, et al. Two membrane forms of guanylyl cyclase found in the eye. Proc Natl Acad Sci U S A. 1995;92:602–606.
234 Lowe DG, Dizhoor AM, Liu K, et al. Cloning and expression of a second photoreceptor-specific membrane retina guanylyl cyclase (RetGC), RetGC-2. Proc Natl Acad Sci U S A. 1995;92:5535–5539.
235 Shyjan AW, de Sauvage FJ, Gillett NA, et al. Molecular cloning of a retina-specific membrane guanylyl cyclase. Neuron. 1992;9:727–737.
236 Goraczniak RM, Duda T, Sitaramayya A, et al. Structural and functional characterization of the rod outer segment membrane guanylate cyclase. Biochem J. 1994;302:455–461.
237 Duda T, Goraczniak R, Surgucheva I, et al. Calcium modulation of bovine photoreceptor guanylate cyclase. Biochemistry. 1996;35:8478–8482.
238 Laura RP, Dizhoor AM, Hurley JB. The membrane guanylyl cyclase, retinal guanylyl cyclase-1, is activated through its intracellular domain. J Biol Chem. 1996;271:11646–11651.
239 Dizhoor AM, Lowe DG, Olshevskaya EV, et al. The human photoreceptor membrane guanylyl cyclase, retGC, is present in outer segments and is regulated by calcium and a soluble activator. Neuron. 1994;12:1345–1352.
240 Yang RB, Garbers DL. Two eye guanylyl cyclases are expressed in the same photoreceptor cells and form homomers in preference to heteromers. J Biol Chem. 1997;272:13738–13742.
241 Gorczyca WA, Polans AS, Surgucheva IG, et al. Guanylyl cyclase activating protein. A calcium-sensitive regulator of phototransduction. J Biol Chem. 1995;270:22029–22036.
242 Yang RB, Robinson SW, Xiong WH, et al. Disruption of a retinal guanylyl cyclase gene leads to cone-specific dystrophy and paradoxical rod behavior. J Neurosci. 1999;19:5889–5897.
243 Semple-Rowland SL, Lee NR, Van Hooser JP, et al. A null mutation in the photoreceptor guanylate cyclase gene causes the retinal degeneration chicken phenotype. Proc Natl Acad Sci U S A. 1998;95:1271–1276.
244 Payne AM, Downes SM, Bessant DA, et al. A mutation in guanylate cyclase activator 1A (GUCA1A) in an autosomal dominant cone dystrophy pedigree mapping to a new locus on chromosome 6p21.1. Hum Mol Genet. 1998;7:273–277.
245 Dizhoor AM, Boikov SG, Olshevskaya EV. Constitutive activation of photoreceptor guanylate cyclase by Y99C mutant of GCAP-1. Possible role in causing human autosomal dominant cone degeneration. J Biol Chem. 1998;273:17311–17314.
246 Sokal I, Li N, Surgucheva I, et al. GCAP1 (Y99C) mutant is constitutively active in autosomal dominant cone dystrophy. Mol Cell. 1998;2:129–133.
247 Paquet-Durand F, Beck S, Michalakis S, et al. A key role for cyclic nucleotide gated (CNG) channels in cGMP-related retinitis pigmentosa. Hum Mol Genet. 2011;20:941–947.
248 Andrade AL, Melich K, Whatley GG, et al. Cyclic nucleotide-gated channel block by hydrolysis-resistant tetracaine derivatives. J Med Chem. 2011;54:4904–4912.
[/level-membership-for-opthalmology-category][not-level-membership-for-opthalmology-category]
Chapter 14 Structure and Function of Rod and Cone Photoreceptors
Introduction
Our visual experience is initiated by rod and cone photoreceptors in the retina. The human eye contains over 100 million rods and about 6 million cones, which are located within the outer nuclear layer of the retina and allow our visual experience to extend over 12 orders of magnitude in light intensity by splitting this range. Rod photoreceptors mediate vision under conditions of dim illumination, and allow our visual system to reach the limit imposed by the absorption of single photons.1,2 Cones are less sensitive by ~100-fold, but their tremendous capacity for adaptation allows them to encode light intensities in the brightest of days.3,4 Over the last decade, our understanding of structure and function of these cells has increased dramatically. Over 150 genes have been cloned or linked to retinal diseases, and, surprisingly, as many as half of these genes are specifically expressed or highly enriched in the photoreceptor cells. Discovery of the molecular constituents of the rods and cones is progressing at an increasing rate, particularly enhanced by the availability of “complete” genomic sequences for both human and mouse. While photoreceptor genes can be identified through association with a retinal disease (linkage), information about their function does not accompany their identification.5 Basic science research must then be undertaken to explain their role in both the normal, healthy photoreceptor as well as in photoreceptor diseases.
Though much of what we know about the structure and function of photoreceptor cells has come from studying animal models of inherited blindness,6,7 more recently, transgenic and knockout animal technologies have established themselves as powerful tools for understanding function and studying disease. After new photoreceptor genes are identified from patients, mutant animals can be engineered to emulate human photoreceptor pathologies. Before these molecular technologies were available, most data about retinal disease were gleaned from the rare “informative” patient or donor retina that had surviving photoreceptors to examine.8 Other major sources of information were studies of animal models that occurred through inbreeding or random inheritance of mutations in photoreceptor genes. Examples of these are the Irish Setter dog,9 the Briard dog,10 the Abyssinian cat,11 the Royal College of Surgeons (RCS) rat,12 and the rd13,14 and rds15,16 mouse models. Now, the ability to engineer transgenic animals has made the search for “informative” patients and naturally occurring animal models less acute, since any single gene of interest can be introduced17–19 or removed20–24 from the photoreceptor. However, the most instructive examples of structure–function relationships occur when there are patients and animal models with analogous mutation(s) and/or disease phenotype.
Photoreceptor fundamentals
The rod and cone photoreceptors are specialized sensory neurons that contain the protein machinery necessary to convert incident light into a signal that can be interpreted by the nervous system. Rod photoreceptors are more numerous than cones in most mammalian retina, and are highly sensitive. In the fully dark-adapted state, rods can reliably report the absorption of single photons to the retinal output, and they permit our scotopic, or night, vision. Cone photoreceptors are morphologically and functionally distinct from rods and express several types of visual pigments, or opsins, whose spectral sensitivity varies based on the cone’s subtype. In humans, three classes of cones confer robust color vision: S, M, and L cones (see Chapter 10, Color vision and night vision). Cones are also less sensitive than rods and generate light responses that are temporally briefer. This allows cones to mediate our photopic, or day, vision with improved temporal resolution. The concerted action of these two types of photoreceptors, and the retinal circuitry that carries their signals to the retinal output, ultimately underlie our rich visual experience.
Both rod and cone photoreceptors are highly polarized elongated cells that can be described as having four subcellular compartments: the outer segment (OS), the inner segment (IS), the nucleus, and the synaptic terminal (Figs 14.1 and 14.2). The OS is where photons are captured and activation of the phototransduction cascade begins. The IS lies immediately proximal to the OS, and contains the cell’s protein synthesis (Golgi apparatus and endoplasmic reticulum) and metabolic (mitochondria) machinery. Light-evoked signals are relayed passively down the photoreceptor axon (up to 75 µm long) to the synaptic terminals in the outer plexiform layer. The structure of photoreceptor terminals is unique in the nervous system, as they contain a specialized structure called a ribbon that facilitates the release of the excitatory neurotransmitter glutamate on to second-order retinal neurons (bipolar and horizontal cells). Thus, the photoreceptor cells transduce the sensory stimulus, light, and pass on a signal to retinal circuits that carry this information to higher visual centers.
Photoreceptor outer-segment structure
The OS compartment contains all components necessary for phototransduction, which is a set of biochemical reactions that convert photon capture to a change in a cationic current at the plasma membrane. Rods have cylindrical OS about 1.3 µm in diameter and length that ranges from 25 to 45 µm,25–28 that depends on numerous factors, including the time of day, the light intensity, their location in the retina, and the animal species. The cone OS is shorter, typically half the length of rods, with a larger diameter at the base that gradually tapers towards the tip (Fig. 14.2). Mouse rod OS contains an average of 810 membranous discs, which occupy two-thirds of the volume of the OS.29 The majority of the protein on these discs is rod opsin (or rhodopsin when the opsin is bound to its chromophore, 11-cis retinal). The density of rhodopsin on the disc membrane has been measured to be about 24 000 molecules/µm2.30 Despite this dense packing, rhodopsin freely diffuses in the membrane,31 which facilitates its encounter with, and activation of, transducin molecules to amplify the light signal. Many G-protein-coupled receptors are known to form dimers and higher-order oligomers. Rhodopsin has been observed to form a paracrystalline array of dimers when native disc membrane is viewed under atomic force microscopy.32 Whether it exists as dimers in vivo is still an area of controversy. However, it is known that rhodopsin is functional as monomers.33
Proteins that stabilize the structure of outer-segment discs
Rhodopsin is not only integral to the phototransduction cascade, it is also required for the formation and maintenance of OS discs; in the absence of rhodopsin, the OS structure is not formed.22 Peripherin and Rom-115,34,35 are two other disc membrane components that contribute to maintaining the flattened structure of the disc. Both of these proteins belong to the tetraspanin family of integral membrane proteins that form large multiprotein complexes known as the tetraspanin web or tetraspanin-enriched microdomains.36 Peripherin/rds is normally found within the edge or “rim” of the disc. It self-associates to form higher-order complexes and also interacts with Rom-1.37 Peripherin/rds and Rom-1 likely function as adhesion molecules that assist in keeping the discs vertically aligned, and may stabilize the disc stack with bridges to the overlying plasma membrane. Peripherin/rds is also thought to function in forming the curvature of the disc rim.38
Deletion or disruption of peripherin/rds results in malformation of discs at the OS base. The naturally occurring retinal degeneration in the mouse named “retinal degeneration, slow” (rds) has been shown to be due to a defect in peripherin/rds. The absence of peripherin/rds in the rds/rds mouse prevents normal development of the photoreceptor OS and leads to photoreceptor cell death.15,39 Transgenic mice that lack peripherin/rds fail to form an OS. When the level is reduced, as in the heterozygous mice, large whorls of disc membrane are formed instead of an organized stack of uniform discs.15,39 Mutations that affect the quaternary structure of peripherin/rds also lead to malformed discs. These observations underscore the importance of peripherin/rds in the formation and maintenance of disc structure in the OS.
Although Rom-1 is analogous to peripherin in structure and function, its absence in rods shows a less severe phenotype than peripherin/rds knockout mice, suggesting that peripherin/rds is more critical for disc formation. Although the discs grow larger than normal sizes, photoreceptor function appears to be retained to a much higher level than when peripherin/rds is absent. Nevertheless, abnormal Rom-1 does lead to slow and progressive photoreceptor degeneration both in mice and in humans.40
Functional differences of peripherin/rds and Rom-1 have been noted in rods and cones. Cones appear to have a lower ratio of Rom-1/peripherin,36 and different peripherin/rds mutations appear to affect rods and cones differentially. For example, the C214S and N244K mutations have a greater effect on rods whereas R172W and N244H tend to cause cone-dominant diseases such as macular dystrophy.36
Transmission electron microscopy has revealed structural elements between the lamellar discs as well as between the discs and the plasma membrane that are mainly localized to the rim region and incisures of discs.41–44 A study of OS structure using cryoelectron tomography of vitrified OS showed previously unobserved spacers distributed throughout the discs.30 These structural elements likely maintain the proper distance between adjacent discs and between discs and the plasma membrane. The protein identities of these spacers are not known, but peripherin/rds and Rom-1 may be candidates.
Another component of the rod disc is ABCR (photoreceptor cell-specific ATP-binding cassette transporter).45–47 Mutations in ABCR are responsible for a large variety of retinal degenerations, including Stargardt macular dystrophy, fundus flavimaculatus, some forms of cone–rod degeneration, and retinitis pigmentosa (RP). Other ABCR mutations are thought to increase the risk of developing age-related macular degeneration (AMD). ABCR is a member of the ABC superfamily. This is a large family of transmembrane proteins involved in energy-dependent transport of many different substrates across membrane “barriers.” The localization of ABCR at the disc rim suggests that it is involved in the movement of molecules from the disc lumen into the cytosol. This hypothesis was supported by studies in ABCR knockout mice, which show delayed dark adaptation, increased all-trans retinal following light exposure, and elevated phosphatidylethanolamine (PE) in the rod OS.48 Biochemical analysis of retinas from ABCR knockout mice revealed accumulation of a novel complex of all-trans retinal and PE, termed N-retinylidene-PE, which is not found in normal retina. Once in the cytosol, the all-trans retinol moves to the retinal pigment epithelium (RPE) for recycling and chromophore (11-cis retinal) regeneration (see Chapter 16, Cell biology of the retinal pigment epithelium). The primary pathologic defect in Stargardt disease, and also in ABCR knockout mice, is accumulation of toxic lipofuscin pigments, such as A2E, in RPE cells. Thus the phenotype is remarkably similar between mice and that observed in the fundus of Stargardt patients.
The animal model and further biochemical investigations49 suggest that ABCR functions as an outwardly directed flippase for N-retinylidene-PE. Delayed dark adaptation is likely due to accumulation (in discs) of the noncovalent complex between opsin and all-trans retinal. ABCR-mediated retinal degeneration in patients may result from “poisoning” of the RPE due to A2E accumulation, with secondary photoreceptor degeneration due to loss of the ABCR support role.
Though ABCR most definitely plays a role in the structure of discs, no mutations have been conclusively associated with abnormal structural features per se. In other words, it appears that mutations do not directly affect folding at the disc rim, and more likely affect the biochemical function of the transporter. In corroboration of these data, Radu and colleagues tested the effects of isotretinoin on lipofuscin accumulation in ABCR knockout mice,50 a model of recessive Stargardt disease. They observed by electron microscopy that isotretinoin blocked both the formation of A2E biochemically, and the accumulation of lipofuscin pigments. Further, no significant visual loss was observed in ABCR null mice by electroretinography (when treated), and isotretinoin also blocked the slower, age-dependent accumulation of lipofuscin in wild-type mice. The results suggest that treatment with isotretinoin may inhibit lipofuscin accumulation and delay the onset of visual loss in patients with Stargardt disease and may be an effective treatment for other forms of retinal or macular degeneration associated with lipofuscin accumulation, though “normal” visual function may be somewhat compromised by such treatment.
The presence of an OS is clearly not optional, as there are many examples of retinal disease in which the photoreceptor cell dies shortly after loss of the OS.51–53 Why loss of the OS triggers rod cell death is not obvious, since all of the required cellular organelles inhabit the IS and cell body; the OS appears devoid of organelles. The necessity to maintain an OS is further surprising since photoreceptors continually replace all the OS components. In fact, the renewal of rod cell OS components is perhaps more readily seen in the photoreceptor than in any other cell in the body. The disc and plasma membrane components of the OS are completely replaced within 2 weeks.54,55 To support this turnover there is an influx of newly synthesized proteins and lipids from the IS. These components are transported to the base of the OS, and from there discs gradually migrate towards the RPE for phagocytosis and recycling. One idea is that the high oxygen tension coming from the choroidal vasculature may interfere with the cellular machinery in the IS and nucleus of the photoreceptor, and the presence of the OS puts a protective distance between the choroid and the photoreceptor nuclei.56
Disc morphogenesis
Although it is known that new discs are formed at the base of the OS, the process by which these discs are formed is not fully resolved (Fig. 14.2). One model was described in the work of Anderson et al. (model 1).57,58 Based on morphologic studies of adult monkey rods, it was observed that the folded membrane stacks at the base of the OS are continuous with the plasma membrane of the connecting cilium, whereas the older discs are closed off and separated from the plasma membrane. From this appearance a model was proposed that the newly arrived rhodopsin-bearing vesicles fuse with the plasma membrane, and the growing membrane evaginates to form open discs. Eventually these discs pinch off and form the mature closed discs. Another model (model 2), the vesicular targeting model, was recently proposed by Chuang and colleagues.59 They provide molecular evidence that rhodopsin’s carboxyl terminus interacts with a protein called SARA (smad anchor for receptor activation), as well as phosphatidylinositol 3-phosphate (PI3P), and syntaxin 3. In their model, the new discs are already closed and enveloped by the OS plasma membrane. Their evidence suggests that the protein–protein and protein–lipid interactions organized by SARA regulate the vesicular targeting of axonemal rhodopsin-bearing vesicles to the newly formed discs. Thus the discs grow by fusing with the rhodopsin-bearing vesicles and not from the evaginated plasma membrane. This model is more consistent with the morphology of their experimental model system, the mouse rods, which do not display open discs at the base of the OS.
Outer-segment plasma membrane
It is clear that, in addition to its obvious role in phototransduction, the OS plasma membrane contains a rich array of specialized proteins, many of which regulate the movement of ions into and out of the OS. The best-characterized proteins of this type are the retinal cGMP-gated (CNG) cation channels. In the dark-adapted rods and cones, Na+ and Ca2+ flow into the OS through these channels in the plasma membrane. Calcium comprises about 10% of the dark current carried by these channels in rods,60 and perhaps 20% or more in cones.61,62 In both rods and cones Na+ is extruded from the IS through the Na+/K+ pump. This flow of ions sets up the circulating dark current, of which the vast majority is carried by the Na+. The probability of the opening of the CNG channel, which in turn determines the size of the circulating current, depends on the amount of free [cGMP], which in the dark is estimated to be 3–4 µM.63 At this concentration, the probability of channel opening is estimated to be only 0.1–0.2.64,65 This underscores the impact that elevated [cGMP] can have on the number of open channels in the diseased state. The influx of Ca2+ through the channel is balanced by an efflux of Ca2+ by the Na+/Ca2+-K+ exchanger (NCKX) in the rod OS plasma membrane, thereby maintaining the intracellular level of Ca2+ at a relatively constant level.66 Kaupp and colleagues67 cloned the cGMP channel from bovine retina, while Pittler and colleagues68 determined the primary structures of the human and mouse retinal rod cGMP-gated cation channel. These studies found that the sequence of the cGMP channel has significant similarity (59%) to the olfactory cAMP-gated channel. The retinal rod CNG channel is a hetero-oligomer composed of three alpha (CNGA1) and one beta (CNGB1) subunits,69,70 each with cytoplasmic amino (N)- and carboxyl (C)-termini, six putative transmembrane domains, and a pore region.71,72 Mutations in CNGA1 and CNGB1 have been linked to disease. A point mutation in CNGB173 and several mutations in CNGA174 have been identified and linked to recessive RP in humans. The CNG channel in cones consists of two CNGA3 and two CNGB3 subunits.75 Together, mutations in either of these genes account for ~75% of complete acromatopsia.76,77
In addition to the CNG channel, there are several other channels and transporters in the plasma membrane that serve to regulate the intracellular contents of the OS. The best studied of these is the NCKX. Rods express NCKX1 whereas cones express NCKX2.78 The exchanger moves with every cycle 4 Na+ into the OS, and in exchange, moves one Ca2+ and one K+ into the subretinal space; exchange is thus electrogenic. Other transmembrane proteins, such as the GLUT-1 glucose transporter, have been shown to be present on both rod and cone OS.79
Outer-segment lipids
Docosahexaenoic acid (DHA, 22:6n-6) is the major fatty acid found in the retinal rod OS, and rod photoreceptors have higher levels of DHA than in any other membrane system examined.80 Numerous studies have established that the high level of DHA in rod OS membranes provides an optimal microenvironment for rhodopsin. DHA belongs to the n-3 family of essential polyunsaturated fatty acids. These fatty acids cannot be synthesized by vertebrates and they, or their shorter-chain precursors, must therefore be obtained in the diet.81
Humans with RP and dogs with progressive rod–cone degeneration (prcd) have lower than normal blood levels of long-chain polyunsaturated fatty acids, including DHA. In addition, prcd-affected dogs have lower levels of DHA in their rod OS than control animals.82 The reason for the reduced level of DHA in rod OS of animals with inherited retinal degeneration is not known. However, the fatty acid composition of the rod OS of these animals suggests that the synthesis of DHA-containing glycerolipids is downregulated in retinas of animals with inherited retinal degenerations.
Phototransduction
Visual transduction is initiated by the phototransduction cascade, which is perhaps the best characterized of all G-protein receptor-coupled signaling pathways. Our knowledge about phototransduction is relatively advanced because the components of this pathway are located selectively in the photoreceptor OS and can be isolated in quantities suitable for biochemical analysis. Photoreceptors can also be isolated easily for electrophysiological measurements, either as single cells using suction electrodes (Fig. 14.3), or en masse using field potentials called electoretinograms (ERG: see Chapter 7, Electrogenesis of the electroretinogram). The phototransduction cascade within rods is so robust that single-photon absorption by rhodopsin gives rise to a change in current that can be monitored by suction electrode recordings. Similarly, the remarkable ability of cone phototransduction to adapt to increases in background light intensity can also be monitored with suction electrodes. The combination of transgenic technology and electrophysiological analysis has greatly enhanced our understanding of signal transduction within these photoreceptor cells. In particular, transgenic technologies have allowed the introduction of targeted changes into specific components of the signaling pathway. These genetically altered intact photoreceptor cells can then be subjected to electrophysiological measurements, and their tissues can subsequently be used in biochemical or immunocytochemical assays to pinpoint the molecular mechanism behind the physiological phenotype. In this section we summarize the current state of our understanding of phototransduction in retinal photoreceptors.
Signal activation and amplification
A hallmark of rod phototransduction is its high sensitivity. Fully dark-adapted rods achieve sensitivity that reaches the theoretical maximum, the ability to detect individual quanta of light.1,2 This high sensitivity is generated through several stages of amplification, resulting in a tremendous increase in the signal gain (Fig. 14.4). This cascade of amplification begins at the light-activated G-protein-coupled receptor rhodopsin (R). Absorption of a photon by R leads to a conformation change (R*), allowing it to interact with the heterotrimeric G-protein transducin (Tα, β, and γ), thereby promoting the exchange of bound GDP for GTP. The complex then dissociates into Tα-GTP and Tβγ, and R* is free to activate many other transducin molecules during its catalytic lifetime.83 In the subsequent step, Tα-GTP binds and removes the inhibitory γ-subunit of the cGMP-PDE so that it can now hydrolyze cGMP to 5’-GMP; the enzymatic activity of PDE (PDE*) in turn degrades thousands of cGMP molecules. The decrease in cytoplasmic cGMP concentration then leads to closure of the cGMP-gated cation conductance channel in the rod plasma membrane, resulting in a reduction in the influx of ~ 1 000 000 Na+ ions.84 This is a graded effect: a slight lowering in the cGMP concentration leads to closure of some channels, while a large decrease will eventually lead to closure of all channels. In this instance, the rod cell is said to be saturated.
In darkness, glutamate, the transmitter used by both rods and cones, is released at a steady rate because the photoreceptor’s membrane potential is in a relatively depolarized state. The closure of CNG channels results in a graded hyperpolarization of the cell, leading to a decrease in glutamate release at the synapse. In this manner, the first sensation of light perception is transmitted from the photoreceptor cell to second-order cells of the retina, where signals are further processed and ultimately conveyed to the retinal ganglion cells (see Chapter 15, Functional anatomy of the mammalian retina).
Signal deactivation
A reversal of the activation steps is required ultimately for the photoreceptor to return to its resting state. First, the catalytically active components of the phototransduction cascade, R* and PDE*, must be quenched, then cGMP needs to be resynthesized (Fig. 14.4). These events must be rapid and reproducible for the photoreceptor to maintain its high sensitivity and respond to subsequent stimuli. Our current understanding of these processes is detailed below.
Quenching R*: phosphorylation and arrestin binding
Since the early 1980s, phosphorylation was recognized to play an important role in deactivation of R*.85,86 To determine how receptor deactivation occurs in vivo, transgenic mouse models were developed to assess the contribution of receptor phosphorylation to R* shutoff. In early experiments, a rhodopsin truncation mutation, S334ter, was expressed in the photoreceptors of transgenic mice17 that removed the terminal 15 amino acid residues and thus all putative Ser and Thr phosphorylation sites. Single-photon responses produced by S334ter R* failed to shut off in a timely and stereotyped manner, indicating that this domain is important for R* quenching. In the 1990s, numerous biochemical experiments suggested that specific Ser residues on the C-terminus are crucial in R* shutoff.87–89 However, single-cell recordings from transgenic mice rods expressing rhodopsin where these phosphorylation sites removed in different combinations showed that all of the Ser and Thr sites at the C-terminus are required for rapid and reproducible R* deactivation; the cluster of Ser and Thr sites at rhodopsin’s C-terminus functions to ensure fast and reproducible R* deactivation.90 It is clear that rhodopsin kinase is solely responsible for phosphorylating R*, since phosphorylation of R* does not occur in mice lacking rhodopsin kinase.91
Subsequent to C-terminus phosphorylation, the catalytic activity of R* is quenched fully by binding of visual arrestin. Arrestins are soluble cytoplasmic proteins that bind to G-protein-coupled receptors, thus switching off activation of the G-protein and terminating the signaling pathway that triggers the cellular response; the most commonly studied arrestin is β-arrestin. Visual arrestin exhibits exquisite specificity to phosphorylated R*. Our understanding of how this specificity is conferred emerges from extensive mutagenesis studies, and the crystal structure of visual arrestin.92–95 These studies reveal that arrestin is constrained into a latent, inactive structure by a network of intramolecular interactions.92 According to the current model of arrestin activation, these intramolecular constraints are released by: (1) interaction with multiple phosphates on rhodopsin’s C-terminus; and (2) interaction with the cytoplasmic loops of R*. The physiological role of visual arrestin in controlling the rod single-photon response was studied by Xu and colleagues,24 who created transgenic mice lacking visual arrestin. Suction electrode recordings of photoresponses from the OS of these transgenic rods displayed a rapid, partial recovery, followed by a prolonged final recovery phase. The timing of the slow phase of shutoff is consistent with the spontaneous decay of the R*. Thus, while R* phosphorylation is required to terminate rapidly its catalytic activity, the full quenching of R* activity requires visual arrestin binding.
Deactivating PDE: control of transducin’s GTPase activity
Quenching of the phototransduction cascade also requires the shutoff of PDE*, which occurs through the deactivation of Tα. The GTPase activity of Tα allows the reassociation of the inhibitory γ-subunit to the catalytic PDE subunits,96 a process which is speeded through the participation of a photoreceptor-specific RGS protein, RGS-9, and its binding partner, Gβ5.97 These proteins act sequentially on Tα to facilitate the hydrolysis of bound GTP. The physiological role of all these proteins in the shutoff of the rod photoresponse was evaluated with transgenic mice that had alterations in each of these components. Rod photoresponses from transgenic mice expressing a PDE-γ with an impaired ability to stimulate GTP hydrolysis98 showed abnormally slowed recovery, demonstrating the necessary role of PDE-γ in Tα deactivation.99 Similarly, mice lacking RGS-9 showed profoundly slowed response recovery,100 as do mice lacking Gβ5.101 RGS-9 is anchored to photoreceptor membranes by the adaptor protein, R9AP.102–104 Interestingly, human patients with mutations in R9AP have difficulties adapting to sudden changes in light levels that are mediated by cones,105 consistent with the higher concentration of RGS-9 complex in cones compared to rods.106 Together, the current data support an important role of PDE-γ, RGS-9/Gβ5, and R9AP in Tα deactivation, and consequently, response recovery, in both rods and cones.
Resynthesis of cGMP: Ca2+ dependence of guanylyl cyclase
Recovery of the dark current also requires that cGMP is resynthesized to allow CNG channels to open. cGMP is synthesized in photoreceptor OS by a retinal guanylyl cyclase (RetGC), which is regulated by the protein GCAPs (GCAP 1 and 2).107 GCAPs is an EF hand Ca2+-binding protein that confers on RetGC a highly cooperative Ca2+ dependence for cGMP synthesis.108 Near the dark resting Ca2+ concentration RetGC’s activity is inhibited, but as OS Ca2+ falls in response to illumination as CNG channels close, RetGC’s activity accelerates to restore the resting cGMP and reopen CNG channels. The physiological actions of GCAPs have been evaluated in physiological recordings. For instance, when GCAP1 is dialyzed into gecko rods, the recovery phase of the light response is accelerated.109 In addition, suction electrode recordings from rods of GCAPs knockout mice show that the Ca2+ dependence of cGMP synthesis is required to set the time course of the rod’s photoresponse, and to suppress noise within the phototransduction cascade.110
Light adaptation
In a normal cycle of day and night, the illumination at the Earth’s surface varies over 11 orders of magnitude, making photoreceptor adaptation fundamentally important to the normal functioning of the vertebrate visual system. Rods cells are able to adjust their sensitivity over 2–3 log units of light intensities, while cone cells exhibit no response saturation over 6–7 log units.3,4 The switch between rod and cone function from night to bright daylight covers a large portion of vision’s functional range, and downstream circuitry accounts for the remainder of the range extension. While the molecular events underlying the amplification cascade following photon capture by rhodopsin are well delineated, the mechanisms underlying photoreceptor adaptation are less well understood; however, Ca2+ is known to play a feedback role in the adaptation process.111,112
The role of Ca2+ feedback
In darkness, Na+ ions enter the OS through the CNG channels and are extruded in the IS through Na+/K+ ATPase pumps. This steady-state influx and efflux of Na+ is largely the basis for the circulating dark current in photoreceptor cells. Similarly, Ca2+ enters through CNG channels in the OS, but is extruded locally by NCKX exchangers. In mouse rods the steady-state influx and efflux of Ca2+ hold concentration near 250 nM,113 but as light closes CNG channels, Ca2+ entry is blocked, Ca2+ efflux continues, and [Ca2+]i falls. This change in [Ca2+]i is graded just like the dark current: the degree of lowered [Ca2+]i is proportional to the intensity of background light.
This decrease in [Ca2+]i triggers a feedback signal that is necessary for light adaptation, since, when the [Ca2+]i decrease is blocked, adaptation to background illumination is greatly compromised.114,115 Existing evidence suggests that this feedback is rather complex, as it orchestrates several pathways directed toward different components of the transduction machinery.116–118 Here we review the role of Ca2+ on each of these components.
Adaptation mediated by Ca2+ feedback to retinal guanylyl cyclase
It has long been recognized that feedback controlling accelerated recovery contributes to light adaptation.108 Recovery of the light response requires the resynthesis of cGMP, which is needed to reopen the CNG channels and re-establish the circulating current. The control of cGMP synthesis by RetGC/GCAPs is central to this process. The contribution of accelerated cGMP synthesis to adaptation in rod photoreceptors was investigated in GCAP knockout mice.23,110 This work showed that the presence of GCAPs extends the sensitivity to background light in rod photoreceptors ~10-fold (Fig. 14.5), and can account for about half of the rod’s capacity to adapt to background light. Recordings from cones in GCAPs knockout mice also show that GCAP contributes to background adaptation, but to a lesser extent than it does in mouse rods.119
Fig. 14.5 Sensitivity as a function of background light was measured from mouse rod photoreceptors using suction electrodes (Fig. 14.3). Sensitivity was calculated from dim flash responses as the peak response amplitude divided by the flash strength. Flash sensitivity (SF) in the presence of increasing background light, background light (IB) was normalized against the flash sensitivity in darkness (SFD) and is plotted for the rods of several transgenic mice with particular elements of the phototransduction cascade altered. These mice include wild-type (•), mice lacking recoverin (), mice where the calmodulin-binding site on the cyclic nucleotide gated (CNG) channels has been eliminated (), mice lacking guanylyl cyclase-activating proteins 1 and 2 (), and mice where both the calmodulin-binding site on the CNG channel and guanylyl cyclase-activating proteins 1 and 2 have been eliminated (). The red line shows the predicted decline in sensitivity as a function of background light intensity if rods displayed no light adaptation. The blue line is best-fitting Weber–Fechner function for wild-type rods, and shows that light adaptation extends the sensitivity of rod photoreceptors to 100-fold higher background light levels. Rods from mice lacking recoverin, or the calmodulin-binding site on the CNG channels, show little deviation from wild-type rods, indicating that these proteins do not play a significant role in light adaptation. However, in mice lacking guanylyl cyclase-activating protein 1 and 2, light adaptation is impaired, but not absent, even when the calmodulin-binding site on the CNG channel is also eliminated (red dashed line). The remaining mechanisms that control light adaptation in rod photoreceptors remain unidentified.
(Data replotted from Chen J, Woodruff ML, Wang T, et al. Channel modulation and the mechanism of light adaptation in mouse rods. J Neurosci 2010;30:16232–40.)
