[level-membership-for-neurology-category]
Chapter 58 Status Epilepticus
The spectrum of seizure events extends from isolated, brief seizures to status epilepticus (SE), incorporating a full range of recurrent unprovoked seizures and prolonged or acute repetitive seizures. Typically, seizures are brief and self-limited. There has been considerable research directed at efforts to explain the mechanisms underlying seizure termination, as well as perpetuation, in SE. SE is a pediatric and neurologic emergency associated with significant morbidity and mortality [Dodson et al., 1993; Mitchell, 1996; Pellock, 1993a, 1994; Towne et al., 1994; Weise and Bleck, 1997; Wilson and Reynolds, 1990]. Its prompt recognition and management lead to the best chance of successful outcome. SE may represent the brain’s reaction to an acute insult, or it may be a manifestation of already existing epilepsy, either as the initial symptom or as a prolonged exacerbation of seizures [DeLorenzo et al., 1992, 1995, 1996].
Although the definition and classification of SE have been changed numerous times, the term refers to seizures that continue for a prolonged period. Studies suggest that SE frequently goes unrecognized, and that its occurrence has been underestimated in the general population [Treiman, 1993]. One study [O’Dell et al., 2005a] documented that 34 percent of cases of febrile SE presenting in emergency departments across five centers were not recognized as such by the hospital personnel assigned to treat them. Based on this finding, febrile SE has only a 2 out of 3 chance of being recognized in an emergency treatment setting.
This chapter reviews the pathophysiology, definition, classification, epidemiology, etiology, treatment, and prognosis of SE as it occurs in children. The mortality associated with SE is greater in adults than in children, but morbidity and mortality in children are considerable without treatment. Accordingly, current thinking about optimal management uses a more aggressive clinical approach to this neurological emergency, including prompt recognition and initiation of therapy and accelerating the progression of treatment for more rapid termination of the episode [Millikan et al., 2009].
Pathophysiology
A distinguishing feature of SE is the self-sustaining seizure condition. The pathophysiology and biochemical changes underlying the evolution from discrete seizure to SE remain unclear [Lowenstein and Alldredge, 1998; Pellock, 1994; Wasterlain et al., 1993]. Pathophysiological changes that accompany SE can be divided into neuronal (cerebral) and systemic effects. The mechanisms involved in the initiation and maintenance of SE may be different. Ultimately, SE results from a failure of inhibitory mechanisms.
Gamma-aminobutyric acid (GABA) is the most prevalent inhibitory neurotransmitter in the brain. GABAA receptors are postsynaptic ionotropic receptors that bind directly to chloride channels, producing a fast inhibitory postsynaptic potential (IPSP). GABAA receptors are the binding sites for the benzodiazepines and it is the activation of this receptor that accounts for its antiseizure effect. A unique feature of SE compared to brief seizures is the time-dependent development of pharmacoresistance to benzodiazepines [Mazarati et al., 1998]. In animal models, investigators studied GABAA receptor currents by whole-cell patch-clamp techniques in CA1 pyramidal neurons acutely dissociated from rats undergoing lithium/pilocarpine-induced limbic SE and from naive rats [Kapur and Coulter, 1995]. The GABAA receptor current was absent in 47 percent of SE neurons and reduced in 55 percent of the remainder, compared with naive neurons, thus aiding in seizure perpetuation. Kapur et al., using a paired-pulse technique in an electrogenic model of experimental SE, showed that a marked deterioration of GABA-mediated inhibition occurs during continuous hippocampal stimulation [Kapur et al., 1989].
Additionally, more recent work has demonstrated the development of benzodiazepine pharmacoresistance shortly after the onset of ictal spike wave activity [Jones et al., 2002] and in young naive rats [Goodkin et al., 2003]. Treiman et al. reported similar loss of inhibition in hippocampal slices obtained during various electroencephalography (EEG) stages in lithium-/pilocarpine-induced SE [Treiman et al., 2006]. Altered receptor function and changes in representation affect both seizure representation and consequences in the neonatal brain. GABAA receptors are heteromeric protein complexes that mediate most fast synaptic inhibition in the forebrain and have many distinct subtypes. Adult rats that develop epilepsy following pilocarpine-induced SE and adult patients with refractory temporal lobe epilepsy demonstrate significant alterations in GABAA receptor properties in hippocampal dentate granule neurons [Gibbs et al., 1997; Brooks-Kayal et al., 1998, 1999]. In rat pups exposed to pilocarpine-induced SE, different changes are seen in α-1 subunit expression and augmentation compared with those in adult rats. This produced the opposite effect and may serve to enhance inhibition; the rat pups did become epileptic, unlike the adult rats who developed recurrent spontaneous seizures [Zhang et al., 2004a]. Nevertheless, changes in the function of the immature GABAA receptor, from the dual role of excitatory-inhibitory to inhibitory as the rat matures in infancy, may contribute to increased excitability and hence more seizure susceptibility in the neonate [(Khazipov et al., 2004]. Additionally, alteration in glutamate receptor representation may alter seizure susceptibility. Alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors are over-represented in the premature brain, initially in the oligodendrocytes; later they shift their main representation to neurons in the cortex and hippocampus. This condition may partly explain increased risk of periventricular leukomalacia in the preterm infant and increased seizure susceptibility in the term infant [Jensen, 2002]. In one study the effect of SE induced by lithium/pilocarpine in 10-day-old rats demonstrated that pilocarpine-induced prolonged SE caused long-term changes in both glutamate receptors and transporters in hippocampal dentate gyrus. These include a decrease in glutamate receptor 2 mRNA expression and protein levels, as well as an increase in protein levels of the excitatory amino acid carrier 1[Zhang et al., 2004b].
Different studies provide evidence suggesting that endocytosis of GABAA receptors takes place as a seizure transitions to SE. This leads to a decrease in the number of GABAA receptors on the synaptic membrane and therefore a decreased response to benzodiazepines [Naylor et al., 2005; Goodkin et al., 2008]. Resistance to other antiseizure medications, such as phenytoin, has also been demonstrated. In addition to GABAA receptor trafficking, changes in receptor expression and phosphorylation have been shown [Brooks-Kayal et al., 1998, 1999; Terunuma et al., 2008]. Functional changes in voltage-dependent sodium channels have also been described after pilocarpine-induced SE in rats [Remy and Beck, 2006].
Glutamate is the primary excitatory amino acid neurotransmitter and binds to several neuronal receptors, including the N-methyl-d-aspartate (NMDA) receptor, which is activated by depolarization, as well as the AMPA receptor, which mediates fast synaptic transmission in the central nervous system. Using a paired-pulse method in a continuous hippocampal stimulation-induced SE model, NMDA receptors become activated during continuous hippocampal stimulation, and NMDA antagonists block the deterioration of GABA-mediated inhibition [Kapur and Lothman, 1990]. During SE, both NMDA and AMPA receptor subunits increase at the synaptic surface [Mazarati et al., 1998; Wasterlain et al., 2002a]. This increase in glutamate receptors further enhances excitability and is proconvulsant in the midst of uninhibited seizures. In a model of self-sustaining SE, seizures are abolished by NMDA receptor antagonists [Wasterlain et al., 2000].
Neuropeptides have emerged as having a role in SE. Substance P agonists facilitate the initiation of SE in animal models. During the course of SE, the de novo expression of substance P occurs in the cells, such as dentate granule cells, that do not usually express it, possibly playing a role in maintenance of SE [Wasterlain et al., 2002b]. In addition, galanin, a 29–30 amino acid peptide, suppresses hippocampal excitability by post- and presynaptic actions [Mazarati et al., 2000]. However, galanin appears to be depleted by SE.
Laminar necrosis and neuronal damage after prolonged seizures are similar to those after cerebral hypoxia. Neuronal injury and cell death from SE are most prominent in areas rich in NMDA glutamate receptors, including the limbic region. The increase in intracellular calcium is critical to cell death, and calcium activates proteases and lipases that degrade intracellular elements, leading to mitochondrial dysfunction and fatal cellular necrosis. Although young animals may be less likely to develop brain damage from SE [Holmes, 1997; Moshè, 1987], studies using alternative models demonstrate hippocampal cellular injury, even in immature rodents [Sankar et al., 1997; Thompson and Wasterlain, 1994]. It is believed that the glutamate-initiated calcium-dependent cascade is mechanistically similar to the NMDA receptor-mediated cell death occurring during cerebral ischemia. In SE, the degree of neuronal injury is related to seizure duration. In a study of limbic SE induced in adult rats using perforant path stimulation, when animals were allowed to recover, there was evidence of mitochondrial injury and dysfunction demonstrated possibly through a free radical mechanism of injury [Cock et al., 2002]. Further evidence suggests that acute and long-term changes in gene expression may occur after prolonged seizures. These changes in the expression of messenger RNA (mRNA) may lead directly to some of the observed hyperexcitability [Rice and DeLorenzo, 1998].
In animal studies using adolescent baboons, induced SE lasting 1.5–5 hours produced neuronal loss in the hippocampus, cerebellum, and neocortex. Significant cell loss continued to occur, although to a lesser extent if the animals were paralyzed and ventilated with maintenance of oxygen, carbon dioxide, serum glucose, body temperature, and blood pressure [Meldrum, 1974, 1983]. Similar changes have been produced in rat models, as well [Cavalheiro et al., 1987; Sperber et al., 1989].
There are limited data in humans. However, evidence from neuropathology, as well as imaging, has been presented in a pediatric case with direct excitotoxic injury in the absence of hypoxia-ischemia [Tsuchida et al., 2007].
Generalized convulsive SE is associated with serious systemic effects resulting from the metabolic demands of prolonged seizures and the autonomic changes that accompany them: alterations in blood pressure and heart rate, incontinence, emesis, acidosis, hypoxia, changes in respiratory function and body temperature, leukocytosis, rhabdomyolysis, and extreme demands on cerebral oxygen and glucose use [Simon et al., 1997]. Circulating catecholamines increase during the initial 30 minutes of SE, resulting in a hypersympathetic state. Tachycardia, sometimes associated with more severe cardiac dysrhythmias, occurs and may be fatal, but this seems more common in adults [Boggs et al., 1993]. Cardiac output also diminishes, and total peripheral resistance increases, along with mean arterial blood pressure, perhaps because of the sympathetic overload. Hyperpyrexia may become significant during the course of SE, even without prior febrile illness in both children and adults, and may persist for some time. Fever may influence the process of neuronal injury [Liu et al., 1993].
Unlike in adults, in whom biology is more static, the effect of growth and development influences the impact of seizures, both in clinical presentation and in the biologic consequences for the developing brain. Most of our knowledge derives from animal models, so its applicability to the human situation is unclear. Experimental models of seizures in immature animals suggest comparatively less vulnerability to seizure-induced brain injury than in mature animals [Wong and Yamada, 2001]. Repetitive or prolonged neonatal seizures may increase the susceptibility of the developing brain to experience subsequent seizure-induced brain injury later in life. This susceptibility appears to be more closely related to alterations in neuronal connectivity and network properties, rather than to increased cell death during the neonatal period [Holmes et al., 1998; Koh et al., 1999; Schmid et al., 1999].
Rats exposed to early-life seizures demonstrate persistent changes in CA1 hippocampal pyramidal cells, possibly leading to long-term changes in behavior and learning and in epileptogenicity [Villeneuve et al., 2000].
Definition
SE is internationally classified as a seizure lasting more than 30 minutes or recurrent seizures producing more than 30 minutes during which the patient does not regain consciousness [ILAE, 1981]. The World Health Organization previously defined SE as “a condition characterized by epileptic seizures that are so frequently repeated or so prolonged as to create a fixed and lasting condition” [Gastaut, 1982]. Lack of recovery for a fixed period, possible frequent repetition, prolongation, and possible propagation of further seizures are inherent in this definition. In the past, the definition of SE required 1 hour of continuous seizures, but more recent studies have used a 30-minute duration of continuous or recurrent seizures without full recovery as the standard clinical and electrographic definition of SE. The recognition that longer duration of seizures increases risk for long-term injury and the risk for fracture during seizures and their treatment has required a definition implying need for expediency in stopping prolonged seizures. Lowenstein and associates proposed an “operational” definition of 5 minutes or more of continuous seizures or “two discrete seizures between which there is incomplete recovery of consciousness” in adults and children older than 5 years of age [Lowenstein et al., 1999]. This definition applies primarily to generalized convulsive SE and may be used to direct treatment to avoid refractory SE, as well as its sequelae. Aggressive early treatment is justified by recent work demonstrating a 10-fold lower rate of mortality for seizures of 10–29 minutes’ duration versus those lasting longer than 30 minutes [DeLorenzo et al., 1999].
Because of the difficulty in diagnosing and quantifying seizures in the neonate, no broadly accepted definition of SE in the neonate exists. A proposed definition is either 30 minutes of continuous electroencephalographic seizures or presence of seizure activity for 50 percent of the EEG recording time, with or without the expression of coincident clinical signs (Scher et al., 1993b). Debate continues regarding what constitutes a neonatal seizure. Neonatal seizures can be broken down into three categories: electroclinical, electrographic, and clinical only. Controversy still exists about whether episodic abnormal movements seen in some infants, not accompanied by simultaneous ictal discharges on the EEG, are true seizures. Many neonatal paroxysmal events classified as “subtle seizures” have no EEG correlate [Mizrahi and Kellaway, 1998]. Typical subtle seizures include movements of progression, such as bicycling, oral-buccal-lingual movements, such as chewing and tongue thrusting, and other movements, such as random eye movements. Other movements that typically have no EEG correlate include generalized tonic posturing. Movements typically demonstrating simultaneous ictal discharges on EEG include focal clonic, multifocal clonic, and focal tonic; myoclonic movements may or may not have an EEG correlate [Mizrahi and Kellaway, 1998].
Classification
Any type of seizure may become prolonged and thus develop into SE (Box 58-1). Classification of SE should be performed by observing the clinical events and combining electrographic information when possible. The fundamental distinction between seizures is that some are generalized from onset, whereas others are partial in onset. The latter type may or may not then secondarily generalize. From a management standpoint, however, it may be more useful to consider whether the event is convulsive or nonconvulsive, as this may impact more directly on ready recognition and intervention.












Generalized tonic-clonic SE is the most dramatic and life-threatening form of SE. Myoclonic, generalized clonic, and generalized tonic SE occur primarily in children. These children usually have encephalopathic epilepsies [Lockman, 1990; Pellock, 1994; Treiman, 1993], and their consciousness may be preserved throughout the attacks. About one-half of the cases of generalized clonic SE occur in normal children, in whom it is associated with prolonged febrile seizures; the remaining half are distributed among those children with acute and chronic encephalopathies [DeLorenzo et al., 1992]. Generalized tonic SE appears most frequently in children, particularly those with the Lennox–Gastaut syndrome. Prolonged generalized tonic convulsions have been precipitated by benzodiazepine administration.
Nonconvulsive SE also may include complex partial, simple partial, and absence seizures that continue for more than 30 minutes [Kaplan, 1996; Scholtes et al., 1996; Stores et al., 1995]. Complex partial SE may be manifested as an epileptic twilight state marked by a cyclic variation between periods of partial responsiveness and episodes of seemingly motionless staring and complete unresponsiveness accompanied, at times, by automatic behavior [Delgado-Escueta and Treiman, 1987; Privitera, 1997; Scher et al., 1993a; Treiman, 1993]. Simple partial SE is characterized by focal seizures that may persist or be repetitive for at least 30 minutes without impairment of consciousness. When this condition lasts for hours or days, it is termed epilepsia partialis continua [Cockerell et al., 1996; Takahashi et al., 1997]. Absence, or petit mal, status also has been referred to as spike-wave stupor. This type of nonconvulsive SE may be extremely difficult to differentiate from complex partial SE without the aid of an EEG. Classically, features of absence status include a continuous alteration of consciousness without the cyclic variations seen with complex partial SE [Grin and DiMario, 1998]. The EEG recording exhibits prolonged, sometimes continuous, generalized synchronous 3-Hz spike-and-wave complexes, rather than focal ictal discharges, which characterize partial SE [Porter and Penry, 1983; Treiman, 1993]. Absence status does not appear to cause permanent neurological damage [Drislane, 1999]. The child presenting with a prolonged confused state, with a fluctuating level of consciousness or with prolonged unconsciousness, may require both clinical and EEG evaluations in addition to other studies.
Nonconvulsive SE is most likely not rare but simply underdiagnosed. A special category of nonconvulsive SE is subtle SE [Treiman, 1990, 1993]. These patients have severe encephalopathies stemming from a variety of intracranial processes or prolonged uncontrolled convulsive seizures. This type of SE is manifested clinically by the occurrence of mild motor movements, such as nystagmus, or by clonic twitches, which may be unilateral and are intermittent, brief, and without a true sequential pattern. These subtle movements are associated with marked impairment of consciousness, usually with continuous bilateral EEG ictal patterns. These continuing electrographic seizures that are not accompanied by clinical manifestations demonstrate a true “electroclinical dissociation,” and may be seen not only in neonates but also in severely ill children and adults [Mizrahi and Kellaway, 1987; Scher et al., 1993a]. The EEG progressively becomes uniform to produce a pattern of continuous ictal discharges, which then becomes interrupted by periods of relative flatness and then severe cortical depression. In a study of children, nonconvulsive SE most commonly followed a bout of convulsive SE or briefer convulsive seizure but with prolonged alteration in mental status [Tay et al., 2006], and is more likely to occur in children with a prior history of epilepsy [Abend et al., 2007] and remote risk factors for seizures [Classen et al., 2004]. In general, these patients respond poorly to traditional treatment with antiepileptics.
Febrile SE, which is unique to children, represents the extreme end of the complex febrile seizure spectrum. Febrile SE has long been suspected of having a relationship to the development of mesial temporal sclerosis. Patients with intractable temporal lobe epilepsy and mesial temporal sclerosis often have histories of severe febrile convulsions as infants. Diagnostic advances made possible by magnetic resonance imaging (MRI) have shown that very prolonged febrile convulsions may produce hippocampal injury [Lewis, 1999]. More recent studies support the link to the development of mesial temporal sclerosis [Provenzale et al., 2008]. Neuroimaging studies generally show hippocampal swelling during the acute stage [Scott et al., 2002, 2006]. A prospective study investigating long-term outcome in febrile SE found that most were partial (67 percent), and SE was unrecognized in the emergency department about a third of the time [Shinnar et al., 2008]. Human herpesvirus-6B appears to be the most common cause of febrile SE and may play an important pathogenic role in the etiology of mesial temporal lobe epilepsy [Theodore et al., 2008].
Epidemiology
It is projected that between 102,000 and 152,000 events occur in the United States annually, an incidence 2–2.5 times greater than that previously proposed by Hauser [1990], who reported that SE occurs annually in 50,000–60,000 persons in the United States [DeLorenzo et al., 1996; Hauser et al., 1990]. More recent work continues to demonstrate a high incidence of SE varying between 20 and 41 patients per year per 100,000 population [Govoni et al., 2008; Chin et al., 2006; Coeytaux et al., 2000].
Approximately one-third of cases manifest as the initial seizure of a developing epilepsy, one-third occur in patients with previously established epilepsy, and one-third occur as the result of an acute isolated brain insult. Among those previously diagnosed as having epilepsy, estimates of SE occurrence range from 0.5 to 6.6 percent [DeLorenzo et al., 1996].
Hauser reported that up to 70 percent of children who have epilepsy that begins before the age of 1 year will experience an episode of SE [Hauser, 1990]. Also, within 5 years of the initial diagnosis of epilepsy, 20 percent of all patients will experience an episode of SE. Although the subsequent development of epilepsy is likely in adults with SE as their first unprovoked seizure [Hauser et al., 1990], a prospective study of children with SE found only a 0.3 probability plotted on a Kaplan–Meier curve that epilepsy will develop after ≥9 months in those who initially presented with SE [Maytal et al., 1989].
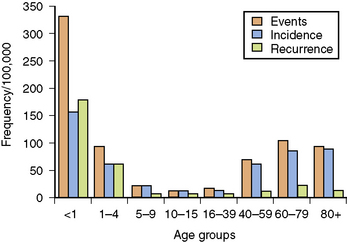
Among children, SE is most common in infants and young toddlers, with more than 50 percent of cases of SE occurring in children younger than 3 years of age [Shinnar et al., 1995]. In a study in Richmond, Virginia, total SE events and incidence per 100,000 population per year demonstrated a bimodal distribution, with the highest values during the first year of life and during the decades beyond 60 years of age [DeLorenzo et al., 1992, 1995, 1996]. Infants younger than 1 year of age represent a subgroup of children with the highest incidence of SE, whether events, total incidents, or recurrences are counted. The recurrence rate for SE in the Richmond study was 10.8 percent [DeLorenzo et al., 1996], but 38 percent of patients younger than 4 years of age had repeat episodes. Children have a much lower mortality rate than adults after adequate treatment [Dunn, 1988; Maytal et al., 1989; Pellock, 1993b; Phillips and Shanahan, 1989; Shinnar et al., 1995]. Age, etiology, and duration directly correlate with mortality [DeLorenzo et al., 1996; Towne et al., 1994].
Etiology
SE usually is a manifestation of an acute precipitating event that affects the central nervous system (CNS) or is an exacerbation of symptomatic epilepsy. Less than 10 percent of cases of SE in adults and children are truly idiopathic in that no precipitating or associated cause can be identified [DeLorenzo et al., 1995, 1996]. Acute symptomatic causes are those most commonly associated with prolonged SE lasting for longer than 1 hour [DeLorenzo et al., 1996]. Thus, a full evaluation for etiology must be undertaken in every case of SE [Dodson et al., 1993; Pellock, 1994]. In patients with pre-existing epilepsy, a precipitating or associated factor may be clearly identified. Identification of this factor may help in treating the episode of SE, preventing further consequences of SE, and perhaps preventing future recurrences.
Although, typically, a precipitant to SE can be identified, a genetic susceptibility may predispose certain persons to develop prolonged seizures in response to an acute insult. Recent work in twins demonstrated a higher incidence in monozygotic twins than in dizygotic twins, providing evidence for a genetic contribution to the risk for SE [Corey et al., 2004]. Seizure type and specific epilepsy syndrome may differ between monozygotic twins, however, and SE may not be a function of the seizure type or syndrome experienced by each person.
A clear difference between causative disorders of SE in adults (Table 58-1) and those in children (Table 58-2) has been identified [Pellock and DeLorenzo, 1997; Riviello et al., 2006]. A major cause of SE in children is that associated with fever secondary to non-CNS infections, an etiology that essentially does not exist in adults. Inadequate antiepileptic drug levels and remote causes, including congenital malformations, also account for a significant number of episodes of SE in children, although some studies have found that patients may have had reasonable drug levels when SE occurred [Maytal et al., 1996]. Of note, many patients with subtherapeutic levels of antiepileptic drugs closely followed the instructions of their physicians and recently had drug-dosage alterations.
Table 58-1 Cause and Mortality Data for Status Epilepticus in Adults
| Etiologic Disorder/Condition | % of Cases | Mortality Rate (%) |
|---|---|---|
| Anoxia | 5 | 71 |
| Hypoxia | 13 | 53 |
| Cerebrovascular accident | 22 | 33 |
| Hemorrhage | 1 | 0 |
| Tumor | 7 | 30 |
| Infection | 7 | 10 |
| Central nervous system infection | 3 | 0 |
| Metabolic | 15 | 30 |
| Low antiepileptic drug level | 34 | 4 |
| Drug overdose | 3 | 25 |
| Alcohol withdrawal | 13 | 20 |
| Trauma | 3 | 25 |
| Remote | 25 | 14 |
| Idiopathic | 3 | 25 |
(From Pellock JM, DeLorenzo RJ. SE. In: Porter RJ, Chadwick D, eds. The epilepsies 2. Boston: Butterworth–Heinemann, 1997;267.)
Table 58-2 Etiologies of Status Epilepticus in Children
| Etiology | Percentage |
|---|---|
| Remote symptomatic epilepsy | 33% |
| Acute symptomatic seizures | 26% |
| Febrile | 22% |
| Cryptogenic | 15% |
| Central nervous system infection | 13% |
| Acute metabolic disorders | 6% |
(From Riviello JJ et al. Practice Parameter: Diagnostic assessment of the child with status epielpticus [an evidence based review]. Neurology 2006;67:1542.)
The distribution of causes associated with SE in children is highly age-dependent [Shinnar et al., 1995]. More than 80 percent of children younger than 2 years of age have SE from a febrile or acute symptomatic cause, whereas cryptogenic or remote symptomatic causes were more common in older children. By contrast, in adults, subtherapeutic levels of antiepileptic drugs, remote causes, and cerebrovascular accidents represent the three most common causes of SE [Pellock and DeLorenzo, 1997]. In adults, SE resulting from remote causes occurred primarily in relation to stroke, so that both acute and previously occurring strokes account for a significant proportion of adult episodes of SE. Stroke is the etiologic disorder in approximately 10 percent of childhood SE episodes, either as the primary acute cause or as a remote event.
Recurrence rates for SE are age-specific, as illustrated in Figure 58-1. Repeat occurrences are much more common in children younger than 1 year of age. In the prospective Richmond study of SE, pediatric, adult, and elderly recurrence rates were 35 percent, 7 percent, and 10 percent, respectively [DeLorenzo et al., 1996]. Recurring SE is more frequent in children with remote symptomatic encephalopathy or progressive degenerative disease [DeLorenzo et al., 1995, 1996]. The extent of clinical and laboratory evaluation that should be performed in each child with recurrent SE will depend on the presentation, signs and symptoms, and underlying medical condition of the patient. For example, a child with recurring SE who has an indwelling shunt for hydrocephalus will almost always need to be evaluated for the possibility of shunt malfunction or infection.
Recently, CNS or systemic autoimmune disorders have been recognized as causes of SE [Shorvon and Tan, 2009]. In some cases, the diseases may be paraneoplastic, but for many, no cause has been determined. Since patients with these etiologies usually are described in case reports or small series, the contribution of the disorders to the epidemiology of SE is difficult to assess, but they appear to represent a fraction of the cases previously diagnosed as infectious, since the patients often have a syndrome qualifying as an encephalitis.
A recently described entity with seizures and persistently altered mentation, including a fluctuating level of consciousness or with prolonged unconsciousness, is anti-NMDA-receptor encephalitis [Dalmau et al., 2008]. Anti-NMDA-receptor encephalitis is a disorder with antibodies against the NR1 subunit of the receptor. This disorder largely affects young people, and its diagnosis is facilitated by the characteristic clinical picture that develops in association with cerebrospinal fluid pleocytosis. Only about half the patients had MRI abnormalities. Recovery from this disorder is typically slow, and symptoms may relapse. The mainstay of treatment is immunotherapy with steroids, plasma exchange, or intravenous immunoglobulin. These immunotherapies may be given individually or in combination [Dalmau et al., 2008]. Other possible autoimmune illnesses include systemic lupus erythematosus, antineuronal antibody syndromes with limbic encephalitis, limbic encephalitis following various systemic viral infections, and Hashimoto’s encephalopathy (autoimmune thyroid encephalopathy).
Management and Therapy
Overview
SE is a neurologic and medical emergency. Therapy includes maintenance of respiration, general medical support, and specific treatment aimed at stopping both electrographic and clinical seizures while the cause of the event is investigated [Epilepsy Foundation of America, 1993]. Prompt diagnosis and management provide the best outcome [DeLorenzo, 1990; Pellock, 1993a, 1993b, 1994; Pellock and DeLorenzo, 1997; Rider and Thapa, 1995; Treiman, 1993].
A single generalized convulsion in a child with a prolonged period of impaired consciousness is much more difficult to diagnose, and an EEG should be obtained urgently. If on-going ictal discharges or electrographic seizures are noted, the patient should be considered to be in electrographic SE, and prompt treatment is indicated. The goals of SE emergency management are listed in Box 58-2. Clinical and electrographic seizure activity must be rapidly terminated while ensuring optimal oxygenation and metabolic balance. Both clinical and electrical seizure activity should be terminated as soon as possible [Treiman, 1990]. The longer an episode of SE continues, the more likely it is to result in permanent neurologic damage and to become refractory to treatment [DeLorenzo, 1990; DeLorenzo et al., 1996; Hauser, 1990; Pellock and DeLorenzo, 1997; Towne et al., 1994; Treiman, 1990; Treiman et al., 1992, 1994].
Box 58-2 Goals of Emergency Management for Status Epilepticus







(From Pellock JM, DeLorenzo RJ. SE. In: Porter RJ, Chadwick D, eds. The epilepsies 2. Boston: Butterworth–Heinemann, 1997:267.)
Rapid initiation of care is essential to ensure the best possible outcome. Increased seizure duration commonly is regarded as an important factor contributing to increased morbidity and mortality. Prolonged seizures of any type are associated with an increased risk of complications [Lowenstein et al., 1999]. In a study in adults and children, the mortality rate in SE was 34.8 percent when seizures lasted longer than 1 hour, compared with 3.7 percent when seizure duration was less than 1 hour [DeLorenzo et al., 1992]. Therefore, limiting the time from seizure onset to initial treatment and attainment of control is essential to minimize the complications of prolonged seizures. In a retrospective study, patients with SE may not arrive at the emergency department for 30 minutes or longer, and treatment initiation may not occur for 40–263 minutes, thereby increasing risk for prolonged seizures [Jordan, 1994]. The Veterans Affairs Cooperative Study reported that the mean delay before treatment of SE was 2.8 hours in generalized convulsive SE and 5.8 hours in subtle SE; this study involved exclusively adult patients [Treiman et al., 1998]. In a prospective study of 889 patients (625 adults, 264 children), 41.5 percent received treatment within 30 minutes and 70.9 percent within 60 minutes of seizure onset; 18.1 percent did not have treatment initiated until after 90 minutes of seizures. This latter group of patients, however, included patients in coma diagnosed with subtle or nonconvulsive SE [Pellock et al., 2004].
Supportive Care
In children in SE, cardiorespiratory function must be assessed immediately by vital sign determination, auscultation, airway inspection, and determination of arterial blood-gas concentrations, with suctioning when necessary. Although these children may appear to be breathing spontaneously on arrival at the emergency department, they may already be hypoxic, with respiratory and metabolic acidosis, apnea, aspiration, or central respiratory depression. The need for ventilatory support depends not only on the respiratory status at the time of presentation but also on the conditions before arrival and the ability to maintain adequate oxygenation throughout on-going seizures and during the intravenous administration of large doses of antiepileptic drugs, all of which may cause respiratory depression. In the neurologically depressed patient, elective intubation and respiratory support are recommended. Placement of an oral airway or nasal oxygen cannula will prove to be insufficient in most patients, because respiratory drive is depressed [Pellock, 1994]. Respiratory arrest and depression are principal factors contributing to morbidity and mortality [DeLorenzo et al., 1996]. Rapidly assessing vital signs and performing a general neurologic examination may provide clues to the etiology of SE.
Laboratory Testing
Blood should be drawn for the determination of blood gases, glucose, calcium, electrolytes, complete blood count, and antiepileptic drug levels, as well as for cultures (bacterial and viral) if indicated. Urine drug and metabolic screening should be performed whenever the etiology cannot be determined. As previously noted, a common etiologic or associated factor found in children with SE is intercurrent infection. In a small number of these children, the clinical presentation will include manifestations similar to the initial symptoms of CNS infection. Lumbar puncture should be done early in the course of management, although not necessarily during the initial phase of stabilization [Rider et al., 1995]. In most children, only rarely should the clinician wait to obtain neuroimaging studies before performing a lumbar puncture. Hyperpyrexia may become significant during the course of SE, even in the absence of a febrile illness. Rectal temperatures should be monitored and fever aggressively treated; significant elevations of temperature may contribute to brain damage [Dodson et al., 1993].
Bradyarrhythmias are commonly noted in children when large doses of antiepileptic drugs are administered. In adults, changes range from evidence of myocardial ischemia to tachyarrhythmia [Boggs et al., 1993, 1994; Tigaran et al., 1997]. These are less frequently noted in children. In children with known congenital heart disease or dysrhythmias, electrocardiographic monitoring must be implemented and continued.
Use of Neuroimaging
The decision to perform neuroimaging studies in the child presenting with SE needs to be individualized according to the available history, general physical and neurologic examination findings, and the possibility of significant structural intracranial pathology or increased intracranial pressure, or the presence of lateralizing findings. A Practice Parameter for imaging the child with SE recommends that, if etiology is unknown or it is clinically indicated, imaging should be performed after SE has been controlled and the patient stabilized [Riviello et al., 2006]. Clinical indications would be similar to the indications for neuroimaging in the emergency patient presenting with a seizure [Practice Parameter, 1996]. The incidence of imaging abnormalities, presumably in patients with clinical indication rather than in all patients, ranges from 29 to 70 percent in class III studies. Common findings were cerebral edema (14 percent), atrophy (12.1 percent), infection (4.6 percent), dysplasia, infarction (2.9 percent), tumor and hematoma (2.3 percent each), and trauma and arteriovenous malformation (1.2 percent each) [Riviello et al., 2006]. MRI is more sensitive and specific than CT. Transient focal findings may be seen [Kramer et al., 1987] and diffusion weighted imaging may reveal vasogenic and cytotoxic edema [Scott et al., 2002]. A recent review of neuroimaging in the emergency department demonstrates that CT of the brain in children with a first seizure changes acute management 3–8 percent of the time [Harden et al., 2007].
A growing literature recognizes peri-ictal changes on neuroimaging and discusses their significance. Local and immediate findings have been described in children. Local swelling of the hippocampus has been described in children with prolonged febrile convulsions [Scott et al., 2002, 2006; VanLandingham et al., 1998]. At follow-up evaluation in some patients, edematous areas later appeared atrophic [Scott et al., 2002]. However, there is some evidence that an underlying, predisposing hippocampal abnormality may be present [Scott et al., 2006]. Prolonged seizure as a potential causative pathophysiological sequence to the development of temporal lobe epilepsy has been extensively investigated in animal models in which SE is induced by chemical [Meldrum, 1997; Ben Ari, 1985; Cavalheiro, 1995] or electrical [Lothman et al., 1989] means. Most recently, a study of 11 children with a history of febrile SE with MRI within 72 hours of SE and follow-up 2 years later found that seven demonstrated MRI signal hyperintensity in the hippocampus. On follow-up imaging, five children had hippocampal volume loss and increased signal intensity, meeting the criteria for mesial temporal sclerosis. Four children had developed focal-onset epilepsy [Provenzale et al., 2008].
Focal edema with effacement of gyri [Silverstein and Alexander, 1998] can develop, with subsequent atrophy [Meierkord et al., 1997]. This finding appears to represent cytotoxic edema; in animal models, increased lactate has been found, and this increase correlates with the degree of histologic damage [Najm et al., 1998].
More confounding is the finding of lesions remote from the ictal focus. The lesions may be remote in time, such as in cases in which transient lesions develop days after SE [Hisano et al., 2000], or remote anatomically. These remote lesions typically are associated with transient focal findings on examination, such as weakness [Hisano et al., 2000] or personality changes. Lesions in the splenium have been associated with psychiatric disease [Cole, 2004].
Use of Electroencephalographic Monitoring
EEG monitoring allows optimal management of SE and should be used whenever available. EEG monitoring is extremely useful, but under-utilized, in initial and subsequent management of SE [Jaitly et al., 1994; Pellock, 1993a, 1993b; Pellock and DeLorenzo, 1997; Treiman, 1993]. A recent prospective study looked at the utility of emergency EEG. The most common indication was diagnosis of convulsive SE or follow-up of SE. In 77.5 percent of the cases, the clinician considered the EEG had contributed to the diagnosis. Overall, the EEG resulted in a modification in treatment in 37.8 percent of the cases [Praline et al., 2007]. Classification of seizure type and evidence regarding etiology and prognosis may be determined from the EEG. Especially useful are the findings in patients with pseudoseizures and those associated with medication overdoses or focal pathologic entities [Dodson et al., 1993; Riviello et al., 2006; Sonnen, 1997].
In children with prolonged coma or confusional states, nonconvulsive SE can sometimes be diagnosed only with the use of EEG recordings. The EEG is essential; motor findings may be subtle facial or limb movement, or isolated eye movements, or the movements may be absent altogether [Drislane et al., 2008]. Researchers in one study used EEG to diagnose SE in 37 percent of patients with altered consciousness whose diagnosis was unclear clinically [Privitera et al., 1994]. In another study, 8 percent of coma patients met the criteria for nonconvulsive SE [Towne et al., 2000]. In a study in children with unexplained decrease in level of consciousness or suspected subclinical seizures, 19 percent were found to be experiencing seizures by continuous EEG monitoring [Classen et al., 2004].
EEG can help to confirm that an episode of SE has ended. When use of neuromuscular blocking agents is contemplated or when recurrence of seizures cannot be documented by clinical observation (i.e., because of coma or large doses of medication), EEG monitoring is mandatory. Electroclinical dissociation may result from the administration of large doses of antiepileptic drugs in newborns and older children. In one study, 50 percent of patients continued to experience electrographic seizures with no clinical correlate in the 24 hours following cessation of SE [DeLorenzo et al., 1998]. Patients with SE who fail to recover rapidly and completely should be monitored with EEG for at least 24 hours after an episode to ensure that recurrent seizures are not missed. The presence of periodic discharges in these patients suggests the possibility of preceding SE, and careful monitoring may clarify the etiology of these discharges and allow detection of recurrent SE. Conversely, patients whose clinical condition has not returned to baseline may have abnormal movements or postures that, without EEG monitoring, may lead to overtreatment [Ross et al., 1999].
The EEG may provide prognostic information as well. The presence of periodic epileptiform discharges during or after SE is associated with a higher risk of poor outcome [Nei et al., 1999]. Additionally, in refractory SE in children, outcome is poorer with multifocal or generalized abnormalities than with a focal abnormality on the initial EEG [Sahin et al., 2001].
There is increasing use of continuous EEG, primarily for the diagnosis and management of nonconvulsive SE. Seizures, predominantly nonconvulsive seizures, are common in the critically ill child, with one study showing in incidence of 44 percent [Jette et al., 2006]. In this study a 1-hour recording only captured seizures in half the patients ultimately diagnosed with on-going seizures, but this rose to 80 percent with a 24-hour recording, demonstrating the importance of continuous EEG in the critically ill child. The incidence of nonconvulsive SE in children has been estimated to be 23–35 percent of patients monitored [Hosain et al., 2005; Tay et al., 2006; Abend et al., 2007], with prior epilepsy, hypoxic-ischemic injury, and stroke identified as the most common etiologies. While we often become concerned about nonconvulsive SE following convulsive SE, this may not occur with any regularity. Undiagnosed nonconvulsive SE often was found only in the case of brief convulsive seizures followed by persistently altered level of consciousness [Tay et al., 2006]. In a study of hypothermia for neurological protection following cardiac arrest in 19 children, continuous EEG was used and found seizures in 47 percent of patients, and nonconvulsive SE in 32 percent [Abend et al., 2009a].
Medical Therapy
Prehospital
The majority of SE begins outside of the hospital. Traditionally, rapid treatment focused on recognition and rapidity of transport to an appropriate emergency department. Over the last few decades, however, the focus has shifted to initiating treatment in the prehospital setting. Early intervention with first-line drug therapy has been associated with an 80 percent response rate; response rates decline progressively the longer treatment is delayed [Lowenstein et al., 1993]. Seizures of longer duration are associated with significantly increased morbidity, which include more intensive treatment in the emergency department, and admission to an intensive care unit. There is evidence that prehospital treatment with diazepam, whether intravenous or rectal, is effective for decreasing the complications of prolonged seizures [Lowenstein et al., 1993]. Treatment outside of the hospital has been associated with decreased emergency department visits [Kriel et al., 1991; O’Dell et al., 2005b]. A review of 45 children indicated that prehospital treatment was associated with a shorter duration of SE (32 minutes versus 60 minutes) and a lower incidence of recurrence of seizures in the emergency department [Allredge et al., 1995].
Initial Therapy in the Emergency Department
Treatment should begin with placement of an intravenous line. Fluid restriction is rarely necessary. In children, immediately after placement of the intravenous line, 25 percent glucose (2–4 mL/kg) should be given by bolus if hypoglycemia is suspected. In adolescents and adults, the intravenous line typically is kept open with normal saline, and the patient is given 100 mg of thiamine, followed by 50 mL of 50 percent glucose by slow intravenous push if hypoglycemia is documented [Pellock and DeLorenzo, 1997; Treiman, 1993]. Whenever the intravenous route cannot be established, the intraosseous route can be used efficiently for fluid and medication administration [Orlowski et al., 1990].
The most frequent error committed in the treatment of SE is the initial administration of inadequate doses of an antiepileptic drug. This suboptimal regimen is then followed by a waiting period until more seizures occur before administration of the necessary total dose [Dodson et al., 1993; Pellock and DeLorenzo, 1997]. Management of SE is best accomplished by use of a predetermined protocol in most settings [Pellock, 1994; Pellock and DeLorenzo, 1997; Treiman, 1990]. In almost all situations in which it is medically possible, drugs should be administered intravenously to ensure the most rapid delivery of maximal doses to the brain. The ideal antiepileptic drug for the treatment of SE should optimally have the following properties:
Rapid onset of action and bioavailability of parenteral forms have made benzodiazepines the preferred initial agent for treatment of SE. Lorazepam has become popular in many centers as the initial agent because of its overall advantages over diazepam of rapid onset of action and longer pharmacologic effect [Appleton et al., 1995; Pellock, 1994; Pellock and DeLorenzo, 1997; Treiman, 1990]. When SE continues after the initial dose of benzodiazepines is administered and persists after a primary antiepileptic drug, such as phenytoin, fosphenytoin, or phenobarbital is given, a second benzodiazepine dose should be administered before changing to an alternative antiepileptic drug. A protocol currently used for the management of SE in children by investigators at the Medical College of Virginia at the Virginia Commonwealth University is outlined in Table 58-3 (modified from Pellock and DeLorenzo [1997]). Similar protocols are becoming universally accepted. Recently, in the United Kingdom a committee investigating ideal therapy for SE in children recommended a protocol using a rapid-onset medication, such as a benzodiazepine after initial stabilization, followed by phenytoin or phenobarbital, and then, if necessary, an anesthetic [Appleton et al., 2000].
Table 58-3 Medical College of Virginia Status Epilepticus Treatment Protocol for Children*
| Elapsed | ||
|---|---|---|
| Step | Time† | Procedure |
| 1 | 0–5 min | Determination of SE. As soon as the diagnosis is made, institute monitoring of temperature, blood pressure, pulse, respirations, ECG, and EEG. Insert oral airway and administer oxygen if necessary. Insert an intravenous catheter and draw venous blood for levels of antiepileptics, glucose (check Dextrostix), electrolytes, calcium, BUN, CBC. Draw arterial antipyretics (acetaminophen). Perform frequent suction |
| 2 | 6–9 min | Place an intravenous line with normal saline. Administer a bolus of 2 mL/kg 50% glucose |
| 3 | 10–30 min | Initial treatment consists of an infusion of intravenous lorazepam given at a rate of 1–2 mg/min (0.1 mg/kg) to a maximum dose of 8 mg. This is followed by intravenous phenytoin (fosphenytoin; PHT [FPHT]), 18–20 mg/kg, infused at a rate not to exceed 1 mg/kg/min or 50 mg/min. Monitor ECG and blood pressure. May repeat phenytoin (FHPT), 10 mg/kg before proceeding to next step. Alternatives to phenytoin includes intravenous valproate, 20–40 mg/kg at rates of 3–6 mg/kg/min or levetiracetam 30–60 mg/kg intravenous over 15 minutes |
| 4 | 31–59 min | If seizures persist, administer a bolus infusion of phenobarbital at a rate not to exceed 50 mg/min until seizures stop or to a loading dose of 20 mg/kg |
| 5 | 60 min | If control is still not achieved, other options include the following: (1) Midazolam with an initial intravenous loading dose of 0.15 mg/kg, followed by a continuous infusion of 1–2 μg/kg/min titrating every 15 minutes to produce seizure control on EEG. Treatment is typically 12–48 hours (2) Pentobarbital with an initial intravenous loading dose of 5 mg/kg with additional amounts given to produce a burst-suppression pattern on EEG. Maintenance of pentobarbital anesthesia is continued for approximately 4 hours by an infusion of 1–5 mg/kg/hr. The patient is then checked for the reappearance of seizure activity by decreasing the infusion rate. If clinical seizures and/or generalized discharges persist on EEG, the procedure is repeated; if not, the pentobarbital is tapered over 12–24 hours |
| 6 | 61–80 min | If seizures are still not controlled, call the anesthesia department to begin general anesthesia with halothane and neuromuscular blockade |
Lumbar puncture should be performed as soon as possible, especially in a febrile child or infant younger than 1 year of age. For infants with a history of neonatal seizures, infantile spasms, or early-onset seizures, pyridoxine 100 mg should be administered intravenously while EEG monitoring is being performed, for both diagnosis and treatment of seizures due to a vitamin B6 deficiency (a rare cause).
BUN, blood urea nitrogen; CBC, complete blood count; D5W, 5% dextrose in water; ECG, electrocardiogram; EEG, electroencephalogram.
* Note: Continuous monitoring of EEG is recommended in an obtunded patient to ensure that SE has not recurred. For management of intractable cases, a neurologist who has expertise in SE should be consulted, and advice from a regional epilepsy center should be sought.
† Time from beginning of intervention.
(Adapted from Pellock JM, DeLorenzo RJ. SE. In: Porter RJ, Chadwick D, eds. The epilepsies 2. Boston: Butterworth–Heinemann, 1997;267.)
The choice of the optimal pharmacologic agent may not be identical for every patient with SE. Benzodiazepines and phenytoin (or fosphenytoin) as initial therapy are preferred by some clinicians, but others may wish to use alternative agents if the patient is known to be on maintenance therapy or already has received smaller doses of phenytoin or phenobarbital [Holmes, 1990; Pellock and DeLorenzo, 1997]. Lorazepam, diazepam, phenytoin or fosphenytoin, and phenobarbital are accepted agents for initial and continued therapy of SE. The large SE treatment study done in adults and sponsored by the U.S. Veterans Administration compared four intravenous drug regimens:
The first three regimens were found to be superior to phenytoin alone for initial management of generalized convulsive SE in adults [Treiman et al., 1994]. The choice of an initial agent may depend on individual patient characteristics, prior antiepileptic drug therapy, and physician preference. Typically, however, lorazepam has become the first-line treatment of choice. Less uniformity exists in the choice of the second-line agent. Currently, there is no evidence to guide the treating physician in choosing among phenytoin plus phenobarbital, valproate, or levetiracetam [Shorvon et al., 2008].
Benzodiazepines
Intravenous lorazepam and intravenous diazepam have been found to be of equal efficacy in children [Qureshi et al., 2002], but intravenous lorazepam has a longer half-life and less respiratory depression [Appleton et al., 1995; Treiman et al., 1998]. The availability of a rectal preparation provides an alternative when intravenous access cannot be obtained. Intubation is less often required with rectal diazepam than with intravenous diazepam, and seizure recurrence is reduced [Diekmann, 1994]. Diazepam rectally may be used in many out-of-hospital settings for patients with recurrent SE, prolonged seizures, or acute repetitive seizures [Camfield et al., 1989; Dooley, 1998; Pellock, 1998]. Based on the long history of use of rectal diazepam in Europe, as well as experience in the United States and Canada, the following dosages are recommended: age 2–5 years, 0.5 mg/kg/dose; age 6–11 years, 0.3 mg/kg/dose; and age 12 years and older, 0.2 mg/kg/dose. These dosages yield an effective minimum plasma concentration of approximately 150–500 ng/mL [Morton et al., 1997b]. Although midazolam is rarely used in the U.S. for initial treatment of SE, it is efficacious and offers alternative administration routes. Unlike other benzodiazepines, midazolam is water-soluble, allowing for intramuscular injection. The intramuscular administration of midazolam may be particularly useful when intravenous access is unavailable or for prehospital treatment. A prospective, randomized study compared intramuscular midazolam with intravenous diazepam in the emergency department. In evaluating time to seizure control after administration, a 4-minute difference favoring midazolam was found [Chamberlain et al., 1997]. Nasal administration is possible, but absorption may be affected by nasal secretions [Wallace, 1997]. Liquid midazolam is rapidly absorbed through the buccal mucosa and is as effective as rectal diazepam [Scott et al., 1999].
Phenytoin/Fosphenytoin
Fosphenytoin is a water-soluble prodrug of phenytoin delivered in a neutral pH solution [Browne, 1997; Leppik et al., 1987; Ramsay and DeToledo, 1996]. This prodrug is replacing the present injectable phenytoin preparation [Pellock, 1996]. Dosing is safe at 150 mg/min (3 mg/kg/min), a rate three times more rapid than that for phenytoin. Fosphenytoin is quickly converted to phenytoin by systemic phosphatases, and higher unbound phenytoin peak levels are more rapidly attained after the intravenous infusion of fosphenytoin than with the older phenytoin preparation. Studies of infants and children furthermore demonstrate no significant difference among conversion rates for infants, children, and adults [Morton et al., 1997a].
Recommended doses for intravenous administration of medications for the treatment of SE are given in Table 58-4 [Pellock, 1993a]. Recommended doses for fosphenytoin are identical to those for phenytoin in mg/kg phenytoin equivalent units. Rectal diazepam is administered according to age and weight.
Table 58-4 Recommended Initial Intravenous Doses of Antiepileptic Drugs for Status Epilepticus
| Drug | Dose | Rate |
|---|---|---|
| Lorazepam | 0.1 mg/kg | IV push |
| Diazepam | 0.3 mg/kg | IV push |
| Phenobarbital | 20 mg/kg | 2 mg/kg/min |
| Phenytoin | 20 mg/kg) | 1 mg/kg/min |
| Fosphenytoin | 20 PE*/kg/min | 3 PE/mg/kg/min |
| Valproate† | 20–40 mg/kg | 3–6 mg/kg/min |
| Levetiracetam | 30–60 mg/kg | Over 5 min |
* PE = phenytoin equivalents, which equals 1 mg.
† Use with caution in children under the age of 2 years as there is a dramatically higher association with hepatic dysfunction.
Valproic Acid/Valproate
Valproate (valproic acid; VPA) is useful in the management of both generalized and partial epilepsy, and particularly useful in absence SE [Holle et al., 1995; Pellock, 1993a]. In the past, administration of valproate by the oral, nasogastric, or rectal route followed benzodiazepine administration. The U.S. Food and Drug Administration approved intravenous valproate for use in 1997. Although the drug is not approved for treatment of SE, reports of effective and rapid seizure control with good tolerability have been published. In one study of 20 patients with generalized and partial motor seizures, 15 mg/kg of valproate achieved control within 30 minutes [Czapinski and Terczynski, 1998]. Several studies have been carried out in children, demonstrating efficacy in a wide variety of seizures using loading doses of 20–40 mg/kg [Campistol et al., 1999; Kian-Ti et al., 2003; Uberall et al., 2000]. The manufacturer recommends administering VPA over 60 minutes at less than 20 mg/min. However, recent work has confirmed that intravenous valproate may be infused rapidly at higher rates. A study of 18 children reported that, when VPA was administered intravenously at 1.5–11 mg/kg/min, there were no severe infusion-site complications and no changes in vital nor other systemic side effects: specifically no arrhythmias, nor hypotension, nor respiratory suppression [Morton et al., 2007]. Its role in the treatment of SE is being defined [Devinsky et al., 1995]. Intravenous valproate conveys some advantages over intravenous phenobarbital due to its lack of sedation, lack of respiratory suppression, and low risk of hypotension. In addition, valproate may be the drug of choice in certain seizures, or second-line following a benzodiazepine.
In a study of adult and children, one study compared intravenous valproate with intravenous phenytoin as first-line treatment. Seizures were controlled in 66 percent of the valproate group, compared to 42 percent in the phenytoin group [Misra et al., 2006]. The valproate group received sodium valproate 30 mg/kg, compared to the phenytoin group, which received phenytoin at 18 mg/kg. A retrospective study of children with acute repetitive seizures and SE found intravenous valproate to be successful in 39 out of 40 patients, with loading doses typically of 25 mg/kg at an infusion rate of approximately 3 mg/kg/min. No adverse effects were appreciated [Yu et al., 2003]. One randomized, open-label study compared intravenous VPA with diazepam in 40 children with refractory SE, failing initial dosing of diazepam followed by phenytoin, and demonstrated that seizures were terminated equally with both medications (80 percent for VPA and 85 percent for diazepam), but the result was more rapid with VPA [Mehta et al., 2007]. In this study, loading doses of 30–40 mg/kg were employed. Most commonly reported doses were 15–45 mg/kg loading dose, followed by a 1 mg/kg/hour continuous infusion. A recent European consensus statement listed intravenous valproate as an option for the treatment of convulsive SE when benzodiazepines have failed [Shorvon et al., 2008].
In one study of patients with recurrent episodes of absence SE as a manifestation of their primary generalized epilepsy, chronic therapy with valproate markedly reduced seizure recurrence [Berkovic et al., 1989]. The intravenous formulation has been used for the treatment of absence SE [Chez et al., 1999; Alehan et al., 1999;Uberall et al., 2000]. Also reported is a case series indicating its utility in myoclonic SE [Sheth et al., 2000; Wheless, 2003].
Levetiracetam
Levetiracetam is a unique antiepileptic drug that is effective in focal and generalized epilepsies. In 2006, the intravenous formulation was approved. It is not licensed for the treatment of SE, however. Levetiracetam has several properties that make its exploration for the treatment of SE tempting. Studies suggest that levetiracetam is safe to administer in critically ill children with absence of destabilization [Abend et al., 2009b]. There is minimal metabolism of the medication and no appreciable pharmacokinetic drug interaction. Clearance of the drug is renal, and therefore dosing adjustments need to be made in patients with evidence of renal impairment. Dialysis will markedly reduce serum levels.
The available studies in pediatric patients focus on patients with refractory SE and/or uncontrolled retrospective studies. One study looked at patients ranging from three weeks to 19 years of age. Initial loading doses up to 40 mg/kg were used, with a mean total loading dose of 50 mg/kg, though doses as high as 118 mg/kg were recorded. Administration was safe and in no case was the drug discontinued because of side effects [Goraya et al., 2008]. An additional study evaluating levetiracetam for refractory SE in children found the drug to be effective 45 percent of the time [Gallentine et al., 2009]. No patients responded at loading doses less than 30 mg/kg and doses up to 70 mg/kg were utilized. A review of all available series of case studies demonstrated a response rate of 64 percent in refractory SE treated with levetiracetam [Trinka et al., 2009].
Ideally, a drug used to treat SE must be effective, safe, and able to be administered with few side effects. Recent work has focused on rapid infusion, giving large doses of up to 2500 mg in adults in as little as 5 minutes [Uges et al., 2009].
Topiramate
There have also been reports of topiramate being efficacious in refractory SE in adults and children. Topiramate is safe and well tolerated, with sedation being the primary adverse effect [Kahriman et al., 2003; Towne et al., 2003]. High doses (e.g., 10 mg/kg/day) have demonstrated efficacy within 1 day, but experience is limited [Perry, 2006]. The role of topiramate in the treatment of SE is unclear, and limited by the lack of a commercially available parenteral formulation. Most likely it will have a role in refractory SE but not as a first- or second-line agent.
Lacosamide
Data regarding the efficacy of lacosamide in seizure emergencies and SE is restricted to single case reports without pediatric experience. Therefore, at this juncture its future role in the management of SE is unclear. However, lacosamide is effective in the pilocarpine, cobalt-homocysteine, and electrical stimulation models of SE [Stöhr et al., 2007; Bialer et al., 2009]. A single case report described an adult in SE who had failed benzodiazepines, levetiracetam, and etomidate, but responded to 300 mg of lacosamide [Tilz et al., 2009].
SE resistant to therapy with first-line agents requires urgent treatment with agents that depress cerebral activity. Current recommendations for the management of refractory SE include the use of intravenous midazolam, pentobarbital, propofol, thiopental, and valproic acid. These medications are not without significant risk, including the need for prolonged mechanical ventilation, hemodynamic instability, and metabolic syndrome. No randomized controlled trials for any of these medications have been performed in either adults or children with refractory SE. In each case, the dose is regulated such as to control all clear-cut clinical or electrographic seizures and to maintain a suppression-burst pattern in the EEG. Therefore, continuous bedside EEG is monitored. There is no consensus as to the duration of “burst” and “flat” periods that signify optimal dosing [Krishnamurthy et al., 1999]. Most consider that establishing and maintaining any degree of burst-suppression is adequate.
Anesthetic Agents
Continuous infusion with high-dose pentobarbital is most commonly used [Pellock and DeLorenzo, 1997; Eriksson and Koivikko, 1997; Kinoshita et al., 1995] (see Table 53-3). Continuous benzodiazepine infusion with diazepam, midazolam [Nordt and Clark, 1997; Lal Koul et al., 1997; Chamberlain et al., 1997], or lorazepam may also be used. Continuous infusion with midazolam has been demonstrated to be as efficacious as pentobarbital infusion but with fewer cardiovascular adverse effects overall [Gilbert et al., 1999; Holmes et al., 1999; Igartua et al., 1999]. The choice depends on the practitioner’s level of comfort with the various agents, as well as their specific treatment-related complications. Propofol anesthesia [Singhi et al., 1998; Stecker et al., 1998; Harrison et al., 1997] has also been effective. However, there have been some concerns that it may be proconvulsant in some patients [Makela et al., 1993], and can lead to metabolic acidosis [Niermeijer et al., 2003]. A recent retrospective study using propofol was noted to have a high mortality rate in refractory SE [Prasad et al., 2001]. However, a study in which 22 children received propofol found that it was successful in 64 percent and there were no safety concerns raised unless the dose exceeded 5 mg/kg/hour. Dosing was typically started with 1–2 mg/kg/hour [van Gestel et al., 2005]. Ketamine, an NMDA antagonist, has not been studied beyond small series [Sheth and Gidal, 1998; Prüss and Holtkamp, 2008]. Ketamine has not been useful early in the treatment of SE, but may become effective later when GABAergic agents generally are no longer effective [Prüss and Holtkamp, 2008].
Nonpharmacological Approaches
In a few extraordinary circumstances, nonpharmacologic treatment options may be available. If a definite seizure focus can be determined, resective surgery may be an option in selected cases (see Chapter 61). In one series of 10 children with pre-existing epilepsy, refractory SE was terminated in all and 7 remained seizure-free at 4 months to 6.5 years of follow-up [Alexopoulos et al., 2005]. Additionally, acute implantation of a vagus nerve stimulator has been reported in refractory SE [De Herdt et al., 2009].
There have been a limited number of cases utilizing the ketogenic diet for the treatment of refractory SE [Francois et al., 2003].
Therapeutic hypothermia is increasingly used in postanoxic patients. Hypothermia may also be useful in the treatment of refractory SE. Hypothermia to 31–35°C for 20–61 hours was reported to terminate SE refractory to multiple agents in 2 adult patients of 4 studied [Corry et al., 2008]. One study demonstrated control of refractory SE in three children with a combination of hypothermia to 30–31°C combined with thiopental [Orlowski et al., 1984].
Medical Complications of Status Epilepticus
The treatment of SE requires close monitoring of physiologic variables and excellent nursing to prevent secondary complications [Pellock, 1993b, 1994; Treiman, 1993]. Besides underlying or precipitating disease states associated with SE, other medical complications are quite common. Pulmonary care, proper positioning, and careful observation of seizures to note possible changes in seizure pattern are mandatory. Constant maintenance of intravenous fluids with adequate glucose and electrolyte administration and appropriate corrections, particularly in neonates and small infants, is essential. Optimal oxygenation, expectant observation, and treatment for hyperthermia and other medical complications lead directly to a lessening of morbidity and mortality. Cardiovascular, respiratory, and renal effects may be severe. Medical complications of SE that may occur in both infants and older children are listed in Box 58-3.






























Prognosis
SE is recognized as a medical and neurologic emergency because of associated significant morbidity and mortality. Common sequelae of SE include intellectual dysfunction, permanent neurologic deficits, and continuing recurrent seizures. Neuropathologic studies in animals have demonstrated that prolonged electrical activity results in irreversible neuronal damage to the CNS [Meldrum, 1983a]. Experimental studies suggest that seizures lasting for less than 30–60 minutes may produce neuronal injuries that are reversible. Seizures lasting longer than 1 hour produce neuronal death [Meldrum, 1983b; Simon, 1985]. As noted previously, prolonged seizures are associated with inadequate blood flow, decreased glucose use and oxygen consumption, and increased excitatory amino acid release, all of which lead to impaired mitochondrial function and neuronal destruction [Dwyer et al., 1986; Lothman, 1990; Wasterlain et al., 1983]. The duration of the SE episode, the patient’s age, and the causative disorder all influence the resultant insult to involved neuronal populations.
In the aforementioned prospective population-based epidemiologic study [DeLorenzo et al., 1996, the annual incidence of SE was 41 cases per 100,000 population, and the annual incidence of total SE episodes was 50 per 100,000. The mortality rate was 22 percent, suggesting that, for the approximately 126,000–195,000 SE events, 22,000–42,000 deaths per year will occur in the United States. A majority of patients with SE have no previous history of epilepsy. The highest mortality rate is among the elderly, and the lowest is seen in healthy children or those with febrile SE [van Esch et al., 1996]. Although the overall mortality is extremely low in pediatric patients, younger children are at highest risk, with a mortality rate of 17.8 percent for those younger than 1 year in one study [Morton et al., 2001]. Mortality in these children typically is associated with specific underlying disorders, such as severe brain damage secondary to hypoxia.
Recurrences of SE are common in young children. These patients usually have chronic neurologic disabilities. In those patients who die, death rarely occurs during the acute episode of SE. Rather, most deaths occur 13–30 days after onset of the SE episode. Children with epilepsy and low antiepileptic drug levels have the lowest mortality rate. One reason for the low mortality rate in children is that they are less likely than adults to have systemic diseases. However, in one study, two independent risk factors for increased mortality in children, after excluding progressive or acute neurological insult, were duration of seizures in excess of 2 hours and a history of asthma [Maegaki et al., 2005]. In the elderly, cardiovascular decompensation occurring during the stress of SE may play an important role in the higher mortality rate [Boggs et al., 1993, 1994].
The morbidity associated with SE in children was examined in a database from Virginia [Fortner et al., 1993]. Before their SE event, 81 percent of children with no prior seizures were neurologically normal, in contrast with only 31 percent of children with seizure histories. Of the neurologically normal children with no prior seizures, more than 25 percent deteriorated after their first SE event, in comparison with less than 15 percent of neurologically normal children with a seizure history. Children who were neurologically abnormal without prior seizures deteriorated less frequently (6.7 percent), compared with 11.3 percent of the children who were neurologically abnormal who had a seizure history. In some children, neurologic deficits, such as mild ataxia, incoordination, or mild motor deficits, are transient. Symptoms usually can be attributed to the acute therapies or clinical changes after prolonged seizures.
Determining whether language deficits or school performance difficulties will be transient or more permanent is much more difficult. A previous retrospective study yielded a high rate of morbidity of nearly 30 percent, suggesting a sampling bias [Fortner et al., 1993]. In a prospective study, only 11–15 percent of affected patients had significant morbidity after an episode of SE [Morton et al., 1998]. These more recent findings suggest that the neurologic morbidity is substantially lower than the “greater than 50 percent” rate previously reported in children with SE [Aicardi and Chevrie, 1987]. The morbidity and mortality rates in sick infants, however, are higher than those in older children [Morton et al., 1998].
Patients with refractory SE represent a special population at particularly high risk for mortality and morbidity. Refractory SE typically is diagnosed when seizures have persisted for longer than 60 minutes and have not responded to use of three or more medications. Several series have demonstrated mortality rates ranging from 16 to 32 percent [Gilbert et al., 1999; Kernitsky et al., 2002; Sahin et al., 2001; Singhi et al., 1998] – much higher than the rate typical for childhood SE of approximately 3 percent [DeLorenzo et al., 1996; Maytal et al., 1989; Towne et al., 1994]. In addition, the morbidity rate was quite high in this population, with 34 percent experiencing developmental deterioration after refractory SE [Sahin et al., 2001].
The consequences of SE for the developing brain are not clearly known. This issue, however, has been difficult to approach in clinical research studies, as multiple factors are all potential contributors to cognitive dysfunction in children with epilepsy. In addition to the seizures themselves, medications, underlying brain malformations, and pre-existing learning problems are all potential contributors to cognitive dysfunction. Studies of laboratory animals experiencing convulsive SE also have revealed an age dependence in vulnerability to neurocognitive impairment [Lui et al., 1994]. While evidence in humans at times is unclear or contradictory, recent work suggests some age-dependent effect. Neonatal SE seems to be an independent risk factor for adverse outcome in full-term newborns compared to similar newborns with seizures [Pisani et al., 2007]. There is some evidence that both recurrent seizures and SE may affect the developing brain in myriad ways, ranging from injury and altered neurogenesis to plasticity leading to epileptogenicity and the development of behavioral and cognitive consequences, even in the absence of readily discernible injury. Using animal models, it is reasonable to estimate that P14–P21 rat pups represent toddlers and young children, and P28 rats represent older children prior to puberty. A 3 mg/kg dose of kainic acid produces very severe seizures in the 15-day-old rats. When subjected to kainic acid again at postnatal day 15, test subjects experienced more severe brain damage and performed worse in spatial learning tasks than their control counterparts [Koh et al., 1999]. Emerging evidence suggests that early-life seizures can alter the function of neurotransmitter systems and intrinsic neuronal properties in the brain, possibly contributing to cognitive and learning impairments. Enhancement of GABAA receptor function with benzodiazepines disrupts long-term potentiation and memory formation [Seabrook et al., 1997] and hippocampal-dependent spatial learning [Rudolph and Mohler, 2004].
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Abend N.S., Dlugos D.J. Nonconvulsive SE in a pediatric intensive care unit. Pediatr Neurol. 2007;37:165.
Abend N.S., Topjian A., Ichord R., et al. Electroencephalographic monitoring during hypothermia after pediatric cardiac arrest. Neurology. 2009;72:1931.
Abend N.S., Monk H.M., Licht D.J., et al. Intravenous levetiracetam in critically ill children with status epilepticus or acute repetitive seizures. Pediatr Crit Care Med. 2009;10(4):505.
Aicardi J.F., Chevrie J.J. Convulsive SE in infants and children: A study of 239 cases. Epilepsia. 1987;11:187.
Alehan F., Morton L.D., Pellock J.M. Treatment of absence status with intravenous valproate. Neurology. 1999;52:889.
Alexopoulos A., Lachhwani D.K., Gupta A., et al. Resective surgery to treat refractory SE in children with focal epileptogenesis. Neurology. 2005;64:567.
Alldredge B.K., Wall D.B., Ferriero D.M. Effect of prehospital treatment on the outcome of SE in children. Pediatr Neurol. 1995;12:213.
Appleton R., Choonara I., Martland T., et al. The treatment of convulsive SE in children. The Status Epilepticus Working Group. Arch Dis Child. 2000;83:415.
Appleton R., Sweeney A., Choonara I., et al. Lorazepam versus diazepam in the acute treatment of epileptic seizures and SE. Dev Med Child Neurol. 1995;37:682.
Ben Ari Y. Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience. 1985;14:375.
Berkovic S.F., Andermann F., Griberman A., et al. Valproate prevents the recurrence of absence status. Neurology. 1989;39:1294.
Bialer M., Johannessen S.I., Levy R.H. Progress report on new antiepileptic drugs: A summary of the Ninth Eilat Conference. Epilepsy Res. 2009;83:1.
Boggs J.G., Painter J.A., DeLorenzo R.J. Analysis of electrocardiographic changes in SE. Epilepsy Res. 1993;14:87.
Boggs J.G., Painter J.A., Wood M.A., et al. Signal-averaged electrocardiograms in patients with SE [abstract]. Neurology. 1994;44(Suppl):A205.
Brooks-Kayal A.R., Shumate M.D., Jin H., et al. Selective changes in single cell GABA(A) receptor subunit expression and function in temporal lobe epilepsy. Nat Med. 1998;4:1166.
Brooks-Kayal A.R., Shumate M.D., Jin H., et al. Human neuronal gamma-aminobutyric acid(A) receptors: Coordinated subunit mRNA expression and functional correlates in individual dentate granule cells. J Neurosci. 1999;19:8312.
Browne T.R. Fosphenytoin (Cerebyx). Clin Neuropharmacol. 1997;20:1.
Camfield C.S., Camfield P.R., Smith E., et al. Home use of rectal diazepam to prevent SE in children with convulsive disorders. J Child Neurol. 1989;4:125.
Campistol J., Fernandez A., Ortega J. SE in children. Experience with intravenous valproate. Update of treatment guidelines. Rev Neurol. 1999;29:359.
Cavalheiro E.A. The pilocarpine model of epilepsy. Ital J Neurol Sci. 1995;16:33.
Cavalheiro E.A., Silva D.F., Turski W.A., et al. The susceptibility of rats to pilocarpine-induced seizures is age-dependent. Dev Brain Res. 1987;37:43.
Chamberlain J.M., Altieri M.A., Futterman C., et al. A prospective, randomized study comparing intramuscular midazolam with intravenous diazepam for the treatment of seizures in children. Pediatr Emerg Care. 1997;13:92.
Chez M.G., Hammer M.S., Loeffel M., et al. Clinical experience of three pediatric and one adult case of spike-and-wave SE treated with injectable valproic acid. J Child Neurol. 1999;14:239.
Chin R.F., Neville B.G., Peckham C., et al. Incidence, cause, and short-term outcome of convulsive SE in childhood: prospective population-based study. Lancet. 2006;368:222.
Classen J., Mayer S.A., Kowalski R.G., et al. Detection of electrographic seizures with continuous EEG monitoring in critically ill patients. Neurology. 2004;62(10):1743.
Cock H.R., Tong X., Hargreaves I.P., et al. Mitochondrial dysfunction associated with neuronal death following SE in rat. Epilepsy Res. 2002;48:157.
Cockerell O.C., Rothwell J., Thompson P.D., et al. Clinical and physiological features of epilepsia partialis continua. Cases ascertained in the UK. Brain. 1996;119:393.
Cole A.J. SE and periictal imaging. Epilepsia. 2004;45(Suppl 4):72.
Corry J.J., Dhar R., Murphy T., et al. Hypothermia for refractory SE. Neurocrit Care. 2008;9:189.
Corey L.A., Pellock J.M., DeLorenzo R.J. SE in a population-based Virginia twin sample. Epilepsia. 2004;45:159.
Coeytaux A., Jallon P., Galobardes B., et al. Incidence of SE in French-speaking Switzerland: (EPISTAR). Neurology. 2000;55:693.
Czapinski P., Terczynski A. Intravenous valproic acid administration in SE. Neurol Neurochir Pol. 1998;32:11.
Dalmau J., Gleichman A.J., Hughes E.G., et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7:1091.
De Herdt V., Waterschoot L., Vonck K., et al. Vagus nerve stimulation for refractory SE. Eur J Paediatr Neurol. 2009;13:286.
Delgado-Escueta A.V., Treiman D.M. Focal SE: Modern concepts. In: Luders H., Lesser R.P., editors. Epilepsy electroclinical syndromes. London: Springer, 1987.
DeLorenzo R.J.. SE. Johnson R.J., editor. Current therapy in neurologic disease, 3. Philadelphia: BC Decker, 1990.
DeLorenzo R.J., Garnett L.K., Towne A.R., et al. Comparison of SE with prolonged seizure episodes lasting from 10 to 29 minutes. Epilepsia. 1999;40:164.
DeLorenzo R.J., Hauser W.A., Towne A.R., et al. A prospective, population-based study of SE in Richmond, Virginia. Neurology. 1996;46:1029.
DeLorenzo R.J., Pellock J.M., Towne A.R., et al. Pathophysiology of SE. J Clin Neurol. 1995;12:316.
DeLorenzo R.J., Towne A.R., Pellock J.M., et al. SE in children, adults, and the elderly. Epilepsia. 1992;33(Suppl 4):S15.
DeLorenzo R.J., Waterhouse E.J., Towne A.R., et al. Persistent nonconvulsive SE after the control of convulsive SE. Epilepsia. 1998;39:833.
Devinsky O., Leppik I., Wilmore L.G., et al. Safety of intravenous valproate. Ann Neurol. 1995;388:670.
Dieckmann R.A. Rectal diazepam for prehospital pediatric status epilepticus. Annals of Emergency Medicine. 1994;23(2):216.
Dodson W.E., DeLorenzo R.J., Pedley T.A., et al. For the Epilepsy Foundation of America’s Working Group on Status Epilepticus. Treatment of convulsive SE. JAMA. 1993;270:854.
Dooley J.M. Rectal use of benzodiazepines. Epilepsia. 1998;39(Suppl 1):S24.
Drislane F.W. Evidence against permanent neurologic damage from nonconvulsive SE. J Clin Neurophysiol. 1999;16:323.
Drislane F.W., Lopez M.R., Blum A.S., et al. Detection and treatment of refractory SE in the intensive care unit. J Clin Neurophysiol. 2008;25:181.
Dunn W. SE in children: Etiology, clinical features, and outcome. J Child Neurol. 1988;3:167.
Dwyer B.E., Wasterlain C.G., Fajikwa D.G., et al. Brain protein metabolism in epilepsy. In: Delgado-Escueta A.V., Ward A.A., Woodburg D.M., et al, editors. Basic mechanisms of the epilepsies: Molecular and cellular approaches. New York: Raven Press, 1986.
Epilepsy Foundation of America. Treatment of convulsive SE. Recommendations of the Epilepsy Foundation of America’s Working Group on Status Epilepticus. JAMA. 1993;7:854.
Eriksson K.J., Koivikko M.J. SE in children: Aetiology, treatment, and outcome. Dev Med Child Neurol. 1997;39:652.
Fortner C., Pellock J.M., Driscoll S.M., et al. Morbidity in children after a first episode of SE. Epilepsia. 1993;34(Suppl 6):55. (abstract)
Francois L.L., Manel V., Rouselle C., et al. Ketogenic regime as antiepileptic treatment: it’s use in 29 epileptic children. Arch Pediatr. 2003;10:300.
Gallentine W.B., Hunnicutt A.S., Husain A.M. Levetiracetam in children with refractory SE. Epilepsy Behav. 2009;14:215.
Gastaut H. Classification of SE. In: Delgado-Escueta A.V., Porter R.J., Wasterlain C.G., editors. SE: Mechanisms of brain damage and treatment. New York: Raven Press, 1982.
Gibbs J.W., Shumate M.D., Coulter D.A. Differential epilepsy-associated alterations in dentate granule and CA1 neurons. J Neurophys. 1997;77:1924.
Gilbert D.L., Gartside P.S., Glauser T.A. Efficacy and mortality in treatment of refractory generalized convulsive SE in children: A meta-analysis. J Child Neurol. 1999;14:602.
Goodkin H.P., Liu X., Holmes G.L. Diazepam terminates brief but not prolonged seizures in young, naïve rats. Epilepsia. 2003;44:1109.
Goodkin H.P., Joshi S., Mtchedlishvilli Z., et al. Subunit specific trafficking of GABAA receptors during SE. J Neurosci. 2008;28:2527.
Goraya J.S., Khurana D.S., Valencia I., et al. Intravenous Levetiracetam in Children With Epilepsy. Pediatr Neurol. 2008;38:177.
Govoni V., Fallica E., Monetti V.C., et al. Incidence of Status Epilepticus in Southern Europe: A Population Study in the Health District of Ferrara, Italy. Eur Neurol. 2008;59:120.
Grin J.M., DiMario F.J.Jr. Absence SE causing a prolonged acute confusional state. Clin Pediatr (Phila). 1998;37:37.
Harden C.L., Huff J.S., Schwartz T.H., et al. Reassessment: Neuroimaging in the emergency patient presenting with seizure (an evidence-based review) Report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2007;69:1772.
Harrison A.M., Lugo R.A., Schunk J.E. Treatment of convulsive SE with propofol: Case report. Pediatr Emerg Care. 1997;13:420.
Hauser W.A. SE: Epidemiologic considerations. Neurology. 1990;40(Suppl 2):9.
Hauser W.A., Rich S.S., Annegars J.F., et al. Seizure recurrence after a first unprovoked seizure: An extended follow-up. Neurology. 1990;40:1163.
Hisano T., Ohno M., Egawa T., et al. Changes in diffusion-weighted MRI after SE. Pediatr Neurol. 2000;22:327.
Holle L.M., Gidal B.E., Collins D.M. Valproate in SE. Ann Pharmacother. 1995;29:1042.
Holmes G.L. Epilepsy in the developing brain: lessons from the laboratory and clinic. Epilepsia. 1997;38:12.
Holmes G.L. Drug of choice for SE. J Epilepsy. 1990;3:1.
Holmes G.L., Gairsa J.L., Chevassus-Au-Louis N., et al. Consequences of neonatal seizures in the rat: Morphological and behavioral effects. Ann Neurol. 1998;44:845.
Holmes G.L., Riviello J.J. Midazolam and pentobarbital for refractory SE. Pediatr Neurol. 1999;20:259.
Hosain S.A., Solomon G.E., Kobylarz E.J. Electroencephalographic patterns in unresponsive pediatric patients. Pediatr Neurol. 2005;32:162.
Igartua J., Silver P., Maytal J., et al. Midazolam coma for refractory SE in children. Crit Care Med. 1999;27:1982.
ILAE Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia. 1981;22:489.
Jaitly R., Sgro J.A., Towne A.R., et al. A prospective study of prolonged (24 hour) EEG monitoring in a large population of SE patients. Neurology. 1994;44(Suppl):A172. (abstract)
Jensen F.E. The role of the glutamate receptor maturation in perinatal seizures and brain injury. Int J Dev Neurosci. 2002;20:339.
Jette N., Claassen J., Emerson R.G., et al. Frequency and Predictors of Nonconvulsive Seizures During Continuous Electroencephalographic Monitoring in Critically Ill Children. Arch Neurol. 2006;63:1750.
Jones D.M., Esmaeil N., Marin S., et al. Characterization of pharmacoresistance to benzodiazepines in the rat Li-pilocarpine model of SE. Epilepsy Res. 2002;50:301.
Jordan K.G. SE: A perspective from the neuroscience intensive care unit. Neurosurg Intensive Care. 1994;5:671.
Kahriman M., Minecan D., Kutluay E., et al. Efficacy of topiramate in children with refractory SE. Epilepsia. 2003;44:1353.
Kaplan P.W. Nonconvulsive SE. Semin Neurol. 1996;16:33.
Kapur J., Stringer J.L., Lothman E.W. Evidence that repetitive seizures in the hippocampus cause a lasting reduction of GABAergic inhibition. J Neurophysiol. 1989;61:417.
Kapur J., Coulter D.A. Experimental SE alters gamma-aminobutyric acid type A receptor function in CA1 pyramidal neurons. Ann Neurol. 1995;38:893.
Kapur J., Lothman E.W. NMDA receptor activation mediates the loss of GABAergic inhibition induced by recurrent seizures. Epilepsy Res. 1990;5:103.
Kernitsky L., Morton L.D., Pellock J.M., et al. Refractory SE in children. Epilepsia. 2002;43(Suppl 7):73.
Khazipov R., Khalilov I., Tyzio R., et al. Developmental changes in GABAergic actions and seizure susceptibility in the rat hippocampus. Eur J Neurosci. 2004;19:590.
Kian-Ti Y., Mills S., Thompson N., et al. Safety and efficacy of intravenous valproate in pediatric SE and acute repetitive seizures. Epilepsia. 2003;44:724.
Kinoshita H., Nakagawa E., Iwasaki Y., et al. Pentobarbital therapy for SE in children: timing of tapering. Pediatr Neurol. 1995;13:164.
Kramer R.E., Luders H., Lesser R.P., et al. Transient focal abnormalities of neuroimaging studies during focal SE. Epilepsia. 1987;28:528.
Kriel R.L., Cloyd J.C., Hadsall R.S., et al. Home use of rectal diazepam for cluster and prolonged seizures: efficacy, adverse reactions, quality of life, and cost analysis. Pediatr Neurol. 1991;7:13.
Krishnamurthy K.B., Drislane F.W. Depth of EEG suppression and outcome in barbiturate anesthetic treatment for refractory SE. Epilepsia. 1999;40:759.
Koh S., Storey T.W., Santos T.C., et al. Early-life seizures in rats increase susceptibility to seizure-induced brain injury in adulthood. Neurology. 1999;53:915.
Lal Koul R., Raj Aithala G., Chacko A., et al. Continuous midazolam infusion as treatment of SE. Arch Dis Child. 1997;76:445.
Leppik I.E., Boucher B.A., Wilder B.J., et al. Pharmacokinetics and safety of phenytoin products given IV or IM in patients. Neurology. 1987;37:500.
Lewis D.V. Febrile convulsions and mesial temporal sclerosis. Curr Opin Neurol. 1999;12(2):197.
Liu Z., Gatt A., Mikati M., et al. Effect of temperature on keyinic acid–induced seizures. Brain Res. 1993;631:51.
Liu Z., Gatt A., Werner S.J., et al. Long-term behavioral deficits following pilocarpine seizures in immature rats. Epilepsy Res. 1994;19:191.
Lockman L.A. Treatment of SE in children. Neurology. 1990;40(Suppl 2):43.
Lothman E. The biochemical basis and pathophysiology of SE. Neurology. 1990;40(Suppl 2):13.
Lothman E.W., Bertram E.H., Bekenstein J.W., et al. Self-sustaining limbic SE induced by ‘continuous’ hippocampal stimulation: electrographic and behavioral characteristics. Epilepsy Res. 1989;3:107-119.
Lowenstein D.H., Alldredge B.K. Status epilepticus at an urban public hospital in the 1980s. Neurology. 1993;43:483-488.
Lowenstein D.H., Alldredge B.K. SE. N Engl J Med. 1998;338:970.
Lowenstein D.H., Bleck T., Macdonald R.L. It’s time to revise the definition of SE. Epilepsia. 1999;40:120.
Maegaki Y., Kurozawa Y., Hanaki K., et al. Risk factors for fatality and neurological sequelae after SE in children. Neuropediatrics. 2005;36:185.
Makela J.P., Iivanainen M., Pieninkeroinen I.P., et al. Seizures associated with propofol anesthesia. Epilepsia. 1993;34:832.
Maytal J., Novak G., Ascher C., et al. SE in children with epilepsy: The role of antiepileptic drug levels in prevention. Pediatrics. 1996;98:1119.
Maytal J., Shinnar S., Moshè S.L., et al. Low morbidity and mortality of SE in children. Pediatrics. 1989;83:323.
Mazarati A.M., Liu H.T., Soomets U., et al. Galanin modulation of seizures and seizure modulation of hippocampal galanin in animal models of SE. J Neurosci. 1998;18:10070.
Mazarati A.M., Hohmann J.G., Bacon A., et al. Modulation of hippocampal excitability and seizures by galanin. J Neurosci. 2000;20:6276-6281.
Mehta V., Singhi P., Singhi S. Intravenous sodium valproate versus diazepam infusion for the control of refractory SE in children: a randomized controlled trial. J Child Neurol. 2007;22:1191.
Meierkord H., Wieshmann U., Niehaus L., et al. Structural consequences of SE demonstrated with serial magnetic resonance imaging. Acta Neurol Scand. 1997;96:127.
Meldrum B.S. Metabolic factors during prolonged seizures and their relation to nerve cell death. Adv Neurol. 1983;34:261.
Meldrum B.S. Metabolic factors during two prolonged seizures and their relationship to nerve cell death. In: Delgado-Escueta A.V., Wasterlain C.G., Treiman D.M., et al, editors. SE: Mechanisms of brain damage and treatment. New York: Raven Press, 1983.
Meldrum B.S. First Alfred Meyer Memorial Lecture. Epileptic brain damage: a consequence and a cause of seizures. Neuropathol Appl Neurobiol. 1997;23:185.
Meldrum B.S., Horton R.W., Brierley J.B. Epileptic brain damage in adolescent baboons following seizures induced by allyl-glycine. Brain. 1974;97:407.
Millikan D., Rice B., Silbergeit R. Emergency treatment of SE: critical thinking. Emerg Med Clin N Amer. 2009;27:101.
Misra U.K., Kalita J., Patel R. Sodium valproate vs phenytoin in SE: A pilot study. Neurology. 2006;67:340.
Mitchell W.G. SE and acute repetitive seizures in children, adolescents, and young adults: etiology, outcome, and treatment. Epilepsia. 1996;37(Suppl 1):S74.
Mizrahi E.M., Kellaway P. Characterization and classification of neonatal seizures. Neurology. 1987;37:1837.
Mizrahi E.M., Kellaway P. Diagnosis and management of neonatal seizures. Philadelphia: Lippincott-Raven; 1998.
Morton L.D., DeLorenzo S., Garnett L., et al. Morbidity and mortality of SE in the first year of life. Neurology. 1998;50(Suppl 4):A444.
Morton L.D., Garnett L.K., Towne A.R., et al. Mortality of SE in the first year of life. Epilepsia. 2001;42(Suppl 7):165.
Morton L.D., O’Hara K.A., Coots B.P., et al. Safety of rapid intravenous valproate infusion in pediatric patients. Pediatr Neurol. 2007;36:81.
Morton L.D., Pellock J.M., Maria B.L., et al. Fosphenytoin safety and pharmacokinetics in children. Epilepsia. 1997;38:194. (abstract)
Morton L.D., Rizkallah E., Pellock J.M. New drug therapy for acute seizure management. Semin Pediatr Neurol. 1997;4(Suppl):60.
Moshè S.L. Epileptogenesis and the immature brain. Epilepsia. 1987;28(Suppl 1):S3.
Najm I.M., Wang Y., Shedid D., et al. MRS metabolic markers of seizures and seizure-induced neuronal damage. Epilepsia. 1998;39:244.
Naylor D.E., Lui H., Wasterlain C.G. Trafficking of GABAA receptors, loss of inhibition, and the mechanism for pharmacoresistance and SE. J Neurosci. 2005;25:7724.
Nei M., Lee J.M., Shanker V.L., et al. The EEG and prognosis in SE. Epilepsia. 1999;40:157.
Niermeijer J.M., Uiterwaal C.S., Van Donselaar C.A. Propofol in SE: little evidence, many dangers? J Neurol. 2003;250(10):1237.
Nordt S.P., Clark R.F. Midazolam: A review of therapeutic uses and toxicity. J Emerg Med. 1997;15:357.
O’Dell C., Nordli D., Pellock J.M., et al. Recognition of febrile SE in the emergency department [abstract]. Ann Neurol. 2005;58(Suppl 9):S112.
O’Dell C., Shinnar S., Ballaban-Gil K.R., et al. Rectal diazepam gel in the home management of seizures in children. Pediatr Neurol. 2005;33:166.
Orlowski J.P., Erenberg G., Lueders H., et al. Hypothermia and barbiturate coma for refractory SE. Crit Care Med. 1984;12:367.
Orlowski J.P., Porembha D.T., Gallagher B.B., et al. Comparison study of intraosseous, central intravenous and peripheral intravenous infusions of emergency drugs. Am J Dis Child. 1990;144:112.
Pellock J.M. SE. In Pellock J.M., Myer E.C., editors: Neurologic emergencies in infancy and childhood, ed 2, New York: Demos, 1993.
Pellock J.M. SE. In: Dodson W.E., Pellock J.M., editors. Pediatric epilepsy, diagnosis and therapy. New York: Demos, 1993.
Pellock J.M. SE in children: Update and review. J Child Neurol. 1994;9(Suppl 2):S27.
Pellock J.M., editor. Formulary report: Issues in the management of acute seizures. Pharmacol Ther. 1996;21:1S. Suppl 5
Pellock J.M. Management of acute seizure episodes. Epilepsia. 1998;39(Suppl 1):S28.
Pellock J.M., DeLorenzo R.J. SE. In: Porter R.J., Chadwick D., editors. The epilepsies 2. Boston: Butterworth-Heinemann, 1997.
Pellock J.M., Marmarou A., DeLorenzo R.J. Time to treatment in prolonged seizure episodes. Epilepsy Behav. 2004;5:192.
Perry M.S., Holt P.J., Sladky J.T. Topiramate Loading for Refractory Status Epilepticus in Children. Epilepsia. 47(6), 2006.
Phillips S.A., Shanahan R.J. Etiology and mortality of SE in children. Arch Neurol. 1989;46:74.
Pisani F., Cerminara C., Fusco C., et al. Neonatal SE vs recurrent neonatal seizures:clinical findings and outcome. Neurology. 2007;69:2177.
Porter R.J., Penry J.K. Petit mal status. In: Delgado-Escueta A.V., Wasterlain C.G., et al, editors. SE. New York: Raven Press, 1983.
Practice Parameter. Neuroimaging in the emergency patient presenting with seizure – summary statement. Quality Standards Subcommittee of the American Academy of Neurology in cooperation with American College of Emergency Physicians, American Association of Neurological Surgeons, and American Society of Neuroradiology. Neurology. 1996;47:288.
Praline J., Grujic J., Corcia P., et al. Emergent EEG in clinical practice. Clin Neurophysiol. 2007;118:2149.
Prasad A., Worrall B.B., Bertram E.H., et al. Propofol and midazolam in the treatment of refractory SE. Epilepsia. 2001;42:380.
Privitera M. Complex partial SE. Neurology. 1997;48:1140.
Privitera M., Hoffman M., Moore J.L., et al. EEG detection of nontonic-clonic SE in patients with altered consciousness. Epilepsy Res. 1994;18:155.
Provenzale J.M., Barboriak D.P., VanLandingham K.E., et al. Hippocampal MRI signal after febrile SE is predictive of subsequent mesial temporal sclerosis. AJR. 2008;190:976.
Prüss H., Holtkamp M. Ketamine successfully terminates malignant SE. Epilepsy Res. 2008;82:219.
Qureshi A., Wassmere E., Davies P., et al. Comparative audit of intravenous lorazepam and diazepam in the emergency treatment of convulsive SE in children. Seizure. 2002;11:141.
Ramsay R.E., DeToledo J. Intravenous administration of fosphenytoin: Options for the management of seizures. Neurology. 1996;46(6 Suppl 1):S17.
Remy S., Beck H. Molecular and cellular mechanism of pharmacoresistance and epilepsy. Brain. 2006;129:18.
Rice A.C., DeLorenzo R.J. Kindling induces long-term changes in gene expression. In: Corcoran M.E., Moshè S.L., editors. Kindling 5. New York: Plenum, 1998.
Rider L.G., Thapa P.B., Del Beccaro M.A., et al. Cerebrospinal fluid analysis in children with seizures. Pediatr Emerg Care. 1995;11:226.
Riviello J.J., Ashwal S., Hirtz D., et al. Practice Parameter: Diagnostic assessment of the child with status epilepticus (an evidence based review). Neurology. 2006;67:1542.
Ross C., Blake A., Whitehouse W.P. SE on the paediatric intensive care unit – the role of EEG monitoring. Seizure. 1999;8:335.
Rudolph U., Mohler H. Analysis of GABAA receptor function and dissection of the pharmacology of benzodiazepines and general anesthetics through mouse genetics. Annu Rev Pharmacol Toxicol. 2004;44:475.
Sahin M., Menache C.C., Holmes G.L., et al. Outcome of severe refractory SE in children. Epilepsia. 2001;42:1467.
Sankar R., Shin D.H., Wasterlain C.G. GABA metabolism during SE in the developing rat brain. Dev Brain Res. 1997;98:60.
Scher M.S., Aso K., Beggarly M.E., et al. Electrographic seizures in preterm and full-term neonates: Clinical correlates, associated brain lesions, and risk for neurological sequelae. Pediatrics. 1993;91:128.
Scher M.S., Hamid M.Y., Steppe D.A., et al. Ictal and interictal durations in preterm and term neonates. Epilepsia. 1993;34:284.
Schmid R., Tandon P., Stafstrom C.E., et al. Effects of neonatal seizures on subsequent seizure-induced brain injury. Neurology. 1999;53:1754.
Scholtes F.B., Renier W.O., Meinardi H. Non-convulsive SE: Causes, treatment, and outcome in 65 patients. J Neurol Neurosurg Psychiatry. 1996;61:93.
Scott R.C., Besag F.M., Neville B.G. Buccal midazolam and rectal midazolam for treatment of prolonged seizures in childhood and adolescence: A randomized trial. Lancet. 1999;353:623.
Scott R.C., Gadian D.G., King M.D., et al. Magnetic resonance imaging findings within 5 days of SE in childhood. Brain. 2002;125:1951.
Scott R.C., King M.D., Gadian D.G., et al. Prolonged febrile seizures are associated with hippocampal vasogenic edema and developmental changes. Epilepsia. 2006;47:1493.
Seabrook G.R., Easter A., Dawson G.R., et al. Modulation of long-term potentiation in CA1 region of mouse hippocampal brain slices by GABAA receptor benzodiazepine site ligands. Neuropharmacology. 1997;36:823.
Sheth R.D., Gidal B.E. Refractory status epilepticus: response to ketamine. Neurology. 1998;51:1765.
Sheth R.D., Gidal B.E. Intravenous valproic acid for myoclonic SE. Neurology. 2000;54(5):1201.
Shinnar S., Pellock J.M., Berg A.T., et al. An inception cohort of children with febrile SE: Cohort characteristics and early outcomes. Epilepsia. 1995;36(Suppl 4):31. (abstract)
Shinnar S., Hesdorffer D.C., Nordli D.R., et al. Phenomenology of prolonged febrile seizures Results of the FEBSTAT study. Neurology. 2008;71:170-176.
Shorvon S., Baulac M., Cross H. The drug treatment of SE in Europe: Consensus document from a workshop at the first London colloquium on SE. Epilepsia. 2008;49:1277.
Shorvon S., Tan R. Uncommon causes of status epilepticus. Epilepsia. 2009;50(Suppl 12):63.
Silverstein A.M., Alexander J.A. Acute postictal cerebral imaging. Am J Neuroradiol. 1998;19:1485.
Simon R.P. Physiologic consequences of SE. Epilepsia. 1985;26(Suppl 1):S58.
Simon R.P., Pellock J.M., DeLorenzo R.J. Acute morbidity and mortality of SE. In: Engel J., Pedley T.A., editors. Epilepsy: A comprehensive textbook. Philadelphia: Lippincott-Raven, 1997.
Singhi S., Banerjee S., Singhi P. Refractory SE in children: Role of continuous diazepam infusion. J Child Neurol. 1998;13:23.
Sonnen A.E. Psychogenic SE. Neurology. 1997;49:637.
Sperber E.F., Wurpel J.N.D., Zhao D.Y., et al. Evidence for the involvement of nigral GABAA receptors in seizures of adult rats. Dev Brain Res. 1989;480:378.
Stecker M.M., Kramer T.H., Raps E.C., et al. Treatment of refractory SE with propofol: Clinical and pharmacokinetic findings. Epilepsia. 1998;39:18.
Stöhr T., Kupferberg H.J., Stables J.P., et al. Lacosamide, a novel anti-convulsant drug, shows efficacy with a wide safety margin in rodent mottles for epilepsy. Epilepsy Res. 2007;74:147.
Stores G., Zaiwalla Z., Styles E., et al. Non-convulsive SE. Arch Dis Child. 1995;73:106.
Takahashi Y., Kubota H., Fujiwara T., et al. Epilepsia partialis continua of childhood involving bilateral brain hemispheres. Acta Neurol Scand. 1997;96:345.
Tay S.K.H., Hirsch L.J., Leary L., et al. Nonconvulsive SE in children: clinical and EEG characteristics. Epilepsia. 2006;47:1504.
Terunuma M., Xu J., Vithlani M., et al. Deficits in phosphorylation of GABAA receptors by intimately associated protein kinase C activity underlie compromised synaptic inhibition during SE. J Neurosci. 2008;28:376.
Theodore W.H., Epstein L., Gaillard W.D., et al. Human herpes virus 6B: A possible role in epilepsy? Epilepsia. 2008;49(11):1828.
Thompson K., Wasterlain C. The model of SE that produces neuronal necrosis in the immature brain. Neurology. 1994;44:A272.
Tigaran S., Rasmussen V., Dam M., et al. ECG changes in epilepsy patients. Acta Neurol Scand. 1997;96:72.
Tilz C., Resch R., Hofer T., et al. Successful treatment for refractory convulsive SE by non-parenteral lacosamide. Epilepsia. 2009. Published Online: 8 Aug 2009
Towne A.R., Garnett L.K., Waterhouse E.J., et al. The use of topiramate in refractory SE. Neurology. 2003;60:332.
Towne A.R., Pellock J.M., Ko D., et al. Determinants of mortality in SE. Epilepsia. 1994;35:27.
Towne A.R., Waterhouse E.J., Boggs J.G., et al. Prevalence of nonconvulsive SE in comatose patients. Neurology. 2000;54:340.
Treiman D.M. SE. In: Laidlaw J., Richens A., Chadwick D., editors. A textbook of epilepsy. Edinburgh: Churchill Livingstone, 1993.
Treiman D.M. The role of benzodiazepines in the management of SE. Neurology. 1990;40(Suppl 2):32.
Treiman D.M., Meyers P.D., Walton N.Y., et al. Duration of generalized convulsive SE: Relationship to clinical symptomatology and response to treatment. Epilepsia. 1992;33(Suppl 3):66. (abstract)
Treiman D.M., Meyers P.D., Walton N.Y., et al. Duration of generalized convulsive SE: A multicenter comparison of four drug regimens. Neurology. 1994;44(Suppl):A219. (abstract)
Treiman D.M., Meyers P.D., Walton N.Y., et al. A comparison of four treatments for generalized convulsive SE. Veterans Affairs Status Epilepticus Cooperative Study Group. N Engl J Med. 1998;339:792.
Treiman D.M., Walton N.Y., Kendrick C. A progressive sequence of electroencephalogenic changes during generalized convulsive SE. Epilepsy Res. 1990;5:49.
Treiman M.A., Wu J., Marsh S.T., et al. EEG stage predicts electrophysiological changes in hippocampal slice recordings during experimental SE. Epilepsia. 2006;47(Suppl 4):326.
Trinka E., Dobesberger J. Review: New treatment options in SE: a critical review on intravenous levetiracetam. Ther Adv Neurol Disord. 2009;2:79.
Tsuchida T.N., Barkovich A.J., Bollen A.W., et al. Childhood Status Epilepticus and Excitotoxic Neuronal Injury. Pediatr Neurol. 2007;36:253.
Uberall M.A., Trollmann R., Wunsiedler U., et al. Intravenous valproate in pediatric epilepsy patients with refractory SE. Neurology. 2000;54:2188.
Uges J.W., van Huizen M.D., Engelsman J., et al. Safety and pharmacokinetics of intravenous levetiracetam infusion as add-on in SE. Epilepsia. 2009;50(3):415.
van Esch A., Ramlal I.R., van Steensel-Moll H.A., et al. Outcome after febrile SE. Dev Med Child Neurol. 1996;38:19.
van Gestel J.P.J., Blussé van Oud-Alblas H.J., Malingré M., et al. Propofol and thiopental for refractory status epilepticus in children. Neurology. 2005;65:591.
VanLandingham K.E., Heinz E.R., Cavazos J.E., et al. Magnetic resonance imaging evidence of hippocampal injury after prolonged focal febrile convulsions. Ann Neurol. 1998;43:413.
Villeneuve N., Ben-Ari Y., Holmes G.L., et al. Neonatal seizures induced persistent changes in the intrinsic properties of CA1 rat hippocampal cells. Ann Neurol. 2000;47:729.
Wallace S.J. Nasal benzodiazepines for management of acute childhood seizures? Lancet. 1997;349:222.
Wasterlain C.G. Glucose and energy metabolism of the immature brain during status epilepticus. In: Delgado-Escueta A.V., Wasterlain C.G., Treiman D.M., et al, editors. Status epilepticus: mechanisms of brain damge and treatment. New York: Raven, 1983.
Wasterlain C.G., Fujikawa D.G., Penix L., et al. Pathophysiological mechanisms of brain damage from SE. Epilepsia. 1993;11:S37.
Wasterlain C.G., Liu H., Mazarati A.M., et al. Self-sustaining SE: A condition maintained by potentiation of glutamate receptors and by plastic changes in substance P and other peptide modulators. Epilepsia. 2000;41(Suppl 6):S134.
Wasterlain C.G., Liu H., Mazarati A.M., et al. NMDA receptor trafficking during the transition from single seizures to SE. Ann Neurol. 2002;52(Suppl 1):S16.
Wasterlain C.G., Mazarati A.M., Naylor D.E., et al. short-term plasticity of hippocampal neuropeptides and neuronal circuitry in experimental SE. Epilepsia. 2002;43(Suppl 5):20.
Wilson J.V.K., Reynolds E.H. Translation and analysis of a cuneiform text forming part of a Babylonian treatise on epilepsy. Med Hist. 1990;34:185.
Weise K.L., Bleck T.P. SE in children and adults. Crit Care Clin. 1997;13:629.
Wheless J.W. Acute management of seizures in the syndromes of idiopathic generalized epilepsies. Epilepsia. 2003;44(Suppl 2):22.
Wong M., Yamada K.A. Developmental characteristics of epileptiform activity in immature rat neocortex: A comparison of four in vitro seizure models. Dev Brain Res. 2001;128:113.
Yu K.T., Mills S., Thompson N., et al. Safety and efficacy of intravenous valproate in pediatric SE and acute repetitive seizures. Epilepsia. 2003;44:724.
Zhang G., Raol Y.H., Hsu F.C., et al. Effects of SE on hippocampal GABAA receptors are age-dependent. Neuroscience. 2004;125:299.
Zhang G., Raol Y.H., Hsu F.C., et al. Long-term alterations in glutamate receptor and transporter expression following early-life seizures are associated with increased seizure susceptibility. J Neurochem. 2004;88:91.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 58 Status Epilepticus
The spectrum of seizure events extends from isolated, brief seizures to status epilepticus (SE), incorporating a full range of recurrent unprovoked seizures and prolonged or acute repetitive seizures. Typically, seizures are brief and self-limited. There has been considerable research directed at efforts to explain the mechanisms underlying seizure termination, as well as perpetuation, in SE. SE is a pediatric and neurologic emergency associated with significant morbidity and mortality [Dodson et al., 1993; Mitchell, 1996; Pellock, 1993a, 1994; Towne et al., 1994; Weise and Bleck, 1997; Wilson and Reynolds, 1990]. Its prompt recognition and management lead to the best chance of successful outcome. SE may represent the brain’s reaction to an acute insult, or it may be a manifestation of already existing epilepsy, either as the initial symptom or as a prolonged exacerbation of seizures [DeLorenzo et al., 1992, 1995, 1996].
Although the definition and classification of SE have been changed numerous times, the term refers to seizures that continue for a prolonged period. Studies suggest that SE frequently goes unrecognized, and that its occurrence has been underestimated in the general population [Treiman, 1993]. One study [O’Dell et al., 2005a] documented that 34 percent of cases of febrile SE presenting in emergency departments across five centers were not recognized as such by the hospital personnel assigned to treat them. Based on this finding, febrile SE has only a 2 out of 3 chance of being recognized in an emergency treatment setting.
This chapter reviews the pathophysiology, definition, classification, epidemiology, etiology, treatment, and prognosis of SE as it occurs in children. The mortality associated with SE is greater in adults than in children, but morbidity and mortality in children are considerable without treatment. Accordingly, current thinking about optimal management uses a more aggressive clinical approach to this neurological emergency, including prompt recognition and initiation of therapy and accelerating the progression of treatment for more rapid termination of the episode [Millikan et al., 2009].
Pathophysiology
A distinguishing feature of SE is the self-sustaining seizure condition. The pathophysiology and biochemical changes underlying the evolution from discrete seizure to SE remain unclear [Lowenstein and Alldredge, 1998; Pellock, 1994; Wasterlain et al., 1993]. Pathophysiological changes that accompany SE can be divided into neuronal (cerebral) and systemic effects. The mechanisms involved in the initiation and maintenance of SE may be different. Ultimately, SE results from a failure of inhibitory mechanisms.
Gamma-aminobutyric acid (GABA) is the most prevalent inhibitory neurotransmitter in the brain. GABAA receptors are postsynaptic ionotropic receptors that bind directly to chloride channels, producing a fast inhibitory postsynaptic potential (IPSP). GABAA receptors are the binding sites for the benzodiazepines and it is the activation of this receptor that accounts for its antiseizure effect. A unique feature of SE compared to brief seizures is the time-dependent development of pharmacoresistance to benzodiazepines [Mazarati et al., 1998]. In animal models, investigators studied GABAA receptor currents by whole-cell patch-clamp techniques in CA1 pyramidal neurons acutely dissociated from rats undergoing lithium/pilocarpine-induced limbic SE and from naive rats [Kapur and Coulter, 1995]. The GABAA receptor current was absent in 47 percent of SE neurons and reduced in 55 percent of the remainder, compared with naive neurons, thus aiding in seizure perpetuation. Kapur et al., using a paired-pulse technique in an electrogenic model of experimental SE, showed that a marked deterioration of GABA-mediated inhibition occurs during continuous hippocampal stimulation [Kapur et al., 1989].
Additionally, more recent work has demonstrated the development of benzodiazepine pharmacoresistance shortly after the onset of ictal spike wave activity [Jones et al., 2002] and in young naive rats [Goodkin et al., 2003]. Treiman et al. reported similar loss of inhibition in hippocampal slices obtained during various electroencephalography (EEG) stages in lithium-/pilocarpine-induced SE [Treiman et al., 2006]. Altered receptor function and changes in representation affect both seizure representation and consequences in the neonatal brain. GABAA receptors are heteromeric protein complexes that mediate most fast synaptic inhibition in the forebrain and have many distinct subtypes. Adult rats that develop epilepsy following pilocarpine-induced SE and adult patients with refractory temporal lobe epilepsy demonstrate significant alterations in GABAA receptor properties in hippocampal dentate granule neurons [Gibbs et al., 1997; Brooks-Kayal et al., 1998, 1999]. In rat pups exposed to pilocarpine-induced SE, different changes are seen in α-1 subunit expression and augmentation compared with those in adult rats. This produced the opposite effect and may serve to enhance inhibition; the rat pups did become epileptic, unlike the adult rats who developed recurrent spontaneous seizures [Zhang et al., 2004a]. Nevertheless, changes in the function of the immature GABAA receptor, from the dual role of excitatory-inhibitory to inhibitory as the rat matures in infancy, may contribute to increased excitability and hence more seizure susceptibility in the neonate [(Khazipov et al., 2004]. Additionally, alteration in glutamate receptor representation may alter seizure susceptibility. Alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors are over-represented in the premature brain, initially in the oligodendrocytes; later they shift their main representation to neurons in the cortex and hippocampus. This condition may partly explain increased risk of periventricular leukomalacia in the preterm infant and increased seizure susceptibility in the term infant [Jensen, 2002]. In one study the effect of SE induced by lithium/pilocarpine in 10-day-old rats demonstrated that pilocarpine-induced prolonged SE caused long-term changes in both glutamate receptors and transporters in hippocampal dentate gyrus. These include a decrease in glutamate receptor 2 mRNA expression and protein levels, as well as an increase in protein levels of the excitatory amino acid carrier 1[Zhang et al., 2004b].
Different studies provide evidence suggesting that endocytosis of GABAA receptors takes place as a seizure transitions to SE. This leads to a decrease in the number of GABAA receptors on the synaptic membrane and therefore a decreased response to benzodiazepines [Naylor et al., 2005; Goodkin et al., 2008]. Resistance to other antiseizure medications, such as phenytoin, has also been demonstrated. In addition to GABAA receptor trafficking, changes in receptor expression and phosphorylation have been shown [Brooks-Kayal et al., 1998, 1999; Terunuma et al., 2008]. Functional changes in voltage-dependent sodium channels have also been described after pilocarpine-induced SE in rats [Remy and Beck, 2006].
Glutamate is the primary excitatory amino acid neurotransmitter and binds to several neuronal receptors, including the N-methyl-d-aspartate (NMDA) receptor, which is activated by depolarization, as well as the AMPA receptor, which mediates fast synaptic transmission in the central nervous system. Using a paired-pulse method in a continuous hippocampal stimulation-induced SE model, NMDA receptors become activated during continuous hippocampal stimulation, and NMDA antagonists block the deterioration of GABA-mediated inhibition [Kapur and Lothman, 1990]. During SE, both NMDA and AMPA receptor subunits increase at the synaptic surface [Mazarati et al., 1998; Wasterlain et al., 2002a]. This increase in glutamate receptors further enhances excitability and is proconvulsant in the midst of uninhibited seizures. In a model of self-sustaining SE, seizures are abolished by NMDA receptor antagonists [Wasterlain et al., 2000].
Neuropeptides have emerged as having a role in SE. Substance P agonists facilitate the initiation of SE in animal models. During the course of SE, the de novo expression of substance P occurs in the cells, such as dentate granule cells, that do not usually express it, possibly playing a role in maintenance of SE [Wasterlain et al., 2002b]. In addition, galanin, a 29–30 amino acid peptide, suppresses hippocampal excitability by post- and presynaptic actions [Mazarati et al., 2000]. However, galanin appears to be depleted by SE.
Laminar necrosis and neuronal damage after prolonged seizures are similar to those after cerebral hypoxia. Neuronal injury and cell death from SE are most prominent in areas rich in NMDA glutamate receptors, including the limbic region. The increase in intracellular calcium is critical to cell death, and calcium activates proteases and lipases that degrade intracellular elements, leading to mitochondrial dysfunction and fatal cellular necrosis. Although young animals may be less likely to develop brain damage from SE [Holmes, 1997; Moshè, 1987], studies using alternative models demonstrate hippocampal cellular injury, even in immature rodents [Sankar et al., 1997; Thompson and Wasterlain, 1994]. It is believed that the glutamate-initiated calcium-dependent cascade is mechanistically similar to the NMDA receptor-mediated cell death occurring during cerebral ischemia. In SE, the degree of neuronal injury is related to seizure duration. In a study of limbic SE induced in adult rats using perforant path stimulation, when animals were allowed to recover, there was evidence of mitochondrial injury and dysfunction demonstrated possibly through a free radical mechanism of injury [Cock et al., 2002]. Further evidence suggests that acute and long-term changes in gene expression may occur after prolonged seizures. These changes in the expression of messenger RNA (mRNA) may lead directly to some of the observed hyperexcitability [Rice and DeLorenzo, 1998].
In animal studies using adolescent baboons, induced SE lasting 1.5–5 hours produced neuronal loss in the hippocampus, cerebellum, and neocortex. Significant cell loss continued to occur, although to a lesser extent if the animals were paralyzed and ventilated with maintenance of oxygen, carbon dioxide, serum glucose, body temperature, and blood pressure [Meldrum, 1974, 1983]. Similar changes have been produced in rat models, as well [Cavalheiro et al., 1987; Sperber et al., 1989].
There are limited data in humans. However, evidence from neuropathology, as well as imaging, has been presented in a pediatric case with direct excitotoxic injury in the absence of hypoxia-ischemia [Tsuchida et al., 2007].
Generalized convulsive SE is associated with serious systemic effects resulting from the metabolic demands of prolonged seizures and the autonomic changes that accompany them: alterations in blood pressure and heart rate, incontinence, emesis, acidosis, hypoxia, changes in respiratory function and body temperature, leukocytosis, rhabdomyolysis, and extreme demands on cerebral oxygen and glucose use [Simon et al., 1997]. Circulating catecholamines increase during the initial 30 minutes of SE, resulting in a hypersympathetic state. Tachycardia, sometimes associated with more severe cardiac dysrhythmias, occurs and may be fatal, but this seems more common in adults [Boggs et al., 1993]. Cardiac output also diminishes, and total peripheral resistance increases, along with mean arterial blood pressure, perhaps because of the sympathetic overload. Hyperpyrexia may become significant during the course of SE, even without prior febrile illness in both children and adults, and may persist for some time. Fever may influence the process of neuronal injury [Liu et al., 1993].
Unlike in adults, in whom biology is more static, the effect of growth and development influences the impact of seizures, both in clinical presentation and in the biologic consequences for the developing brain. Most of our knowledge derives from animal models, so its applicability to the human situation is unclear. Experimental models of seizures in immature animals suggest comparatively less vulnerability to seizure-induced brain injury than in mature animals [Wong and Yamada, 2001]. Repetitive or prolonged neonatal seizures may increase the susceptibility of the developing brain to experience subsequent seizure-induced brain injury later in life. This susceptibility appears to be more closely related to alterations in neuronal connectivity and network properties, rather than to increased cell death during the neonatal period [Holmes et al., 1998; Koh et al., 1999; Schmid et al., 1999].
Rats exposed to early-life seizures demonstrate persistent changes in CA1 hippocampal pyramidal cells, possibly leading to long-term changes in behavior and learning and in epileptogenicity [Villeneuve et al., 2000].
Definition
SE is internationally classified as a seizure lasting more than 30 minutes or recurrent seizures producing more than 30 minutes during which the patient does not regain consciousness [ILAE, 1981]. The World Health Organization previously defined SE as “a condition characterized by epileptic seizures that are so frequently repeated or so prolonged as to create a fixed and lasting condition” [Gastaut, 1982]. Lack of recovery for a fixed period, possible frequent repetition, prolongation, and possible propagation of further seizures are inherent in this definition. In the past, the definition of SE required 1 hour of continuous seizures, but more recent studies have used a 30-minute duration of continuous or recurrent seizures without full recovery as the standard clinical and electrographic definition of SE. The recognition that longer duration of seizures increases risk for long-term injury and the risk for fracture during seizures and their treatment has required a definition implying need for expediency in stopping prolonged seizures. Lowenstein and associates proposed an “operational” definition of 5 minutes or more of continuous seizures or “two discrete seizures between which there is incomplete recovery of consciousness” in adults and children older than 5 years of age [Lowenstein et al., 1999]. This definition applies primarily to generalized convulsive SE and may be used to direct treatment to avoid refractory SE, as well as its sequelae. Aggressive early treatment is justified by recent work demonstrating a 10-fold lower rate of mortality for seizures of 10–29 minutes’ duration versus those lasting longer than 30 minutes [DeLorenzo et al., 1999].
Because of the difficulty in diagnosing and quantifying seizures in the neonate, no broadly accepted definition of SE in the neonate exists. A proposed definition is either 30 minutes of continuous electroencephalographic seizures or presence of seizure activity for 50 percent of the EEG recording time, with or without the expression of coincident clinical signs (Scher et al., 1993b). Debate continues regarding what constitutes a neonatal seizure. Neonatal seizures can be broken down into three categories: electroclinical, electrographic, and clinical only. Controversy still exists about whether episodic abnormal movements seen in some infants, not accompanied by simultaneous ictal discharges on the EEG, are true seizures. Many neonatal paroxysmal events classified as “subtle seizures” have no EEG correlate [Mizrahi and Kellaway, 1998]. Typical subtle seizures include movements of progression, such as bicycling, oral-buccal-lingual movements, such as chewing and tongue thrusting, and other movements, such as random eye movements. Other movements that typically have no EEG correlate include generalized tonic posturing. Movements typically demonstrating simultaneous ictal discharges on EEG include focal clonic, multifocal clonic, and focal tonic; myoclonic movements may or may not have an EEG correlate [Mizrahi and Kellaway, 1998].
Classification
Any type of seizure may become prolonged and thus develop into SE (Box 58-1). Classification of SE should be performed by observing the clinical events and combining electrographic information when possible. The fundamental distinction between seizures is that some are generalized from onset, whereas others are partial in onset. The latter type may or may not then secondarily generalize. From a management standpoint, however, it may be more useful to consider whether the event is convulsive or nonconvulsive, as this may impact more directly on ready recognition and intervention.
Generalized tonic-clonic SE is the most dramatic and life-threatening form of SE. Myoclonic, generalized clonic, and generalized tonic SE occur primarily in children. These children usually have encephalopathic epilepsies [Lockman, 1990; Pellock, 1994; Treiman, 1993], and their consciousness may be preserved throughout the attacks. About one-half of the cases of generalized clonic SE occur in normal children, in whom it is associated with prolonged febrile seizures; the remaining half are distributed among those children with acute and chronic encephalopathies [DeLorenzo et al., 1992]. Generalized tonic SE appears most frequently in children, particularly those with the Lennox–Gastaut syndrome. Prolonged generalized tonic convulsions have been precipitated by benzodiazepine administration.
Nonconvulsive SE also may include complex partial, simple partial, and absence seizures that continue for more than 30 minutes [Kaplan, 1996; Scholtes et al., 1996; Stores et al., 1995]. Complex partial SE may be manifested as an epileptic twilight state marked by a cyclic variation between periods of partial responsiveness and episodes of seemingly motionless staring and complete unresponsiveness accompanied, at times, by automatic behavior [Delgado-Escueta and Treiman, 1987; Privitera, 1997; Scher et al., 1993a; Treiman, 1993]. Simple partial SE is characterized by focal seizures that may persist or be repetitive for at least 30 minutes without impairment of consciousness. When this condition lasts for hours or days, it is termed epilepsia partialis continua [Cockerell et al., 1996; Takahashi et al., 1997]. Absence, or petit mal, status also has been referred to as spike-wave stupor. This type of nonconvulsive SE may be extremely difficult to differentiate from complex partial SE without the aid of an EEG. Classically, features of absence status include a continuous alteration of consciousness without the cyclic variations seen with complex partial SE [Grin and DiMario, 1998]. The EEG recording exhibits prolonged, sometimes continuous, generalized synchronous 3-Hz spike-and-wave complexes, rather than focal ictal discharges, which characterize partial SE [Porter and Penry, 1983; Treiman, 1993]. Absence status does not appear to cause permanent neurological damage [Drislane, 1999]. The child presenting with a prolonged confused state, with a fluctuating level of consciousness or with prolonged unconsciousness, may require both clinical and EEG evaluations in addition to other studies.
Nonconvulsive SE is most likely not rare but simply underdiagnosed. A special category of nonconvulsive SE is subtle SE [Treiman, 1990, 1993]. These patients have severe encephalopathies stemming from a variety of intracranial processes or prolonged uncontrolled convulsive seizures. This type of SE is manifested clinically by the occurrence of mild motor movements, such as nystagmus, or by clonic twitches, which may be unilateral and are intermittent, brief, and without a true sequential pattern. These subtle movements are associated with marked impairment of consciousness, usually with continuous bilateral EEG ictal patterns. These continuing electrographic seizures that are not accompanied by clinical manifestations demonstrate a true “electroclinical dissociation,” and may be seen not only in neonates but also in severely ill children and adults [Mizrahi and Kellaway, 1987; Scher et al., 1993a]. The EEG progressively becomes uniform to produce a pattern of continuous ictal discharges, which then becomes interrupted by periods of relative flatness and then severe cortical depression. In a study of children, nonconvulsive SE most commonly followed a bout of convulsive SE or briefer convulsive seizure but with prolonged alteration in mental status [Tay et al., 2006], and is more likely to occur in children with a prior history of epilepsy [Abend et al., 2007] and remote risk factors for seizures [Classen et al., 2004]. In general, these patients respond poorly to traditional treatment with antiepileptics.
Febrile SE, which is unique to children, represents the extreme end of the complex febrile seizure spectrum. Febrile SE has long been suspected of having a relationship to the development of mesial temporal sclerosis. Patients with intractable temporal lobe epilepsy and mesial temporal sclerosis often have histories of severe febrile convulsions as infants. Diagnostic advances made possible by magnetic resonance imaging (MRI) have shown that very prolonged febrile convulsions may produce hippocampal injury [Lewis, 1999]. More recent studies support the link to the development of mesial temporal sclerosis [Provenzale et al., 2008]. Neuroimaging studies generally show hippocampal swelling during the acute stage [Scott et al., 2002, 2006]. A prospective study investigating long-term outcome in febrile SE found that most were partial (67 percent), and SE was unrecognized in the emergency department about a third of the time [Shinnar et al., 2008]. Human herpesvirus-6B appears to be the most common cause of febrile SE and may play an important pathogenic role in the etiology of mesial temporal lobe epilepsy [Theodore et al., 2008].
Epidemiology
It is projected that between 102,000 and 152,000 events occur in the United States annually, an incidence 2–2.5 times greater than that previously proposed by Hauser [1990], who reported that SE occurs annually in 50,000–60,000 persons in the United States [DeLorenzo et al., 1996; Hauser et al., 1990]. More recent work continues to demonstrate a high incidence of SE varying between 20 and 41 patients per year per 100,000 population [Govoni et al., 2008; Chin et al., 2006; Coeytaux et al., 2000].
Approximately one-third of cases manifest as the initial seizure of a developing epilepsy, one-third occur in patients with previously established epilepsy, and one-third occur as the result of an acute isolated brain insult. Among those previously diagnosed as having epilepsy, estimates of SE occurrence range from 0.5 to 6.6 percent [DeLorenzo et al., 1996].
Hauser reported that up to 70 percent of children who have epilepsy that begins before the age of 1 year will experience an episode of SE [Hauser, 1990]. Also, within 5 years of the initial diagnosis of epilepsy, 20 percent of all patients will experience an episode of SE. Although the subsequent development of epilepsy is likely in adults with SE as their first unprovoked seizure [Hauser et al., 1990], a prospective study of children with SE found only a 0.3 probability plotted on a Kaplan–Meier curve that epilepsy will develop after ≥9 months in those who initially presented with SE [Maytal et al., 1989].
Among children, SE is most common in infants and young toddlers, with more than 50 percent of cases of SE occurring in children younger than 3 years of age [Shinnar et al., 1995]. In a study in Richmond, Virginia, total SE events and incidence per 100,000 population per year demonstrated a bimodal distribution, with the highest values during the first year of life and during the decades beyond 60 years of age [DeLorenzo et al., 1992, 1995, 1996]. Infants younger than 1 year of age represent a subgroup of children with the highest incidence of SE, whether events, total incidents, or recurrences are counted. The recurrence rate for SE in the Richmond study was 10.8 percent [DeLorenzo et al., 1996], but 38 percent of patients younger than 4 years of age had repeat episodes. Children have a much lower mortality rate than adults after adequate treatment [Dunn, 1988; Maytal et al., 1989; Pellock, 1993b; Phillips and Shanahan, 1989; Shinnar et al., 1995]. Age, etiology, and duration directly correlate with mortality [DeLorenzo et al., 1996; Towne et al., 1994].
Etiology
SE usually is a manifestation of an acute precipitating event that affects the central nervous system (CNS) or is an exacerbation of symptomatic epilepsy. Less than 10 percent of cases of SE in adults and children are truly idiopathic in that no precipitating or associated cause can be identified [DeLorenzo et al., 1995, 1996]. Acute symptomatic causes are those most commonly associated with prolonged SE lasting for longer than 1 hour [DeLorenzo et al., 1996]. Thus, a full evaluation for etiology must be undertaken in every case of SE [Dodson et al., 1993; Pellock, 1994]. In patients with pre-existing epilepsy, a precipitating or associated factor may be clearly identified. Identification of this factor may help in treating the episode of SE, preventing further consequences of SE, and perhaps preventing future recurrences.
Although, typically, a precipitant to SE can be identified, a genetic susceptibility may predispose certain persons to develop prolonged seizures in response to an acute insult. Recent work in twins demonstrated a higher incidence in monozygotic twins than in dizygotic twins, providing evidence for a genetic contribution to the risk for SE [Corey et al., 2004]. Seizure type and specific epilepsy syndrome may differ between monozygotic twins, however, and SE may not be a function of the seizure type or syndrome experienced by each person.
A clear difference between causative disorders of SE in adults (Table 58-1) and those in children (Table 58-2) has been identified [Pellock and DeLorenzo, 1997; Riviello et al., 2006]. A major cause of SE in children is that associated with fever secondary to non-CNS infections, an etiology that essentially does not exist in adults. Inadequate antiepileptic drug levels and remote causes, including congenital malformations, also account for a significant number of episodes of SE in children, although some studies have found that patients may have had reasonable drug levels when SE occurred [Maytal et al., 1996]. Of note, many patients with subtherapeutic levels of antiepileptic drugs closely followed the instructions of their physicians and recently had drug-dosage alterations.
Table 58-1 Cause and Mortality Data for Status Epilepticus in Adults
| Etiologic Disorder/Condition | % of Cases | Mortality Rate (%) |
|---|---|---|
| Anoxia | 5 | 71 |
| Hypoxia | 13 | 53 |
| Cerebrovascular accident | 22 | 33 |
| Hemorrhage | 1 | 0 |
| Tumor | 7 | 30 |
| Infection | 7 | 10 |
| Central nervous system infection | 3 | 0 |
| Metabolic | 15 | 30 |
| Low antiepileptic drug level | 34 | 4 |
| Drug overdose | 3 | 25 |
| Alcohol withdrawal | 13 | 20 |
| Trauma | 3 | 25 |
| Remote | 25 | 14 |
| Idiopathic | 3 | 25 |
(From Pellock JM, DeLorenzo RJ. SE. In: Porter RJ, Chadwick D, eds. The epilepsies 2. Boston: Butterworth–Heinemann, 1997;267.)
Table 58-2 Etiologies of Status Epilepticus in Children
| Etiology | Percentage |
|---|---|
| Remote symptomatic epilepsy | 33% |
| Acute symptomatic seizures | 26% |
| Febrile | 22% |
| Cryptogenic | 15% |
| Central nervous system infection | 13% |
| Acute metabolic disorders | 6% |
(From Riviello JJ et al. Practice Parameter: Diagnostic assessment of the child with status epielpticus [an evidence based review]. Neurology 2006;67:1542.)
The distribution of causes associated with SE in children is highly age-dependent [Shinnar et al., 1995]. More than 80 percent of children younger than 2 years of age have SE from a febrile or acute symptomatic cause, whereas cryptogenic or remote symptomatic causes were more common in older children. By contrast, in adults, subtherapeutic levels of antiepileptic drugs, remote causes, and cerebrovascular accidents represent the three most common causes of SE [Pellock and DeLorenzo, 1997]. In adults, SE resulting from remote causes occurred primarily in relation to stroke, so that both acute and previously occurring strokes account for a significant proportion of adult episodes of SE. Stroke is the etiologic disorder in approximately 10 percent of childhood SE episodes, either as the primary acute cause or as a remote event.
Recurrence rates for SE are age-specific, as illustrated in Figure 58-1. Repeat occurrences are much more common in children younger than 1 year of age. In the prospective Richmond study of SE, pediatric, adult, and elderly recurrence rates were 35 percent, 7 percent, and 10 percent, respectively [DeLorenzo et al., 1996]. Recurring SE is more frequent in children with remote symptomatic encephalopathy or progressive degenerative disease [DeLorenzo et al., 1995, 1996]. The extent of clinical and laboratory evaluation that should be performed in each child with recurrent SE will depend on the presentation, signs and symptoms, and underlying medical condition of the patient. For example, a child with recurring SE who has an indwelling shunt for hydrocephalus will almost always need to be evaluated for the possibility of shunt malfunction or infection.
Recently, CNS or systemic autoimmune disorders have been recognized as causes of SE [Shorvon and Tan, 2009]. In some cases, the diseases may be paraneoplastic, but for many, no cause has been determined. Since patients with these etiologies usually are described in case reports or small series, the contribution of the disorders to the epidemiology of SE is difficult to assess, but they appear to represent a fraction of the cases previously diagnosed as infectious, since the patients often have a syndrome qualifying as an encephalitis.
A recently described entity with seizures and persistently altered mentation, including a fluctuating level of consciousness or with prolonged unconsciousness, is anti-NMDA-receptor encephalitis [Dalmau et al., 2008]. Anti-NMDA-receptor encephalitis is a disorder with antibodies against the NR1 subunit of the receptor. This disorder largely affects young people, and its diagnosis is facilitated by the characteristic clinical picture that develops in association with cerebrospinal fluid pleocytosis. Only about half the patients had MRI abnormalities. Recovery from this disorder is typically slow, and symptoms may relapse. The mainstay of treatment is immunotherapy with steroids, plasma exchange, or intravenous immunoglobulin. These immunotherapies may be given individually or in combination [Dalmau et al., 2008]. Other possible autoimmune illnesses include systemic lupus erythematosus, antineuronal antibody syndromes with limbic encephalitis, limbic encephalitis following various systemic viral infections, and Hashimoto’s encephalopathy (autoimmune thyroid encephalopathy).
Management and Therapy
Overview
SE is a neurologic and medical emergency. Therapy includes maintenance of respiration, general medical support, and specific treatment aimed at stopping both electrographic and clinical seizures while the cause of the event is investigated [Epilepsy Foundation of America, 1993]. Prompt diagnosis and management provide the best outcome [DeLorenzo, 1990; Pellock, 1993a, 1993b, 1994; Pellock and DeLorenzo, 1997; Rider and Thapa, 1995; Treiman, 1993].
A single generalized convulsion in a child with a prolonged period of impaired consciousness is much more difficult to diagnose, and an EEG should be obtained urgently. If on-going ictal discharges or electrographic seizures are noted, the patient should be considered to be in electrographic SE, and prompt treatment is indicated. The goals of SE emergency management are listed in Box 58-2. Clinical and electrographic seizure activity must be rapidly terminated while ensuring optimal oxygenation and metabolic balance. Both clinical and electrical seizure activity should be terminated as soon as possible [Treiman, 1990]. The longer an episode of SE continues, the more likely it is to result in permanent neurologic damage and to become refractory to treatment [DeLorenzo, 1990; DeLorenzo et al., 1996; Hauser, 1990; Pellock and DeLorenzo, 1997; Towne et al., 1994; Treiman, 1990; Treiman et al., 1992, 1994].
Box 58-2 Goals of Emergency Management for Status Epilepticus
(From Pellock JM, DeLorenzo RJ. SE. In: Porter RJ, Chadwick D, eds. The epilepsies 2. Boston: Butterworth–Heinemann, 1997:267.)
Rapid initiation of care is essential to ensure the best possible outcome. Increased seizure duration commonly is regarded as an important factor contributing to increased morbidity and mortality. Prolonged seizures of any type are associated with an increased risk of complications [Lowenstein et al., 1999]. In a study in adults and children, the mortality rate in SE was 34.8 percent when seizures lasted longer than 1 hour, compared with 3.7 percent when seizure duration was less than 1 hour [DeLorenzo et al., 1992]. Therefore, limiting the time from seizure onset to initial treatment and attainment of control is essential to minimize the complications of prolonged seizures. In a retrospective study, patients with SE may not arrive at the emergency department for 30 minutes or longer, and treatment initiation may not occur for 40–263 minutes, thereby increasing risk for prolonged seizures [Jordan, 1994]. The Veterans Affairs Cooperative Study reported that the mean delay before treatment of SE was 2.8 hours in generalized convulsive SE and 5.8 hours in subtle SE; this study involved exclusively adult patients [Treiman et al., 1998]. In a prospective study of 889 patients (625 adults, 264 children), 41.5 percent received treatment within 30 minutes and 70.9 percent within 60 minutes of seizure onset; 18.1 percent did not have treatment initiated until after 90 minutes of seizures. This latter group of patients, however, included patients in coma diagnosed with subtle or nonconvulsive SE [Pellock et al., 2004].
Supportive Care
In children in SE, cardiorespiratory function must be assessed immediately by vital sign determination, auscultation, airway inspection, and determination of arterial blood-gas concentrations, with suctioning when necessary. Although these children may appear to be breathing spontaneously on arrival at the emergency department, they may already be hypoxic, with respiratory and metabolic acidosis, apnea, aspiration, or central respiratory depression. The need for ventilatory support depends not only on the respiratory status at the time of presentation but also on the conditions before arrival and the ability to maintain adequate oxygenation throughout on-going seizures and during the intravenous administration of large doses of antiepileptic drugs, all of which may cause respiratory depression. In the neurologically depressed patient, elective intubation and respiratory support are recommended. Placement of an oral airway or nasal oxygen cannula will prove to be insufficient in most patients, because respiratory drive is depressed [Pellock, 1994]. Respiratory arrest and depression are principal factors contributing to morbidity and mortality [DeLorenzo et al., 1996]. Rapidly assessing vital signs and performing a general neurologic examination may provide clues to the etiology of SE.
Laboratory Testing
Blood should be drawn for the determination of blood gases, glucose, calcium, electrolytes, complete blood count, and antiepileptic drug levels, as well as for cultures (bacterial and viral) if indicated. Urine drug and metabolic screening should be performed whenever the etiology cannot be determined. As previously noted, a common etiologic or associated factor found in children with SE is intercurrent infection. In a small number of these children, the clinical presentation will include manifestations similar to the initial symptoms of CNS infection. Lumbar puncture should be done early in the course of management, although not necessarily during the initial phase of stabilization [Rider et al., 1995]. In most children, only rarely should the clinician wait to obtain neuroimaging studies before performing a lumbar puncture. Hyperpyrexia may become significant during the course of SE, even in the absence of a febrile illness. Rectal temperatures should be monitored and fever aggressively treated; significant elevations of temperature may contribute to brain damage [Dodson et al., 1993].
[/not-level-membership-for-neurology-category]
