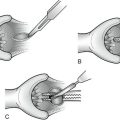
CHAPTER 18 Splenectomy
INDICATIONS FOR SPLENECTOMY
I. Platelet-Associated: ITP is the most common indication for elective splenectomy. ITP is caused by circulating autoantibodies that bind to platelet membrane antigens and facilitate phagocytosis in the spleen and elsewhere.
A. Patients with ITP commonly present with a history of bruising, epistaxis, and gingival bleeding. Life-threatening complications, such as gastrointestinal or intracranial hemorrhage, are much less common. Women are affected more often than are men; however, in children, ITP affects both sexes equally. ITP in children is typically characterized by a more rapid onset and a more severe course than in adults. Spontaneous remission occurs in approximately 80% of affected children, and splenectomy should be reserved for cases of severe refractory ITP lasting longer than 1 year in this patient population.
B. Therapy for ITP is aimed at achieving sustained remission. In patients with asymptomatic disease, treatment is indicated for platelet counts between 20,000/mm3 and 30,000/mm3 or for platelet counts less than 50,000/mm3 with additional risk factors for bleeding, such as hypertension and peptic ulcer disease. Initial treatment consists of glucocorticoids. In patients with platelet counts less than 20,000/mm3 or with life-threatening bleeding, hospitalization is indicated. IVIG plays an important role in the management of acute bleeding as well as in the surgical preparation of patients with platelet counts of less than 20,000/mm3. Splenectomy is indicated in patients with thrombocytopenia refractory to steroids and IVIG, steroid-associated side effects, or recurrent thrombocytopenia after steroid treatment.
II. Erythrocyte-Associated
A. Hereditary spherocytosis (HS): HS is an autosomal dominant hemolytic anemia caused by a deficiency of spectrin, a protein that gives red blood cells their shape, strength, and flexibility. Erythrocytes that are deficient in spectrin lose surface area and deformability and have a characteristic spherical shape. Spherocytes are sequestered in the microcirculation of the spleen and destroyed. Patients with HS most commonly present with anemia, jaundice, and splenomegaly. Splenectomy significantly improves the anemia by eliminating the predominant site of red cell destruction. Splenectomy should be delayed until after 4 years of age to preserve immunologic function and reduce the risk of overwhelming postsplenectomy infection (see Complications). Given the high incidence of bilirubin gallstones in patients with this disorder, an abdominal ultrasound should be performed before splenectomy. If cholelithiasis is present, cholecystectomy should be performed at the time of splenectomy.
B. Hemoglobinopathies: Inherited disorders of hemoglobin synthesis, such as sickle cell disease and the thalassemias, are associated with increased erythrocyte destruction and anemia.
1. Sickle cell disease results from a genetic mutation in the beta globin chain that causes an abnormal hemoglobin tetramer (hemoglobin S). Deoxygenated hemoglobin S polymerizes within erythrocytes, leading to the characteristic sickle shape. These red blood cells can occlude capillaries and lead to microinfarction within various organ systems. Repeated vaso-occlusive insults cause the spleen to infarct and atrophy. Splenic sequestration crisis is characterized by a marked decrease in the hemoglobin concentration, hypovolemia, and splenomegaly. Patients may experience severe pain and require multiple blood transfusions. Splenectomy is indicated after a first episode of acute splenic sequestration and in patients with hypersplenism.
2. The thalassemias are a group of disorders resulting from abnormal synthesis of one of the hemoglobin chains. Patients with these disorders present with varying degrees of anemia and splenomegaly. Transfusions and iron chelation are the hallmarks of long-term treatment. Splenectomy is indicated in patients with increasing transfusion requirements and symptomatic splenomegaly.
C. Autoimmune hemolytic anemia (AIHA): AIHA is a term that describes a group of disorders characterized by the presence of autoantibodies that bind to the surface of erythrocytes and lead to hemolysis.
1. In warm-antibody AIHA, so named because the predominant IgG antibodies preferentially bind at 37°C, tissue macrophages within the spleen recognize antibody-bound erythrocytes and remove them from the circulation. Initial treatment consists of steroid therapy or other immunosuppressive agents. Splenectomy eliminates the major site of hemolysis and should be performed in patients in whom medical therapy is unsuccessful and in those who cannot receive corticosteroids.
III. Leukocyte-Associated
A. Hodgkin’s lymphoma: Hodgkin’s lymphoma is a malignancy that predominantly affects young adults. Historically, the staging of Hodgkin’s lymphoma required laparotomy and splenectomy. Improvements in computed tomography (CT) scanning and other imaging modalities have made surgical staging far less important in directing treatment. Splenectomy is indicated for symptomatic splenomegaly.
B. Non-Hodgkin’s lymphoma (NHL): NHL constitutes a diverse group of primary malignancies of the lymphoreticular tissue. Splenectomy is indicated in patients with hypersplenism resulting in anemia, thrombocytopenia, neutropenia, or symptomatic splenomegaly. NHL limited to the spleen is an additional indication for splenectomy.
C. Hairy cell leukemia (HCL): HCL is characterized by splenomegaly, pancytopenia, and evidence of cell membrane projections on microscopy. Purine analogues constitute first-line treatment. Splenectomy is reserved for patients with disease refractory to medical therapy and those with symptomatic splenomegaly.
D. Chronic lymphocytic leukemia: Chronic lymphocytic leukemia is a lymphoproliferative disorder characterized by the accumulation of functionally incompetent lymphocytes of monoclonal origin. Splenectomy is indicated for symptomatic splenomegaly and for hypersplenism.
E. Chronic myelogenous leukemia (CML): CML is a myeloproliferative disorder characterized by clonal proliferation of myeloid cells at all stages of maturation. A chromosomal translocation between chromosomes 9 and 22 (Philadelphia chromosome), which leads to the elaboration of a novel fusion protein (Bcr-Abl), is critical to the pathogenesis of this disease. Treatment of CML includes tyrosine kinase inhibitors and hematopoietic stem cell transplantation. Splenectomy may be performed for symptomatic splenomegaly or hypersplenism.
IV. Nonhematologic Abnormalities of the Spleen
A. Tumors: Primary nonlymphoid tumors of the spleen are uncommon. Hemangiomas are the most common benign primary tumor of the spleen and are sometimes associated with splenic rupture and thrombocytopenia when large. Splenectomy is indicated for symptomatic lesions. Splenic hamartomas and lymphangiomas are benign tumors and are rarely associated with symptoms. Splenectomy is sometimes performed for diagnostic purposes. Angiosarcomas are highly aggressive tumors with a poor prognosis. Patients may present with splenomegaly, hemolytic anemia, or spontaneous splenic rupture. Splenectomy is indicated, but is most often palliative. Metastases to the spleen are uncommon and are usually indicative of disseminated disease. Splenectomy may be appropriate if the spleen is the site of an isolated metastasis.
B. Splenic cysts
1. True (epithelium-lined) splenic cysts may be parasitic or nonparasitic. In the United States, nonparasitic cysts are much more common. These are typically discovered incidentally, but may cause symptoms such as abdominal pain, nausea, and early satiety when large. Splenectomy is indicated for patients with symptomatic cysts. Parasitic cysts are rare in the United States and are most commonly due to infection with Echinococcus granulosus. Associated liver involvement is common. Cyst rupture and spillage of scoleces can be catastrophic; therefore, splenectomy is indicated for the treatment of all parasitic cysts.
C. Splenic abscess: Splenic abscesses may result from hematogenous spread of infection from a remote site, such as the heart (endocarditis) or bone (osteomyelitis), or from bacteremia (most commonly, in the setting of intravenous drug use). Other risk factors include malignancy, previous trauma, poor nutritional status, and immunocompromise. Patients may present with fever, vague abdominal pain, or pleuritic pain. Treatment consists of intravenous antibiotic therapy and control of the infectious source. Percutaneous drainage may be attempted for a unilocular splenic abscess. Multilocular abscesses typically require splenectomy. Failure of percutaneous drainage and clinical deterioration are additional indications for splenectomy.
D. Sinistral hypertension: Pancreatitis is sometimes associated with splenic vein thrombosis and the development of sinistral (left-sided) portal hypertension, which may be complicated by gastric varix formation. Symptomatic hypersplenism and gastrointestinal bleeding from varices are indications for splenectomy.
V. Trauma
A. Splenic injuries are most commonly caused by rapid deceleration, puncture from adjacent rib fractures, penetrating trauma, or high-energy transfer through the posterolateral aspect of the chest wall. Given the extensive arterial supply of the spleen, it is not surprising that such injuries can result in extensive hemorrhage. Current modalities for evaluating splenic injury include helical CT scanning and ultrasound. CT scanning is particularly useful in that it allows for the rapid evaluation of multiple abdominal organs and the retroperitoneum. The Focused Abdominal Sonography for Trauma (FAST) examination can identify fluid in the peritoneum. A positive finding on FAST examination in a hemodynamically unstable patient warrants emergent laparotomy.
B. Nonoperative management of splenic injuries is appropriate in certain situations. If a patient is hemodynamically stable without additional abdominal injuries requiring exploration and there is no blush (i.e., extravasation of contrast) from the spleen on CT scan, splenic injuries may be observed without immediate operative intervention (Fig. 18-1). Evidence of a hilar injury, hemodynamic instability after resuscitation, and persistent transfusion requirements are indications for operative exploration in patients with evidence of a splenic injury.
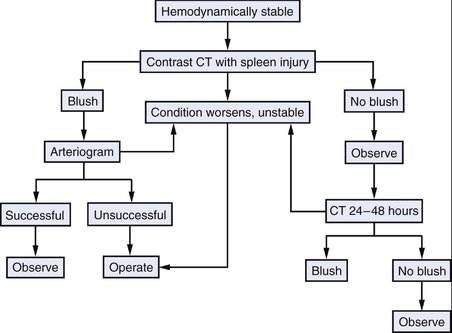
PREOPERATIVE EVALUATION
I. Imaging Studies: Preoperative imaging of the abdomen commonly includes CT. The need for additional imaging is largely dictated by the diagnosis.
II. Vaccination: All patients undergoing elective splenectomy should receive vaccines for the encapsulated organisms Streptoccus pneumoniae, Neisseria meningiditis, and Haemophilus influenzae type B 2 or more weeks before the planned operation. After urgent or emergent splenectomy, patients should be vaccinated before discharge.
COMPONENTS OF THE PROCEDURE AND APPLIED ANATOMY
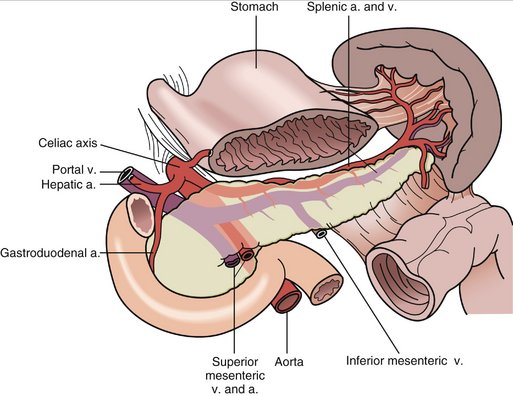
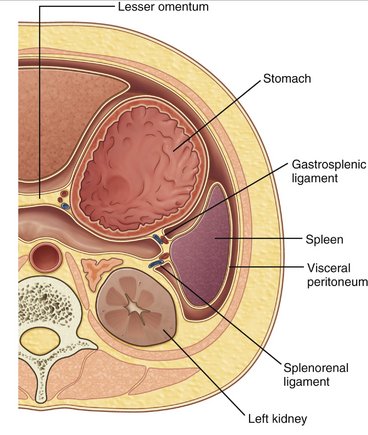
see Figures 18-2 and 18-3.
Open Splenectomy
II. Preoperative Considerations
III. Incision: A left subcostal incision can be used if the spleen is of normal size. A midline approach should be used in patients with splenomegaly and for the exploration of traumatic injuries.
IV. Exposure and Splenectomy
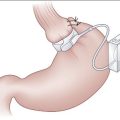
B. The gastrosplenic ligament, which extends from the greater curvature of the stomach to the spleen and contains the short gastric vessels, is then divided. The most superior short gastric vessels are divided first, followed by the more inferior vessels, until a connection is made with the gastrocolic window.
C. Medial traction is applied to the spleen to expose the lateral peritoneal attachments, which are divided with electrocautery. The plane posterior to the spleen and the tail of the pancreas is then developed bluntly.
D. The splenocolic ligament is ligated and divided. The spleen is mobilized toward the midline and delivered to the level of the subcutaneous tissues.
E. The splenic artery and vein are identified at the splenic hilum and are individually ligated and divided.
Laparoscopic Splenectomy
I. Patient Positioning and Preparation
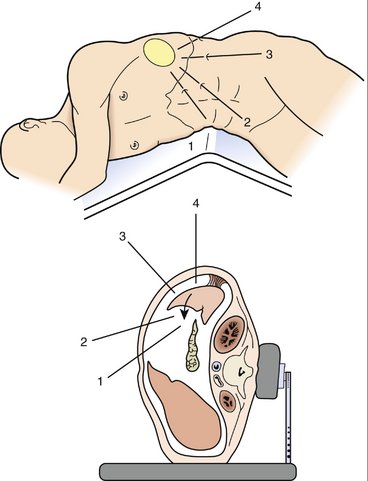
A. The patient is placed supine on the operating table in the modified lithotomy position. Alternatively, the patient can be placed in the right lateral decubitus position, with the iliac crest over the break in the table (Fig. 18-4).
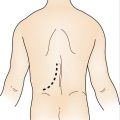
II. Port Placement
III. Technical Points
A. The abdomen is insufflated to a pressure of approximately 15 mm Hg. The camera is inserted, and a thorough exploration of the abdomen is undertaken to exclude the presence of accessory spleens.
B. The head of the table is tilted up, shifting the abdominal contents away from the left upper quadrant.
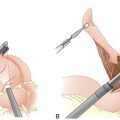
D. The short gastric vessels are divided and the stomach is rotated to the right, exposing the splenic hilum.
F. Remaining peritoneal attachments are divided, and the spleen is placed in an endoscopic bag. The spleen is then removed through one of the larger port sites, after morcellation with ring forceps. Alternatively, one of the port incisions can be extended to allow for the intact removal of the spleen.
Additional Operative Considerations
I. Splenorrhaphy: Splenic salvage is sometimes possible after low-grade splenic injuries. Topical hemostatic agents, such as Gelfoam or Surgicel, as well as argon beam coagulation can be useful in achieving hemostasis. Higher-grade injuries may sometimes be repaired with pledgeted sutures or wrapped with absorbable mesh.
COMPLICATIONS
I. Hemorrhage: Patients should be closely monitored for signs of postoperative bleeding; however, hemorrhage requiring reoperation is unusual.
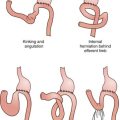
II. Iatrogenic Injuries: Injury to the pancreatic tail may result from dissection around the splenic hilum. If recognized in the operating room, a closed-suction drain can be placed in the surgical bed. Alternatively, some surgeons leave a drain in place routinely. Unrecognized pancreatic injury may lead to a pancreatic leak, which can manifest with abdominal pain, nausea, vomiting, fever, or intra-abdominal abscess. CT of the abdomen may show a collection in the region of the pancreatic tail. Percutaneous drainage is typically sufficient to control pancreatic leaks, which are usually self-limited. Injury to the diaphragm is an uncommon complication of laparoscopic splenectomy and can lead to an effusion or pneumothorax.
III. Infection: Postoperative infections can include pneumonia, surgical site infections, urinary tract infection, and subphrenic abscess. Overwhelming postsplenectomy infection (OPSI) describes the onset of fulminant sepsis in asplenic patients. OPSI is caused by bacteremia from encapsulated organisms, most commonly S. pneumoniae. The risk is lifelong, but is highest in the first several years after splenectomy. The estimated incidence is 0.2% in adults and slightly higher in children. OPSI should be suspected in any asplenic patient presenting with fever. Broad-spectrum antibiotics should be initiated without delay because patients can deteriorate rapidly. Immunizations against encapsulated organisms are crucial in preventing OPSI. Daily antibiotic prophylaxis is recommended in children, although the optimal duration of therapy is unclear.
Beauchamp RD, Holzman MD, Fabian TC. Spleen. In: Townsend CM, Beauchamp RD, Evers BM, Mattox KL, editors. Sabiston Textbook of Surgery: The Biological Basis of Modern Surgical Practice. 17th ed. Philadelphia: Saunders; 2004:1679-1704.
Dente CJ, Parry NG, Rozycki GS. Splenic salvage procedures: therapeutic options. In: Cameron JL, editor. Current Surgical Therapy. 8th ed. Philadelphia: Mosby; 2004:539-543.
McKinlay R, Park AE. Splenectomy for hematologic diseases. In: Cameron JL, editor. Current Surgical Therapy. 8th ed. Philadelphia: Elsevier Mosby; 2004:533-536.