CHAPTER 309 Spinal Cord Tumors in Adults
Spinal cord tumors account for about 15% of central nervous system neoplasms,1 and most intradural tumors arise from the cellular constituents of the spinal cord and filum terminale, nerve roots, or meninges. Metastatic involvement of the spinal intradural compartment is rarely manifested as a mass lesion. Intradural spinal cord tumors are broadly categorized according to their relationship to the spinal cord: intramedullary tumors arise within the substance of the spinal cord, whereas extramedullary tumors are extrinsic to the spinal cord. However, a small number of neoplasms may have both intramedullary and extramedullary components that usually communicate either through a nerve root entry zone or through the conus medullaris–filum terminale transition. Similarly, some intradural tumors may extend through the nerve root sleeve into the extradural compartment.
Extramedullary Tumors
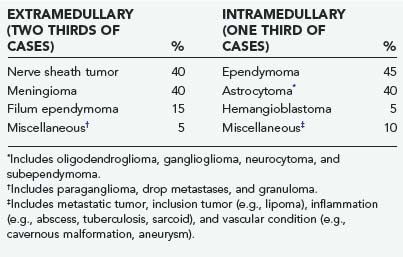
About two thirds of spinal cord tumors in adults are extramedullary (Table 309-1). Nerve sheath tumors, meningiomas, and filum terminale ependymomas account for most extramedullary neoplasms,2 with metastases, inclusion tumors and cysts, paragangliomas, and melanocytic neoplasms being rare. With few exceptions, extramedullary tumors are histologically benign and amenable to complete surgical resection.
Incidence and Etiology
Nerve Sheath Tumors
The histologic appearance of neurofibromas consists of an abundance of fibrous tissue and the conspicuous presence of nerve fibers within the tumor stroma.3 Grossly, the tumor produces fusiform (plexiform) enlargement of the involved nerve, which makes it impossible to distinguish between tumor and nerve tissue. Multiple neurofibromas establish the diagnosis of neurofibromatosis (NF), but this syndrome should be considered even in patients with apparent solitary involvement. Both NF1 and NF2 are associated with nerve sheath tumors. Although neurofibromas predominate in NF1, schwannomas are more common in NF2.4
Schwannomas appear grossly as smooth globoid masses that do not enlarge the nerve but are suspended eccentrically from it with a discrete attachment. Their histologic appearance consists of elongated bipolar cells with fusiform, darkly staining nuclei arranged in compact interlacing fascicles with a tendency toward palisade formation (Antoni-A). A loosely arranged pattern of stellate-shaped cells (Antoni-B) is less common.5 Multiple schwannomas, or “schwannomatosis,” can occur in patients without NF, and these tumors currently have no known genetic basis.6,7
Nerve sheath tumors account for about 25% of intradural spinal cord tumors in adults2,8 with an annual incidence of 0.3 to 0.4 per 100,000.7 Most are solitary schwannomas that occur proportionally throughout the spinal canal (Fig. 309-1). The fourth through sixth decades of life represent the peak incidence of occurrence. Men and women are affected equally.
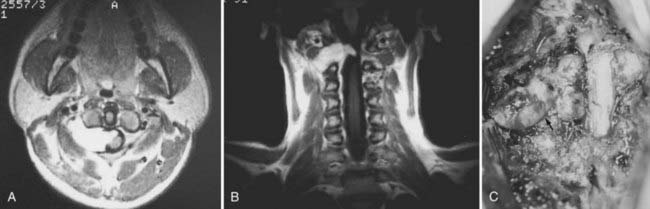
Most nerve sheath tumors arise from a dorsal nerve root, although neurofibromas represent a higher proportion of ventral root tumors.9 The majority of nerve sheath tumors are entirely intradural, but 30% extend through the dural root sleeve, which results in a “dumbbell”-shaped tumor with both intradural and extradural components (Fig. 309-1).7 About 10% of nerve sheath tumors are epidural or paraspinal in location. Transdural growth is common in cervical tumors because the intradural root segment is short.7 One percent of nerve sheath tumors are intramedullary and are thought to arise from the perivascular nerve sheaths that accompany penetrating spinal cord vessels.7 Centripetal growth of a nerve sheath tumor may also result in subpial extension, most often with plexiform neurofibromas. In these cases, both intramedullary and extramedullary tumor components are apparent. Brachial or lumbar plexus neurofibromas may extend centrally into the intradural space along multiple nerve roots. Conversely, retrograde intraspinal extension of a paraspinal schwannoma usually remains epidural.
About 2.5% of intradural spinal nerve sheath tumors are malignant.10 At least half occur in patients with NF. Malignant nerve sheath tumors carry a poor prognosis; survival rarely extends beyond 1 year. These tumors must be distinguished from the rare cellular schwannoma that displays aggressive histologic features but is associated with a favorable prognosis.
Meningiomas
Meningiomas and nerve sheath tumors occur with about equal frequency in adults. They usually arise from arachnoid cap cells embedded in the dura near the root sleeve, which accounts for their predominantly lateral location. Meningiomas may also arise from pia or dural fibroblasts, probably as a result of their mesodermal origin.3
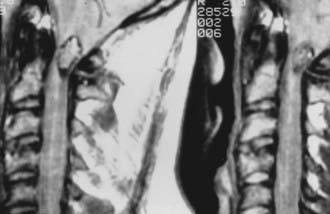
Meningiomas can develop in any age group, but the majority occur in individuals between the fifth and seventh decades of life. Seventy-five percent to 85% occur in women, and about 80% are thoracic (Fig. 309-2).2,11–14 The upper cervical spine and foramen magnum are also common sites.15 Here, meningiomas often occupy a ventral or ventrolateral position and may adhere to the vertebral artery near its intradural entry and initial intracranial course (Fig. 309-3). Low cervical and lumbar meningiomas are rare. Most spinal meningiomas are entirely intradural, but about 10% may be both intradural and extradural or entirely extradural.2
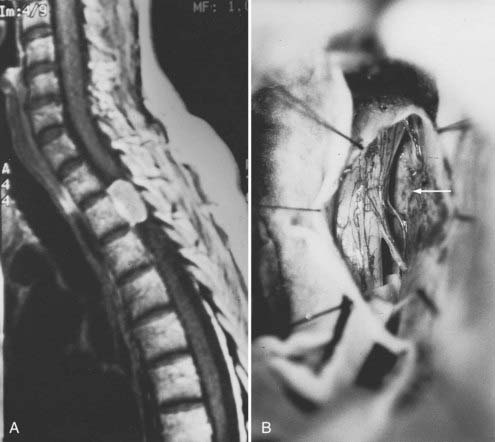
Their gross characteristics range from smooth and fibrous to the more frequent variegated, fleshy, and friable appearance. Microscopic calcification may occur. The dural attachment is often broader than expected. En plaque examples are unusual but have been described.16 The well-defined epidural space of the spine precludes bony involvement. Unlike intracranial meningiomas, spinal meningiomas do not penetrate the pia. This feature simplifies surgical resection and has been attributed to the presence of an “intermediate leptomeningeal layer” between the pia and arachnoid.17 Another explanation is that spinal meningiomas manifest signs of spinal cord compression early in their course and are therefore treated surgically before they have a chance to penetrate the pia.17
Filum Terminale Ependymomas
About 40% of spinal canal ependymomas arise within the filum terminale,1 most in its proximal intradural portion. Astrocytomas, oligodendrogliomas, and paragangliomas can also originate in the filum terminale but are rare. Filum terminale ependymomas occur throughout life but are most common in the third to fifth decades. They occur in men slightly more often than in women. Filum ependymomas and cauda equina nerve sheath tumors occur with about equal frequency.
Myxopapillary ependymomas are by far the most common histologic type encountered in the filum terminale. Their histologic appearance consists of a papillary arrangement of cuboidal or columnar tumor cells surrounding a vascularized core of hyalinized and poorly cellular connective tissue.5 Almost all are histologically benign.18 These tumors, however, tend to be more biologically aggressive in younger age groups.19
Miscellaneous Pathologic Processes
Numerous neoplastic and non-neoplastic processes are occasionally manifested as an extramedullary mass lesion. Dermoids, epidermoids, lipomas, teratomas, and neurenteric cysts are inclusion tumors and cysts that result from disordered embryogenesis.20,21 These lesions can occur throughout the spinal canal but are more common in the thoracolumbar and lumbar spine. They can be intramedullary or extramedullary. Associated anomalies such as metameric cutaneous lesions, sinus tracts, occult anterior or posterior rachischisis, or split cord malformations may be present.20,22 Inclusion tumors and cysts are most often manifested as a mass lesion, but recurrent meningitis, tethered cord syndrome, or congenital deformity may represent the dominant clinical feature. In most cases, treatment consists of excision. A tethered spinal cord is released or a sinus tract is excised if necessary. In some cases, dense adherence of the lesion to neural structures precludes total extirpation.
Paragangliomas are rare tumors of neural crest origin and may arise from the filum terminale or cauda equina.23 They are benign, nonfunctioning tumors and histologically resemble extra-adrenal paraganglia (i.e., carotid body and glomus jugulare). Grossly, they appear as well-circumscribed vascular tumors that are indistinguishable clinically or radiographically from filum terminale ependymomas. Identification of dense core neurosecretory granules on electron microscopy establishes the diagnosis. Complete resection is usually possible. Cavernous malformations, hemangioblastomas, and ganglioneuromas may involve an intradural nerve root and appear as an extramedullary mass lesion. Clinically, these tumors can occur as nerve sheath tumors with early radicular symptoms. Ganglioneuromas often take the form of dumbbell tumors in pediatric patients. Subarachnoid hemorrhage has been associated with nerve root cavernous malformations. These lesions are benign and are removed surgically. The involved nerve root is usually sacrificed, although it can occasionally be spared with small tumors.
Non-neoplastic lesions are also manifested as extramedullary masses. Arachnoid cysts are a well-known example. They are most commonly found in the thoracic spine dorsal to the spinal cord.24 Intraspinal aneurysms are exceedingly rare. They generally occur in conjunction with arteriovenous malformations or coarctations of the aorta. Most isolated cases occur in the region of the foramen magnum and arise from the vertebral or posterior inferior cerebellar arteries. An anomalous vessel origin or course (i.e., kinking) is frequently described. Isolated spinal aneurysms have also been reported to arise from the anterior spinal artery, posterior spinal artery, and medullary arteries.25 Most involve the anterior spinal artery. Patients with a spinal aneurysm may initially be evaluated for subarachnoid hemorrhage or compressive myelopathy. A definitive diagnosis is achieved with selective spinal angiography. Rarely, a herniated intervertebral disk transgresses the dura to occupy an intradural location.26
An inflammatory pathologic process such as sarcoidosis, tuberculoma, or subdural empyema is occasionally manifested as an intradural mass lesion.26,27 Although spinal carcinomatous meningitis often complicates systemic cancer, secondary metastatic involvement of the spinal canal is rarely manifested as a mass lesion. Malignant intracranial neoplasms that appose the subarachnoid space or ventricles are the most likely intracranial tumors to demonstrate cerebrospinal fluid (CSF) drop metastases into the spinal subarachnoid space.28 Systemic cancer gains access to the subarachnoid space by direct penetration of the dural root sleeve or, more commonly, through the choroid plexus.29,30 Surgical removal of a significant secondary neoplastic deposit may be appropriate in some cases.
Clinical Features
The clinical features of most extramedullary tumors reflect a slow-growing intraspinal mass. Specific clinical signs and symptoms are variable and largely determined by tumor location (i.e., local pain and signs of compression of adjacent neural structures). Most spinal tumors cause radicular pain or a dull axial pain, depending on whether the initial neural compression or infiltration involves the nerve roots or the spinal cord. Neurological symptoms gradually arise. Upper cervical and foramen magnum tumors are often located ventrally and cause symptoms of suboccipital pain and distal arm weakness with atrophy and clumsiness of the intrinsic hand muscles.15 The cause of this well-known syndrome is uncertain but it most likely results from venous insufficiency. Increased intracranial pressure and hydrocephalus rarely occur with extramedullary tumors at any level but are most common with upper cervical tumors.31 This syndrome is probably caused by elevated levels of CSF protein, which impairs CSF flow and absorption. Segmental motor weakness and long-tract signs are hallmarks of midlevel and lower cervical tumors. Asymmetric early signs and symptoms are typical and reflect the predominantly lateral location of most intradural tumors. A Brown-Séquard type of syndrome characterized by dysfunction of the corticospinal tract, posterior column, and contralateral spinothalamic tract is common. Rarely, schwannomas are accompanied by subarachnoid hemorrhage.32
Diagnostic Imaging
On T1-weighted images, most intradural tumors are isointense or slightly hypointense with respect to the spinal cord. On T2-weighted images, nerve sheath tumors are more likely than meningiomas to be hyperintense with respect to the spinal cord, but exceptions exist. On both T1- and T2-weighted images, the signal intensity of cauda equina tumors is usually greater than that of CSF. Small cauda equina tumors, however, are easily overlooked on nonenhanced images.33
Almost all spinal cord tumors demonstrate some degree of contrast enhancement. Meningiomas (see Figs. 309-2 and 309-3) typically exhibit intense uniform enhancement, although nonenhanced calcifications or intratumoral cysts occur occasionally. Enhancement of the adjacent dura (i.e., dural tail) strongly supports the diagnosis of meningioma.34 Although most nerve sheath tumors and filum ependymomas demonstrate uniform uptake of contrast media, heterogeneous enhancement from intratumoral cysts, hemorrhage, or necrosis is common. A peritumoral hypointense rim is often present around meningiomas and corresponds to a well-formed peritumoral CSF space.17
Treatment
Nerve Sheath Tumors
Treatment of benign nerve sheath tumors is complete surgical excision. In almost all cases, resection can be accomplished through a standard posterior laminectomy with partial or complete unilateral facetectomy as needed.35,36 Hemilaminectomy and unilateral facetectomy are options that can reduce postoperative pain and preserve spinal stability.37 Recurrences are rare after gross total resection. Because these tumors grow slowly, complete resection should not be pursued if it carries a high risk of incurring a significant neurological deficit. This is particularly relevant for neurofibromas, which can be admixed with functional neural tissue. The rate of clinical progression after subtotal resection is 50%.7 Recurrences can be treated by reoperation or radiosurgery.
Once exposure is adequate, the correct plane of dissection (directly on the tumor surface) must be identified. The arachnoid membrane generally adheres to the tumor surface tightly. This is the fenestrated arachnoid layer, which separately ensheathes each dorsal and ventral nerve root within the subarachnoid space.38 This layer is incised sharply and reflected off the surface of the tumor. The tumor capsule is cauterized to diminish its vascularity and to shrink the volume of the tumor. Removal of the tumor requires identification and division of the proximal and distal nerve root where the origin of the tumor attaches. With large tumors, this site may not be immediately apparent. Internal decompression with a laser or ultrasonic aspirator is used in such cases. The nerve root of origin must usually be sacrificed to remove the tumor. Occasionally, some fascicles of the nerve root may be preserved, especially with smaller tumors. Neurostimulation is a useful tool to establish the functionality of tumor-associated nerve roots before sacrifice. Once identified, the corresponding intradural nerve root can be preserved because the fenestrated arachnoid sheaths allow anatomic separation of the dorsal and ventral nerve roots to a point just distal to the dorsal root ganglion. In a typical tumor of dorsal root origin, it is possible to preserve the ventral root, which is tightly adherent to the ventral tumor surface, but this may not be feasible with larger tumors.
Dumbbell extension through the nerve root sleeve, however, generally necessitates resection of both nerve roots because the tumor has invaded the corresponding spinal nerve where sensory and motor fibers are admixed.38 Such resection rarely causes a significant deficit, even at the cervical and lumbar enlargement levels. Presumably, the adjacent roots have already compensated for the function of this root. A tumor with a very proximal origin may be partially embedded in the epipial tissue or may elevate the pia to occupy a subpial location. In such cases, the interface between the tumor and spinal cord may be difficult to develop. It may then be necessary to resect a segment of the pia to remove the tumor completely.
Significant tumor extension into the paraspinal region through an enlarged foramen increases the surgical considerations (see Fig. 309-1C). Surgical approaches are influenced by the surgeon’s preference, size and location of the paraspinal tumor component, and intradural tumor extension. Preoperative determination of intradural tumor extension is particularly important. Although MRI adequately identifies various tumor components and relationships to both intraspinal and paraspinal structures, myelography-CT provides greater spatial resolution and more sensitive identification of intradural involvement of dumbbell tumors.
The cervical paraspinal region is difficult to access anteriorly because of the narrow confines of the neck and the numerous neurovascular structures, such as the brachial plexus, descending lower cranial nerves, and vertebral artery. The mandible and skull base musculoskeletal attachments further limit upper cervical exposure. Fortunately, most cervical dumbbell tumors can be removed completely through an extended posterior exposure. A midline incision and standard laminectomy permit safe removal of both intradural and epidural intraspinal tumor components. Complete unilateral facetectomy allows paraspinal access up to 4 cm from the lateral dural margin.36 The vertebral artery is consistently displaced anteromedially and is separated from the tumor capsule by periosteum and an extensive venous plexus. Some authors, however, prefer the anterolateral approach for cervical schwannomas below C2 because the facet is preserved and the vertebral artery can be controlled early.39
When complete unilateral cervical facetectomy is required, tumor resection should be accompanied by lateral mass arthrodesis.36 In cases in which extensive partial facetectomy is required, a unilateral laminectomy may reduce the risk for instability.37 This may be performed after a traditional unilateral subperiosteal muscle dissection or with a muscle-dilating approach and a tubular retractor.40
Paraspinal extension from thoracic tumors can cause a large mass in the cavity. Standard posterior approaches provide inadequate exposure to the anterior paraspinal region, but an anterior transpleural or extrapleural thoracotomy permits excellent visualization of the paraspinal region. Intraspinal access is more limited, however, and the spinal cord is not visualized until most of the tumor has been removed. Because of negative intrathoracic pressure and chest tube drainage, postoperative CSF pleural fistulas can occur if intradural exposure is required. A combination of both anterior and posterior exposure, either staged or consecutive, can be used. Thoracoscopic procedures may be useful for tumors located peripherally within the intercostal nerves or those that extend from the neural foramen into the chest without intradural extension.41
The lateral extracavitary approach is useful as a single-stage operation for tumors that require concomitant complex exposure of the intraspinal and paraspinal compartments.42,43 This exposure is achieved through a hockey stick incision and allows the surgeon to work on either side of the mobilized paraspinal muscles. The superficial thoracic scapular muscles (i.e., trapezius, rhomboid) are detached at the midline and rotated laterally with the skin flap to expose the longitudinally oriented paraspinal muscles. These muscles are mobilized off the posterior spinal elements and ribs. Rib resection and depression of the pleura provide extensive extrapleural paraspinal exposure lateral to the paraspinal muscles. Intraspinal exposure is achieved through a standard laminectomy medial to the paraspinal muscles. Circumferential stabilization, although rarely necessary after removal of dumbbell thoracic tumors, can also be performed through this exposure. CSF fistulas are not a significant problem because the pleural cavity is not entered.
Lumbar dumbbell tumors are also well exposed through the lateral extracavitary approach.42,43 At this level, the thoracodorsal fascia is incised in line with the skin incision and elevated laterally with the skin flap. Paraspinal tumor components in the lumbar spine are buried deep within the psoas muscle. Safe removal of such components through a retroperitoneal exposure is complicated because it is difficult to distinguish the tumor margins from the overlying elongated and blanched fibers of the psoas muscle. The lumbar nerve roots and their branches, including the femoral nerve, course through the psoas muscle. They are difficult to identify and subject to injury during retroperitoneal dissection of the psoas muscle.
Dumbbell sacral tumors generally require both anterior and posterior exposure, which can be staged or performed simultaneously with the patient in a lateral position.44
Severe long-term adverse sequelae from surgical excision of schwannomas are rare. Unlike patients with neurofibromas, the life expectancy of patients with schwannomas parallels that of the general population.7 Symptomatic arachnoiditis and cystic myelopathy may occur in 6% of patients within a few years after tumor removal.7 More than half report some long-term local or radiating pain, but less than 10% seek medical attention.7
Meningiomas
Complete surgical removal is the treatment of choice for spinal meningiomas and can be achieved more than 90% of the time.11 Favorable features relative to intracranial meningiomas include less difficult requirements for ventral exposure, absence of bony involvement because of the well-defined spinal epidural space, lack of venous sinus or major blood vessel involvement, and the presence of a peritumoral hypointense rim on MRI.17,38 Despite these favorable features, the recurrence rate of spinal meningiomas 10 years after gross total or nearly total removal is 10% to 15%.45
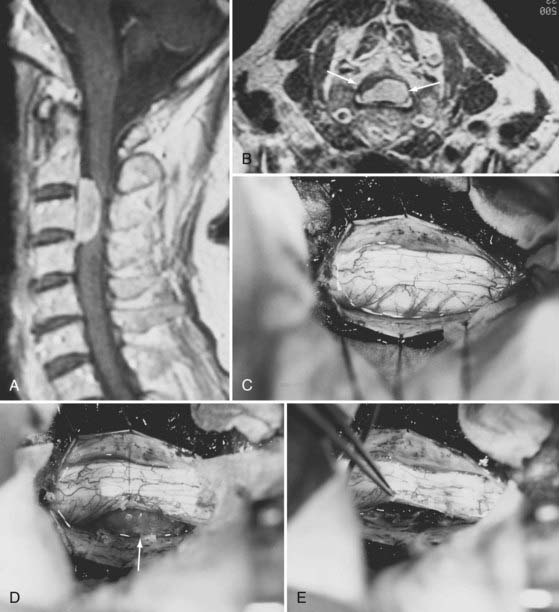
Posterior laminectomy provides adequate exposure in most cases. Unilateral laminectomy and facetectomy can be used for eccentrically located or ventral tumors. Large ventral tumors may also be approached satisfactorily through standard posterior exposures because the tumors have already retracted the spinal cord. Suture retraction on a divided dentate ligament or a noncritical dorsal root provides additional ventral exposure (see Fig. 309-3D). Depression of the paraspinal muscle mass with table-mounted retractors further facilitates ventral access. Alternatively, a costotransversectomy or lateral extracavitary approach can be used for ventral thoracic tumors.42 The extreme lateral approach described by Sen and Sekhar46 is used when the tumor has a significant ventral component above the foramen magnum. Anterior approaches for meningiomas are appropriate for purely ventral tumors that are blocked from posterolateral approaches by the spinal cord. Intradural extramedullary tumors, as well as some intramedullary tumors, can be safely accessed and resected through a standard anterior cervical approach,47,48 whereas upper thoracic lesions may require transthoracic exposure. Transoral approaches for foramen magnum tumors have also been described.49
Frequently, direct visualization of the entire tumor–spinal cord interface of ventrally located tumors may not be possible. Nevertheless, an arachnoid layer is almost invariably reflected over the central surface of the tumor. This plane is easily developed by gentle traction on the tumor away from the spinal cord. If peritumoral hypointensity is not seen on MRI, blood vessels may be adherent to the surface of the tumor, but the pia is always intact.17 En plaque meningiomas tend to be associated with a significant amount of arachnoid scarring, which renders surgery more difficult.16
Management of the dural base is the most controversial aspect of treating spinal meningiomas. Options include excision of the dural origin with patch graft reconstruction or extensive in situ coagulation. Solero and colleagues found no significant difference in recurrence rates between these two maneuvers.13 After complete resection, long-term recurrence rates have ranged between 3% and 23%.8,11,13,16 Therefore, management of the dural base is determined by practical considerations. Removal of dorsal and dorsolateral meningiomas is facilitated by excision of the dural base. Tumors of the ventral half of the canal, however, are amputated flush with the dura. The dural origin is then coagulated extensively.
Filum Terminale Ependymomas
Large ependymomas of the filum terminale, however, can be difficult to resect completely. Typically, they have been present for many years and may have widely disseminated via CSF. When a large cauda equina mass is identified, the entire neuraxis should be evaluated by MRI before surgical treatment. These tumors may become enormous within the capacious thecal sac before they are diagnosed. These unencapsulated, pliable neoplasms can insinuate among the nerve roots and within the arachnoid sheaths of the cauda equina, where they are compartmentalized by innumerable arachnoid septa. They can also spread as contiguous tumor sheaths along the arachnoid septa, which act as a scaffolding for surface growth. CSF dissemination may have already occurred because of their location in the subarachnoid space. In such cases, tumor removal is necessarily piecemeal and almost always subtotal. Indeed, dense tumor attachments to the roots of the cauda equina are associated with a significant risk for postoperative deficits because so much manipulation would be required for tumor resection. In these cases, subtotal removal aimed at diminishing the tumor bulk is all that can be accomplished. Even when piecemeal gross total removal is achieved, a recurrence rate of at least 20% can be anticipated.18,19,50
If total or nearly total piecemeal removal has been achieved, patients can be monitored with serial gadolinium-enhanced MRI, which provides some insight into the natural history of the tumor. Biologically aggressive tumors, more common in the younger population, demonstrate early tumor recurrence and can be treated with radiation therapy. If significant tumor burden is present after the initial surgery, however, particularly in the case of known CSF dissemination, postoperative radiation therapy is the primary adjunctive treatment modality. Postoperative radiation therapy is delayed if piecemeal total or nearly total removal has been accomplished. In these cases, tumor recurrences can be treated with repeat surgery, followed by radiation therapy. Although the response of spinal cord ependymomas to radiation therapy is unpredictable, some evidence suggests that it offers long-term control.51 This response, however, cannot be predicted individually. Because prior radiation therapy markedly increases the morbidity of future surgical prospects, it is typically delayed if further surgery may be contemplated.
Intramedullary Tumors
A wide variety of pathologic processes can arise from or secondarily involve the spinal cord as mass lesions. Primary glial tumors account for at least 80% of intramedullary tumors in most series52–56 and include astrocytomas, ependymomas, and less common glial neoplasms such as gangliogliomas, oligodendrogliomas, and subependymomas (see Table 309-1). Hemangioblastomas account for 3% to 8% of intramedullary neoplasms.57 Inclusion tumors and cysts, metastases, nerve sheath tumors, neurocytomas, and melanocytomas account for most of the remaining intramedullary mass lesions. Metastatic involvement of the spinal cord accounts for less than 5% of intramedullary spinal cord tumors. The lung and breast are the most common primary tumor sites.
Clinically and radiographically, non-neoplastic processes may be manifested as intramedullary mass lesions. Examples include inflammatory conditions such as bacterial abscess, tuberculoma, inflammatory pseudotumor, sarcoidosis, multiple sclerosis, viral or parainfectious myelitis, paraneoplastic involvement, or an entity intermediate between multiple sclerosis and acute disseminated encephalomyelitis.26,58–60 An acute or subacute clinical course, characteristic of systemic involvement, further suggests the diagnosis. These conditions are associated with an acute or subacute myelopathy that advances rapidly over a period of several hours to a few days but rarely longer. With demyelinating disease, the course is occasionally chronic and progressive or recurring. Operative intervention in such patients should be undertaken with caution because tiny biopsy specimens tend to yield a nonspecific, inflammatory response and rarely provide the diagnosis or determine the medical treatment.
Incidence and Etiology
Astrocytomas
About 3% of central nervous system astrocytomas arise within the spinal cord (Fig. 309-4).1 These tumors occur at any age but seem most prevalent in the first 3 decades of life. By far, they are the most common pediatric intramedullary spinal cord tumor. They account for about 90% of intramedullary tumors in patients younger than 10 years and about 60% of adolescent intramedullary neoplasms. Almost 60% of these tumors occur in the cervical and cervicothoracic region,53 and 20% have associated syringes.61 Thoracic, lumbosacral, or conus medullaris locations are less common. Filum terminale examples are rare. There is an association between NF1 and intramedullary astrocytomas.62
Spinal cord astrocytomas are a heterogeneous group with respect to their histologic findings, gross characteristics, biologic features, and natural history. They include low-grade fibrillary and pilocytic astrocytomas, malignant astrocytomas and glioblastomas, gangliogliomas, and the rare oligodendrogliomas. Most are grade I or II fibrillary astrocytomas. Juvenile pilocytic astrocytomas and gangliogliomas are more common in the pediatric population. The designation of a tumor as a pilocytic astrocytoma in an adult usually reflects an abundance of pilocytic features that occur as secondary structures in an otherwise typical fibrillary astrocytoma.3 Whether these pilocytic features have prognostic significance is unclear. High-grade (III or IV) spinal astrocytomas are rare (Fig. 309-5).53,63
Ependymomas
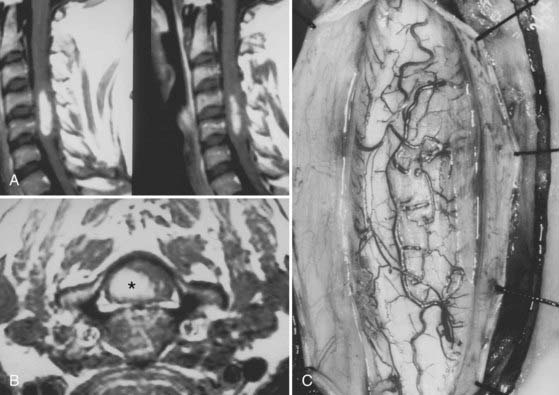
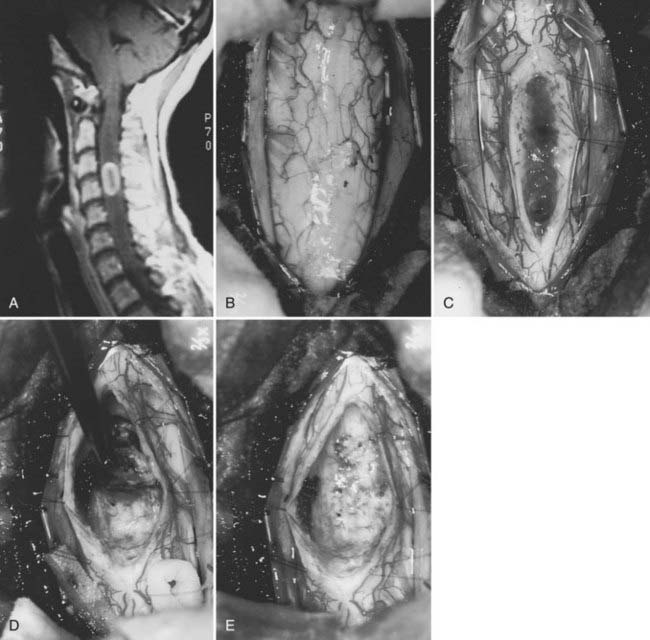
Ependymomas are the most common intramedullary tumors in adults.64 They occur throughout life but are most common in the middle adult years. Men and women are equally affected. Approximately 65% have associated cysts, particularly when cervical locations are involved.61 There is an association between NF2 and intramedullary ependymomas,59 and most sporadic ependymomas also feature mutations in the NF2 gene.65 A variety of histologic subtypes may be encountered. Cellular ependymoma is the most common variety, but epithelial, tanycytic (fibrillar), subependymomal, myxopapillary, or mixed examples can occur. Almost all are histologically benign.3,53,55,63,66 Although unencapsulated, these glial-derived tumors are usually well circumscribed and do not infiltrate adjacent spinal cord tissue (Fig. 309-6).
Hemangioblastomas
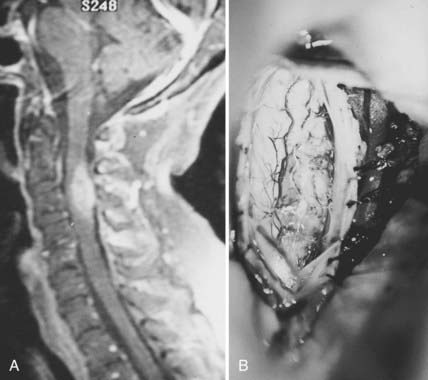
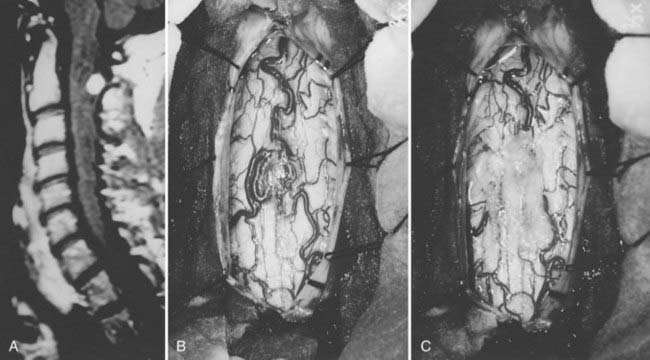
Hemangioblastomas account for 3% to 8% of intramedullary tumors.54 Fifteen percent to 25% are associated with von Hippel-Lindau syndrome, an autosomal dominant condition with incomplete penetrance and incomplete expression.57 These tumors arise at any age but are rare in early childhood. Associated syringes are common.61 Hemangioblastomas are benign tumors of vascular origin. They are sharply circumscribed but not encapsulated, and almost all have a pial attachment. Most are located dorsally or dorsolaterally (Fig. 309-7).
Miscellaneous Tumors and Other Pathologic Processes
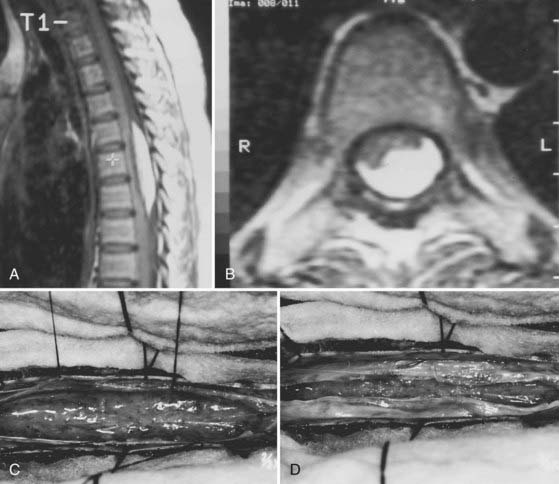
Inclusion tumors and cysts are rarely intramedullary. Lipomas are the most common dysembryogenic lesion and account for about 1% of intramedullary tumors (Fig. 309-8). They enlarge and produce symptoms in the early and middle adult years through increased fat deposition in metabolically normal fat cells. Because they occupy a subpial location, they are considered juxtamedullary. Metastases account for less than 5% of intramedullary tumors, probably because of the small size of the spinal cord and its remote vascular accessibility to hematogenous tumor emboli.67 The lung and breast are the most common primary tumor sites. Melanocytomas, melanomas, fibrosarcomas, and primitive neuroectodermal tumors can also arise in an intramedullary location.26,68 The appearance of radiation necrosis can mimic an intramedullary tumor, but a history of radiation therapy at the appropriate level is needed to confirm its diagnosis.69 Cavernous malformations, amyloid angiopathy, and other unusual intramedullary vascular lesions can also be manifested as an intramedullary mass (Fig. 309-9).59,70
Clinical Features
The clinical features of intramedullary spinal cord tumors are variable. In adults, the most common finding is a nonspecific axial pain followed by slow, progressive neurological decline.52,53,56,71,72 Symptoms are often present 3 to 4 years before the diagnosis.52,56 The course of malignant or metastatic neoplasms is much briefer, in the range of several weeks to a few months.52,56 Intratumoral hemorrhage can cause abrupt deterioration, a feature most often associated with ependymomas. The pain typically localizes to the level of the tumor and is rarely radicular. The distribution and progression of symptoms are related to the tumor’s location. Upper extremity symptoms predominate with cervical neoplasms. Thoracic tumors produce spasticity and sensory disturbances. Numbness is a common complaint and typically begins distally in the legs and progresses proximally. Tumors of the lumbar enlargement and conus medullaris often become symptomatic with back and leg pain. The leg pain may be radicular. Urogenital and anorectal dysfunction tends to occur early.
Diagnostic Imaging
Gadolinium-enhanced MRI is the procedure of choice for preoperative evaluation of an intramedullary tumor.73 Spinal cord enlargement and tumor enhancement are the characteristic findings. On T1-weighted images, most intramedullary tumors are isointense or slightly hypointense with respect to the surrounding spinal cord.73 On T1-weighted images, the spinal cord enlargement is often ill defined. Because most tumors are hyperintense in comparison to the spinal cord on T2-weighted images, they are the most sensitive for tumor identification.73 Almost all intramedullary neoplasms demonstrate uptake of contrast material. Ependymomas usually exhibit uniform contrast enhancement and are symmetrically located within the spinal cord (see Fig. 309-6A). Polar cysts are found in most cases, particularly in cervical and cervicothoracic locations.61 Heterogeneous enhancement from intratumoral cysts or necrosis can be seen with ependymomas.
The appearance of astrocytomas on MRI is much more variable. On contrast-enhanced MRI, these tumors tend to be less well defined than ependymomas because of their irregular margins. Uptake of contrast may be minimal, uniform, or patchy (see Fig. 309-4A and B).73 Heterogeneous uptake and patchy irregular margins are common with astrocytomas because of intratumoral cysts or necrosis. Despite the characteristic pattern of astrocytomas on MRI, their appearance on MRI overlaps with that of other tumors, thus precluding competent histologic diagnosis of tumor type based on MRI characteristics alone.
The appearance of inflammatory conditions on MRI is variable and probably related to their etiology.59 Acute multiple sclerosis plaque, for example, usually demonstrates focal homogeneous contrast enhancement confined to the white matter. There is minimal, if any spinal cord enlargement. Patchy contrast enhancement over several spinal cord segments is more characteristic of viral or parainfectious myelitis. Leptomeningeal enhancement can be seen with lymphomas, metastases, and bacterial, fungal, or tuberculous myelitis.73 Radiation necrosis may appear as a ring-enhancing lesion on T1-weighted images. High-intensity signal on T2-weighted images and spinal cord enlargement make differentiation of radiation necrosis from intramedullary tumor difficult.69
Treatment
Surgery is the most effective treatment of the majority of intramedullary tumors,52,56,63,71 including most ependymomas and hemangioblastomas. The benefits of aggressive surgical resection of astrocytomas are more controversial. Because most intramedullary spinal cord neoplasms are low-grade lesions and well circumscribed, long-term tumor control or cure with preservation of neurological function can be achieved in most patients by microsurgical removal alone.52,56,63,71
The most important factor in determining the degree of surgical removal is the plane between the tumor and the spinal cord. This interface can be assessed accurately only through an adequate myelotomy that extends over the entire rostrocaudal extent of the tumor (see Fig. 309-6C). Although the presence of a syrinx may improve the chances of achieving gross total resection, it cannot be used as an independent predictor of outcome.61 Benign tumors such as ependymomas and hemangioblastomas, although unencapsulated, are noninfiltrative lesions that typically display a distinct plane. Gross total removal is the treatment of choice in such cases (see Fig. 309-6E). Astrocytomas are more variable. Some benign astrocytomas possess a clear gross morphologic boundary with respect to the cord parenchyma, thereby permitting safe, gross total radiographic resection. Most, however, are infiltrative, (see Fig. 309-6E), with unclear boundaries that reflect a gradual transition zone between the tumor and functional tissue. Dissection beyond what is clearly tumor tissue risks neurological injury. Furthermore, the literature is conflicting regarding correlation between the extent of resection and tumor control.53,61,72 Accordingly, preservation of neurological function rather than complete tumor resection is paramount in such cases, and the resection should be limited to tissue that is clearly distinguishable from the surrounding spinal cord.
Management of less common intramedullary mass lesions is also dictated by the nature of the interface between the tumor and spinal cord. Metastatic spinal cord tumors, for example, are usually well-circumscribed focal masses amenable to gross total resection. Intramedullary inclusion tumors such as lipomas, dermoids/epidermoids, and teratomas result from disordered embryogenesis—probably during cleavage of the germ cell layers. They are not true neoplasms but probably enlarge slowly through continued fat deposition in metabolically normal cells. Gross total resection is impossible because these lesions insinuate into functional spinal cord tissue at their margins. In most cases, conservative internal decompression results in long-term clinical stabilization (see Fig. 309-8D).
Intraoperative biopsy can be useful in certain circumstances but should not be used as the sole criterion dictating the surgical objective. First, interpretation of tiny biopsy fragments on a poor-quality frozen section is often inaccurate or nondiagnostic and may consist of only peritumoral gliosis that can be erroneously interpreted as an infiltrating astrocytoma. Second, it is difficult, if not impossible, to assess the nature of the tumor–spinal cord interface accurately through a tiny myelotomy. However, intraoperative frozen sections can be helpful when they indicate positive histologic features. Malignant features such as vascular proliferation or necrosis, for example, usually signal an end to the procedure because most surgeons believe that aggressive resection of malignant intramedullary neoplasms is not beneficial.53,74,75 Ependymal rosettes or Rosenthal fibers, in contrast, may compel a surgeon to look more diligently for an anatomic plane to effect a more complete resection.
Surgical Technique
After intubation and perioperative administration of steroids and antibiotics, the patient is turned to the prone position. A Mayfield skull clamp is used for cervical and upper thoracic lesions. Neck flexion and head elevation (i.e., military prone) reduce the spinal curvature at these levels. Sensory and motor evoked potentials are monitored throughout the procedure.76,77 Sensory potentials may decrease in amplitude or disappear after a midline myelotomy and do not predict postoperative motor function. Subdural and epidural motor evoked potentials can be monitored in 60% of patients and provide real-time feedback. A 50% decline in the amplitude of motor evoked potentials can be an indication of a new, permanent postoperative weakness.77
A midline incision and subperiosteal bony dissection are carried out. A standard laminectomy, which should extend at least one segment above and below the solid tumor component, is performed. The facets are preserved. In adults, delayed instability rarely follows laminectomy for intramedullary tumor removal. For tumors spanning high-risk regions such as the cervicothoracic junction, laminoplasty or hemilaminar exposures may be reasonable options, particularly in children.52,71 Fusion can be always be contemplated at a later date. Strict hemostasis must be secured before the dura is opened to prevent ongoing blood contamination of the dependent microsurgical field. Wide, moist, cottonoid wall-offs cover the exposed muscles. A hemostatic agent such as thrombin-soaked Gelfoam is spread generously over the lateral gutters to prevent contamination of blood in the operative field.
The dura is opened in the midline and tented laterally to the muscles with sutures, and the operating microscope is brought into the field (see Fig. 309-4C). The arachnoid is opened separately and the spinal cord inspected for surface abnormalities. Most glial tumors cause only localized spinal cord enlargement. The spinal cord may be rotated. Occasionally, the overlying spinal cord may be thinned or even made transparent by a large or eccentrically located tumor or polar cyst. Ultrasonography is useful to localize the tumor and ensure adequate bony exposure.78 Malignant neoplasms can replace surface spinal cord tissue or fungate through the pia into the subarachnoid space, or both (see Fig. 309-5B). Most hemangioblastomas arise from the dorsal half of the spinal cord and have a visible pial attachment (see Fig. 309-7B).
The tumor is first encountered in the area where the spinal cord is enlarged maximally. Dissection continues on the surface of the tumor until the entire rostrocaudal extent has been identified. Polar cysts are entered and drained when present. Once the entire dorsal extent of the tumor has been identified, 6-0 pial traction sutures are placed (see Fig. 309-6C).
The technique of tumor removal is determined by the surgical objective and by the gross and histologic characteristics and size of the tumor. If no plane is apparent between the tumor and surrounding spinal cord, the tumor is probably infiltrative. A biopsy specimen is obtained to provide some additional information for intraoperative decision making. If an infiltrating or malignant astrocytoma is identified and is consistent with the intraoperative findings, further tumor removal is probably unwarranted. In most cases, however, a reasonably well-defined benign glial tumor is identified. Ependymomas have a smooth, reddish gray glistening tumor surface that is sharply demarcated from the surrounding spinal cord (see Fig. 309-6C). Variable blood vessels cross the surface of the tumor, thus distinguishing these tumors from astrocytomas, which rarely display these surface characteristics. Traction on the surface of the tumor is used against the countertraction provided by the pial sutures to allow the dissection plane to be developed (see Fig. 309-6D). Small feeding blood vessels and fibrous adhesions between the spinal cord and tumor are cauterized and divided. Although en bloc resection is preferred, large tumors may require internal decompression with an ultrasonic aspirator or laser. Once the tumor has been debulked significantly, the lateral and ventral margins can be dissected. The ventral tumor margin is developed by applying traction to a tumor pole, perpendicular to the long axis of the spinal cord. Feeding arteries from the anterior spinal artery are easily identified, cauterized, and divided.
Outcome of Surgical Treatment
The immediate results of surgery are related primarily to the patient’s preoperative status and tumor location.52,53,56,63,71 Most patients note a degree of sensory compromise immediately after surgery, which is most likely a result of manipulation of the dorsal columns. These complaints can be significant even when little or no objective deficit is present on neurological examination. Sensory deficits usually improve over the first year after surgery but almost always persist to a degree.71 Additional surgical morbidity is directly related to the patient’s preoperative status, the location of the tumor, and the presence of spinal cord atrophy and arachnoid scarring.52,53,56,61,71 Patients with major or long-standing deficits rarely recover significantly, and their condition is likely to worsen after surgery. If preoperative symptoms have been present only a brief time, patients are more likely to improve, even if they had a significant preoperative deficit, particularly those with ependymomas.71 Thoracic lesions have been correlated with a decline in postoperative function,52,54,71,79 perhaps because of the relatively tenuous blood supply in this region. Spinal cord atrophy and arachnoid scarring may indicate chronic spinal cord compression and predict a poor functional outcome.61,71 Preservation rather than restoration of neurological function is the reasonable expectation for intramedullary tumor surgery. Minimally symptomatic patients with intramedullary tumors derive the greatest benefit and the least risk from surgery.53,56,71 This relationship underscores the importance of early diagnosis and aggressive initial treatment before an objective deficit appears. It is equally important during follow-up because periodic evaluation with MRI will most likely demonstrate evidence of tumor recurrence before clinical recurrence becomes apparent.
Long-term outcome and risk for recurrence primarily depend on the tumor’s histologic characteristics and, with the exception of malignant neoplasms and many low-grade astrocytomas, on the completeness of the original resection. Gross total removal of benign intramedullary ependymomas more consistently cures or controls tumors in the long term than does subtotal resection and radiation therapy.52,55,63,71,80 Nevertheless, these tumors are friable and often adhere to the spinal cord, particularly at their polar regions. These features may preclude total microscopic resection. Long-term follow-up with periodic clinical evaluation and gadolinium-enhanced MRI is mandatory because of the continued risk for tumor recurrence. Depending on the patient’s age and critical circumstances, reoperation may be undertaken if tumor recurrence is clearly established on MRI.
The optimal treatment strategy for astrocytomas is less clear. The influence of the extent of resection on outcome remains controversial. Although some authors have found that gross total resection influences outcome,52,54 other authors find no such relationship.53,72 Most authors agree that aggressive debulking is of no benefit for high-grade lesions. The appropriate therapy for low-grade intramedullary spinal cord astrocytomas has been difficult to evaluate, in part because of their biologic variability. Age appears to be the most significant prognostic factor. Pediatric astrocytomas are associated with a particularly indolent behavior and are more likely to exhibit clearer tumor margins, which may make them more amenable to surgical resection. This can be partly explained by their predominantly benign histologic characteristics (90%) and the high percentage of juvenile pilocytic astrocytomas and gangliogliomas.72,81,82 In adults, however, even low-grade astrocytomas tend to be diffusely infiltrative and generally pursue a progressive course.
Parameters that predict which patients will benefit from aggressive resection have not been identified. We strive for gross total resection if pilocytic features are seen on frozen section or there is a clear demarcation in color and consistency between tumor and normal tissue because surgical cure is safe and achievable in a small subgroup of patients.54,72 If resection is subtotal only, radiation therapy may be given, although data on its efficacy for low-grade tumors are inconclusive. Recurrence within the irradiated volume after radiation therapy is seen in approximately 50% of patients, and there is little agreement about the dose-response relationship.53,72,81,83,84
We do not routinely administer radiation therapy after subtotal resection for several reasons. It complicates the prospects for future surgery, efficacious doses may be higher than the accepted tolerance of the spinal cord, and it has been associated with an increased incidence of spinal deformity and secondary malignancy.79,83,85 Instead, we monitor patients clinically and with serial MRI. Depending on the clinical circumstances, repeat surgery is offered at the time of clinical recurrence, which is often several years after radiographic recurrence. Radiation therapy may then be considered, depending on the time of recurrence and the degree of resection accomplished. Surgery plays only a diagnostic role in patients who harbor malignant intramedullary astrocytic neoplasms. Radical resection does not prolong survival and is associated with a high rate of surgical morbidity.53,54,56 Radiation therapy is recommended for these patients, but their average survival is poor—6 to 12 months.52,54,75,84
Epstein FJ, Farmer JP, Freed D. Adult intramedullary astrocytomas of the spinal cord. J Neurosurg. 1992;77:355-359.
Garcia DM. Primary spinal cord tumors treated with surgery and postoperative irradiation. Int J Radiat Oncol Biol Phys. 1985;11:1933-1939.
Helseth A, Mørk SJ. Primary intraspinal neoplasms in Norway, 1955 to 1986: a population-based survey of 467 patients. J Neurosurg. 1989;71:842-845.
Klekamp J, Samii M. Surgical results of spinal meningiomas. Acta Neurochir Suppl. 1996;65:77-81.
Levy WJ, Latchaw J, Hahn JF, et al. Spinal neurofibromas: A report of 66 cases and a comparison with meningiomas. Neurosurgery. 1986;18:331-334.
Linstadt DE, Wara WM, Leibel SA, et al. Postoperative radiotherapy of primary spinal cord tumors. Int J Radiat Oncol Biol Phys. 1989;16:1397-1403.
McCormick PC. Surgical management of dumbbell and paraspinal tumors of the thoracic and lumbar spine. Neurosurgery. 1996;38:67-75.
McCormick PC. Surgical management of dumbbell tumors of the cervical spine. Neurosurgery. 1996;38:294-300.
McCormick PC, Torres R, Post KD, et al. Intramedullary ependymoma of the spinal cord. J Neurosurg. 1990;72:523-532.
Mirimanoff RO, Dosoretz DE, Linggood RM, et al. Meningioma: analysis of recurrence and progression following neurosurgical resection. J Neurosurg. 1985;62:18-24.
Neumann HP, Eggert HR, Weigel K, et al. Hemangioblastomas of the central nervous system: a 10-year study with special reference to von Hippel-Lindau syndrome. J Neurosurg. 1989;70:24-30.
Ogden AT, Feldstein NA, McCormick PC. Anterior approach to cervical intramedullary pilocytic astrocytoma. J Neurosurg Spine. 2008;9:253-257.
O’Sullivan C, Jenkin RD, Doherty MA, et al. Spinal cord tumors in children: long-term results of combined surgical and radiation treatment. J Neurosurg. 1994;81:507-512.
O’Toole JE, McCormick PC. Midline ventral intradural schwannoma of the cervical cord resected via anterior corpectomy with reconstruction. Neurosurgery. 2003;52:1482-1485.
Samii M, Klekamp J. Surgical results of 100 intramedullary tumors in relation to accompanying syringomyelia. Neurosurgery. 1994;35:865-873.
Seppala MT, Haltia MJ, Sankila RJ, et al. Long-term outcome after removal of spinal neurofibroma. J Neurosurg. 1995;82:572-577.
Seppala MT, Haltia MJ, Sankila RJ, et al. Long-term outcome after removal of spinal schwannoma: a clinicopathological study of 187 cases. J Neurosurg. 1995;83:621-626.
Sloof JL, Kernohan JW, McCarthy CS. Primary Intramedullary Tumors of the Spinal Cord and Filum Terminale. Philadelphia: WB Saunders; 1964.
Sonneland PR, Scheithauer BW, Onofrio BM. Myxopapillary ependymoma: a clinicopathologic and immunocytochemical study of 77 cases. Cancer. 1985;56:883-893.
Tredway TL, Santiago P, Hrubes MR, et al. Minimally invasive resection of intradural-extramedullary spinal neoplasms. Neurosurgery. 2006;58(1 Suppl):ONS52-ONS58.
1 Sloof JL, Kernohan JW, McCarthy CS. Primary Intramedullary Tumors of the Spinal Cord and Filum Terminale. Philadelphia: WB Saunders; 1964.
2 Nittner K. Spinal meningiomas, neurinomas and neurofibromas, and hourglass tumours. In: Vinken PH, Bruyn GW, editors. Handbook of Clinical Neurology. New York: Elsevier; 1976:177-322.
3 Russell DS, Rubenstein LJ. Pathology of Tumors of the Nervous System. Baltimore: Williams & Wilkins; 1989.
4 Halliday AL, Sobel RA, Martuza RL. Benign spinal nerve sheath tumors: their occurrence sporadically and in neurofibromatosis types 1 and 2. J Neurosurg. 1991;74:248-253.
5 Kernohan JW, Sayre GP. Tumors of the Central Nervous System, Fascicle 35. Washington DC: Armed Forces Institute of Pathology; 1952.
6 Purcell SM, Dixon SL. Schwannomatosis: an unusual variant of neurofibromatosis or a distinct clinical entity? Arch Dermatol. 1989;125:390-393.
7 Seppala MT, Haltia MJ, Sankila RJ, et al. Long-term outcome after removal of spinal schwannoma: a clinicopathological study of 187 cases. J Neurosurg. 1995;83:621-626.
8 Levy WJ, Latchaw J, Hahn JF, et al. Spinal neurofibromas: a report of 66 cases and a comparison with meningiomas. Neurosurgery. 1986;18:331-334.
9 Seppala MT, Haltia MJ, Sankila RJ, et al. Long-term outcome after removal of spinal neurofibroma. J Neurosurg. 1995;82:572-577.
10 Seppala MT, Haltia MJ. Spinal malignant nerve-sheath tumor or cellular schwannoma? A striking difference in prognosis. J Neurosurg. 1993;79:528-532.
11 Roux F-X, Nataf F, Pinaudeau M, et al. Intraspinal meningiomas: review of 54 cases with discussion of poor prognosis factors and modern therapeutic management. Surg Neurol. 1996;46:458-464.
12 Levy WJJr, Bay J, Dohn D. Spinal cord meningioma. J Neurosurg. 1982;57:804-812.
13 Solero CL, Fornari M, Giombini S, et al. Spinal meningiomas: review of 174 operated cases. Neurosurgery. 1989;25:153-160.
14 Preston-Martin S, Monroe K, Lee PJ, et al. Spinal meningiomas in women in Los Angeles County: investigation of an etiological hypothesis. Cancer Epidemiol Biomarkers Prev. 1995;4:333-339.
15 Stein BM, Leeds NE, Taveras JM, et al. Meningiomas of the foramen magnum. J Neurosurg. 1963;20:740-751.
16 Klekamp J, Samii M. Surgical results of spinal meningiomas. Acta Neurochir Suppl. 1996;65:77-81.
17 Salpietro FM, Alafaci C, Lucerna S, et al. Do spinal meningiomas penetrate the pial layer? Correlation between magnetic resonance imaging and microsurgical findings and intracranial tumor interfaces. Neurosurgery. 1997;41:254-258.
18 Sonneland PR, Scheithauer BW, Onofrio BM. Myxopapillary ependymoma: a clinicopathologic and immunocytochemical study of 77 cases. Cancer. 1985;56:883-893.
19 Davis C, Barnard RO. Malignant behavior of myxopapillary ependymoma: report of three cases. J Neurosurg. 1985;62:925-929.
20 Pang D. Split cord malformation. Part II. Clinical syndrome. Neurosurgery. 1992;31:481-500.
21 Agnoli AL, Laun A, Schonmayr R. Enterogenous intraspinal cysts. J Neurosurg. 1984;61:834-840.
22 Gregorios JB, Green B, Page L, et al. Spinal cord tumors presenting with neural tube defects. Neurosurgery. 1986;19:962-966.
23 Reyes MG, Torres H. Intrathecal paraganglioma of the cauda equina. Neurosurgery. 1984;15:578-582.
24 Nabors MW, Pait TG, Byrd EB, et al. Updated assessment and current classification of spinal meningeal cysts. J Neurosurg. 1988;68:366-377.
25 Moore DW, Hunt WE, Zimmerman JE. Ruptured anterior spinal artery aneurysm: repair via a posterior approach. Neurosurgery. 1982;10:626-630.
26 McCormick PC, Stein BM. Miscellaneous intradural pathology. Neurosurg Clin N Am. 1990;1:687-699.
27 Fraser RA, Ratzan K, Wolpert SM, et al. Spinal subdural empyema. Arch Neurol. 1973;28:235-238.
28 Calvo FA, Hornedo J, de la Torre A. Intracranial tumors with risk of dissemination in neuroaxis. Int J Radiat Oncol Biol Phys. 1983;9:1297-1301.
29 Perrin RG, Livingston KE, Aarabi B. Intradural extramedullary metastasis: a report of 10 cases. J Neurosurg. 1982;56:835-837.
30 Olson ME, Chernik NL, Posner JB. Infiltration of the leptomeninges by systemic cancer: a clinical and pathologic study. Arch Neurol. 1974;30:122-137.
31 Feldmann E, Bromfield E, Navia B. Hydrocephalic dementia and spinal cord tumor: report of a case and review of the literature. Arch Neurol. 1986;43:714-718.
32 Mills B, Marks PV, Nixon JM. Spinal subarachnoid haemorrhage from an “ancient” schwannoma of the cervical spine. Br J Neurosurg. 1993;7:557-579.
33 Epstein NE, Bhuchar S, Gavin R, et al. Failure to diagnose conus ependymomas by magnetic resonance imaging. Spine. 1989;14:134-137.
34 Quekel LG, Versteege CW. The “dural tail sign” in MRI of spinal meningioma. J Comput Assist Tomogr. 1995;19:890-892.
35 McCormick PC, Post KD, Stein BM. Intradural extramedullary tumors in adults. Neurosurg Clin N Am. 1990;1:591-608.
36 McCormick PC. Surgical management of dumbbell tumors of the cervical spine. Neurosurgery. 1996;38:294-300.
37 Sridhar K, Ramamurthi R, Vasudevan MC, et al. Limited unilateral approach for extramedullary spinal tumours. Br J Neurosurg. 1998;12:430-433.
38 McCormick PC. Anatomic principles of intradural spinal surgery. Clin Neurosurg. 1994;41:204-223.
39 Lot G, George B. Cervical neuromas with extradural components: surgical management in a series of 57 patients. Neurosurgery. 1997;41:813-822.
40 Tredway TL, Santiago P, Hrubes MR, et al. Minimally invasive resection of intradural-extramedullary spinal neoplasms. Neurosurgery. 2006;58(1 Suppl):ONS52-ONS58.
41 Dickman CA, Apfelbaum RI. Thoracoscopic microsurgical excision of thoracic schwannoma: case report. J Neurosurg. 1998;88:898-902.
42 Steck JC, Dietze DD, Fessler RG. Posterolateral approach to intradural extramedullary thoracic tumors. J Neurosurg. 1994;81:202-205.
43 McCormick PC. Surgical management of dumbbell and paraspinal tumors of the thoracic and lumbar spine. Neurosurgery. 1996;38:67-75.
44 McCormick PC, Post KD. Surgical approaches to the sacrum. In: Doty JR, Rengachary SS, editors. Surgical Disorders of the Sacrum. New York: Thieme; 1994:257-265.
45 Mirimanoff RO, Dosoretz DE, Linggood RM, et al. Meningioma: analysis of recurrence and progression following neurosurgical resection. J Neurosurg. 1985;62:18-24.
46 Sen CN, Sekhar LN. An extreme lateral approach to intradural lesions of the cervical spine and foramen magnum. Neurosurgery. 1990;27:197-204.
47 O’Toole JE, McCormick PC. Midline ventral intradural schwannoma of the cervical cord resected via anterior corpectomy with reconstruction. Neurosurgery. 2003;52:1482-1485.
48 Ogden AT, Feldstein NA, McCormick PC. Anterior approach to cervical intramedullary pilocytic astrocytoma. J Neurosurg Spine. 2008;9:253-257.
49 Miller E, Crockard HA. Transoral transclival removal of anteriorly placed meningiomas at the foramen magnum. Neurosurgery. 1987;20:966-968.
50 Ilgren EB, Stiller CA, Hughes JT, et al. Ependymomas: a clinical and pathological study. Part II. Survival features. Clin Neuropathol. 1984;3:122-127.
51 Whitaker SJ, Bessell EM, Ashley SE, et al. Postoperative radiotherapy in the management of spinal cord ependymoma. J Neurosurg. 1991;74:720-728.
52 Cristante L, Herrmann HD. Surgical management of intramedullary spinal cord tumors: functional outcome and sources of morbidity. Neurosurgery. 1994;35:69-76.
53 Cooper PR. Outcome after operative treatment of intramedullary spinal cord tumors in adults: intermediate and long-term results in 51 patients. Neurosurgery. 1989;25:855-859.
54 Epstein FJ, Farmer JP, Freed D. Adult intramedullary astrocytomas of the spinal cord. J Neurosurg. 1992;77:355-359.
55 Epstein FJ, Farmer JP, Freed D. Adult intramedullary spinal cord ependymomas: the result of surgery in 38 patients. J Neurosurg. 1993;79:204-209.
56 McCormick PC, Stein BM. Intramedullary tumors in adults. Neurosurg Clin N Am. 1990;1:609-630.
57 Neumann HP, Eggert HR, Weigel K, et al. Hemangioblastomas of the central nervous system: a 10-year study with special reference to von Hippel-Lindau syndrome. J Neurosurg. 1989;70:24-30.
58 Jallo GI, Zagzag D, Lee M, et al. Intraspinal sarcoidosis: diagnosis and management. Surg Neurol. 1997;48:514-521.
59 Lee M, Epstein FJ, Rezai AR, et al. Nonneoplastic intramedullary spinal cord lesions mimicking tumors. Neurosurgery. 1998;43:788-795.
60 Kepes JJ. Large focal tumor-like demyelinating lesions of the brain: intermediate entity between multiple sclerosis and acute disseminated encephalomyelitis? A study of 31 patients. Ann Neurol. 1993;33:18-27.
61 Samii M, Klekamp J. Surgical results of 100 intramedullary tumors in relation to accompanying syringomyelia. Neurosurgery. 1994;35:865-873.
62 Lee M, Rezai AR, Freed D, et al. Intramedullary spinal cord tumors in neurofibromatosis. Neurosurgery. 1996;38:32-37.
63 McCormick PC, Torres R, Post KD, et al. Intramedullary ependymoma of the spinal cord. J Neurosurg. 1990;72:523-532.
64 Helseth A, Mørk SJ. Primary intraspinal neoplasms in Norway, 1955 to 1986: a population-based survey of 467 patients. J Neurosurg. 1989;71:842-845.
65 Birch BD, Johnson JP, Parsa A, et al. Frequent type 2 neurofibromatosis gene transcript mutations in sporadic intramedullary spinal cord ependymomas. Neurosurgery. 1996;39:135-140.
66 Mørk SJ, Løken AC. Ependymoma: a follow-up study of 101 cases. Cancer. 1977;40:907-915.
67 Costigan DA, Winkelman MD. Intramedullary spinal cord metastases: a clinicopathological study of 13 cases. J Neurosurg. 1985;62:227-233.
68 Deme S, Ang LC, Skaf G, et al. Primary intramedullary primitive neuroectodermal tumor of the spinal cord: case report and review of the literature. Neurosurgery. 1997;41:1417-1420.
69 Yasui T, Yagura H, Komiyama M, et al. Significance of gadolinium-enhanced magnetic resonance imaging in differentiating spinal cord radiation myelopathy from tumor: Case report. J Neurosurg. 1992;77:628-631.
70 Schwartz TH, Chang Y, Stein BM. Unusual intramedullary vascular lesions: report of two cases. Neurosurgery. 1997;40:1295-1301.
71 Hoshimaru M, Koyama T, Hashimoto N, et al. Results of microsurgical treatment for intramedullary spinal cord ependymomas: analysis of 36 cases. Neurosurgery. 1999;44:264-269.
72 Sandler HM, Papadopoulos SM, Thornton AFJr, et al. Spinal cord astrocytomas: results of therapy. Neurosurgery. 1992;30:490-493.
73 Bourgouin PM, Lesage J, Fontaine S, et al. A pattern approach to the differential diagnosis of intramedullary spinal cord lesions on MR imaging. AJR Am J Roentgenol. 1998;170:1645-1649.
74 Cohen AR, Wisoff JH, Allen JC, et al. Malignant astrocytomas of the spinal cord. J Neurosurg. 1989;70:50-54.
75 Kopelson G, Linggood RM. Intramedullary spinal cord astrocytoma versus glioblastoma: the prognostic importance of histologic grade. Cancer. 1982;50:732-735.
76 Adams DC, Emerson RG, Heyer EJ, et al. Monitoring of intraoperative motor-evoked potentials under conditions of controlled neuromuscular blockade. Anesth Analg. 1993;77:913-918.
77 Morota N, Deletis V, Constantini S, et al. The role of motor evoked potentials during surgery for intramedullary spinal cord tumors. Neurosurgery. 1997;41:1327-1336.
78 Epstein FJ, Farmer JP, Schneider SJ. Intraoperative ultrasonography: an important surgical adjunct for intramedullary tumors. J Neurosurg. 1991;74:729-733.
79 Brotchi J, Dewitte O, Levivier M, et al. A survey of 65 tumors within the spinal cord: surgical results and the importance of preoperative magnetic resonance imaging. Neurosurgery. 1991;29:651-657.
80 Fischer G, Mansuy L. Total removal of intramedullary ependymomas: follow-up study of 16 cases. Surg Neurol. 1980;14:243-249.
81 Rossitch EJr, Zeidman SM, Burger PC, et al. Clinical and pathological analysis of spinal cord astrocytomas in children. Neurosurgery. 1990;27:193-196.
82 Epstein FJ, Farmer JP. Pediatric spinal cord tumor surgery. Neurosurg Clin N Am. 1990;1:569-590.
83 Garcia DM. Primary spinal cord tumors treated with surgery and postoperative irradiation. Int J Radiat Oncol Biol Phys. 1985;11:1933-1939.
84 Linstadt DE, Wara WM, Leibel SA, et al. Postoperative radiotherapy of primary spinal cord tumors. Int J Radiat Oncol Biol Phys. 1989;16:1397-1403.
85 O’Sullivan C, Jenkin RD, Doherty MA, et al. Spinal cord tumors in children: long-term results of combined surgical and radiation treatment. J Neurosurg. 1994;81:507-512.