[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 28 Spinal Cord Tumors
Epidemiology and Etiology
Primary spinal cord tumors comprise 3% to 4% of all primary central nervous system (CNS) tumors in adults.1 Age-adjusted incidence rates are slightly higher for men than women, 0.68 versus 0.61 per 100,000 person-years, respectively.1 Most of these tumors present as extramedullary tumors (70%), whereas intramedullary tumors are less common (30%). Approximately 15% to 25% of spinal tumors involve the cervical spine, including the foramen magnum; 50% to 55% involve the thoracic spinal canal; and 25% to 30% involve the lumbosacral spine.
In the pediatric population, spinal cord tumors are slightly more common, 5% to 6% of all primary CNS tumors.1 Although ependymomas and nerve sheath tumors are the most common histologic types, gliomas make up a larger percentage of tumors in children as compared with adults1 (Table 28-1).
TABLE 28-1 Distribution of Primary Spinal Cord Tumors by Histologic Type (CBTRUS 2010)
| Histologic Type | Adult Distribution | Pediatric Distribution |
|---|---|---|
| Nerve sheath tumor | 40.5% | 15.7% |
| Glial origin | ||
| Ependymoma | 33.6% | 24.1% |
| Pilocytic astrocytoma | 1.4% | 12.6% |
| Other glioma | 4.1% | 13.3% |
| Meningeal tumor | 6.1% | 10.1% |
| Hemangioma | 2.6% | NA |
NA, not available.
Biologic Characteristics and Molecular Biology
The diversity of primary spinal axis tumors partly results from the large spectrum of phenotypically distinct cells in the axis that are capable of neoplastic transformation.2 The genetic and molecular bases of spinal cord tumors are not yet well understood and vary depending on histologic findings.3 Most tumors are histologically benign but can cause significant morbidity by direct compression of important neural structures.
Anatomy, Pathology, and Pathways of Spread
Anatomy
The spinal cord is responsible for transmitting motor and sensory neural signals between the peripheral nervous system and the brain and contains its own independent pathways for coordinating certain reflexes (Fig. 28-1). Functionally, the cord is divided into 31 segments. At each level, a pair of spinal nerves exits: 8 cervical, 12 thoracic, 5 lumbar, and 5 sacral pairs and 1 coccygeal pair. In the cervical region, the spinal nerves exit above the corresponding vertebrae, whereas from the thoracic region and downward, the spinal nerves exit below the equivalent named vertebrae. Although the vertebral column continues to lengthen until adulthood, the canal and the cord are actually much shorter than the column because both stop lengthening at approximately 4 years of age. This growth differential is responsible for the formation of the cauda equina, a collection of the lower lumbar, sacral, and coccygeal nerves that fills the lower spinal column beyond the conus medullaris.
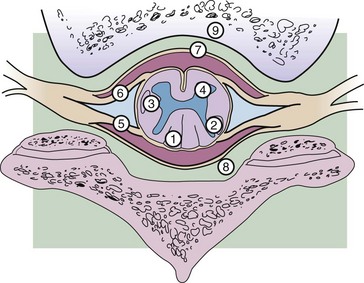
Tumors are classified based on their location (Fig. 28-2). Intradural tumors include both intramedullary and extramedullary tumors. Intramedullary tumors develop from the cells that make up the spinal cord and cauda equina, and extramedullary tumors arise from the supportive tissues that lie outside the spinal cord and cauda equina, such as the meninges, vascular supply, and connective tissue. Extradural tumors arise from the vertebral canal itself or supportive tissues surrounding the dura.
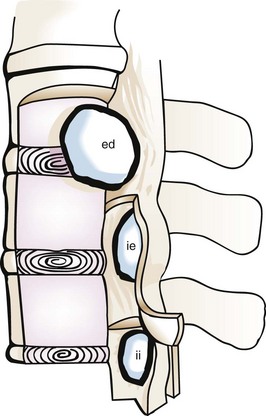
Pathology and Pathways of Spread
The overall and anatomic distributions of histologic types are listed in Tables 28-1 and 28-2. Intradural extramedullary schwannoma (neurilemmoma) and meningioma are the most common intradural tumors, although occasionally they may present as extradural tumors. Other intradural extramedullary tumors include epidermoid and drop metastases of intracranial primary tumors such as medulloblastoma. The most common intramedullary tumors are those derived from glial precursors, most frequently, ependymomas and astrocytomas of low to intermediate grade. Nonneoplastic conditions may mimic spinal cord tumors (Fig. 28-3), including acute transverse myelitis, Angiostrongylus cantonensis infection, infectious meningitis, intradural disc hernia, pseudotumor, sarcoidosis, and tuberculosis.4–8
TABLE 28-2 Anatomic Distribution of Spinal Tumors by Histologic Type
| Extradural Tumors |
Intradural Extramedullary Tumors | Intradural Intramedullary Tumors |
|---|---|---|
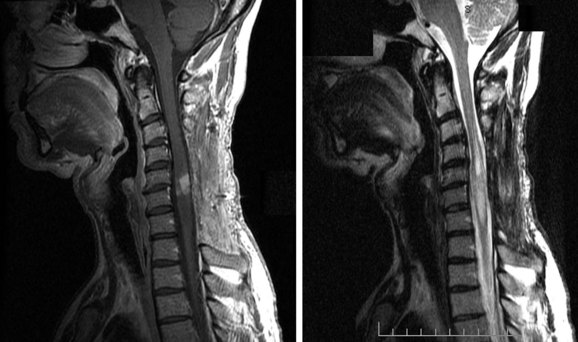
The predominant mode of spread of primary tumors is by direct extension. Spread along the subarachnoid space with spinal or cranial involvement of the meninges may also occur. One should not always assume that the spinal cord tumor found on imaging is the primary lesion. Although CSF seeding is uncommon, supratentorial tumors that show a tendency toward leptomeningeal metastases include 1.2% of glioblastomas and 1.5% of anaplastic gliomas.9 There are no lymphatics in the spinal cord, and hematogenous spread is rare.
Clinical Manifestations, Patient Evaluation, and Staging
The clinical presentation of tumors of the spinal axis is a function of the local anatomy (see Fig. 28-1). Within the spinal canal, there exists a well-defined extradural space containing epidural fat and blood vessels. The extradural space communicates with adjacent extraspinal compartments via the intervertebral foramina.
Clinical Workup
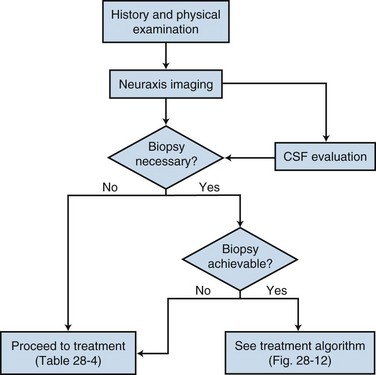
The diagnostic approach to primary spinal cord tumors is shown in Figure 28-4. In addition to focusing on neurologic symptoms, the history should include an evaluation of the patient’s functional status and symptoms, which may suggest metastatic disease or nonmalignant causes of spinal cord tumors.
Sensory impairment often helps to localize the level of the tumor based on loss of dermatome function, although the upper level of impaired long-tract function may be several segments below the actual tumor (Fig. 28-5). A lesion involving the lateral spinothalamic tract causes numbness, paresthesias, and decreased temperature sensation over the contralateral limb or trunk below the lesion. This produces the classic finding of a sensory level, which is best demonstrated with a pin or a large metal or cold object to test temperature sensation and with observation for absence of perspiration. A lesion in the posterior column causes an ataxic or tabetic gait. Standing posture is affected with the eyes closed (Romberg’s sign). Paresthesias can occur below the level of the lesion.
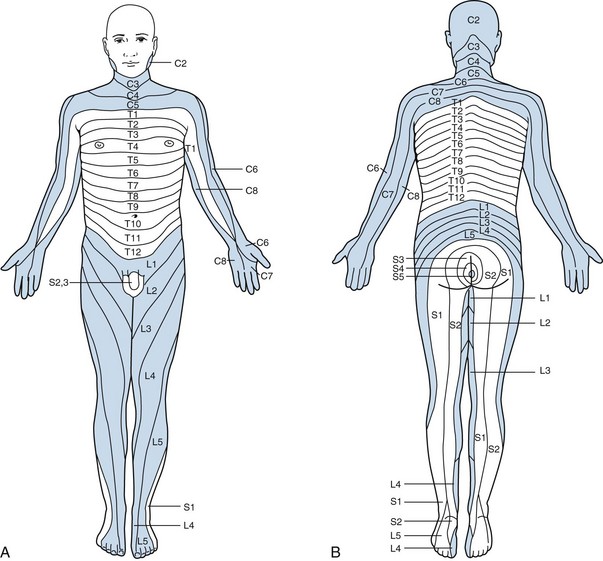
Figure 28-5 Anterior (A) and posterior (B) views of the dermatomes. C, cervical; T, thoracic; L, lumbar; S, sacral.
CREDIT LINE: Adapted from Netter FH, Freidberg SR, Baker RA: Disorders of spinal cord, nerve root, and plexus. In Netter FH, Jones HR Jr, Dingle RV (eds): The Ciba Collection of Medical Illustrations, Vol 1. Nervous System, Part II. West Caldwell, New Jersey, Ciba-Geigy, 1986, pp 181-189; Keegan JJ, Garrett FD: The segmental distribution of cutaneous nerves in the limbs of man. Anat Rec 102:409, 1948.
Primary Therapy
General Principles of Surgery
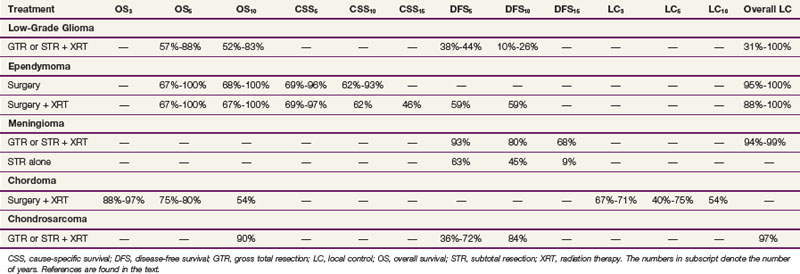
Surgery has traditionally been the mainstay of therapy for spinal cord tumors.10 As modern diagnostic and therapeutic techniques such as MRI, the operating microscope, microsurgical tools, and intraoperative neurophysiologic assessment have become available, more aggressive surgical intervention has become a more prevalent treatment strategy.11 This section will discuss general principles of surgery for CNS lesions, depending on anatomic location within the spinal cord. The outcome of surgery and its role in multidisciplinary management are described in the sections for each individual type of tumor. Table 28-3 shows an overview of the outcomes of the most common spinal cord tumors by treatment modality.
Surgical Techniques, Postoperative Management, and Complications
Surgery of Intramedullary Tumors
The most common intramedullary tumors are ependymomas and astrocytomas. Modern series advocate maximum safe resection of intramedullary spinal tumors. The ability to resect these tumors completely varies widely, from less than 10% to as much as 86%, depending on the histologic type.12,13–19 Most completely resectable lesions are WHO grade I tumors because higher-grade tumors are defined by their infiltrative potential.16,20–24 The gross total resection (GTR) rates for ependymomas range from 19% to 100%, whereas 0% to 43 % of astrocytomas are resectable.25–28 For infiltrative astrocytomas, the role of surgery is most frequently limited to biopsy for diagnosis. Laminectomy offers limited functional benefit by decompressing the spinal cord in the case of bulky infiltrative tumors. Functional postoperative outcomes may be excellent in selected cases, with the majority of patients (72% to 79%) experiencing either improvement or stabilization of their preoperative presenting symptoms, often after transient postsurgical worsening.12,19,29,30 The incidence of total, permanent postoperative paralysis is reported at 1%.11 Paralysis is often the result of unpredictable, sporadic vascular events.
Intramedullary tumors are almost always approached through a laminectomy exposure with the patient in the prone position.11 After the dura is opened, a longitudinal myelotomy is made over the widened region of the spinal cord, where imaging demonstrates the closest approximation of the tumor to the surface and the incision is deepened several millimeters until the tumor surface is reached. Infiltrative tumors with poorly defined dissection planes cannot be removed completely and, therefore, biopsy for diagnosis is often the safest course of action. When there is an exophytic component to the tumor, maximum safe debulking is typically performed.
Complications of Surgery
In patients who are neurologically intact before surgery, the incidence of significant acute morbidity is generally below 5% for extramedullary tumors and as low as 5% for carefully selected patients with intramedullary tumors.11 Spinal deformities, including scoliosis and kyphosis, as well as infection and CSF leakage, can occur.31,32 The development of postlaminectomy spinal deformities is a serious postoperative complication, occurring in up to 40% of pediatric and adolescent patients, and may be observed as soon as 8 months postoperatively.33
General Principles of Radiation Therapy
Because of the potential for growth and developmental side effects, the use of irradiation in children and infants with spinal cord tumors is controversial. Some authors do not recommend postoperative radiation therapy, and suggest that observation with close imaging follow-up and re-resection at the time of second occurrence is a better approach.12 With this strategy, radiation therapy may be avoided or used only for children with multiply recurrent tumors.
General Principles of Chemotherapy
The efficacy of chemotherapy may be limited by the intact blood–nervous system barrier. Regional drug delivery may be in the form of intracerebrospinal fluid therapy, intra-arterial infusion, and intratumoral therapy. Extramedullary spinal tumors gain their blood supply from meningeal blood vessels that are significantly more permeable than those of the brain, and chemotherapy may be of more use for these tumors. Drugs that have some efficacy in treating primary CNS malignant tumors and that are thought to cross the blood-brain barrier have been used in the treatment of malignant glial tumors of the spinal cord; however, no clear benefit from the use of chemotherapy has been demonstrated thus far.*
Irradiation Techniques and Tolerance
Definitions of Treatment Volumes
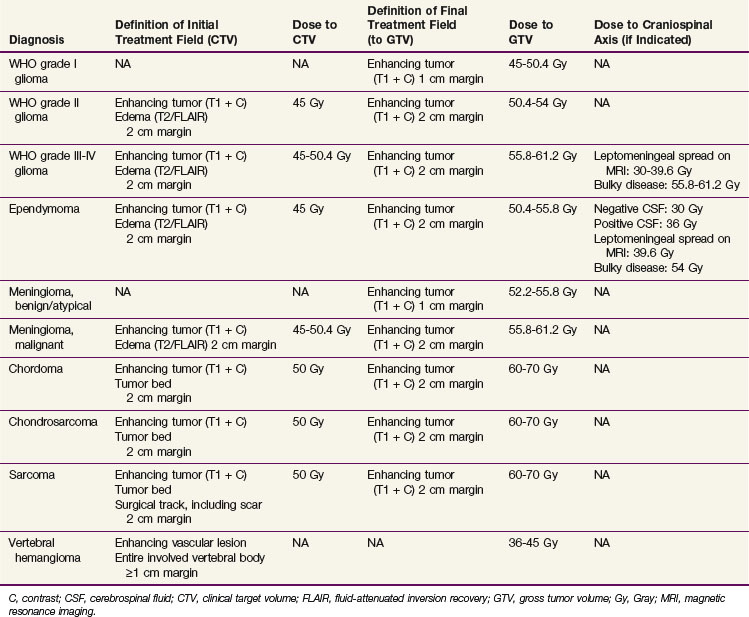
Three-dimensional treatment planning based on International Commission on Radiation Units (ICRU) volume definitions is the norm.39,40 The gross target volume (GTV) represents the grossly visible disease burden. Typically, this is the T1-enhancing abnormality on MRI or the nonenhancing tumor seen on T2 or FLAIR images. If there is no residual abnormality after a surgical resection, the tumor resection cavity is defined to be the GTV. Surrounding edema is not considered part of the GTV. The clinical target volume (CTV) is the T2 or FLAIR abnormality (which does include edema) on MRI as well as any areas potentially containing microscopic disease. Suggested definitions and doses to be delivered to the GTV and CTV are listed by histologic diagnosis in Table 28-4. The planning target volume (PTV) adds a dosimetric margin that takes into account uncertainties in daily treatment setup and physiologic variations that are difficult or impossible to control, such as (potential) fluctuations in the mass effect from cord edema that may occur over the course of treatment. Organs at risk typically include the thyroid and salivary glands, esophagus, lungs, heart, stomach, small bowel, liver, kidneys, bladders, ovaries, testicles, and uninvolved portions of the spinal cord itself.
Treatment Techniques
The most common approaches include a single posterior field (PA), opposed lateral fields, a PA field with opposed laterals, and oblique wedge-pair fields.41 They are typically designed to treat the CTV and GTV. For tumors in the cervicothoracic region, a split-beam approach is sometimes used. The central axis is placed just above the shoulders. Opposed lateral fields are used to treat the upper spine, whereas a PA field is used for the area of the spine below. Tumors in the thoracic region are often treated with a three-field approach using a PA field and opposed lateral beams. In the lumbar region, care must be taken to minimize the dose to the kidneys; a four-field approach using AP/PA and opposed lateral beams with the AP/PA beams preferentially weighted may be useful.
Certain neoplasms require treatment of the entire craniospinal axis. Several modifications of this approach are used in clinical practice (Fig. 28-6). Patients may be treated either in the supine or prone position, often in an immobilization cast to ensure daily positional reproducibility.42,43 The intracranial contents, including the upper one or two segments of the cervical cord, are treated through opposed lateral fields. Customized blocks protect the normal head and neck tissues from the primary radiation beam. The spine is treated through one or two posterior fields, depending on the size of the patient. In one method, the collimator for the lateral cranial fields is angled to match the divergence of the upper border of the adjacent spinal field, and the treatment couch is angled so that the inferior border of the cranial field is perpendicular to the superior edge of the spinal field. Alternatively, one may dispense with collimator and couch angles by calculating appropriate gaps. The gap is calculated so that the 50% isodose lines meet at the level of the anterior spinal cord. All junction lines are moved 0.5 to 1 cm every 8 to 10 Gy to avoid overdosing or underdosing segments of the cord. This is accomplished by shortening the inferior margin of the lateral cranial fields, symmetrically lengthening the superior and inferior margins of the posterior spinal field, and shortening the cranial margin of the caudal spinal field; a fixed block is placed at the inferior margin of the caudal spinal field to keep the lower margin of the irradiated volume at the same location.

IMRT is a treatment delivery method that may be used to further optimize the dose distribution. Because multiple critical sensitive organs are located near the spinal cord, improved dose distribution should allow the dose to these structures to be minimized. Because most spinal cord tumors typically recur locally, IMRT should allow the exploration of anatomic/biologic treatment planning and delivery in order to optimize different doses to different cell populations within heterogeneous volumes. The use of image guidance may also allow for reduced margins for sparing of critical structures. The use of IMRT is currently under investigation at multiple institutions.44,45
Stereotactic radiosurgery (SRS) and body radiotherapy (SBRT) deliver a highly conformal high dose of radiotherapy with stereotactic guidance. Doses may be delivered as a single dose (SRS) or as a short fractionated course (SBRT). Potential advantages include patient convenience with a shorter treatment course and the steep dose gradient at the edge of the field, which may allow for greater tissue sparing. SRS for primary spinal cord tumors remains experimental at this time but is an active area of research.46–48
Tolerance of the Spinal Cord and Lumbosacral Nerve Roots
A dose of 45 to 50.4 Gy in 25 to 28 fractions over 5 or 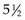 weeks is usually considered to be safe. The risk of myelopathy is less than 1%, well below the steep portion of the dose-response curve.46,49,50 It is estimated that with conventionally fractionated irradiation (1.8 to 2 Gy per fraction in five fractions per week), at 5 years the incidence of myelopathy is 5% for doses in the range of 57 to 61 Gy (tolerance dose TD5/5) and 50% for doses of 68 to 73 Gy (TD50/5).49 There is no convincing evidence that the cervical and thoracic cord differ in their radiosensitivity and there appears to be little change in tolerance with variations in the length of cord irradiated.49 Table 28-5 shows a range of isomorbid fractionation schemes, all of which carry a 5% risk of radiation myelopathy.
weeks is usually considered to be safe. The risk of myelopathy is less than 1%, well below the steep portion of the dose-response curve.46,49,50 It is estimated that with conventionally fractionated irradiation (1.8 to 2 Gy per fraction in five fractions per week), at 5 years the incidence of myelopathy is 5% for doses in the range of 57 to 61 Gy (tolerance dose TD5/5) and 50% for doses of 68 to 73 Gy (TD50/5).49 There is no convincing evidence that the cervical and thoracic cord differ in their radiosensitivity and there appears to be little change in tolerance with variations in the length of cord irradiated.49 Table 28-5 shows a range of isomorbid fractionation schemes, all of which carry a 5% risk of radiation myelopathy.
TABLE 28-5 Fractionation Schemes with a 5% Risk of Radiation-Induced Spinal Cord Myelopathy
| Dose per Fraction (Gy) | No. Fractions | Total Dose (Gy) |
|---|---|---|
| 2 | 29 | 58 |
| 3 | 13 | 39 |
| 3.3 | 11 | 33 |
| 4 | 7 | 28 |
| 5 | 5 | 25 |
| 10 | 1 | 10 |
Data from references 50, 98, 116, 206–209, 210, 211–214.
The tolerance of the lumbosacral nerve roots appears to be somewhat higher than that of the spinal cord. Most series report a 0% complication rate if patients are treated to doses of 70 Gy (or equivalent) as long as fraction sizes are kept at or below 2 Gy.51–53
The Quantitative Analysis of Normal Tissue Effects in the Clinic (QUANTEC) reviewed data from preclinical, conventional, reirradiation, and SBRT studies in order to refine normal tissue dose/volume tolerance guidelines.54 Based on this, a total dose of 54, 60, or 69 Gy with conventional 2-Gy once-daily fractionation was associated with a myelopathy rate of less than 1%, 6%, and 50%, with a calculated strong dependence on dose/fraction (α/β = 0.87 Gy). With reirradiation of the full cord cross section at conventional fractionation, cord tolerance appears to increase at least 2% at 6 months after an initial course of radiation therapy. For partial cord irradiation as part of spinal SRS, a maximum cord dose of 13 Gy in a single fraction or 20 Gy in three fractions appeared to have a minimal (<1%) risk of injury.55
Primary Therapy By Tumor Type
Astrocytoma
Astrocytomas represent 7% to 11% of all primary spinal tumors. They are the most common intramedullary spinal cord tumors and comprise 35% to 45% of all reported cases of intramedullary tumors. The median age at presentation is approximately 35 years.16,56–58 There does not appear to be a difference in the age of presentation between low- and high-grade tumors.16 In children, 75% to 90% of intramedullary spinal cord tumors are astrocytomas and 85% to 95% of these are low-grade, fibrillary, or juvenile pilocytic astrocytomas (Fig. 28-7). Fewer than 10% of pediatric and 25% of adult spinal cord astrocytomas are malignant.*
Motor weakness is the most common presenting symptom in astrocytomas, occurring in up to 87% of patients. The median time from onset of symptoms to diagnosis ranges from 6 months to 2 years.16,41,57,60 In general, high-grade astrocytomas tend to have a shorter prodrome (median duration, 1.6 to 7 months) than low-grade tumors (median duration, 2 years).16,29,56 Tumor dissemination by spread along the subarachnoid space is reported in up to 58% of patients with high-grade astrocytomas.56,61 Approximately 1% of WHO grade III to IV spinal cord astrocytomas have multifocal disease at presentation.10
Prognostic Factors
The most significant prognostic factors in patients with primary spinal cord astrocytomas are tumor histology, grade, and performance status.† High histopathologic grade is typically associated with a high risk of mortality and short median survival time. The 10-year overall survival (OS) of patients with pilocytic astrocytomas is 81%, in contrast to 15% for patients with diffuse fibrillary astrocytic tumors.69 For patients with WHO grade I to II spinal cord astrocytomas, the 5-year OS are 48% to 89%, but they are only 0% to 33% for WHO grade III to IV lesions; the latter have a median survival time of approximately 6 months.56,58,60,65,70 Other factors thought to be prognostic include young age (patients <18 years old live the longest, whereas those >40 years of age do poorly); the pattern of failure (local vs. disseminated, with the latter pattern developing in approximately 60% of patients with high-grade tumors); and duration of symptoms prior to diagnosis (<6 months vs. >6 months, with patients with a short duration doing worse).‡ In most series, neither extent of resection nor adjuvant radiation therapy appears to be prognostic, although this remains controversial.§
Treatment and Outcomes
The treatment of choice for intramedullary astrocytomas is complete excision of the tumor, when it can be safely accomplished without neurologic compromise.75 Otherwise, an incomplete excision is typically performed for grade I lesions and biopsy alone is the surgical strategy for the nonexophytic component of an infiltrative glioma. Gross total resection (GTR) is typically extremely difficult to achieve due to the infiltrative nature of all but the pilocytic lesions, with most authors reporting a 0% to 50% likelihood of GTR for spinal cord astrocytoma.* In patients with favorable prognostic factors (low-grade histologic findings, good performance status, and young age), observation with serial imaging studies, reserving irradiation for local recurrence, is an appropriate management option, particularly for young children.11,65,70,77 In the remainder of patients, adjuvant irradiation (RT) is usually recommended because progression of tumor in the spinal cord may lead to significant neurologic impairment.† Doses have traditionally been based on the experience with cerebral tumors, with lower-grade tumors typically receiving 45 to 54 Gy and high-grade tumors receiving higher doses.41,77,79
The overall outcomes are similar for patients with low-grade gliomas of the spinal cord treated either by GTR or subtotal resection (STR) or biopsy followed by external beam irradiation (EBRT), with most series reporting OS at 5 and 10 years in the ranges of 55% to 100% and 39% to 83%, respectively, and disease-free survival (DFS) of 38% to 93% and 26% to 80% at 5 and 10 years, respectively. Postoperative EBRT has shown a survival benefit for patients with infiltrative gliomas (24 months vs. 3 months) but not with pilocytic tumors.41,66 However, postoperative EBRT may improve progression-free survival (PFS) in low-grade and intermediate-grade astrocytomas.66 Overall long-term local control rates for gliomas range from 31% to 100%, with grade being the strongest predictor.‡ A comparison of survival and local control rates is shown in Table 28-3.
With high-grade tumors in adults and children, the median survival time is quite poor (4 to 10 months) despite surgery and EBRT; approximately two thirds of patients die of both local and disseminated disease.§ One small series suggested a survival benefit for patients with high-grade tumors treated with craniospinal axis irradiation.82
Data specific to adjuvant chemotherapy (after surgery and/or radiation therapy) for spinal cord astrocytomas are scant and composed mostly of small retrospective series.23,35,37,72 In two small trials, children with high-grade spinal astrocytoma had a 5-year PFS of 46% to 50% and a 5-year OS of 54% to 88% when chemotherapy was given either in the adjuvant setting or at the time of relapse.37,72 Overall, although there is no clear evidence of a difference in outcomes with or without chemotherapy for spinal cord tumors, chemotherapy is usually given for patients with malignant astrocytoma.34,83 Temozolomide has shown modest activity in patients with recurrent disease.84
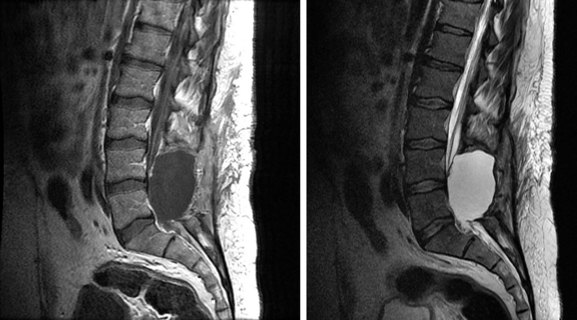
Ependymoma
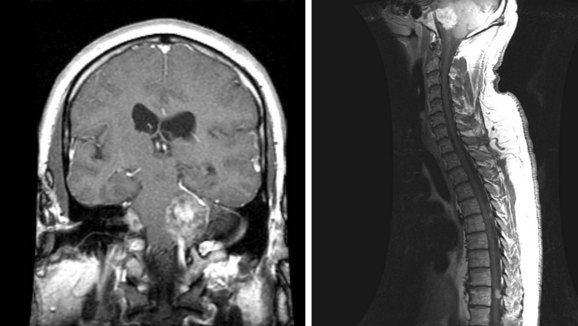
Ependymomas, which are glial tumors, are approximately equal in incidence to astrocytomas. The mean age at presentation is 30 to 39 years. These tumors are more common in adults than in children, and in males than in females.85,86 The median duration of symptoms prior to presentation is 2 to 4 years.16,28,57,86 Two-thirds occur in the lumbosacral region, 40% from the filum terminale28,87 (Fig. 28-8). Because of the propensity of these tumors for seeding the craniospinal axis, CSF evaluation and craniospinal MRI are strongly recommended for patients diagnosed with ependymoma. It is also important to note that 11% of patients with intracranial ependymomas have documented spinal metastases, so imaging of the brain may be indicated to rule out a primary brain tumor88 (Fig. 28-9).
Pathology
Ependymomas arise from ependymal cells and typically occur in the central canal of the spinal cord, the filum terminale, and the white matter adjacent to a ventricular surface. They are either low-grade tumors or anaplastic tumors, the latter being more likely to disseminate via the CSF. The majority of tumors are of a low grade.14,27,57,89 Myxopapillary ependymomas are low-grade tumors that typically occur in the lumbosacral region (filum terminale), are well-differentiated, and are often encapsulated but can seed the CSF, typically going to the thecal sac.90,91 Myxopapillary ependymomas often progress slowly and cause milder-than-expected neurologic deficits for their size. Some published series have included ependymoblastomas; however, these are primitive neuroectodermal tumors (PNETs), which have a high propensity to disseminate throughout the CNS and therefore should be considered in the medulloblastoma-PNET family of tumors.
Prognostic Factors
Multiple factors have been reviewed in the literature as being prognostic for local recurrence and survival. Factors prognostic for a favorable outcome include patient age less than 40 years; tumors with a lumbosacral location, myxopapillary histologic findings, and/or a grade of World Health Organization (WHO) grade I; tumors amenable to GTR or STR; and good preoperative function of the patient.* Some authors believe that the volume of residual disease appears to correlate with a worse outcome after EBRT, whereas other authors have found no differences in outcomes when comparing complete excision with STR followed by EBRT.27,87,94–96 Myxopapillary subtypes appear to be associated with a favorable prognosis, potentially because of ease of resection due to their anatomic location.92 Five-year OS for the myxopapillary subtype and for WHO grade I tumors are 97% to 100%, and the 5-year disease-specific survival (DSS) is 97%.93,94 Recently, the expression of erb-B2 and erb-B4 has been identified as a negative prognostic marker, especially in intracranial ependymomas, but the prognostic significance of these substances in spinal ependymomas is not clear.
Treatment and Outcomes
Most intradural extramedullary ependymomas are myxopapillary and are often amenable to complete surgical resection if they are not multifocal.86,90,97,98 The goal of surgery is GTR. Every attempt should be made to remove myxopapillary tumors as a whole as opposed to piecemeal removal, because of the risk of seeding, including upward seeding to the cranial nerves. The potential for cure may increase the severity of a hopefully transient deficit that surgeon and patient may be willing to incur and illustrates the importance of prospective multidisciplinary management of these tumors. Typically, complete resection is achievable in 79% to 100% of modern series.* Some authors report complete resection rates as low as 19% to 50%, but these data tend to be from older series or those with a significant number of high-grade tumors.16,57,76,87,92 Five- and 10-year OS for all spinal cord ependymomas is 67% to 100% and 68% to 100%, respectively.† DSS at 5 and 10 years is 69% to 96% and 62% to 93%, respectively.16,89,92 Local control rates after GTR are 95% to 100%.14,26,99 Late failures may occur more than 4 years after curative surgery, particularly with the myxopapillary subtype, so long-term follow-up is warranted.101,102
Postoperative EBRT appears to improve local control in patients with subtotally resected ependymomas, with all high-grade lesions, and with craniospinal axis dissemination (positive CSF or MRI scan); not all authors agree on this, with many series showing increasing age to be the most significant predictor.‡ In most series, the outcome for STR followed by EBRT appears to be similar to that of complete excision. Typically, the dose given to the tumor bed is 49 to 56 Gy, whereas the craniospinal axis (if indicated) receives 30 to 36 Gy.27,93,99 Low-grade lesions with a low risk of seeding are typically treated with limited fields to 50.4 to 55.8 Gy in 1.8-Gy daily fractions. In patients with tumors at high risk of seeding, when pretreatment CSF cytologic studies reveal malignant cells, or if the spinal MRI scan shows evidence of leptomeningeal disease, the craniospinal axis should be treated to 36 Gy in 1.5- to 1.8-Gy daily fractions. Subsequently, the primary tumor site is boosted to a total dose of 50.4 to 55.8 Gy. If gross leptomeningeal spread is evident, the craniospinal axis dose should be 39.6 Gy (1.8 Gy/fraction) or 40.5 Gy (1.5 Gy/fraction), with the same boost dose to the primary tumor as previously discussed.
In patients undergoing incomplete resection followed by EBRT, OS at 5, 10, and 15 years is 67% to 100%, 67% to 100%, and 75%, respectively.26,27,66,93,94 Cause-specific survival (CSS) at 5, 10, and 15 years for all tumors is 74% to 93%, 50% to 93%, and 35% to 46%, respectively.66,89,92,108 CSS at 5 years is 87% to 97% for myxopapillary and low-grade lesions and 27% to 71% for high-grade tumors.93,94 DFS at 5 and 10 years is 59% and 59%, respectively.92 Local control rates are 88% to 100%.27,94,99,109
There is no strong body of evidence thus far demonstrating that the addition of chemotherapy to EBRT improves the outcome.36,38,110–112 A single trial of etoposide for recurrent spinal ependymoma has shown a median response duration of 15 months, with a median survival time of 16 months.38 Patients who were responders had a median survival time of 20 months, whereas those who did not respond lived only 4 months; the results approached statistical significance. Pediatric patients with anaplastic ependymoma or ependymoblastoma are routinely given chemotherapy.113,114 A recent trial has started evaluating cytotoxic agents in combination with erb-B inhibitors, but further clinical research in this area is necessary.
Meningioma
Spinal meningiomas make up only 8% of all meningiomas but 25% to 46% of all spinal neoplasms.115,116,117 They tend to occur in females three to seven times more commonly than in males.§ Most patients are over the age of 40 years, with a mean age at diagnosis of 49 to 63 years.118,119 The incidence appears to be bimodal, the first peak being in the third decade of life, with a second larger peak seen after age 50 years, especially in the sixth and seventh decades of life.118,119 In younger patients, 13% have neurofibromatosis type 2.119 Younger patients (<50 years of age) have a recurrence rate of 23% versus 5% in older patients, though not all authors agree that young age in and of itself carries a worse prognosis.115,119
The most common site is the thoracic region, where 55% to 80% of tumors occur, whereas cervical lesions make up approximately one-third of all lesions.115,116,118,119,120 The incidence by location varies with age: before age 50 years, 39%, 56%, and 5% of lesions occur in the cervical, thoracic, or lumbar spine, respectively; in patients over age 50 years, 16%, 80%, and 5% occur in the cervical, thoracic, or lumbar spine, respectively.119
Treatment and Outcomes
Functional improvement of neurologic deficits is seen in 66% to 100% of patients.31,118,120,121 Complete resection is achievable in 82% to 100% of cases, with a 1% to 6% late local failure rate.116,120,121 With STR, late local recurrence rates range from 17% to 100%.31,116,121 Because local failures can take place even decades after surgery, the actual late failure rate may be even higher, similar to that seen in patients with intracranial meningiomas. In one large series, after complete resection, recurrence-free rates at 5, 10, and 15 years were 93%, 80%, and 68%, respectively, and after STR, recurrence-free rates at 5, 10, and 15 years were 63%, 45%, and 9%, respectively.115 Therefore, the necessity for life-long follow-up cannot be overemphasized. Because local failure rates in the spine are higher with STR, some authors recommend radiation therapy.
The role of irradiation is not well understood due to the small number of cases reported in the literature, and most clinicians extrapolate from the data on irradiation of intracranial meningiomas.118,120,122 The expected outcome of radiation therapy is long-term stabilization of disease.123 Patients who undergo complete resection should be observed. Even those with STR who either have improvement of their neurologic status or are asymptomatic postoperatively can be closely followed. When patients are irradiated, the dose delivered is typically 52.2 to 54 Gy, based on the data from the treatment of intracranial meninigoma.123 Single-fraction SRS must be considered investigational at this time.124,125
Hydroxyurea chemotherapy has been used but has minimal activity.126
Chordoma
Chordomas are slow-growing neoplasms thought to be remnants of the embryonic notochord. They make up approximately 4% of spinal tumors. Approximately 24% to 35% arise in the region of the base of the skull; as many as 17% are thought to have originated in the cervical spine; 11% to 15% are known as chordomas of the mobile spine and arise in the true vertebral region (usually, the lumbar region); and 50% to 58% arise in the sacrococcygeal region.127,128,129 Most chordomas occur during the fourth decade of life, with a male to female ratio of 2 to 1, though sacrococcygeal chordomas tend to arise in the fifth and sixth decades.53,130,131–134
Prognostic Factors
Several prognostic factors have been identified. Location is prognostic for survival, with a 5-year OS of 66% for sacrococcygeal tumors and 50% for vertebral lesions.135 Overall, 14% to 39% of patients develop metastases at a median of 5 years, most commonly to the lungs.* The incidence of metastases varies by location of the primary tumor: 26% to 44% of patients with sacrococcygeal lesions develop metastatic disease (usually to the thecal sac), whereas 61% of those with vertebral primaries can metastasize, typically in a caudal direction. In some series, skull base lesions have rarely been reported to metastasize.53,132 Gender may also be prognostic: 5-year and 8-year local control rates are 81% and 75% for men and 65% and 17% for women, respectively.139,140 The prognostic importance of age is controversial; in some series, children have a higher incidence of metastases, but in other series, patients over the age of 52 years have a worse local control rate.127,141,142 Finally, residual microscopic disease after resection (either tumor spillage or positive margins) increases the risk of local relapse to 60% to 64% from 25% to 28% after GTR.134,143 Primary tumor size less than 8 cm has also been suggested as a good prognostic factor.142
Treatment and Outcomes
Although en bloc GTR has traditionally been advocated for these lesions, it is rarely possible. As such, most patients undergo STR or biopsy followed by EBRT. However, surgery is an important component of treatment despite potential morbidity; a recent large database review showed that patients who had undergone resection had significantly prolonged survival times.142 In most series, patients have been treated with conventional photon radiotherapy, typically to doses of 55 to 70 Gy in 2-Gy fractions, with 5-year OS of 38% to 58%.† Although EBRT prolongs the disease-free interval, most tumors recur unless they have also been completely resected; local progression is the most common cause of death.130 In one older series, the local control rate of sacrococcygeal tumors was 0% with either surgery or EBRT as monotherapy but 40% when the two were combined.129
Some treatment series using conventional photon radiation therapy suggest that local control rates improve with increasing radiation doses. With doses of less than 40 Gy, more than 48 Gy, more than 50 Gy, and more than 55 Gy, 5-year DFS is approximately 0%, 13%, 31%, and 41%, respectively.53,145 However, not all authors have found that increased dose improves outcome.147,148
In order to improve the therapeutic ratio when treating residual or recurrent disease, most authors now advocate a combination of photon and charged particle therapy (typically, with protons).127,135,149–156 When patients are treated either immediately after maximum resection or at the time of recurrence, with median doses of 65 to 76 Gy equivalents using charged particle radiation therapy (with or without photons), local control rates at 3, 5, and 10 years are 67% to 71%, 40% to 75%, and 54%, respectively, with OS at 3, 5, and 10 years at 88% to 97%, 75% to 80%, and 54%, respectively.52,127,152,153,157
SRS has also been used in the treatment of residual and recurrent cervical spine chordoma. A mean dose of 19 Gy in a single fraction prescribed at the tumor margin resulted in a 4-year local control rate of 80% without neurologic sequelae in one series.158
The experience with interstitial irradiation is minimal. This modality has fallen out of favor with the advent of charged particle therapy and radiosurgery.159–161
Chondrosarcoma
Primary chondrosarcoma of the spine is a rare, slowly growing tumor thought to originate from primitive mesenchymal cells or from the embryonal rests of the cranial cartilaginous matrix. Spinal chondrosarcomas are slightly more common in men than in women.162 The mean and median ages of presentation are 45 years and 51 years, respectively.163,164 Of the axial skeleton chondrosarcomas, 33% occur in the mobile spine, most often in the thoracic region.163,164 Another 33% arise in the sacrum.165
Patients with spinal chondrosarcomas typically have a long-term OS of 25% to 54%.162–164166 Adverse prognostic factors include positive surgical margins, high tumor grade, and older patient age.165,166 Twelve percent to 19% of lesions are grade 4 tumors, which carry a poor prognosis (3-year OS of only 14%; median survival time of 2.7 years; 75% to 86% incidence of metastases).165,166
Treatment and Outcomes
Surgical resection is the mainstay of treatment of these lesions. OS after any degree of resection is 55% to 72%, 67%, and 63% at 5, 10, and 15 years, respectively.164,165 The median survival time is typically 6 years, with local control rates ranging from 36% to 72%.163–166 Patients with GTR have a better outcome than those with STR. After complete resection, the local control rate is 97%, with a 90% 10-year OS and an 84% DFS.165 Many chondrosarcomas have indolent courses, but others behave more aggressively. Radiation therapy after incomplete resection has been shown to lengthen the disease-free interval from 16 to 44 months in one series.163 Chemotherapy has not been shown to improve outcomes.163
Miscellaneous Primary Spinal Tumors
Chloroma
The most effective treatment appears to be multimodality therapy coupled with early diagnosis. Options include surgical decompression, intravenous and/or intrathecal chemotherapy, radiation therapy at doses of 20 to 30 Gy or any combination of these treatments. Patient survival times range from 18 days to 9.5 years after diagnosis. In a review of the literature, no clear relationship could be found between the specific treatment modality used and the length of survival. Those receiving systemic therapy soon after diagnosis experienced the longest disease-free episodes, however.167
Hemangioblastoma
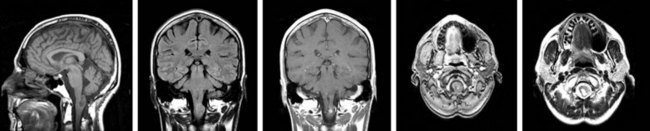
Intramedullary hemangioblastomas are slowly growing vascular tumors composed of endothelial and stromal components and account for 3% to 13% of intramedullary spinal tumors.61,64 The male to female ratio is 1.8 to 1.168 The median age at presentation is 36 years for sporadic lesions, whereas patients with von Hippel-Lindau syndrome typically present a decade earlier and have multiple tumors. Multiple CNS hemangioblastomas are diagnostic of von Hippel-Lindau syndrome. Multiply recurrent hemangioblastoma may also raise the suspicion of von Hippel-Lindau syndrome. Patients with isolated spinal hemangioblastoma should be screened for other manifestations of von Hippel-Lindau syndrome by ophthalmologic evaluation, brain MRI, and abdominal CT scanning to rule out retinal and cranial hemangioblastoma and renal tumors, respectively. In patients with von Hippel-Lindau syndrome, 51% of neuraxis lesions are located in the spinal cord and only 29% are completely intramedullary, with the remainder having an extramedullary component169 (Fig. 28-10).
Factors prognostic for favorable outcome (defined as improvement or stabilization of neurologic function) are minimal or no preoperative neurologic deficits, lesions smaller than 0.5 cm2, dorsally located lesions, and total surgical removal of the lesion.169,170 Progressive syringomyelia is a negative prognostic factor with respect to preservation of neurologic function and is often recalcitrant to treatment attempts.
Surgical resection is the mainstay of treatment for symptomatic lesions. Hemangioblastomas are extremely vascular tumors occurring in the spinal intramedullary compartment. The dorsal location of most of these lesions and the commonly associated cyst help to simplify the removal somewhat because the cyst provides a safe route for approaching the tumor. Removal of the enhancing tumor is satisfactory to prevent reaccumulation of the cyst, and the remainder of the cyst wall may, therefore, be left intact. Tumor margins are addressed first and feeding arteries are coagulated. Then the tumor is dissected and removed en bloc. After surgical resection, 41% to 68% of patients experience improvement of neurologic function and another 32% to 84% have stabilization of their preoperative function.168,169,171
Although SRS is an option for cerebellar hemangioblastoma, it has not been properly evaluated for spinal hemangioblastoma because of concerns regarding the radiation tolerance of the spinal cord. A small series suggests safety and efficacy.172,173
Lipoma
Early surgical decompression is mandatory for preservation of existing neurologic function, but complete surgical debulking is almost impossible due to the infiltrative nature of the tumors.174–176 There is no known role for radiation therapy or chemotherapy.
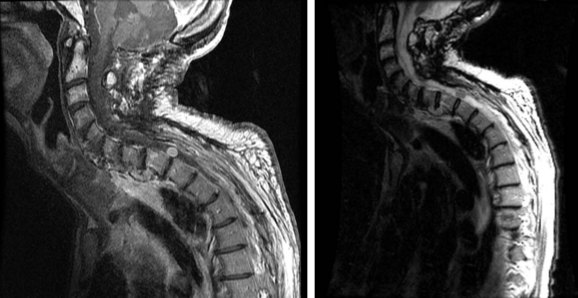
Lymphoma
Primary spinal cord lymphomas are uncommon lesions. Only 4% of lymphomas are primary epidural tumors of which 90% are of B-cell origin177,178 (Fig. 28-11). Typically, lymphomas involving the spinal cord are seen in the context of primary CNS non-Hodgkin’s lymphoma, with 27% having positive CSF findings and 16% eventually developing leptomeningeal disease involving the spine.178
If the initial presentation is one of spinal cord compression, management is similar to that of other metastatic tumors described below, although rapid responses may be seen with steroids, chemotherapy, and irradiation to a median dose of 38 Gy.177 Lengthy administration of preoperative steroids may result in disappearance of the tumor by the time of surgery (ghost-tumor effect). Otherwise, management is similar to that of cerebral non-Hodgkin’s lymphoma. After decompressive laminectomy, subtotal tumor resection, and spinal irradiation, the local control rate is 60%, with a median duration of survival of 42 months.177
Neurocytoma
Central neurocytoma has been reported to arise in the spine.179,180 Staining for synaptophysin and neurospecific enolase allows these tumors to be reliably distinguished. As in the brain, their behavior is variable. Because this tumor arises centrally in the spinal cord, observation or irradiation may be the primary management strategy, with resection reserved for tumors with progressive neurologic deficits.
Sarcoma
Primary sarcomas of the spinal cord are exceedingly rare, accounting for 0.7% of CNS malignant tumors; 5.6% of these are found in the spinal canal.181 Malignant fibrous histiocytoma represents the most common histologic diagnosis; tumors arise from the nerve roots of the cauda equina, the spinal cord, or the spinal blood vessels.181,182 Surgical debulking and postoperative EBRT to approximately 60 Gy in 1.8- to 2-Gy fractions represents the typical treatment approach described in the literature.181,182 The 5-year OS of patients with high-grade lesions is significantly worse at 28% than the 83% reported with low-grade lesions. The role of chemotherapy is not known.
Schwannoma
Spinal schwannoma may present simultaneously in both the spinal canal and the thoracic cavity. These dumbbell-shaped tumors are typically benign and may involve the spinal cord and spinal nerves via the neural foramina. Surgical resection is the treatment of choice.32,183,184 Schwannomas arise from spinal rootlets (most often, the dorsal rootlets) and may grow along the nerve root in a dumbbell fashion through a neural foramen. Therefore, resection requires removal of sections of the involved rootlets. Only 3% of these tumors are malignant; adjuvant EBRT has been used in such cases.185 Outcomes are mixed.183,185
Teratoma
True intramedullary teratoma is an extremely rare tumor, with only nine cases reported in the literature. Early surgical resection is recommended to preserve neurologic status. Because teratomas may adhere to the functional neural tissue of the spinal cord in such a way that no dissection plane is available, complete resection is often not possible. A low-grade histopathologic status predominates, so the symptomatic recurrence of incompletely resected mature teratomas is often slow and may eventually require a second surgical procedure.186 Radiation therapy is generally reserved for multiply recurrent, subtotally resected disease.
Vertebral Hemangioma
Vertebral hemangiomas are benign growths of endothelial origin typically located in the thoracic and upper lumbar spine. They are the most common benign spinal column tumors, with an incidence of approximately 11%. Because vertebral body hemangiomas are most frequently stable over long periods of time, some authors consider them to be nonneoplastic. Growth causing progressive symptoms has been documented. Only 1% to 2% of patients develop clinically significant pain. New onset of back pain followed by subacute progression of a thoracic myelopathy, typically over a period of 4 to 5 months, is the most common presentation for patients with neurologic deficit.187 These tumors also occasionally cause root and cord compression syndromes. A preoperative diagnosis of compressive vertebral hemangioma can be made from 65% of radiographs and 80% of CT examinations, whereas asymptomatic vertebral hemangioma is much more easily recognized with this imaging combination.188
Treatment is initiated only when progressive symptoms, including focal pain or progressive neurologic deficit, develop. Historically, standard treatment consisted of surgical removal of the lesion. Endovascular embolization of these very vascular growths may also provide pain control. Vertebroplasty has high reported rates of success. The local control rate at 3 years is 70%. Of lesions that regrow, 90% recur within the first 2 years.189
Irradiation is an effective treatment alternative. A meta-analysis demonstrated that a total dose of 30 Gy given in 2-Gy fractions resulted in a 57% complete amelioration of pain and 32% partial improvement of pain.190 A second, smaller analysis showed an apparent dose-response effect. Patients treated with the biologic equivalent 36 to 44 Gy in 2-Gy fractions had significantly better complete pain relief than those treated with 20 to 34 Gy (82% vs. 39%, respectively).191
Treatment Complications
Radiation myelopathy may present as a transient early delayed or late delayed reaction. Transient radiation myelopathy is clinically manifested by momentary, electrical shock-like paresthesias or numbness radiating from the neck to the extremities, precipitated by neck flexion (Lhermitte’s sign).192 The syndrome typically develops 3 to 4 months after treatment and spontaneously resolves over the following 3 to 6 months without therapy. It is attributed to transient demyelination caused by radiation-induced inhibition of myelin-producing oligodendroglial cells in the irradiated spinal cord segment.192–194
Irreversible radiation myelopathy usually is not seen earlier than 6 to 12 months after completion of treatment. Typically, half of the patients who develop radiation-induced myelopathy in the cervical or thoracic cord region will do so within 20 months of treatment and 75% of cases will occur within 30 months.195 The signs and symptoms are typically progressive over several months, but acute onset of plegia over several hours or a few days is possible. It is thought to be multifactorial in origin, involving demyelination and white matter necrosis ultimately due to oligodendroglial cell depletion and microvascular injury. The diagnosis of radiation myelopathy is one of exclusion; a history of radiation therapy in doses sufficient to result in injury must be present. The region of the irradiated cord must lie slightly above the dermatome level of expression of the lesion; the latent period from the completion of treatment to the onset of injury must be consistent with that observed in radiation myelopathy; and local tumor progression must be ruled out.
There are no pathognomonic laboratory tests or imaging studies that conclusively diagnose radiation myelopathy. MRI findings include swelling of the spinal cord with hyperintensity on the T2-weighted images with or without areas of contrast enhancement.193,196 There is no known consistently effective treatment for radiation myelitis.197,198 The probability of dying from radiation myelopathy is approximately 70% with cervical lesions and 30% with thoracic spinal cord injury.199
Radiation side effects in children include growth abnormalities such as decreased vertebral height, kyphosis, and scoliosis.200 Secondary malignant disease, including bone or soft tissue sarcomas and glioblastoma multiforme lesions, has been reported after irradiation of spinal cord tumors.201–203
Treatment Algorithm, Challenges, and Future Possibilities
Treatment Algorithm
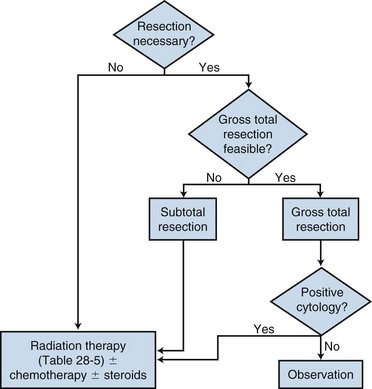
Primary tumors of the spinal cord are uncommon lesions. Much of the literature groups these tumors together by location (intramedullary vs. extramedullary) when reporting outcomes. However, spinal cord tumors clearly behave differently depending upon the histologic type, and this needs to be taken into account when making management decisions. Nonetheless, the primary treatment for most spinal cord tumors continues to be surgical resection, with irradiation recommended for patients in whom a complete resection cannot be achieved and for those at high risk of recurrence. The role of chemotherapy in the management of spinal cord tumors remains unclear. A general simplified treatment algorithm is shown in Figure 28-12.
Challenges and Future Possibilities
The most significant future advances one may anticipate are the possibilities of noninvasively obtaining an accurate tissue diagnosis of a suspicious spinal cord lesion and accurately measuring treatment response. To this extent, novel MRI contrast agents could serve as biochemical reporters of various cellular functions.204 These newer contrast agents could be tailored to report on the physiologic status or metabolic activity of biologic systems: enzyme-activated agents correlate developmental biologic events with gene expression; calcium-activated agents can measure intracellular signal transduction; pH-activated agents provide a blood oxygen level-dependent (BOLD) signal that might be used for noninvasive mapping of human nervous system function; and T2-activated agents can detect specific oligonucleotide sequences. The agents of interest are currently under investigation in the setting of primary brain tumor treatment, and it remains to be seen how the results will be applied to spinal cord therapy. Regarding toxicity, better refinements in dose and volume tolerance are necessary, with improvements presented in the recent QUANTEC study.55 Long-term data on patients treated with hypofractionated regimens and SBRT are awaited.
1 CBTRUS. Statistical report. Primary brain and central nervous system tumors diagnosed in the United States in 2004-2006. Hinsdale, Illinois: Central Brain Tumor Registry of the United States; 2010.
2 Cancer International Agency of Research on Cancer (IARC): World Health Organization Classifiction of Tumours: Pathology and Genetics: Tumours of the Nervous System, ed 2. Oxford University Press, Lyon, France, 2000..
12 Constantini S, Miller DC, Allen JC, et al. Radical excision of intramedullary spinal cord tumors. Surgical morbidity and long-term follow-up evaluation in 164 children and young adults. J Neurosurg. 2000;93:183-193.
32 Seppala MT, Haltia MJ, Sankila RJ, et al. Long-term outcome after removal of spinal schwannoma. A clinicopathological study of 187 cases. J Neurosurg. 1995;83:621-626.
34 Stewart LA. Chemotherapy in adult high-grade glioma. A systematic review and meta-analysis of individual patient data from 12 randomised trials. Lancet. 2002;359:1011-1018.
36 Robertson PL, Zeltzer PM, Boyett JM, et al. Survival and prognostic factors following radiation therapy and chemotherapy for ependymomas in children. A report of the Children’s Cancer Group. J Neurosurg. 1998;88:695-703.
37 Doireau V, Grill J, Zerah M, et al. Chemotherapy for unresectable and recurrent intramedullary glial tumours in children. Brain tumours subcommittee of the french society of paediatric oncology (SFOP). Br J Cancer. 1999;81:835-840.
40 ICRU Report 62. Prescribing, recording and reporting photon beam therapy (supplement to ICRU Report 50). Besthesda, Maryland, Nuclear Technology, 1999.
41 Minehan KJ, Brown PD, Scheithauer BW, et al. Prognosis and treatment of spinal cord astrocytoma. Int J Radiat Oncol Biol Phys. 2009;73:727-733.
50 Marcus RBJr, Million RR. The incidence of myelitis after irradiation of the cervical spinal cord. Int J Radiat Oncol Biol Phys. 1990;19:3-8.
66 Abdel-Wahab M, Etuk B, Palermo J, et al. Spinal cord gliomas. A multi-institutional retrospective analysis. Int J Radiat Oncol Biol Phys. 2006;64:1060-1071.
68 Garces-Ambrossi GL, McGirt MJ, Mehta VA, et al. Factors associated with progression-free survival and long-term neurological outcome after resection of intramedullary spinal cord tumors: analysis of 101 consecutive cases. J Neurosurg Spine. 2009;11:591-599.
105 McGuire CS, Sainani KL, Fisher PG. Both location and age predict survival in ependymoma. A SEER study. Pediatr Blood Cancer. 2009;52:65-69.
107 Pica A, Miller R, Villa S, et al. The results of surgery, with or without radiotherapy, for primary spinal myxopapillary ependymoma. A retrospective study from the rare cancer network. Int J Radiat Oncol Biol Phys. 2009;74:1114-1120.
117 Helseth A, Mork SJ. Primary intraspinal neoplasms in Norway, 1955 to 1986. A population-based survey of 467 patients. J Neurosurg. 1989;71:842-845.
119 Cohen-Gadol AA, Zikel OM, Koch CA, et al. Spinal meningiomas in patients younger than 50 years of age. A 21-year experience. J Neurosurg. 2003;98:258-263.
127 Noel G, Habrand JL, Jauffret E, et al. Radiation therapy for chordoma and chondrosarcoma of the skull base and the cervical spine. Prognostic factors and patterns of failure. Strahlenther Onkol. 2003;179:241-248.
130 Klekamp J, Samii M. Spinal chordomas—results of treatment over a 17-year period. Acta Neurochir (Wien). 1996;138:514-519.
146 Amichetti M, Cianchetti M, Amelio D, et al. Proton therapy in chordoma of the base of the skull. A systematic review. Neurosurg Rev. 2009;32:403-416.
190 Heyd R, Strassmann G, Filipowicz I, et al. [Radiotherapy in vertebral hemangioma.]. Rontgenpraxis. 2001;53:208-220.
210 Macbeth FR, Wheldon TE, Girling DJ, et al. Radiation myelopathy. Estimates of risk in 1048 patients in three randomized trials of palliative radiotherapy for non-small cell lung cancer. The medical research council lung cancer working party. Clin Oncol (R Coll Radiol). 1996;8:176-181.
1 CBTRUS. Statistical report. Primary brain and central nervous system tumors diagnosed in the United States in 2004-2006. Hinsdale, Illinois: Central Brain Tumor Registry of the United States; 2010.
2 Cancer International Agency of Research on Cancer (IARC): World Health Organization Classifiction of Tumours: Pathology and Genetics: Tumours of the Nervous System, ed 2. Oxford University Press, Lyon, France, 2000..
3 Parsa AT, Fiore AJ, McCormick PC, Bruce JN. Genetic basis of intramedullary spinal cord tumors and therapeutic implications. J Neurooncol. 2000;47:239-251.
4 Aydin MV, Ozel S, Sen O, et al. Intradural disc mimicking. A spinal tumor lesion. Spinal Cord. 2004;42:52-54.
5 Dhiwakar M, Buxton N. Acute transverse myelitis mimicking an intramedullary neoplasm. Br J Neurosurg. 2004;18:72-73.
6 Yukawa Y, Kato F. Isolated spinal cord sarcoidosis mimicking an intramedullary tumor. J Spinal Disord. 1999;12:530-533.
7 Fouladi M, Heideman R, Langston JW, et al. Infectious meningitis mimicking recurrent medulloblastoma on magnetic resonance imaging. Case report. J Neurosurg. 1999;91:499-502.
8 Kilic G, Blankstein J, Kadanoff R. Vertebral tuberculosis presenting with elevated CA-125 and weight loss mimicking ovarian malignancy. Case report. Eur J Gynaecol Oncol. 2003;24:561-562.
9 Choucair AK, Levin VA, Gutin PH, et al. Development of multiple lesions during radiation therapy and chemotherapy in patients with gliomas. J Neurosurg. 1986;65:654-658.
10 Gowers WR, Horsley V. A case of tumour of the spinal cord. Removal and recovery. Med Chir Trans. 1888;53:377-428.
11 Jallo GI, Freed D, Epstein F. Intramedullary spinal cord tumors in children. Childs Nerv Syst. 2003;19:641-649.
12 Constantini S, Miller DC, Allen JC, et al. Radical excision of intramedullary spinal cord tumors. Surgical morbidity and long-term follow-up evaluation in 164 children and young adults. J Neurosurg. 2000;93:183-193.
13 Zileli M, Coskun E, Ozdamar N, et al. Surgery of intramedullary spinal cord tumors. Eur Spine J. 1996;5:243-250.
14 Goh KY, Velasquez L, Epstein FJ. Pediatric intramedullary spinal cord tumors. Is surgery alone enough? Pediatr Neurosurg. 1997;27:34-39.
15 Abdel-Wahab M, Corn B, Wolfson A, et al. Prognostic factors and survival in patients with spinal cord gliomas after radiation therapy. Am J Clin Oncol. 1999;22:344-351.
16 Shirato H, Kamada T, Hida K, et al. The role of radiotherapy in the management of spinal cord glioma. Int J Radiat Oncol Biol Phys. 1995;33:323-328.
17 Rodrigues GB, Waldron JN, Wong CS, Laperriere NJ. A retrospective analysis of 52 cases of spinal cord glioma managed with radiation therapy. Int J Radiat Oncol Biol Phys. 2000;48:837-842.
18 Weiner HL, Freed D, Woo HH, et al. Intra-axial tumors of the cervicomedullary junction. Surgical results and long-term outcome. Pediatr Neurosurg. 1997;27:12-18.
19 Xu QW, Bao WM, Mao RL, Yang GY. Aggressive surgery for intramedullary tumor of cervical spinal cord. Surg Neurol. 1996;46:322-328.
20 Nishio S, Morioka T, Fujii K, et al. Spinal cord gliomas. Management and outcome with reference to adjuvant therapy. J Clin Neurosci. 2000;7:20-23.
21 Santi M, Mena H, Wong K, et al. Spinal cord malignant astrocytomas. Clinicopathologic features in 36 cases. Cancer. 2003;98:554-561.
22 Bouffet E, Pierre-Kahn A, Marchal JC, et al. Prognostic factors in pediatric spinal cord astrocytoma. Cancer. 1998;83:2391-2399.
23 Merchant TE, Nguyen D, Thompson SJ, et al. High-grade pediatric spinal cord tumors. Pediatr Neurosurg. 1999;30:1-5.
24 Kim MS, Chung CK, Choe G, et al. Intramedullary spinal cord astrocytoma in adults. Postoperative outcome. J Neurooncol. 2001;52:85-94.
25 Ohata K, Takami T, Gotou T, et al. Surgical outcome of intramedullary spinal cord ependymoma. Acta Neurochir (Wien). 1999;141:341-346. discussion Acta Neurochir (Wien) 141:346-347, 1999
26 Hanbali F, Fourney DR, Marmor E, et al. Spinal cord ependymoma. Radical surgical resection and outcome. Neurosurgery. 2002;51:1162-1172. discussion Neurosurgery 51:1172-1174, 2002
27 McLaughlin MP, Marcus RBJr, Buatti JM, et al. Ependymoma. Results, prognostic factors and treatment recommendations. Int J Radiat Oncol Biol Phys. 1998;40:845-850.
28 McCormick PC, Torres R, Post KD, Stein BM. Intramedullary ependymoma of the spinal cord. J Neurosurg. 1990;72:523-532.
29 Cristante L, Herrmann HD. Surgical management of intramedullary spinal cord tumors. Functional outcome and sources of morbidity. Neurosurgery. 1994;35:69-74. discussion Neurosurgery 35:76, 1994
30 Hejazi N, Hassler W. Microsurgical treatment of intramedullary spinal cord tumors. Neurol Med Chir (Tokyo). 1998;38:266-271. discussion Neurol Med Chir (Tokyo) 38:271-273, 1998
31 Klekamp J, Samii M. Surgical results for spinal meningiomas. Surg Neurol. 1999;52:552-562.
32 Seppala MT, Haltia MJ, Sankila RJ, et al. Long-term outcome after removal of spinal schwannoma. A clinicopathological study of 187 cases. J Neurosurg. 1995;83:621-626.
33 Reimer R, Onofrio BM. Astrocytomas of the spinal cord in children and adolescents. J Neurosurg. 1985;63:669-675.
34 Stewart LA. Chemotherapy in adult high-grade glioma. A systematic review and meta-analysis of individual patient data from 12 randomised trials. Lancet. 2002;359:1011-1018.
35 Hassall TE, Mitchell AE, Ashley DM. Carboplatin chemotherapy for progressive intramedullary spinal cord low-grade gliomas in children. Three case studies and a review of the literature. Neurooncology. 2001;3:251-257.
36 Robertson PL, Zeltzer PM, Boyett JM, et al. Survival and prognostic factors following radiation therapy and chemotherapy for ependymomas in children. A report of the Children’s Cancer Group. J Neurosurg. 1998;88:695-703.
37 Doireau V, Grill J, Zerah M, et al. Chemotherapy for unresectable and recurrent intramedullary glial tumours in children. Brain tumours subcommittee of the french society of paediatric oncology (SFOP). Br J Cancer. 1999;81:835-840.
38 Chamberlain MC. Etoposide for recurrent spinal cord ependymoma. Neurology. 2002;58:1310-1311.
39 ICRU Report 50. Prescribing, Recording, and Reporting Photon Beam Therapy. Bethesda, Maryland, Nuclear Technology, 1993.
40 ICRU Report 62. Prescribing, recording and reporting photon beam therapy (supplement to ICRU Report 50). Besthesda, Maryland, Nuclear Technology, 1999.
41 Minehan KJ, Brown PD, Scheithauer BW, et al. Prognosis and treatment of spinal cord astrocytoma. Int J Radiat Oncol Biol Phys. 2009;73:727-733.
42 Rades D, Holtzhauer R, Baumann R, et al. Craniospinal axis irradiation in children. Treatment in supine position including field verification as a prerequisite for anaesthesia without intubation. Strahlenther Onkol. 1999;175:409-412.
43 Parker WA, Freeman CR. A simple technique for craniospinal radiotherapy in the supine position. Radiother Oncol. 2006;78:217-222.
44 Pai Panandiker A, Ning H, Likhacheva A, et al. Craniospinal irradiation with spinal IMRT to improve target homogeneity. Int J Radiat Oncol Biol Phys. 2007;68:1402-1409.
45 Yamada Y, Lovelock M, Bilsky MH. Image-guided intensity-modulated radiation therapy of spine tumors. Curr Neurol Neurosci Rep. 2006;6:207-211.
46 Ryu SI, Chang SD, Kim DH, et al. Image-guided hypo-fractionated stereotactic radiosurgery to spinal lesions. Neurosurgery. 2001;49:838-846.
47 Yin FF, Ryu S, Ajlouni M, et al. A technique of intensity-modulated radiosurgery (IMRS) for spinal tumors. Med Phys. 2002;29:2815-2822.
48 Rock JP, Ryu S, Yin FF, et al. The evolving role of stereotactic radiosurgery and stereotactic radiation therapy for patients with spine tumors. J Neurooncol. 2004;69:319-334.
49 Schultheiss TE, Kun LE, Ang KK, Stephens LC. Radiation response of the central nervous system. Int J Radiat Oncol Biol Phys. 1995;31:1093-1112.
50 Marcus RBJr, Million RR. The incidence of myelitis after irradiation of the cervical spinal cord. Int J Radiat Oncol Biol Phys. 1990;19:3-8.
51 Pieters RS, Niemierko A, Fullerton BC, Munzenrider JE. Cauda equina tolerance to high-dose fractionated irradiation. Int J Radiat Oncol Biol Phys. 2006;64:251-257.
52 Schoenthaler R, Castro JR, Petti PL, et al. Charged particle irradiation of sacral chordomas. Int J Radiat Oncol Biol Phys. 1993;26:291-298.
53 Fuller DB, Bloom JG. Radiotherapy for chordoma. Int J Radiat Oncol Biol Phys. 1988;15:331-339.
54 Marks LB, Yorke ED, Jackson A, et al. Use of normal tissue complication probability models in the clinic. Int J Radiat Oncol Biol Phys. 2010;76:S10-S19.
55 Kirkpatrick JP, van der Kogel AJ, Schultheiss TE. Radiation dose-volume effects in the spinal cord. Int J Radiat Oncol Biol Phys. 2010;76:S42-S49.
56 Cohen AR, Wisoff JH, Allen JC, Epstein F. Malignant astrocytomas of the spinal cord. J Neurosurg. 1989;70:50-54.
57 Hulshof MC, Menten J, Dito JJ, et al. Treatment results in primary intraspinal gliomas. Radiother Oncol. 1993;29:294-300.
58 Huddart R, Traish D, Ashley S, et al. Management of spinal astrocytoma with conservative surgery and radiotherapy. Br J Neurosurg. 1993;7:473-481.
59 Miller DC. Surgical pathology of intramedullary spinal cord neoplasms. J Neurooncol. 2000;47:189-194.
60 Jyothirmayi R, Madhavan J, Nair MK, Rajan B. Conservative surgery and radiotherapy in the treatment of spinal cord astrocytoma. J Neurooncol. 1997;33:205-211.
61 Chun HC, Schmidt-Ullrich RK, Wolfson A, et al. External beam radiotherapy for primary spinal cord tumors. J Neurooncol. 1990;9:211-217.
62 Lee HK, Chang EL, Fuller GN, et al. The prognostic value of neurologic function in astrocytic spinal cord glioma. Neuro Oncol. 2003;5:208-213.
63 Innocenzi G, Salvati M, Cervoni L, et al. Prognostic factors in intramedullary astrocytomas. Clin Neurol Neurosurg. 1997;99:1-5.
64 McLaughlin MP, Buatti JM, Marcus RBJr, et al. Outcome after radiotherapy of primary spinal cord glial tumors. Radiat Oncol Investig. 1998;6:276-280.
65 Milano MT, Johnson MD, Sul J, et al. Primary spinal cord glioma. A surveillance, epidemiology, and end results database study. J Neurooncol. 2009;98:83-92.
66 Abdel-Wahab M, Etuk B, Palermo J, et al. Spinal cord gliomas. A multi-institutional retrospective analysis. Int J Radiat Oncol Biol Phys. 2006;64:1060-1071.
67 Eroes CA, Zausinger S, Kreth FW, et al. Intramedullary low grade astrocytoma and ependymoma. Surgical results and predicting factors for clinical outcome. Acta Neurochir (Wien). 2010;152:611-618.
68 Garces-Ambrossi GL, McGirt MJ, Mehta VA, et al. Factors associated with progression-free survival and long-term neurological outcome after resection of intramedullary spinal cord tumors: analysis of 101 consecutive cases. J Neurosurg Spine. 2009;11:591-599.
69 Minehan KJ, Shaw EG, Scheithauer BW, et al. Spinal cord astrocytoma. Pathological and treatment considerations. J Neurosurg. 1995;83:590-595.
70 Scheinemann K, Bartels U, Huang A, et al. Survival and functional outcome of childhood spinal cord low-grade gliomas. Clinical article. J Neurosurg Pediatr. 2009;4:254-261.
71 Sandler HM, Papadopoulos SM, Thornton AFJr, Ross DA. Spinal cord astrocytomas. Results of therapy. Neurosurgery. 1992;30:490-493.
72 Allen JC, Aviner S, Yates AJ, et al. Treatment of high-grade spinal cord astrocytoma of childhood with “8-in-1” chemotherapy and radiotherapy. A pilot study of CCG-945. Children’s cancer group. J Neurosurg. 1998;88:215-220.
73 Epstein FJ, Farmer JP. Pediatric spinal cord tumor surgery. Neurosurg Clin North Am. 1990;1:569-590.
74 Przybylski GJ, Albright AL, Martinez AJ. Spinal cord astrocytomas. Long-term results comparing treatments in children. Childs Nerv Syst. 1997;13:375-382.
75 McGirt MJ, Goldstein IM, Chaichana KL, et al. Extent of surgical resection of malignant astrocytomas of the spinal cord: outcome analysis of 35 patients. Neurosurgery. 2008;63:55-60. discussion Neurosurgery 63:-61, 2008
76 Lunardi P, Licastro G, Missori P, et al. Management of intramedullary tumours in children. Acta Neurochir (Wien). 1993;120:59-65.
77 Robinson CG, Prayson RA, Hahn JF, et al. Long-term survival and functional status of patients with low-grade astrocytoma of spinal cord. Int J Radiat Oncol Biol Phys. 2005;63:91-100.
78 Harrop JS, Ganju A, Groff M, Bilsky M. Primary intramedullary tumors of the spinal cord. Spine (Phila Pa 1976). 2009;34:S69-S77.
79 Katoh N, Shirato H, Aoyama H, et al. Hypofractionated radiotherapy boost for dose escalation as a treatment option for high-grade spinal cord astrocytic tumor. J Neurooncol. 2006;78:63-69.
80 Zorlu F, Ozyigit G, Gurkaynak M, et al. Postoperative radiotherapy results in primary spinal cord astrocytomas. Radiother Oncol. 2005;74:45-48.
81 Epstein FJ, Farmer JP, Freed D. Adult intramedullary astrocytomas of the spinal cord. J Neurosurg. 1992;77:355-359.
82 Ciappetta P, Salvati M, Capoccia G, et al. Spinal glioblastomas. Report of seven cases and review of the literature. Neurosurgery. 1991;28:302-306.
83 Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987-996.
84 Chamberlain MC. Temozolomide for recurrent low-grade spinal cord gliomas in adults. Cancer. 2008;113:1019-1024.
85 Fine MJ, Kricheff II, Freed D, Epstein FJ. Spinal cord ependymomas. MR imaging features. Radiology. 1995;197:655-658.
86 Shaw EG, Evans RG, Scheithauer BW, et al. Radiotherapeutic management of adult intraspinal ependymomas. Int J Radiat Oncol Biol Phys. 1986;12:323-327.
87 Sgouros S, Malluci CL, Jackowski A. Spinal ependymomas—the value of postoperative radiotherapy for residual disease control. Br J Neurosurg. 1996;10:559-566.
88 Marks JE, Adler SJ. A comparative study of ependymomas by site of origin. Int J Radiat Oncol Biol Phys. 1982;8:37-43.
89 Linstadt DE, Wara WM, Leibel SA, et al. Postoperative radiotherapy of primary spinal cord tumors. Int J Radiat Oncol Biol Phys. 1989;16:1397-1403.
90 Sonneland PR, Scheithauer BW, Onofrio BM. Myxopapillary ependymoma. A clinicopathologic and immunocytochemical study of 77 cases. Cancer. 1985;56:883-893.
91 Rezai AR, Woo HH, Lee M, et al. Disseminated ependymomas of the central nervous system. J Neurosurg. 1996;85:618-624.
92 Whitaker SJ, Bessell EM, Ashley SE, et al. Postoperative radiotherapy in the management of spinal cord ependymoma. J Neurosurg. 1991;74:720-728.
93 Waldron JN, Laperriere NJ, Jaakkimainen L, et al. Spinal cord ependymomas. A retrospective analysis of 59 cases. Int J Radiat Oncol Biol Phys. 1993;27:223-229.
94 Schild SE, Nisi K, Scheithauer BW, et al. The results of radiotherapy for ependymomas. The Mayo Clinic experience. Int J Radiat Oncol Biol Phys. 1998;42:953-958.
95 Cervoni L, Celli P, Fortuna A, Cantore G. Recurrence of spinal ependymoma. Risk factors and long-term survival. Spine (Phila Pa 1976). 1994;19:2838-2841.
96 Healey EA, Barnes PD, Kupsky WJ, et al. The prognostic significance of postoperative residual tumor in ependymoma. Neurosurgery. 1991;28:666-671. discussion Neurosurgery 28:671-672, 1991
97 Clover LL, Hazuka MB, Kinzie JJ. Spinal cord ependymomas treated with surgery and radiation therapy. A review of 11 cases. Am J Clin Oncol. 1993;16:350-353.
98 Schiffer D, Chio A, Giordana MT, et al. Histologic prognostic factors in ependymoma. Childs Nerv Syst. 1991;7:177-182.
99 Lee TT, Gromelski EB, Green BA. Surgical treatment of spinal ependymoma and post-operative radiotherapy. Acta Neurochir (Wien). 1998;140:309-313.
100 Fornari M, Pluchino F, Solero CL, et al. Microsurgical treatment of intramedullary spinal cord tumours. Acta Neurochir Suppl (Wien). 1988;43:3-8.
101 Chan HS, Becker LE, Hoffman HJ, et al. Myxopapillary ependymoma of the filum terminale and cauda equina in childhood. Rreport of seven cases and review of the literature. Neurosurgery. 1984;14:204-210.
102 Schweitzer JS, Batzdorf U. Ependymoma of the cauda equina region. Diagnosis, treatment, and outcome in 15 patients. Neurosurgery. 1992;30:202-207.
103 Akyurek S, Chang EL, Yu TK, et al. Spinal myxopapillary ependymoma outcomes in patients treated with surgery and radiotherapy at M.D. Anderson cancer center. J Neurooncol. 2006;80:177-183.
104 Lin YH, Huang CI, Wong TT, et al. Treatment of spinal cord ependymomas by surgery with or without postoperative radiotherapy. J Neurooncol. 2005;71:205-210.
105 McGuire CS, Sainani KL, Fisher PG. Both location and age predict survival in ependymoma. A SEER study. Pediatr Blood Cancer. 2009;52:65-69.
106 Wahab SH, Simpson JR, Michalski JM, Mansur DB. Long term outcome with post-operative radiation therapy for spinal canal ependymoma. J Neurooncol. 2007;83:85-89.
107 Pica A, Miller R, Villa S, et al. The results of surgery, with or without radiotherapy, for primary spinal myxopapillary ependymoma. A retrospective study from the rare cancer network. Int J Radiat Oncol Biol Phys. 2009;74:1114-1120.
108 Gomez DR, Missett BT, Wara WM, et al. High failure rate in spinal ependymomas with long-term follow-up. Neurooncology. 2005;7:254-259.
109 Wen BC, Hussey DH, Hitchon PW, et al. The role of radiation therapy in the management of ependymomas of the spinal cord. Int J Radiat Oncol Biol Phys. 1991;20:781-786.
110 Evans AE, Anderson JR, Lefkowitz-Boudreaux IB, Finlay JL. Adjuvant chemotherapy of childhood posterior fossa ependymoma. Cranio-spinal irradiation with or without adjuvant CCNU, vincristine, and prednisone. A Children’s Cancer Group study. Med Pediatr Oncol. 1996;27:8-14.
111 Goldwein JW, Leahy JM, Packer RJ, et al. Intracranial ependymomas in children. Int J Radiat Oncol Biol Phys. 1990;19:1497-1502.
112 Goldwein JW, Glauser TA, Packer RJ, et al. Recurrent intracranial ependymomas in children. Survival, patterns of failure, and prognostic factors. Cancer. 1990;66:557-563.
113 Duffner PK, Horowitz ME, Krischer JP, et al. Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors. N Engl J Med. 1993;328:1725-1731.
114 Grill J, Le Deley MC, Gambarelli D, et al. Postoperative chemotherapy without irradiation for ependymoma in children under 5 years of age. A multicenter trial of the French Society of Pediatric Oncology. J Clin Oncol. 2001;19:1288-1296.
115 Mirimanoff RO, Dosoretz DE, Linggood RM, et al. Meningioma: analysis of recurrence and progression following neurosurgical resection. J Neurosurg. 1985;62:18-24.
116 Solero CL, Fornari M, Giombini S, et al. Spinal meningiomas. Review of 174 operated cases. Neurosurgery. 1989;25:153-160.
117 Helseth A, Mork SJ. Primary intraspinal neoplasms in Norway, 1955 to 1986. A population-based survey of 467 patients. J Neurosurg. 1989;71:842-845.
118 Gezen F, Kahraman S, Canakci Z, Beduk A. Review of 36 cases of spinal cord meningioma. Spine (Phila Pa 1976). 2000;25:727-731.
119 Cohen-Gadol AA, Zikel OM, Koch CA, et al. Spinal meningiomas in patients younger than 50 years of age. A 21-year experience. J Neurosurg. 2003;98:258-263.
120 Roux FX, Nataf F, Pinaudeau M, et al. Intraspinal meningiomas. Review of 54 cases with discussion of poor prognosis factors and modern therapeutic management. Surg Neurol. 1996;46:458-463. discussion Surg Neurol 46:463-464, 1996
121 Levy WJJr, Bay J, Dohn D. Spinal cord meningioma. J Neurosurg. 1982;57:804-812.
122 Schiebe ME, Hoffmann W, Kortmann RD, Bamberg M. Radiotherapy in recurrent malignant meningiomas with multiple spinal manifestations. Acta Oncol. 1997;36:88-90.
123 Goldsmith BJ, Wara WM, Wilson CB, Larson DA. Postoperative irradiation for subtotally resected meningiomas. A retrospective analysis of 140 patients treated from 1967 to 1990. J Neurosurg. 1994;80:195-201.
124 Ryu S, Fang Yin F, Rock J, et al. Image-guided and intensity-modulated radiosurgery for patients with spinal metastasis. Cancer. 2003;97:2013-2018.
125 Gerszten PC, Burton SA, Ozhasoglu C, et al. Radiosurgery for benign intradural spinal tumors. Neurosurgery. 2008;62:887-895. discussion Neurosurgery 62:895-896, 2008
126 Schrell UM, Rittig MG, Anders M, et al. Hydroxyurea for treatment of unresectable and recurrent meningiomas. II. Decrease in the size of meningiomas in patients treated with hydroxyurea. J Neurosurg. 1997;86:840-844.
127 Noel G, Habrand JL, Jauffret E, et al. Radiation therapy for chordoma and chondrosarcoma of the skull base and the cervical spine. Prognostic factors and patterns of failure. Strahlenther Onkol. 2003;179:241-248.
128 Chetiyawardana AD. Chordoma. Results of treatment. Clin Radiol. 1984;35:159-161.
129 Saxton JP. Chordoma. Int J Radiat Oncol Biol Phys. 1981;7:913-915.
130 Klekamp J, Samii M. Spinal chordomas—results of treatment over a 17-year period. Acta Neurochir (Wien). 1996;138:514-519.
131 O’Neill P, Bell BA, Miller JD, et al. Fifty years of experience with chordomas in southeast Scotland. Neurosurgery. 1985;16:166-170.
132 Rich TA, Schiller A, Suit HD, Mankin HJ. Clinical and pathologic review of 48 cases of chordoma. Cancer. 1985;56:182-187.
133 Bethke KP, Neifeld JP, Lawrence WJr. Diagnosis and management of sacrococcygeal chordoma. J Surg Oncol. 1991;48:232-238.
134 Kaiser TE, Pritchard DJ, Unni KK. Clinicopathologic study of sacrococcygeal chordoma. Cancer. 1984;53:2574-2578.
135 Benk V, Liebsch NJ, Munzenrider JE, et al. Base of skull and cervical spine chordomas in children treated by high-dose irradiation. Int J Radiat Oncol Biol Phys. 1995;31:577-581.
136 Chetty R, Levin CV, Kalan MR. Chordoma. A 20-year clinicopathologic review of the experience at Groote Schuur Hospital, Cape Town. J Surg Oncol. 1991;46:261-264.
137 Markwalder TM, Markwalder RV, Robert JL, Krneta A. Metastatic chordoma. Surg Neurol. 1979;12:473-478.
138 Higinbotham NL, Phillips RF, Farr HW, Hustu HO. Chordoma. Thirty-five-year study at Memorial Hospital. Cancer. 1967;20:1841-1850.
139 Suit HD, Goitein M, Munzenrider J, et al. Definitive radiation therapy for chordoma and chondrosarcoma of base of skull and cervical spine. J Neurosurg. 1982;56:377-385.
140 Terahara A, Niemierko A, Goitein M, et al. Analysis of the relationship between tumor dose inhomogeneity and local control in patients with skull base chordoma. Int J Radiat Oncol Biol Phys. 1999;45:351-358.
141 Borba LA, Al-Mefty O, Mrak RE, Suen J. Cranial chordomas in children and adolescents. J Neurosurg. 1996;84:584-591.
142 Jawad MU, Scully SP. Surgery significantly improves survival in patients with chordoma. Spine (Phila Pa 1976). 2010;35:117-123.
143 Ozaki T, Hillmann A, Winkelmann W. Surgical Treatment of sacrococcygeal chordoma. J Surg Oncol. 1997;64:274-279.
144 Magrini SM, Papi MG, Marletta F, et al. Chordoma-natural history, treatment and prognosis. The Florence Radiotherapy Department experience (1956-1990) and a critical review of the literature. Acta Oncol. 1992;31:847-851.
145 Romero J, Cardenes H, la Torre A, et al. Chordoma: results of radiation therapy in eighteen patients. Radiother Oncol. 1993;29:27-32.
146 Amichetti M, Cianchetti M, Amelio D, et al. Proton therapy in chordoma of the base of the skull. A systematic review. Neurosurg Rev. 2009;32:403-416.
147 Cummings BJ, Hodson DI, Bush RS. Chordoma. The results of megavoltage radiation therapy. Int J Radiat Oncol Biol Phys. 1983;9:633-642.
148 Catton C, O’Sullivan B, Bell R, et al. Chordoma. Long-term follow-up after radical photon irradiation. Radiother Oncol. 1996;41:67-72.
149 Park L, Delaney TF, Liebsch NJ, et al. Sacral chordomas. Impact of high-dose proton/photon-beam radiation therapy combined with or without surgery for primary versus recurrent tumor. Int J Radiat Oncol Biol Phys. 2006;65:1514-1521.
150 Austin-Seymour M, Munzenrider JE, Goitein M, et al. Progress in low-LET heavy particle therapy. Intracranial and paracranial tumors and uveal melanomas. Radiat Res Suppl. 1985;8:S219-S226.
151 Berson AM, Castro JR, Petti P, et al. Charged particle irradiation of chordoma and chondrosarcoma of the base of skull and cervical spine. The Lawrence Berkeley Laboratory experience. Int J Radiat Oncol Biol Phys. 1988;15:559-565.
152 Hug EB, Loredo LN, Slater JD, et al. Proton radiation therapy for chordomas and chondrosarcomas of the skull base. J Neurosurg. 1999;91:432-439.
153 Munzenrider JE, Liebsch NJ. Proton therapy for tumors of the skull base. Strahlenther Onkol. 1999;175(Suppl 2):57-63.
154 Noel G, Habrand JL, Mammar H, et al. Combination of photon and proton radiation therapy for chordomas and chondrosarcomas of the skull base. The Centre de Protontherapie D’Orsay experience. Int J Radiat Oncol Biol Phys. 2001;51:392-398.
155 Rutz HP, Weber DC, Sugahara S, et al. Extracranial chordoma. Outcome in patients treated with function-preserving surgery followed by spot-scanning proton beam irradiation. Int J Radiat Oncol Biol Phys. 2007;67:512-520.
156 Torres MA, Chang EL, Mahajan A, et al. Optimal treatment planning for skull base chordoma. Photons, protons, or a combination of both? Int J Radiat Oncol Biol Phys. 2009;74:1033-1039.
157 Castro JR, Linstadt DE, Bahary JP, et al. Experience in charged particle irradiation of tumors of the skull base. 1977-1992. Int J Radiat Oncol Biol Phys. 1994;29:647-655.
158 Chang SD, Martin DP, Lee E, Adler JRJr. Stereotactic radiosurgery and hypofractionated stereotactic radiotherapy for residual or recurrent cranial base and cervical chordomas. Neurosurg Focus. 2001;10:E5.
159 Bernstein M, Gutin PH. Interstitial irradiation of skull base tumours. Can J Neurol Sci. 1985;12:366-370.
160 Gutin PH, Leibel SA, Hosobuchi Y, et al. Brachytherapy of recurrent tumors of the skull base and spine with iodine-125 sources. Neurosurgery. 1987;20:938-945.
161 Kumar PP, Good RR, Skultety FM, Leibrock LG. Local control of recurrent clival and sacral chordoma after interstitial irradiation with iodine-125. New techniques for treatment of recurrent or unresectable chordomas. Neurosurgery. 1988;22:479-483.
162 Camins MB, Duncan AW, Smith J, Marcove RC. Chondrosarcoma of the spine. Spine (Phila Pa 1976). 1978;3:202-209.
163 York JE, Berk RH, Fuller GN, et al. Chondrosarcoma of the spine. 1954 to 1997. J Neurosurg. 1999;90:73-78.
164 Shives TC, McLeod RA, Unni KK, Schray MF. Chondrosarcoma of the spine. J Bone Joint Surg Am. 1989;71:1158-1165.
165 Bergh P, Gunterberg B, Meis-Kindblom JM, Kindblom LG. Prognostic factors and outcome of pelvic, sacral, and spinal chondrosarcomas. A center-based study of 69 cases. Cancer. 2001;91:1201-1212.
166 Sheth DS, Yasko AW, Johnson ME, et al. Chondrosarcoma of the pelvis. Prognostic factors for 67 patients treated with definitive surgery. Cancer. 1996;78:745-750.
167 Mostafavi H, Lennarson PJ, Traynelis VC. Granulocytic sarcoma of the spine. Neurosurgery. 2000;46:78-83. discussion Neurosurgery 46:84, 2000
168 Roonprapunt C, Silvera VM, Setton A, et al. Surgical management of isolated hemangioblastomas of the spinal cord. Neurosurgery. 2001;49:321-327. discussion Neurosurgery 49:327-328, 2001
169 Lonser RR, Weil RJ, Wanebo JE, et al. Surgical management of spinal cord hemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg. 2003;98:106-116.
170 Lee DK, Choe WJ, Chung CK, Kim HJ. Spinal cord hemangioblastoma. Surgical strategy and clinical outcome. J Neurooncol. 2003;61:27-34.
171 Cristante L, Herrmann HD. Surgical management of intramedullary hemangioblastoma of the spinal cord. Acta Neurochir (Wien). 1999;141:333-339. discussion Acta Neurochir (Wien) 141:339-340, 1999
172 Jawahar A, Kondziolka D, Garces YI, et al. Stereotactic radiosurgery for hemangioblastomas of the brain. Acta Neurochir (Wien). 2000;142:641-644. discussion Acta Neurochir (Wien) 142:644-645, 2000
173 Moss JM, Choi CY, Adler JRJr, et al. Stereotactic radiosurgical treatment of cranial and spinal hemangioblastomas. Neurosurgery. 2009;65:79-85.
174 Dyck P. Intramedullary lipoma. Diagnosis and treatment. Spine (Phila Pa 1976). 1992;17:979-981.
175 Lee M, Rezai AR, Abbott R, et al. Intramedullary spinal cord lipomas. J Neurosurg. 1995;82:394-400.
176 Razack N, Jimenez OF, Aldana P, Ragheb J. Intramedullary holocord lipoma in an athlete: case report. Neurosurgery. 1998;42:394-396. discussion Neurosurgery 42:396-397, 1998
177 Lyons MK, O’Neill BP, Kurtin PJ, Marsh WR. Diagnosis and management of primary spinal epidural non-Hodgkin’s lymphoma. Mayo Clin Proc. 1996;71:453-457.
178 Ishikawa H, Hasegawa M, Tamaki Y, et al. Comparable outcomes of radiation therapy without high-dose methotrexate for patients with primary central nervous system lymphoma. Jpn J Clin Oncol. 2003;33:443-449.
179 Martin AJ, Sharr MM, Teddy PJ, et al. Neurocytoma of the thoracic spinal cord. Acta Neurochir (Wien). 2002;144:823-828.
180 Tatter SB, Borges LF, Louis DN. Central neurocytomas of the cervical spinal cord. Report of two cases. J Neurosurg. 1994;81:288-293.
181 Oliveira AM, Scheithauer BW, Salomao DR, et al. Primary sarcomas of the brain and spinal cord. A study of 18 cases. Am J Surg Pathol. 2002;26:1056-1063.
182 Merimsky O, Lepechoux C, Terrier P, et al. Primary sarcomas of the central nervous system. Oncology. 2000;58:210-214.
183 Shadmehr MB, Gaissert HA, Wain JC, et al. The surgical approach to “dumbbell tumors” of the mediastinum. Ann Thorac Surg. 2003;76:1650-1654.
184 Konno S, Yabuki S, Kinoshita T, Kikuchi S. Combined laminectomy and thoracoscopic resection of dumbbell-type thoracic cord tumor. Spine (Phila Pa 1976). 2001;26:E130-E134.
185 Seppala MT, Haltia MJ. Spinal malignant nerve-sheath tumor or cellular schwannoma? A striking difference in prognosis. J Neurosurg. 1993;79:528-532.
186 Hejazi N, Witzmann A. Spinal intramedullary teratoma with exophytic components. Report of two cases and review of the literature. Neurosurg Rev. 2003;26:113-116.
187 Fox MW, Onofrio BM. The natural history and management of symptomatic and asymptomatic vertebral hemangiomas. J Neurosurg. 1993;78:36-45.
188 Cross JJ, Antoun NM, Laing RJ, Xuereb J. Imaging of compressive vertebral haemangiomas. Eur Radiol. 2000;10:997-1002.
189 Djindjian M, Nguyen JP, Gaston A, et al. Multiple vertebral hemangiomas with neurological signs. Case report. J Neurosurg. 1992;76:1025-1028.
190 Heyd R, Strassmann G, Filipowicz I, et al. [Radiotherapy in vertebral hemangioma.]. Rontgenpraxis. 2001;53:208-220.
191 Rades D, Bajrovic A, Alberti W, Rudat V. Is there a dose-effect relationship for the treatment of symptomatic vertebral hemangioma? Int J Radiat Oncol Biol Phys. 2003;55:178-181.
192 Esik O, Csere T, Stefanits K, et al. A review on radiogenic Lhermitte’s sign. Pathol Oncol Res. 2003;9:115-120.
193 Nieder C, Ataman F, Price RE, Ang KK. Radiation myelopathy. New perspective on an old problem. Radiat Oncol Investig. 1999;7:193-203.
194 Okada S, Okeda R. Pathology of radiation myelopathy. Neuropathology. 2001;21:247-265.
195 Schultheiss TE, Higgins EM, El-Mahdi AM. The latent period in clinical radiation myelopathy. Int J Radiat Oncol Biol Phys. 1984;10:1109-1115.
196 Wang PY, Shen WC, Jan JS. MR imaging in radiation myelopathy. AJNR Am J Neuroradiol. 1992;13:1049-1055. discussion AJNR Am J Neuroradiol 13:1056-1058, 1992
197 Liu CY, Yim BT, Wozniak AJ. Anticoagulation therapy for radiation-induced myelopathy. Ann Pharmacother. 2001;35:188-191.
198 Feldmeier JJ, Lange JD, Cox SD, et al. Hyperbaric oxygen as prophylaxis or treatment for radiation myelitis. Undersea Hyperb Med. 1993;20:249-255.
199 Schultheiss TE, Stephens LC, Peters LJ. Survival in radiation myelopathy. Int J Radiat Oncol Biol Phys. 1986;12:1765-1769.
200 Mayfield JK. Postradiation spinal deformity. Orthop Clin North Am. 1979;10:829-844.
201 Garcia DM. Primary spinal cord tumors treated with surgery and postoperative irradiation. Int J Radiat Oncol Biol Phys. 1985;11:1933-1939.
202 Nadeem SQ, Feun LG, Bruce-Gregorios JH, Green B. Post radiation sarcoma (malignant fibrous histiocytoma) of the cervical spine following ependymoma (a case report). J Neurooncol. 1991;11:263-268.
203 Rappaport ZH, Loven D, Ben-Aharon U. Radiation-induced cerebellar glioblastoma multiforme subsequent to treatment of an astrocytoma of the cervical spinal cord. Neurosurgery. 1991;29:606-608.
204 Meade TJ, Taylor AK, Bull SR. New magnetic resonance contrast agents as biochemical reporters. Curr Opin Neurobiol. 2003;13:597-602.
205 Ang KK, Jiang GL, Feng Y, et al. Extent and kinetics of recovery of occult spinal cord injury. Int J Radiat Oncol Biol Phys. 2001;50:1013-1020.
206 Benzil DL, Saboori M, Mogilner AY, et al. Safety and efficacy of stereotactic radiosurgery for tumors of the spine. J Neurosurg. 2004;101(Suppl 3):413-418.
207 Jeremic B, Djuric L, Mijatovic L. Incidence of radiation myelitis of the cervical spinal cord at doses of 5500 cGy or greater. Cancer. 1991;68:2138-2141.
208 Cohen L, Creditor M. An iso-effect table for radiation tolerance of the human spinal cord. Int J Radiat Oncol Biol Phys. 1981;7:961-966.
209 Leber KA, Bergloff J, Pendl G. Dose-response tolerance of the visual pathways and cranial nerves of the cavernous sinus to stereotactic radiosurgery. J Neurosurg. 1998;88:43-50.
210 Macbeth FR, Wheldon TE, Girling DJ, et al. Radiation myelopathy. Estimates of risk in 1048 patients in three randomized trials of palliative radiotherapy for non-small cell lung cancer. The Medical Research Council Lung Cancer Working Party. Clin Oncol (R Coll Radiol). 1996;8:176-181.
211 McCunniff AJ, Liang MJ. Radiation tolerance of the cervical spinal cord. Int J Radiat Oncol Biol Phys. 1989;16:675-678.
212 Nieder C, Milas L, Ang KK. Tissue tolerance to reirradiation. Semin Radiat Oncol. 2000;10:200-209.
213 Niewald M, Feldmann U, Feiden W, et al. Multivariate logistic analysis of dose-effect relationship and latency of radiomyelopathy after hyperfractionated and conventionally fractionated radiotherapy in animal experiments. Int J Radiat Oncol Biol Phys. 1998;41:681-688.
214 Schultheiss TE. The radiation dose-response of the human spinal cord. Int J Radiat Oncol Biol Phys. 2008;71:1455-1459.
* See references 23, 34, 35, 36, 37, 38.
* References 11, 12, 13–16, 29, 30, 57, 59.
† References 12, 14, 17, 41, 58, 62–65, 66, 67, 68.
‡ References 17, 22, 23, 41, 65, 71–73.
§ References 11, 22, 24, 41, 58, 63, 70, 71, 74.
* References 17, 18, 20, 22–24, 57, 58, 76.
† References 12, 16, 17, 20, 41, 64, 66, 78.
‡ References 22, 41, 58, 60, 62, 66, 70, 74, 77, 80.
§ References 21, 23, 24, 41, 56, 58, 72, 81.
* References 26, 27, 67, 68, 86, 87, 90, 92–95.
* References 19, 20, 25, 26, 28, 30, 99, 100.
† References 15, 26, 27, 57, 87, 93, 94.
‡ References 66, 86, 87, 92, 103, 104, 105, 106, 107.
§ References 31, 115, 116, 117, 118, 119.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 28 Spinal Cord Tumors
Epidemiology and Etiology
Primary spinal cord tumors comprise 3% to 4% of all primary central nervous system (CNS) tumors in adults.1 Age-adjusted incidence rates are slightly higher for men than women, 0.68 versus 0.61 per 100,000 person-years, respectively.1 Most of these tumors present as extramedullary tumors (70%), whereas intramedullary tumors are less common (30%). Approximately 15% to 25% of spinal tumors involve the cervical spine, including the foramen magnum; 50% to 55% involve the thoracic spinal canal; and 25% to 30% involve the lumbosacral spine.
In the pediatric population, spinal cord tumors are slightly more common, 5% to 6% of all primary CNS tumors.1 Although ependymomas and nerve sheath tumors are the most common histologic types, gliomas make up a larger percentage of tumors in children as compared with adults1 (Table 28-1).
TABLE 28-1 Distribution of Primary Spinal Cord Tumors by Histologic Type (CBTRUS 2010)
| Histologic Type | Adult Distribution | Pediatric Distribution |
|---|---|---|
| Nerve sheath tumor | 40.5% | 15.7% |
| Glial origin | ||
| Ependymoma | 33.6% | 24.1% |
| Pilocytic astrocytoma | 1.4% | 12.6% |
| Other glioma | 4.1% | 13.3% |
| Meningeal tumor | 6.1% | 10.1% |
| Hemangioma | 2.6% | NA |
NA, not available.
Biologic Characteristics and Molecular Biology
The diversity of primary spinal axis tumors partly results from the large spectrum of phenotypically distinct cells in the axis that are capable of neoplastic transformation.2 The genetic and molecular bases of spinal cord tumors are not yet well understood and vary depending on histologic findings.3 Most tumors are histologically benign but can cause significant morbidity by direct compression of important neural structures.
Anatomy, Pathology, and Pathways of Spread
Anatomy
The spinal cord is responsible for transmitting motor and sensory neural signals between the peripheral nervous system and the brain and contains its own independent pathways for coordinating certain reflexes (Fig. 28-1). Functionally, the cord is divided into 31 segments. At each level, a pair of spinal nerves exits: 8 cervical, 12 thoracic, 5 lumbar, and 5 sacral pairs and 1 coccygeal pair. In the cervical region, the spinal nerves exit above the corresponding vertebrae, whereas from the thoracic region and downward, the spinal nerves exit below the equivalent named vertebrae. Although the vertebral column continues to lengthen until adulthood, the canal and the cord are actually much shorter than the column because both stop lengthening at approximately 4 years of age. This growth differential is responsible for the formation of the cauda equina, a collection of the lower lumbar, sacral, and coccygeal nerves that fills the lower spinal column beyond the conus medullaris.
Tumors are classified based on their location (Fig. 28-2). Intradural tumors include both intramedullary and extramedullary tumors. Intramedullary tumors develop from the cells that make up the spinal cord and cauda equina, and extramedullary tumors arise from the supportive tissues that lie outside the spinal cord and cauda equina, such as the meninges, vascular supply, and connective tissue. Extradural tumors arise from the vertebral canal itself or supportive tissues surrounding the dura.
Pathology and Pathways of Spread
The overall and anatomic distributions of histologic types are listed in Tables 28-1 and 28-2. Intradural extramedullary schwannoma (neurilemmoma) and meningioma are the most common intradural tumors, although occasionally they may present as extradural tumors. Other intradural extramedullary tumors include epidermoid and drop metastases of intracranial primary tumors such as medulloblastoma. The most common intramedullary tumors are those derived from glial precursors, most frequently, ependymomas and astrocytomas of low to intermediate grade. Nonneoplastic conditions may mimic spinal cord tumors (Fig. 28-3), including acute transverse myelitis, Angiostrongylus cantonensis infection, infectious meningitis, intradural disc hernia, pseudotumor, sarcoidosis, and tuberculosis.4–8
TABLE 28-2 Anatomic Distribution of Spinal Tumors by Histologic Type
| Extradural Tumors |
Intradural Extramedullary Tumors | Intradural Intramedullary Tumors |
|---|---|---|
The predominant mode of spread of primary tumors is by direct extension. Spread along the subarachnoid space with spinal or cranial involvement of the meninges may also occur. One should not always assume that the spinal cord tumor found on imaging is the primary lesion. Although CSF seeding is uncommon, supratentorial tumors that show a tendency toward leptomeningeal metastases include 1.2% of glioblastomas and 1.5% of anaplastic gliomas.9 There are no lymphatics in the spinal cord, and hematogenous spread is rare.
Clinical Manifestations, Patient Evaluation, and Staging
The clinical presentation of tumors of the spinal axis is a function of the local anatomy (see Fig. 28-1). Within the spinal canal, there exists a well-defined extradural space containing epidural fat and blood vessels. The extradural space communicates with adjacent extraspinal compartments via the intervertebral foramina.
Clinical Workup
The diagnostic approach to primary spinal cord tumors is shown in Figure 28-4. In addition to focusing on neurologic symptoms, the history should include an evaluation of the patient’s functional status and symptoms, which may suggest metastatic disease or nonmalignant causes of spinal cord tumors.
Sensory impairment often helps to localize the level of the tumor based on loss of dermatome function, although the upper level of impaired long-tract function may be several segments below the actual tumor (Fig. 28-5). A lesion involving the lateral spinothalamic tract causes numbness, paresthesias, and decreased temperature sensation over the contralateral limb or trunk below the lesion. This produces the classic finding of a sensory level, which is best demonstrated with a pin or a large metal or cold object to test temperature sensation and with observation for absence of perspiration. A lesion in the posterior column causes an ataxic or tabetic gait. Standing posture is affected with the eyes closed (Romberg’s sign). Paresthesias can occur below the level of the lesion.
Figure 28-5 Anterior (A) and posterior (B) views of the dermatomes. C, cervical; T, thoracic; L, lumbar; S, sacral.
CREDIT LINE: Adapted from Netter FH, Freidberg SR, Baker RA: Disorders of spinal cord, nerve root, and plexus. In Netter FH, Jones HR Jr, Dingle RV (eds): The Ciba Collection of Medical Illustrations, Vol 1. Nervous System, Part II. West Caldwell, New Jersey, Ciba-Geigy, 1986, pp 181-189; Keegan JJ, Garrett FD: The segmental distribution of cutaneous nerves in the limbs of man. Anat Rec 102:409, 1948.
Primary Therapy
General Principles of Surgery
Surgery has traditionally been the mainstay of therapy for spinal cord tumors.10 As modern diagnostic and therapeutic techniques such as MRI, the operating microscope, microsurgical tools, and intraoperative neurophysiologic assessment have become available, more aggressive surgical intervention has become a more prevalent treatment strategy.11 This section will discuss general principles of surgery for CNS lesions, depending on anatomic location within the spinal cord. The outcome of surgery and its role in multidisciplinary management are described in the sections for each individual type of tumor. Table 28-3 shows an overview of the outcomes of the most common spinal cord tumors by treatment modality.
Surgical Techniques, Postoperative Management, and Complications
Surgery of Intramedullary Tumors
The most common intramedullary tumors are ependymomas and astrocytomas. Modern series advocate maximum safe resection of intramedullary spinal tumors. The ability to resect these tumors completely varies widely, from less than 10% to as much as 86%, depending on the histologic type.12,13–19 Most completely resectable lesions are WHO grade I tumors because higher-grade tumors are defined by their infiltrative potential.16,20–24 The gross total resection (GTR) rates for ependymomas range from 19% to 100%, whereas 0% to 43 % of astrocytomas are resectable.25–28 For infiltrative astrocytomas, the role of surgery is most frequently limited to biopsy for diagnosis. Laminectomy offers limited functional benefit by decompressing the spinal cord in the case of bulky infiltrative tumors. Functional postoperative outcomes may be excellent in selected cases, with the majority of patients (72% to 79%) experiencing either improvement or stabilization of their preoperative presenting symptoms, often after transient postsurgical worsening.12,19,29,30 The incidence of total, permanent postoperative paralysis is reported at 1%.11 Paralysis is often the result of unpredictable, sporadic vascular events.
Intramedullary tumors are almost always approached through a laminectomy exposure with the patient in the prone position.11 After the dura is opened, a longitudinal myelotomy is made over the widened region of the spinal cord, where imaging demonstrates the closest approximation of the tumor to the surface and the incision is deepened several millimeters until the tumor surface is reached. Infiltrative tumors with poorly defined dissection planes cannot be removed completely and, therefore, biopsy for diagnosis is often the safest course of action. When there is an exophytic component to the tumor, maximum safe debulking is typically performed.
Complications of Surgery
In patients who are neurologically intact before surgery, the incidence of significant acute morbidity is generally below 5% for extramedullary tumors and as low as 5% for carefully selected patients with intramedullary tumors.11 Spinal deformities, including scoliosis and kyphosis, as well as infection and CSF leakage, can occur.31,32 The development of postlaminectomy spinal deformities is a serious postoperative complication, occurring in up to 40% of pediatric and adolescent patients, and may be observed as soon as 8 months postoperatively.33
General Principles of Radiation Therapy
Because of the potential for growth and developmental side effects, the use of irradiation in children and infants with spinal cord tumors is controversial. Some authors do not recommend postoperative radiation therapy, and suggest that observation with close imaging follow-up and re-resection at the time of second occurrence is a better approach.12 With this strategy, radiation therapy may be avoided or used only for children with multiply recurrent tumors.
General Principles of Chemotherapy
The efficacy of chemotherapy may be limited by the intact blood–nervous system barrier. Regional drug delivery may be in the form of intracerebrospinal fluid therapy, intra-arterial infusion, and intratumoral therapy. Extramedullary spinal tumors gain their blood supply from meningeal blood vessels that are significantly more permeable than those of the brain, and chemotherapy may be of more use for these tumors. Drugs that have some efficacy in treating primary CNS malignant tumors and that are thought to cross the blood-brain barrier have been used in the treatment of malignant glial tumors of the spinal cord; however, no clear benefit from the use of chemotherapy has been demonstrated thus far.*
Irradiation Techniques and Tolerance
Definitions of Treatment Volumes
Three-dimensional treatment planning based on International Commission on Radiation Units (ICRU) volume definitions is the norm.39,40 The gross target volume (GTV) represents the grossly visible disease burden. Typically, this is the T1-enhancing abnormality on MRI or the nonenhancing tumor seen on T2 or FLAIR images. If there is no residual abnormality after a surgical resection, the tumor resection cavity is defined to be the GTV. Surrounding edema is not considered part of the GTV. The clinical target volume (CTV) is the T2 or FLAIR abnormality (which does include edema) on MRI as well as any areas potentially containing microscopic disease. Suggested definitions and doses to be delivered to the GTV and CTV are listed by histologic diagnosis in Table 28-4. The planning target volume (PTV) adds a dosimetric margin that takes into account uncertainties in daily treatment setup and physiologic variations that are difficult or impossible to control, such as (potential) fluctuations in the mass effect from cord edema that may occur over the course of treatment. Organs at risk typically include the thyroid and salivary glands, esophagus, lungs, heart, stomach, small bowel, liver, kidneys, bladders, ovaries, testicles, and uninvolved portions of the spinal cord itself.
Treatment Techniques
The most common approaches include a single posterior field (PA), opposed lateral fields, a PA field with opposed laterals, and oblique wedge-pair fields.41 They are typically designed to treat the CTV and GTV. For tumors in the cervicothoracic region, a split-beam approach is sometimes used. The central axis is placed just above the shoulders. Opposed lateral fields are used to treat the upper spine, whereas a PA field is used for the area of the spine below. Tumors in the thoracic region are often treated with a three-field approach using a PA field and opposed lateral beams. In the lumbar region, care must be taken to minimize the dose to the kidneys; a four-field approach using AP/PA and opposed lateral beams with the AP/PA beams preferentially weighted may be useful.
Certain neoplasms require treatment of the entire craniospinal axis. Several modifications of this approach are used in clinical practice (Fig. 28-6). Patients may be treated either in the supine or prone position, often in an immobilization cast to ensure daily positional reproducibility.42,43 The intracranial contents, including the upper one or two segments of the cervical cord, are treated through opposed lateral fields. Customized blocks protect the normal head and neck tissues from the primary radiation beam. The spine is treated through one or two posterior fields, depending on the size of the patient. In one method, the collimator for the lateral cranial fields is angled to match the divergence of the upper border of the adjacent spinal field, and the treatment couch is angled so that the inferior border of the cranial field is perpendicular to the superior edge of the spinal field. Alternatively, one may dispense with collimator and couch angles by calculating appropriate gaps. The gap is calculated so that the 50% isodose lines meet at the level of the anterior spinal cord. All junction lines are moved 0.5 to 1 cm every 8 to 10 Gy to avoid overdosing or underdosing segments of the cord. This is accomplished by shortening the inferior margin of the lateral cranial fields, symmetrically lengthening the superior and inferior margins of the posterior spinal field, and shortening the cranial margin of the caudal spinal field; a fixed block is placed at the inferior margin of the caudal spinal field to keep the lower margin of the irradiated volume at the same location.
IMRT is a treatment delivery method that may be used to further optimize the dose distribution. Because multiple critical sensitive organs are located near the spinal cord, improved dose distribution should allow the dose to these structures to be minimized. Because most spinal cord tumors typically recur locally, IMRT should allow the exploration of anatomic/biologic treatment planning and delivery in order to optimize different doses to different cell populations within heterogeneous volumes. The use of image guidance may also allow for reduced margins for sparing of critical structures. The use of IMRT is currently under investigation at multiple institutions.44,45
Stereotactic radiosurgery (SRS) and body radiotherapy (SBRT) deliver a highly conformal high dose of radiotherapy with stereotactic guidance. Doses may be delivered as a single dose (SRS) or as a short fractionated course (SBRT). Potential advantages include patient convenience with a shorter treatment course and the steep dose gradient at the edge of the field, which may allow for greater tissue sparing. SRS for primary spinal cord tumors remains experimental at this time but is an active area of research.46–48
Tolerance of the Spinal Cord and Lumbosacral Nerve Roots
A dose of 45 to 50.4 Gy in 25 to 28 fractions over 5 or weeks is usually considered to be safe. The risk of myelopathy is less than 1%, well below the steep portion of the dose-response curve.46,49,50 It is estimated that with conventionally fractionated irradiation (1.8 to 2 Gy per fraction in five fractions per week), at 5 years the incidence of myelopathy is 5% for doses in the range of 57 to 61 Gy (tolerance dose TD5/5) and 50% for doses of 68 to 73 Gy (TD50/5).49 There is no convincing evidence that the cervical and thoracic cord differ in their radiosensitivity and there appears to be little change in tolerance with variations in the length of cord irradiated.49 Table 28-5 shows a range of isomorbid fractionation schemes, all of which carry a 5% risk of radiation myelopathy.
TABLE 28-5 Fractionation Schemes with a 5% Risk of Radiation-Induced Spinal Cord Myelopathy
| Dose per Fraction (Gy) | No. Fractions | Total Dose (Gy) |
|---|---|---|
| 2 | 29 | 58 |
| 3 | 13 | 39 |
| 3.3 | 11 | 33 |
| 4 | 7 | 28 |
| 5 | 5 | 25 |
| 10 | 1 | 10 |
Data from references 50, 98, 116, 206–209, 210, 211–214.
The tolerance of the lumbosacral nerve roots appears to be somewhat higher than that of the spinal cord. Most series report a 0% complication rate if patients are treated to doses of 70 Gy (or equivalent) as long as fraction sizes are kept at or below 2 Gy.51–53
The Quantitative Analysis of Normal Tissue Effects in the Clinic (QUANTEC) reviewed data from preclinical, conventional, reirradiation, and SBRT studies in order to refine normal tissue dose/volume tolerance guidelines.54 Based on this, a total dose of 54, 60, or 69 Gy with conventional 2-Gy once-daily fractionation was associated with a myelopathy rate of less than 1%, 6%, and 50%, with a calculated strong dependence on dose/fraction (α/β = 0.87 Gy). With reirradiation of the full cord cross section at conventional fractionation, cord tolerance appears to increase at least 2% at 6 months after an initial course of radiation therapy. For partial cord irradiation as part of spinal SRS, a maximum cord dose of 13 Gy in a single fraction or 20 Gy in three fractions appeared to have a minimal (<1%) risk of injury.55
Primary Therapy By Tumor Type
Astrocytoma
Astrocytomas represent 7% to 11% of all primary spinal tumors. They are the most common intramedullary spinal cord tumors and comprise 35% to 45% of all reported cases of intramedullary tumors. The median age at presentation is approximately 35 years.16,56–58 There does not appear to be a difference in the age of presentation between low- and high-grade tumors.16 In children, 75% to 90% of intramedullary spinal cord tumors are astrocytomas and 85% to 95% of these are low-grade, fibrillary, or juvenile pilocytic astrocytomas (Fig. 28-7). Fewer than 10% of pediatric and 25% of adult spinal cord astrocytomas are malignant.*
Motor weakness is the most common presenting symptom in astrocytomas, occurring in up to 87% of patients. The median time from onset of symptoms to diagnosis ranges from 6 months to 2 years.16,41,57,60 In general, high-grade astrocytomas tend to have a shorter prodrome (median duration, 1.6 to 7 months) than low-grade tumors (median duration, 2 years).16,29,56
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
