Solid-state properties
Graham Buckton
Chapter contents
Key points
Solid state
The three states of matter are solid, liquid and gas (or vapour). In a sealed container, vapours will diffuse to occupy the total space, liquids will flow to fill part of the container completely, whereas solids will retain their original shape unless a compressive force is applied to them. From this simple consideration it becomes clear that solids are unique. Importantly their physical form (the packing of the molecules and the size and shape of the particles) can have an influence on the way the material will behave. At normal room temperature and pressure, the majority of drugs and excipients exist as solids, thus the study of solid-state properties is of enormous pharmaceutical importance.
Solid particles are made up of molecules that are held in close proximity to each other by intermolecular forces. The strength of interaction between two molecules is due to the individual atoms within the molecular structure. For example, hydrogen bonds occur due to an electrostatic attraction involving one hydrogen atom and one electronegative atom, such as oxygen. For molecules which cannot hydrogen bond, attraction is due to van der Waals forces. The term van der Waals forces is generally taken to include dipole-dipole (Keesom), dipole-induced dipole (Debye) and induced dipole-induced dipole (London) forces. In this context a dipole is where the molecule has a small imbalance of charge from one end to the other, making it behave like a small bar magnet. When the molecules pack together to form a solid, these dipoles align and give attraction between the positive pole of one and the negative pole on the next. Induced dipoles are where the free molecule does not have an imbalance of charge, but an imbalance is caused by bringing a second molecule into close proximity with the first.
Crystallization
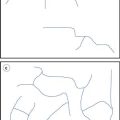
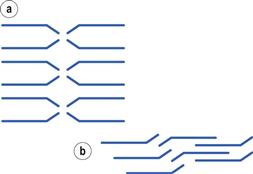
Materials in the solid state can be crystalline or amorphous (or a combination of both). Crystalline materials are those in which the molecules are packed in a defined order, and this same order repeats over and over again throughout the particle. In Figure 8.1a, an ordered packing of a molecule is shown; here the shape of the molecule is shown as a ‘hockey stick’ style image, which is representing a planar structure with a functional group pointing up at the end. This is not a real molecule – it has been drawn to provide an easy representation of a possible crystal packing arrangement. A characteristic property of a crystal is that it has a melting point. The melting point is the temperature at which the crystal lattice breaks down, due to the molecules having gained sufficient energy from the heating process to overcome the attractive forces that hold the crystal together. It follows that crystals with weak forces holding the molecules together (such as paraffins, which only have London van der Waals interactions) have low melting points, whereas crystals with strong lattices (i.e. those held together with strong attractive forces) have high melting points.
Crystals are produced by inducing a change from the liquid to the solid state. There are two options: one is to cool a molten sample to below the melting point. Pharmaceutical examples of crystallizing through cooling include the formation of suppositories, creams and semi-solid matrix oral dosage forms (although these will not always yield crystalline material). The other method of crystallization is to have a solution of the material and to change the system so that the solid is formed. At a given temperature and pressure, any solute (where the solute is the material that has been dissolved and the liquid is the solvent) has a certain maximum amount that can be dissolved in any liquid (called a saturated solution). If crystals are to be formed from a solution, it is necessary to have more solute present than can be dissolved, which is known as a supersaturated solution. As crystals form from a supersaturated solution, the systems will progress until there are solid particles in equilibrium with a saturated solution. In order to make a solid precipitate out of solution one can:
Many drugs are crystallized by adding water as an anti-solvent to a solution of the drug in an organic liquid. For example, if a drug is almost insoluble in water but freely soluble in ethanol, the drug could be crystallized by adding water to a near-saturated solution of the drug in ethanol.
The processes by which a crystal forms are called nucleation and growth. Nucleation is the formation of a small mass onto which a crystal can grow. Growth is the addition of more solute molecules onto the nucleation site. In order to achieve nucleation and growth, it is necessary to have a supersaturated solution. As mentioned above, a supersaturated solution is one where the amount of solute dissolved in the liquid is greater than the true solubility. Supersaturated solutions are not thermodynamically stable, so in these circumstances the system will adjust in order to move back to the true solubility, and to do this, the excess solute will precipitate. However, in some circumstances the process of nucleation can be slow. Many students will at some stage have had a supersaturated solution which has not crystallized but on simply scratching the side of the beaker with a glass rod, crystallization was induced. The scratching action produces a small amount of rough surface that acts as a nucleation site and causes the supersaturated solute to precipitate rapidly.
Polymorphism
If the crystallization conditions are changed in any way, it is possible that the molecules may start to form crystals with a different packing pattern to that which occurred when the original conditions were used. The change in conditions could be a different solvent, a change in the stirring, or different impurities being present. In Figure 8.1b, an alternative packing arrangement is shown to that which occurred for the same molecule in Figure 8.1a. As both the packing arrangements in Figure 8.1 are repeating ordered systems, they are both crystals; these would be called polymorphic forms.
By looking at the packing arrangements in Figure 8.1, it can be seen that the molecules in (a) are more spaced out than those in (b), which means that the two crystal forms would have different densities (i.e. the same mass of material would occupy different volumes). It looks as though it would be easier to physically pull a molecule off structure (a) than (b), as the molecules in (b) are more interwoven into the structure. If this were the case, then (a) would have a lower melting point than (b), and (a) may dissolve more easily. Also if an attempt were made to mill the two crystals, it looks like (a) would break easily, as there are natural break lines (either vertically or horizontally), whereas (b) does not seem to have an obvious weak line to allow easy breakage. This could mean that the milling and compaction (tableting) properties of the two forms will differ. In summary, a change in the packing arrangement of the same molecule, giving two different crystal forms, could result in significant changes in the properties of the solid.
Many organic molecules, including drugs and excipients, exhibit polymorphism. Often this is of a form called monotropic polymorphism, which means that only one polymorphic form is stable and any other polymorph that is formed will eventually convert to the stable form. However, some materials exhibit enantropic polymorphism, which means that under different conditions (temperature and pressure) the material can reversibly transform between alternative stable forms; this type of behaviour will not be considered further here. Considering monotropic polymorphism, the true stable form has the highest melting point and all other forms are described as metastable. This means that the other forms exist for a period of time, and thus appear stable, but given a chance they will convert to the true stable form. Different metastable forms can exist for very short times or many months before they convert to the stable form, depending upon the conditions under which they are stored.
In general, there will be a correlation between the melting point of the different polymorphs and the rate of dissolution, because the one with the lowest melting point will most easily give up molecules to dissolve, whereas the most stable form (highest melting point) will not give up molecules to the solvent so readily.
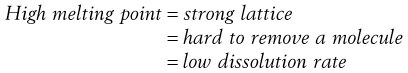
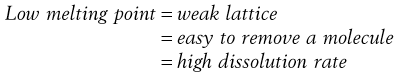
It is relatively easy to understand that changes in polymorphic form can cause changes in the rate at which a drug will dissolve. However, it is less easy to understand why this can lead to a change in the apparent solubility. Nonetheless, it is true that when a metastable polymorphic form is dissolved, it can give a greater amount of material in solution than the saturated solubility. In other words, metastable forms can dissolve to give supersaturated solutions. These supersaturated solutions will eventually return to the equilibrium solubility, due to the stable crystal form precipitating from solution, but that process may not be instantaneous. In fact, the supersaturated solution can often exist long enough to cause an increase in bioavailability of a poorly soluble drug. In Figure 8.2 the solubility of two different polymorphs of sulphamethoxydiazine is shown. It can be seen that Form II, a metastable form, has a higher solubility than Form III, a stable form, and that this lasts throughout the 90-minute experiment. However, if crystals of Form III are added to the solution of Form II then the solubility reverts rapidly to that of Form III, because the excess solute in the supersaturated solution will have seed crystals of Form III on which to precipitate.
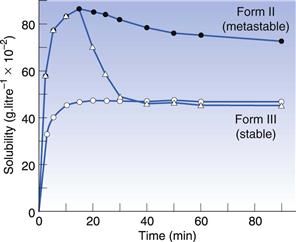
Fig. 8.2 The solubility time relationship for sulphamethoxydiazine. Open circles: solubility of polymorphic Form III, which rises to the drug’s equilibrium solubility and plateaux. Filled circles: solubility of polymorphic form II, which dissolves to twice the extent of Form III and then shows a gradual decline with time, as the stable form crystallizes from solution. Triangles: the effect of adding crystals of Form III to the solution of Form II at the peak of solubility. It can be seen that the amount dissolved falls rapidly from the supersaturated level to the true equilibrium solubility because the added crystals of Form III act as nucleation sites. Reproduced from Ebian et al 1973, with permission.
Polymorphism and bioavailability
Many drugs are hydrophobic and have very limited solubility in water. For drugs of this type, the rate at which they dissolve will be slow (slow dissolution rate), due to their limited aqueous solubility, and this can result in only a small percentage of the administered drug actually being available to the patient (low bioavailability). A classic example of the importance of polymorphism in bioavailability is that of chloramphenicol palmitate suspensions. In Figure 8.3 the blood serum level is plotted as a function of time after dosing. It can be seen that the stable α-polymorph produces low serum levels, whereas the metastable β-polymorph yields much higher serum levels when the same dose is administered.
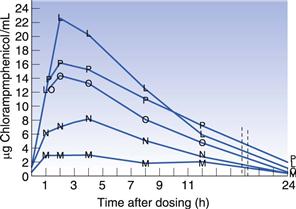
Fig. 8.3 Comparison of mean blood serum levels after administration of chloramphenicol palmitate suspensions using varying ratios of the stable (α) and the metastable (β) polymorphs. M = 100% α polymorph, N = 25 : 75 β:α, O = 50 : 50 β:α, P = 75 : 25 β:α, L = 100% β-polymorph. Reproduced from Aguiar et al 1976, with permission.
For drugs that are freely soluble in water the bioavailability is not likely to be limited by the dissolution, so it would be surprising for polymorphism to influence bioavailability in this way. However, for drugs with low aqueous solubility, the polymorphic form must be well controlled in order to ensure that the bioavailability is the same each time the product is made, and throughout the shelf-life of the product. It would be risky to deliberately make a product using anything other than the stable form of a drug, as other polymorphic forms could convert to the stable form during the shelf-life of the product, which could result in a reduction in bioavailability and thus therapeutic effect of certain products. This strategy is occasionally followed if the most-soluble metastable form is ‘stable enough’ to survive the agreed shelf-life of the product with insignificant change. The impact of polymorphism on drug dissolution and bioavailability is discussed further in Chapter 20.
In conclusion, the stable polymorphic form will have the slowest dissolution rate, so there may be occasions when it would be desirable to speed the dissolution by using a metastable form. However, the risk associated with using the metastable form is that it will convert back to the stable form during the product life, and give a consequent change in properties.
As polymorphism can have such serious consequences for bioavailability of drugs with low aqueous solubility, it is essential that manufacturers check for the existence of polymorphism and ensure that they use the same appropriate polymorphic form every time they make a product. New drugs are therefore screened to see how many polymorphs (and solvates and hydrates – see below) exist, and then to identify which one is the most stable. The screening process requires many crystallizations from numerous different solvent systems, with variations in method and conditions, in order to try to induce different polymorphs to form. The products are then checked with spectroscopy (e.g. Raman) and X-ray diffraction to see if they have different internal packing (see also Chapter 23). Sadly, there are examples of products being taken to market with what was believed to be the stable form, only for the stable form to be produced at a later stage. In these circumstances, the stable form may have been inhibited from being formed by a certain impurity, which may have been lost due to an alteration in the method of chemical synthesis of the drug, so the stable form suddenly was produced. Having produced the stable form, if the drug is poorly soluble it would be probable that the bioavailability would reduce. Also, having made the stable form, it is often then very hard to stabilize the metastable form again. This can result in products having to be recalled from the market and reformulated and retested clinically. The fact that major pharmaceutical companies, all of whom take the study of physical form very seriously, have seen the stable form arrive after product launch shows that it is difficult to be sure that you are working with the most stable form of the drug.
As was mentioned above, many properties other than rate of solution can change when a material is in a different polymorphic form. For example, paracetamol is a high-dose drug with poor compression properties, which can make it difficult to form into tablets. This is because there is an upper limit on the size of tablet that can be swallowed easily, so for high-dose drugs the amount of compressible excipient that can be added is modest. Consequently, researchers have tried to experiment with different polymorphic forms of paracetamol in order to find one that is more compressible.
Hydrates and solvates
It is possible for materials to crystallize and in so doing to trap individual molecules of the solvent within the lattice. If the solvent used is water, the material will be described as a hydrate. This entrapment is often in an exact molar ratio with the crystallizing material; for example, a monohydrate will have one molecule of water for each molecule of the crystallizing material. It is possible to have different levels of hydrate; for example, some drugs can exist as a monohydrate, dihydrate and trihydrate (respectively one, two and three molecules of water to each molecule of drug). Morris (1999) notes that about 11% (over 16 000 compounds) of all structures recorded on the Cambridge Structural Database exist as hydrates. Of the classes of hydrate materials that were similar to drugs, about 50% were monohydrates, over 20% were dihydrates, 8% were trihydrates and 8% were hemihydrates (1 water molecule:2 host); other hydrate levels (up to 10 water:1 host) became progressively less common.
If solvents other than water are present in a crystal lattice, the material is called a solvate. For example, if ethanol is present it would be an ethanolate. In general, it is undesirable to use solvates for pharmaceuticals as the presence of retained organic material would be regarded as an unnecessary impurity in the product, unless it was seen to possess advantageous properties and be safe for pharmaceutical use. If the organic solvent were toxic in any way it would obviously be inappropriate for pharmaceuticals. For this reason discussion will be limited to hydrates.
Hydrates often have very different properties from the anhydrous form, in the same way as two different polymorphs have different properties from each other. For this reason, the difference between hydrates and anhydrous forms is sometimes described inelegantly as pseudopolymorphism. With polymorphism, the stable form will have the highest melting point and the slowest dissolution rate (see above). However, with hydrates it is possible for the hydrate form to have either a faster or slower dissolution rate than the anhydrous form. The most usual situation is for the anhydrous form to have a faster dissolution rate than the hydrate; an example of this is shown in Figure 8.4 for theophylline. In this situation, water could hydrogen bond between two drug molecules and tie the lattice together; this would give a much stronger, more stable lattice and thus a slower dissolution rate. It can be seen from Figure 8.4 that the anhydrous theophylline rises to a high concentration in solution and then falls again until the amount dissolved is the same as that recorded for the hydrate. The reason for this is that the hydrate has come to the true equilibrium solubility, whereas the anhydrous form had initially formed a supersaturated solution (as has been described for metastable polymorphic forms above).
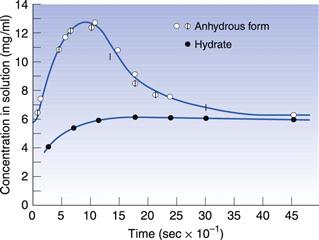
Fig. 8.4 The dissolution of theophylline monohydrate rising to an equilibrium solubility, compared with that for theophylline anhydrous which forms a supersaturated solution with a peak over twice that of the dissolving hydrate, before crystallizing to form the true equilibrium solubility. Reproduced from Shefter & Higuchi 1963, with permission.
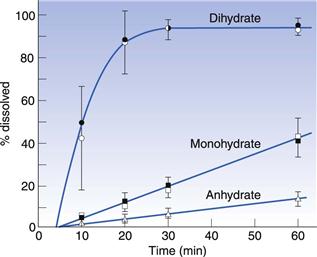
Although anhydrous forms are usually more rapidly soluble than the hydrate, there are examples of the opposite being true. In such circumstances one could think of water as a wedge pushing two molecules apart and preventing the optimum interaction between the molecules in the lattice. Here water would be weakening the lattice and would result in a more rapid dissolution rate. An example of the hydrate form speeding up dissolution is shown in Figure 8.5 for erythromycin.
Amorphous state
When a material is in the solid state but the molecules are not packed in a repeating long-range ordered fashion, it is said to be amorphous. Amorphous solids have very different properties from the crystal form of the same material. For example, crystals have a melting point (the break-up of the crystal lattice), whereas the amorphous form does not (as it does not have a crystal lattice to break!).
Polymeric materials (or other large molecular weight species) have molecules that are so large and flexible that it is not possible for them to align perfectly to form crystals. For these materials it will be usual to have ordered regions within the structure surrounded by disorder, so they are described as semi-crystalline. For materials such as these, it will not be possible to produce a completely crystalline sample; however, the degree of crystallinity can vary depending upon processing conditions. This can affect the properties of the material and thus how it functions in pharmaceutical products.
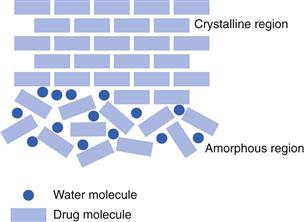
For low molecular weight materials, the amorphous form may be produced if the solidification process was too fast for the molecules to have a chance to align in the correct way to form a crystal (this could happen, for example, when a solution is spray-dried). Alternatively, a crystal may be formed but then may be broken. This could happen if a crystal were exposed to energy, such as milling. A simple analogy is that a crystal is like a brick wall, which has ordered long-range packing. If the wall is hit hard, perhaps as during demolition, the bricks will separate (Fig. 8.6). Unlike the brick wall, however, a disrupted crystal will be thermodynamically unstable and will revert back to the crystal form. This conversion may be rapid or very slow and, as with polymorphism, its pharmaceutical significance will depend on how long the partially amorphous form survives.
Amorphous forms have a characteristic temperature at which there is a major change in properties. This is called the glass transition temperature (Tg). If the sample is stored below the Tg, the amorphous form will be brittle, described as being in the glassy state. If the sample is above its Tg, it becomes rubbery. The Tg, although not well understood, is a point at which the molecules in the glass exhibit a major change in mobility. The lack of mobility when the sample is glassy allows the amorphous form to exist for a longer time, whereas when Tg is below the storage temperature, the increased molecular mobility allows rapid conversion to the crystalline form.
The glass transition temperature of an amorphous material can be lowered by adding a small molecule, called a plasticizer, that fits between the glassy molecules, giving them greater mobility. Water has a good plasticizing effect on many materials, so the glass transition temperature will usually reduce when in the presence of water vapour.
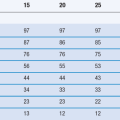
Most amorphous materials are able to absorb large quantities of water vapour. Absorption is a process whereby one molecule passes into the bulk of another material and should not be confused with adsorption, which is where something concentrates at the surface of another material. In Figure 8.6, the way in which water can access amorphous regions is shown. Figure 8.7 shows the amount of water that is adsorbed to a crystalline material (Fig. 8.7a) in comparison with that absorbed into an amorphous form of the same material (Fig. 8.7b). It can be seen that the amount absorbed is many times greater than that adsorbed. This large difference in water content at any selected relative humidity is important in many materials. For example, it is possible that certain drugs can degrade by hydrolysis when amorphous but remain stable when crystalline. The extent of hydrolysis of an antibiotic which had been processed to yield different levels of crystalline : amorphous forms is shown in Table 8.1; the extent of degradation is greater when the amorphous content is increased. This concept is also discussed in Chapter 4 of this book.
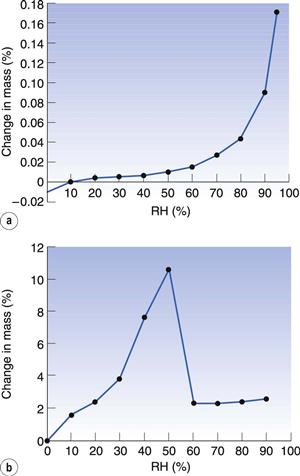
Table 8.1
The chemical stability of cephalothin sodium related to the amorphous content of the sample
| Sample | % amorphous | % stable drug after storage at 31% RH 50 °C |
| Crystalline | 0 | 100 |
| Freeze-dried | 12 | 100 |
| Freeze-dried | 46 | 85 |
| Spray-dried | 53 | 44 |
(Data derived from Pikal et al 1978.)
In Figure 8.7 it can be seen that the amorphous form absorbs a very large amount of water until 50% RH, after which there is a weight loss. The reason for the loss is that the sample has crystallized. Crystallization occurs because the absorbed water has plasticized the sample to such an extent that the Tg has dropped below room temperature and allowed sufficient molecular mobility that the molecules are able to align and crystallize. The water is lost during this process as absorption can only occur in the amorphous form, so it cannot endure into the crystalline state. However, some water is retained in this example (Fig. 8.7a, b), because lactose is able to form a monohydrate. The amount of water required to form a monohydrate with lactose is 5% w/w (calculated from the molecular weight of lactose and water), which is much less than the 11% that was present in the amorphous form (Fig. 8.7b).
In Figure 8.8 the amorphous content of lactose is seen to increase in proportion to the length of time it was left in an air-jet mill (micronizer). In Figure 8.9 it can be seen that a drug substance became partially amorphous when treated in a simple ball mill, and extensively amorphous when micronized. Although the example in Figure 8.9 is an extreme behaviour, it is not unusual for highly processed materials to become partially amorphous. Although milling does not necessarily make all materials partially amorphous, the chance of seeing disruption to the crystalline lattice will increase with the amount of energy used in the milling.
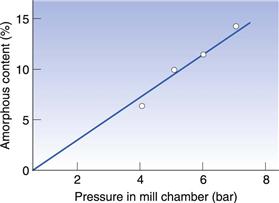
Fig. 8.8 The amorphous content induced in crystalline lactose as a consequence of milling in an air-jet mill at different air pressures. Redrawn from Briggner et al 1994, with permission.
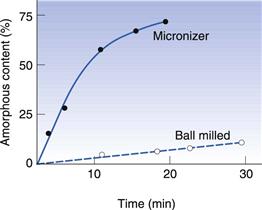
Fig. 8.9 The amorphous content of a model drug substance following milling in a ball mill and a micronizer Redrawn from Ahmed et al 1996, with permission.
The fact that processing can make crystalline materials partially amorphous means that it is possible that very complex materials can be formed that contain different metastable states. For example, in Figure 8.3 the plasma levels of two polymorphs of chloramphenicol palmitate were shown; if the β-polymorph were milled it is possible that it may also become partially amorphous, which could make the plasma level even higher than when the crystalline form was used. However, milling the β-polymorph could also provide the necessary energy to convert it to the stable α-polymorph, which would reduce the effective plasma level. Equally, milling could disrupt the α-polymorph giving a partially amorphous form that may have a higher bioavailability than the crystal. In other words, the effect of processing on the physical form can be very complicated, and often unpredictable. It is possible to produce a physical form that is partially amorphous and partially crystalline. The crystalline component could then be stable or metastable. Inevitably, with time (for low molecular weight species) the sample will revert to only contain the stable crystalline form, with no amorphous content and none of the metastable polymorph(s), but as this does not necessarily happen instantly, the physical form and its complexity are of great importance.
Crystal habit
All the discussion above has related to the internal packing of molecules. It has been shown that they may have no long-range order (amorphous) or different repeating packing arrangements (polymorphic crystals) or have solvent molecules included in the crystal (solvates and hydrates). Each of these changes in internal packing of a solid will give rise to changes in properties. However, it is also possible to change the external shape of a crystal. The external shape is called the crystal habit and this is a consequence of the rate at which different faces grow. Changes in internal packing usually (but not always) give an easily distinguishable change in habit. However, for the same crystal packing, it is possible to change the external appearance by changes in the crystallization conditions.
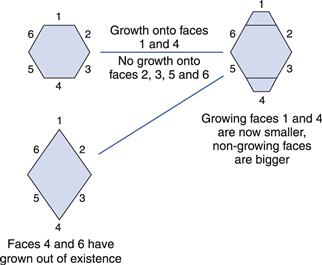
With any crystalline material, the largest face is always the slowest growing. The reason for this is shown in Figure 8.10, where it can be seen that if drug is deposited on two faces of the hexagonal crystal habit, then the first consequence is that the face where drug is deposited actually becomes a smaller part of the crystal, whereas the other faces get larger. Eventually, the fastest growing faces will no longer exist (Fig. 8.10). The growth on different faces will depend on the relative affinities of the solute for the solvent and the growing faces of the crystal. Every molecule is made up of different functional groups – some are relatively polar (such as carboxylic acid groups) whereas others are non-polar (such as a methyl group). Depending on the packing geometry of the molecules into the lattice, some crystal faces may have more exposed polar groups and others may be relatively non-polar. If the crystal were growing from an aqueous solution, drug would deposit on the faces that make the crystal more polar (i.e. the non-polar faces would grow, making the more polar faces dominate). If, however, the same crystal form were growing from a non-polar solvent, then the opposite would be true.
Obviously the external shape can alter the properties of drugs and excipients. For example, the dissolution rate of a drug can change if the surface area to volume ratio is altered. An extreme difference would be between a long needle and a sphere (Fig. 8. 11). A sphere of 20 µm radius has approximately the same volume (mass) as a needle of 335 × 10 × 10 µm; however, the surface area of the needle is 2.7 times greater than that of the sphere. As dissolution rate is directly proportional to surface area, the needle would dissolve much faster than the sphere. Crystals do not grow to make spheres, although through milling, crystals can develop rounded geometries, the closest to a sphere would be a cube, which would still have under half the surface area of the needle shown in Figure 8.11.
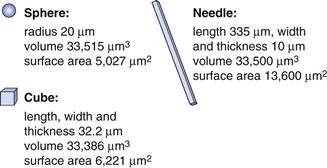
Fig. 8.11 The relative surface areas of a sphere, cube and needle that have similar volumes of material.
As well as changes in dissolution rate, different crystal habits can cause changes in powder flow (which is important as, for example, the die of a tableting machine is filled by volume and requires good powder flow in order to guarantee content uniformity of the product) and sedimentation and caking of suspensions.
It is technically possible to engineer changes in crystal habit, by deliberately manipulating the rate of growth of different faces of the crystal. This is done by intentionally adding a small amount of impurity to the solution. The impurity must preferentially interact with one face of the growing crystal, and in so doing it will stop growth on that face, so the remaining face(s) grow more rapidly. The impurity would either be a very similar molecule to that of the crystallizing material, so that part of the molecule is included in the lattice but the remainder of the molecule blocks further layers from attaching, or it may be a surfactant that adsorbs to one growing face.
Surface nature of particles
Dry powder inhalers
Dry powder inhalers (Chapter 37) often have a micronized drug, which has to be small enough to be inhaled, mixed with a larger carrier particle which is often lactose. The carrier particle is there to make the powder suitable for handling and dosing, as micronized particles have poor flow properties. The shape and surface properties of the drug and/or carrier particles can be critical parameters in controlling the dose of drug that is delivered. It may be necessary to adjust the surface roughness of carrier particles. Figure 8.12a is a cartoon of a rough carrier particle; this would hold the micronized drug too strongly, essentially trapped within the rough regions of the carrier, so the inhaled dose would be very low. In Figure 8.12b, a smooth particle of the same drug is seen. Here the drug will easily be displaced from the carrier during inhalation but it may not stay mixed with the carrier during filling of the inhaler and dosing. In Figure 8.12c, a rough carrier particle has first been mixed with micronized carrier and then with micronized drug. Using this approach, the drug is free to detach, as the micronized carrier is trapped in all the crevices on the carrier surface.
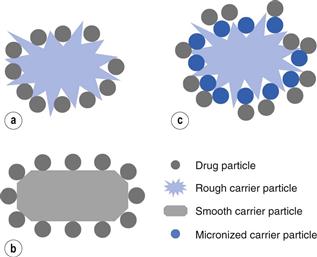
The hypothesis relating to the use of fine carrier particles to enhance the delivery of micronized drug from large carrier particles is not proved beyond doubt. It remains possible that interactions between the fine carrier and fine drug may be the reason for the enhanced delivery.
It should of course be noted that the diagrams in Figure 8.12 simplify the real situation greatly. In Figure 8.13 a real lactose particle is shown along with added micronized particles. It can be seen that the large lactose particle (lactose particles are often described as ‘tomahawk’ shaped) has rough ridges on its surface and there are some very fine particles aligned to some extent in the rough areas. It is also clear that many fine particles are not on the surface of the lactose and that some fine particles are on smooth regions of the lactose.

Fig. 8.13 An electron micrograph showing a large lactose carrier particle with added fine lactose, some of which is seen to be at rough spots on the large carrier, but also there is a lot of non-adsorbed fine lactose content. This shows that Figure 8.12 is an enormous simplification of the real system.
For products such as these (discussed more fully in Chapter 37), it is becomingly increasingly important to first measure the surface nature of samples and then to control the form in order to achieve the desired delivery of drug. The shape of the carrier is an important consideration for the design of this type of product. A further concern is the surface energy as this can influence the way in which the drug and carrier are attached to each other.
Surface energy
Surfaces and surface energy are discussed in Chapter 4, a summary of those aspects relevant to the solid state are presented here. Molecules at the surface of a material have a net inward force exerted on them from the molecules in the bulk; this is the basis of surface energy. Surface energy is important as every interaction (except the mixing of two gasses) starts by an initial contact between two surfaces. If this surface interaction is favoured then the process will probably proceed, whereas if it is not favoured then the process will be limited. A good example of the role of surface energy is the wetting of a powder by a liquid; here the powder cannot dissolve until the liquid makes good contact with it. A practical example is instant coffee, where some brands are hard to wet and dissolve whereas others dissolve easily. Changes in the wetting of powders can affect the processes of wet granulation, suspension formation, film coating and drug dissolution.
The measurement and understanding of surface energy for solid powders is complex. Even on the same crystal form, it would be expected that every crystal face, edge and defect could experience different forces pulling from the bulk and thus could have a different surface energy. It would be reasonable to assume that different physical forms of the same drug could have quite different surface energies. Thus, for the same drug, it is possible that changes in habit and/or polymorphic form and/or the presence of a solvate or hydrate would change the surface energy. For amorphous forms, the molecules at the surface have greater freedom to move and reorientate than do molecules in crystal surfaces, so the amorphous form could have changes in surface energy with time (and with physical state in relation to Tg).
The conventional way of determining the surface energy of a solid is to place a drop of liquid onto the solid surface and measure the contact angle as discussed in Chapter 4. Perfect wetting of a solid by a liquid will result in a contact angle of 0 degrees.
For smooth solid surfaces, contact angles are an ideal way of assessing surface energy. However, powders present problems as it is not possible to place a drop of liquid on the surface. Consequently a compromise will always be required when measuring a contact angle for powdered systems. An example of such a compromise would be to make a compact of the powder in order to produce a smooth flat surface. However, the disadvantage of this is that the process of compaction may well change the surface energy of the powder.
A preferred option by which to assess the surface energy of powders would be vapour sorption.
Vapour sorption
Adsorption, absorption and deliquescence are also discussed in Chapter 4. When a powder is exposed to a vapour, or gas, the interaction will take one of the following forms:
Absorption into the bulk can occur if the sample is amorphous, whereas the interaction will be limited to adsorption if the powder is crystalline. The extent and energetics of interaction between vapours and powder surfaces allow the surface energy to be calculated. The other processes listed are deliquescence, which is where the powder dissolves in the vapour, and hydrate formation which has been discussed in Chapter 4.
It is possible therefore to use adsorption and/or absorption behaviour as a method by which the powder surface energy can be determined. There are three basic approaches to this: gravimetric (measuring weight change), calorimetric (measuring heat change) and chromatographic (measuring retention to a solid using analysis such as flame ionization of the eluted carrier from a column). Each of these techniques has found application in studies of batch-to-batch variability of materials. An example of a critical case could be that a certain drug shows extensive variability in respirable dose from a dry powder inhaler. Assuming that the size distribution was acceptable in all cases, it would be necessary to understand why some batches yielded unacceptable doses. These vapour sorption techniques could then be used to assess the surface energy and then define values that would be acceptable in order to get good drug dosing, and equally to define batches of drug that will give unacceptable products.
Gravimetric methods use sensitive microbalances as a means of determining the extent of vapour sorption to a powder surface. The calorimetric approaches measure the enthalpy change associated with vapour/powder interaction, which gives clear information on the nature of the powder surface. By use of the principles of gas chromatography it is possible to pack the powder, for which the surface energy is required, into a column and then to inject different vapours into the column with a carrier gas. Obviously, the time taken for the vapour to come out of the other end of the column is a measure of how favourable was the interaction between the powder and the vapour. Inverse gas chromatography, as this is called, is described in Chapter 4.
References
1. Aguiar AJ, Krc Jr J, Kinkel AW, Samyn JC. Journal of Pharmaceutical Sciences. 1967;56:847–853.
2. Ahmed H, Buckton G, Rawlins DA. International Journal of Pharmaceutics. 1996;130:195–201.
3. Allen PV, Rahn PD, Sarapu AC, Vanderwielen AJ. Journal of Pharmaceutical Sciences. 1978;67:1087–1093.
4. Briggner L-E, Buckton G, Bystrom K, Darcy P. International Journal of Pharmaceutics. 1994;105:125–135.
5. Ebian AR, Moustafa MA, Khalil SA, Motawi MM. Journal of Pharmacy and Pharmacology. 1973;25:13–20.
6. Morris KR. Structural aspects of hydrates and solvates. In: Brittain HG, ed. Polymorphism in Pharmaceutical Solids. New York: Marcel Dekker; 1999.
7. Pikal MJ, Lukes AL, Lang JE, Gaines K. Journal of Pharmaceutical Sciences. 1978;67:767–773.
8. Shefter E, Higuchi T. Journal of Pharmaceutical Sciences. 1963;52:781–791.
Bibliography
1. Brittain HG, ed. Polymorphism in Pharmaceutical Solids. New York: Marcel Dekker; 1999.
2. Buckton G. Interfacial Phenomena in Drug Delivery and Targeting. Amsterdam: Harwood Academic Press; 1995.
3. Florence AT, Attwood D. Physicochemical Principles of Pharmacy. 5th edn London: Pharmaceutical Press; 2011.
4. Mersmann A, ed. Crystallization Technology Handbook. New York: Marcel Dekker; 1994.