Chapter 12 Skeletal Muscle Relaxants
| Abbreviations | |
|---|---|
| ACh | Acetylcholine |
| AChE | Acetylcholinesterase |
| CNS | Central nervous system |
| GABA | γ-Aminobutyric acid |
Therapeutic Overview
long-lasting muscle paralysis by blocking the release of acetylcholine (ACh) from motor nerves.
Clinical uses of these compounds are listed in the Therapeutic Overview Box.
| Therapeutic Overview |
|---|
| Neuromuscular blocking drugs |
| Endotracheal intubation |
| Reduce muscle contractility and depth of anesthesia required for surgery |
| In the intensive care unit to prevent high airway pressures, decrease O2 consumption, and abolish muscle rigidity in patients on mechanical ventilation |
| Prevent bone fractures during electroconvulsive therapy |
| Antispasticity drugs |
| Reduce muscle cramping and tightness in neurological disorders and spinal cord injury and disease |
| Antispasm drugs |
| Prevent use-related minor muscle spasms |
| Motor nerve blocker (Botulinum toxin) |
| Blepharospasm and strabismus |
| Elective cosmetic purposes |
Mechanisms of Action
Skeletal muscles are innervated by somatic motor nerves, which originate in the spinal cord, terminate at muscle cells, and release ACh as their neurotransmitter (see Chapters 9 10). Upon arrival of an action potential, ACh is released from synaptic vesicles by exocytosis, crosses the synapse, and interacts with skeletal muscle nicotinic cholinergic receptors to depolarize the postsynaptic membrane (see Chapter 1). When the membrane reaches threshold, a muscle action potential is generated and propagates along the fiber to initiate excitation-contraction coupling. The action of ACh is terminated very rapidly by hydrolysis by acetylcholinesterase (AChE) located in the synaptic junction. Neuromuscular transmission is depicted in Figure 12-1.
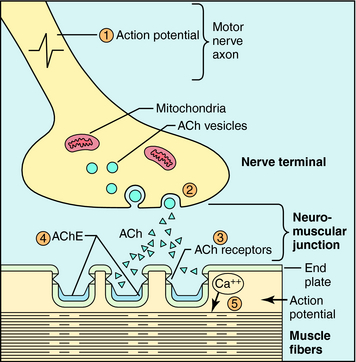
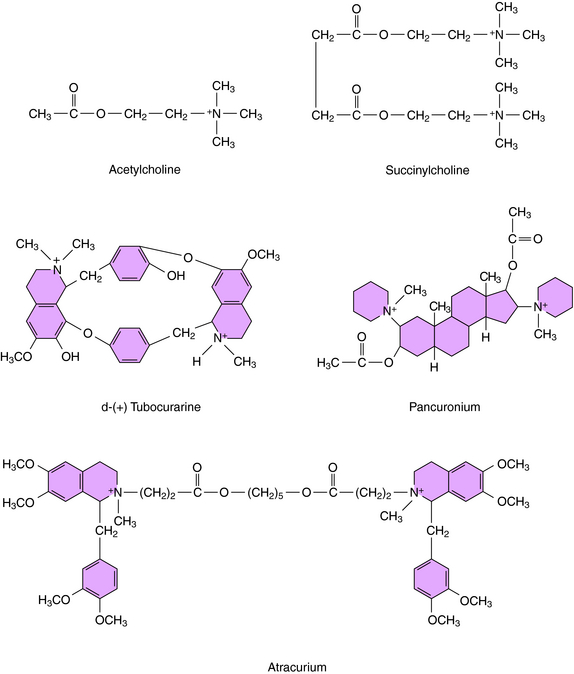
Neuromuscular blocking agents interfere with neurotransmission by either: (1) occupying and activating the nicotinic receptor for a prolonged period of time, leading to blockade, which occurs with the depolarizing agents; or (2) competitively antagonizing the actions of ACh at nicotinic acetylcholine receptors, which occurs with the nondepolarizing agents. Not surprisingly, the structures of the depolarizing agents resemble that of ACh, whereas the nondepolarizing agents are bulky, rigid molecules. A comparison of the structure of ACh with prototypical depolarizing (succinylcholine) and nondepolarizing (tubocurarine and pancuronium) neuromuscular blockers is shown in Figure 12-2.
Because nondepolarizing blockers compete with ACh, the blockade can be reversed by increasing the concentration of ACh. This is done by inhibiting AChE, which hydrolyzes ACh (Chapter 9 10). Neostigmine, edrophonium, and pyridostigmine are AChE inhibitors used clinically to reverse neuromuscular block caused by nondepolarizing blockers. However, if the concentration of the competitive blocking agent is greater than that needed for blockade of 95% of the receptors, AChE inhibitors will be unable to increase ACh sufficiently to reverse the block.
There is only one depolarizing agent currently in clinical use, succinylcholine (see Fig. 12-2). This compound binds to and activates muscle nicotinic receptors in the same manner as ACh. However, succinylcholine is not metabolized by AChE, resulting in receptor occupation for a prolonged period. Succinylcholine is hydrolyzed primarily by butyrylcholinesterase, which is present in the plasma but not in high concentrations at the neuromuscular junction, resulting in continuing muscle depolarization. The neuromuscular block resulting from succinylcholine is characterized by two phases. The first, termed phase I block, is a consequence of prolonged depolarization, rendering the membrane unresponsive to further stimuli. It is characterized by initial muscle fasciculations followed by a flaccid paralysis that is not reversed, but intensified, by administration of AChE inhibitors. With continued exposure to succinylcholine, phase II block occurs, during which the membrane repolarizes but is still unresponsive, reflecting a desensitized state of the nicotinic cholinergic receptor. This phase progresses to a state in which the block appears similar to that produced by nondepolarizing agents, that is, it becomes responsive to high concentrations of ACh and can be reversed by AChE inhibitors.
The antispasticity agents include baclofen, which is a structural analog of γ-aminobutyric acid (GABA). Baclofen decreases spasticity by binding to GABAB receptors on presynaptic terminals of spinal interneurons (Fig. 12-3). Binding to presynaptic GABAB receptors results in hyperpolarization of the membrane, which reduces Ca++ influx and decreases the release of the excitatory neurotransmitters, glutamate, and aspartate. Postsynaptic interactions with sensory afferent terminals cause membrane hyperpolarization via a G-protein-coupled receptor that leads to increases in K+ conductance, enhancing inhibition. Baclofen may also inhibit γ-motor neuron activity and reduce muscle spindle sensitivity, leading to inhibition of monosynaptic and polysynaptic spinal reflexes.
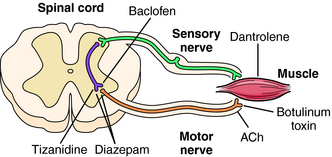
Benzodiazepines such as diazepam have an antispasticity effect by acting on GABAA receptors to increase their affinity for GABA in the brain and in the spinal cord (see Chapter 31 and Fig. 12-3).
Dantrolene has direct effects on skeletal muscle to inhibit ryanodine receptor Ca++ release channels on the sarcoplasmic reticulum of skeletal muscle, thereby uncoupling motor nerve excitation and muscle contraction (see Fig. 12-3). Dantrolene may also have actions on the CNS that contribute to its antispasticity effects, although the cellular mechanisms have not been elucidated.
The antispasm agents do not act on motor neurons or on the muscle itself. Rather, these compounds act primarily on the brain and perhaps spinal reflexes to relax skeletal muscle by unknown mechanisms. Cyclobenzaprine, methocarbamol, and metaxalone depress the CNS and induce sedation, which may be counter-productive for the active physical therapy currently recommended to treat certain types of muscle spasms. Diazepam exerts both antispasm and antispasticity actions by enhancing the effects of GABA at GABAA receptors in the brain (see Chapter 31) and spinal cord (see Fig. 12-3), where it may have both presynaptic and postsynaptic effects.
Botulinum neurotoxins are produced by the anaerobic bacterium Clostridium botulinum. There are seven types of the toxin (A-G), all of which block ACh release. Botulinum toxin interferes with specific synaptic proteins involved in exocytotic release of synaptic vesicles containing ACh. This results in a long-duration (3 months) flaccid paralysis of the muscle into which it is injected (see Fig. 12-3). Botulinum toxin will also block autonomic synapses in the vicinity of the injection. Botulism is a serious form of food poisoning from improperly processed foods, and outbreaks of this problem occur sporadically.
Pharmacokinetics
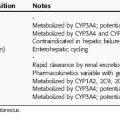
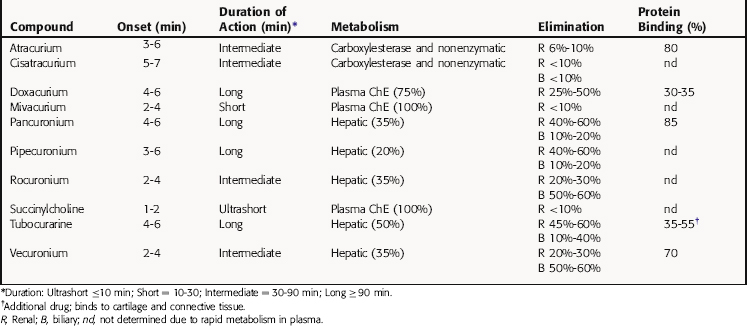
The neuromuscular blocking drugs differ considerably in their pharmacokinetic properties, and the choice of a particular compound is determined in part by the duration of action of the agent needed for the particular procedure (Table 12-1). Because these drugs are positively charged, they cross membranes poorly and are generally limited in distribution to the extracellular space. However, small amounts of pancuronium, vecuronium, and pipecuronium cross membranes. Pancuronium also crosses the placenta but not in sufficient amounts to cause problems in the fetus when used during a cesarean section.
Relationship of Mechanisms of Action to Clinical Response
The actions of nondepolarizing blocking agents are often potentiated by inhalational anesthetics (see Chapter 35) and also by low concentrations of extracellular K+ or Ca++, as may occur after use of diuretics or in renal dysfunction. Elevated K+ or Ca++ and reduced Mg++ concentrations, however, may counteract drug actions through changes in ACh release in response to depolarization or changes in membrane potential at the muscle endplate.
Dantrolene is the drug of choice for the treatment of malignant hyperthermia, which is a rare and potentially lethal disorder characterized by hypermetabolism, tachycardia, hypertension, premature ventricular contractions, rigidity, cyanosis, and rapid temperature increase. The hyperthermic response is not ameliorated by typical antipyretic drugs such as aspirin or acetaminophen. Malignant hyperthermia may develop in susceptible patients exposed to halogenated anesthetic gases with or without succinylcholine (see Chapter 35).
Botulinum toxin A is approved for treatment of muscle disorders of the eye, including blepharospasm and strabismus, characterized by excessive neuromuscular contractility, and for elective cosmetic purposes. Botulinum toxin is also injected into muscles characterized with “repetitive use” disorders such as “tennis elbow” and “violinist wrist” and to treat chronic spasticity of skeletal muscle in cerebral palsy, and cervical dystonia, primary axillary hyperhidrosis (severe underarm sweating); it is also being investigated for the treatment of severe migraine and overactive bladder. In spasticity disorders the objective is to permit the contralateral muscle to grow while the spastic muscle is relaxed.
Pharmacovigilance: Side Effects, Clinical Problems, and Toxicity
Neuromuscular blocking agents must be used with caution in patients with underlying neuromuscular, hepatic, or renal disease or electrolyte imbalance. Patients with neuromuscular disorders such as myasthenia gravis may be resistant to succinylcholine because of a decrease in the number of ACh receptors; the dose of muscle relaxants must be reduced by 50% to 75% in such patients. Patients with myasthenia gravis are also more likely than healthy patients to develop a phase II block in response to succinylcholine, particularly when repeated doses have been administered. Patients with myasthenia gravis exhibit much greater sensitivity to nondepolarizing agents, and the use of long-acting muscle relaxants such as pancuronium, pipecuronium, and doxacurium must be avoided in these patients. Intermediate- and short-acting nondepolarizing drugs can be administered carefully in lower doses with close monitoring of neuromuscular transmission. Lambert-Eaton Myasthenic syndrome, an autoimmune presynaptic neuromuscular disorder in which the stimulated release of ACh is reduced at the neuromuscular junction, is another disease in which patients are very sensitive to muscle relaxants.
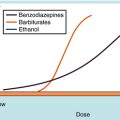
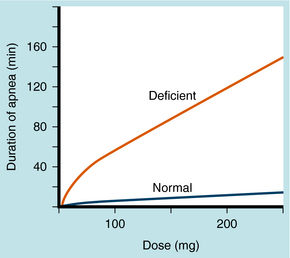
Genetic variations in butyrylcholinesterase activity result in either lower concentrations of normal enzyme or an abnormal enzyme. A dose of 1 to 2 mg/kg succinylcholine in healthy patients produces neuromuscular blockade lasting <15 minutes; in a patient with a variant of this enzyme with decreased effectiveness, the same dose may last much longer. This is illustrated in Figure 12-4, with block defined as the duration of apnea. Trauma, alcoholism, pregnancy, use of oral contraceptives, and other conditions in which butyrylcholinesterase activity is changed can alter the duration of neuromuscular block produced by succinylcholine.
Bowman WC. Neuromuscular block. Br J Pharmacol. 2006;147(Suppl 1):S277-S286.
Lee C. Conformation, action, and mechanism of action of neuromuscular blocking muscle relaxants. Pharmacol Ther. 2003;98:143-169.
Nicholson WT, Sprung J, Jankowski CJ. Sugammadex: A novel agent for the reversal of neuromuscular blockade. Pharmacotherapy. 2007;27:1181-1188.
Papapetropoulos S, Singer C. Botulinum toxin in movement disorders. Semin Neurol. 2007;27:183-194.