CHAPTER 13 Sexual differentiation
normal and abnormal
Normal Embryological Development of the Reproductive System
Although chromosomal sex is determined at the time of fertilization, gonadal sex results from the differentiation of the indifferent undifferentiated gonad which becomes either a testis or an ovary. This begins during the fifth week of embryological development; at this time, an area of coelemic epithelium develops on the medial aspect of the urogenital ridge, and proliferation leads to the establishment of the gonadal ridge. Epithelial cords then grow into the mesenchyme (primary sex cords), and the gonad now possesses an outer cortex and an inner medulla. In XY individuals, the medulla becomes the testis and the cortex regresses. In embryos with an XX complement, the cortex differentiates to become an ovary and the medulla regresses. The primordial germ cells develop by the fourth week in the endodermal cells of the yolk sac, and during the fifth week, they migrate along the dorsal mesentery of the hindgut to the gonadal ridges, eventually becoming incorporated into the mesenchyme and the primary sex cords by the end of the sixth week (Figure 13.1).
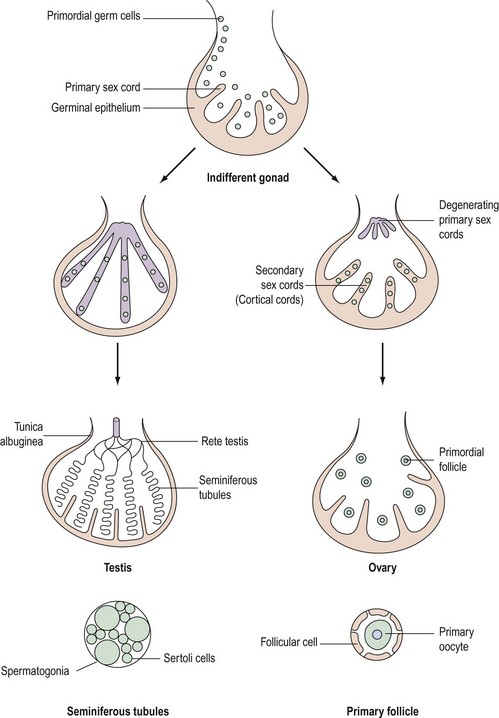
Development of the testis
The primary sex cords become concentrated on the medulla of the gonad and proliferate, and their ends anastomose to form the rete testis. The sex cords become isolated by the development of a capsule called the ‘tunica albuginea’ and the developing sex cords become the seminiferous tubules. Mesenchyme grows between the tubes to separate them (Leydig cells). The seminiferous tubules are composed of two layers of cells: supporting cells (Sertoli cells) derived from the germinal epithelium, and spermatogonia derived from the primordial germ cells (Figure 13.1).
Genetic control of gonadal development (Figure 13.2)
The pathway which controls the genetic interaction which transforms intermediate mesoderm to the bipotential gonad is beginning to be understood. It is now possible to identify with certainty two genes that are associated with this process. These two genes are Wilms’ tumour gene (WT1) and FTZ1. WT1 has been found on chromosome 11, p13 and regulates DNA transcriptase (Hastie 1994, Dong et al 1997). FTZ1 produces steroidogenic factor 1 (SF-1), and this has been found to be required for differentiation to the bipotential gonad (Wilhelm et al 2007).
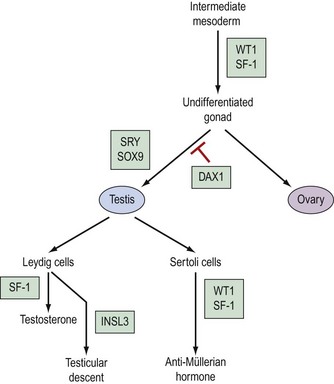
As the presence of a Y chromosome is essential for testicular differentiation, the localization of testicular-determining factor (TDF) which was localized at the short arm of the Y chromosome using cloning and DNA sequencing techniques, and the gene determining TDF was sequenced and designated SRY (sex-determining region Y gene) (Guellaen et al 1984, Behlke et al 1993). The second gene which is involved in the differentiation of the testis is known as SOX9, and this gene coexists with the SRY gene in its homeobox and is a regulator of DNA transcription. SOX9 gene expression is increased in male gonadal differentiation and decreased in female gonadal differentiation. SOX9 has also recently been shown to be a regulator of type II collagen which is involved in the formation of cartilage (Lefebvre et al 1997). The DAX1 gene is responsible for development of the receptor on the surface of the undifferentiated gonad, which allows SRY and SOX9 to differentiate the gonad to become a testis. Following the development of Leydig cells and Sertoli cells, SF-1 is involved in the control of steroid hydroxylases, and P450 aromatase causes an increase in the synthesis of testosterone and a reduction in the conversion of androgens to oestrogens, leading to a contribution to testicular regulation (Ogata 2008). In combination with ST1, it is also responsible for Sertoli cells producing anti-Müllerian hormone. Following differentiation, Leydig cells produce insulin-like 3 (INSL3) gene, expression of which leads to testicular descent and shortening of the gubernaculums (Foresta and Ferlin 2004).
The genes involved in ovarian differentiation are much more difficult to define, although DAX1 expression continues during ovarian differentiation. Therefore, it is suggested that DAX1 antagonizes the actions of the SRY gene (Swain et al 1998).
Sex chromosome anomalies
Variations may occur in the number or the structure of chromosomes in a mosaic or a non-mosaic form. The non-mosiac forms are summarized in Box 13.1.
Aneuploidy
Although there are a wide range of clinical effects, some generalizations can be made:
Structural abnormalities of X chromosomes
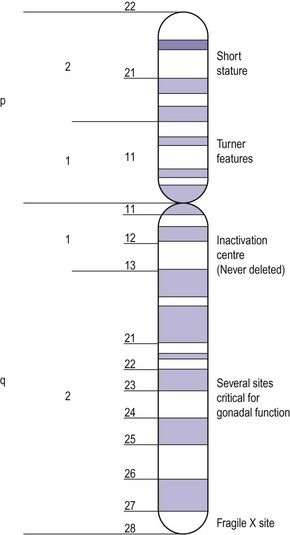
Apart from the number of X chromosomes in mosaicism, there are a number of possible variations within the X chromosome in the female. The X chromosome carries a large number of genes, not all of which have been delineated but some of which are responsible for metabolic and development disorders, and the gene map for the X chromosome is constantly being updated. If there is one normal X chromosome and the other is abnormal, inactivation of the abnormal X chromosome usually occurs in the somatic cells and this tends to diminish the effect of the abnormality, except in the ovary. Although only one X chromosome is sufficient for early ovarian development, germ cell maintenance requires several loci on the second X chromosome and on autosomes (Figure 13.3) (Simpson 1999).
Intersex Disorders
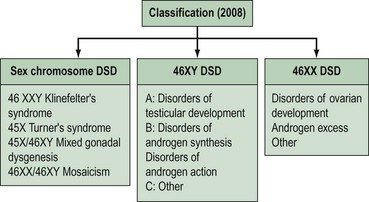
Intersex disorders are best classified into three groups (see Figure 13.4). This classification was suggested by Hughes (2008) as a new simple classification which includes all disorders of intersex.
Female (46XX) disorders of sexual development
Congenital adrenal hyperplasia
Pathophysiology
This is the most common cause of female pseudohermaphroditism and is an autosomal-recessive disorder resulting in enzyme deficiency in the biosynthesis of cortisol in the adrenal gland. Cortisol production occurs in the zona fasciculata and zona reticularis (Figure 13.5), and is controlled by adrenocorticotrophic hormone (ACTH) secreted by the pituitary gland. Adrenal androgen production occurs in the same area and is influenced by ACTH. A deficiency in any enzyme in the pathway results in decreased production of cortisol with resultant elevated levels of ACTH. This leads to increased steroid production by the adrenal reticularis and consequent hyperplasia. The stimulation by ACTH elevates the levels of circulating androgens, and this results in virilization of the female fetus.
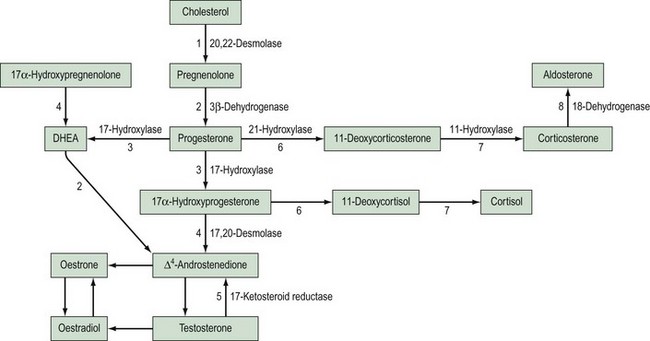
21-Hydroxylase deficiency
Aetiology
Deficiency of 21-hydroxylase is an autosomal-recessive disorder. The link between human leukocyte antigen (HLA) type and 21-hydroxylase deficiency was established by Dupont et al (1977), and this allowed mapping of the gene which was located on the short arm of chromosome 6. It is located between the HLA-B and HLA-DR loci, and subgroups of HLA-B have been closely linked to congenital adrenal hyperplasia type: HLA-BW47 is linked to salt-losing congenital adrenal hyperplasia and HLA-BW51 is linked to the simple virilizing form. Studies by Donohoue et al (1986) have shown that there are two 21-hydroxylase genes: 21-OHA and 21-OHB. Only one is active (21-OHB) and they both lie between the fourth components of complement C4A and C4B. A variety of mutations have been reported, including gene deletions of 21-OHB, gene conversions and point mutations.
Epidemiology
The incidence of 21-hydroxylase deficiency is between one in 5000 and one in 15,000, based on neonatal screening programmes (Cacciari et al 1983), although a higher incidence (one in 700) has been reported in specific populations of Eskimos (Pang et al 1982). The incidence of the non-classic form of the disease, when androgenization fails to appear before late childhood or puberty, is much more common; approximately one in 300 in the White population and one in 30 in European Jews (Pang et al 1988).
Presentation
Affected females are born with an enlarged clitoris, fused labioscrotal folds and a urogenital sinus which may become a phallic urethra. There is often great variation in the degree of masculinization of the external genitalia; this is classified according to Prader (1958).
The internal genitalia develop normally as they are not influenced by androgens.
Treatment
This is divided into four parts.
Long-term cortisol replacement therapy
Although previous reports suggested that mineralocorticoid therapy could be discontinued after infancy (Newns 1974), more recent data suggest that it is essential to continue therapy for life (Hughes et al 1979).
Surgical correction
Once the sex of rearing has been established as female, some attempt to feminize the genitalia may be made, usually within the first 18 months of life. If the clitoris is enlarged, a reduction clitoridectomy can be performed and the perineal region modified (Edmonds 1989). There are major surgical problems associated with severe virilization in congenital adrenal hyperplasia. The urogenital sinus has been formed by labial fold fusion and the folds are usually thin, but may be thick with associated narrowing of the lower vagina, especially in salt-losers. If the labial folds are thin, division by a simple posterior incision may be performed around 3 or 4 years of age. However, thick perineal tissue should be left until after puberty, and no attempt at surgery should be made until the girl is physically and mentally sexually mature. The operation, when it is performed, involves a flap vaginoplasty with a pedicle graft of labia used to recreate the vagina. Alternatively, a Williams vaginoplasty may be used.
Mulaikal et al (1987) reviewed the fertility rates in 80 women with 21-hydroxylase deficiency; of 25 with the simple form who attempted pregnancy, 15 were successful, whereas only one of the salt-losers who tried to become pregnant succeeded. There is no doubt that a major reason for the failure of the salt-losing group is the disappointing results of surgery and subsequent lack of adequate sexual function.
11-Hydroxylase deficiency
This is the hypertensive form of congenital adrenal hyperplasia which accounts for approximately 5–8% of all cases (Zachmann et al 1983). The absence of 11-hydroxylase leads to elevated levels of 11-deoxycorticosterone (DOC), and although this means a decreased amount of aldosterone, DOC has salt-retaining properties, leading to hypertension. Androstenedione levels are also elevated and this can result in ambiguous genitalia.
The genetics, however, are rather different. There is no HLA association with 11-hydroxylase deficiency, but the use of a DNA probe has located the gene on the long arm of chromosome 8 (White et al 1985).
Androgen-secreting tumours
Ovary
A number of androgen-secreting tumours have been reported, including luteoma (Hensleigh and Woodruff 1978, Cohen et al 1982), polycystic ovaries (Fayez et al 1974), mucinous cystadenoma (Novak et al 1970, Post et al 1978), arrhenoblastoma (Barkan et al 1984) and Krukenberg tumours (Connor et al 1968, Forest et al 1978). Not all female fetuses will be affected and there is no association with gestation and exposure. The fetus may be partly protected by the conversion of the maternally derived androgen to oestrogen in the placenta, and thus the degree of virilization is variable.
Adrenal gland
There are only two reports of adrenal adenomas causing fetal masculinization (Murset et al 1970, Fuller et al 1983). These tumours may be responsive to human chorionic gonadotrophin, and thus levels of androgen may be higher in pregnancy than in the non-pregnant state, leading to androgenization of the fetus.
Drugs
The association between the use of progestogens and masculinization of the female fetus has received much publicity, but the only progestogen proven to have such an effect is 17-ethinyl testosterone (Ishizuka et al 1962). These infants had clitoromegaly and, in some cases, labioscrotal fusion, but the risk is very small (one in 50). Gestogens which are derived from testosterone should be avoided in the pregnant woman. There have been two case reports of androgenization of female fetuses from exposure to danazol during pregnancy (Castro-Magana et al 1981, Duck and Katamaya 1981), and both babies were born with external genitalia similar to those with adrenogenital syndrome.
Associated multiple congenital abnormalities
Female intersex has been described in association with a number of multiple abnormality states, most commonly those in association with the urinary and gastrointestinal tracts. It has also been described in association with VATER (Vertebrae, Anus, Trachea,Oesophagus and Renal) syndrome (Say and Carpentier 1979).
46XY Disorders of Sexual Development
Failure of testicular development
46XY true hermaphroditism
These individuals have the presence of both testicular and ovarian tissue with a 46XY karyotype. It has been suggested that mutation of the SRY gene may be the aetiology of this (Berkovitz and Seeherunvong 1998).
46XX sex reversal
This nomenclature is used to describe the situation in which testicular tissue develops in someone with a 46XX karyotype. If testicular development is normal, they are referred to as ‘46XX males’, but when testicular determination is incomplete, they are referred to as ‘46XX true hermaphrodites’. In 46XX males, the external genitalia are usually normal or a small percentage may have hypospadias. Clinically, they do not present until puberty, when this is delayed due to failure to be able to produce testosterone and due to the degeneration of seminiferous tubules and Leydig cell hyperplasia, which seems to occur just prior to puberty. It seems that in the majority of these individuals, the sex reversal is related to the translocation of Y chromosome sequences, including the SRY gene from the paternal Y chromosome to the paternal X chromosome (Tomomasa et al 1999). However, one-third of individuals are not found to have Y chromosome sequences in their DNA, and this means that there must be a mutation in the genomic DNA which permits testicular determination to occur in the absence of TDF. These gene defects remain to be explained.
Errors in testosterone biosynthesis
This type of disorder accounts for only 4% of XY females, and results from deficiency of an enzyme involved in testosterone synthesis (see Figure 13.5).
Androgen insensitivity at the target site (Table 13.1)
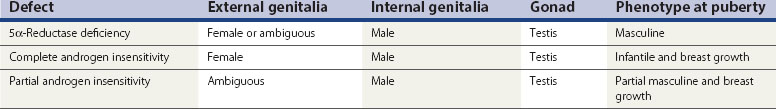
5α-Reductase deficiency
This results in failure of the conversion of testosterone to DHT in target tissues and thus a failure of masculinization of the site. In infancy, there is usually a small phallus, some degree of hypospadias, a bifid scrotum and a blind vaginal pouch. The testes are found either in the inguinal canal or in the labioscrotal folds, and Müllerian structures are absent. At puberty, elevated levels of androgen lead to masculinization, including an increase in phallic growth, although this remains smaller than normal. Seminal production has been reported (Petersen et al 1977).
Complete androgen insensitivity
These patients may lack the androgen receptor and may be shown to lack the gene located on the X chromosome between Xp11 and Xq13. Work by Brown et al (1982), however, suggests that there may be a variety of defects, ranging from absence of receptors to presence of a normal number of receptors which are inactive. The exact mechanism of the defects in patients with androgen receptors awaits definition.
Anomalous Vaginal Development
Aetiology
Three mechanisms explain most vaginal anomalies. They may be familial; for example, XY females who have a hereditary disorder as described previously. Congenital absence of the vagina has been very rarely reported in XX siblings (Jones and Mermut 1974), and also in monozygotic twins with only one child affected (Lischke et al 1973).
The case of a female-limited autosomal-dominant trait was first reported by Shokeir (1978) who studied 16 Saskatchewan families in which there was a proband with vaginal agenesis. However, Carson et al (1983), in a study of 23 probands, disputed Shokeir’s theory. The previous evidence with regard to monozygotic twins also makes this mode of inheritance unlikely. Polygenic or multifactorial inheritance does, however, offer some explanation that families may exhibit the trait as reported. The recurrent risk of a polygenic multifactorial trait in first-degree relatives is reported to be between 1% and 5%.
Epidemiology
The incidence of vaginal malformations has been variously estimated at between one in 4000 and one in 10,000 female births (Evans et al 1981). The infrequency of this anomaly makes accurate estimates of the true incidence very difficult to obtain, but when considered as a cause of primary amenorrhoea, vaginal malformation ranks second to gonadal dysgenesis.
Pathophysiology
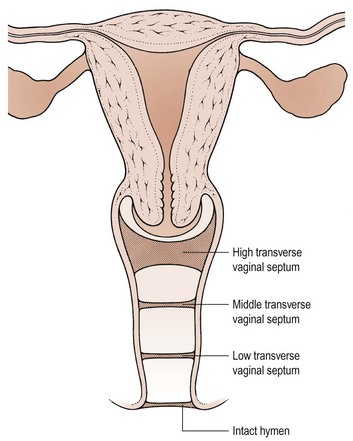
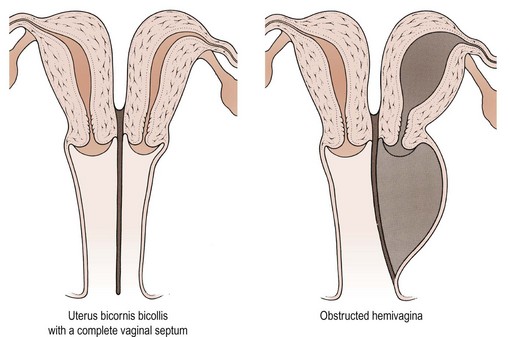
The pathophysiology of vaginal absence may be either as a result of failure of the vaginal plate to form or failure of cavitation. Absence of the uterus and fallopian tubes indicates total failure of Müllerian duct development, but in the Rokitansky syndrome, the uterus is often present, although rudimentary, and therefore it must be failure of vaginal plate formation and subsequent vaginal development which leads to the absent vagina. Vertical fusion defects (Figure 13.6) may result from failure of fusion of the Müllerian system with the urogenital sinus, or may be due to incomplete canalization of the vagina. Disorders of lateral fusion are due to the failure of the Müllerian ducts to unite, and may create a duplicated uterovaginal septum which may be obstructive or non-obstructive, depending on the mode of development (Figure 13.7).
Presentation
Vertical fusion defects
Here, the transverse vaginal septum prevents loss of menstrual blood and therefore cryptomenorrhoea results. Most patients present as teenagers with cyclical abdominal pain, and a haematocolpos will be palpable within the pelvis on rectal examination. The patient may also present with associated pressure symptoms of urinary frequency and/or retention. The incidence of vertical fusion defects is reported as 46% high, 35% mid and 19% low septae in the vagina (Rock et al 1982).
Investigation
Vaginal atresia
A patient presenting with a clinically absent vagina and no cyclical abdominal pain requires an ultrasound examination of the pelvis to determine the presence or absence of a uterus and/or a haematometra. Laparoscopy in these patients is unnecessary. Some 15% of patients with the Rokitansky syndrome suffer major defects of the urinary system, including congenital absence of a kidney; 40% of patients also have trivial urinary abnormalities (d’Alberton et al 1981). It is therefore important to perform an intravenous urogram in order to establish any abnormalities in the renal system or the presence of a pelvic kidney, which may alert the surgeon to take extra care if abdominal surgery becomes necessary.
Treatment
Vaginal atresia
Occasionally, cultural problems arise which make management much more difficult, especially in ethnic groups where the ability to procreate is fundamental to marriage and social acceptance. These patients and their parents can be very difficult to console and often refuse to accept the situation, questioning the diagnosis of their sterility over a number of years. The immediate reaction of most patients is to request surgical correction of the abnormality to return them to ‘normal’. Unfortunately, opting for surgical treatment without adequate psychological and physical preparation inevitably leads to disaster. The author recommends a minimum of 6 months of preparation before any surgical procedure is performed, and during this time, psychological support can be implemented for the patient and her parents. There are two major areas of support required: first, the correction of the loss of esteem and inevitable depression; repeated counselling sessions by trained personnel are required if these symptoms are to be overcome. The second problem involves the psychological aspect; again, prolonged counselling sessions will be required if an adequate and fulfilling sex life is going to be possible in the future. The sexual life achieved by well-managed patients can be excellent and has been reported to be comparable to that of the normal population (Raboch and Horejsi 1982, Poland and Evans 1985).
Management of the absent vagina with a non-functioning uterus
In a patient with an absent or rudimentary uterus, the creation of a vaginal passage may be non-surgical or surgical. The non-surgical technique involves the repeated use of graduated vaginal dilators over a period of 6–12 months. A vaginal dimple of at least 1 cm is necessary for this technique to succeed, and patients require support and encouragement during this time. Patients are instructed to begin with a small vaginal dilator which is pressed firmly against the vaginal dimple for a period of some 20 minutes twice a day. Pressure is exerted but pain should be avoided, and repeated use of these vaginal dilators will meet with success in approximately 90% of cases with appropriate selection (Broadbent et al 1984, Edmonds 2003). This technique was first described by Frank in 1938, and in view of its undoubted success, with no complications, this method must be attempted in all girls with an absent vagina and a 1 cm dimple before any surgical procedure is considered.
The McIndoe–Reed procedure was first described in 1938 (McIndoe and Banister 1938) and involved the use of a split-thickness skin graft over a solid mould; this mould is placed in a surgically created space between the urethra and the rectum. This space is created digitally following a transverse incision in the vaginal dimple. Digital exploration of the space must be performed with great care as damage to the bladder or the rectum may occur. The space created must reach the peritoneum of the pouch of Douglas if an adequate length of a vagina is to be created. A split-thickness skin graft is then taken from a donor site and an appropriately sized mould is chosen. The skin graft is then fashioned over the mould with the external skin surface in apposition to the mould. The skin-covered mould is then placed in the neovaginal space, and the labia are sutured together to hold the mould in situ. McIndoe reported his own series of 105 patients in 1959 and had a satisfactory outcome in 80% of patients. Cali and Pratt (1968) reported on their series of 123 patients; 90% had good sexual function but 6% had major complications which were primarily fistulae, and subsequent reconstructive surgery was necessary in 8%. These complications resulted in modifications of the technique and the use of a soft material for the mould to prevent fistula formation due to pressure necrosis.
The search for an alternative material to line the neovagina and avoid the scarring of the donor skin site led to the use of amnion for the vaginoplasty procedure (Ashworth et al 1986). The technique involves the creation of a neovaginal space in the same way as described for the McIndoe–Reed procedure, but amnion obtained at the time of elective caesarean section is used to line the neovagina. The mesenchymal surface of the amnion is placed against the new vaginal surface to promote epithelialization, and the mould is kept in situ for 7 days and then replaced with a new amnion graft for a further 7 days. Subsequently, patients are encouraged to use dilators on a frequent basis to maintain the vaginal passage. Again, reported success of approximately 90% has been achieved by these authors.
The Williams vulvovaginoplasty (Williams 1976) has almost no place in the management of these disorders, and is much less frequently used than in the past. In patients in whom dissection of a neovagina is impossible but the labia majora are normal, this technique is valuable. The principle of the operation is to create a vaginal pouch from the full-thickness skin flaps created from the labia majora, which are united in the midline. Following surgery and adequate healing, the patient is taught to use vaginal dilators in the same way as described in the Frank technique. There is no doubt that this does allow the patient and her partner to enjoy a sex life with mutual orgasm, but the angle of the vagina is unnatural and unsatisfactory for some patients. Although the operation is simple to perform, the psychological problems of the distorted external genitalia can be considerable.
Complications
Some 25% of women will have some degree of dyspareunia following a vaginoplasty (Smith 1983). This is most commonly due to scarring at the upper margin of the vagina, involving the peritoneum. Similar incidences of dyspareunia occur in nearly all series and it is difficult to know how best to avoid this. It seems to occur regardless of technique, but contraction of the upper part of the vagina is difficult to avoid. The artificial vagina created in these ways acquires all the characteristics of normal vaginal epithelium, and the exposure of the grafted epithelium to a new environment means that care has to be taken to ensure it remains healthy. Four cases of intraepithelial neoplasia in neovaginas have been reported (Jackson 1959, Duckler 1972, Rotmensch et al 1983, Imrie et al 1986).
Results
The results of surgery are extremely good when judged by sexual satisfaction. However, Rock et al (1982) reported pregnancy success following surgical correction of transverse vaginal septa, and noted that patients with a transverse vaginal septum had only a 47% pregnancy rate when the site of the septum was taken into account. If the obstruction was in the lower third, all patients achieved a pregnancy, compared with 43% in the middle third and 25% in the upper third. It is suggested that the difficulties in conceiving may be secondary to the development of endometriosis, and the higher the site of the septum, the more likely the development of this disorder may be. Thus prompt diagnosis and surgical correction are important in an attempt to preserve the maximum reproductive capacity in these patients.
Complications
Complications are primarily those of dyspareunia and failure of pregnancy, as described above.
Malformations of the Uterus
Classification and pathophysiology
There have been numerous classification systems for uterine malformations, varying from those based on embryological development to those based on obstetric performance. However, the most widely used classification is that of Buttram and Gibbons (1979), which is based upon the degree of failure of normal development. Six categories are described:
Many of these uterine malformations are shown in Figure 13.8.
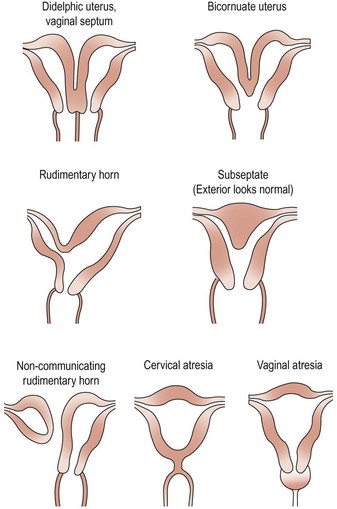
Incidence
The incidence of uterine anomalies is difficult to define as it depends entirely upon the interest of the investigator and the diligence with which investigation is pursued. Obstetric series show an incidence of uterine abnormalities ranging from one in 100 to one in 1000 (Semmens 1962). In an infertile population, the incidence increases to approximately 3% (Sanfillippo et al 1978). It is likely that the incidence of uterine malformations is greatly overestimated, as no gynaecological or reproductive problems are ever experienced in the vast majority of patients.
Recurrent pregnancy loss
Recurrent pregnancy wastage in the form of abortion or premature labour is a common way in which uterine anomalies will be discovered. The role of uterine anomalies in pregnancy wastage is discussed in Chapter 21 on female infertility and Chapter 23 on recurrent miscarriage.
Urological abnormalities
Not uncommonly, urologists who discover malformations of the urinary system investigate the genital system and find abnormalities. Thompson and Lynn (1966) reported that 66% of patients with a maldevelopment of the renal tract had an associated Müllerian duct abnormality. However, only 13.5% of women with anomalous renal development had anomalous uterine development. In patients with a single kidney, the most common uterine abnormality is uterus didelphis with a vaginal septum which is associated with unilateral occlusion.
Menstrual disorders
Uterine abnormalities may be responsible for a number of menstrual disorders including oligomenorrhoea, dysmenorrhoea and menorrhagia. The specific menstrual symptoms will depend on the anomaly. In an interesting study by Sorensen (1981), investigating infertile women with oligomenorrhoea, 56% of patients were found to have mild uterine abnormalities. Sorensen suggested that this oligomenorrhoea might be due to poor vascularization or steroid receptor development in the malformed uterus. With regard to dysmenorrhoea, uterine abnormalities seem to be associated with a higher incidence of primary dysmenorrhoea, although this may also be associated with obstructed outflow problems. Rudimentary hemiuteri may also be the cause of dysmenorrhoea in some women.
Diethylstilboestrol exposure
The malformations associated with DES exposure have been well described (Kaufman et al 1980). These abnormalities include a classic T-shaped uterus with a widening of the interstitial and isthmic portions of the fallopian tubes and narrowing of the lower two-thirds of the uterus, as well as non-specific uterine abnormalities with changes of cavity seen on hysterosalpingography. These patients may present with impaired reproductive function, and pregnancy wastage may be as high as 50–60%. However, as DES exposure was terminated in 1970, almost all of these individuals will now have finished their reproductive performance.
Investigations and treatment
When uterine anomalies are suspected, a number of imaging techniques can be used to identify the abnormality. These include ultrasound, magnetic resonance imaging and hysterosalpingography. For a review, the reader is referred to Troiano and McCarthy (2004).
The value of treatment of uterine malformations is discussed in Chapter 21.
KEY POINTS
Ashworth MF, Morton KE, Dewhurst CJ, et al. Vaginoplasty using amnion. Obstetrics and Gynecology. 1986;67:443-444.
Barkan A, Cassorla F, Loriaux D, Marshall JC. Pregnancy in a patient with virilising arrhenoblastoma. American Journal of Obstetrics and Gynecology. 1984;149:909-910.
Behlke MA, Bogan JS, Beer-Romero P, Page DC. Evidence that the SRY protein is encoded by a single exon on the human Y chromosome. Genomics. 1993;17:736-739.
Berkovitz GD, Seeherunvong T. Abnormalities of gonadal differentiation. Baillière’s Clinical Endocrinology and Metabolism. 1998;12:133-142.
Broadbent RT, Woolf RM, Herbertson R. Non-operative construction of the vagina. Plastic and Reconstructive Surgery. 1984;73:117-122.
Brown TR, Maes M, Rothwell SW, Migeon CJ. Human complete androgen insensitivity with normal dihydrotestosterone receptor binding capacity in cultured genital skin biopsies. Journal of Clinical Endocrinology and Metabolism. 1982;55:61-69.
Buttram VS, Gibbons WE. Müllerian anomalies — a proposed classification. Fertility and Sterility. 1979;32:40-48.
Cacciari E, Balsamo A, Cassio A, et al. Neonatal screening for congenital adrenal hyperplasia. Archives of Disease in Childhood. 1983;58:803-806.
Cali RW, Pratt JH. Congenital absence of the vagina. American Journal of Obstetrics and Gynecology. 1968;100:752-754.
Carson SA, Simpson JL, Malinak LR, et al. Heritable aspects of uterine anomalies II. Genetic analysis of Müllerian aplasia. Fertility and Sterility. 1983;40:86-91.
Castro-Magana M, Chervanky T, Collipp PJ, Ghavami-Maibadi Z, Angulo M, Stewart C. Transient adrenogenital syndrome due to exposure to danazol in utero. American Journal of Diseases of Childhood. 1981;135:1032-1034.
Cohen VA, Daughaday WH, Weldon V. Fetal and maternal virilization association with pregnancy. American Journal of Diseases of Childhood. 1982;136:353-356.
Connor TB, Ganis FM, Levin HS, Migeon CJ, Martin LG. Gonadotrophin dependent Krukenberg tumour causing virilization during pregnancy. Journal of Clinical Endocrinology and Metabolism. 1968;28:198-201.
D’Alberton A, Reschini E, Ferrari N, Candiani P. Prevalence of urinary tract abnormalities in a large series of patients with uterovaginal atresia. Journal of Urology. 1981;126:623-627.
Dong WF, Heng HH, Lowsky R. Cloning, expression and chromosomal localisation to 11p12-13 of a human LIM homeobox gene, hLIM-1. DNA and Cell Biology. 1997;16:671-678.
Donohoue PA, van Dop C, McLean RH, et al. Gene conversion in salt-losing congenital adrenal hyperplasia with absent complement C4B protein. Journal of Clinical Endocrinology and Metabolism. 1986;62:995-1002.
Duck SC, Katamaya KP. Danazol may cause female pseudohermaphroditism. Fertility and Sterility. 1981;35:230-231.
Duckler L. Squamous cell carcinoma developing in an artificial vagina. Obstetrics and Gynecology. 1972;40:35.
Dupont B, Oberfield SE, Smithwick EM, et al. Close genetic linkage between HLA and congenital adrenal hyperplasia. The Lancet. 1977;ii:1309-1312.
Edmonds DK. Intersexuality. Dewhurst’s Practical Paediatric and Adolescent Gynaecology, 2nd edn. Butterworths, London, 1989.
Edmonds DK. Congenital malformation of the genital tract. Best Practice and Research in Clinical Obstetrics and Gynaecology. 2003;17:19-40.
Evans TN, Poland ML, Boving RL. Vaginal malformations. American Journal of Obstetrics and Gynecology. 1981;141:910-916.
Fayez JA, Bunch TR, Miller GL. Virilization in pregnancy associated with polycystic ovary disease. Obstetrics and Gynecology. 1974;44:511-521.
Forest MG, Orgiazzi J, Tranchant D, Mornex R, Bertrand J. Approach to the mechanism of androgen production in a case of Krukenberg tumor responsible for virilization during pregnancy. Journal of Clinical Endocrinology and Metabolism. 1978;47:428-434.
Foresta C, Ferlin A. Role of INSL3 and LGR8 in cryptorchidism and testicular function. Reproductive Biomedicine Online. 2004;9:294-298.
Frank RT. The formation of an artificial vagina without operation. American Journal of Obstetrics and Gynecology. 1938;35:1053-1055.
Fuller PJ, Pettigrew IG, Pike JW, Stockigt JR. An adrenal adenoma causing virilization of mother and infant. Clinical Endocrinology. 1983;18:143-153.
Guellaen G, Casanova M, Bishop C. Human XX males with Y single copy DNA fragments. Nature. 1984;307:172-173.
Hastie ND. The genetics of Wilm’s tumour. Annual Review of Genetics. 1994;28:523-558.
Hensleigh PA, Woodruff DA. Differential maternal fetal response to androgenizing luteoma or hyperreactio luteinalis. Obstetrical and Gynaecological Surgery. 1978;33:262-271.
Hughes IA, Wilton A, Lole CA, Glay OP. Continuing need for mineralocorticoid therapy in salt-losing congenital adrenal hyperplasia. Archives of Disease in Childhood. 1979;54:350-358.
Hughes IA. Disorders of sex development. A new definition and classification. Best Practice in Research in Clinical Endocrinology and Metabolism. 2008;22:119-134.
Imrie JEA, Kennedy JH, Holmes JD, et al. Intraepithelial neoplasia arising in an artificial vagina. British Journal of Obstetrics and Gynaecology. 1986;93:886-887.
Ishizuka SC, Kawashima Y, Nakanishi T, et al. Statistical observations on genital anomalies of newborns following the administration of progestins to their mothers. Journal of the Japanese Obstetrical and Gynaecological Society. 1962;9:271-282.
Jackson GW. Primary carcinoma of an artificial vagina. Obstetrics and Gynaecology. 1959;14:534.
Jones HW, Mermut S. Familial occurrence of congenital absence of the vagina. Obstetrics and Gynecology. 1974;42:38-40.
Kaufman RH, Adam E, Binder GL, Gerthoffer E. Upper genital tract changes and pregnancy outcome in offspring exposed in utero to DES. American Journal of Obstetrics and Gynecology. 1980;137:299-306.
Lefebvre V, Huang W, Harley VR. SOX9 is a potent activator of the chondrocyte-specific enhancer of the proalpha1(II) collagen gene. Molecular Cellular and Biology. 17, 1997. 2336–2246
Lischke JH, Curtis CH, Lamb EJ. Discordance of vaginal agenesis in monozygotic twins. Obstetrics and Gynecology. 1973;41:902-922.
McIndoe AH. Discussion on treatment of congenital absence of the vagina with emphasis on long term results. Proceedings of the Royal Society of Medicine. 1959;52:952-953.
McIndoe AH, Banister JB. An operation for the cure of congenital absence of the vagina. Journal of Obstetrics and Gynaecology of the British Commonwealth. 1938;45:490-495.
Mulaikal RM, Migeon CJ, Rock JA. Fertility rates in female patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. New England Journal of Medicine. 1987;315:178-181.
Murset G, Zachmann M, Prader A, Fischer J, Labhart A. Male external genitalia of a girl caused by a virilizing adrenal tumour in the mother. Acta Endocrinologica. 1970;65:627-638.
Newns GH. Congenital adrenal hyperplasia. Archives of Disease in Childhood. 1974;49:716-724.
Novak DJ, Lauchlan SC, McCauley JC. Virilization during pregnancy: case report and review of literature. American Journal of Medicine. 1970;49:281-286.
Ogata T. Progress in analyzing disorders of sexual development. Sexual Development. 2008;2:167-178.
Pang S, Murphey W, Levine LS, et al. A pilot newborn screening for congenital adrenal hyperplasia in Alaska. Journal of Clinical Endocrinology and Metabolism. 1982;55:413-420.
Pang SY, Wallace MA, Hofman L, et al. Worldwide experience in newborn screening for congenital adrenal hyperplasia. Pediatrics. 1988;81:866-874.
Petersen RE, Imperato-McGinley J, Gautier T, Sturla E. Male pseudohermaphroditism due to 5α reductase deficiency. American Journal of Medicine. 1977;62:170-191.
Poland ML, Evans TN. Psychologic aspects of vaginal agenesis. Journal of Reproductive Medicine. 1985;30:340-348.
Post WD, Steele HD, Gorwill H. Mucinous cystadenoma and virilization during pregnancy. Canadian Medical Association Journal. 1978;118:948-953.
Prader A. Vollkommen männlichie aussere Genitalentwicklung und Salzverlustsyndrom bei Mädchen mit kongenitalem adrenogenitalem Syndrom. Helvetica Paediatrica Acta. 1958;13:5-14.
Raboch J, Horejsi J. Sexual life of women with Kuster–Rokitansky syndrome. Archives of Sexual Behavior. 1982;11:215-219.
Rock JA, Zacur HA, Dlugi AM, et al. Pregnancy success following surgical correction of imperforate hymen and complete transverse vaginal septum. Obstetrics and Gynecology. 1982;59:448-454.
Rotmensch J, Rosenheim N, Dillon M, et al. Carcinoma arising in a neovagina. Obstetrics and Gynecology. 1983;61:534.
Sanfillippo JS, Yussman MA, Smith O. Hysterosalpingography in the evaluation of infertility. Fertility and Sterility. 1978;30:636-639.
Say B, Carpentier NJ. Genital malformations in a child with VATER association. American Journal of Diseases of Childhood. 1979;133:438-439.
Semmens JP. Congenital anomalies of the female genital tract. Obstetrics and Gynecology. 1962;19:328-333.
Shokeir MHK. Aplasia of the Müllerian system. Evidence of probable sex limited autosomal dominant inheritance. Birth Defects. 1978;14:147-151.
Simpson JL. Genetics of human reproduction. Journal of Medical Genetics. 1999;89:224-239.
Smith MR. Vaginal aplasia: therapeutic options. American Journal of Obstetrics and Gynecology. 1983;146:534-538.
Sorensen SS. Minor Müllerian anomalies and oligomenorrhoea in infertile women. American Journal of Obstetrics and Gynecology. 1981;140:636-640.
Swain A, Narvaez V, Burgoyne P. Dax 1 antagonises SRY action in mammalian sex determination. Nature. 1998;391:761-767.
Therman E, Susman M. Abnormal human sex chromosome constitution. In Human Chromosomes: Structure, Behavior and Effects, 3rd edn, New York: Springer Verlag; 1993:220-227.
Thompson DP, Lynn HB. Genital abnormalities associated with solitary kidney. Mayo Clinic Proceedings. 1966;41:438-442.
Tomomasa H, Adachi Y, Iwabuchi M. XX-male syndrome bearing the sex-determining region Y. Archives of Andrology. 1999;42:89-96.
Troiano RN, McCarthy SM. Müllerian duct anomalies: imaging and clinical issues. Radiology. 2004;233:19-34.
White R, Leppert M, Bishop DT, et al. Construction of linkage maps with DNA markers for human chromosomes. Nature. 1985;313:101-105.
Wilhelm D, Palmer S, Koopman P. Sex determination and gonadal development in mammals. Physiology Review. 2007;87:1-28.
Williams EA. Uterovaginal agenesis. Annals of the Royal College of Surgeons of England. 1976;58:266-277.
Zachmann M, Tassinari D, Prader A. Clinical and biochemical variability of congenital adrenal hyperplasia due to 11-hydroxylase deficiency. Journal of Clinical Endocrinology and Metabolism. 1983;56:222-229.






