[level-membership-for-critical-care-medicine-category]29
Severe Heart Failure
DEFINITION, EPIDEMIOLOGY, AND STAGING OF HEART FAILURE
PROGNOSIS IN ACUTE HEART FAILURE
PHARMACOLOGIC MANAGEMENT OF ACUTE HEART FAILURE
TRANSITION TO CHRONIC PHARMACOLOGIC THERAPY FOR SEVERE HEART FAILURE
CORONARY HEART DISEASE AND HEART FAILURE: SPECIAL CONSIDERATIONS
HEART FAILURE WITH PRESERVED LEFT VENTRICULAR EJECTION FRACTION (DIASTOLIC HEART FAILURE)
ACUTE MYOCARDITIS AND HEART FAILURE
DEVICE THERAPY: IMPLANTED CARDIOVERTER-DEFIBRILLATORS AND CARDIAC RESYNCHRONIZATION THERAPY
MECHANICALLY ASSISTED CIRCULATORY SUPPORT: VENTRICULAR ASSIST DEVICES
Definition, Epidemiology, and Staging of Heart Failure
Heart failure is a very common illness; 5.8 million Americans are affected.1,2 It is estimated that 1% of the population of Americans over the age of 65 are affected by heart failure, and 20% of hospital admissions in patients over age 65 are due to heart failure.3 Men and black Americans are affected more frequently. In 2008 670,000 new cases of heart failure were diagnosed in the United States, where there are nearly 1 million hospital discharges, 658,000 visits to the emergency department, over 3.4 million ambulatory outpatient visits, and 6.5 million hospital days annually for patients with a primary diagnosis of heart failure.1,2 Over 56,000 people in the United States died in 2007 with heart failure as the primary cause. The treatment of heart failure incurs a very large economic burden on the U.S. health care system. Estimated direct and indirect cost of heart failure in 2010 was $29 billion. The majority of this cost was related to the treatment of patients hospitalized with heart failure.
Epidemiologic data showed significant increases in both the incidence and prevalence of heart failure in the U.S. population in the 1990s,3 likely influenced by the aging of the population, a high prevalence of hypertension, and improved treatment and survival of patients with ischemic heart disease.4 However, there is evidence that hospitalizations for heart failure have declined during the past decade. In the United States, the rate of hospitalization for heart failure declined from 2845 per 100,000 patient-years in 1998 to 2007 per 100,000 person-years in 2008, a decline of 29%.5
Patients hospitalized with acute heart failure are usually elderly, with a mean age in the early 70s. In the United States, roughly 80% of patients will have a previous history of heart failure,6 and in European studies one third of patients had a new diagnosis of heart failure.7 From 40% to 55% of patients will have normal or relatively normal left ventricular systolic function. This occurs more commonly in women than in men. Coronary heart disease is present in 50% to 60% of patients and hypertension in 72% of patients. Comorbid conditions are common, including renal disease in 30% of patients, diabetes mellitus in 43%, and chronic obstructive pulmonary disease (COPD) in roughly 30%.6–8
In the American College of Cardiology/American Heart Association Guidelines for the diagnosis and management of chronic heart failure, four stages in the development of heart failure are recognized (Fig. 29.1). This staging system emphasizes the progressive nature of left ventricular dysfunction and heart failure, and describes evidence-based guidelines for therapy for each stage.4 It is important to realize that heart failure may be preventable. Several common medical conditions place patients at high risk for developing left ventricular dysfunction and the heart failure syndrome. Attention to and appropriate management of these conditions may prevent the development of heart failure. In addition, in patients who have left ventricular dysfunction but who have not yet developed heart failure, appropriate therapy can improve prognosis and prevent the development of severe heart failure.
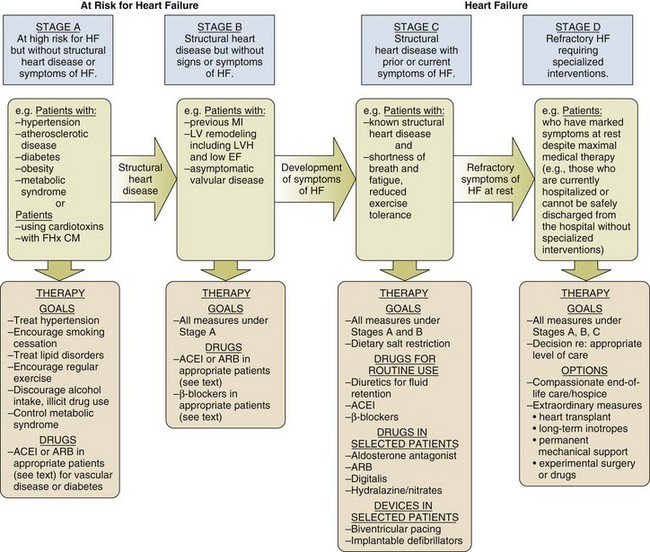
Stage A represents patients at high risk for heart failure but without structural heart disease or symptoms of heart failure. These patients include those with hypertension, atherosclerotic heart disease, diabetes, obesity, or the metabolic syndrome; those exposed to cardiotoxic medications; and those with a family history of cardiomyopathy. About 75% of patients who develop heart failure have antecedent hypertension.2 Stage B patients have structural heart disease but without signs or symptoms of heart failure. This group includes patients who have suffered a myocardial infarction, those with abnormal left ventricular ejection fraction but no symptoms of heart failure, and those patients with asymptomatic valvular heart disease. Stage C patients have structural heart disease with prior or current symptoms of heart failure, such as shortness of breath, fatigue, and reduced exercise tolerance. Lastly, stage D patients have refractory symptoms of heart failure at rest despite maximal medical therapy. They require specialized or extraordinary interventions such as cardiac transplantation, mechanical circulatory support, or end-of-life care.
Pathophysiology
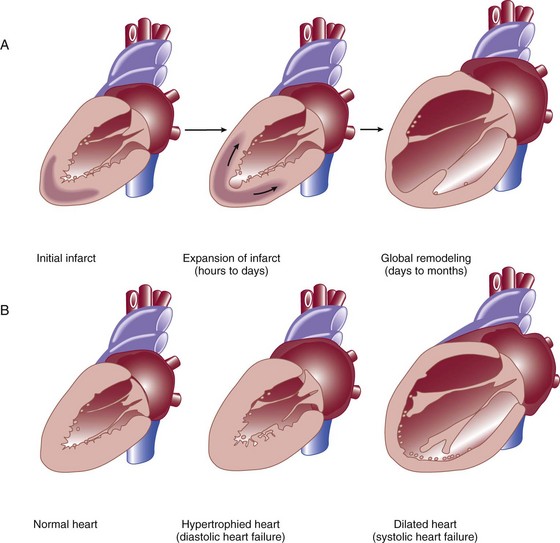
Heart failure can be due primarily to left ventricular systolic dysfunction or diastolic dysfunction, although both abnormalities are often present together. Right ventricular dysfunction may accompany left ventricular dysfunction or may be the primary problem. Systolic heart failure results from inability of the heart to expel blood normally owing to depressed left ventricular contraction. The left ventricle is often dilated. There is loss of myocytes and fibrosis, resulting in reduced left ventricular ejection fraction. Diastolic heart failure is caused by a reduction in left ventricular compliance, which leads to impaired diastolic filling, higher left ventricular diastolic pressure, and elevated pulmonary capillary wedge pressure (PCWP). Diastolic heart failure is also called heart failure with preserved ejection fraction (HFpEF) or heart failure with normal ejection fraction (HFnEF) (Fig. 29.2A and B).
Numerous conditions may cause damage to left ventricular myocardium and result in systolic heart failure. The most common of these conditions is atherosclerotic coronary artery disease with myocardial infarction and chronic myocardial ischemia. Other common causes include hypertension; familial and idiopathic cardiomyopathy; viral myocarditis; valvular heart disease, such as aortic stenosis, aortic insufficiency, and mitral regurgitation; peripartum cardiomyopathy; transient apical ballooning syndrome (takotsubo cardiomyopathy); and cardiomyopathy due to cardiotoxic cancer chemotherapy. Less common causes are alcoholic cardiomyopathy, diabetic cardiomyopathy, and a cardiomyopathy seen in some patients with hyperthyroidism or severe obesity.9–11
After myocardial damage and loss of myocytes occur, a process known as ventricular remodeling is initiated. This remodeling results in dilation of the ventricular chamber, a change in ventricular geometry from ellipsoid-like to a more spherical shape, worsening of left ventricular contractile force, and reduction in ventricular ejection fraction.3,12 In addition, the change in left ventricular geometry often leads to an increase in the size of the mitral annulus and altered physical relationships of the mitral valve structures. This results in increasing mitral regurgitation and worsening of the heart failure syndrome (see Fig. 29.2).
Remodeling is mediated by a series of maladaptive systemic responses, commonly termed neurohormonal activation. Two major systems involved in neurohormonal activation are the renin-angiotensin-aldosterone system and the sympathetic nervous system. Activation of the renin-angiotensin-aldosterone system leads to elevated levels of renin, angiotensin II, and aldosterone, which has deleterious consequences on cardiac function and hemodynamics.3,12 These effects include salt and fluid retention, endothelial dysfunction, vasoconstriction, myocyte hypertrophy, myocardial fibrosis, and myocyte apoptosis (programmed cell death). Sympathetic nervous system activation is in part mediated via decreased cardiac output, which results in tachycardia, increased myocardial oxygen consumption, and peripheral vasoconstriction.13 Renal effects of sympathetic nervous system activation lead to further activation of the renin-angiotensin-aldosterone system. Increased circulating norepinephrine levels also contribute to myocyte injury and death. Arrhythmias become common. A detrimental positive feedback loop is established, causing progressive deterioration in left ventricular structure and performance over time with progressive worsening of chronic heart failure.
Diagnosis
Heart failure is a clinical diagnosis, determined after evaluation of a patient’s symptoms and physical examination and supported by results of ancillary testing, such as chest x-ray and echocardiography. Typical symptoms of heart failure include shortness of breath at rest or with exertion, cough, swelling, orthopnea, and paroxysmal nocturnal dyspnea, which are due to pulmonary or systemic venous congestion. Fatigue, anorexia, and change in mental status are symptoms that may be caused by low cardiac output. Physical examination often reveals pulmonary rales; elevated jugular venous pressure; signs of cardiomegaly; cardiac murmurs, especially mitral regurgitation and third heart sound (S3) gallop; hepatic enlargement; and ascites and edema. An S3 gallop and elevated jugular venous pressure are the most specific signs of heart failure; absence of rales is not infrequent in patients with acute heart failure, and chest radiograph may not show obvious congestion.14 Manifestations of more severe heart failure include marked dyspnea at rest, possibly resulting in respiratory failure, cyanosis, cool extremities, reduced urine output and altered mental state. A careful evaluation for the presence of pulsus paradoxus is important if cardiac tamponade is suspected. However, these signs and symptoms are not specific for heart failure, and coexisting conditions such as obesity, chronic lung disease, and deconditioning can add uncertainty to the clinical diagnosis. Heart failure is often both underdiagnosed and overdiagnosed in outpatient and emergency department settings.
Studies have shown that serum assays of B-natriuretic peptide (BNP) and N-terminal pro-BNP are very helpful in improving the accuracy of diagnosing heart failure. BNP is a 32–amino acid peptide produced by cardiac myocytes in response to pressure-induced wall stretch and tension.15 Physiologic actions of BNP include arterial and venous dilation and natriuresis. A study of 1530 patients presenting to the emergency department with dyspnea showed that knowledge of BNP serum levels resulted in improved accuracy of the diagnosis of heart failure when compared to clinical judgment alone, from an initial range of 65% to 74%, up to 81% accuracy.16 BNP levels above 100 pg/mL had a sensitivity of 90% and a specificity of 76% for the diagnosis of heart failure and were very useful for discriminating patients with dyspnea due to uncomplicated lung disease who had BNP values below 100 pg/mL.17 Patients with BNP values below 100 pg/mL were very unlikely to have heart failure as the cause of dyspnea. Levels between 100 and 400 pg/mL can be seen in dyspneic patients with cor pulmonale, pulmonary hypertension not due to left ventricular failure, and acute pulmonary embolism. Heart failure is the likely diagnosis when levels are above 400 pg/mL. It should be emphasized that BNP levels should always be used in conjunction with all other clinical data to arrive at the correct diagnosis.
Evaluating BNP levels in heart failure patients in the emergency department was shown to decrease the need for hospitalization and decrease the need for intensive care unit (ICU) admissions, without affecting 30-day mortality rates.18 Total hospital stay was shortened by 3 days, cost of treatment was significantly reduced, and time to initiation of definitive therapy in the emergency department was shortened by 30 minutes.
BNP levels correlate with disease severity, as values are higher in patients with more severe heart failure and with worse left ventricular systolic function. Higher levels also have been correlated with a poorer prognosis, and can predict an increased rate of functional deterioration and a higher mortality rate.19 In a study of 114 patients admitted to the hospital with class IV heart failure, of all variables evaluated, predischarge BNP level was most strongly associated with death or readmission within 6 months, with BNP levels greater than 350 pg/mL having impressive sensitivity and specificity.20 BNP levels may also be elevated in patients with diastolic heart failure, those with renal failure, and stable patients with chronic left ventricular systolic dysfunction with compensated heart failure. BNP levels may initially be low in patients with “flash” pulmonary edema, as they may present to the hospital more quickly than the time required for significant rises in serum BNP to occur.
Serum levels of N-terminal (NT) pro-BNP can also be used for the diagnosis of heart failure. A precursor hormone, pro-BNP, is cleaved to form BNP and NTpro-BNP, which is physiologically inactive. NTpro-BNP has a longer half-life than BNP. It is cleared from the serum by the kidneys, so levels are higher in patients with coexisting renal disease. Levels of NTpro-BNP rise significantly in older populations. A cut off level of NTpro-BNP below 300 pg/mL yielded a negative predictive value of 98% for exclusion of the diagnosis of heart failure.21,22 The diagnosis of heart failure is very likely at levels over 450 pg/mL for patients under age 50, above 900 pg/mL in patients between 50 and 75 years old, and above 1800 pg/mL in patients over the age of 75.
Prognosis in Acute Heart Failure
Hospitalization for acute heart failure is associated with a poor prognosis. In-hospital mortality rate is high, reported at 4% to 8%. There is a 9% mortality rate at 60 to 90 days, and a 1-year mortality rate of 29%.1,6,7,23–25 The 90-day rehospitalization rate is around 30%, although only half of these rehospitalizations are caused by heart failure. In a European heart failure database, in-hospital mortality rate was 6.9%, 12-week readmission rate was 24%, and total mortality rate at 12 weeks was 13.5%.8 Randomized trials of pharmacologic therapy reported an annual mortality rate of 10% in patients with class II-III symptoms and 20% to 50% in class IV patients. In the most severe chronic heart failure group, patients awaiting cardiac transplantation, 1-year mortality rate was 75% with 2-year mortality rate of 92%.26 More recent data indicate a decline in the risk-adjusted inpatient mortality rate, from 5.5% in 2000 to 2.8% in 2007. Similar reductions were seen in both sexes and across all age groups.27 The prognosis for patients with cardiogenic shock due to acute myocardial infarction continues to be poor. Mortality rate remains 40% to 50% even with aggressive supportive therapy and emergency revascularization strategies.28
Several clinical factors have been shown to identify patients with a poorer prognosis and include lower left ventricular ejection fraction, low blood pressure on admission, and higher PCWP. In the United States ADHERE database registry of 62,275 admissions for heart failure, blood urea nitrogen (BUN) greater than 43 mg/dL, admission systolic blood pressure under 115 mm Hg, and creatinine level greater than 2.75 mg/dL were the three factors that indicated a poor prognosis for patients admitted with acute heart failure. The presence of an elevated BUN was associated with a fourfold increase in hospital mortality rates to 8.35%. The presence of all three factors yielded an in-hospital mortality rate of 19.8%24 (Box 29.1).
Other factors associated with increased mortality rates include hyponatremia, higher serum BNP level, and elevation of serum troponin.6,29,30 In ADHERE, higher levels of serum BNP on admission were associated with a higher in-hospital mortality rate, ranging from 1.9% in the lowest quartile to 6.0% in the highest quartile (BNP over 1730 pg/mL). The ability of BNP to predict prognosis was present even after multivariate adjustment for coexisting conditions, and was also true for patients with normal and abnormal left ventricular ejection fraction.31 Serum troponin is elevated in 6% to 10.4% of patients admitted with acute heart failure (with serum creatinine <2.0 mg/dL). Troponin elevation was associated with a lower blood pressure, lower ejection fraction, and longer hospital length of stay. Hypothetical mechanisms of troponin release in chronic heart failure include ischemia, cytokine activation, oxidative stress, and apoptosis. Hospital mortality rate was 8.0% in troponin-positive patients and 2.7% if troponin-negative. The ability of troponin elevation to predict mortality rate also was independent of other variables and was true even in patients with nonischemic causes of heart failure.32
The coexistence of kidney disease significantly worsens the prognosis of patients with heart failure. Kidney disease aggravates the tendency to volume overload and heart failure decompensation, and heart failure often worsens renal function. This complex interaction of severe heart failure and worsening kidney function is called the cardiorenal syndrome. In addition, high-dose diuretic therapy may also temporarily worsen renal function. There is increasing evidence that elevated systemic venous pressure causing renal venous congestion plays an important role in the pathogenesis.33 In a retrospective study of patients with acute heart failure who had right-sided heart catheterization, elevated central venous pressure was associated with reduced glomerular filtration rate (GFR) and higher all-cause mortality rate. These findings were independent of the measured cardiac output.34 Up to 50% of patients hospitalized with heart failure demonstrate a GFR less than 60 mL/minute/m2, and renal function may worsen in up to 30% of patients admitted with heart failure.35 Patients at particular risk for developing worsening renal function are those with lower left ventricular ejection fractions, lower blood pressure, diabetes mellitus, a history of hypertension, and older age. These patients have longer hospital stays and higher readmission and mortality rates. A meta-analysis of 16 large studies of heart failure patients revealed that 29% of heart failure patients had moderate to severe impairment of renal function. These patients had more than 100% increased relative mortality risk. Any degree of renal impairment had an approximately 50% increased relative mortality risk.36 The best treatment strategy for these patients is not clear, as patients with significant renal impairment have generally been excluded from large randomized pharmacologic heart failure trials.
Acute Heart Failure Syndromes
Initial Evaluation and Therapy
Acute heart failure is defined as the rapid onset of severe symptoms of heart failure, usually within hours to several days. Acute heart failure can occur with predominant systolic or diastolic dysfunction. Acute heart failure is often life-threatening and requires urgent diagnostic and therapeutic interventions, often simultaneously.23 Acute myocardial ischemia is a common cause and should always be considered in the differential diagnosis of this syndrome.
Several distinct clinical syndromes of acute heart failure can be identified23 (Table 29.1).
Table 29.1
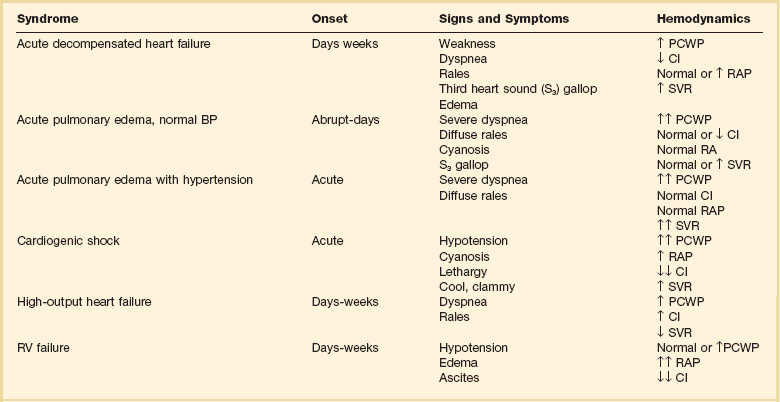
Adapted from Nieminen MS, Bohm M, Cowie MR, et al: Executive summary of the guidelines on the diagnosis and treatment of acute heart failure. The Task Force on Acute Heart Failure of the European Society of Cardiology. Eur Heart J 2005;26:384-416; and Gheorghiade M, Zannad F, Sopko G: Acute heart failure syndromes. Circulation 2005;112:3958-3968.
1. Acute worsening or decompensation of chronic heart failure symptoms, either in the setting of known chronic cardiovascular illness or de novo. These patients do not have shock or pulmonary edema. This is the most common presentation of acute heart failure requiring admission to hospital, occurring in approximately 70% of patients with acute heart failure.
2. Acute pulmonary edema with normal blood pressure, often caused by acute myocardial infarction or acute coronary ischemia.
3. Acute pulmonary edema associated with elevated blood pressure, often in the setting of chronic severe hypertension and chronic kidney disease. Pulmonary edema accounts for roughly 25% of acute heart failure admissions.
4. Cardiogenic shock with heart failure, usually due to acute myocardial infarction. This syndrome is the most severe presentation of acute heart failure and is associated with high in-hospital mortality rates. This accounts for about 5% of acute heart failure cases.
5. “High cardiac output heart failure” often induced by sepsis, hyperthyroidism, or cardiac arrhythmia. This type is the least common presentation, occurring in a small percentage of patients.
6. Acute right ventricular failure, occurring with acute right ventricular myocardial infarction, massive pulmonary embolism, or cardiac tamponade.
Physical examination often shows pulmonary rales and wheezes, an S3 gallop, and elevation of jugular venous pressure. Peripheral pulses are weak and thready with diminished cardiac output states. A low cardiac output is reliably predicted by a low “proportional pulse pressure,” which is calculated by the pulse pressure (systolic blood pressure minus diastolic blood pressure) divided by systolic blood pressure. A ratio below 0.25 predicts a cardiac index below 2.2 L/minute/m2.37,38 The skin may be cool and clammy and there may be evidence of cyanosis. Peripheral edema and ascites may indicate concomitant right ventricular failure of longer duration. Chest radiography is done urgently. An electrocardiogram (ECG) is needed to assess for signs of ischemia and infarction, and to evaluate for arrhythmia. Cardiac rhythm needs to be monitored continuously. Laboratory examination includes evaluation of hemoglobin and hematocrit, electrolytes, renal and liver function, thyroid profile, and cardiac biomarkers (troponin I or troponin T) to look for evidence of myocardial necrosis. BNP level assists in the diagnosis of heart failure in patients presenting with dyspnea, and can be followed serially to assess effectiveness of therapy. Pulse oximetry helps to assess oxygenation and pulmonary function. An arterial line is helpful in managing patients with hypotension or cardiogenic shock. Urgent two-dimensional echocardiography is essential to evaluate left ventricular size and function, right ventricular function, valve function, and the presence of pericardial effusion. Doppler echocardiographic assessment of valve stenosis and regurgitation and of hemodynamics is invaluable.
When patients are admitted to hospital with acute decompensation of chronic systolic heart failure, the specific reason for a patient’s deterioration must be searched for and corrected when possible (Box 29.2). Environmental factors such as excessive salt and fluid intake or alcohol consumption are common. Patient adherence to outpatient therapies must be assessed, as heart failure regimens often involve numerous medications. Emotional and physical stressors should be corrected when feasible.
Concomitant administration of medications for noncardiac conditions can have detrimental effects. A partial listing includes corticosteroids and nonsteroidal anti-inflammatory drugs (NSAIDs). These drugs can cause fluid retention and can aggravate hypertension. NSAIDs also interfere with the beneficial renal effects of angiotensin-converting enzyme (ACE) inhibitors and can interfere with the action of loop diuretics.39 The use of NSAIDs has been reported to increase the risk of hospitalization for heart failure by tenfold in patients with a history of heart failure.8 Metformin and thiazolindinediones can contribute to water retention and aggravate the symptoms and signs of heart failure. Cancer chemotherapies can cause myocardial damage. Cardiac toxicity due to anthracycline chemotherapy is well described. Tyrosine-kinase inhibitors, a newer class of cancer chemotherapy, are also being recognized as agents that can aggravate heart failure and cardiomyopathy.40 Cardiac medications such as calcium channel blockers and antiarrhythmic drugs can also have direct negative effects on left ventricular contractility. Calcium channel blockers are generally contraindicated in patients admitted with acute decompensated heart failure.23 Type 1A and 1C antiarrhythmic drugs are also contraindicated in patients with abnormal left ventricular systolic function.
There are few controlled-trial data to arrive at evidence-based guidelines for the treatment of acute heart failure. Many recommendations are based on small studies, experience, observation, and a general consensus of opinion.41 The goals of initial therapy are to improve symptoms, optimize blood pressure, lower PCWP, and improve cardiac output. Treatments to reverse or prevent myocardial injury are instituted, and a search for reversible causes of heart failure needs to occur. Optimization of other comorbid conditions is important, including hyperglycemia, renal disease, and pulmonary function.
Initial therapy includes supplemental oxygen and assessment of the need for ventilatory assistance with noninvasive positive airway pressure ventilation or endotracheal intubation. Noninvasive ventilation improves oxygenation and pulmonary compliance and decreases work of breathing. Endotracheal intubation may be required for patients with severe hypercapnia, acidosis, and respiratory muscle fatigue. The use of continuous oxygen administration alone was prospectively compared with the use of continuous positive airway pressure ventilation (CPAP) and noninvasive intermittent positive pressure ventilation (NIPPV) in 1069 patients who presented with acute cardiogenic pulmonary edema. No difference among these therapies was noted in the primary end point of death at 7 days, or in the secondary end point of death plus endotracheal intubation at 7 days. Noninvasive ventilation did result in more rapid improvement in dyspnea, tachycardia, hypercapnia, and acidosis. There was no difference in safety or efficacy between CPAP and NIPPV.42
Treatment of arrhythmias is essential. Rapid atrial fibrillation is a common problem in these patients. In the Euroheart Failure Study, 9% of patients hospitalized with acute heart failure had atrial fibrillation during the hospitalization, and 42% had a history of paroxysmal atrial fibrillation.43 Other studies report atrial fibrillation in 25% to 30% of hospitalized heart failure patients.36 Control of the ventricular response to atrial fibrillation is vitally important, especially in patients with diastolic heart failure. This control can be achieved rapidly with the use of intravenous beta blockers such as metoprolol or esmolol, parenteral digoxin, or intravenous amiodarone. Intravenous diltiazem can be used in patients whose left ventricular systolic function is known to be normal or near normal.
Indications for Invasive Hemodynamic Monitoring
Placement of a pulmonary artery (PA) catheter enables the clinician to accurately measure PCWP, cardiac output, and mixed venous oxygen saturation. It can also help assess the effectiveness of therapy. Whether to routinely use PA catheters to assess and manage patients with acute heart failure has been long debated. The ESCAPE trial evaluated the routine use of PA catheterization in patients hospitalized with acute exacerbation of chronic heart failure and left ventricular systolic dysfunction.44 There was no difference in the primary end point of days alive out of the hospital during 6 months after discharge in groups managed with or without a PA catheter. There were no significant adverse effects with PA catheter use in this study. There were no subgroups identified in which use of the PA catheter was beneficial. However, there was a trend noted in improving the initial diuresis, with less deterioration of renal function, in the PA catheter group. The authors concluded that there was no indication for the routine use of PA catheters in the setting of acute heart failure.
However, a PA catheter is often essential for the management of patients with acute severe heart failure. Findings on physical examination are often insensitive indicators of hemodynamic status. Indications for PA catheter use include cardiogenic shock, differentiating pulmonary from cardiac causes of dyspnea, hemodynamic assessment if one is unsure of the diagnosis or severity of heart failure by clinical assessment, worsening renal function, guiding parenteral vasodilator therapy, and in patients who are not improving with initially prescribed therapy.37 PA catheter placement is also necessary as part of the evaluation for cardiac transplantation or the implantation of a ventricular assist device (VAD) (Box 29.3).
Consecutive patients with severe heart failure were evaluated and classified according to hemodynamic measurements of PCWP and cardiac index. Patients were described as “dry” with average PCWP less than 17 mm Hg, or “wet” with PCWP reading of 29 mm Hg on average. The patients were also described as “warm” versus “cold,” based on a cardiac index of greater than 2.1 L/minute/m2 versus an index of less than 1.6 L/minute/m2. The severity of symptoms and findings on physical examination did not predict the hemodynamic status as defined by invasive monitoring. In addition, the hemodynamic picture did not predict the response to therapy and survival was similar in all four groups, except that patients with higher cardiac output and lower PCWP had slightly better outcomes than patients with low cardiac output and high PCWP.45
Tailoring pharmacologic therapy to hemodynamic measurements is often helpful and necessary to determine precise measurements of cardiac output and left ventricular filling pressure, in order to guide intensive intravenous drug therapy. Aggressive therapy tailored to the response in hemodynamic measurements has been advocated as an effective method to obtain more rapid and sustained improvement in patients with the most severe heart failure.46 When PCWP is reduced to less than 16 mm Hg, and right atrial pressure is reduced to less than 8 mm Hg, most patients will improve acutely and for the remainder of their hospitalization. Additional hemodynamic goals include reducing systemic vascular resistance to less than 1200 dynes × sec/cm5, raising cardiac index to greater than 2.6 L/minute/m2, and maintaining systolic blood pressure over 80 mm Hg. PCWP can be lowered to a normal value of 10 to 12 mm Hg in many patients with significant left ventricular dysfunction without untoward effects.46,47 In a group of patients referred for cardiac transplantation, a combination of aggressive parenteral therapy, targeted to optimal hemodynamics, followed by conversion to appropriate oral therapy, resulted in clinical improvement so that 30% of these patients were able to be removed from transplant lists.46
Pharmacologic Management of Acute Heart Failure
Goals of treatment of heart failure can be defined as short-term, to relieve dyspnea and reverse acute hemodynamic decompensation, and long-term, to prevent rehospitalization, improve functional status, and prolong survival. Additional goals include preserving renal function, preventing arrhythmias, and preventing myocardial necrosis in patients with ischemic and nonischemic disease. Pharmacologic therapies to prevent or attenuate chronic remodeling should be instituted or strengthened prior to discharge from the hospital, as these have been shown to improve long-term survival (Box 29.4).
Intravenous Diuretics
Although no randomized clinical trials exist, the use of loop diuretics is supported by a long history of clinical success. These agents increase renal excretion of salt and water. The onset of action of intravenous bolus furosemide is 30 minutes, and the drug peaks at 1 to 2 hours. The half-life of the medication is 6 hours, so twice daily dosing is usually required.39 Other loop diuretics often used are bumetanide and torsemide (Tables 29.2 and 29.3). Several small clinical trials suggested that a constant infusion of a loop diuretic resulted in superior diuresis when compared to intermittent bolus dosing,48,49 although other studies did not confirm this.50 A large, randomized controlled study was performed in patients hospitalized with acute decompensated heart failure who were taking a high dose of furosemide prior to admission. Patients were randomized within 24 hours of admission to bolus dosing or constant infusion, and usual daily outpatient dose (given intravenously) versus high dose (2.5 times their usual daily dose). No significant differences were noted in predefined outcomes between bolus and infusion administration. However, the high-dose group had better relief of dyspnea and more fluid and weight loss than the lower-dose group; also, 23% of the high-dose group had a significant deterioration in renal function, but at 60 days there was no difference in renal function between the two groups.51
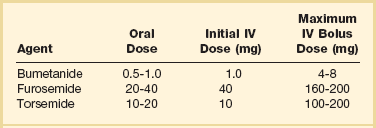
Table 29.3
Treatment of Refractory, Diuretic-Resistant Heart Failure
| To Loop Diuretic, add: | Dose |
| Hydrochlorothiazide | 25-50 mg once or twice daily |
| or metolazone | 2.5-5.0 mg once or twice daily |
| or spironolactone | 12.5-50 mg once daily |
An association between high-dose loop diuretics and a worse prognosis has been observed. There is concern that this may be mediated by further activation of the renin-angiotensin-aldosterone system by loop diuretics. However, in a retrospective analysis utilizing propensity matching, hospital mortality rate was not different between low-dose and high-dose diuretic groups.52 It is likely that poorer outcomes associated with high-dose diuretics is not due to the drug itself but reflects a greater severity of heart failure illness with concomitant renal disease.53
Patients with chronic heart failure with or without renal dysfunction may exhibit resistance to loop diuretics, defined as an acute reduction in diuretic efficacy after repeated loop diuretic dosing. With chronic loop diuretic use, there is an increase in sodium reabsorption in the distal nephron and stimulation of aldosterone release. In edematous states there is delayed oral absorption of the drug. With renal dysfunction there may be reduced levels of drug delivered to the renal tubule. This resistance is associated with a poorer prognosis.23 Diuretics that act distally in the renal tubule, such as metolazone or hydrochlorothiazide, or aldosterone blockers such as spironolactone can be added.4 Combination diuretic therapy induces a greater diuresis than simply increasing the dose of loop diuretic further. The response may be delayed for 48 to 72 hours. Combining diuretics often augments diuresis even in the setting of significant chronic kidney disease, and a good response can be expected in over 70% of patients. Particular attention must be given to following potassium, sodium, chloride, and magnesium levels when combination diuretic therapy is prescribed54 (Table 29.4).
Table 29.4
Continuous Intravenous (IV) Infusion of Loop Diuretics
| Diuretic | Dose |
| Bumetanide | 1 mg IV load, then 0.5-2 mg/hr infusion |
| Furosemide | 40 mg IV load, then 10-40 mg/hr infusion |
| Torsemide | 20 mg IV load, then 5-20 mg/hr infusion |
Adapted from Nieminen MS, Bohm M, Cowie MR, et al: Executive summary of the guidelines on the diagnosis and treatment of acute heart failure. The Task Force on Acute Heart Failure of the European Society of Cardiology. Eur Heart J 2005;26:384-416.
Vasopressin Inhibitors
The use of novel diuretics, vasopressin inhibitors, has been evaluated in clinical trials of treatment of acute heart failure.55 Vasopressin is a hormone synthesized in the hypothalamus; its major effect is to control free water clearance. It acts through V1a receptors in vascular smooth muscle and myocardium, leading to peripheral and coronary vasoconstriction, myocyte hypertrophy, and positive inotropy. Vasopressin also acts through V2 receptors at the renal tubule collecting ducts to cause free water retention and hyponatremia. Levels of vasopressin are increased in patients with chronic heart failure, and higher vasopressin levels correlate with worse heart failure severity. Vasopressin release is stimulated by changes in serum osmolality and cardiac output and leads to further vasoconstriction and retention of free water.56 Inhibition of vasopressin’s effects would have theoretic benefits in patients with heart failure.57 In contrast to loop diuretics, inhibition of vasopressin theoretically would not cause hypotension or neurohormonal activation, and would not aggravate cardiac arrhythmias due to electrolyte depletion.
Conivaptan is a vasopressin antagonist that inhibits V1a and V2 receptors. Tolvaptan and lixivaptan are antagonists selective for the V2 receptor. These medications increase urine volume and free water excretion, with a rise in the serum sodium concentration. The use of conivaptan in patients with class III-IV heart failure was associated with increased urine output, and decreases in PCWP and right atrial pressure, without changes in cardiac output.8 Oral use of tolvaptan was associated with fluid loss and diuresis without change in heart rate, blood pressure, or serum creatinine.
In a large, multicenter, placebo-controlled randomized trial called EVEREST, tolvaptan was administered to patients hospitalized with heart failure. There were no adverse consequences on heart rate, blood pressure, or serum electrolytes and there was more rapid improvement in dyspnea and signs of heart failure when compared to usual therapy.58 Hyponatremia improved. Hemodynamic effects included rapid reduction in PCWP and right atrial pressure.59 However, at 10-months follow-up after hospitalization, there was no improvement in mortality rates or readmission rates.60 Vasopressin inhibitors are approved for treatment of severe hyponatremic states but are not approved for use in heart failure.
Parenteral Vasodilators (Table 29.5)
Intravenous vasodilator therapy is often added to diuretic therapy to obtain more rapid improvement in severe heart failure. A clear indication for vasodilators is in patients with severe hypertension and pulmonary edema. Their use should also be considered in patients who are not responding to intravenous diuretics combined with standard oral therapies. Blood pressure response to these medications needs to be carefully monitored because hypotension is a common effect. Improvement in hemodynamics has been obtained with aggressive intravenous vasodilator therapy using intravenous nitroprusside, intravenous nitroglycerin, or nesiritide. The choice of agents depends on matching the patient’s clinical picture and hemodynamics with the predicted effects of each vasodilator.61,62
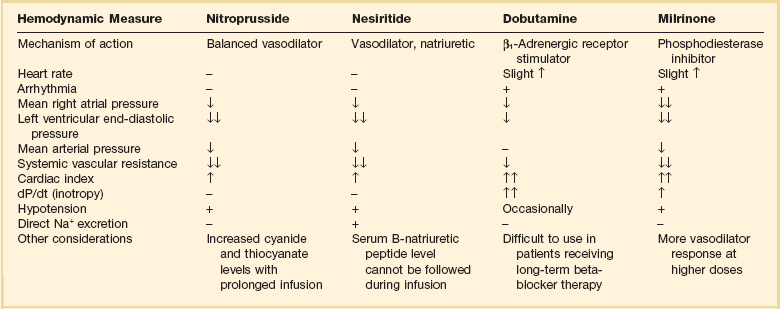
Nitroprusside
Intravenous sodium nitroprusside is a powerful venous and arterial dilator. It is a drug of choice in treating hypertension-related heart failure with pulmonary edema and severe heart failure due to acute mitral regurgitation. The use of nitroprusside requires hospitalization in the ICU and invasive monitoring with a PA catheter and arterial line. This drug causes a significant reduction of afterload and preload, leading to decreased right atrial pressure, decreased systemic vascular resistance, decreased mean systemic blood pressure, decreased PCWP, and increased cardiac index in patients with heart failure and left ventricular dysfunction. Limitations of nitroprusside use include inducing a coronary “steal” syndrome in patients with active coronary ischemia.39 In addition, toxic metabolites can accumulate with more prolonged administration. In patients with significant hepatic dysfunction, thiocyanate levels rise, and in patients with renal dysfunction, cyanide is generated. Dosage range is 0.3 to 5.0 µg/kg/minute.
Nesiritide (B-natriuretic Peptide)
Human BNP can be manufactured by recombinant DNA technology and is available as an intravenous medication, nesiritide, for heart failure therapy. BNP is a hormone produced by ventricular and atrial myocytes in response to stretch from cardiac chamber dilatation. Hemodynamic effects include venous and arterial dilation, coronary vasodilation, and natriuresis. Reduction in PCWP and right atrial pressure exceeding the effects of intravenous nitroglycerin, when compared directly, was reported.61 It is not proarrhythmic and does not induce tolerance.63 It may potentiate the effects of loop diuretics. Significant hypotension may limit its use in some patients.39 Because the hypotensive effects of nesiritide are less marked than nitroprusside, nesiritide can be used without invasive hemodynamic monitoring and can be initiated in emergency department settings. Nesiritide is initiated as an intravenous bolus dose of 2 µg/kg followed by infusion of 0.01 µg/kg/minute.
Nesiritide was compared to dobutamine in patients with severe heart failure. Nesiritide infusion was associated with less tachycardia and ventricular arrhythmia.64 Other nonrandomized studies suggested a trend toward improved survival and lower rehospitalization rates with nesiritide.63 A meta-analysis of three randomized trials of nesiritide suggested a slight increase in mortality rates in patients given nesiritide versus a placebo control group, possibly mediated through an adverse effect on renal function.65,66 A retrospective review of patients receiving intravenous vasoactive medications for acute heart failure indicated mortality rates, adjusted for clinical variables, were equivalent for patients receiving nitroglycerin or nesiritide. Patients who received intravenous nesiritide or intravenous nitroglycerin had a lower in-hospital mortality rate compared with patients who received dobutamine or milrinone.67
In view of these conflicting results of retrospective analyses, a randomized, placebo-controlled international study, called ASCEND-HF, was performed to evaluate the effect of nesiritide on relief of dyspnea, mortality rate, and renal function in patients hospitalized with acute heart failure; 7141 patients were enrolled. Over 90% of patients received loop diuretics and 15% received vasodilator therapy. Patients who received nesiritide had slightly better relief of dyspnea at 6 and 24 hours, although this did not meet a prespecified significant improvement in dyspnea score. There was no significant difference in hospital mortality rates, 30-day mortality rate, or death or rehospitalization at 30 days. There was no significant worsening of renal function in the nesiritide group. Hypotension was significantly more common in the nesiritide goup (26.6%) compared with control group (15.3%). The authors concluded that nesiritide was not useful for routine use in the management of patients with severe heart failure.68
Inotropic Drugs (see Table 29.5)
Dobutamine
In patients with heart failure, β-receptors may be chronically downregulated. Therefore, the effects of dobutamine may be attenuated in chronic heart failure patients. Dobutamine may be detrimental in patients with active coronary ischemia or following myocardial infarction due to increased myocardial oxygen demand and oxygen consumption. Ventricular arrhythmias are associated with dobutamine use.64 Tolerance to the effects of dobutamine has been demonstrated in patients with infusions lasting more than 24 hours, due theoretically to induction of β-receptor downregulation.69
Considerations Regarding the Use of Inotropes
Inotropic drugs are used in patients with reduced LV systolic function and low cardiac output with persistent symptomatic hypotension and signs of end-organ hypoperfusion or cardiogenic shock. Clinically, this is often manifest as systolic blood pressure less than 90 mm Hg, narrow pulse pressure, cool and clammy extremities, anorexia, obtundation, and oliguria. Hemodynamic findings that may lead to use of inotropes include cardiac index less than 2.0 L/minute/m2, PCWP greater than 20 mm Hg, and right atrial pressure greater than 10 mm Hg.70
The choice of milrinone versus dobutamine depends on the specific clinical circumstances.71 Dobutamine tends to cause a slight rise in heart rate and has little effect on mean arterial pressure, whereas milrinone often lowers systemic arterial pressure due to more prominent lowering of systemic vascular resistance.72,73 Patients who do not respond to dobutamine may have a favorable response to milrinone. In the setting of acute heart failure, milrinone is used more often then dobutamine in view of its more potent vasodilator properties. In addition, its effects are not primarily mediated through β-receptors, which is an important consideration in patients receiving concomitant beta-blocker therapy. However, dobutamine has a much shorter half-life than milrinone, so dobutamine-induced hypotension can be more rapidly reversed by discontinuing the drug, making dobutamine a somewhat safer drug in the acute setting. Several studies have been done evaluating the usefulness of routine inotropic therapy, comparing milrinone or dobutamine with placebo. The consistent conclusion has been that inotropic agents are not useful for routine use in patients with decompensated heart failure, and in fact may worsen short-term prognosis. The use of a 48-hour infusion of milrinone was evaluated as routine therapy in patients admitted with class III-IV heart failure, when inotropic therapy was not felt to be essential. When compared to standard therapy without milrinone, no improvement in symptom relief, hospital length of stay, or rehospitalization rate within 60 days was demonstrated.74 Milrinone was associated with an increased incidence of hypotension and atrial arrhythmias. In the FIRST study of class III-IV heart failure patients, an average 14-day infusion of dobutamine was associated with an increased risk of morbid events and higher short-term mortality rates.75 No clinical studies have shown improved short-term or medium-term outcomes with inotropic therapy. The use of inotropic agents has been consistently associated with a worse prognosis for survival.39 These negative outcomes with inotropic agents are felt to be related to their propensity to stimulate sympathetic nervous system activation, increasing myocardial oxygen demand, exacerbating serious cardiac arrhythmias, increasing myocardial ischemia, and furthering myocyte loss. Stimulation of chronic hibernating myocardium may also result in myonecrosis.
If use of an inotrope is necessary, the shortest duration of therapy should be attempted (i.e., less than 72 hours). Current ACC/AHA Guidelines for evaluation and management of chronic heart failure indicate that long-term intermittent infusions of a positive inotropic drug as therapy for symptomatic systolic dysfunction is contraindicated.4 Continuous intravenous infusion of an inotropic drug can be used as a bridge to therapy with mechanical circulatory assist devices or cardiac transplantation. Continuous inotrope infusion can also be recommended for palliation of symptoms in patients with refractory end-stage heart failure (stage D). These patients will have been deemed poor candidates for more advanced therapies. In this setting, quality of remaining life takes precedence over prolonging life. In one report of patients with refractory end-stage heart failure, median survival of patient on continuous inotrope infusion was 3.4 months, with 26% of patients surviving to 6 months.76 The decision to use inotropic agents in this circumstance is one that should be carefully individualized.
Vasopressors
Dopamine
Dopamine effects include increased renal blood flow (at low doses, 1-5 µg/kg/minute), increased myocardial contractility and chronotropy through stimulation of β-receptors (doses of 3-7 µg/kg/minute), and vasoconstriction at higher doses (5-20 µg/kg/minute). Dopamine is a less useful agent for treatment of heart failure because its effects result in tachycardia, coronary vasoconstriction, increased afterload, and increased oxygen consumption. Dobutamine generally will lead to a greater rise in cardiac output than dopamine. Dopamine can be used when significant hypotension is part of the hemodynamic picture, when it is necessary to restore adequate arterial pressure for end-organ perfusion. Although dopamine at low doses is frequently used as add-on therapy to inotropic agents in an attempt to increase renal blood flow and augment diuresis, no controlled trials have demonstrated dopamine’s usefulness in this setting. No significant benefit of “renal dose dopamine” has been shown in preventing acute renal failure in high-risk patients or in the treatment of established renal failure.69
Norepinephrine
Norepinephrine is a sympathomimetic agent with strong α-agonist and weak β-agonist effects. In patients with heart failure, norepinephrine’s main effect is to raise blood pressure by increasing systemic vascular resistance with little effect on cardiac output. It will increase myocardial oxygen demand. Its use in the setting of heart failure is restricted to patients with the most severe hypotension, unresponsive to dopamine, or in patients with complicating illnesses such as sepsis.69 Norepinephrine should be weaned and discontinued as early as possible. Dosage range is 0.2 to 1 µg/kg/minute.
Norepinephrine and dopamine were compared in a randomized controlled trial of 1679 patients with shock, 280 (16.7%) of whom had cardiogenic shock. In the entire group, there was no difference in survival at 28 days if patients were treated with norepinephrine or dopamine. However, in the subgroup with cardiogenic shock (a predefined subgroup analysis), dopamine use was associated with a significantly increased mortality rate. In addition, in the entire group, dopamine was associated with significantly higher rates of arrhythmia (24% versus 12.4% with norepinephrine), especially atrial fibrillation, which led to stopping the vasopressor more frequently. There was also a higher rate of severe arrhythmia with dopamine (6.1%) compared with norepinephrine (1.6%).77 Based on the results of this study, norepinephrine is the preferred vasopressor in patients with cardiogenic shock.
An algorithm for the approach to the evaluation and therapy of acute heart failure is presented in Figure 29.3.
Ultrafiltration
A new approach to the treatment of acute heart failure is venovenous ultrafiltration. This process removes iso-osmolar extracellular fluid via a convection process and is not associated with changes in serum electrolytes.78 Anticoagulation is utilized during the process. Newer ultrafiltration systems utilize peripheral arm veins, and central venous access is not required. In a study of 40 patients admitted with heart failure, usual care for heart failure with diuretic therapy was compared with usual care combined with ultrafiltration. At 24 hours, average fluid loss with diuretic therapy was 2838 mL, compared with 4650 mL with ultrafiltration. Weight loss and improvement in dyspnea was similar with the two therapies. Ultrafiltration was not associated with significant changes in heart rate or blood pressure.79 In another study, 20 patients with acute decompensated heart failure with renal insufficiency and diuretic resistance were treated with an 8-hour course of ultrafiltration. Over 24 hours, an average of 8650 mL of fluid was removed, with an average weight loss of 6 kg during hospitalization. Renal function remained stable, and there was no associated hypotension.78
The UNLOAD trial investigated the use of ultrafiltration compared to intravenous diuretic therapy in 200 patients admitted with acute decompensated heart failure. Ultrafiltration was associated with more rapid weight loss and fluid loss. At 90 days, there were fewer rehospitalizations for heart failure in the ultrafiltration group, without change in mortality rates. There was no difference with respect to renal function in the two groups.80
A trial was conducted that compared ultrafiltration with standard intravenous diuretic therapy in patients admitted to hospital with acute decompensated heart failure, persistent congestion, and worsening renal function. Fluid was removed with UF at a rate of 200 mL per hour. This trial showed worse outcomes with ultrafiltration at 96 hours of therapy. Renal function worsened with UF and did not with diuretics. Total weight loss was unchanged between the two groups. Serious adverse effects were more common in the UF group, mainly renal failure, bleeding, and catheter-related complications. There was no difference between the two groups in symptom relief, mortality, or rehospitalization at 60 days.80a At this time, ultrafiltration is reserved for the relief of severe congestion in patients who are refractory to aggressive diuretic therapy and should not be used to replace diuretics.81 Further studies will determine if this therapy should be used more routinely in the management of acute heart failure.
Transition to Chronic Pharmacologic Therapy for Severe Heart Failure
The medical treatment of chronic systolic heart failure is based on results of many large, randomized, placebo-controlled trials (see later) and is indicated for almost all causes of chronic left ventricular dysfunction. Pharmacologic agents should be started when left ventricular dysfunction is first diagnosed. Therapy is aimed at optimizing fluid balance and reversing the neurohormonal activation responsible for left ventricular remodeling and progressive decline in left ventricular function.82 Long-term prognosis is directly related to the process of “reverse remodeling.” After patients have improved with acute therapies, medical treatments are instituted to address the long-term goals of improvements in functional status, exercise tolerance, and survival. Standard drug regimens combine several classes of medication, all of which have been shown in large, randomized controlled trials to reduce mortality rate, reduce the rate of rehospitalization, and decrease the risk of sudden arrhythmic death. Doses of these medications are optimized during hospitalization. Chronic adherence with these medications is more consistent when they are initiated in-hospital.
Diuretics
Loop diuretics are routinely used in patients with signs or symptoms of fluid retention.4 Diuretics are continued once patients are euvolemic to prevent reaccumulation of fluid. A flexible dosing schedule, based on daily weights and close telephone contact with a heart failure treatment team member, can be very effective in maintaining a euvolemic state while reducing the frequency of side effects.
Furosemide is the most common loop diuretic used. Bumetanide or torsemide may be helpful in patients with suboptimal responses to furosemide, due to their more consistent absorption after oral administration. Metolazone or a thiazide diuretic can be used in addition to a loop diuretic in patients with more severe heart failure due to their synergistic effects. Patients must be periodically monitored for side effects of these agents including azotemia, hypokalemia, alkalosis, hyponatremia, and hypomagnesemia (Table 29.6).
Table 29.6
Oral Diuretics Recommended for Use in the Treatment of Fluid Retention in Chronic Heart Failure
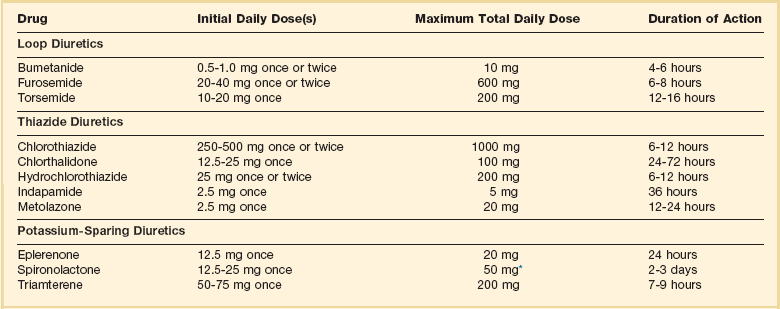
*Higher doses may occasionally be used with close monitoring of serum creatinine and potassium levels.
Adapted from Hunt S, Abraham WT, Chin M, et al: ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult—Summary Article. A report on the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for Evaluation and Management of Heart Failure): Developed in collaboration with the American College of Chest Physicians and the International Society of Heart and Lung Transplantation: Endorsed by the Heart Rhythm Society. Circulation 2005;112:1825-1852.
Beta Blockers (Table 29.7)
Catecholamine levels are increased in heart failure, and higher levels correlate with worse disease severity. Catecholamines have direct negative effects on the myocardium including induction of myocyte hypertrophy and apoptosis.13 Clinically, these effects are evident as left ventricular dilatation, increased ischemia, increased peripheral vasoconstriction, and cardiac arrhythmia.
Table 29.7
Adapted from Hunt S, Abraham WT, Chin M, et al: ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult—Summary Article. A report on the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for Evaluation and Management of Heart Failure): Developed in collaboration with the American College of Chest Physicians and the International Society of Heart and Lung Transplantation: Endorsed by the Heart Rhythm Society. Circulation 2005;112:1825-1852.
Multiple large trials using beta blockers in thousands of patients with chronic heart failure, or in post–myocardial infarction patients with ejection fractions below 40%, have demonstrated significant and consistent reductions in the need for repeat rehospitalization for heart failure. Mortality rates are also significantly improved. The BHAT study showed a relative 26% reduction in mortality rates at 2 years in post–myocardial infarction patients treated with propranolol.83,84 The CIBIS II trial using bisoprolol showed a 34% relative risk reduction in hospitalizations and mortality rates at 16 months of therapy.85 The MERIT-HF study showed a relative reduction of 33% in these end points at 12 months using metoprolol succinate.86 Patients with more severe heart failure, symptom class III-IV, with severely reduced ejection fractions below 25% were studied in the COPERNICUS trial. These patients began therapy with carvedilol during hospitalization. At mean follow-up of 10.4 months, a 35% relative mortality rate reduction was seen, with improvement in mortality rates beginning as early as 3 weeks after initiation of therapy87 (Fig. 29.4). In these trials, beta blockers were not discontinued more frequently than placebo for perceived side effects, and there was no increased risk of heart failure exacerbation due to beta-blocker therapy when compared to placebo, even in the early phases of drug administration.88 Several trials with carvedilol have shown an approximate 6% absolute increase in left ventricular ejection fraction after a minimum of 2 years of therapy.89
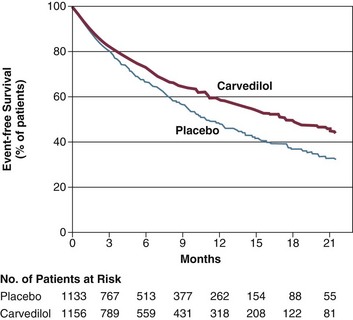
For patients who are already taking beta-blocker therapy and who are hospitalized with acute decompensation of heart failure, beta blockers should be continued. Withdrawal of this medication was associated with higher short-term mortality rates and more frequent rehospitalizations for heart failure in several observational studies.90 In patients with a severe heart failure exacerbation, reduction of the chronic dose by 50% can be considered. Beta blockers should only be stopped in the presence of symptomatic hypotension, cardiogenic shock, severe bradycardia, or heart block. If beta blockers are temporarily withheld, they should be reinstituted prior to discharge unless there are ongoing specific contraindications.91,92
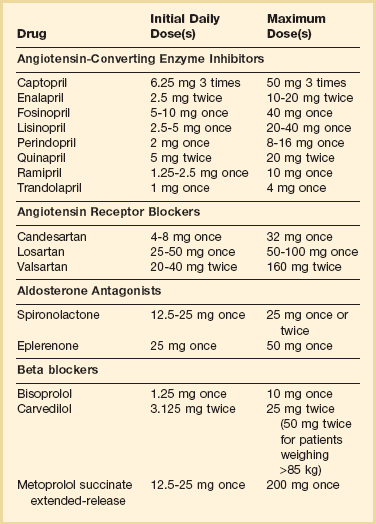
Angiotensin-Converting Enzyme Inhibitors (see Table 29.7)
These agents act to counteract the effects of activation of the renin-angiotensin system by blocking the conversion of angiotensin I to angiotensin II, inhibiting the deleterious effects of angiotensin II and aldosterone. Many studies have shown benefits of ACE inhibitor therapy in post–myocardial infarction patients as well as patients with cardiomyopathy and heart failure. The SOLVD trial using enalapril in patients with class II-III heart failure and ejection fractions below 35% showed a 10% relative risk reduction in mortality rate at 3.5 years.93 Enalapril given to patients less ill, with asymptomatic left ventricular dysfunction (ejection fractions under 35%) in the companion SOLVD trial showed a reduction in the clinical diagnosis of heart failure and a statistically significant reduction in heart failure hospitalizations at 3 years.94 A meta-analysis performed of 32 trials involving 7105 patients, using captopril, enalapril, ramipril, quinapril, or lisinopril, found that ACE inhibitors reduce the risk of death and hospitalization due to heart failure.95 These results indicate that the positive effects of ACE inhibitors are likely to be a class effect, and not specific to a particular agent.
Side effects of these agents include cough, worsening renal function in patients with underlying renal disease or renal artery stenosis, angioneurotic edema, and hyperkalemia. The dose of ACE inhibitors should be increased as renal function and blood pressure allow. Studies have shown that medium doses of ACE inhibitors, when compared to low doses, significantly reduce hospitalization rates for heart failure. However, higher doses given routinely do not significantly reduce cardiovascular events further.96 Additional improvement in symptoms and mortality rates is thus best achieved by adding on beta blocker and other heart failure therapy rather than increasing ACE inhibitors to the highest doses.97
In a study of class II-III chronic heart failure patients over age 65, with an ejection fraction of less than 35%, the importance of first beginning heart failure therapy with an ACE inhibitor or a beta blocker was studied. The primary end point was time to death or hospitalization for heart failure. Initiation of the beta blocker bisoprolol was not inferior to the strategy of starting therapy with the ACE inhibitor enalapril. There was also no difference in safety.98
Angiotensin Receptor Blockers (see Table 29.7)
The RESOLVD trial, using candesartan in heart failure patients with a mean ejection fraction of 27%, showed equivalent mortality rates and similar exercise tolerance and functional class to patients treated with enalapril at 3.5 years.99 ELITE II, a study using losartan compared with captopril in patients with ejection fractions under 40%, showed no difference in mortality rates or congestive heart failure admissions. Losartan was better tolerated because of the lower incidence of problematic cough.100 The ValHeft 2001 trial showed that valsartan as a substitute for ACE inhibitor therapy was associated with a relative 33% risk reduction in mortality rate when compared with placebo.101 In CHARM, candesartan was prescribed for patients with ejection fractions under 40%. A 17.5% relative risk reduction in cardiovascular death and congestive heart failure admissions was seen when this ARB was used as a substitute for ACE inhibitors.102 When candersartan was added to therapy with ACE inhibitors and beta blockers, a small 10% relative risk reduction was seen, without increased mortality rates.103
ARBs are an appropriate choice for patients who cannot be maintained on ACE inhibitors because of side effects such as cough. Patients with angioneurotic edema during ACE inhibitor therapy have often been successfully treated with ARBs without developing this complication.104,105 Adding ARB therapy onto ACE inhibitors and beta blockers may achieve a small additional benefit, at the risk of more renal dysfunction and hyperkalemia.
Aldosterone Antagonists (see Table 29.7)
Aldosterone antagonists counteract the salt and water retention caused by aldosterone. In addition, aldosterone is felt to be involved in the progressive myocardial fibrosis that occurs as part of the remodeling process. In the RALES trial, the aldosterone antagonist spironolactone was given to patients with severe, class III-IV heart failure with ejection fraction below 35%. A 24% relative risk reduction in mortality was seen in treated patients over 2 years, with reduced cardiovascular death and reduced need for rehospitalization106 (Fig. 29.5). Thus, these agents are effective at improving outcomes in patients with the most severe chronic heart failure. Aldosterone antagonists are contraindicated in patients with renal insufficiency and creatinine levels over 2.5 mg/dL (or GFR under 30 mL/minute) or in patients with baseline potassium levels over 5.0 mmol/L. Spironolactone should be initiated at low doses, such as 12.5 mg daily or every other day, especially in elderly patients.
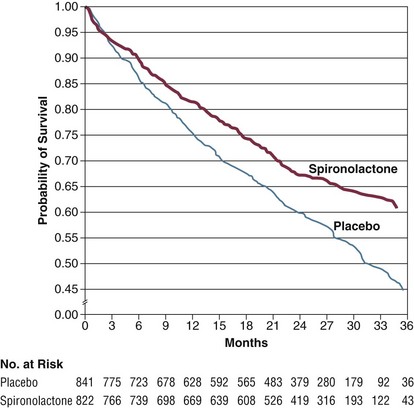
An analysis of a large Canadian health care database showed a substantial increase in the frequency of spironolactone use after the RALES study was published. This increased use was temporally associated with a two- to threefold increased rate of hospitalization for hyperkalemia.107 This highlights the need for careful monitoring of serum electrolytes after aldosterone blockers are initiated.
Eplerenone is an aldosterone blocker similar to spironolactone except that it has less antiandrogen effects and thus is free of the side effect of gynecomastia in men. It has been found to be effective in treating heart failure following acute myocardial infarction (see later). More recently, it was also found to be very effective in patients with chronic heart failure. In EMPHASIS-HF, a placebo-controlled randomized study of 2737 patients with LV ejection fraction below 35% and class II heart failure, eplerenone was added to therapy with ACE inhibitors and beta blockers, at a target dose of 25 to 50 mg daily. Mean follow-up was 21 months. Eplerenone reduced the occurrence of the primary end point of cardiovascular death plus hospitalization for heart failure from 25.9% to 18.3% (P < 0.001), and with improvement in other clinical end points. Serious hyperkalemia that necessitated drug discontinuation occurred in only 1.1% of patients.108
Aldosterone blockade appears to result in beneficial effects in heart failure independent of its potassium sparing and diuretic effects.109 Aldosterone blockers are an important therapy for heart failure and should be routinely added to ACE inhibitors and beta blockers in patients with symptomatic heart failure with low left ventricular ejection fraction.
Combination Hydralazine/Isosorbide Dinitrate
Combination therapy with the vasodilators hydralazine and isosorbide dinitrate (ISDN) was associated with improvement in mortality rate when compared to placebo in one study in the era prior to the advent of ACE inhibitors and beta-blocker therapy for heart failure.110 A retrospective analysis of this study suggested that African-American patients may have benefited preferentially. To evaluate this finding prospectively, the A-Heft study enrolled over 1000 self-described African-American patients with class II-IV heart failure and ejection fractions below 45%. The use of the combination of hydralazine with ISDN, titrated to a dose of 75 mg hydralazine plus 40 mg ISDN given three times daily, was associated with a 40% relative risk reduction in mortality at 10 months and a 33% relative risk reduction in first hospitalization for heart failure. This therapy was added on to treatment with beta blockers, ACE inhibitors, and spironolactone.111 Hydralazine-ISDN is approved for treatment of African-American patients with heart failure and left ventricular systolic dysfunction.
Digoxin
Digoxin works by inhibiting the myocyte sodium-potassium pump, leading to increased intracellular calcium levels and increased inotropy. It also has vagotonic effects. However, digoxin is a relatively weak inotropic agent. The use of digoxin in heart failure was studied in large numbers of patients in the Digitalis Investigation Group (DIG) trial. A decreased need for rehospitalization for heart failure was seen with this therapy. However, there was no improvement in overall mortality rate.112 A post-hoc analysis of this data concluded that patients with lower serum digoxin levels (0.5-0.9 ng/dL) did have lower mortality rates and lower rates of heart failure hospitalization when compared to those with levels greater than 0.9 ng/mL.113
Digoxin is indicated only for patients with symptomatic heart failure, stages C and D.4 It is also useful in heart failure patients with atrial fibrillation to help control the ventricular rate. Digoxin is not useful in the setting of acute heart failure, and it should be avoided in patients with hyopkalemia, bradycardia, or heart block.
Coronary Heart Disease and Heart Failure: Special Considerations
Coronary artery disease can lead to acute heart failure via several different mechanisms. Acute ischemia leads to impaired myocardial relaxation, acute diastolic dysfunction, and sudden elevation of left ventricular filling pressure. Acute ischemia can also cause “stunning,” defined as myocardial dysfunction due to severe and prolonged ischemia without infarction, which may persist for days or weeks after normal blood flow is restored but which eventually recovers function. Acute myocardial infarction may cause myocardial necrosis and acute left ventricular systolic dysfunction due to loss of contractile tissue. Papillary muscle ischemia, infarction, and rupture result in acute, severe mitral regurgitation and acute heart failure. Infarction, necrosis, and rupture of the intraventricular septum result in left-to-right shunting and acute heart failure with cardiogenic shock. Chronic ischemia can cause myocardial dysfunction and systolic heart failure without infarction, a process termed “hibernation.” Often, acute myocardial ischemia is superimposed on a ventricle impaired by chronic ischemia and infarction, so multiple mechanisms are usually present in patients with chronic coronary disease presenting with acute heart failure.23,114
In a European observational study of acute heart failure complicating acute coronary syndromes (ACS) in patients without previous history of heart failure, 13% of ACS patients presented with heart failure on admission.115 Heart failure developed later during hospitalization in an additional 5.6% of patients. The incidence of acute heart failure of 15.6% was identical in patients with ST-segment elevation myocardial infarction and non–ST-segment elevation myocardial infarction. Eight percent of patients with unstable angina developed heart failure. Prognosis for these patients was poor, with in-hospital mortality rates of 12% for patients with heart failure on admission and 17.8% if heart failure developed during the hospitalization. This represents a three- to fourfold increase in mortality rates compared with patients with ACS without heart failure.
Several important points need to be stressed regarding acute heart failure and ACS. Acute heart failure may develop even without evidence of significant left ventricular systolic dysfunction on echocardiography and without acute necrosis documented by myocardial enzyme determinations. The majority of patients with ACS and heart failure do not have left ventricular systolic dysfunction at discharge, and only a minority of these patients develop chronic heart failure. These patients not only have high in-hospital mortality rates but also have a high rate of morbidity and mortality after discharge, with 8.5% additional mortality rate at 6 months and 6-month rehospitalization rate of 24%.114
Early aggressive pharmacologic and interventional reperfusion strategies are indicated and should always be considered in these patients. Acute treatment should include intravenous diuretics, intravenous nitroglycerin, and beta-blocker therapy. Intra-aortic balloon counterpulsation should be used in patients with signs and symptoms of continued ischemia despite aggressive medical therapies or who have cardiogenic shock. Urgent coronary angiography is indicated to determine the most appropriate reperfusion strategy.114
Acute Heart Failure Following Myocardial Infarction
In a large registry of 5573 consecutive patients with acute myocardial infarction, 42% of patients had heart failure or left ventricular systolic dysfunction during hospitalization.116 These patients tended to be older, were more commonly women, and were more likely to have had previous myocardial infarction or coronary bypass surgery. Comorbid conditions were present more commonly, including peripheral arterial disease, hypertension, diabetes mellitus, or previous stroke. In-hospital mortality rate for these patients was 13%, versus 2.3% for patients with acute myocardial infarction but without heart failure or left ventricular dysfunction. Mortality rate ranged 13% to 21% for patients with lung congestion and left ventricular ejection fraction below 40%. Other complications also occurred more commonly in these patients, including atrial and ventricular arrhythmias, reinfarction, and stroke (Figs. 29.6 and 29.7).
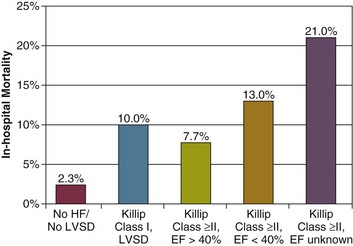
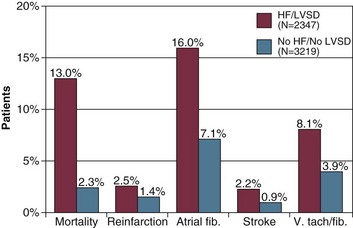
In patients with acute heart failure due to acute myocardial infarction, rapid reperfusion is the cornerstone of therapy and may be achieved with thrombolytic therapy, acute coronary angioplasty, or urgent coronary artery bypass surgery. In the GRACE Study, revascularization therapies were associated with a lower mortality rate in patients with ACS and acute heart failure.115
In patients who develop heart failure or significant left ventricular systolic dysfunction following myocardial infarction, a number of pharmacologic therapies have been shown in large placebo-controlled randomized studies to improve mortality rate and reduce repeat hospitalization. Beta blockers should be part of standard post–myocardial infarction therapy in these patients. In the CAPRICORN study, the use of carvedilol led to a significant 23% relative risk reduction.117 SAVE was the first trial to show that the use of an ACE inhibitor, captopril, was beneficial in post–myocardial infarction patients with ejection fractions below 40%.118 There was a 5% absolute mortality rate reduction after 42 months of follow-up. In AIRE, treatment with the ACE inhibitor ramipril, started 3 to 10 days after myocardial infarction in patients with heart failure, resulted in a significant mortality rate benefit at an average of 15 months of follow-up.119 Another trial showed that treatment with the ACE inhibitor trandolapril following myocardial infarction and an ejection fraction below 35% resulted in a significantly improved survival at 2 to 4 years of follow-up.120 In VALIANT, a high dose of the ARB valsartan was as effective as an ACE inhibitor in improving survival and reducing cardiovascular morbidity.121
Aldosterone blockers have also been shown to be effective in improving prognosis in patients with heart failure or left ventricular dysfunction following myocardial infarction and are recommended for these patients. The EPHESUS study showed that eplerenone significantly reduced all-cause mortality rates and repeat hospitalizations at 16-month follow-up.122 Reduction in mortality rate and sudden cardiac death was noted as early as 30 days after initiation of eplerenone.123
Heart Failure with Preserved Left Ventricular Ejection Fraction (Diastolic Heart Failure)
Heart failure can occur in patients with normal or relatively normal left ventricular systolic function. It is now recognized that in up to 50% of patients hospitalized with acute heart failure, the primary cardiac abnormality is diastolic dysfunction. There is no consistent definition or diagnostic test for diastolic heart failure. The European Society of Cardiology has proposed that this diagnosis can be applied to patients who present with clinical signs and symptoms of heart failure, who have normal left ventricular systolic function, and who have abnormal parameters of diastolic filling as demonstrated on Doppler echocardiography or invasive evaluation of diastolic function. A more practical definition includes the presence of heart failure and normal systolic function in the absence of primary valve disease.82,124
Diastolic heart failure occurs due to impairment of left ventricular filling and abnormal left ventricular relaxation. Pathophysiology includes myocyte hypertrophy, increased amounts of collagen in extracellular matrix, increased wall stiffness and wall thickness, abnormal left ventricular geometry with increased left ventricular mass-to-volume ratio, fibrosis, and impaired compliance. There is impaired left ventricular filling at normal left atrial pressures, and thus an increase in left ventricular filling pressure is necessary to maintain cardiac output. This leads to chronic pulmonary venous hypertension and pulmonary congestion. Patients with diastolic heart failure usually demonstrate normal indices of left ventricular systolic performance and contractility.125–128
Diastolic heart failure is a common entity. The reported frequency of diastolic heart failure in the general heart failure population varies according to the definition of diastolic heart failure used. Some reports include patients with mildly abnormal systolic function (i.e., ejection fraction over 40%), whereas others restrict inclusion to patients with ejection fraction above 50%. Prevalence is also affected by the demographics of study populations, including age of patients, inpatient and outpatient status, the proportion of African Americans and women studied, and whether patients were evaluated at an academic referral center or a community-based setting.124 In the general population of Olmsted County, Minnesota, moderate to severe diastolic dysfunction was seen in 7% of echocardiogram studies.2 The prevalence of diastolic heart failure in population-based studies was 3.1% to 5.5% of patients over age 65.124 The prevalence of diastolic heart failure may be increasing.129 As compared with patients with systolic heart failure, patients with diastolic heart failure tend to be older, are more commonly women, and have a higher prevalence of hypertension and a lower incidence of coronary artery disease. In various studies, up to 50% of hospital admissions for heart failure in the United States are for patients with diastolic heart failure. In an international observational database of 4953 hospitalizations for heart failure, 25% of patients had a left ventricular ejection fraction over 45%.130 Up to 73% of patients with diastolic heart failure are women, with an incidence of hypertension or hypertensive heart disease of 64% to 78%. Reported incidences of concomitant diabetes (33-46%) and coronary artery disease (26-43%) are also high. Other common comorbid conditions include atrial fibrillation, abnormal renal function, and obesity.131–134
Patients who present with heart failure due to diastolic dysfunction have a history and physical examination indistinguishable from patients with systolic heart failure. Presenting blood pressure may be higher, and acute “flash” pulmonary edema may be more common. Common exacerbating factors include severe hypertension, medication noncompliance, myocardial ischemia, and valve dysfunction. In one study, no precipitating factors could be identified in 50% of these patients.129
The prognosis of patients with diastolic heart failure is serious. Mortality rate is probably not as high as in patients with systolic left ventricular dysfunction and heart failure, although one study found similar adjusted and nonadjusted mortality rates at 30 days and 1 year for hospitalized patients with heart failure and left ventricular ejection fraction less than 40% or greater than 50%.135 In-hospital mortality rate is around 4%,106 or four times age-matched control subjects.131,136 Annual mortality rate is variable and likely depends on the frequency of comorbid conditions in the cohort of patients studied; it has been reported in the range of 1.3% to 17.5%.127 Readmission rates are as high as 50% at 1 year. Factors that identify patients with a worse prognosis include renal dysfunction, worse functional class, male gender, and advanced age.136
Unlike systolic heart failure, there are no placebo-controlled randomized studies that have demonstrated effective therapy for diastolic heart failure. The CHARM-Preserved Study compared the use of the ARB candesartan with placebo in treating patients with heart failure and preserved systolic function. After an average 3-year follow-up, the mortality rate from cardiovascular cause or admission for heart failure was similar in the two groups. There was a modest impact on preventing hospitalizations due to heart failure in patients treated with candesartan.137 The I-PRESERVE trial utilizing irbesartan in over 4000 patients with heart failure and left ventricular ejection fraction over 45% showed no beneficial effect on mortality rates or heart failure rehospitalizations.138
Beta blockers can be useful to reduce heart rate and thus improve diastolic filling time. They are also effective medications for hypertension and coronary ischemia. Similarly, calcium channel blockers may improve symptoms of diastolic heart failure by treating hypertension and ischemia and improving diastolic relaxation. The use of ACE inhibitors and ARBs is helpful to lower blood pressure, reduce myocardial fibrosis, and block the adverse effects of the activation of the renin-angiotensin system. In a retrospective study of patients hospitalized with heart failure and ejection fraction greater than 40%, patients who were prescribed an ACE inhibitor had better quality of life scores, improved functional class, and lower adjusted mortality rate.133 Aldosterone antagonists such as spironolactone or eplerenone treat hypertension and may reduce left ventricular hypertrophy and fibrosis.113 Therapy with positive inotropic drugs or digoxin are not useful in patients with diastolic heart failure4,139,140 (Table 29.8).
Table 29.8
| Recommendation | Class* | Level of Evidence† |
| Physicians should control systolic and diastolic hypertension in accordance with published guidelines. | I | A |
| Physicians should control ventricular rate in patients with atrial fibrillation. | I | C |
| Physicians should use diuretics to control pulmonary congestion and peripheral edema. | I | C |
| Coronary revascularization is reasonable in patients with coronary artery disease in whom symptomatic or demonstrable myocardial ischemia is judged to be having an adverse effect on cardiac function. | IIa | C |
| Restoration and maintenance of sinus rhythm in patients with atrial fibrillation might be useful to improve symptoms. | IIb | C |
| The use of β-adrenergic blocking agents, angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, or calcium antagonists in patients with controlled hypertension might be effective to minimize symptoms of heart failure. | IIb | C |
| The use of digitalis to minimize symptoms of heart failure is not well established. | IIb | C |
*I: There is evidence or general agreement that therapy is beneficial, useful, and effective; IIa: there is conflicting evidence about the usefulness of therapy, but the weight of the evidence is in favor of efficacy; IIb: there is conflicting evidence about the usefulness of therapy, and use is less well established by evidence/opinion.
†A: Data are derived from multiple randomized clinical trials or meta-analysis; C: only consensus opinion of experts, case studies, or standard of care.
Adapted from Hunt S, Abraham WT, Chin M, et al: ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult—Summary Article. A report on the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for Evaluation and Management of Heart Failure): Developed in collaboration with the American College of Chest Physicians and the International Society of Heart and Lung Transplantation: Endorsed by the Heart Rhythm Society. Circulation 2005;112:1825-1852.
Acute Myocarditis and Heart Failure
Myocarditis is defined as inflammation of heart muscle and is an uncommon cause of acute heart failure. Patients with acute myocarditis can develop severe left ventricular dysfunction and acute heart failure. In a minority of cases, myocarditis can present with fulminant heart failure and cardiogenic shock, with a high mortality rate.141,142 A large body of experimental animal data indicates that viral myocarditis results in activation of immune mechanisms, which can also result in chronic dilated cardiomyopathy and chronic heart failure.141
Myocarditis is a diagnosis made on clinical grounds and should be suspected in patients who present with new-onset heart failure, with or without antecedent flu-like symptoms. Chest pain may be present. Elevated leukocyte count, elevated erythrocyte sedimentation rate, elevated creatinine kinase and troponin levels, and ECG changes suggestive of myocardial ischemia or infarction may be seen but are not always present. Endomyocardial biopsy may be used to aid in the diagnosis of myocarditis. However, histologic findings of both inflammation and myocyte necrosis in biopsy specimens have been very insensitive in making the diagnosis and have a high degree of interobserver variability. In patients suspected of having myocarditis on clinical grounds, only 10% to 67% of patients have had positive biopsies in reported series.141,142 Cardiac magnetic resonance imaging has more recently been shown to be an important tool in the diagnosis of myocarditis.143,144
Pharmacologic management of heart failure due to myocarditis is similar to the management of heart failure from other causes. Diuretics, ACE inhibitors, and beta blockers should be prescribed. Many patients will experience significant spontaneous improvement in left ventricular function during the first 6 months after diagnosis. The use of corticosteroids and other immunosuppressive drugs is controversial. Early studies suggested a small improvement in left ventricular ejection fraction with the use of corticosteroids.145 Other studies suggest targeting immunosuppressive drugs to patients with signs of immune activation.146 The Myocarditis Treatment Trial showed no significant benefit with immunosuppressive therapy, although there are several procedural problems with this study.147 We generally recommend a 1- to 3-month trial of corticosteroids and azathioprine in patients with myocarditis and left ventricular dysfunction who are not improving spontaneously after 1 to 2 months with conventional heart failure therapies or who continue to worsen acutely. If the immunosuppressive regimen produces an improvement in ejection fraction, it can then be tapered over a 6- to 12-month period.142
Myocarditis with acute, severe heart failure and cardiogenic shock is termed fulminant myocarditis. These patients often require treatment with intravenous vasodilators and inotropes and may be candidates for implantation of a VAD as a bridge to recovery or transplantation. When supported aggressively, most patients will recover fully with normal ventricular function, and fulminant myocarditis has a good late prognosis. Therefore, aggressive supportive therapy, including the use of VADs, is indicated, even in gravely ill patients.148 Cardiac transplantation may be necessary for patients who do not improve.
Device Therapy: Implanted Cardioverter-Defibrillators and Cardiac Resynchronization Therapy
Prophylactic antiarrhythmic drug therapy aimed at preventing sudden arrhythmic cardiac death has been shown to be ineffective and may even worsen prognosis. In the Cardiac Arrhythmia Suppression Trial (CAST), patients with coronary artery disease, a history of myocardial infarction, and frequent premature ventricular contractions on baseline Holter monitoring had an increased mortality rate when treated with the antiarrhythmic drugs encainide or flecainide, despite good suppression in frequency of arrhythmia on follow-up Holter monitoring.149 This adverse effect on mortality rate is presumed due to the known potential for these drugs to worsen arrhythmia (“proarrhythmia”). Therefore, class IA and IC antiarrhythmic drugs quinidine, procainamide, disopyramide, flecainide, and propafenone are not useful for prevention of sudden death and are contraindicated in patients with heart failure and left ventricular systolic dsyfunction.
Amiodarone was also studied for primary prevention of sudden death, as the propensity for proarrhythmia is less than with type I antiarrhythmics; 675 patients with heart failure and ejection fractions below 40% were randomized to treatment with amiodarone or placebo, with no survival difference between the two groups at 45 months.150 There was a trend toward reducing mortality rates with amiodarone in patients with nonischemic cardiomyopathy. In another study, 1486 post–myocardial infarction patients with ejection fractions under 40% were randomized to amiodarone or placebo with no difference in mortality rates in 21 months, although there was a suggestion of a reduction in deaths due to arrhythmia.151 Unlike the findings in CAST, there was no observed increased risk of death with amiodarone. Thus, this medication is not helpful in the prevention of sudden cardiac death, but it is considered safe for treatment of supraventricular arrhythmias in heart failure patients.
Implanted Cardioverter-Defibrillator Therapy
The ICD was compared to antiarrhythmic drugs for secondary prevention of sudden cardiac death in the AVID study. These patients had been resuscitated from cardiac arrest or survived ventricular tachycardia associated with syncope and had ejection fractions under 40%. In the study 1016 patients were randomized to receive an ICD or antiarrhythmic drugs (over 90% received amiodarone). There was a statistically improved survival rate with ICD therapy at 1, 2, and 3 years of follow-up.152 In two other studies of similar patients, a nonsignificant reduction in rates of all-cause mortality and arrhythmic death was seen with the ICD when compared to amiodarone,153,154 and increased mortality rate was seen with propafenone therapy (a class IC antiarrhythmic).154 Published guidelines recommend ICDs as first-line therapy, in preference to antiarrhythmic drugs, in patients with ischemic and nonischemic cardiomyopathy who have survived a cardiac arrest or an episode of sustained ventricular tachycardia.82
ICD therapy has also been evaluated as primary prevention of arrhythmic death in patients with coronary heart disease and previous myocardial infarction with left ventricular dysfunction, without symptomatic clinical arrhythmias, and also in patients with nonischemic cardiomyopathy. In MADIT I, post–myocardial infarction patients with ejection fractions below 35% and asymptomatic nonsustained ventricular tachycardia on monitoring were studied with electrophysiologic testing. Patients were enrolled in this study if they had inducible ventricular tachycardia not suppressed with antiarrhythmic therapy. At 2 years of follow-up, total mortality rate was significantly reduced from 39% in the medically treated group (standard medical therapy with or without amiodarone) to 15% in the ICD group.155 In the MUSTT study, a similar cohort of patients showed an improvement in mortality rate from 55% in patients treated with medication to 24% in patients treated with ICDs at 5 years.156 In MADIT II, the patient population was extended to include patients with a history of myocardial infarction and ejection fractions of less than 30%. Importantly, in this study, neither ventricular arrhythmias seen on monitoring nor those induced at electrophysiologic study were necessary for inclusion. A statistically significant improvement in total mortality rate was seen in patients treated with ICDs, from 19.8% to 14.2% at 4 years157. It should be noted that class IV heart failure patients were not included in any of these studies.
The Sudden Cardiac Death Heart Failure Trial (SCD-HeFT) enrolled patients with functional class II and III heart failure with left ventricular ejection fractions under 35%. This study differed from MADIT I and II in that a greater number of patients (48%) had nonischemic cardiomyopathy. Primary prevention of sudden cardiac death was compared among standard heart failure pharmacologic therapy, standard therapy combined with amiodarone, and standard therapy plus an ICD. After a mean follow-up of 45.5 months, mortality rate was improved from 29% in the medical group and 28% in the amiodarone group to 22% in the ICD group, which was statistically significant. This improvement was particularly notable in class II heart failure patients.158 Benefits of ICD therapy were seen in patients with both ischemic and nonischemic cardiomyopathy. In the DEFINITE study, the use of the ICD as primary prevention was evaluated in 458 patients with nonischemic cardiomyopathy who had ventricular arrhythmia seen on routine monitoring. After a mean follow-up of 29 months, total mortality rate was not significantly reduced (17.5% with placebo vs. 12% in ICD, P = 0.08) but prevention of sudden death was significantly reduced.159 A meta-analysis of all studies in which ICDs were used as primary prevention in patients with nonischemic cardiomyopathy indicated a statistically significant 31% relative risk reduction in favor of ICDs.160
Thus, the approved use of ICD therapy for prevention of sudden cardiac death has been extended to prophylactic primary prevention therapy in patients with symptomatic heart failure, class II-III and left ventricular ejection fractions under 35% due to both ischemic as well as nonischemic cardiomyopathy.82
Cardiac Resynchronization Therapy
In many patients with heart failure, there is abnormal timing and coordination of systolic motion of the intraventricular septum and left ventricular free wall, termed dyssynchrony. This often coexists with His-Purkinje conduction system disease, with marked QRS prolongation on ECG.161 In fact, left bundle branch block pattern with long QRS duration is associated with an increase in all-cause mortality rate in heart failure patients. Pacemaker therapies have been developed to correct left ventricular dyssynchrony, delivering timed pacing to both intraventricular septum and left ventricular free wall. This is termed biventricular pacing or cardiac resynchronization therapy (CRT). Standard dual-chamber transvenous leads are placed in the right atrium (in the absence of chronic atrial fibrillation) and right ventricle. The left ventricular free wall is paced via a third electrode passed through the coronary sinus into an epicardial lateral cardiac vein. Alternatively, a left ventricular lead can be placed directly on the epicardium of the lateral wall of the left ventricle via thoracoscopy. The pacemaker is programmed to coordinate timing of septal stimulation via the right ventricle with left ventricular lateral wall stimulation161,162 (Fig. 29.8).
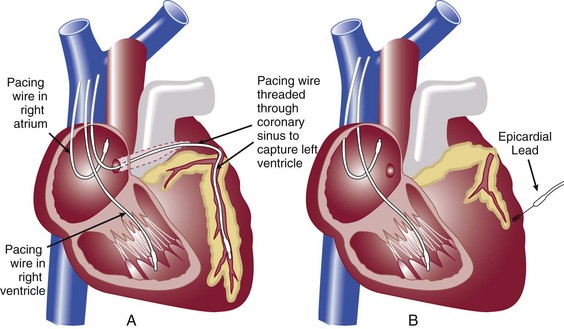
In the MIRACLE trial, 453 patients with ejection fractions under 35% and class III-IV heart failure and QRS duration greater than 130 ms were treated with standard medical therapy or resynchronization therapy plus medical therapy. At 6 months of follow-up, CRT resulted in a statistically significant improvement in 6-minute walking distance, quality of life score, and functional class, with fewer hospitalizations for recurrent heart failure.161 No significant mortality rate improvement was noted during a relatively short follow-up period. There was an 8% rate of unsuccessful left ventricular lead placement and a 1.2% incidence of serious complications of implantation of the pacemaker device, including coronary sinus dissection or perforation. A meta-analysis of three major resynchronization trials in 1634 patients concluded that chronic resynchronization therapy was associated with a statistically significant 51% reduction in death from progressive heart failure.163
In the CARE-HF study, resynchronization therapy (without ICD) was evaluated in 813 patients with class III-IV heart failure due to left ventricular systolic dysfunction, with ejection fraction under 35% and QRS duration greater than 150 ms or QRS duration greater than 120 ms with signs of dyssynchrony on echocardiography. The primary end point of all-cause mortality rate plus unplanned hospitalization from cardiovascular causes was reduced from 55% in medically treated patients to 39% in resynchronization patients (P < 0.001). All-cause mortality rate was reduced from 30% to 20% with resynchronization therapy (P < 0.002). Mean follow-up was 29.4 months. Measures of quality of life were improved, and ejection fractions improved.164 Thus, resynchronization therapy alone, without an ICD, can reduce mortality rates as well as improve symptoms in chronic heart failure patients.
Devices combining biventricular pacing and ICD capabilities were evaluated in patients with systolic heart failure and low ejection fraction. These patients often have indications for both devices. The rationale is to decrease morbidity and mortality rates associated with progressive heart failure as well as decrease mortality rates associated with sudden life-threatening ventricular arrhythmia. Biventricular pacing does not interfere with appropriate ICD detection and termination of ventricular arrhythmias.165 The COMPANION trial enrolled 1520 patients with advanced heart failure, functional class III-IV, in sinus rhythm, who had the ECG finding of QRS duration greater than 120 ms. Therapies compared were best pharmacologic heart failure therapy, best therapy along with biventricular pacing, and best therapy along with combination biventricular pacing and ICD. Successful implantation occurred in 87% to 91% of patients in the latter two groups, with a procedural mortality rate of 0.5% to 0.8%. The primary end points of death from any cause or hospitalization for any cause, followed over 12 months, were 68% in the drug therapy group and 56% in both device groups, a significant difference. One-year rates of death or hospitalization due to cardiovascular cause were 60% in the drug group, with a relative risk reduction of 25% in the biventricular pacing group and 28% in the biventricular pacing/ICD group. The authors concluded that there was a significant reduction in heart failure hospitalizations with biventricular pacing, and there was an additional significant reduction in mortality rate when ICD therapy was added. Improvement occurred in both patients with ischemic and those with nonischemic cardiomyopathy.166
CRT was then studied in class II and III heart failure patients with ejection fraction below 30%; 1798 patients with QRS duration greater than 120 ms during native conduction, or 200 ms during ventricular pacing, were treated with an ICD alone or with ICD plus CRT. At a mean follow-up of 40 months, there was a significant improvement with CRT in the primary end point of all-cause mortality rate plus hospitalization for heart failure (33.2% with CRT-ICD versus 40.3% with CRT alone, P < 0.001). Therefore, CRT has been successfully extended to class II heart failure patients with low ejection fraction.167
Mechanically Assisted Circulatory Support: Ventricular Assist Devices
It is estimated that over 100,000 patients in the United States have advanced functional class IV heart failure. These patients have a very poor short-term prognosis, even with appropriate pharmacologic and device therapy.168 Cardiac transplantation offers an effective therapy for many patients with end-stage heart failure and is currently associated with a 1-year survival rate of over 80%. However, the pool of donor hearts is currently less than 3000 per year in the United States, and it is not increasing.169,170 There is a role for mechanically assisted circulatory support in these patients utilizing VADs, which support or replace the function of the failing heart. VADs provide normal cardiac output and flow to vital organs.
Patients with heart failure who may benefit from VAD therapy are those with the worst prognosis, such as patients with refractory cardiogenic shock. In patients with chronic heart failure, factors that indicate a very poor prognosis include inability to wean from parenteral inotropic drugs, persistent class IV symptoms despite optimal medical and device therapy, progressive cardiorenal dysfunction precluding the use of ACE inhibitors, and hypotension preventing the use of ACE inhibitors or beta blockers.168 Peak oxygen consumption of less than 12 mL/kg/minute on cardiopulmonary stress testing is also a poor prognostic indicator. Specific acute clinical circumstances in which VADs can be considered include cardiogenic shock following acute myocardial infarction or cardiac surgery, fulminant myocarditis, or severe postpartum cardiomyopathy.171 A classification system to define the severity of heart failure illness and the urgency of the need for mechanically assisted circulatory support has been adopted (Fig. 29.9).

There are five clinical scenarios for the use of mechanical circulatory assistance:
1. VADs can be used as a bridge to cardiac transplantation. A mortality rate of 30% has been reported in patients listed for and awaiting heart transplantation, so VADs may enable many patients to survive to obtain transplantation.
2. VADs can be used as a “bridge to decision.” These patients have urgent need for support but have severe medical conditions exacerbated by heart failure that make transplantation an initial poor option. These medical conditions may improve after a period of improved circulation with a VAD so that transplantation may become feasible.
3. VADs may be used as a bridge to definitive surgical therapy, such as in patients with severe ischemic heart disease who require coronary revascularization, which may lead to recovery of myocardial function.
4. A small minority of patients may require VAD therapy as a bridge to recovery, when improvement in cardiac function with nonsurgical approaches is expected. An example of this is patients with acute fulminant myocarditis.148 In addition, there have been case reports of patients with subacute cardiomyopathy and severe heart failure who received VAD support for months and had improvement in left ventricular function such that the VAD was removed and patients survived without cardiac transplantation.172 However, only 5% to 10% of patients with chronic heart disease will improve sufficiently for VAD removal.
5. Finally, current VADs have a low incidence of pump failure and can be used as long-term support, or “destination therapy.”
Catheter-based pumps can be used for acute hemodynamic support and can provide cardiac output up to 3.5 L/minute.171 These systems are implanted percutaneously and provide circulatory support for several days. The TandemHeart device (CardiacAssist, Pittsburgh, PA) requires placement of a catheter via a transatrial septal puncture. The Impella pump (Abiomed, Danvers, MA) is entirely intracorporeal and pulls blood from the left ventricle and pumps through the catheter tip in the aorta at a flow of 2.5 L/minute.173
The first VADs to be used were large extracorporeal devices, inserted via a midline sternotomy, that provided pulsatile flow. Because there were many moving parts, there was a high incidence of pump failure. Current VADs are smaller, are placed intracorporeally, and provide continuous, nonpulsatile flow. These have significantly improved long-term durability and are suitable for long-term support. Battery packs are small and wearable, so patients with VADs have freedom of movement and can participate in rehabilitation and normal daily activities (see Fig. 29.10). These devices do require systemic anticoagulation and antiplatelet therapy. A study comparing continuous flow VADs with pulsatile flow devices was reported in 2009; 200 patients were studied who had advanced heart failure and were ineligible for cardiac transplantation. The continuous flow device was associated with improved 2-year survival, fewer adverse events, fewer rehospitalizations, and improved quality of life. The incidence of pump failure requiring VAD replacement was reduced by 87%.174 The Heart Mate II continuous flow VAD was approved for use in 2008 and was approved to be used as destination therapy in 2010.
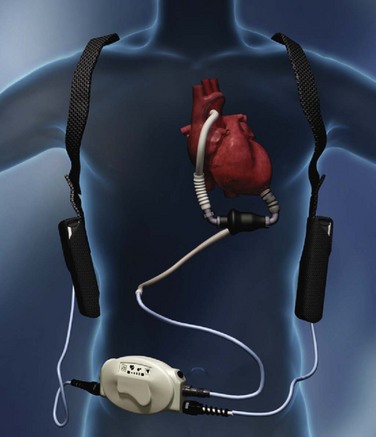
A very important issue in VAD therapy is appropriate patient selection. Hemodynamic indications for use include persistent hypotension with systolic blood pressure less than 80 mm Hg, PCWP greater than 20 mm Hg, and cardiac index less than 2 L/minute/m2 despite maximal pharmacologic support. Patient characteristics that define a worse prognosis with VAD therapy include older age, malnourishment (low albumin), renal dysfunction (i.e., serum creatinine >3.0 mg/dL or BUN >51 mg/dL), hepatic dysfunction (elevated transaminase, bilirubin, and international normalized ratio), coaogulopathy, neurologic deficits, low mean pulmonary artery pressure (reflecting right ventricular dysfunction), and anemia.168,175 Contraindications to the use of VADs include severe chronic obstructive pulmonary disease, need for hemodialysis, aortic insufficiency, or aortic valve mechanical prosthesis.168 Risk scores have been developed to attempt to define appropriate patient selection but need prospective validation. The major complications of VAD therapy include right ventricular failure, sepsis, thromboembolic complications including stroke, and bleeding often associated with an acquired von Willebrand factor deficiency. In patients who were deemed at lower risk, there was a 93.7% survival rate to hospital discharge and 81.2% 1-year survival rate. The highest risk patients had only 13.7% hospital survival rate and 10.7% 1-year survival rate.175
VADs are more frequently being used as long-term or “destination therapy.” The REMATCH trial compared the pulsatile flow VAD as destination therapy with standard optimal heart failure medical therapy in 129 patients with class IV heart failure, ejection fraction less than 25%, and initial dependence on intravenous inotropes. This study showed improved survival and quality of life in patients treated with first-generation VADs. Survival rate at 1 year was 52% with a VAD as compared with 25% with medical therapy, but 2-year survival rate with VAD support was only 23% (compared with 8% for medical therapy).176 The prognosis with current VAD technology is significantly better. In 2011, there were approximately 1500 VAD implants in the United States, virtually all of which were continuous flow devices: 23.7% were used as a bridge to transplant, 40.5% were used to determine transplant candidacy, and 34% were implanted as destination therapy. A classification system can be used to define short-term prognosis and suitability for VAD implantation177 (see Fig. 29.9). Of patients receiving VADs, 14% had critical cardiogenic shock at the time of implant, 41.4% were failing continuous intravenous inotrope therapy, 27.7% were stable on inotrope therapy, and 12.1% had severe class IV symptoms at rest but were out of hospital. Current actuarial 1-year survival rate in all VAD recipients is 80%, with 2-year survival rate of 70%.178
References
1. Weintraub, NL, Collins, SP, Pang, PS, et al. Acute heart failure syndromes: Emergency department presentation, treatment, and disposition: Current approaches and future aims. A scientific statement from the American Heart Association. on behalf of the American Heart Association Council on Clinical Cardiology and Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation. Circulation. 2010; 122:1975–1996.
2. Roger, VL, Go, AS, Lloyd-Jones, DM, et al. AHA statistical update. Heart disease and stroke statistics—2011 update. on behalf of the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2011; 123:e18–e209.
3. Jessup, M, Brozena, S. Heart failure. N Engl J Med. 2003; 348:2007–2018.
4. Jessup, M, Abraham, WT, Casey, DE, et al. 2009 focused update: ACCF/AHA guidelines for the diagnosis and management of heart failure in the adult. J Am Coll Cardiol. 2009; 53:1343–1382.
5. Chen, J, Normand, ST, Wang, Y, Krumholz, HM. National and regional trends in heart failure hospitalization and mortality rates for Medicare beneficiaries, 1998-2008. JAMA. 2011; 306:1669–1678.
6. Gheorghiade, M, Zannad, F, Sopko, G. Acute heart failure syndromes. Current state and framework for future research. Circulation. 2005; 112:3958–3968.
7. Rudiger, T, Harjola, V, Muller, A, et al. Acute heart failure: Clinical presentation. One year mortality and prognostic factors. Eur J Heart Fail. 2005; 7:662–670.
8. Sharma, M, Teerlink, JR. A rational approach for the treatment of acute heart failure: Current strategies and future options. Curr Opin Cardiol. 2004; 19:254–263.
9. Fadel, BM, Ellahham, S, Ringe, MD. Hyperthyroid heart disease. Clin Cardiol. 2000; 23:402–408.
10. Asbun, J, Villarreal, F. The pathogenesis of myocardial fibrosis in the setting of diabetic cardiomyopathy. J Am Coll Cardiol. 2006; 47:693–700.
11. Alpert, MA. Obesity cardiomyopathy: Pathophysiology and evolution of the clinical syndrome. Am J Med Sci. 2001; 321:225–236.
12. Wu, A, Cody, R. Medical and surgical treatment of chronic heart failure. Curr Probl Cardiol. 2003; 28:225–260.
13. Packer, M. Current role of beta-adrenergic blockers in the management of chronic heart failure. Am J Med. 2001; 110(7A):81S–84S.
14. Pang, P. Acute heart failure syndromes: Initial management. Emerg Med Clin North Am. 2011; 29:675–688.
15. Cowie, M, Mendez, G. B-natriuretic peptide and congestive heart failure. Curr Probl Cardiol. 2003; 44:264–311.
16. McCullough, PA, Nowack, PM, McCord, J, et al. B-type natriuretic peptide and clinical judgment in emergency diagnosis of heart failure: Analysis from breathing not properly (BNP) multinational study. Circulation. 2002; 106:416–422.
17. Morrison, LK, Harrison, A, Krishnaswamy, P, et al. Utility of a rapid BNP assay in differentiating congestive heart failure from lung disease in patients presenting with dyspnea. J Am Coll Cardiol. 2002; 39:202–209.
18. Muller, C, Scholer, A, Lawle-Kilian, K, et al. Use of B-type natriuretic peptide in the evaluation and management of acute dyspnea. N Engl J Med. 2004; 350:647–654.
19. Maisel, A. B-type natriuretic peptide levels. Diagnosis and prognosis in congestive heart failure: What’s next? Circulation. 2002; 105:2328–2331.
20. Logeart, D, Thabut, G, Jourdian, P, et al. Predischarge B-type natriuretic peptide assay for identifying patients at high risk of readmission after decompensated heart failure. J Am Coll Cardiol. 2004; 43:635–641.
21. Januzzi, JL, van Kimmenade, R, Lainchbury, J, et al. NT-proBNP testing for diagnosis and short-term prognosis in acute destabilized heart failure: An international pooled analysis of 1256 patients. The International Collaborative of NT-proBNP Study. Eur Heart J. 2006; 27:330–337.
22. O’Donoghue, M, Chen, A, Baggish, AL, et al. The effects of ejection fraction on N-Terminal ProBNP and BNP levels in patients with acute CHF: Analysis from the ProBNP Investigation of Dyspnea in the Emergency Department (PRIDE) Study. J Card Fail (Suppl). 2005; 11:S9–S14.
23. Nieminen, MS, Bohm, M, Cowie, MR, et al. Executive summary of the guidelines on the diagnosis and treatment of acute heart failure. The Task Force on Acute Heart Failure of the European Society of Cardiology. Eur Heart J. 2005; 26:384–416.
24. Fonarow, GC, Adams, KF, Abraham, WT, et al. Risk stratification for in-hospital mortality in acutely decompensated heart failure. JAMA. 2005; 293:572–580.
25. O’Connor, CM, Abraham, WT, Albert, NM, et al. Predictors of mortality after discharge in patients hospitalized with heart failure: An analysis from the Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients with Heart Failure (OPTIMIZE-HF). Am Heart J. 2008; 156:662–673.
26. Rose, E, Gelijns, A, Moskowitz, A, et al. Long-term use of a left ventricular assist device for end-stage heart failure. N Engl J Med. 2001; 345:1435–1443.
27. Collins, SP, Lindsell, CJ, Storrow, AB, et al. Early changes in clinical characteristics after emergency department therapy for acute heart failure syndromes: Identifying patients who do not respond to standard therapy. Heart Fail Rev. 2012; 17:387–394.
28. Hochman, J, Sleeper, L, Webb, J, et al. Early revascularization in acute myocardial infarction complicated by cardiogenic shock. SHOCK investigators: SHould we emergently revascularize Occluced Coronaries for cardiogenic shocK? N Engl J Med. 1999; 341:625–634.
29. Jemtel, TH, Alt, EU. Are hemodynamic goals viable in tailoring heart failure therapy? Hemodynamic goals are outdated. Circulation. 2006; 113:1027–1032.
30. Chin, M, Goldman, L. Correlates of major complications or death in patients admitted to the hospital with congestive heart failure. Arch Intern Med. 1996; 156:1814–1820.
31. Fonarow, GC, Peacock, WP, Phillips, CO, et al. Admission B-type natriuretic peptide levels and in-hospital mortality in acute decompensated heart failure. for the ADHERE Scientific Advisory Committee and Investigators. J Am Coll Cardiol. 2007; 49:1943–1950.
32. Peacock, WF, DeMarco, T, Fonarow, GC, et al. Cardiac troponin and outcome in acute heart failure. N Engl J Med. 2008; 358:2117–2126.
33. Stevenson, LW. Are hemodynamic goals viable in tailoring heart failure therapy? Hemodynamic goals are relevant. Circulation. 2006; 113:1020–1027.
34. Damman, K, van Deursen, VM, Navis, G, et al. Increased central venous pressure is associated with impaired renal function and mortality in a broad spectrum of patients with cardiovascular disease. J Am Coll Cardiol. 2009; 53:582–588.
35. Mullens, W, Abrahams, Z, Francis, GS, et al. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J Am Coll Cardiol. 2009; 53:589–596.
36. Smith, GL, Lichtman, JH, Bracken, MB, et al. Renal impairment and outcomes in heart failure. Systematic review and meta-analysis. J Am Coll Cardiol. 2006; 47:1987–1996.
37. Harinstein, ME, Flaherty, JD, Fonarow, GC, et al. Clinical assessment of acute heart failure syndromes: Emergency department through the early post-discharge period. Heart. 2011; 97:1607–1618.
38. Thomas, SS, Nohria, A. Hemodynamic classifications of acute heart failure and their clinical application. An update. Circ J. 2012; 76:278–286.
39. Jain, P, Massie, B, Gattis, W, et al. Current medical treatment for the exacerbation of chronic heart failure resulting in hospitalization. Am Heart J. 2003; 145:S3–17.
40. Floyd, JD, Nguyen, DT, Lobins, RL, et al. Cardiotoxicity of cancer therapy. J Clin Oncol. 2005; 23:7685–7696.
41. Gheorghiade, M, Pang, PS. Acute heart failure syndromes. J Am Coll Cardiol. 2009; 53:557–573.
42. Gray, A, Goodcare, S, Newby, DE, et al. Noninvasive ventilation in acute cardiogenic pulmonary edema. N Engl J Med. 2008; 359:142–151.
43. Nieminen, MS, Brutsaert, D, Dickstein, K, et al. EuroHeart Failure Survey II (EHFS II): A survey on hospitalized acute heart failure patients: Description of population. Eur Heart J. 2006; 27:2725–2736.
44. The ESCAPE Investigators and ESCAPE Study Coordinators. Evaluation study of congestive heart failure and pulmonary artery catheterization effectiveness. JAMA. 2005; 294:1625–1633.
45. Stevenson, L. Tailored therapy for hemodynamic goals for advanced heart failure. Eur J Heart Fail. 1999; 1:251–257.
46. Steimle, A, Stevenson, L, Chelimsky-Fallick, C, et al. Sustained hemodynamic efficacy of therapy tailored to reduce filling pressures in survivors with advanced heart failure. Circulation. 1997; 96:1165–1172.
47. Stevenson, L, Tillisch, J. Maintenance of cardiac output with normal filling pressures in patients with dilated heart failure. Circulation. 1986; 74:1303–1308.
48. Dormans, T, van Meyel, J, Gerlag, P, et al. Diuretic efficacy of high dose furosemide in severe heart failure: Bolus injection versus continuous infusion. J Am Coll Cardiol. 1996; 28:376–382.
49. Thompson, MR, Nappi, JM, Dunn, JP, et al. Continuous versus intermittent infusion of furosemide in acute decompensated heart failure. J Card Fail. 2010; 16:188–193.
50. Allen, LA, Turer, AT, DeWald, T, et al. Continuous versus bolus dosing of furosemide for patients hospitalized with heart failure. Am J Cardiol. 2010; 105:1794–1797.
51. Felker, GM, Lee, KL, Bull, DA, et al. Diuretic strategies in patients with acute decompensated heart failure. for the NHLBI Heart Failure Clinical Research Network. N Engl J Med. 2011; 364:797–805.
52. Yilmaz, MB, Gayat, E, Salem, R, et al. Impact of diuretic dosing on mortality in acute heart failure using a propensity-matched analysis. Eur J Heart Fail. 2011; 13:1244–1252.
53. Testani, JM, Cappola, TP, Brensinger, CM, et al. Interaction between loop diuretic-associated mortality and blood urea nitrogen concentration in chronic heart failure. J Am Coll Cardiol. 2011; 58:375–382.
54. Jentzer, JC, DeWald, TA, Hernandez, AF. Combination of loop diuretics with thiazide-type diuretics in heart failure. J Am Coll Cardiol. 2010; 56:1527–1534.
55. Gheorghiade, M, Gattis, W, O’Connor, CM, et al. Effects of tolvaptan, a vasopressin antagonist, in patients hospitalized with worsening heart failure. JAMA. 2004; 291:1963–1971.
56. Goldsmith, SR, Gheorghiade, M. Vasopressin antagonism in heart failure. J Am Coll Cardiol. 2005; 46:1785–1791.
57. Orlandi, C, Zimmer, CA, Gheroghiade, M. Role of vasopressin antagonists in the management of acute decompensated heart failure. Curr Heart Fail Rep. 2005; 2:131–139.
58. Gheorghiade, M, Konstam, MA, Burnett, JC, et al. Short term clinical effects of tolvaptan, an oral vasopressin antagonist in patients hospitalized for heart failure: Results of the EVEREST clinical status trials. JAMA. 2007; 297:1332–1343.
59. Udelson, JE, Orlandi, C, Ouyang, J, et al. Acute hemodynamic effects of tolvaptan, a vasopressin V2 receptor blocker, in patients with symptomatic heart failure and systolic dysfunction: An international, multicenter, randomized, placebo-controlled trial. J Am Coll Cardiol. 2008; 52:1540–1545.
60. Konstam, MA, Gheorghiade, M, Burnett, JC, et al. JAMA. 2007; 297:1319–1331.
61. Publication Committee for the VMAC Investigators. Intravenous nesiritide versus nitroglycerin for treatment of decompensated congestive heart failure: A randomized controlled trial. JAMA. 2000; 287:1531–1540.
62. Colucci, W, Elkayam, U, Horton, D, et al. Intravenous nesiritide, a natriuetic in the treatment of decompensated congestive heart failure. N Engl J Med. 2002; 343:246–253.
63. Silver, M, Horton, D, Ghali, J, et al. Effect of nesiritide versus dobutamine on short-term outcomes in the treatment of patients with acute decompensated heart failure. J Am Coll Cardiol. 2002; 39:798–803.
64. Burger, A, Horton, D, LeGemtel, T, et al. Effect of neseritide (B-type natriuretic peptide) and dobutamine on ventricular arrhythmias in the treatment of patients with acutely decompensated congestive heart failure: The PRECEDENT Study. Am Heart J. 2002; 144:1102–1108.
65. Sackner-Bernstein, JD, Kowalski, M, Fox, M, Aaronson, K. Short-term risk of death after treatment with nesiritide for decompensated heart failure. JAMA. 2005; 293:1900–1905.
66. Sackner-Bernstein, JD, Skopicki, HA, Aarronson, KD. Risk of worsening renal function with nesiritide in patients with acutely decompensated heart failure. Circulation. 2005; 111:1487–1491.
67. Abraham, WT, Adams, KF, Fonarow, GC, et al. In-hospital mortality in patients with acute decompensated heart failure requiring intravenous vasoactive medications. J Am Coll Cardiol. 2005; 4:57–64.
68. O’Connor, CM, Starling, RC, Hernandez, AF, et al. Effect of nesiritide in patients with acute decompensated heart failure. N Engl J Med. 2011; 365:32–43.
69. Chattergee, K, DeMarco, T. Role of nonglycosidic inotropic agents. Indications, ethics, and limitations. Med Clin North Am. 2003; 87:391–418.
70. Teerlink, JR, Metra, M, Zaca, V, et al. Agents with inotropic properties for the management of acute heart failure syndromes. Traditional agents and beyond. Heart Fail Rev. 2009; 14:243–253.
71. Shah, M, Hasselblad, V, Stinnett, S, et al. Hemodynamic profiles of advanced heart failure: Association with clinical characteristics and long-term outcomes. J Card Fail. 2001; 7:105–113.
72. Jaski, B, Fifer, M, Wright, R, et al. Positive inotropic and vasodilator actions of milrinone in patients with severe congestive heart failure. J Clin Inves. 1985; 75:643–649.
73. Colucci, W, Wright, R, Jaski, B, et al. Milrinone and dobutamine in severe heart failure: Differing hemodynamic effects and individual patient responsiveness. Circulation. 1986; 73:175–183.
74. Cuffe, M, Califf, R, Adams, K, et al. Short-term intravenous milrinone for acute exacerbation of chronic heart failure: A randomized controlled trial. JAMA. 2002; 287:1541–1547.
75. O’Connor, C, Gattis, W, Uretsky, B, et al. Continuous intravenous dobutamine is associated with an increased risk of death in patients with advanced heart failure: Insights from the Flolan International Randomized Survival Trial (FIRST). Am Heart J. 1999; 138:78–86.
76. Hershberger, RE, Nauman, D, Walter, TL, et al. Care processes and clinical outcomes of Continuous Outpatient Support with Inotropes (COSI) in patients with refractory end stage heart failure. J Card Fail. 2003; 9:180–187.
77. De Backer, D, Biston, P, Devriendt, J, et al. Comparison of dopamine and norepinephrine in the treatment of shock. for the SOAP II Investigators. N Engl J Med. 2010; 362:779–789.
78. Jaski, BE, Ha, J, Denys, BG, et al. Peripherally inserted veno-venous ultrafiltration for rapid treatment of volume overloaded patients. J Card Fail. 2003; 9:227–231.
79. Bart, BA, Boyle, A, Bank, AJ, et al. Ultrafiltration versus usual care for hospitalized patients with heart failure. J Am Coll Cardiol. 2005; 46:2043–2053.
80. Constanzo, MK, Guglin, ME, Saltzberg, MT, et al. Ultrafiltration versus intravenous diuretics for patients hospitalized for acute decompensated heart failure. J Am Coll Cardiol. 2007; 49:675–683.
80a. Bart, BA, Goldsmith, SR, Lee, KL, et al. Ultrafiltration in decompensated heart failure with vardiorenal syndrome. for the Heart Failure Clinical Research Network. N Engl J Med. 2012; 367:2296–2304.
81. Fiaccadori, E, Regolisti, G, Maggiore, M, et al. Ultrafiltration in heart failure. Am Heart J. 2011; 161:439–449.
82. Lindenfeld, J, Albert, NM, Boehmer, JP, et al. Executive summary: HFSA 2010 comprehensive heart failure practice guideline. J Card Fail. 2010; 16:475–539.
83. BHAT Trial Research Group. A randomized trial of propranolol in patients with acute myocardial infarction. I: Mortality results. JAMA. 1982; 747:1707–1714.
84. BHAT Trial Research Group. A randomized trial of propranolol in patients with acute myocardial infarction. II: Morbidity results. JAMA. 1983; 250:2814–2819.
85. CIBIS II Investigators. The Cardiac Insufficiency Bisoprolol Study II: A randomized trial. Lancet. 1999; 353:9–13.
86. The MERIT-HF Study Group. Effects of controlled release metoprolol on total mortality, hospitalizations and well-being in patients with heart failure. JAMA. 2000; 283:1295–1302.
87. Packer, M, Coats, A, Fowler, M, et al. Effect of carvedilol on survival in severe chronic heart failure. N Engl J Med. 2001; 344:1651–1658.
88. Krum, H, Roecker, E, Mohacsi, P, et al. Effects on initiating carvedilol in patients with severe chronic heart failure. Results from the COPERNICUS Study. JAMA. 2003; 289:712–718.
89. Bristow, M, Gilbert, E, Abraham, W, et al. Carvedilol produces dose related improvements in left ventricular function and survival in subjects with chronic heart failure. Circulation. 1996; 94:2807–2816.
90. Yilmaz, MB, Laribi, S, Mebazaa, A. Managing beta-blockers in acute heart failure: When to start and when to stop? Curr Heart Fail Rep. 2010; 7:110–115.
91. Bohm, M, Link, A, Cai, D, et al. Beneficial association of beta-blocker therapy on recovery from severe acute heart failure treatment: Data from the Survival of Patients with Acute Heart Failure in Need of Intravenous Inotropic Support Trial. Crit Care Med. 2011; 39:940–944.
92. Ginsberg, FL. Beta-blockers: Essential heart failure therapy. Crit Care Med. 2011; 39:1198–1199.
93. The SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med. 1991; 325:293–302.
94. The SOLVD Investigators. Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fraction. N Engl J Med. 1992; 327:685–691.
95. Garg, R, Yusuf, S. Overview of randomized trials of angiotensin converting enzyme inhibitors on mortality and morbidity in patients with heart failure. JAMA. 1995; 273:1450–1456.
96. Packer, M, Poole-Wilson, PA, Armstrong, PW, et al. Comparative effects of low and high doses of the angiotensin-converting enzyme inhibitor lisinopril on morbidity and mortality in chronic heart failure. Atlas Study Group. Circulation. 1999; 100:2312–2318.
97. Aronow, WS. Epideminology, pathophysiology, prognosis, and treatment of systolic and diastolic heart failure. Card Rev. 2006; 14:108–124.
98. Willenheimer, R, van Veldhuisen, DJ, Silke, B, et al. Effect on survival and hospitalization of initiating treatment for chronic heart failure with bisoprolol followed by enalapril, as compared with the opposite sequence. Circulation. 2005; 112:2426–2435.
99. The RESOLVD Pilot Study Investigators. Comparison of candesartan, enalapril, and their combination in congestive heart failure. Circulation. 1999; 100:1056–1064.
100. Pitt, B, Poole-Wilson, P, Segal, R, et al. Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure: Randomized trial—The Losartan Heart Failure Survival Study (ELITE II). Lancet. 2000; 355:1582–1587.
101. Cohn, J, Tognoni, G. A randomized trial of the angiotensin receptor blocker valsartan in chronic heart failure. N Engl J Med. 2001; 345:1667–1675.
102. Granger, C, McMurray, J, Yusuf, S, et al. Effects of candesartan in patients with chronic heart failure and reduced left ventricular systolic function intolerant to angiotensin-converting enzyme inhibitors. The CHARM Alternative Trial. Lancet. 2003; 362:772–776.
103. McMurray, J, Ostergren, J, Swedberg, K, et al. Effects of candesartan in patients with chronic heart failure and reduced left ventricular systolic function taking angiotensin-converting enzyme inhibitors. The CHARM-Added Trial. Lancet. 2003; 362:759–766.
104. Manohair, P, Pina, I. Therapeutic role of angiotensin II receptor blockers in the treatment of heart failure. Mayo Clin Proc. 2003; 78:334–338.
105. Sharma, D, Buyse, M, Pitt, B, et al. Meta-analysis of observed mortality data from all controlled double-blind multiple dose studies of losartan in heart failure. Am J Cardiol. 2000; 85:187–192.
106. Pitt, B, Zannad, F, Remme, W, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med. 1999; 341:709–717.
107. Juurlink, DN, Mamdani, NM, Lee, DS, et al. Rates of hyperkalemia after publication of the randomized aldactone study. N Engl J Med. 2004; 351:543–551.
108. Zannad, F, McMurray, JJV, Krum, H, et al. Eplerenone in patients with systolic heart failure and mild symptoms. for the EMPHASIS-HF Study Group. N Engl J Med. 2011; 364:11–21.
109. Rossignol, P, Ménard, J, Fay, R, et al. Eplerenone survival benefits in heart failure patients post-myocardial infarction are independent from its diuretic and potassium-sparing effects: Insights from an EPHESUS (Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study) substudy. J Am Coll Cardiol. 2011; 58:1958–1966.
110. Cohn, J, Archibald, D, Ziesche, S, et al. Effect of vasodilator therapy on mortality in chronic congestive heart failure. Results of a Veteran Administration Cooperative Study. N Engl J Med. 1986; 314:1547–1552.
111. Taylor, AL, Zieche, S, Yancy, C, et al. Combination of isosorbide dinitrate and hydralazine in blacks with heart failure. N Engl J Med. 2004; 351:2049–2057.
112. The Digitalis Investigation Group. The effect of digoxin on mortality and morbidity in patients with heart failure. N Engl J Med. 1997; 336:525–533.
113. Ahmed, A, Rich, MW, Love, TE, et al. Digoxin and reduction in mortality and hospitalization in heart failure: A comprehensive post hoc analysis of the DIG trial. Eur Heart J. 2006; 27:178–186.
114. Velazquez, EJ, Pfeffer, MA. Acute heart failure complicating acute coronary syndromes. Circulation. 2004; 109:440–442.
115. Steg, PG, Dabbous, DH, Feldman, LJ, et al. Determinants and prognostic impact of heart failure complicating acute coronary syndromes. Circulation. 2004; 109:494–499.
116. Velazquez, EJ, Francis, GS, Armstrong, PW, et al. An international perspective on heart failure and left ventricular systolic dysfunction complicating myocardial infarction: The VALIANT registry. Eur Heart J. 2004; 25:1911–1919.
117. The CAPRICORN Investigators. Effect of carvedilol on outcome after myocardial infarction in patients with left ventricular dysfunction: The CAPRICORN Randomized Trial. Lancet. 2001; 357:1385–1390.
118. Pfeffer, M, Braunwald, E, Moye, L, et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 1992; 327:669–677.
119. The Acute Infarction Ramipril Efficacy (AIRE) Study Investigators. Effect of ramipril on mortality and morbidity of survivors of acute myocardial infarction with clinical evidence of heart failure. Lancet. 1993; 342:821–828.
120. Kober, L, Torp-Pedersen, C, Carlsen, JE, et al. A clinical trial of the angiotensin-converting-enzyme inhibitor trandolapril in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 1995; 333:670–676.
121. Pfeffer, MA, McMurray, JJ, Velazquez, EJ. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med. 2003; 349:1893–1903.
122. Pitt, B, Remme, W, Zannad, F, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003; 348:1309–1321.
123. Pitt, B, White, H, Nicolau, J. Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. J Am Coll Cardiol. 2005; 46:424–430.
124. Owan, TE, Redfield, MM. Epideminology of diastolic heart failure. Prog Cardiovasc Dis. 2005; 47:320–332.
125. Leite-Moreira, AF. Current perspectives in diastolic dysfunction and diastolic heart failure. Heart. 2006; 92:712–718.
126. Gaasch, WH. Diagnosis and treatment of heart failure based on left ventricular systolic or diastolic dysfunction. JAMA. 1994; 271:1276–1280.
127. Vasan, RS, Benjamin, EJ, Levy, D. Prevalence, clinical features and prognosis of diastolic heart failure: An epidemiologic perspective. J Am Coll Cardiol. 1995; 26:1565–1574.
128. Aurigemma, GP, Zile, MR, Gaasch, WH. Contractile behavior of the left ventricle in diastolic heart failure. Circulation. 2006; 113:296–304.
129. Owan, TE, Hodge, DO, Herges, RM, et al. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med. 2006; 355:251–259.
130. Parissis, JT, Ikonomidis, I, Rafouli-Stergiou, P, et al. Clinical characteristics and predictors of in-hospital mortality in acute heart failure with preserved left ventricular ejection fraction. Am J Cardiol. 2011; 107:79–84.
131. Klapholz, M, Maurer, M, Lowe, AM, et al. Hospitalization for heart failure in the presence of a normal left ventricular ejection fraction. J Am Coll Cardiol. 2004; 43:1432–1438.
132. Devereux, RB, Roman, MJ, Liu, JE, et al. Congestive heart failure despite normal left ventricular systolic function in a population-based sample: The strong heart study. Am J Cardiol. 2000; 86:1090–1096.
133. Philbin, EF, Rocco, TA, Jr., Lindenmuth, NW. Systolic versus diastolic heart failure in community practice: Clinical features, outcomes, and the use of angiotensin-converting enzyme inhibitors. Am J Med. 2000; 1009:605–613.
134. Yancy, CW, Lopatin, M, Stevenson, LW, et al. Clinical presentation, management, and in-hospital outcomes of patients admitted with acute decompensated heart failure with preserved systolic function. J Am Coll Cardiol. 2006; 47:76–84.
135. Bhatia, RS, Tu, JV, Lee, DL, et al. Outcome of heart failure with preserved ejection fraction in a population-based study. N Engl J Med. 2006; 355:260–269.
136. Franklin, KM, Aurigemma, GP. Prognosis in diastolic heart failure. Prog Cardiovasc Dis. 2005; 47:333–339.
137. Yusuf, S, Pfeffer, MA, Swedberg, K, et al. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: The CHARM-Preserved Trial. Lancet. 2003; 362:777–781.
138. Massie, BM, Carson, PE, McMurray, JJ, et al. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med. 2008; 359:2456–2467.
139. Little, WC, Brucks, S. Therapy for diastolic heart failure. Prog Cardiovasc Dis. 2005; 47:380–388.
140. Zile, M, Baica, CF. Alterations in ventricular function: Diastolic heart failure. In: Mann DL, et al, eds. Heart Failure. A Companion to Braunwald’s Heart Disease. Philadelphia: WB Saunders; 2004:222–227.
141. Feldman, A, McNamara, D. Myocarditis. N Engl J Med. 2000; 19:1388–1398.
142. Parrillo, JE. Inflammatory cardiomyopathy (myocarditis): Which patients should be treated with anti-inflammatory therapy? Circulation. 2001; 104:4–6.
143. Nelson, KH, Li, T, Afonso, L. Diagnostic approach and the role of MRI in the assessment of acute myocarditis. Cardiol Rev. 2009; 17:24–30.
144. Friedrich, MG, Sechtem, U, Shulz-Menger, J, et al. Cardiovascular magnetic resonance in myocarditis: A JACC white paper. J Am Coll Cardiol. 2009; 53:1475–1487.
145. Parrillo, JE, Cunnion, R, Epstein, S, et al. A prospective randomized controlled trial of prednisone for dilated cardiomyopathy. N Engl J Med. 1989; 321:1061–1068.
146. Wojnicz, R, Nowalany-Kozielska, E, Wojciechowska, C, et al. Randomized, placebo-controlled study for immunosuppressive treatment of inflammatory dilated cardiomyopathy: Two-year follow-up results. Circulation. 2001; 104:39–45.
147. Mason, J, O’Connell, J, Herskowitz, A, et al. A clinical trial of immunosuppressive therapy for myocarditis. N Engl J Med. 1995; 333:269–275.
148. McCarthy, R, Boehmer, J, Hrubianr, F, et al. Long-term outcome of fulminant myocarditis as compared with acute (non-fulminant) myocarditis. N Engl J Med. 2002; 342:690–695.
149. The Cardiac Arrhythmia Suppression Trial Investigators Preliminary Report. Effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med. 1989; 321:406–412.
150. Singh, S, Fletcher, R, Fisher, S, et al. Amiodarone in patients with congestive heart failure and asymptomatic ventricular arrhythmia. N Engl J Med. 1995; 333:77–82.
151. Julian, D, Camm, A, Frangin, G, et al. Randomized trial of effect of amiodarone on mortality in patients with left ventricular dysfunction after recent myocardial infarction: EMIAT. Lancet. 1997; 349:667–674.
152. The AVID Investigators. A comparison of antiarrhythmic drug therapy with implantable defibrillators in patients resuscitated from near-fatal ventricular arrhythmias. N Engl J Med. 1997; 337:1576–1583.
153. Connoly, S, Gent, M, Roberts, R, et al. Canadian Implantable Defibrillator Study (CIDS): A randomized trial of the implantable cardioverter-defibrillator against amiodarone. Circulation. 2000; 101:1297–1302.
154. Kuck, K, Cappato, R, Siebels, J, et al. Randomized comparison of antiarrhythmic drug therapy with implantable defibrillators in patients resuscitated from cardiac arrest: The Cardiac Arrest Study Hamburg (CASH). Circulation. 2000; 102:748–754.
155. MADIT Investigators. Improved survival with an implanted defibrillator in patients with coronary disease at high risk for ventricular arrhythmia. N Engl J Med. 1995; 335:1933–1940.
156. Buxton, A, Lee, K, Fisher, J, et al. A randomized study of the prevention of sudden death in patients with coronary artery disease. N Engl J Med. 1999; 341:1882–1890.
157. MADIT II Investigators. Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med. 2001; 346:877–883.
158. Bardy, GH, Lee, KL, Mark, DB, et al. Amiodarone or an implantable cardioverter defibrillator for congestive heart failure (SCD-HeFT). N Engl J Med. 2005; 352:225–237.
159. Kadish, A, Dyer, A, Daubert, JP, et al. Prophylactic defibrillator implantation in patients with nonischemic dilated cardiomyopathy. N Eng J Med. 2004; 350:2151–2158.
160. Desai, AS, Farg, JC, Maisel, WH, Baughman, KL. Implantable defibrillators for the prevention of mortality in patients with non-ischemic cardiomyopathy. A meta-analysis of randomized controlled trials. JAMA. 2004; 292:2874–2879.
161. Abraham, W, Fisher, W, Smith, A, et al. Cardiac resynchronization in chronic heart failure. N Engl J Med. 2002; 346:1845–1853.
162. Jarcho, JA. Biventricular pacing. N Engl J Med. 2006; 355:288–294.
163. Bradley, D, Bradley, E, Baughman, K, et al. Cardiac resynchronization and death from progressive heart failure. A meta-analysis of randomized controlled trials. JAMA. 2003; 289:730–740.
164. Cleland, JAF, Danbert, J, Erdmann, E, et al. The effect of cardiac resynchronization on morbidity and mortality in heart failure. N Engl J Med. 2005; 352:1539–1549.
165. Young, J, Abraham, W, Smith, A, et al. Combined cardiac resynchronization and implantable cardioversion defibrillation in advanced chronic heart failure. The MIRACLE ICD trial. JAMA. 2003; 289:2685–2694.
166. Bristow, MR, Saxon, LA, Boehmer, J, et al. Comparison of medical therapy, pacing and defibrillation in heart failure (COMPANION). N Engl J Med. 2004; 350:2140–2150.
167. Tang, ASL, Wells, GA, Talajic, M, et al. Cardiac-resynchronization therapy for mild-to-moderate heart failure. for the Resynchronization-Defibrillation for Ambulatory Heart failure Trial (RAFT) Investigators. N Engl J Med. 2010; 363:2385–2395.
168. Wilson, SR, Mudge, GH, Jr., Stewart, JC, et al. Evaluation for a ventricular assist device: Selecting the appropriate candidate. Circulation. 2009; 119:2225–2232.
169. Stevenson, LW, Rose, EA. Left ventricular assist devices: Bridges to transplantation, recovery and destination therapy for whom? Circulation. 2003; 108:3059–3063.
170. Felker, GM, Rogers, JG. Same bridge, new destinations. Rethinking paradigms for mechanical cardiac support in heart failure. J Am Coll Cardiol. 2006; 47:930–932.
171. Mancini, D, Burkoff, D. Mechanical device-based methods of managing and treating heart failure. Circulation. 2005; 12:438–448.
172. Muller, J, Wallukat, G, Weng, Y, et al. Weaning from mechanical cardiac support in patients with idiopathic dilated cardiomyopathy. Circulation. 1997; 96:542–549.
173. Terraciano, CM, Miller, LW, Yacoub, MH. Contemporary use of ventricular assist devices. Ann Rev Med. 2010; 24:184–189.
174. Slaughter, MS, Rogers, JG, Milano, CA, et al. Advanced heart failure treated with continuous-flow left ventricular assist device. for the HeartMate II Investigators. N Engl J Med. 2009; 361:2241–2251.
175. Shreenivas, SS, Rame, JE, Jessup, M. Mechanical circulatory support as a bridge to transplantation or for destination therapy. Curr Heart Fail Rep. 2010; 7:159–166.
176. Rose, E, Gelijas, A, Moskowitz, A, et al. Long-term use of a left ventricular assist device for end-stage heart failure. N Engl J Med. 2001; 345:1435–1443.
177. Stevenson, LW, Pagani, FD, Young, JB, et al. INTERMACS profiles of advanced heart failure: The current picture. J Heart Lung Transplant. 2009; 28:535–541.
178. Kirklin, JK, Naftel, DC, Formos, RL, et al. The 24th INTERMACS Annual Report. 4,000 implants and counting. J Heart Lung Transplant. 2012; 31:117–126.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]29
Severe Heart Failure
DEFINITION, EPIDEMIOLOGY, AND STAGING OF HEART FAILURE
PROGNOSIS IN ACUTE HEART FAILURE
PHARMACOLOGIC MANAGEMENT OF ACUTE HEART FAILURE
TRANSITION TO CHRONIC PHARMACOLOGIC THERAPY FOR SEVERE HEART FAILURE
CORONARY HEART DISEASE AND HEART FAILURE: SPECIAL CONSIDERATIONS
HEART FAILURE WITH PRESERVED LEFT VENTRICULAR EJECTION FRACTION (DIASTOLIC HEART FAILURE)
ACUTE MYOCARDITIS AND HEART FAILURE
DEVICE THERAPY: IMPLANTED CARDIOVERTER-DEFIBRILLATORS AND CARDIAC RESYNCHRONIZATION THERAPY
MECHANICALLY ASSISTED CIRCULATORY SUPPORT: VENTRICULAR ASSIST DEVICES
Definition, Epidemiology, and Staging of Heart Failure
Heart failure is a very common illness; 5.8 million Americans are affected.1,2 It is estimated that 1% of the population of Americans over the age of 65 are affected by heart failure, and 20% of hospital admissions in patients over age 65 are due to heart failure.3 Men and black Americans are affected more frequently. In 2008 670,000 new cases of heart failure were diagnosed in the United States, where there are nearly 1 million hospital discharges, 658,000 visits to the emergency department, over 3.4 million ambulatory outpatient visits, and 6.5 million hospital days annually for patients with a primary diagnosis of heart failure.1,2 Over 56,000 people in the United States died in 2007 with heart failure as the primary cause. The treatment of heart failure incurs a very large economic burden on the U.S. health care system. Estimated direct and indirect cost of heart failure in 2010 was $29 billion. The majority of this cost was related to the treatment of patients hospitalized with heart failure.
Epidemiologic data showed significant increases in both the incidence and prevalence of heart failure in the U.S. population in the 1990s,3 likely influenced by the aging of the population, a high prevalence of hypertension, and improved treatment and survival of patients with ischemic heart disease.4 However, there is evidence that hospitalizations for heart failure have declined during the past decade. In the United States, the rate of hospitalization for heart failure declined from 2845 per 100,000 patient-years in 1998 to 2007 per 100,000 person-years in 2008, a decline of 29%.5
Patients hospitalized with acute heart failure are usually elderly, with a mean age in the early 70s. In the United States, roughly 80% of patients will have a previous history of heart failure,6 and in European studies one third of patients had a new diagnosis of heart failure.7 From 40% to 55% of patients will have normal or relatively normal left ventricular systolic function. This occurs more commonly in women than in men. Coronary heart disease is present in 50% to 60% of patients and hypertension in 72% of patients. Comorbid conditions are common, including renal disease in 30% of patients, diabetes mellitus in 43%, and chronic obstructive pulmonary disease (COPD) in roughly 30%.6–8
In the American College of Cardiology/American Heart Association Guidelines for the diagnosis and management of chronic heart failure, four stages in the development of heart failure are recognized (Fig. 29.1). This staging system emphasizes the progressive nature of left ventricular dysfunction and heart failure, and describes evidence-based guidelines for therapy for each stage.4 It is important to realize that heart failure may be preventable. Several common medical conditions place patients at high risk for developing left ventricular dysfunction and the heart failure syndrome. Attention to and appropriate management of these conditions may prevent the development of heart failure. In addition, in patients who have left ventricular dysfunction but who have not yet developed heart failure, appropriate therapy can improve prognosis and prevent the development of severe heart failure.
Stage A represents patients at high risk for heart failure but without structural heart disease or symptoms of heart failure. These patients include those with hypertension, atherosclerotic heart disease, diabetes, obesity, or the metabolic syndrome; those exposed to cardiotoxic medications; and those with a family history of cardiomyopathy. About 75% of patients who develop heart failure have antecedent hypertension.2 Stage B patients have structural heart disease but without signs or symptoms of heart failure. This group includes patients who have suffered a myocardial infarction, those with abnormal left ventricular ejection fraction but no symptoms of heart failure, and those patients with asymptomatic valvular heart disease. Stage C patients have structural heart disease with prior or current symptoms of heart failure, such as shortness of breath, fatigue, and reduced exercise tolerance. Lastly, stage D patients have refractory symptoms of heart failure at rest despite maximal medical therapy. They require specialized or extraordinary interventions such as cardiac transplantation, mechanical circulatory support, or end-of-life care.
Pathophysiology
Heart failure can be due primarily to left ventricular systolic dysfunction or diastolic dysfunction, although both abnormalities are often present together. Right ventricular dysfunction may accompany left ventricular dysfunction or may be the primary problem. Systolic heart failure results from inability of the heart to expel blood normally owing to depressed left ventricular contraction. The left ventricle is often dilated. There is loss of myocytes and fibrosis, resulting in reduced left ventricular ejection fraction. Diastolic heart failure is caused by a reduction in left ventricular compliance, which leads to impaired diastolic filling, higher left ventricular diastolic pressure, and elevated pulmonary capillary wedge pressure (PCWP). Diastolic heart failure is also called heart failure with preserved ejection fraction (HFpEF) or heart failure with normal ejection fraction (HFnEF) (Fig. 29.2A and B).
Numerous conditions may cause damage to left ventricular myocardium and result in systolic heart failure. The most common of these conditions is atherosclerotic coronary artery disease with myocardial infarction and chronic myocardial ischemia. Other common causes include hypertension; familial and idiopathic cardiomyopathy; viral myocarditis; valvular heart disease, such as aortic stenosis, aortic insufficiency, and mitral regurgitation; peripartum cardiomyopathy; transient apical ballooning syndrome (takotsubo cardiomyopathy); and cardiomyopathy due to cardiotoxic cancer chemotherapy. Less common causes are alcoholic cardiomyopathy, diabetic cardiomyopathy, and a cardiomyopathy seen in some patients with hyperthyroidism or severe obesity.9–11
After myocardial damage and loss of myocytes occur, a process known as ventricular remodeling is initiated. This remodeling results in dilation of the ventricular chamber, a change in ventricular geometry from ellipsoid-like to a more spherical shape, worsening of left ventricular contractile force, and reduction in ventricular ejection fraction.3,12 In addition, the change in left ventricular geometry often leads to an increase in the size of the mitral annulus and altered physical relationships of the mitral valve structures. This results in increasing mitral regurgitation and worsening of the heart failure syndrome (see Fig. 29.2).
Remodeling is mediated by a series of maladaptive systemic responses, commonly termed neurohormonal activation. Two major systems involved in neurohormonal activation are the renin-angiotensin-aldosterone system and the sympathetic nervous system. Activation of the renin-angiotensin-aldosterone system leads to elevated levels of renin, angiotensin II, and aldosterone, which has deleterious consequences on cardiac function and hemodynamics.3,12 These effects include salt and fluid retention, endothelial dysfunction, vasoconstriction, myocyte hypertrophy, myocardial fibrosis, and myocyte apoptosis (programmed cell death). Sympathetic nervous system activation is in part mediated via decreased cardiac output, which results in tachycardia, increased myocardial oxygen consumption, and peripheral vasoconstriction.13 Renal effects of sympathetic nervous system activation lead to further activation of the renin-angiotensin-aldosterone system. Increased circulating norepinephrine levels also contribute to myocyte injury and death. Arrhythmias become common. A detrimental positive feedback loop is established, causing progressive deterioration in left ventricular structure and performance over time with progressive worsening of chronic heart failure.
Diagnosis
Heart failure is a clinical diagnosis, determined after evaluation of a patient’s symptoms and physical examination and supported by results of ancillary testing, such as chest x-ray and echocardiography. Typical symptoms of heart failure include shortness of breath at rest or with exertion, cough, swelling, orthopnea, and paroxysmal nocturnal dyspnea, which are due to pulmonary or systemic venous congestion. Fatigue, anorexia, and change in mental status are symptoms that may be caused by low cardiac output. Physical examination often reveals pulmonary rales; elevated jugular venous pressure; signs of cardiomegaly; cardiac murmurs, especially mitral regurgitation and third heart sound (S3) gallop; hepatic enlargement; and ascites and edema. An S3 gallop and elevated jugular venous pressure are the most specific signs of heart failure; absence of rales is not infrequent in patients with acute heart failure, and chest radiograph may not show obvious congestion.14 Manifestations of more severe heart failure include marked dyspnea at rest, possibly resulting in respiratory failure, cyanosis, cool extremities, reduced urine output and altered mental state. A careful evaluation for the presence of pulsus paradoxus is important if cardiac tamponade is suspected. However, these signs and symptoms are not specific for heart failure, and coexisting conditions such as obesity, chronic lung disease, and deconditioning can add uncertainty to the clinical diagnosis. Heart failure is often both underdiagnosed and overdiagnosed in outpatient and emergency department settings.
Studies have shown that serum assays of B-natriuretic peptide (BNP) and N-terminal pro-BNP are very helpful in improving the accuracy of diagnosing heart failure. BNP is a 32–amino acid peptide produced by cardiac myocytes in response to pressure-induced wall stretch and tension.15 Physiologic actions of BNP include arterial and venous dilation and natriuresis. A study of 1530 patients presenting to the emergency department with dyspnea showed that knowledge of BNP serum levels resulted in improved accuracy of the diagnosis of heart failure when compared to clinical judgment alone, from an initial range of 65% to 74%, up to 81% accuracy.16 BNP levels above 100 pg/mL had a sensitivity of 90% and a specificity of 76% for the diagnosis of heart failure and were very useful for discriminating patients with dyspnea due to uncomplicated lung disease who had BNP values below 100 pg/mL.17 Patients with BNP values below 100 pg/mL were very unlikely to have heart failure as the cause of dyspnea. Levels between 100 and 400 pg/mL can be seen in dyspneic patients with cor pulmonale, pulmonary hypertension not due to left ventricular failure, and acute pulmonary embolism. Heart failure is the likely diagnosis when levels are above 400 pg/mL. It should be emphasized that BNP levels should always be used in conjunction with all other clinical data to arrive at the correct diagnosis.
Evaluating BNP levels in heart failure patients in the emergency department was shown to decrease the need for hospitalization and decrease the need for intensive care unit (ICU) admissions, without affecting 30-day mortality rates.18 Total hospital stay was shortened by 3 days, cost of treatment was significantly reduced, and time to initiation of definitive therapy in the emergency department was shortened by 30 minutes.
BNP levels correlate with disease severity, as values are higher in patients with more severe heart failure and with worse left ventricular systolic function. Higher levels also have been correlated with a poorer prognosis, and can predict an increased rate of functional deterioration and a higher mortality rate.19 In a study of 114 patients admitted to the hospital with class IV heart failure, of all variables evaluated, predischarge BNP level was most strongly associated with death or readmission within 6 months, with BNP levels greater than 350 pg/mL having impressive sensitivity and specificity.20 BNP levels may also be elevated in patients with diastolic heart failure, those with renal failure, and stable patients with chronic left ventricular systolic dysfunction with compensated heart failure. BNP levels may initially be low in patients with “flash” pulmonary edema, as they may present to the hospital more quickly than the time required for significant rises in serum BNP to occur.
Serum levels of N-terminal (NT) pro-BNP can also be used for the diagnosis of heart failure. A precursor hormone, pro-BNP, is cleaved to form BNP and NTpro-BNP, which is physiologically inactive. NTpro-BNP has a longer half-life than BNP. It is cleared from the serum by the kidneys, so levels are higher in patients with coexisting renal disease. Levels of NTpro-BNP rise significantly in older populations. A cut off level of NTpro-BNP below 300 pg/mL yielded a negative predictive value of 98% for exclusion of the diagnosis of heart failure.21,22 The diagnosis of heart failure is very likely at levels over 450 pg/mL for patients under age 50, above 900 pg/mL in patients between 50 and 75 years old, and above 1800 pg/mL in patients over the age of 75.
Prognosis in Acute Heart Failure
Hospitalization for acute heart failure is associated with a poor prognosis. In-hospital mortality rate is high, reported at 4% to 8%. There is a 9% mortality rate at 60 to 90 days, and a 1-year mortality rate of 29%.1,6,7,23–25 The 90-day rehospitalization rate is around 30%, although only half of these rehospitalizations are caused by heart failure. In a European heart failure database, in-hospital mortality rate was 6.9%, 12-week readmission rate was 24%, and total mortality rate at 12 weeks was 13.5%.8 Randomized trials of pharmacologic therapy reported an annual mortality rate of 10% in patients with class II-III symptoms and 20% to 50% in class IV patients. In the most severe chronic heart failure group, patients awaiting cardiac transplantation, 1-year mortality rate was 75% with 2-year mortality rate of 92%.26 More recent data indicate a decline in the risk-adjusted inpatient mortality rate, from 5.5% in 2000 to 2.8% in 2007. Similar reductions were seen in both sexes and across all age groups.27 The prognosis for patients with cardiogenic shock due to acute myocardial infarction continues to be poor. Mortality rate remains 40% to 50% even with aggressive supportive therapy and emergency revascularization strategies.28
Several clinical factors have been shown to identify patients with a poorer prognosis and include lower left ventricular ejection fraction, low blood pressure on admission, and higher PCWP. In the United States ADHERE database registry of 62,275 admissions for heart failure, blood urea nitrogen (BUN) greater than 43 mg/dL, admission systolic blood pressure under 115 mm Hg, and creatinine level greater than 2.75 mg/dL were the three factors that indicated a poor prognosis for patients admitted with acute heart failure. The presence of an elevated BUN was associated with a fourfold increase in hospital mortality rates to 8.35%. The presence of all three factors yielded an in-hospital mortality rate of 19.8%24 (Box 29.1).
Other factors associated with increased mortality rates include hyponatremia, higher serum BNP level, and elevation of serum troponin.6,29,30 In ADHERE, higher levels of serum BNP on admission were associated with a higher in-hospital mortality rate, ranging from 1.9% in the lowest quartile to 6.0% in the highest quartile (BNP over 1730 pg/mL). The ability of BNP to predict prognosis was present even after multivariate adjustment for coexisting conditions, and was also true for patients with normal and abnormal left ventricular ejection fraction.31 Serum troponin is elevated in 6% to 10.4% of patients admitted with acute heart failure (with serum creatinine <2.0 mg/dL). Troponin elevation was associated with a lower blood pressure, lower ejection fraction, and longer hospital length of stay. Hypothetical mechanisms of troponin release in chronic heart failure include ischemia, cytokine activation, oxidative stress, and apoptosis. Hospital mortality rate was 8.0% in troponin-positive patients and 2.7% if troponin-negative. The ability of troponin elevation to predict mortality rate also was independent of other variables and was true even in patients with nonischemic causes of heart failure.32
The coexistence of kidney disease significantly worsens the prognosis of patients with heart failure. Kidney disease aggravates the tendency to volume overload and heart failure decompensation, and heart failure often worsens renal function. This complex interaction of severe heart failure and worsening kidney function is called the cardiorenal syndrome. In addition, high-dose diuretic therapy may also temporarily worsen renal function. There is increasing evidence that elevated systemic venous pressure causing renal venous congestion plays an important role in the pathogenesis.33 In a retrospective study of patients with acute heart failure who had right-sided heart catheterization, elevated central venous pressure was associated with reduced glomerular filtration rate (GFR) and higher all-cause mortality rate. These findings were independent of the measured cardiac output.34 Up to 50% of patients hospitalized with heart failure demonstrate a GFR less than 60 mL/minute/m2, and renal function may worsen in up to 30% of patients admitted with heart failure.35 Patients at particular risk for developing worsening renal function are those with lower left ventricular ejection fractions, lower blood pressure, diabetes mellitus, a history of hypertension, and older age. These patients have longer hospital stays and higher readmission and mortality rates. A meta-analysis of 16 large studies of heart failure patients revealed that 29% of heart failure patients had moderate to severe impairment of renal function. These patients had more than 100% increased relative mortality risk. Any degree of renal impairment had an approximately 50% increased relative mortality risk.36 The best treatment strategy for these patients is not clear, as patients with significant renal impairment have generally been excluded from large randomized pharmacologic heart failure trials.
Acute Heart Failure Syndromes
Initial Evaluation and Therapy
Acute heart failure is defined as the rapid onset of severe symptoms of heart failure, usually within hours to several days. Acute heart failure can occur with predominant systolic or diastolic dysfunction. Acute heart failure is often life-threatening and requires urgent diagnostic and therapeutic interventions, often simultaneously.23 Acute myocardial ischemia is a common cause and should always be considered in the differential diagnosis of this syndrome.
Several distinct clinical syndromes of acute heart failure can be identified23 (Table 29.1).
Table 29.1
Adapted from Nieminen MS, Bohm M, Cowie MR, et al: Executive summary of the guidelines on the diagnosis and treatment of acute heart failure. The Task Force on Acute Heart Failure of the European Society of Cardiology. Eur Heart J 2005;26:384-416; and Gheorghiade M, Zannad F, Sopko G: Acute heart failure syndromes. Circulation 2005;112:3958-3968.
1. Acute worsening or decompensation of chronic heart failure symptoms, either in the setting of known chronic cardiovascular illness or de novo. These patients do not have shock or pulmonary edema. This is the most common presentation of acute heart failure requiring admission to hospital, occurring in approximately 70% of patients with acute heart failure.
2. Acute pulmonary edema with normal blood pressure, often caused by acute myocardial infarction or acute coronary ischemia.
3. Acute pulmonary edema associated with elevated blood pressure, often in the setting of chronic severe hypertension and chronic kidney disease. Pulmonary edema accounts for roughly 25% of acute heart failure admissions.
4. Cardiogenic shock with heart failure, usually due to acute myocardial infarction. This syndrome is the most severe presentation of acute heart failure and is associated with high in-hospital mortality rates. This accounts for about 5% of acute heart failure cases.
5. “High cardiac output heart failure” often induced by sepsis, hyperthyroidism, or cardiac arrhythmia. This type is the least common presentation, occurring in a small percentage of patients.
6. Acute right ventricular failure, occurring with acute right ventricular myocardial infarction, massive pulmonary embolism, or cardiac tamponade.
Physical examination often shows pulmonary rales and wheezes, an S3 gallop, and elevation of jugular venous pressure. Peripheral pulses are weak and thready with diminished cardiac output states. A low cardiac output is reliably predicted by a low “proportional pulse pressure,” which is calculated by the pulse pressure (systolic blood pressure minus diastolic blood pressure) divided by systolic blood pressure. A ratio below 0.25 predicts a cardiac index below 2.2 L/minute/m2.37,38 The skin may be cool and clammy and there may be evidence of cyanosis. Peripheral edema and ascites may indicate concomitant right ventricular failure of longer duration. Chest radiography is done urgently. An electrocardiogram (ECG) is needed to assess for signs of ischemia and infarction, and to evaluate for arrhythmia. Cardiac rhythm needs to be monitored continuously. Laboratory examination includes evaluation of hemoglobin and hematocrit, electrolytes, renal and liver function, thyroid profile, and cardiac biomarkers (troponin I or troponin T) to look for evidence of myocardial necrosis. BNP level assists in the diagnosis of heart failure in patients presenting with dyspnea, and can be followed serially to assess effectiveness of therapy. Pulse oximetry helps to assess oxygenation and pulmonary function. An arterial line is helpful in managing patients with hypotension or cardiogenic shock. Urgent two-dimensional echocardiography is essential to evaluate left ventricular size and function, right ventricular function, valve function, and the presence of pericardial effusion. Doppler echocardiographic assessment of valve stenosis and regurgitation and of hemodynamics is invaluable.
When patients are admitted to hospital with acute decompensation of chronic systolic heart failure, the specific reason for a patient’s deterioration must be searched for and corrected when possible (Box 29.2). Environmental factors such as excessive salt and fluid intake or alcohol consumption are common. Patient adherence to outpatient therapies must be assessed, as heart failure regimens often involve numerous medications. Emotional and physical stressors should be corrected when feasible.
Concomitant administration of medications for noncardiac conditions can have detrimental effects. A partial listing includes corticosteroids and nonsteroidal anti-inflammatory drugs (NSAIDs). These drugs can cause fluid retention and can aggravate hypertension. NSAIDs also interfere with the beneficial renal effects of angiotensin-converting enzyme (ACE) inhibitors and can interfere with the action of loop diuretics.39 The use of NSAIDs has been reported to increase the risk of hospitalization for heart failure by tenfold in patients with a history of heart failure.8 Metformin and thiazolindinediones can contribute to water retention and aggravate the symptoms and signs of heart failure. Cancer chemotherapies can cause myocardial damage. Cardiac toxicity due to anthracycline chemotherapy is well described. Tyrosine-kinase inhibitors, a newer class of cancer chemotherapy, are also being recognized as agents that can aggravate heart failure and cardiomyopathy.40 Cardiac medications such as calcium channel blockers and antiarrhythmic drugs can also have direct negative effects on left ventricular contractility. Calcium channel blockers are generally contraindicated in patients admitted with acute decompensated heart failure.23 Type 1A and 1C antiarrhythmic drugs are also contraindicated in patients with abnormal left ventricular systolic function.
There are few controlled-trial data to arrive at evidence-based guidelines for the treatment of acute heart failure. Many recommendations are based on small studies, experience, observation, and a general consensus of opinion.41 The goals of initial therapy are to improve symptoms, optimize blood pressure, lower PCWP, and improve cardiac output. Treatments to reverse or prevent myocardial injury are instituted, and a search for reversible causes of heart failure needs to occur. Optimization of other comorbid conditions is important, including hyperglycemia, renal disease, and pulmonary function.
Initial therapy includes supplemental oxygen and assessment of the need for ventilatory assistance with noninvasive positive airway pressure ventilation or endotracheal intubation. Noninvasive ventilation improves oxygenation and pulmonary compliance and decreases work of breathing. Endotracheal intubation may be required for patients with severe hypercapnia, acidosis, and respiratory muscle fatigue. The use of continuous oxygen administration alone was prospectively compared with the use of continuous positive airway pressure ventilation (CPAP) and noninvasive intermittent positive pressure ventilation (NIPPV) in 1069 patients who presented with acute cardiogenic pulmonary edema. No difference among these therapies was noted in the primary end point of death at 7 days, or in the secondary end point of death plus endotracheal intubation at 7 days. Noninvasive ventilation did result in more rapid improvement in dyspnea, tachycardia, hypercapnia, and acidosis. There was no difference in safety or efficacy between CPAP and NIPPV.42
Treatment of arrhythmias is essential. Rapid atrial fibrillation is a common problem in these patients. In the Euroheart Failure Study, 9% of patients hospitalized with acute heart failure had atrial fibrillation during the hospitalization, and 42% had a history of paroxysmal atrial fibrillation.43 Other studies report atrial fibrillation in 25% to 30% of hospitalized heart failure patients.36 Control of the ventricular response to atrial fibrillation is vitally important, especially in patients with diastolic heart failure. This control can be achieved rapidly with the use of intravenous beta blockers such as metoprolol or esmolol, parenteral digoxin, or intravenous amiodarone. Intravenous diltiazem can be used in patients whose left ventricular systolic function is known to be normal or near normal.
Indications for Invasive Hemodynamic Monitoring
Placement of a pulmonary artery (PA) catheter enables the clinician to accurately measure PCWP, cardiac output, and mixed venous oxygen saturation. It can also help assess the effectiveness of therapy. Whether to routinely use PA catheters to assess and manage patients with acute heart failure has been long debated. The ESCAPE trial evaluated the routine use of PA catheterization in patients hospitalized with acute exacerbation of chronic heart failure and left ventricular systolic dysfunction.44 There was no difference in the primary end point of days alive out of the hospital during 6 months after discharge in groups managed with or without a PA catheter. There were no significant adverse effects with PA catheter use in this study. There were no subgroups identified in which use of the PA catheter was beneficial. However, there was a trend noted in improving the initial diuresis, with less deterioration of renal function, in the PA catheter group. The authors concluded that there was no indication for the routine use of PA catheters in the setting of acute heart failure.
However, a PA catheter is often essential for the management of patients with acute severe heart failure. Findings on physical examination are often insensitive indicators of hemodynamic status. Indications for PA catheter use include cardiogenic shock, differentiating pulmonary from cardiac causes of dyspnea, hemodynamic assessment if one is unsure of the diagnosis or severity of heart failure by clinical assessment, worsening renal function, guiding parenteral vasodilator therapy, and in patients who are not improving with initially prescribed therapy.37 PA catheter placement is also necessary as part of the evaluation for cardiac transplantation or the implantation of a ventricular assist device (VAD) (Box 29.3).
Consecutive patients with severe heart failure were evaluated and classified according to hemodynamic measurements of PCWP and cardiac index. Patients were described as “dry” with average PCWP less than 17 mm Hg, or “wet” with PCWP reading of 29 mm Hg on average. The patients were also described as “warm” versus “cold,” based on a cardiac index of greater than 2.1 L/minute/m2 versus an index of less than 1.6 L/minute/m2. The severity of symptoms and findings on physical examination did not predict the hemodynamic status as defined by invasive monitoring. In addition, the hemodynamic picture did not predict the response to therapy and survival was similar in all four groups, except that patients with higher cardiac output and lower PCWP had slightly better outcomes than patients with low cardiac output and high PCWP.45
Tailoring pharmacologic therapy to hemodynamic measurements is often helpful and necessary to determine precise measurements of cardiac output and left ventricular filling pressure, in order to guide intensive intravenous drug therapy. Aggressive therapy tailored to the response in hemodynamic measurements has been advocated as an effective method to obtain more rapid and sustained improvement in patients with the most severe heart failure.46 When PCWP is reduced to less than 16 mm Hg, and right atrial pressure is reduced to less than 8 mm Hg, most patients will improve acutely and for the remainder of their hospitalization. Additional hemodynamic goals include reducing systemic vascular resistance to less than 1200 dynes × sec/cm5, raising cardiac index to greater than 2.6 L/minute/m2, and maintaining systolic blood pressure over 80 mm Hg. PCWP can be lowered to a normal value of 10 to 12 mm Hg in many patients with significant left ventricular dysfunction without untoward effects.46,47 In a group of patients referred for cardiac transplantation, a combination of aggressive parenteral therapy, targeted to optimal hemodynamics, followed by conversion to appropriate oral therapy, resulted in clinical improvement so that 30% of these patients were able to be removed from transplant lists.46
Pharmacologic Management of Acute Heart Failure
Goals of treatment of heart failure can be defined as short-term, to relieve dyspnea and reverse acute hemodynamic decompensation, and long-term, to prevent rehospitalization, improve functional status, and prolong survival. Additional goals include preserving renal function, preventing arrhythmias, and preventing myocardial necrosis in patients with ischemic and nonischemic disease. Pharmacologic therapies to prevent or attenuate chronic remodeling should be instituted or strengthened prior to discharge from the hospital, as these have been shown to improve long-term survival (Box 29.4).
Intravenous Diuretics
Although no randomized clinical trials exist, the use of loop diuretics is supported by a long history of clinical success. These agents increase renal excretion of salt and water. The onset of action of intravenous bolus furosemide is 30 minutes, and the drug peaks at 1 to 2 hours. The half-life of the medication is 6 hours, so twice daily dosing is usually required.39 Other loop diuretics often used are bumetanide and torsemide (Tables 29.2 and 29.3). Several small clinical trials suggested that a constant infusion of a loop diuretic resulted in superior diuresis when compared to intermittent bolus dosing,48,49 although other studies did not confirm this.50 A large, randomized controlled study was performed in patients hospitalized with acute decompensated heart failure who were taking a high dose of furosemide prior to admission. Patients were randomized within 24 hours of admission to bolus dosing or constant infusion, and usual daily outpatient dose (given intravenously) versus high dose (2.5 times their usual daily dose). No significant differences were noted in predefined outcomes between bolus and infusion administration. However, the high-dose group had better relief of dyspnea and more fluid and weight loss than the lower-dose group; also, 23% of the high-dose group had a significant deterioration in renal function, but at 60 days there was no difference in renal function between the two groups.51
Table 29.3
Treatment of Refractory, Diuretic-Resistant Heart Failure
| To Loop Diuretic, add: | Dose |
| Hydrochlorothiazide | 25-50 mg once or twice daily |
| or metolazone | 2.5-5.0 mg once or twice daily |
| or spironolactone | 12.5-50 mg once daily |
An association between high-dose loop diuretics and a worse prognosis has been observed. There is concern that this may be mediated by further activation of the renin-angiotensin-aldosterone system by loop diuretics. However, in a retrospective analysis utilizing propensity matching, hospital mortality rate was not different between low-dose and high-dose diuretic groups.52 It is likely that poorer outcomes associated with high-dose diuretics is not due to the drug itself but reflects a greater severity of heart failure illness with concomitant renal disease.53
Patients with chronic heart failure with or without renal dysfunction may exhibit resistance to loop diuretics, defined as an acute reduction in diuretic efficacy after repeated loop diuretic dosing. With chronic loop diuretic use, there is an increase in sodium reabsorption in the distal nephron and stimulation of aldosterone release. In edematous states there is delayed oral absorption of the drug. With renal dysfunction there may be reduced levels of drug delivered to the renal tubule. This resistance is associated with a poorer prognosis.23 Diuretics that act distally in the renal tubule, such as metolazone or hydrochlorothiazide, or aldosterone blockers such as spironolactone can be added.4 Combination diuretic therapy induces a greater diuresis than simply increasing the dose of loop diuretic further. The response may be delayed for 48 to 72 hours. Combining diuretics often augments diuresis even in the setting of significant chronic kidney disease, and a good response can be expected in over 70% of patients. Particular attention must be given to following potassium, sodium, chloride, and magnesium levels when combination diuretic therapy is prescribed54 (Table 29.4).
Table 29.4
Continuous Intravenous (IV) Infusion of Loop Diuretics
| Diuretic | Dose |
| Bumetanide | 1 mg IV load, then 0.5-2 mg/hr infusion |
| Furosemide | 40 mg IV load, then 10-40 mg/hr infusion |
| Torsemide | 20 mg IV load, then 5-20 mg/hr infusion |
Adapted from Nieminen MS, Bohm M, Cowie MR, et al: Executive summary of the guidelines on the diagnosis and treatment of acute heart failure. The Task Force on Acute Heart Failure of the European Society of Cardiology. Eur Heart J 2005;26:384-416.
Vasopressin Inhibitors
The use of novel diuretics, vasopressin inhibitors, has been evaluated in clinical trials of treatment of acute heart failure.55 Vasopressin is a hormone synthesized in the hypothalamus; its major effect is to control free water clearance. It acts through V1a receptors in vascular smooth muscle and myocardium, leading to peripheral and coronary vasoconstriction, myocyte hypertrophy, and positive inotropy. Vasopressin also acts through V2 receptors at the renal tubule collecting ducts to cause free water retention and hyponatremia. Levels of vasopressin are increased in patients with chronic heart failure, and higher vasopressin levels correlate with worse heart failure severity. Vasopressin release is stimulated by changes in serum osmolality and cardiac output and leads to further vasoconstriction and retention of free water.56 Inhibition of vasopressin’s effects would have theoretic benefits in patients with heart failure.57 In contrast to loop diuretics, inhibition of vasopressin theoretically would not cause hypotension or neurohormonal activation, and would not aggravate cardiac arrhythmias due to electrolyte depletion.
Conivaptan is a vasopressin antagonist that inhibits V1a and V2 receptors. Tolvaptan and lixivaptan are antagonists selective for the V2 receptor. These medications increase urine volume and free water excretion, with a rise in the serum sodium concentration. The use of conivaptan in patients with class III-IV heart failure was associated with increased urine output, and decreases in PCWP and right atrial pressure, without changes in cardiac output.8 Oral use of tolvaptan was associated with fluid loss and diuresis without change in heart rate, blood pressure, or serum creatinine.
In a large, multicenter, placebo-controlled randomized trial called EVEREST, tolvaptan was administered to patients hospitalized with heart failure. There were no adverse consequences on heart rate, blood pressure, or serum electrolytes and there was more rapid improvement in dyspnea and signs of heart failure when compared to usual therapy.58 Hyponatremia improved. Hemodynamic effects included rapid reduction in PCWP and right atrial pressure.59 However, at 10-months follow-up after hospitalization, there was no improvement in mortality rates or readmission rates.60 Vasopressin inhibitors are approved for treatment of severe hyponatremic states but are not approved for use in heart failure.
Parenteral Vasodilators (Table 29.5)
Intravenous vasodilator therapy is often added to diuretic therapy to obtain more rapid improvement in severe heart failure. A clear indication for vasodilators is in patients with severe hypertension and pulmonary edema. Their use should also be considered in patients who are not responding to intravenous diuretics combined with standard oral therapies. Blood pressure response to these medications needs to be carefully monitored because hypotension is a common effect. Improvement in hemodynamics has been obtained with aggressive intravenous vasodilator therapy using intravenous nitroprusside, intravenous nitroglycerin, or nesiritide. The choice of agents depends on matching the patient’s clinical picture and hemodynamics with the predicted effects of each vasodilator.61,62
Nitroprusside
Intravenous sodium nitroprusside is a powerful venous and arterial dilator. It is a drug of choice in treating hypertension-related heart failure with pulmonary edema and severe heart failure due to acute mitral regurgitation. The use of nitroprusside requires hospitalization in the ICU and invasive monitoring with a PA catheter and arterial line. This drug causes a significant reduction of afterload and preload, leading to decreased right atrial pressure, decreased systemic vascular resistance, decreased mean systemic blood pressure, decreased PCWP, and increased cardiac index in patients with heart failure and left ventricular dysfunction. Limitations of nitroprusside use include inducing a coronary “steal” syndrome in patients with active coronary ischemia.39 In addition, toxic metabolites can accumulate with more prolonged administration. In patients with significant hepatic dysfunction, thiocyanate levels rise, and in patients with renal dysfunction, cyanide is generated. Dosage range is 0.3 to 5.0 µg/kg/minute.
Nesiritide (B-natriuretic Peptide)
Human BNP can be manufactured by recombinant DNA technology and is available as an intravenous medication, nesiritide, for heart failure therapy. BNP is a hormone produced by ventricular and atrial myocytes in response to stretch from cardiac chamber dilatation. Hemodynamic effects include venous and arterial dilation, coronary vasodilation, and natriuresis. Reduction in PCWP and right atrial pressure exceeding the effects of intravenous nitroglycerin, when compared directly, was reported.61 It is not proarrhythmic and does not induce tolerance.63 It may potentiate the effects of loop diuretics. Significant hypotension may limit its use in some patients.39 Because the hypotensive effects of nesiritide are less marked than nitroprusside, nesiritide can be used without invasive hemodynamic monitoring and can be initiated in emergency department settings. Nesiritide is initiated as an intravenous bolus dose of 2 µg/kg followed by infusion of 0.01 µg/kg/minute.
Nesiritide was compared to dobutamine in patients with severe heart failure. Nesiritide infusion was associated with less tachycardia and ventricular arrhythmia.64 Other nonrandomized studies suggested a trend toward improved survival and lower rehospitalization rates with nesiritide.63 A meta-analysis of three randomized trials of nesiritide suggested a slight increase in mortality rates in patients given nesiritide versus a placebo control group, possibly mediated through an adverse effect on renal function.65,66
[/not-level-membership-for-critical-care-medicine-category]
