[level-membership-for-critical-care-medicine-category]23
Septic Shock
Overview
This chapter pertains to pathophysiology, assessment, and management of septic shock, the most severe and overt manifestation of the septic condition. This discussion will specifically focus on cardiovascular and hemodynamic aspects. Other critically important elements of sepsis pathophysiology, assessment, and management (i.e., beyond the cardiovascular and hemodynamic aspects) will be addressed in a separate chapter (see Chapter 25, Sepsis and Multiple Organ Dysfunction). This chapter is also focused specifically on the adult patient with septic shock, as principles and evidence may differ in important ways in the pediatric population.
Historical Perspective
The word sepsis originated from the Greek language. Sepsis was synonymous with putrefaction and pertained to the bacteria-mediated decomposition of organic matter.1 The term persisted for more than 2700 years with essentially unchanged meaning.2 In the twentieth century, our modern understanding of the term sepsis became rooted in a disease in which the clinical manifestations were attributed to severe infection and the release of pathogenic bacterial products into the patient’s bloodstream.3,4
The term shock comes from the French word “choquer,” meaning “to collide with.” This is particularly appropriate terminology for shock due to sepsis, given our modern understanding of the sepsis pathophysiology, whereby the body’s host defenses essentially collide with the invading microorganism, triggering a profound proinflammatory host response.1
Contemporary Definitions
Shock is defined as a failure of the cardiovascular system to maintain effective tissue perfusion. If effective tissue perfusion is not promptly restored, cellular dysfunction and acute organ failure may occur and may become irreversible, leading to acute organ system failure. When shock develops because of a systemic inflammatory response to infection, it is termed septic shock. The American College of Chest Physicians (ACCP) and the Society of Critical Care Medicine (SCCM) first published consensus conference definitions for sepsis syndromes more than 20 years ago,5 and these definitions were revisited and further developed by international consensus in 2003.6 Septic shock was defined as infection-induced hypotension (systolic blood pressure <90 mm Hg [or a drop of >40 mm Hg] plus signs of tissue hypoperfusion despite adequate fluid resuscitation). The concurrent presence of clinical signs of tissue hypoperfusion (e.g., metabolic acidosis, encephalopathy, acute lung injury, oliguria, acute kidney injury, peripheral extremity discoloration, or impaired capillary refill) is an integral component of making the diagnosis of septic shock, because baseline blood pressure can vary among patients, and patients with lower baseline blood pressure may tolerate an arterial pressure lower than the values stated here without being in circulatory shock. The overarching purpose and major impact of the efforts to establish the contemporary definitions given here was the promotion of uniformity in inclusion criteria for sepsis clinical trials.7
Epidemiology
Severe sepsis (sepsis plus acute organ system dysfunction) is a common and deadly disease with major public health implications. Although heterogeneity of definitions of sepsis has historically made the incidence of severe sepsis and septic shock difficult to precisely measure, estimates of the incidence have been possible. Using the International Classification of Diseases (ICD)-9 codes for infection and organ dysfunction, Angus and coworkers estimated that 751,000 cases of severe sepsis occur in the United States every year.8 Figure 23.1 displays the incidence of severe sepsis in the United States compared to other common diseases. The incidence of severe sepsis currently exceeds the incidence of lung and colon cancer, venous thromboembolic disease, and acquired immune deficiency syndrome (AIDS),8–11 and the incidence is projected to increase by 1.5% per year, resulting in more than 1 million cases of severe sepsis annually by the year 2020.8 The incidence of sepsis and septic shock is known to be increasing because of a longer lifespan for patients with severe chronic medical conditions that predispose them to acquiring sepsis. This includes an increase in the number of immunocompromised patients in the community, number of infections caused by resistant organisms, increased use of intravascular catheters, and aging of the population.8
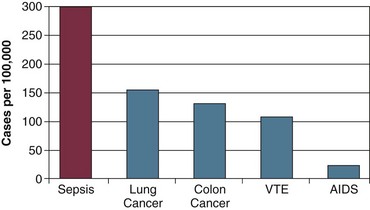
Figure 23.1 Incidence (cases per 100,000 population) of severe sepsis in the United States compared to four high-profile diseases.8–11 AIDS, acquired immune deficiency syndrome; VTE, venous thromboembolic disease.
Sepsis is the leading cause of death among critically ill patients12 and is responsible for as many deaths annually in the United States as acute myocardial infarction.8 Figure 23.2 displays control arm mortality rates in septic shock clinical trials.1 In a recent large multicenter registry study, septic patients with both arterial hypotension and severe lactic acidosis experienced a 46% mortality rate, whereas the mortality rate for arterial hypotension or severe lactic acidosis alone was 37% and 30%, respectively.13 Overall, severe sepsis in general ranks as the tenth leading cause of death in the United States, with 215,000 deaths annually and an estimated 30% in-hospital mortality rate.8,14 Figure 23.3 displays the mortality rate for severe sepsis compared to other high-profile diseases that may require critical care (acute ischemic stroke, acute myocardial infarction, and trauma).8,15–17 The apparent disparity in mortality rates across these diseases may be explained in part by differences in the conventional approach to treatment, as acute ischemic stroke, acute myocardial infarction, and trauma are all typically treated with aggressive interventions in a time-sensitive fashion. Similar to the “golden hour” concept for trauma care that was first recognized more than 30 years ago18 we are now beginning to understand that early aggressive interventions for sepsis can also have an impact on outcome.
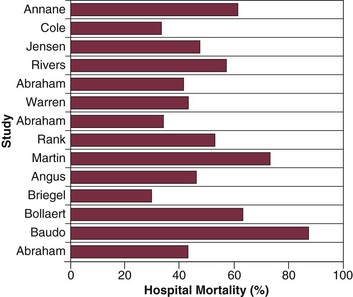
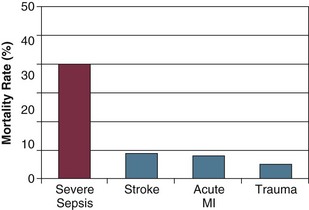
Figure 23.3 Mortality rate of severe sepsis in the United States compared to three diseases that are treated aggressively with time-sensitive interventions.8,15–17 MI, myocardial infarction.
It is also important to recognize that, in addition to a high mortality rate, severe sepsis and septic shock are associated with serious risk of morbidity among survivors.19,20 A systematic review of the literature found that sepsis survivors had substantially diminished quality of life and a sharply reduced long-term survival after typical short-term (i.e., 28-day) outcomes are assessed.19 Among older adults, severe sepsis has been associated with major persistent cognitive impairment and functional disability that could have a substantial impact on those patients’ ability to live independently.20 Taken together, even among patients who survive the sepsis insult, the development of severe sepsis or septic shock can represent a pivotal event in the trajectory of a patient’s life.
Pathogenesis
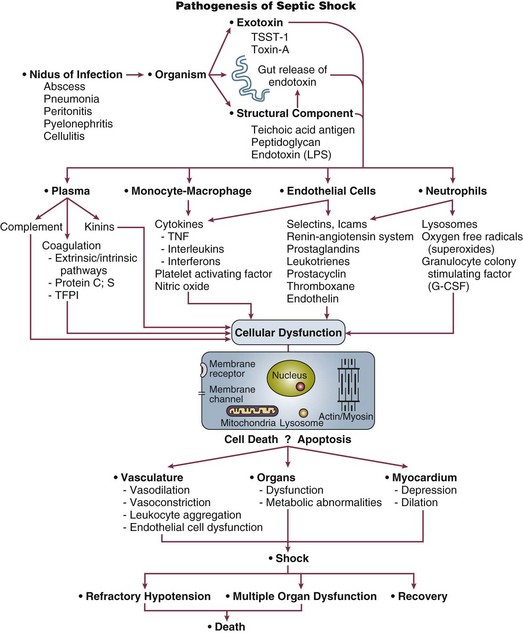
Septic shock results when infectious microorganisms in the bloodstream induce a profound inflammatory response causing hemodynamic decompensation. The pathogenesis involves a complex response of cellular activation that triggers the release of a multitude of proinflammatory mediators. This inflammatory response causes activation of leukocytes and endothelial cells, as well as activation of the coagulation system. The excessive inflammatory response that characterizes septic shock is driven primarily by the cytokines tumor necrosis factor-alpha (TNF-α) and interleukin 1 (IL-1) that are produced by monocytes in response to an infection. Although TNF-α and IL-1 are central to the pathophysiology of septic shock and act synergistically to induce hypotension in experimental models, a number of other vital mediators are also known to play a major role, including high-mobility group box 1 (HMGB1) protein.21 Another important recent advance in our understanding of septic shock pathophysiology has been identification of the close link that exists between the proinflammatory response of septic shock and activation of the coagulation system (e.g., clinical or subclinical disseminated intravascular coagulation [DIC]).22 Although the systemic inflammatory response of sepsis triggers profound macrocirculatory and microcirculatory changes that impair tissue perfusion, another important mechanism playing a role in the development of acute organ dysfunction in septic shock is apoptosis (programmed cell death). Accelerated apoptosis is known to be a critical pathogenic event in this disease. In addition, certain genetic polymorphisms are becoming recognized as major determinants of susceptibility to infection, as well as risk of death from septic shock. Key steps in the pathogenesis of septic shock are shown in Figure 23.4.
Clinical Presentation
Patients with septic shock will typically manifest signs of systemic inflammation including fever or hypothermia, tachycardia, tachypnea, and elevation or reduction of the white blood cell count. Although the absence of arterial hypotension does not necessarily exclude the possibility of subclinical tissue hypoperfusion,23 the hallmark of septic shock is arterial hypotension despite adequate volume resuscitation requiring vasoactive drugs for hemodynamic support. Other signs of potential tissue hypoperfusion may include lactic acidosis, oliguria, encephalopathy, or diminished capillary refill in the extremities. Patients with septic shock typically have multiple organ system dysfunctions; clinical evidence of other organ system dysfunction may range from subtle abnormalities to overt organ failure. Multiorgan system involvement in sepsis may include cardiovascular, respiratory, renal, central nervous system, hepatic, metabolic, or hematologic dysfunction. Respiratory system dysfunction manifests as acute lung injury or, in the most extreme cases, the acute respiratory distress syndrome (ARDS). Sepsis-induced renal dysfunction typically manifests with oliguria and may progress to acute renal failure requiring dialysis. Central nervous system dysfunction will manifest as encephalopathy, which may range from mild cognitive impairment to overt coma. Cholestasis is a common manifestation of hepatic dysfunction in sepsis, but in the presence of severe shock, ischemic hepatitis (“shock liver”) may occur. Metabolic derangements of septic shock include a loss of glycemic control (hyper- or hypoglycemia) as well as metabolic acidosis. Septic shock is commonly associated with a consumptive coagulopathy, which is likely present in almost all patients at least subclinically,24 but may also manifest clinically with thrombocytopenia, prolongation of the prothrombin time, or in the most severe cases, overt DIC.
The multiple organ dysfunction associated with septic shock is not only a critical event in the pathogenesis of this disease, but is also closely linked with mortality rate.8,25,26 There is an approximate 20% increase in septic shock mortality rate with each additional organ system that fails.8 Early evidence of organ failure is an especially strong predictor of death.26,27 Early improvement in organ function (e.g., 0-24 hour improvement in the Sequential Organ Failure Assessment [SOFA] score28,29) is closely related to sepsis survival, whereas later improvement after the first 24 hours has little predictive value.27 These data, garnered largely from observational studies as well as placebo arms of interventional trials, support the concept that aggressive therapy for sepsis to reverse (or prevent the development of) acute organ system failure within the first 24 hours is closely associated with eventual outcome.
Hemodynamic Profile of Septic Shock
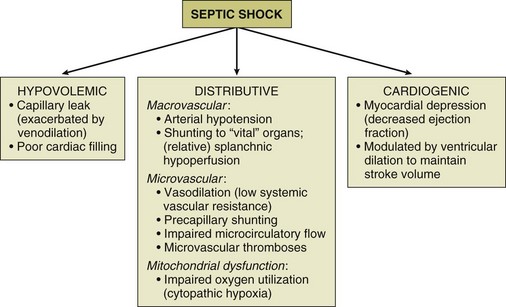
The hemodynamic profile of septic shock is the most complex hemodynamic profile of all shock etiologies (Fig. 23.5). What sets septic shock apart from other causes of circulatory shock is the fact that there may be multiple different mechanisms of circulatory shock occurring simultaneously.1,30 Septic shock may have features of (1) hypovolemic shock (poor cardiac filling secondary to severe systemic capillary leak and increased venous capacitance), (2) cardiogenic shock (infection-induced myocardial depression), and (3) distributive shock (arteriolar vasodilation with tissue hypoperfusion in the face of an adequate cardiac output).1
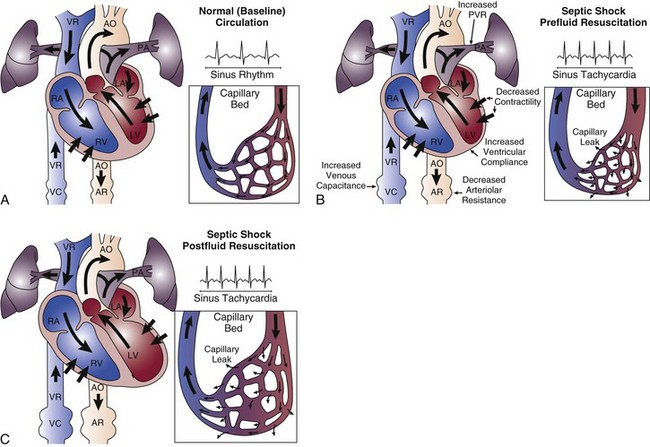
Hypovolemia
The release of proinflammatory mediators into the circulation causes injury to the integrity of the endothelial cell surface throughout the systemic microvasculature, resulting in severe capillary leak and extravasation of fluid into tissues. Venodilation also compromises venous return. These are major factors in producing hypovolemia in the patient with septic shock. The septic shock patient may have a markedly decreased cardiac preload, especially in the initial phase of therapy. Aggressive resuscitation with intravenous volume expansion modulates the hemodynamic profile of septic shock and allows the patient to achieve a hyperdynamic (i.e., high cardiac output) state.1 The combination of a decreased preload and myocardial depression means that in the early phase of sepsis resuscitation, patients may initially be hypodynamic (i.e., low cardiac output) prior to receiving adequate volume resuscitation. Capillary leak is an ongoing process in the course of septic shock therapy, and therefore hypovolemia may recur later in the course of the disease, even after adequate cardiac filling has been initially achieved. Fluid balance (input of intravenous fluids and output of urine) is an unreliable parameter for assessing adequacy of fluid resuscitation in septic shock.
Myocardial Dysfunction
Septic shock is associated with depression of biventricular function with a decrease in the ejection fraction. Ventricular dilation occurs as a compensatory mechanism and raises end-diastolic volume so that stroke volume can be preserved, taking advantage of the Starling principle. When myocardial dysfunction occurs, a high cardiac output can still be achieved in many circumstances because of biventricular dilation, tachycardia, and arteriolar dilatation, as long as the patient is adequately volume resuscitated and does not have a severe cardiac suppression (related either to previously existing cardiac dysfunction or overwhelming sepsis-induced suppression of cardiac systolic function).30 The most important inflammatory mediators that induce myocardial depression are TNF-α, IL-1, and perhaps nitric oxide.31,32 Coronary blood flow is typically normal or increased in septic shock.33 Although coronary blood flow can be diminished by severe arterial hypotension that compromises coronary perfusion pressure (especially if there is preexisting coronary artery disease), myocardial ischemia does not appear to be the causative factor of the depression in myocardial performance. It has been reported that nearly half of patients with septic shock will have echocardiographic evidence of some degree of depression of systolic function, even in the absence of preexisting cardiac disease.34 However, myocardial depression is typically not the predominant feature of the septic shock hemodynamic profile.30 For the majority of patients, aggressive intravascular volume expansion to restore adequate cardiac filling pressures will be enough to achieve a reasonable cardiac output.
Distributive Shock
Septic shock is characterized by peripheral maldistribution of blood flow to tissues such that tissue hypoperfusion abnormalities can persist despite a normal or high cardiac output. This is called “distributive shock.”30 This maldistribution of blood flow may occur at both microcirculatory and macrocirculatory levels. The role of microcirculatory dysfunction is discussed in detail in the next section of this chapter. At the level of the macrocirculation, the autoregulation of blood flow within any single organ system in a normal host can typically maintain effective tissue perfusion over a wide range of systemic pressures (usually ranging from a mean arterial pressure [MAP] of 50 mm Hg to 150 mm Hg). However, there is heterogeneity of blood flow distribution throughout the body in septic shock due to preferential shunting of blood flow to vital organs (e.g., the brain and myocardium). The gastrointestinal tract may be the earliest organ system to experience tissue hypoperfusion in septic shock, as blood is shunted away from the splanchnic circulation in order to preserve blood flow to the brain, myocardium, and skeletal muscles. Ischemic injury to the gastrointestinal tract may be a source of ongoing systemic inflammation in septic shock.
The three components of the hemodynamic profile of septic shock are displayed in Figure 23.6.
Microcirculatory and Mitochondrial Dysfunction
Microcirculatory Dysfunction
Microcirculatory dysfunction is a pivotal element of the pathogenesis of septic shock.35–38 Although the macrocirculation (heart and large arteries) regulates the global distribution of blood flow throughout the body, it is the microcirculation that controls the delivery of blood flow to tissues. Using intravital videomicroscopy, experimental models of sepsis have demonstrated impaired microcirculatory flow velocity, “stopped-flow” microvessels, increased heterogeneity of regional perfusion, and low density of perfused capillaries.39–42 These derangements can cause marked alterations of oxygen transport including impaired tissue oxygen extraction.43 With the advent of new investigational videomicroscopy techniques, it is now possible to study the microcirculatory network in human subjects with septic shock. Microcirculatory failure appears to be one of the critical pathogenic events in sepsis that is associated with acute multiorgan dysfunction and death.35–38 As these alterations of microcirculatory flow in sepsis can occur in the absence of global hemodynamic perturbations (i.e., absence of low arterial pressure or low cardiac output),36,42,44 derangements of small vessel perfusion are largely a function of intrinsic events in the microcirculation.
The causes of microcirculatory flow alterations in sepsis (Fig. 23.7) are multifactorial and include endothelial cell dysfunction, increased leukocyte adhesion, microthrombi formation, rheologic abnormalities, altered local perfusion pressures due to regional redistribution of blood flow, and functional shunting.39,45 The proinflammatory cytokines released in sepsis cause diffuse endothelial cell activation, which is associated with neutrophil activation, expression of endothelial adhesion molecules (i.e., integrins and selectins), and localization of white blood cells to areas of microvascular injury. Pan-endothelial cell injury increases microvascular permeability with the influx of proinflammatory cells into the tissues; this is hypothesized to be an important pathogenic step in the development of acute system organ dysfunction in sepsis. Leukocyte adhesion of white blood cells to the microvessel endothelial surface (primarily in the postcapillary venule) further impedes microcirculatory blood flow. The endothelial injury also triggers the activation of the coagulation cascade via expression of tissue factor on the microvascular endothelium, resulting in fibrin deposition and microvascular thrombosis that may further impair microcirculatory flow. All of these mechanisms collectively contribute to microcirculatory failure in septic shock.37,39
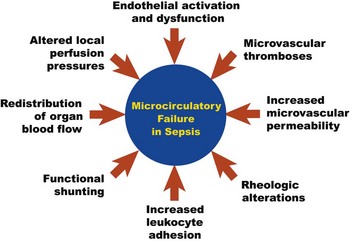
Although septic shock research has classically been focused on macrocirculatory hemodynamic parameters that reflect the distribution of blood flow globally throughout the body, a functional microcirculation is another critical component of the cardiovascular system that is necessary for effective blood flow to tissues. This conceptual framework is depicted in Figure 23.8. Although a shift of research focus from global hemodynamic parameters to indices of microvascular perfusion could potentially be viewed as a major change of direction for septic shock research, the microcirculation likely represents a logical next frontier in the evolution of our understanding of circulatory failure in shock states.37,46 Although there are currently no therapies to specifically target microcirculatory dysfunction in sepsis, going beyond optimization of macrocirculatory hemodynamics and developing new innovative strategies to reverse microcirculatory failure could (in the future) potentially represent a cutting edge method to augment tissue perfusion in sepsis.
 , mixed venous oxygen saturation; SVR, systemic vascular resistance; V
, mixed venous oxygen saturation; SVR, systemic vascular resistance; VMitochondrial Dysfunction
There is strong evidence that cellular utilization of oxygen can be markedly impaired in septic shock.47 Bioenergetic failure can occur even after effective restoration of blood flow to tissues has been achieved, and this has been termed “cytopathic hypoxia.” Despite the current absence of therapies to reverse cytopathic hypoxia, this phenomenon does have some relevance for clinical practice, as impaired cellular oxygen extraction and utilization can manifest clinically with acute organ system failure in the setting of markedly elevated values for mixed (or central) venous oxygen saturation. This venous hyperoxia likely reflects bioenergetic failure and identifies a population at exceptionally high risk of death.48 Cytopathic hypoxia has been associated with acute organ dysfunction, but the extent to which this does or does not represent a cause-and-effect relationship has not yet been fully elucidated.
Management of Septic Shock
Overview and Management Guidelines
The Surviving Sepsis Campaign (SSC) first published comprehensive international consensus guidelines for sepsis management in 2004.49,50 The SSC guidelines have been updated over time as the best evidence for sepsis management continues to evolve. The critical care practitioner should be familiar with the concepts in the SSC guidelines and is referred to the most recent update for a comprehensive review including evaluation of the strength of evidence for each recommendation.50a The SSC guidelines writing committee comprised representatives from numerous medical professional societies that relate to the care of the septic patient, and these medical professional societies have endorsed the guidelines.
General Principles
The patient with septic shock should be brought to a critical care area as quickly as possible to facilitate rapid resuscitation and optimal hemodynamic support. Continuous electrocardiographic monitoring and pulse oximetry are useful tools in the management of critically ill patients with sepsis.51,52 In addition, a variety of more invasive devices may be of use. The arterial catheter has two functions: It allows frequent blood sampling and continuous assessment of arterial pressure. The pulmonary artery catheter (PAC) can provide data such as cardiac filling pressures, cardiac index, and systemic vascular resistance. The data gathered from the PAC can be useful for titrating vasoactive medications in septic shock. Although indications for PAC utilization are controversial and are often debated, it is important to recognize that the PAC represents a tool for guiding therapy rather than being a therapeutic intervention in itself. Monitoring venous oxygen saturation (either mixed venous oxygen saturation [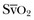 ] or central venous oxygen saturation [ScvO2]) can yield information on the oxygen supply/demand relationship, especially in the early resuscitation phase of septic shock therapy.23 A markedly low value for either
] or central venous oxygen saturation [ScvO2]) can yield information on the oxygen supply/demand relationship, especially in the early resuscitation phase of septic shock therapy.23 A markedly low value for either 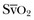 or ScvO2 indicates a significant imbalance in the oxygen supply/demand relationship, and likely indicates a need for augmenting global oxygen delivery.
or ScvO2 indicates a significant imbalance in the oxygen supply/demand relationship, and likely indicates a need for augmenting global oxygen delivery.
Metabolic parameters to monitor the effectiveness of resuscitation and cardiovascular support are limited; however, measurement of blood lactate can provide important information. In 1964, Weil first proposed the utilization of blood lactate levels as a surrogate of adequacy of tissue perfusion.53 It is important to realize, however, that elevation of blood lactate does not necessarily indicate ineffective tissue perfusion, as metabolic derangements and altered cellular metabolism may cause hyperlactatemia and can be responsible for the elevation of blood lactate observed in sepsis. Despite this, blood lactate levels still have prognostic value in septic patients. Regardless of the cause of lactate elevation in sepsis, markedly elevated blood lactate (e.g., lactate ≥4 mmol/L) signals an increased risk of death.54–58
Antibiotic Therapy and Source Control
Early administration of empiric antibiotic therapy and expeditious source control to eliminate any nidus of infection are imperative in the management of septic shock. Appropriate antibiotics given early may substantially improve the likelihood of survival.59,60 A choice of antibiotics is usually empiric because the organism is not yet identified when antibiotics must be delivered. Failure to include antibiotic coverage for what is later identified to be the offending organism has been associated with increased risk of death;61 therefore, broad-spectrum antibiotics are necessary as soon as septic shock is identified. Kumar and associates performed a large-scale multicenter retrospective study of patients with septic shock and found a linear association between the duration of hypotension prior to first dose of antibiotic administration and risk of death.62 One recent prospective emergency department (ED)–based study from Puskarich and colleagues found higher survival rates if antibiotics were administered prior to shock onset compared to after shock onset, but in contrast to the Kumar data the authors did not find a measurable effect of incremental time to administration of antibiotics on survival.63 One potential reason to explain these results is that the Kumar study was a heterogeneous population and the therapeutic interventions (e.g., early resuscitation and hemodynamic support) that the patients received were not standardized, whereas all the patients in the Puskarich study were ED patients treated according to a standardized early resuscitation protocol.
The SSC currently recommends that intravenous antimicrobial therapy be started as soon as possible, preferably within an hour of recognition of septic shock. Even though a 1-hour time window is deemed desirable, the SSC acknowledges that longer time frames are common in real-world clinical practice, and practice surveys verify that a 1-hour window is currently not standard of care.13 One reason for this could be the fact that sepsis often mimics other disorders, and the diagnosis of sepsis as the cause of the illness is often not obvious at the time of initial presentation. As such, the need for antimicrobial agents in the treatment of the patient may also not be immediately obvious. Once the diagnosis is made (or strongly suspected), antimicrobial therapy should be started promptly. Initial empiric antimicrobial selection should be broad enough to cover all likely pathogens based on clinical circumstances. In patients with septic shock, de-escalation or restriction of antibiotic therapy as a strategy to reduce the development of antimicrobial resistance is not recommended until after a causative organism has been identified or after the patient’s condition has markedly improved.
Early Resuscitation
One of the initial goals in the early management of a patient with septic shock is effective resuscitation to restore adequate tissue perfusion and decrease the risk of organ system injury. A number of hypotheses have been developed to explain the relationship between shock and the development of organ failure in critical illness. One hypothesis suggests that organ failure during critical care occurs as a consequence of inadequate oxygen delivery. Based on this hypothesis, a number of investigators have suggested that patients should be resuscitated to supranormal goals of systemic oxygen delivery in an attempt to prevent organ failure and improve outcome. The concept of supranormal oxygen delivery refers to the use of fluid resuscitation and inotropic drugs to drive up the oxygen delivery to achieve a predefined target. Several studies have examined this concept, although it is important to recognize that some studies have been performed in heterogeneous populations of critically ill patients rather than sepsis populations. The earliest clinical trials in perioperative high-risk surgery patients demonstrated an outcome benefit.64,65 Subsequently, however, numerous trials of supranormal oxygen delivery in critically ill patients failed to demonstrate any benefit. In the largest of these studies, Gattinoni and coworkers found no difference in survival or organ failure in a large number of critically ill patients when comparing patients resuscitated to supranormal end points to those receiving standard care.66 In a study by Hayes and associates, increasing oxygen delivery to supranormal levels with the use of high-dose dobutamine was associated with a reduction in survival.67 A meta-analysis concluded that supranormal oxygen delivery in critically ill patients was not beneficial68 and this concept largely fell out of favor in the 1990s.
For goal-oriented hemodynamic optimization to be beneficial, it has become clear that timing is critical. In contrast to the trials in perioperative high-risk surgery patients, subjects in the Gattinoni study were randomized much later, up to 72 hours after initial presentation.66 In a meta-analysis of critically ill patients that stratified studies by severity and the timing of interventions (early versus late), an outcome benefit was identified in the subset of patients with a high severity of illness and early initiation of interventions.69 A recent meta-analysis, this time specifically focused on patients with sepsis, found that quantitative resuscitation (i.e., early hemodynamic optimization targeting predefined quantitative end points of resuscitation) was associated with lower mortality rate in sepsis patients, but only if applied early, defined as less than 24 hours after presentation.70 These data suggest that quantitative resuscitation in the treatment of severe sepsis and septic shock can in fact be beneficial—in the right patient.
This early intervention concept was the rationale behind the study of early goal-directed therapy (EGDT) for severe sepsis and septic shock by Rivers and colleagues.23 EGDT is a type of quantitative resuscitation for septic patients that involves targeting central venous oxygen saturation (ScvO2) as a monitor of the adequacy of oxygen delivery. In a single-center randomized controlled trial of 263 ED patients with severe sepsis and septic shock, Rivers and colleagues targeted predefined end points of resuscitation including central venous pressure (CVP) 8 to 12 mm Hg, MAP 65 mm Hg or greater, and ScvO2 70% or greater in the ED. The authors reported that the EGDT protocol was associated with a 16% absolute risk reduction for mortality rate (30.5% vs. 46.5%). This study was an important contribution to the literature in showing that early interventions in the resuscitation phase of therapy can be associated with a significant improvement in long-term survival for patients with sepsis.
Recently, a multicenter ED-based randomized trial from Jones and associates compared lactate clearance (defined as a decrease by ≥10% in the serum lactate concentration) versus ScvO2 as an end point of sepsis resuscitation.71 Among 300 patients with sepsis-induced tissue hypoperfusion, the authors found that lactate clearance was noninferior to ScvO2 for the primary outcome of all-cause in-hospital deaths. These data suggest that, in addition to ensuring adequate cardiac preload and arterial blood pressure, lactate clearance has potential as a resuscitation target in sepsis-induced tissue hypoperfusion.
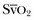 ] ≥ 65%). Although arterial pressure and urine output are routinely monitored in critical care practice, targeting CVP and central or mixed venous oxygen saturation necessitates invasive hemodynamic monitoring. If invasive hemodynamic monitoring is not yet in place (or is not established for any reason), aggressive empiric resuscitation should still be performed because it is possible that empiric resuscitation can optimize cardiac filling pressure and oxygen delivery even if the specific values for CVP or central/mixed venous oxygen saturation are not recorded. The SSC also recommends targeting resuscitation to lactate normalization as soon as possible in patients with elevated serum lactate levels, especially if Scv
] ≥ 65%). Although arterial pressure and urine output are routinely monitored in critical care practice, targeting CVP and central or mixed venous oxygen saturation necessitates invasive hemodynamic monitoring. If invasive hemodynamic monitoring is not yet in place (or is not established for any reason), aggressive empiric resuscitation should still be performed because it is possible that empiric resuscitation can optimize cardiac filling pressure and oxygen delivery even if the specific values for CVP or central/mixed venous oxygen saturation are not recorded. The SSC also recommends targeting resuscitation to lactate normalization as soon as possible in patients with elevated serum lactate levels, especially if ScvThe SSC also acknowledges that achievement of quantitative resuscitation goals can be challenging in routine clinical practice. Although some centers have been successful in implementing programs for quantitative resuscitation,72 a recent large multicenter observational study of the translation of SSC recommendations to clinical practice found that clinicians currently achieve all recommended end points of resuscitation less than 50% of the time.13 The reasons for this are likely multifactorial but may include the fact that from a practical standpoint the provision of quantitative resuscitation at the bedside can be relatively resource intensive, and some institutions may not have the necessary infrastructure to provide this service consistently at the present time.
Cardiovascular Support
Volume Resuscitation
Because there is no proven benefit of colloid therapy over crystalloids in resuscitation,73 the SSC currently recommends initiating volume resuscitation with crystalloid for patients with septic shock and suspicion of hypovolemia. The SSC-recommended volume of crystalloid is a minimum of 30 mL/kg fluid challenge. If there is hemodynamic improvement with this initial fluid challenge, clinicians may continue repeated fluid challenges to see if further hemodynamic improvement occurs. The SSC further suggests consideration of the addition of albumin infusion to initial crystalloid resuscitation if the initial crystalloids are judged to be ineffective. The SSC recommends against the use of synthetic hydroxyethyl starches in volume resuscitation because these agents have been associated with increased risk of acute kidney injury.
If a PAC is in place, the target for pulmonary capillary wedge pressure in a patient without preexisting cardiopulmonary disease is likely in the range of 12 to 15 mm Hg;74 however, it is imperative to remember that the “optimal” cardiac filling pressure may vary widely from patient to patient. One prudent strategy of volume resuscitation (rather than targeting a predefined cardiac filling pressure) would be to continue fluid bolus administration until the cardiac index fails to rise with additional intravascular volume expansion, indicating optimization of cardiac preload. An extremely high left ventricular filling pressure should be avoided because it could contribute to pulmonary capillary leak and cause impairment of oxygenation if the patient has concomitant acute lung injury. In the absence of a PAC to guide therapy, and if a patient has persistent hypotension refractory to an initial 30 mL/kg crystalloid intravascular volume infusion, it would be prudent to continue administering fluid boluses in attempts to raise the arterial pressure (unless the patient is manifesting clinical signs that pulmonary edema is developing, [e.g., increasing supplemental oxygen requirement]).75 Decisions on aggressiveness of fluid resuscitation should be made with consideration of oxygenation status. Patients with minimal supplemental oxygen requirements can be more aggressively fluid resuscitated with minimal concern for deleterious effects of intravascular volume expansion, but more cautious fluid administration is required in patients requiring higher FIO2 to maintain adequate oxygenation. Because the intravascular volume that optimizes stroke volume may produce worsening of oxygenation in patients with acute lung injury, intubation and mechanical ventilation may be required in order to assure adequate tissue perfusion.
Vasopressor Therapy
In addition to fluid administration, pharmacologic support of blood pressure is frequently necessary in both the initial resuscitation and subsequent support of patients with septic shock. These agents are, after fluids, the next most important interventions for the initial management of the hemodynamically unstable patient. Restoration of adequate arterial pressure is the end point of vasopressor therapy. The SSC recommends targeting a MAP of 65 mm Hg; however, blood pressure does not always equate to systemic blood flow, and the precise MAP to target may not necessarily be the same for all patients. LeDoux, Astiz, and coworkers demonstrated that, in septic shock patients treated with norepinephrine to maintain target MAP, MAPs of 65, 75, and 85 mm Hg achieved equivalent indices of tissue perfusion.76 In an observational study of patients with septic shock Varpula and associates found that an area under the curve of 65 mm Hg was the best predictor of positive outcome and showed that among multiple hemodynamic variables, a MAP above 65 mm Hg was the best predictor of a favorable outcome.77 It is notable that a MAP of 65 mm Hg may be inadequate for a patient with preexisting poorly controlled essential hypertension and associated vascular disease. Similarly, it should be recognized that in some patients it is possible to have arterial pressures lower than 65 mm Hg without tissue hypoperfusion. It is hypotension in the presence of tissue hypoperfusion that merits therapy with vasopressor agents. End points of resuscitation such as arterial pressure should be combined with assessment of regional and global perfusion. Other bedside indicators of persistent tissue hypoperfusion (besides hypotension) include oliguria, encephalopathy, poor capillary refill, and metabolic acidosis. Thus, even though the SSC recommends targeting a MAP of 65 mm Hg for most patients, the optimal MAP should be individualized based on the clinical considerations noted here.
Individual Vasoactive Agents
Vasoactive agents and their characteristics are summarized in Table 23.1. The selection of one of these drugs over the other as a first-line agent is a controversial and an often debated subject in the field of critical care medicine. Both norepinephrine and dopamine will effectively raise the blood pressure and the cardiac index, but the rise in cardiac index will be greater with dopamine. Dopamine, however, may cause or exacerbate tachycardia or dysrythmias.78 Norepinephrine is a more potent drug than dopamine in achieving a target MAP.1 Information from five randomized trials (n = 1993 patients with septic shock) comparing norepinephrine and dopamine does not support the routine use of dopamine in the management of septic shock.79
Table 23.1
Vasoactive Agents Commonly Used for Hemodynamic Support in Sepsis

From Trzeciak S, Parrillo JE. Septic shock. In Society of Critical Care Medicine, 8th Adult Critical Care Refresher Course. Chicago: Society of Critical Care Medicine, 2004, used with permission.
Epinephrine is a potent α- and β-adrenergic agent that increases MAP by vasoconstriction and also increases the cardiac index. Although epinephrine is a potent agent in raising the arterial pressure, the chief concern with the use of epinephrine has been the potential for impaired splanchnic perfusion.80–82 A large-scale randomized controlled trial comparing epinephrine to norepinephrine plus dobutamine reported no difference in vasopressor withdrawal, organ failure, and mortality rate. There was also no difference in the rates of serious adverse events. The authors concluded that there is no difference in efficacy and safety for epinephrine versus norepinephrine plus dobutamine in the management of septic shock.83
Vasopressin is an agent that has both vasoconstriction and antidiuretic properties. Vasopressin constricts vascular smooth muscle directly via V1 receptors, and may also increase the responsiveness of the vasculature to endogenous or exogenous catecholamines.84,85 Normally, endogenous vasopressin levels are very low, and there is essentially no vasoconstriction effect in a normal host. However, in septic shock vasopressin levels are initially extremely elevated. In prolonged septic shock, a relative vasopressin deficiency can develop. It has been postulated that this relative vasopressin deficiency may be the result of the depletion of the pituitary stores or the downregulation of vasopressin production by the pituitary via the effects of nitric oxide.84 Exogenous administration of low-dose vasopressin can have a dramatic hemodynamic response in this scenario, rapidly restoring arterial pressure.86,87 A large randomized clinical trial compared vasopressin to norepinephrine in 776 subjects with vasopressor-dependent septic shock. Patients were randomized to vasopressin (0.03 U/minute) or norepinephrine (15 µg/minute). For the group as a whole (intent-to-treat analysis) there was no difference in the primary end point of 28-day mortality rate. It appears that vasopressin (up to 0.03 U/minute) may be equally safe and effective as norepinephrine in patients with septic shock after fluid resuscitation.88 Doses of vasopressin higher than 0.04 U/minute are not recommended due to concerns of coronary, digital, and mesenteric ischemia.
Corticosteroids
Administering high doses of steroids (30 mg/kg of methylprednisolone) failed to show an outcome benefit in septic shock in large-scale randomized controlled trials in the 1980s.89,90 These studies used large doses of steroids over a short time period in an attempt to blunt the proinflammatory response of sepsis. In contrast, an alternative strategy of administering low-dose (i.e., “stress” or “physiologic” dose) steroids appeared to be promising in multiple small studies in the 1990s.91,92 Despite the fact that septic shock patients typically have elevated serum cortisol levels, it was identified that some patients with septic shock may have “relative adrenal insufficiency,” as evidenced by failure to mount a significant elevation of serum cortisol in response to intravenous adrenocorticotropic hormone (ACTH) stimulation. Relative adrenal insufficiency in the context of septic shock may predispose a patient to persistent cardiovascular failure that is refractory to conventional hemodynamic support therapies, and administration of exogenous low-dose steroids could help achieve shock reversal. On the other hand, administration of exogenous steroids could be associated with deleterious effects such as immunosuppression or myopathy.
In 2000, Annane and colleagues performed an observational study focusing on the ability to respond to an ACTH stimulation test in septic shock.93 The highest 28-day mortality rate (75%) was observed in patients who did not increase serum cortisol level greater than 9 µg/dL. Being a “nonresponder” was a better predictor of death than an initially low cortisol value. In a randomized controlled trial by the same investigators in 2002, 300 severely ill (persistent hypotension despite fluid resuscitation and vasopressor initiation) septic shock patients were randomized to 7 days of hydrocortisone plus fludrocortisone versus placebo.94 The study found that in the 229 nonresponders administration of low-dose steroids was associated with an improvement in time to shock reversal and mortality rate. Patients who responded appropriately to ACTH stimulation test did not demonstrate a benefit with low-dose steroids.
The concept of low-dose steroid administration was further tested in a multicenter randomized controlled trial (CORTICUS).95 This study found no difference in the primary outcome measure of mortality rate between those treated with steroids compared to placebo. However, it is notable that (1) in contrast to the Annane study in which all subjects had vasopressor-refractory septic shock, the CORTICUS study tested a more diverse patient population with overall lower severity sepsis, and (2) randomization in the Annane study occurred within 8 hours of developing shock as opposed to CORTICUS, which randomized subjects up to 72 hours after shock onset. Despite the fact that low-dose steroids do not appear to improve outcome in diverse, less severely ill populations of patients with sepsis, patients with vasopressor-unresponsive septic shock likely benefit. In summary, although steroid therapy should not be used in all patients with septic shock, it could be considered in those with persistent circulatory shock despite the administration of vasopressor agents.
References
1. Dellinger, RP. Cardiovascular management of septic shock. Crit Care Med. 2003; 31:946–955.
2. Geroulanos, S, Douka, ET. Historical perspective of the word “sepsis. ”. Intensive Care Med. 2006; 32:2077.
3. Schottmueller, H. Wesen und Behandlung der Sepsis. Inn Med. 1914; 31:257–280.
4. Vincent, JL, Abraham, E. The last 100 years of sepsis. Am J Respir Crit Care Med. 2006; 173:256–263.
5. American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med. 1992; 20:864–874.
6. Levy, MM, Fink, MP, Marshall, JC, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003; 31:1250–1256.
7. Trzeciak, S, Zanotti-Cavazzoni, S, Parrillo, JE, Dellinger, RP. Inclusion criteria for clinical trials in sepsis: Did the American College of Chest Physicians/Society of Critical Care Medicine consensus conference definitions of sepsis have an impact? Chest. 2005; 127:242–245.
8. Angus, DC, Linde-Zwirble, WT, Lidicker, J, et al. Epidemiology of severe sepsis in the United States: Analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001; 29:1303–1310.
9. American Heart Association. Heart disease and stroke statistics—2004 update. Dallas: American Heart Association; 2004.
10. Center for Disease Control and Prevention. Cases of HIV infection and AIDS in the United States by race/ethnicity, 1998-2002. Rockville, MD: CDC; 2003.
11. American Cancer Society. Cancer facts and figures 2003. Atlanta: ACA; 2003.
12. Hotchkiss, RS, Karl, IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003; 348:138–150.
13. Levy, MM, Dellinger, RP, Townsend, SR, et al. The surviving sepsis campaign: Results of an international guideline-based performance improvement program targeting severe sepsis. Crit Care Med. 2010; 38:367–374.
14. Kochanek, KD, Smith, B, National Vital Statistics Report Deaths, Preliminary data for 2002. CDC, Hyattsville, MD, 2004.
15. Lee, KL, Woodlief, LH, Topol, EJ, et al. Predictors of 30-day mortality in the era of reperfusion for acute myocardial infarction. Results from an international trial of 41,021 patients. Gusto-I Investigators. Circulation. 1995; 91:1659–1668.
16. Rosamond, WD, Folsom, AR, Chambless, LE, et al. Stroke incidence and survival among middle-aged adults: 9-year follow-up of the Atherosclerosis Risk in Communities (ARIC) cohort. Stroke. 1999; 30:736–743.
17. American College of Surgeons. National Trauma Data Bank Report 2006. Chicago: ACS; 2006.
18. Cowley, RA. Trauma center. A new concept for the delivery of critical care. J Med Soc N J. 1977; 74:979–987.
19. Winters, BD, Eberlein, M, Leung, J, et al. Long-term mortality and quality of life in sepsis: A systematic review. Crit Care Med. 2010; 38:1276–1283.
20. Iwashyna, TJ, Ely, EW, Smith, DM, Langa, KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA. 2010; 304:1787–1794.
21. Sama, AE, D’Amore, J, Ward, MF, et al. Bench to bedside: HMGB1—a novel proinflammatory cytokine and potential therapeutic target for septic patients in the emergency department. Acad Emerg Med. 2004; 11:867–873.
22. Dellinger, RP. Inflammation and coagulation: Implications for the septic patient. Clin Infect Dis. 2003; 36:1259–1265.
23. Rivers, E, Nguyen, B, Havstad, S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001; 345:1368–1377.
24. Yan, SB, Helterbrand, JD, Hartman, DL, et al. Low levels of protein C are associated with poor outcome in severe sepsis. Chest. 2001; 120:915–922.
25. Martin, GS, Mannino, DM, Eaton, S, Moss, M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003; 348:1546–1554.
26. Shapiro, N, Howell, MD, Bates, DW, et al. The association of sepsis syndrome and organ dysfunction with mortality in emergency department patients with suspected infection. Ann Emerg Med. 2006; 48:583–590.
27. Levy, MM, Macias, WL, Vincent, JL, et al. Early changes in organ function predict eventual survival in severe sepsis. Crit Care Med. 2005; 33:2194–2201.
28. Ferreira, FL, Bota, DP, Bross, A, et al. Serial evaluation of the SOFA score to predict outcome in critically ill patients. JAMA. 2001; 286:1754–1758.
29. Vincent, JL, Moreno, R, Takala, J, et al. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med. 1996; 22:707–710.
30. Parrillo, JE. Pathogenetic mechanisms of septic shock. N Engl J Med. 1993; 328:1471–1477.
31. Kumar, A, Haery, C, Parrillo, JE. Myocardial dysfunction in septic shock. Crit Care Clin. 2000; 16:251–287.
32. Kumar, A, Short, J, Parrillo, JE. Genetic factors in septic shock. JAMA. 1999; 282:579–581.
33. Cunnion, RE, Schaer, GL, Parker, MM, et al. The coronary circulation in human septic shock. Circulation. 1986; 73:637–644.
34. Charpentier, J, Luyt, CE, Fulla, Y, et al. Brain natriuretic peptide: A marker of myocardial dysfunction and prognosis during severe sepsis. Crit Care Med. 2004; 32:660–665.
35. De Backer, D, Creteur, J, Preiser, JC, et al. Microvascular blood flow is altered in patients with sepsis. Am J Respir Crit Care Med. 2002; 166:98–104.
36. Sakr, YL, Dubois, MJ, De Backer, D, et al. Persistent microcirculatory alterations are associated with organ failure and death in septic shock (abstract). Intensive Care Med. 2003; 29:S66.
37. Spronk, PE, Zandstra, DF, Ince, C. Bench-to-bedside review: Sepsis is a disease of the microcirculation. Crit Care. 2004; 8:462–468.
38. Trzeciak, S, Dellinger, RP, Parrillo, JE, et al. Early microcirculatory perfusion derangements in patients with severe sepsis and septic shock: Relationship to hemodynamics, oxygen transport, and survival. Ann Emerg Med. 2007; 49:88–98.
39. Bateman, RM, Sharpe, MD, Ellis, CG. Bench-to-bedside review: Microvascular dysfunction in sepsis—Hemodynamics, oxygen transport, and nitric oxide. Crit Care. 2003; 7:359–373.
40. Farquhar, I, Martin, CM, Lam, C, et al. Decreased capillary density in vivo in bowel mucosa of rats with normotensive sepsis. J Surg Res. 1996; 61:190–196.
41. Fries, M, Weil, MH, Sun, S, et al. Increases in tissue PCO2 during circulatory shock reflect selective decreases in capillary blood flow. Crit Care Med. 2006; 34:446–452.
42. Lam, C, Tyml, K, Martin, C, Sibbald, W. Microvascular perfusion is impaired in a rat model of normotensive sepsis. J Clin Invest. 1994; 94:2077–2083.
43. Ellis, CG, Bateman, RM, Sharpe, MD, et al. Effect of a maldistribution of microvascular blood flow on capillary O2 extraction in sepsis. Am J Physiol Heart Circ Physiol. 2002; 282:H156–H164.
44. Trzeciak, S, Rivers, EP. Clinical manifestations of disordered microcirculatory perfusion in severe sepsis. Crit Care. 2005; 9(Suppl 4):S20–S26.
45. Ince, C, Sinaasappel, M. Microcirculatory oxygenation and shunting in sepsis and shock. Crit Care Med. 1999; 27:1369–1377.
46. Abate, NL, Trzeciak, S. Is impaired capillary perfusion a marker of tissue hypoxia and a hallmark of incipient circulatory shock? Crit Care Med. 2006; 34:566–567.
47. Fink, MP. Cytopathic hypoxia. Mitochondrial dysfunction as mechanism contributing to organ dysfunction in sepsis. Crit Care Clin. 2001; 17:219–237.
48. Pope, JV, Jones, AE, Gaieski, DF, et al. Multicenter study of central venous oxygen saturation (ScvO2) as a predictor of mortality in patients with sepsis. Ann Emerg Med. 2010; 55:40–46.
49. Dellinger, RP, Carlet, JM, Masur, H, et al. Surviving sepsis campaign guidelines for management of severe sepsis and septic shock. Crit Care Med. 2004; 32:858–873.
50. Dellinger, RP, Carlet, JM, Masur, H, et al. Surviving sepsis campaign guidelines for management of severe sepsis and septic shock. Intensive Care Med. 2004; 30:536–555.
50a. Dellinger, RP, Levy, MM, Rhodes, A, et al. Surviving Sepsis Campaign Guidelines Committee including the Pediatrics Surviving Sepsis Campaign: International guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med. 2013; 41:580–637.
51. Wiedemann, HP, Matthay, MA, Matthay, RA. Cardiovascular-pulmonary monitoring in the intensive care unit (Part 2). Chest. 1984; 85:656–668.
52. Wiedemann, HP, Matthay, MA, Matthay, RA. Cardiovascular-pulmonary monitoring in the intensive care unit (Part 1). Chest. 1984; 85:537–549.
53. Broder, G, Weil, MH. Excess lactate: An index of reversibility of shock in human patients. Science. 1964; 143:1457–1459.
54. Aduen, J, Bernstein, WK, Khastgir, T, et al. The use and clinical importance of a substrate-specific electrode for rapid determination of blood lactate concentrations. JAMA. 1994; 272:1678–1685.
55. Bakker, J, Coffernils, M, Leon, M, et al. Blood lactate levels are superior to oxygen-derived variables in predicting outcome in human septic shock. Chest. 1991; 99:956–962.
56. Bakker, J, Gris, P, Coffernils, M, et al. Serial blood lactate levels can predict the development of multiple organ failure following septic shock. Am J Surg. 1996; 171:221–226.
57. Shapiro, NI, Howell, MD, Talmor, D, et al. Serum lactate as a predictor of mortality in emergency department patients with infection. Ann Emerg Med. 2005; 45:524–528.
58. Trzeciak, S, Dellinger, RP, Chansky, ME, et al. Serum lactate as a predictor of mortality in patients with infection. Intensive Care Med. 2007; 33:970–977.
59. Kreger, BE, Craven, DE, McCabe, WR. Gram-negative bacteremia. IV. Re-evaluation of clinical features and treatment in 612 patients. Am J Med. 1980; 68:344–355.
60. Natanson, C, Danner, RL, Reilly, JM, et al. Antibiotics versus cardiovascular support in a canine model of human septic shock. Am J Physiol. 1990; 259:H1440–H1447.
61. Kollef, MH, Sherman, G, Ward, S, Fraser, VJ. Inadequate antimicrobial treatment of infections: A risk factor for hospital mortality among critically ill patients. Chest. 1999; 115:462–474.
62. Kumar, A, Roberts, D, Wood, KE, et al. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit Care Med. 2006; 34:1589–1596.
63. Puskarich, MA, Trzeciak, S, Shapiro, NI, et al. Association between timing of antibiotic administration and mortality from septic shock in patients treated with a quantitative resuscitation protocol. Crit Care Med. 2011; 39:2066–2071.
64. Boyd, O, Grounds, RM, Bennett, ED. A randomized clinical trial of the effect of deliberate perioperative increase of oxygen delivery on mortality in high-risk surgical patients. JAMA. 1993; 270:2699–2707.
65. Shoemaker, WC, Appel, PL, Kram, HB, et al. Prospective trial of supranormal values of survivors as therapeutic goals in high-risk surgical patients. Chest. 1988; 94:1176–1186.
66. Gattinoni, L, Brazzi, L, Pelosi, P, et al. A trial of goal-oriented hemodynamic therapy in critically ill patients. SvO2 collaborative group. N Engl J Med. 1995; 333:1025–1032.
67. Hayes, MA, Timmins, AC, Yau, EH, et al. Elevation of systemic oxygen delivery in the treatment of critically ill patients. N Engl J Med. 1994; 330:1717–1722.
68. Heyland, DK, Cook, DJ, King, D, et al. Maximizing oxygen delivery in critically ill patients: A methodologic appraisal of the evidence. Crit Care Med. 1996; 24:517–524.
69. Kern, JW, Shoemaker, WC. Meta-analysis of hemodynamic optimization in high-risk patients. Crit Care Med. 2002; 30:1686–1692.
70. Jones, AE, Brown, MD, Trzeciak, S, et al. The effect of a quantitative resuscitation strategy on mortality in patients with sepsis: A meta-analysis. Crit Care Med. 2008; 36:2734–2739.
71. Jones, AE, Shapiro, NI, Trzeciak, S, et al. Lactate clearance vs. central venous oxygen saturation as goals of early sepsis therapy: A randomized clinical trial. JAMA. 2010; 303:739–746.
72. Trzeciak, S, Dellinger, RP, Abate, NL, et al. Translating research to clinical practice: A 1-year experience with implementing early goal-directed therapy for septic shock in the emergency department. Chest. 2006; 129:225–232.
73. Choi, PT, Yip, G, Quinonez, LG, Cook, DJ. Crystalloids vs. colloids in fluid resuscitation: A systematic review. Crit Care Med. 1999; 27:200–210.
74. Packman, MI, Rackow, EC. Optimum left heart filling pressure during fluid resuscitation of patients with hypovolemic and septic shock. Crit Care Med. 1983; 11:165–169.
75. Vincent, JL, Weil, MH. Fluid challenge revisited. Crit Care Med. 2006; 34:1333–1337.
76. LeDoux, D, Astiz, ME, Carpati, CM, Rackow, EC. Effects of perfusion pressure on tissue perfusion in septic shock. Crit Care Med. 2000; 28:2729–2732.
77. Varpula, M, Tallgren, M, Saukkonen, K, et al. Hemodynamic variables related to outcome in septic shock. Intensive Care Med. 2005; 31:1066–1071.
78. De Backer, D, Biston, P, Devriendt, J, et al. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med. 2010; 362:779–789.
79. De Backer, D, Aldecoa, C, Njimi, H, Vincent, JL. Dopamine versus norepinephrine in the treatment of septic shock: A meta-analysis. Crit Care Med. 2012; 40:725–730.
80. Levy, B, Bollaert, PE, Charpentier, C, et al. Comparison of norepinephrine and dobutamine to epinephrine for hemodynamics, lactate metabolism, and gastric tonometric variables in septic shock: A prospective, randomized study. Intensive Care Med. 1997; 23:282–287.
81. Martikainen, TJ, Tenhunen, JJ, Giovannini, I, et al. Epinephrine induces tissue perfusion deficit in porcine endotoxin shock: Evaluation by regional CO2 content gradients and lactate-to-pyruvate ratios. Am J Physiol Gastrointest Liver Physiol. 2005; 288:G586–G592.
82. Meier-Hellmann, A, Reinhart, K, Bredle, DL, et al. Epinephrine impairs splanchnic perfusion in septic shock. Crit Care Med. 1997; 25:399–404.
83. Martin, C. Norepinephrine plus dobutamine versus epinephrine alone for the management of septic shock. Barcelona, Spain, European Society of Intensive Care Medicine. 2006.
84. Holmes, CL, Patel, BM, Russell, JA, Walley, KR. Physiology of vasopressin relevant to management of septic shock. Chest. 2001; 120:989–1002.
85. Landry, DW, Oliver, JA. The pathogenesis of vasodilatory shock. N Engl J Med. 2001; 345:588–595.
86. Landry, DW, Levin, HR, Gallant, EM, et al. Vasopressin deficiency contributes to the vasodilation of septic shock. Circulation. 1997; 95:1122–1125.
87. Landry, DW, Levin, HR, Gallant, EM, et al. Vasopressin pressor hypersensitivity in vasodilatory septic shock. Crit Care Med. 1997; 25:1279–1282.
88. Russel, J, Walley, K. Vasopressin and Septic Shock (VASST) Trial: Study results. Barcelona, Spain: European Society of Intensive Care Medicine; 2006.
89. Bone, RC, Fisher, CJ, Jr., Clemmer, TP, et al. A controlled clinical trial of high-dose methylprednisolone in the treatment of severe sepsis and septic shock. N Engl J Med. 1987; 317:653–658.
90. Sprung, CL, Caralis, PV, Marcial, EH, et al. The effects of high-dose corticosteroids in patients with septic shock. A prospective, controlled study. N Engl J Med. 1984; 311:1137–1143.
91. Bollaert, PE, Charpentier, C, Levy, B, et al. Reversal of late septic shock with supraphysiologic doses of hydrocortisone. Crit Care Med. 1998; 26:645–650.
92. Briegel, J, Forst, H, Haller, M, et al. Stress doses of hydrocortisone reverse hyperdynamic septic shock: A prospective, randomized, double-blind, single-center study. Crit Care Med. 1999; 27:723–732.
93. Annane, D, Sebille, V, Troche, G, et al. A 3-level prognostic classification in septic shock based on cortisol levels and cortisol response to corticotropin. JAMA. 2000; 283:1038–1045.
94. Annane, D, Sebille, V, Charpentier, C, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. 2002; 288:862–871.
95. Sprung, CL, Annane, D, Keh, D, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med. 2008; 358:111–124.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]23
Septic Shock
Overview
This chapter pertains to pathophysiology, assessment, and management of septic shock, the most severe and overt manifestation of the septic condition. This discussion will specifically focus on cardiovascular and hemodynamic aspects. Other critically important elements of sepsis pathophysiology, assessment, and management (i.e., beyond the cardiovascular and hemodynamic aspects) will be addressed in a separate chapter (see Chapter 25, Sepsis and Multiple Organ Dysfunction). This chapter is also focused specifically on the adult patient with septic shock, as principles and evidence may differ in important ways in the pediatric population.
Historical Perspective
The word sepsis originated from the Greek language. Sepsis was synonymous with putrefaction and pertained to the bacteria-mediated decomposition of organic matter.1 The term persisted for more than 2700 years with essentially unchanged meaning.2 In the twentieth century, our modern understanding of the term sepsis became rooted in a disease in which the clinical manifestations were attributed to severe infection and the release of pathogenic bacterial products into the patient’s bloodstream.3,4
The term shock comes from the French word “choquer,” meaning “to collide with.” This is particularly appropriate terminology for shock due to sepsis, given our modern understanding of the sepsis pathophysiology, whereby the body’s host defenses essentially collide with the invading microorganism, triggering a profound proinflammatory host response.1
Contemporary Definitions
Shock is defined as a failure of the cardiovascular system to maintain effective tissue perfusion. If effective tissue perfusion is not promptly restored, cellular dysfunction and acute organ failure may occur and may become irreversible, leading to acute organ system failure. When shock develops because of a systemic inflammatory response to infection, it is termed septic shock. The American College of Chest Physicians (ACCP) and the Society of Critical Care Medicine (SCCM) first published consensus conference definitions for sepsis syndromes more than 20 years ago,5 and these definitions were revisited and further developed by international consensus in 2003.6 Septic shock was defined as infection-induced hypotension (systolic blood pressure <90 mm Hg [or a drop of >40 mm Hg] plus signs of tissue hypoperfusion despite adequate fluid resuscitation). The concurrent presence of clinical signs of tissue hypoperfusion (e.g., metabolic acidosis, encephalopathy, acute lung injury, oliguria, acute kidney injury, peripheral extremity discoloration, or impaired capillary refill) is an integral component of making the diagnosis of septic shock, because baseline blood pressure can vary among patients, and patients with lower baseline blood pressure may tolerate an arterial pressure lower than the values stated here without being in circulatory shock. The overarching purpose and major impact of the efforts to establish the contemporary definitions given here was the promotion of uniformity in inclusion criteria for sepsis clinical trials.7
Epidemiology
Severe sepsis (sepsis plus acute organ system dysfunction) is a common and deadly disease with major public health implications. Although heterogeneity of definitions of sepsis has historically made the incidence of severe sepsis and septic shock difficult to precisely measure, estimates of the incidence have been possible. Using the International Classification of Diseases (ICD)-9 codes for infection and organ dysfunction, Angus and coworkers estimated that 751,000 cases of severe sepsis occur in the United States every year.8 Figure 23.1 displays the incidence of severe sepsis in the United States compared to other common diseases. The incidence of severe sepsis currently exceeds the incidence of lung and colon cancer, venous thromboembolic disease, and acquired immune deficiency syndrome (AIDS),8–11 and the incidence is projected to increase by 1.5% per year, resulting in more than 1 million cases of severe sepsis annually by the year 2020.8 The incidence of sepsis and septic shock is known to be increasing because of a longer lifespan for patients with severe chronic medical conditions that predispose them to acquiring sepsis. This includes an increase in the number of immunocompromised patients in the community, number of infections caused by resistant organisms, increased use of intravascular catheters, and aging of the population.8
Figure 23.1 Incidence (cases per 100,000 population) of severe sepsis in the United States compared to four high-profile diseases.8–11 AIDS, acquired immune deficiency syndrome; VTE, venous thromboembolic disease.
Sepsis is the leading cause of death among critically ill patients12 and is responsible for as many deaths annually in the United States as acute myocardial infarction.8 Figure 23.2 displays control arm mortality rates in septic shock clinical trials.1 In a recent large multicenter registry study, septic patients with both arterial hypotension and severe lactic acidosis experienced a 46% mortality rate, whereas the mortality rate for arterial hypotension or severe lactic acidosis alone was 37% and 30%, respectively.13 Overall, severe sepsis in general ranks as the tenth leading cause of death in the United States, with 215,000 deaths annually and an estimated 30% in-hospital mortality rate.8,14 Figure 23.3 displays the mortality rate for severe sepsis compared to other high-profile diseases that may require critical care (acute ischemic stroke, acute myocardial infarction, and trauma).8,15–17 The apparent disparity in mortality rates across these diseases may be explained in part by differences in the conventional approach to treatment, as acute ischemic stroke, acute myocardial infarction, and trauma are all typically treated with aggressive interventions in a time-sensitive fashion. Similar to the “golden hour” concept for trauma care that was first recognized more than 30 years ago18 we are now beginning to understand that early aggressive interventions for sepsis can also have an impact on outcome.
Figure 23.3 Mortality rate of severe sepsis in the United States compared to three diseases that are treated aggressively with time-sensitive interventions.8,15–17 MI, myocardial infarction.
It is also important to recognize that, in addition to a high mortality rate, severe sepsis and septic shock are associated with serious risk of morbidity among survivors.19,20 A systematic review of the literature found that sepsis survivors had substantially diminished quality of life and a sharply reduced long-term survival after typical short-term (i.e., 28-day) outcomes are assessed.19 Among older adults, severe sepsis has been associated with major persistent cognitive impairment and functional disability that could have a substantial impact on those patients’ ability to live independently.20 Taken together, even among patients who survive the sepsis insult, the development of severe sepsis or septic shock can represent a pivotal event in the trajectory of a patient’s life.
Pathogenesis
Septic shock results when infectious microorganisms in the bloodstream induce a profound inflammatory response causing hemodynamic decompensation. The pathogenesis involves a complex response of cellular activation that triggers the release of a multitude of proinflammatory mediators. This inflammatory response causes activation of leukocytes and endothelial cells, as well as activation of the coagulation system. The excessive inflammatory response that characterizes septic shock is driven primarily by the cytokines tumor necrosis factor-alpha (TNF-α) and interleukin 1 (IL-1) that are produced by monocytes in response to an infection. Although TNF-α and IL-1 are central to the pathophysiology of septic shock and act synergistically to induce hypotension in experimental models, a number of other vital mediators are also known to play a major role, including high-mobility group box 1 (HMGB1) protein.21 Another important recent advance in our understanding of septic shock pathophysiology has been identification of the close link that exists between the proinflammatory response of septic shock and activation of the coagulation system (e.g., clinical or subclinical disseminated intravascular coagulation [DIC]).22 Although the systemic inflammatory response of sepsis triggers profound macrocirculatory and microcirculatory changes that impair tissue perfusion, another important mechanism playing a role in the development of acute organ dysfunction in septic shock is apoptosis (programmed cell death). Accelerated apoptosis is known to be a critical pathogenic event in this disease. In addition, certain genetic polymorphisms are becoming recognized as major determinants of susceptibility to infection, as well as risk of death from septic shock. Key steps in the pathogenesis of septic shock are shown in Figure 23.4.
Clinical Presentation
Patients with septic shock will typically manifest signs of systemic inflammation including fever or hypothermia, tachycardia, tachypnea, and elevation or reduction of the white blood cell count. Although the absence of arterial hypotension does not necessarily exclude the possibility of subclinical tissue hypoperfusion,23 the hallmark of septic shock is arterial hypotension despite adequate volume resuscitation requiring vasoactive drugs for hemodynamic support. Other signs of potential tissue hypoperfusion may include lactic acidosis, oliguria, encephalopathy, or diminished capillary refill in the extremities. Patients with septic shock typically have multiple organ system dysfunctions; clinical evidence of other organ system dysfunction may range from subtle abnormalities to overt organ failure. Multiorgan system involvement in sepsis may include cardiovascular, respiratory, renal, central nervous system, hepatic, metabolic, or hematologic dysfunction. Respiratory system dysfunction manifests as acute lung injury or, in the most extreme cases, the acute respiratory distress syndrome (ARDS). Sepsis-induced renal dysfunction typically manifests with oliguria and may progress to acute renal failure requiring dialysis. Central nervous system dysfunction will manifest as encephalopathy, which may range from mild cognitive impairment to overt coma. Cholestasis is a common manifestation of hepatic dysfunction in sepsis, but in the presence of severe shock, ischemic hepatitis (“shock liver”) may occur. Metabolic derangements of septic shock include a loss of glycemic control (hyper- or hypoglycemia) as well as metabolic acidosis. Septic shock is commonly associated with a consumptive coagulopathy, which is likely present in almost all patients at least subclinically,24 but may also manifest clinically with thrombocytopenia, prolongation of the prothrombin time, or in the most severe cases, overt DIC.
The multiple organ dysfunction associated with septic shock is not only a critical event in the pathogenesis of this disease, but is also closely linked with mortality rate.8,25,26 There is an approximate 20% increase in septic shock mortality rate with each additional organ system that fails.8 Early evidence of organ failure is an especially strong predictor of death.26,27 Early improvement in organ function (e.g., 0-24 hour improvement in the Sequential Organ Failure Assessment [SOFA] score28,29) is closely related to sepsis survival, whereas later improvement after the first 24 hours has little predictive value.27 These data, garnered largely from observational studies as well as placebo arms of interventional trials, support the concept that aggressive therapy for sepsis to reverse (or prevent the development of) acute organ system failure within the first 24 hours is closely associated with eventual outcome.
Hemodynamic Profile of Septic Shock
The hemodynamic profile of septic shock is the most complex hemodynamic profile of all shock etiologies (Fig. 23.5). What sets septic shock apart from other causes of circulatory shock is the fact that there may be multiple different mechanisms of circulatory shock occurring simultaneously.1,30 Septic shock may have features of (1) hypovolemic shock (poor cardiac filling secondary to severe systemic capillary leak and increased venous capacitance), (2) cardiogenic shock (infection-induced myocardial depression), and (3) distributive shock (arteriolar vasodilation with tissue hypoperfusion in the face of an adequate cardiac output).1
Hypovolemia
The release of proinflammatory mediators into the circulation causes injury to the integrity of the endothelial cell surface throughout the systemic microvasculature, resulting in severe capillary leak and extravasation of fluid into tissues. Venodilation also compromises venous return. These are major factors in producing hypovolemia in the patient with septic shock. The septic shock patient may have a markedly decreased cardiac preload, especially in the initial phase of therapy. Aggressive resuscitation with intravenous volume expansion modulates the hemodynamic profile of septic shock and allows the patient to achieve a hyperdynamic (i.e., high cardiac output) state.1 The combination of a decreased preload and myocardial depression means that in the early phase of sepsis resuscitation, patients may initially be hypodynamic (i.e., low cardiac output) prior to receiving adequate volume resuscitation. Capillary leak is an ongoing process in the course of septic shock therapy, and therefore hypovolemia may recur later in the course of the disease, even after adequate cardiac filling has been initially achieved. Fluid balance (input of intravenous fluids and output of urine) is an unreliable parameter for assessing adequacy of fluid resuscitation in septic shock.
Myocardial Dysfunction
Septic shock is associated with depression of biventricular function with a decrease in the ejection fraction. Ventricular dilation occurs as a compensatory mechanism and raises end-diastolic volume so that stroke volume can be preserved, taking advantage of the Starling principle. When myocardial dysfunction occurs, a high cardiac output can still be achieved in many circumstances because of biventricular dilation, tachycardia, and arteriolar dilatation, as long as the patient is adequately volume resuscitated and does not have a severe cardiac suppression (related either to previously existing cardiac dysfunction or overwhelming sepsis-induced suppression of cardiac systolic function).30 The most important inflammatory mediators that induce myocardial depression are TNF-α, IL-1, and perhaps nitric oxide.31,32 Coronary blood flow is typically normal or increased in septic shock.33 Although coronary blood flow can be diminished by severe arterial hypotension that compromises coronary perfusion pressure (especially if there is preexisting coronary artery disease), myocardial ischemia does not appear to be the causative factor of the depression in myocardial performance. It has been reported that nearly half of patients with septic shock will have echocardiographic evidence of some degree of depression of systolic function, even in the absence of preexisting cardiac disease.34 However, myocardial depression is typically not the predominant feature of the septic shock hemodynamic profile.30 For the majority of patients, aggressive intravascular volume expansion to restore adequate cardiac filling pressures will be enough to achieve a reasonable cardiac output.
Distributive Shock
Septic shock is characterized by peripheral maldistribution of blood flow to tissues such that tissue hypoperfusion abnormalities can persist despite a normal or high cardiac output. This is called “distributive shock.”30 This maldistribution of blood flow may occur at both microcirculatory and macrocirculatory levels. The role of microcirculatory dysfunction is discussed in detail in the next section of this chapter. At the level of the macrocirculation, the autoregulation of blood flow within any single organ system in a normal host can typically maintain effective tissue perfusion over a wide range of systemic pressures (usually ranging from a mean arterial pressure [MAP] of 50 mm Hg to 150 mm Hg). However, there is heterogeneity of blood flow distribution throughout the body in septic shock due to preferential shunting of blood flow to vital organs (e.g., the brain and myocardium). The gastrointestinal tract may be the earliest organ system to experience tissue hypoperfusion in septic shock, as blood is shunted away from the splanchnic circulation in order to preserve blood flow to the brain, myocardium, and skeletal muscles. Ischemic injury to the gastrointestinal tract may be a source of ongoing systemic inflammation in septic shock.
The three components of the hemodynamic profile of septic shock are displayed in Figure 23.6.
Microcirculatory and Mitochondrial Dysfunction
Microcirculatory Dysfunction
Microcirculatory dysfunction is a pivotal element of the pathogenesis of septic shock.35–38 Although the macrocirculation (heart and large arteries) regulates the global distribution of blood flow throughout the body, it is the microcirculation that controls the delivery of blood flow to tissues. Using intravital videomicroscopy, experimental models of sepsis have demonstrated impaired microcirculatory flow velocity, “stopped-flow” microvessels, increased heterogeneity of regional perfusion, and low density of perfused capillaries.39–42 These derangements can cause marked alterations of oxygen transport including impaired tissue oxygen extraction.43 With the advent of new investigational videomicroscopy techniques, it is now possible to study the microcirculatory network in human subjects with septic shock. Microcirculatory failure appears to be one of the critical pathogenic events in sepsis that is associated with acute multiorgan dysfunction and death.35–38 As these alterations of microcirculatory flow in sepsis can occur in the absence of global hemodynamic perturbations (i.e., absence of low arterial pressure or low cardiac output),36,42,44 derangements of small vessel perfusion are largely a function of intrinsic events in the microcirculation.
The causes of microcirculatory flow alterations in sepsis (Fig. 23.7) are multifactorial and include endothelial cell dysfunction, increased leukocyte adhesion, microthrombi formation, rheologic abnormalities, altered local perfusion pressures due to regional redistribution of blood flow, and functional shunting.39,45 The proinflammatory cytokines released in sepsis cause diffuse endothelial cell activation, which is associated with neutrophil activation, expression of endothelial adhesion molecules (i.e., integrins and selectins), and localization of white blood cells to areas of microvascular injury. Pan-endothelial cell injury increases microvascular permeability with the influx of proinflammatory cells into the tissues; this is hypothesized to be an important pathogenic step in the development of acute system organ dysfunction in sepsis. Leukocyte adhesion of white blood cells to the microvessel endothelial surface (primarily in the postcapillary venule) further impedes microcirculatory blood flow. The endothelial injury also triggers the activation of the coagulation cascade via expression of tissue factor on the microvascular endothelium, resulting in fibrin deposition and microvascular thrombosis that may further impair microcirculatory flow. All of these mechanisms collectively contribute to microcirculatory failure in septic shock.37,39
Although septic shock research has classically been focused on macrocirculatory hemodynamic parameters that reflect the distribution of blood flow globally throughout the body, a functional microcirculation is another critical component of the cardiovascular system that is necessary for effective blood flow to tissues. This conceptual framework is depicted in Figure 23.8. Although a shift of research focus from global hemodynamic parameters to indices of microvascular perfusion could potentially be viewed as a major change of direction for septic shock research, the microcirculation likely represents a logical next frontier in the evolution of our understanding of circulatory failure in shock states.37,46 Although there are currently no therapies to specifically target microcirculatory dysfunction in sepsis, going beyond optimization of macrocirculatory hemodynamics and developing new innovative strategies to reverse microcirculatory failure could (in the future) potentially represent a cutting edge method to augment tissue perfusion in sepsis.
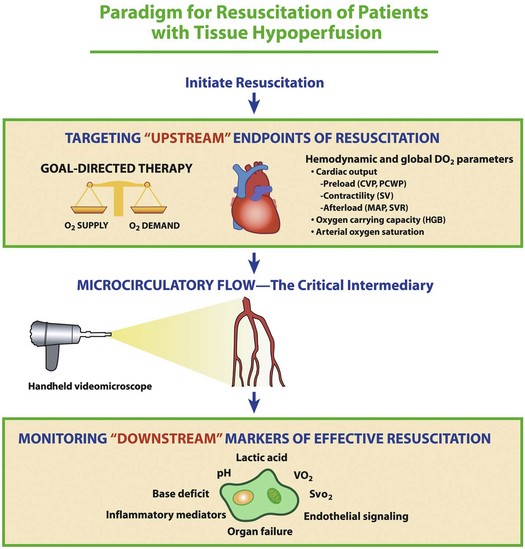
Figure 23.8
[/not-level-membership-for-critical-care-medicine-category]
