[level-membership-for-internal-medicine-category]
STEM CELLS AND TRANSPLANTATION
The nervous system is traditionally considered to be a nonmitotic organ, in particular with respect to neurons. These concepts have been challenged by the finding that neural progenitor or stem cells exist in the adult CNS that are capable of differentiation, migration over long distances, and extensive axonal arborization and synapse formation with appropriate targets. These capabilities also indicate that the repertoire of factors required for growth, survival, differentiation, and migration of these cells exists in the mature nervous system. In rodents, neural stem cells, defined as progenitor cells capable of differentiating into mature cells of neural or glial lineage, have been experimentally propagated from fetal CNS and neuroectodermal tissues and also from adult germinal matrix and ependyma regions. Human fetal CNS tissue is also capable of differentiation into cells with neuronal, astrocyte, and oligodendrocyte morphology when cultured in the presence of growth factors.
Once the repertoire of signals required for cell type specification is better understood, differentiation into specific neural or glial subpopulations can be directed in vitro; such cells could also be engineered to express therapeutic molecules. Another promising approach is to use growth factors, such as BDNF, to stimulate endogenous stem cells to proliferate and migrate to areas of neuronal damage.
A major advance has been the development of induced pluripotent stem cells. Using this technique, adult somatic cells such as skin fibroblasts are treated with four pluripotency factors (SOX2, KLF4, cMYC, and Oct4), and this generates induced pluripotent stem cells (iPSCs). These adult-derived stem cells sidestep the ethical issues of using stem cells derived from human embryos. The development of these cells has tremendous promise for both studying disease mechanisms and testing therapeutics. As yet there is no consensus on the best way to generate and differentiate the iPSCs; however, techniques to avoid using viral vectors and use of Cre-lox systems to remove reprogramming factors result in a better match of gene expression profiles with those of embryonic stem cells. Over the years, the field of directed differentiation has used three main strategies to specify neural lineages from human pluripotent stem cells. These strategies are embryoid body formation, coculture on neural-inducing feeders, and direct neural induction. Thus far, iPSCs have been made from patients with all of the major human neurodegenerative diseases, and studies using them are under way.
Although stem cells hold tremendous promise for the treatment of debilitating neurologic diseases, such as Parkinson’s disease and spinal cord injury, it should be emphasized that medical application is in its infancy. Major obstacles are the generation of position- and neurotransmitter-defined subtypes of neurons and their isolation as pure populations of the desired cells. This is crucial to avoid persistence of undifferentiated embryonic stem (ES) cells, which can generate tumors. The establishment of appropriate neural connections and afferent control is also critical. For instance, human ES motor neurons will need to be introduced at multiple segments in the neuraxis, and then their axons will need to regenerate from the spinal cord to distal musculature.
Experimental transplantation of human fetal dopaminergic neurons in patients with Parkinson’s disease has shown that these transplanted cells can survive within the host striatum; however, some patients developed disabling dyskinesias, and this approach is no longer in clinical development. The possibility that iPSCs will be used in Parkinson’s disease was strengthened by studies showing that they can be differentiated into dopaminergic neurons. The dopaminergic neurons were then shown to rescue the parkinsonian phenotype in a MPTP-induced primate model with excellent dopaminergic neuron survival function and lack of neural overgrowth. The correction of tau mutations in iPSC-derived neurons has been shown to reverse the toxic phenotype in dendrite retraction and cell death.
Another new use for iPSCs is to screen drugs as potential treatments for neurodegenerative and other diseases. The feasibility of this has been shown using iPSC-induced macrophages from patients with Gaucher’s disease, and verifying the efficacy of protein chaperones in these cells as a means of stabilizing the mutant glucocerebrosidase and increasing its activity and the duration of its effects. Other approaches are to attempt to reduce expression of proteins, such as amyloid, tau, and α-synuclein, implicated in the pathogenesis of neurodegenerative diseases. One difficulty has been that reprogramming cells to iPSCs resets their identity back to an embryonic age, which is a hurdle in modeling of late-onset diseases. One approach to this has been to express a fragment of the mutated gene, such as a portion of lamin A, which causes premature aging in progeria. This approach showed that dendrite degeneration and progressive loss of tyrosine hydroxylase expression, as well as enlarged mitochondria and Lewy body precursor inclusions, were induced in iPSC-derived dopaminergic neurons with progerin-induced aging.
Studies of transplantation for patients with Huntington’s disease have also reported encouraging, although very preliminary, results. OPCs transplanted into mice with a dysmyelinating disorder effectively migrated in the new environment, interacted with axons, and mediated myelination; such experiments raise hope that similar transplantation strategies may be feasible in human disorders of myelin such as MS. The promise of stem cells for treatment of both neurodegenerative diseases and neural injury is great, but development has been slowed by unresolved concerns over safety (including the theoretical risk of malignant transformation of transplanted cells), ethics (particularly with respect to use of fetal tissue), and efficacy.
In developing brain, the extracellular matrix provides stimulatory and inhibitory signals that promote neuronal migration, neurite outgrowth, and axonal extension. After neuronal damage, reexpression of inhibitory molecules such as chondroitin sulfate proteoglycans may prevent tissue regeneration. Chondroitinase degraded these inhibitory molecules and enhanced axonal regeneration and motor recovery in a rat model of spinal cord injury. Several myelin proteins, specifically Nogo, oligodendrocyte myelin glycoprotein (OMGP), and myelin-associated glycoprotein (MAG), may also interfere with axon regeneration. Sialidase, which cleaves one class of receptors for MAG, enhances axonal outgrowth. Antibodies against Nogo promote regeneration after experimental focal ischemia or spinal cord injury. Nogo, OMGP, and MAG all bind to the same neural receptor, the Nogo receptor, which mediates its inhibitory function via the p75 neurotrophin receptor signaling.
CELL DEATH: EXCITOTOXICITY AND APOPTOSIS
Excitotoxicity refers to neuronal cell death caused by activation of excitatory amino acid receptors (Fig. 444e-4). Compelling evidence for a role of excitotoxicity, especially in ischemic neuronal injury, is derived from experiments in animal models. Experimental models of stroke are associated with increased extracellular concentrations of the excitatory amino acid neurotransmitter glutamate, and neuronal damage is attenuated by denervation of glutamate-containing neurons or the administration of glutamate receptor antagonists. The distribution of cells sensitive to ischemia corresponds closely with that of N-methyl-D-aspartate (NMDA) receptors (except for cerebellar Purkinje cells, which are vulnerable to hypoxia-ischemia but lack NMDA receptors); and competitive and noncompetitive NMDA antagonists are effective in preventing focal ischemia. In global cerebral ischemia, non-NMDA receptors (kainic acid and α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate [AMPA]) are activated, and antagonists to these receptors are protective. Experimental brain damage induced by hypoglycemia is also attenuated by NMDA antagonists.
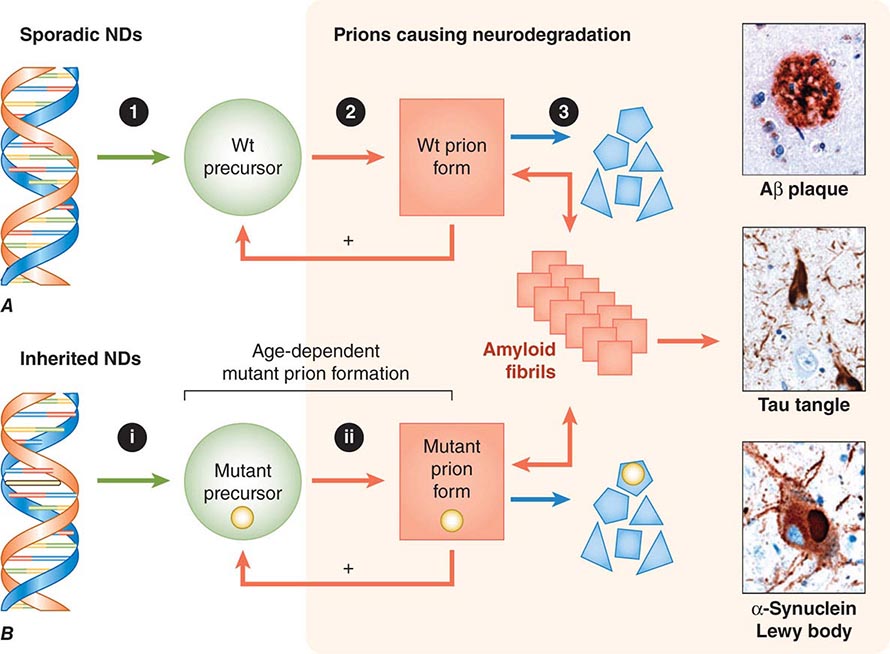
FIGURE 444e-4 Neurodegeneration caused by prions. A. In sporadic neurodegenerative diseases (NDs), wild-type (Wt) prions multiply through self-propagating cycles of posttranslational modification, during which the precursor protein (green circle) is converted into the prion form (red square), which generally is high in β-sheet content. Pathogenic prions are most toxic as oligomers and less toxic after polymerization into amyloid fibrils. The small polygons (blue) represent proteolytic cleavage products of the prion. Depending on the protein, the fibrils coalesce into Aβ amyloid plaques in Alzheimer’s disease (AD), neurofibrillary tangles in AD and Pick’s disease, or Lewy bodies in Parkinson’s disease and Lewy body dementia. Drug targets for the development of therapeutics include: (1) lowering the precursor protein, (2) inhibiting prion formation, and (3) enhancing prion clearance. B. Late-onset heritable neurodegeneration argues for two discrete events: The (i) first event is the synthesis of mutant precursor protein (green circle), and the (ii) second event is the age-dependent formation of mutant prions (red square). The highlighted yellow bar in the DNA structure represents mutation of a base pair within an exon, and the small yellow circles signify the corresponding mutant amino acid substitution. Green arrows represent a normal process; red arrows, a pathogenic process; and blue arrows, a process that is known to occur but unknown whether it is normal or pathogenic. (Micrographs prepared by Stephen J. DeArmond. Reprinted with permission from SB Prusiner: Biology and genetics of prions causing neurodegeneration. Annu Rev Genet 47:601, 2013.)
Excitotoxicity is not a single event but rather a cascade of cell injury. Excitotoxicity causes influx of calcium into cells, and much of the calcium is sequestered in mitochondria rather than in the cytoplasm. Increased cytoplasmic calcium causes metabolic dysfunction and free radical generation; activates protein kinases, phospholipases, nitric oxide synthase, proteases, and endonucleases; and inhibits protein synthesis. Activation of nitric oxide synthase generates nitric oxide (NO•), which can react with superoxide (O•2) to generate peroxynitrite (ONOO–), which may play a direct role in neuronal injury. Another critical pathway is activation of poly-ADP-ribose polymerase, which occurs in response to free radical–mediated DNA damage. Experimentally, mice with knockout mutations of neuronal nitric oxide synthase or poly-ADP-ribose polymerase, or those that overexpress superoxide dismutase, are resistant to focal ischemia.
Another aspect of excitotoxicity is that it has been demonstrated that stimulation of extrasynaptic NMDA receptors mediates cell death, where as stimulation of synaptic receptors is protective. This has been shown to play a role in excitotoxicity in transgenic mouse models of Huntington’s disease, in which using low-dose memantine to selectively block the extrasynaptic receptors is beneficial.
Although excitotoxicity is clearly implicated in the pathogenesis of cell death in stroke, to date treatment with NMDA antagonists has not proven to be clinically useful. One approach has been to use an inhibitor of the postsynaptic density-95 protein that uncouples NMDA receptors from neurotoxic pathways, including the generation of nitric oxide. This approach was effective in a primate stroke model and in a phase 2 clinical trial of stroke associated with endovascular repair of cerebral aneurysms. Transient receptor potentials (TRPs) are calcium channels that are activated by oxidative stress in parallel with excitotoxic signal pathways. In addition, glutamate-independent pathways of calcium influx via acid-sensing ion channels have been identified. These channels transport calcium in the setting of acidosis and substrate depletion, and pharmacologic blockade of these channels markedly attenuates stroke injury. These channels offer a potential new therapeutic target for stroke.
Apoptosis, or programmed cell death, plays an important role in both physiologic and pathologic conditions. During embryogenesis, apoptotic pathways operate to destroy neurons that fail to differentiate appropriately or reach their intended targets. There is mounting evidence for an increased rate of apoptotic cell death in a variety of acute and chronic neurologic diseases. Apoptosis is characterized by neuronal shrinkage, chromatin condensation, and DNA fragmentation, whereas necrotic cell death is associated with cytoplasmic and mitochondrial swelling followed by dissolution of the cell membrane. Apoptotic and necrotic cell death can coexist or be sequential events, depending on the severity of the initiating insult. Cellular energy reserves appear to have an important role in these two forms of cell death, with apoptosis favored under conditions in which ATP levels are preserved. Evidence of DNA fragmentation has been found in a number of degenerative neurologic disorders, including AD, Huntington’s disease, and ALS. The best characterized genetic neurologic disorder related to apoptosis is infantile spinal muscular atrophy (Werdnig-Hoffmann disease), in which two genes thought to be involved in the apoptosis pathways are causative.
Mitochondria are essential in controlling specific apoptosis pathways. The redistribution of cytochrome c, as well as apoptosis-inducing factor (AIF), from mitochondria during apoptosis leads to the activation of a cascade of intracellular proteases known as caspases. Caspase-independent apoptosis occurs after DNA damage, activation of poly-ADP-ribose polymerase, and translocation of AIF into the nucleus. Redistribution of cytochrome c is prevented by overproduction of the apoptotic protein BCL2 and is promoted by the proapoptotic protein BAX. These pathways may be triggered by activation of a large pore in the mitochondrial inner membrane known as the permeability transition pore, although in other circumstances, they occur independently. The permeability transition pore is made up of dimers of ATP synthase and is activated by cyclophilin D, leading to large calcium fluxes across the inner mitochondrial membrane. Certain forms of congenital muscular dystrophies are caused by mutations in collagen VI, which leads to increased activation of the permeability transition pore. Recent studies suggest that blocking the mitochondrial pore reduces both hypoglycemic and ischemic cell death. Mice deficient in cyclophilin D, a key protein involved in opening the permeability transition pore, are resistant to necrosis produced by focal cerebral ischemia.
PROTEIN AGGREGATION AND NEURODEGENERATION
The possibility that protein aggregation plays a role in the pathogenesis of neurodegenerative diseases is a major focus of current research. Protein aggregation is a major histopathologic hallmark of neurodegenerative diseases. Deposition of β-amyloid is strongly implicated in the pathogenesis of AD. Genetic mutations in familial AD cause increased production of β-amyloid with 42 amino acids, which has an increased propensity to aggregate, as compared to β-amyloid with 40 amino acids. Furthermore, mutations in the amyloid precursor protein (APP), which reduce the production of β-amyloid, protect against the development of AD and are associated with preserved cognition in the elderly. Mutations in genes encoding the MAPT lead to altered splicing of tau and the production of neurofibrillary tangles in frontotemporal dementia and progressive supranuclear palsy. Familial Parkinson’s disease is associated with mutations in leucine-rich repeat kinase 2 (LRRK2), α-synuclein, parkin, PINK1, and DJ-1. PINK1 is a mitochondrial kinase (see below), and DJ-1 is a protein involved in protection from oxidative stress. Parkin, which causes autosomal recessive early-onset Parkinson’s disease, is a ubiquitin ligase. The characteristic histopathologic feature of Parkinson’s disease is the Lewy body, an eosinophilic cytoplasmic inclusion that contains both neurofilaments and α-synuclein. Huntington’s disease and cerebellar degenerations are associated with expansions of polyglutamine repeats in proteins, which aggregate to produce neuronal intranuclear inclusions. Familial ALS is associated with superoxide dismutase mutations and cytoplasmic inclusions containing superoxide dismutase. An important finding was the discovery that ubiquinated inclusions observed in most cases of ALS and the most common form of frontotemporal dementia are composed of TAR DNA binding protein 43 (TDP-43). Subsequently, mutations in the TDP-43 gene, and in the fused in sarcoma gene (FUS), were found in familial ALS. Both of these proteins are involved in transcription regulation as well as RNA metabolism. In autosomal dominant neurohypophyseal diabetes insipidus, mutations in vasopressin result in abnormal protein processing, accumulation in the endoplasmic reticulum, and cell death.
Another key mechanism linked to cell death is mitochondrial dynamics, which refers to the processes involved in movement of mitochondria, as well as in mitochondrial fission and fusion, which play a critical role mitochondrial turnover and in replenishment of damaged mitochondria. Mitochondrial dysfunction is strongly linked to the pathogenesis of a number of neurodegenerative diseases such as Friedreich’s ataxia, which is caused by mutations in an iron-binding protein that plays an important role in transferring iron to iron-sulfur clusters in aconitase and complex I and II of the electron transport chain. Mitochondrial fission is dependent on the dynamin-related proteins (Drp1), which bind to its receptor Fis, whereas mitofuscins 1 and 2 (MF 1/2) and optic atrophy protein 1 (OPA1) are responsible for fusion of the outer and inner mitochondrial membrane, respectively. Mutations in MFN2 cause Charcot-Marie-Tooth neuropathy type 2A, and mutations in OPA1 cause autosomal dominant optic atrophy. Both β-amyloid and mutant huntingtin protein induce mitochondrial fragmentation and neuronal cell death associated with increased activity of Drp1. In addition, mutations in genes causing autosomal recessive Parkinson’s disease, parkin and PINK1, cause abnormal mitochondrial morphology and result in impairment of the ability of the cell to remove damaged mitochondria by autophagy.
One major scientific question is whether protein aggregates directly contribute to neuronal death or whether they are merely secondary bystanders. A current focus in all the neurodegenerative diseases is on small protein aggregates termed oligomers. These may be the toxic species of β-amyloid, α-synuclein, and proteins with expanded polyglutamines such as are associated with Huntington’s disease. Protein aggregates are usually ubiquinated, which targets them for degradation by the 26S component of the proteasome. An inability to degrade protein aggregates could lead to cellular dysfunction, impaired axonal transport, and cell death by apoptotic mechanisms.
Autophagy is the degradation of cystolic components in lysosomes. There is increasing evidence that autophagy plays an important role in degradation of protein aggregates in the neurodegenerative diseases, and it is impaired in AD, Parkinson’s disease, and Huntington’s disease. Autophagy is particularly important to the health of neurons, and failure of autophagy contributes to cell death. In Huntington’s disease, a failure of cargo recognition occurs, contributing to protein aggregates and cell death. Rapamycin, which induces autophagy, exerts beneficial therapeutic effects in transgenic mouse models of AD, Parkinson’s disease, and Huntington’s disease.
There is other evidence for lysosomal dysfunction and impaired autophagy in Parkinson’s disease. Mutations in glucocerebrosidase are associated with 5% of all Parkinson’s disease cases as well as 8–9% of patients with dementia with Lewy bodies. Therefore, this is the most important genetic cause of both disorders thus far identified. There appear to be reciprocal interactions between glucocerebrosidase and α-synuclein. It has been shown that glucocerebrosidase concentrations and enzymatic activity are reduced in the substantia nigra of sporadic Parkinson’s disease patients. Furthermore, α-synuclein is degraded by chaperone-mediated and macro autophagy. The degradation of α-synuclein has been shown to be impaired in transgenic mice deficient in glucocerebrosidase as well as in mice in which the enzyme has been inhibited. Furthermore, it is known that α-synuclein inhibits the activity of glucocerebrosidase. Therefore, there is bidirectional feedback between α-synuclein and glucocerebrosidase. An attractive therapeutic intervention could be to use protein chaperones to increase the activity and duration of action of glucocerebrosidase. This would also reduce α-synuclein levels and block the degeneration of dopaminergic neurons.
The retromer complex is a conserved membrane-associated protein complex that functions in the endosome-to-Golgi complex. The retromer complex contains a cargo selective complex consisting of VPS35, VPS26, and VPS29, along with a sorting nexin dimer. Recently, mutations in VPS35 were shown to be a cause of late-onset autosomal dominant Parkinson’s disease. The retromer also traffics APP away from endosomes, where it is cleaved to generate β-amyloid. Deficiencies of VPS35 and VPS26 were also identified in hippocampal brain tissue from AD. A new therapeutic approach to these diseases might therefore be to use chaperones to stabilize the retromer and reduce the generation of β-amyloid and α-synuclein.
The LRRK2 mutations were shown to have effects on clearance of Golgi-derived vesicles through the autophagy-lysosome system both in vitro and in vivo. LRRK2 mutations also are linked to elevated protein synthesis mediated by ribosomal protein s15 phosphorylation. Blocking this phosphorylation reduces LRRK2-mediated neurite loss and cell death in human dopamine and cortical neurons.
Interestingly, in experimental models of Huntington’s disease and cerebellar degeneration, protein aggregates are not well correlated with neuronal death and may be protective. A substantial body of evidence suggests that the mutant proteins with polyglutamine expansions in these diseases bind to transcription factors and that this contributes to disease pathogenesis. In Huntington’s disease, there is dysfunction of the transcriptional co-regulator, PGC-1α, a key regulator of mitochondrial biogenesis. There is evidence that impaired function of PGC-1α is also important in both Parkinson’s disease and AD, making it an attractive target for treatments. Agents that upregulate gene transcription are neuroprotective in animal models of these diseases. A number of compounds have been developed to block β-amyloid production and/or aggregation, and these agents are being studied in early clinical trials in humans. Another approach under investigation is immunotherapy with antibodies that bind β-amyloid, tau, or α-synuclein. These studies have shown efficacy in preventing the spread of amyloid, tau, and α-synuclein in animal studies, raising hopes that this could lead to effective therapies by blocking neuron-to-neuron propagation. Two large clinical trials of β-amyloid immunotherapy, however, did not show efficacy, although this therapeutic strategy is still being studied.
PRIONS AND NEURODEGENERATIVE DISEASES
As we have learned more about the etiology and pathogenesis of the neurodegenerative diseases, it has become clear that the histologic abnormalities that were once curiosities, in fact, are likely to reflect the etiologies. For example, the amyloid plaques in kuru and Creutzfeldt-Jakob disease (CJD) are filled with the PrPSc prions that have assembled into fibrils. The past three decades have witnessed an explosion of new knowledge about prions. For many years, kuru, CJD, and scrapie of sheep were thought to be caused by slow-acting viruses, but a large body of experimental evidence argues that the infectious pathogens causing these diseases are devoid of nucleic acid. Such pathogens are called prions, which are composed of host-encoded proteins that adopt alternative conformations (Chap. 453e). Prions are self-propagating by imposing their conformations on the normal, precursor protein; most prions are enriched for β-sheet and can assemble into amyloid fibrils.
Similar to the plaques in kuru and CJD that are composed of PrP prions, the amyloid plaques in AD are filled with Aβ prions that have polymerized into fibrils. This relationship between the neuropathologic findings and the etiologic prion was strengthened by the genetic linkage between familial CJD and mutations in the PrP gene, as well as (as noted above) between familial AD and mutations in the APP gene. Moreover, a mutation in the APP gene that prevents Aβ peptide formation was correlated with a decreased incidence of AD in Iceland.
The heritable neurodegenerative diseases offer an important insight into the pathogenesis of the more common, sporadic ones. Although the mutant proteins that cause these disorders are expressed in the brains of people early in life, the diseases do not occur for many decades. Many explanations for the late onset of familial neurodegenerative diseases have been offered, but none are supported by substantial experimental evidence. The late onset might be due to a second event in which a mutant protein, after its conversion into a prion, begins to accumulate at some rather advanced age (Fig. 444e-5). Such a formulation is also consistent with data showing that the protein quality-control mechanisms diminish in efficiency with age. Thus, the prion forms of both wild-type and mutant proteins are likely to be efficiently degraded in younger people but are less well handled in older individuals. This explanation is consistent with the view that neurodegenerative diseases are disorders of the aging nervous system.
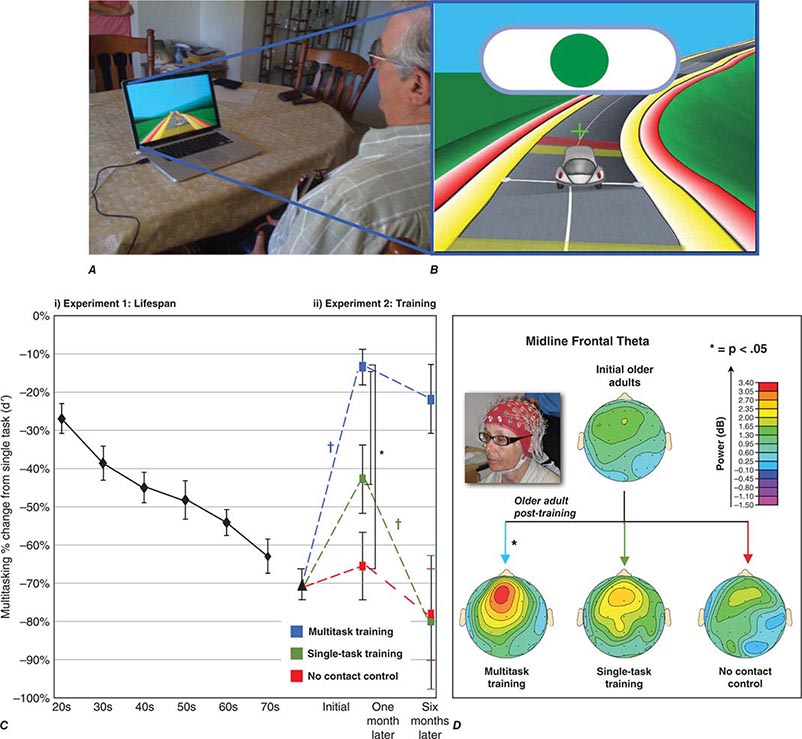
FIGURE 444e-5 Video game training can enhance cognitive performance. A. An older participant engaging in NeuroRacer training (driving while responding to target signs), with (B) a screen shot of the experimental training session. C. NeuroRacer multitasking costs for target discrimination increased (i.e., a larger percentage decrease from single task performance when multitasking) in (i) a linear fashion across the lifespan and with (ii) costs before training 1 month after training and 6 months after training, showing a differential benefit of multitasking training compared to a no-contact control group and a single-task training group. D. Midline frontal theta activity obtained with electroencephalogram showed significantly enhanced activity only following multitasking training, mimicking the pattern of change in the behavioral data as well as performance improvements on untrained tests of working memory and sustained attention (not presented). For details, see JA Anguera et al: Nature 501:97, 2013.
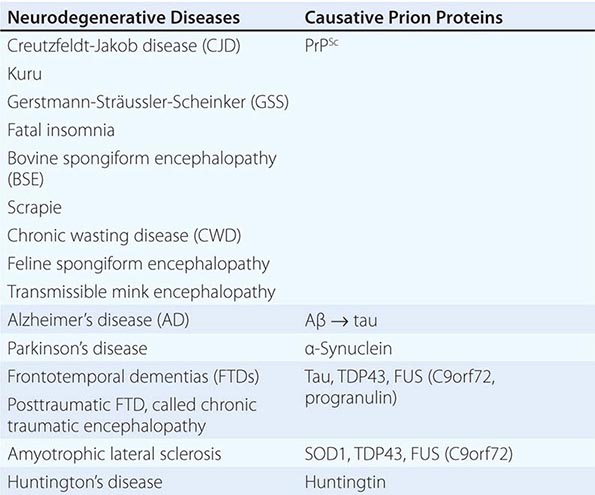
A new classification for neurodegenerative diseases can be proposed based on not only the traditional phenotypic presentation and neuropathology, but also the prion etiology (Table 444e-4). Over the past decade, an expanding body of experimental data has accumulated implicating prions in each of these illnesses. In addition to kuru and CJD, Gerstmann-Sträussler-Scheinker disease (GSS) and fatal insomnia in humans are caused by PrPSc prions. In animals, PrPSc prions cause scrapie of sheep and goats, bovine spongiform encephalopathy (BSE), chronic wasting disease (CWD) of deer and elk, feline spongiform encephalopathy, and transmissible mink encephalopathy (TME). Similar to PrP, Aβ, tau, α-synuclein, superoxide dismutase 1 (SOD1), and possibly huntingtin all adopt alternative conformations that become self-propagating, and thus, each protein can become a prion and be transferred to synaptically connected neurons. Moreover, each of these prions causes a distinct constellation of neurodegenerative diseases.
|
A PRION-BASED CLASSIFICATION OF NEURODEGENERATIVE DISEASES |
Evidence for a prion etiology of AD comes from a series of transmission experiments initially performed in marmosets and more recently in transgenic (Tg) mice inoculated with a synthetic Aβ peptide folded into a prion. Studies with the tau protein have shown that it not only features in the pathogenesis of AD, but also causes such illnesses as the frontotemporal dementias including chronic traumatic encephalopathy, which has been reported in both contact sport athletes and military personnel who have suffered traumatic brain injuries. A series of incisive studies using cultured cells and Tg mice has demonstrated that tau can become a prion and multiply in the brain. In contrast to the Aβ and tau prions, a strain of α-synuclein prions found in the brains of patients who died of multiple system atrophy (MSA) killed the Tg mouse host ~90 days after intracerebral inoculation, whereas α-synuclein prions formed spontaneously in Tg mouse brains killed recipient mice in ~200 days.
For many years, the most frequently cited argument against prions was the existence of strains that produced distinct clinical presentations and different patterns of neuropathologic lesions. Some investigators argued that the biologic information carried in different prion strains could only be encoded within a nucleic acid. Subsequently, many studies demonstrated that strain-specified variation is enciphered in the conformation of PrPSc, but the molecular mechanisms responsible for the storage of biologic information remains enigmatic. The neuroanatomical patterns of prion deposition have been shown to be dependent on the particular strain of prion. Convincing evidence in support of this proposition has been accumulated for PrP, Aβ, tau, and α-synuclein prions.
Although the number of prions identified in mammals and in fungi continues to expand, the existence of prions in other phylogeny remains undetermined. Some mammalian prions perform vital functions and do not cause disease; such nonpathogenic prions include the cytoplasmic polyadenylation element binding (CPEB) protein, the mitochondrial antiviral-signaling (MAVS) protein, and T cell–restricted intracellular antigen 1 (TIA-1).
All mammalian prion proteins adopt a β-sheet-rich conformation and appear to readily oligomerize as this process becomes self-propagating. Control of the self-propagating state of benign mammalian prions is not well understood but is critical for the well-being of the host. In contrast, pathogenic mammalian prions appear to multiply exponentially, but the mechanisms by which they cause disease are poorly defined. We do not know if prions multiply as monomers or as oligomers; notably, the ionizing radiation target size of PrPSc prions seems to suggest it is a trimer. The oligomeric states of pathogenic mammalian prions are thought to be the toxic forms, and assembly into larger polymers, such as amyloid fibrils, seems to be a mechanism for minimizing toxicity.
To date, there is no medication that halts or even slows a human neurodegenerative disease. The development of drugs designed to inhibit the conversion of the normal precursor proteins into prions or to enhance the degradation of prions focuses on the initial step in prion accumulation. Although several drugs that cross the blood-brain barrier have been identified that prolong the lives of mice infected with scrapie prions, none have been identified that extend the lives of Tg mice that replicate human CJD prions. Despite doubling the length of incubation times in mice inoculated with scrapie prions, all of the mice eventually succumb to illness. Because all of the treated mice develop neurologic dysfunction at the same time, the mutation rate as judged by drug resistance is likely to approach 100%, which is much higher than mutation rates recorded for bacteria and viruses. Mutations in prions seem likely to represent conformational variants that are selected for in mammals where survival becomes limited by the fastest-replicating prions. The results of these studies make it likely that cocktails of drugs that attack a variety of prion conformers will be required for the development of effective therapeutics.
SYSTEMS NEUROSCIENCE
Systems neuroscience refers to study of the functions of neural circuits and how they relate to brain function, behavior, motor activity, and cognition. Brain imaging techniques, primarily functional magnetic resonance imaging (fMRI) and position emission tomography (PET), have made it possible to investigate, noninvasively and in awake individuals, cognitive processes such as perception, making judgments, paying attention, and thinking. This has allowed insights into how networks of neurons operate to produce behavior. Many of these studies at present are based on determining the connectivity of neural circuits and how they operate, and how this can be then modeled to produce improved understanding of physiologic processes. fMRI uses contrast mechanisms related to physiologic changes in tissue, and brain perfusion can be studied by observing the time course of changes in brain water signal as a bolus of injected paramagnetic gadolinium contrast moves through the brain. More recently, to study intrinsic contrast-related local changes in blood oxygenation with brain activity, blood-oxygen-level-dependent (BOLD) contrast has been used to provide a rapid noninvasive approach for functional assessment. These techniques have been reliably used in the field of both behavior and cognitive sciences. One example is the use of fMRI to demonstrate mirror neuron systems, imitative pathways activated when observing actions of others. Mirror neurons are thought to be important for social conditioning and for many forms of learning, and abnormalities in mirror neurons may underlie some autism disorders.
Both structural and functional connectivity methods show large-scale network dysfunction in AD and frontotemporal dementia. The networks targeted have been defined as the default network in AD and the salience network in frontotemporal dementia. The default network is characterized by an area of reduced glucose metabolism in the temporoparietal cortex, which precedes the onset of dementia and which is an area preferentially affected by amyloid deposition. These networks are now thought to be pathways accounting for the spread of abnormally templated proteins (prions; see above), including β-amyloid, tau, and α-synuclein.
Other examples of the use of fMRI include the study of memory, revealing that not only is hippocampal activity correlated with declarative memory consolidation, but it also involves activation in the ventral medial prefrontal cortex. Consolidation of memory over time results in decreased activity of the hippocampus and progressively stronger activation in the ventral medial prefrontal region associated with retrieval of consolidated memories. fMRI has also been used to identify sequences of brain activation involved in normal movements and alterations in their activation associated with both injury and recovery, to plan neurosurgical operations, and remarkably, to reconstruct actual visual images from the occipital cortex. Noninvasive brain-computer interfaces have extraordinary potential to advance the development of robotics and exoskeleton devices guided by brain activity for patients with a variety of nervous system afflictions. Diffusion tensor imaging is a recently developed MRI technique that can measure macroscopic axonal organization in nervous system tissues; it appears to be useful in assessing myelin and axonal injuries as well as brain development. Advances in understanding neural processing have led to the development of the ability to demonstrate that humans have online voluntary control of human temporal lobe neurons.
Multitasking capabilities, including attention to tasks when faced with distractions, decline as we age due to a decline in medial prefrontal cognitive control system. When faced with a multitasking challenge, video game training can improve cognitive control capabilities by augmenting prefrontal suppression of the default network and, as measured by electroencephalography and fMRI, result in improved performance that is sustained and, importantly, transfers to other cognitive tasks not associated with the training paradigm.
A significant recent advance in neuropathology has discovered that sodium dodecyl sulfate (SDS) detergent treatment can render the brain transparent (CLARITY), removing lipids while preserving most protein and structural elements and providing opportunities to identify brain structures and neural networks with unprecedented detail.
A therapeutic technology that has long-reaching implications for the development of novel interventions for neurologic, including behavioral, conditions has been the development of deep-brain stimulation as a highly effective therapeutic intervention for treating excessively firing neurons in the subthalamic nucleus of patients with Parkinson’s disease and the precingulate cortex in patients with depression.
BRAIN RESEARCH THROUGH ADVANCING INNOVATIVE NEUROTECHNOLOGIES (BRAIN) INITIATIVE
The BRAIN initiative, grand in scope, was launched in 2013 to speed development of advances to understand, treat, repair, and prevent common neurologic disorders that, in aggregate, affect more than 1 billion people worldwide. The initial goal of BRAIN is to bring together experts in neurobiology (including optogenetics), engineering, information technology, and other fields to develop novel visualization and electrophysiologic methods to better define and understand neural circuits and all the connections among individual neurons. The announcement of the BRAIN initiative followed just weeks after a similarly ambitious program, the Human Brain Project (HBP), was unveiled by the European Union. The HBP seeks to model individual neurons, neural circuits, and ultimately the entire brain using computer technologies. Its architects also envision layering clinical and biomarker data from large health care databases to identify biosignatures associated with human phenotypes, possibly leading to a fundamental reclassification of disease, a concept also proposed by others, including in a 2011 report of the National Academy of Sciences (Toward Precision Medicine: Building a Knowledge Network for Biomedical Research and a New Taxonomy of Disease). These two ambitious projects are expected to be complementary and over time will hopefully become increasingly integrated. Emerging discoveries are also certain to stimulate a range of ethical questions, many of which are not unique to the neurosciences but do come into sharpest focus in this area. These include possible risks to the privacy of personal information about our health, cognitive capabilities, or behavioral attributes; the sanctity of our private thoughts; as well as concerns regarding neuroenhancement technologies, including their potential military use. Both the BRAIN and HBP initiatives have put into place strong ethical components to ensure that these programs are carried out, from the outset and to the fullest extent possible, consistent with guiding ethical principles that include respect for individuals, public beneficence, justice and fairness, democratic deliberation, and transparency.
SECTION 2 |
DISEASES OF THE CENTRAL NERVOUS SYSTEM |
445 |
Seizures and Epilepsy |
A seizure (from the Latin sacire, “to take possession of”) is a paroxysmal event due to abnormal excessive or synchronous neuronal activity in the brain. Depending on the distribution of discharges, this abnormal brain activity can have various manifestations, ranging from dramatic convulsive activity to experiential phenomena not readily discernible by an observer. Although a variety of factors influence the incidence and prevalence of seizures, ~5–10% of the population will have at least one seizure, with the highest incidence occurring in early childhood and late adulthood.
The meaning of the term seizure needs to be carefully distinguished from that of epilepsy. Epilepsy describes a condition in which a person has recurrent seizures due to a chronic, underlying process. This definition implies that a person with a single seizure, or recurrent seizures due to correctable or avoidable circumstances, does not necessarily have epilepsy. Epilepsy refers to a clinical phenomenon rather than a single disease entity, because there are many forms and causes of epilepsy. However, among the many causes of epilepsy there are various epilepsy syndromes in which the clinical and pathologic characteristics are distinctive and suggest a specific underlying etiology.
Using the definition of epilepsy as two or more unprovoked seizures, the incidence of epilepsy is ~0.3–0.5% in different populations throughout the world, and the prevalence of epilepsy has been estimated at 5–30 persons per 1000.
CLASSIFICATION OF SEIZURES
Determining the type of seizure that has occurred is essential for focusing the diagnostic approach on particular etiologies, selecting the appropriate therapy, and providing potentially vital information regarding prognosis. The International League against Epilepsy (ILAE) Commission on Classification and Terminology, 2005–2009 has provided an updated approach to classification of seizures (Table 445-1). This system is based on the clinical features of seizures and associated electroencephalographic findings. Other potentially distinctive features such as etiology or cellular substrate are not considered in this classification system, although this will undoubtedly change in the future as more is learned about the pathophysiologic mechanisms that underlie specific seizure types.
|
CLASSIFICATION OF SEIZURES |
A fundamental principle is that seizures may be either focal or generalized. Focal seizures originate within networks limited to one cerebral hemisphere (note that the term partial seizures is no longer used). Generalized seizures arise within and rapidly engage networks distributed across both cerebral hemispheres. Focal seizures are usually associated with structural abnormalities of the brain. In contrast, generalized seizures may result from cellular, biochemical, or structural abnormalities that have a more widespread distribution. There are clear exceptions in both cases, however.
FOCAL SEIZURES
Focal seizures arise from a neuronal network either discretely localized within one cerebral hemisphere or more broadly distributed but still within the hemisphere. With the new classification system, the subcategories of “simple focal seizures” and “complex focal seizures” have been eliminated. Instead, depending on the presence of cognitive impairment, they can be described as focal seizures with or without dyscognitive features. Focal seizures can also evolve into generalized seizures. In the past this was referred to as focal seizures with secondary generalization, but the new system relies on specific descriptions of the type of generalized seizures that evolve from the focal seizure.
The routine interictal (i.e., between seizures) electroencephalogram (EEG) in patients with focal seizures is often normal or may show brief discharges termed epileptiform spikes, or sharp waves. Because focal seizures can arise from the medial temporal lobe or inferior frontal lobe (i.e., regions distant from the scalp), the EEG recorded during the seizure may be nonlocalizing. However, the seizure focus is often detected using sphenoidal or surgically placed intracranial electrodes.
Focal Seizures Without Dyscognitive Features Focal seizures can cause motor, sensory, autonomic, or psychic symptoms without impairment of cognition. For example, a patient having a focal motor seizure arising from the right primary motor cortex near the area controlling hand movement will note the onset of involuntary movements of the contralateral, left hand. These movements are typically clonic (i.e., repetitive, flexion/extension movements) at a frequency of ~2–3 Hz; pure tonic posturing may be seen as well. Since the cortical region controlling hand movement is immediately adjacent to the region for facial expression, the seizure may also cause abnormal movements of the face synchronous with the movements of the hand. The EEG recorded with scalp electrodes during the seizure (i.e., an ictal EEG) may show abnormal discharges in a very limited region over the appropriate area of cerebral cortex if the seizure focus involves the cerebral convexity. Seizure activity occurring within deeper brain structures is sometimes not detected by the standard EEG, however, and may require intracranial electrodes for its detection.
Three additional features of focal motor seizures are worth noting. First, in some patients, the abnormal motor movements may begin in a very restricted region such as the fingers and gradually progress (over seconds to minutes) to include a larger portion of the extremity. This phenomenon, described by Hughlings Jackson and known as a “Jacksonian march,” represents the spread of seizure activity over a progressively larger region of motor cortex. Second, patients may experience a localized paresis (Todd’s paralysis) for minutes to many hours in the involved region following the seizure. Third, in rare instances, the seizure may continue for hours or days. This condition, termed epilepsia partialis continua, is often refractory to medical therapy.
Focal seizures may also manifest as changes in somatic sensation (e.g., paresthesias), vision (flashing lights or formed hallucinations), equilibrium (sensation of falling or vertigo), or autonomic function (flushing, sweating, piloerection). Focal seizures arising from the temporal or frontal cortex may also cause alterations in hearing, olfaction, or higher cortical function (psychic symptoms). This includes the sensation of unusual, intense odors (e.g., burning rubber or kerosene) or sounds (crude or highly complex sounds), or an epigastric sensation that rises from the stomach or chest to the head. Some patients describe odd, internal feelings such as fear, a sense of impending change, detachment, depersonalization, déjá vu, or illusions that objects are growing smaller (micropsia) or larger (macropsia). These subjective, “internal” events that are not directly observable by someone else are referred to as auras.
Focal Seizures with Dyscognitive Features Focal seizures may also be accompanied by a transient impairment of the patient’s ability to maintain normal contact with the environment. The patient is unable to respond appropriately to visual or verbal commands during the seizure and has impaired recollection or awareness of the ictal phase. The seizures frequently begin with an aura (i.e., a focal seizure without cognitive disturbance) that is stereotypic for the patient. The start of the ictal phase is often a sudden behavioral arrest or motionless stare, which marks the onset of the period of impaired awareness. The behavioral arrest is usually accompanied by automatisms, which are involuntary, automatic behaviors that have a wide range of manifestations. Automatisms may consist of very basic behaviors such as chewing, lip smacking, swallowing, or “picking” movements of the hands, or more elaborate behaviors such as a display of emotion or running. The patient is typically confused following the seizure, and the transition to full recovery of consciousness may range from seconds up to an hour. Examination immediately following the seizure may show an anterograde amnesia or, in cases involving the dominant hemisphere, a postictal aphasia.
The range of potential clinical behaviors linked to focal seizures is so broad that extreme caution is advised before concluding that stereotypic episodes of bizarre or atypical behavior are not due to seizure activity. In such cases additional, detailed EEG studies may be helpful.
EVOLUTION OF FOCAL SEIZURES TO GENERALIZED SEIZURES
Focal seizures can spread to involve both cerebral hemispheres and produce a generalized seizure, usually of the tonic-clonic variety (discussed below). This evolution is observed frequently following focal seizures arising from a focus in the frontal lobe, but may also be associated with focal seizures occurring elsewhere in the brain. A focal seizure that evolves into a generalized seizure is often difficult to distinguish from a primary generalized-onset tonic-clonic seizure, because bystanders tend to emphasize the more dramatic, generalized convulsive phase of the seizure and overlook the more subtle, focal symptoms present at onset. In some cases, the focal onset of the seizure becomes apparent only when a careful history identifies a preceding aura. Often, however, the focal onset is not clinically evident and may be established only through careful EEG analysis. Nonetheless, distinguishing between these two entities is extremely important, because there may be substantial differences in the evaluation and treatment of epilepsies associated with focal versus generalized seizures.
GENERALIZED SEIZURES
Generalized seizures are thought to arise at some point in the brain but immediately and rapidly engage neuronal networks in both cerebral hemispheres. Several types of generalized seizures have features that place them in distinctive categories and facilitate clinical diagnosis.
Typical Absence Seizures Typical absence seizures are characterized by sudden, brief lapses of consciousness without loss of postural control. The seizure typically lasts for only seconds, consciousness returns as suddenly as it was lost, and there is no postictal confusion. Although the brief loss of consciousness may be clinically inapparent or the sole manifestation of the seizure discharge, absence seizures are usually accompanied by subtle, bilateral motor signs such as rapid blinking of the eyelids, chewing movements, or small-amplitude, clonic movements of the hands.
Typical absence seizures are associated with a group of genetically determined epilepsies with onset usually in childhood (ages 4–8 years) or early adolescence and are the main seizure type in 15–20% of children with epilepsy. The seizures can occur hundreds of times per day, but the child may be unaware of or unable to convey their existence. Because the clinical signs of the seizures are subtle, especially to parents who may not have had previous experience with seizures, it is not surprising that the first clue to absence epilepsy is often unexplained “daydreaming” and a decline in school performance recognized by a teacher.
The electrophysiologic hallmark of typical absence seizures is a generalized, symmetric, 3-Hz spike-and-wave discharge that begins and ends suddenly, superimposed on a normal EEG background. Periods of spike-and-wave discharges lasting more than a few seconds usually correlate with clinical signs, but the EEG often shows many more brief bursts of abnormal cortical activity than were suspected clinically. Hyperventilation tends to provoke these electrographic discharges and even the seizures themselves and is routinely used when recording the EEG.
Atypical Absence Seizures Atypical absence seizures have features that deviate both clinically and electrophysiologically from typical absence seizures. For example, the lapse of consciousness is usually of longer duration and less abrupt in onset and cessation, and the seizure is accompanied by more obvious motor signs that may include focal or lateralizing features. The EEG shows a generalized, slow spike-and-wave pattern with a frequency of ≤2.5 per second, as well as other abnormal activity. Atypical absence seizures are usually associated with diffuse or multifocal structural abnormalities of the brain and therefore may accompany other signs of neurologic dysfunction such as mental retardation. Furthermore, the seizures are less responsive to anticonvulsants compared to typical absence seizures.
Generalized, Tonic-Clonic Seizures Generalized-onset tonic-clonic seizures are the main seizure type in ~10% of all persons with epilepsy. They are also the most common seizure type resulting from metabolic derangements and are therefore frequently encountered in many different clinical settings. The seizure usually begins abruptly without warning, although some patients describe vague premonitory symptoms in the hours leading up to the seizure. This prodrome is distinct from the stereotypic auras associated with focal seizures that generalize. The initial phase of the seizure is usually tonic contraction of muscles throughout the body, accounting for a number of the classic features of the event. Tonic contraction of the muscles of expiration and the larynx at the onset will produce a loud moan or “ictal cry.” Respirations are impaired, secretions pool in the oropharynx, and cyanosis develops. Contraction of the jaw muscles may cause biting of the tongue. A marked enhancement of sympathetic tone leads to increases in heart rate, blood pressure, and pupillary size. After 10–20 s, the tonic phase of the seizure typically evolves into the clonic phase, produced by the superimposition of periods of muscle relaxation on the tonic muscle contraction. The periods of relaxation progressively increase until the end of the ictal phase, which usually lasts no more than 1 min. The postictal phase is characterized by unresponsiveness, muscular flaccidity, and excessive salivation that can cause stridorous breathing and partial airway obstruction. Bladder or bowel incontinence may occur at this point. Patients gradually regain consciousness over minutes to hours, and during this transition, there is typically a period of postictal confusion. Patients subsequently complain of headache, fatigue, and muscle ache that can last for many hours. The duration of impaired consciousness in the postictal phase can be extremely long (i.e., many hours) in patients with prolonged seizures or underlying central nervous system (CNS) diseases such as alcoholic cerebral atrophy.
The EEG during the tonic phase of the seizure shows a progressive increase in generalized low-voltage fast activity, followed by generalized high-amplitude, polyspike discharges. In the clonic phase, the high-amplitude activity is typically interrupted by slow waves to create a spike-and-wave pattern. The postictal EEG shows diffuse slowing that gradually recovers as the patient awakens.
There are a number of variants of the generalized tonic-clonic seizure, including pure tonic and pure clonic seizures. Brief tonic seizures lasting only a few seconds are especially noteworthy since they are usually associated with specific epileptic syndromes having mixed seizure phenotypes, such as the Lennox-Gastaut syndrome (discussed below).
Atonic Seizures Atonic seizures are characterized by sudden loss of postural muscle tone lasting 1–2 s. Consciousness is briefly impaired, but there is usually no postictal confusion. A very brief seizure may cause only a quick head drop or nodding movement, whereas a longer seizure will cause the patient to collapse. This can be extremely dangerous, because there is a substantial risk of direct head injury with the fall. The EEG shows brief, generalized spike-and-wave discharges followed immediately by diffuse slow waves that correlate with the loss of muscle tone. Similar to pure tonic seizures, atonic seizures are usually seen in association with known epilepsy syndromes.
Myoclonic Seizures Myoclonus is a sudden and brief muscle contraction that may involve one part of the body or the entire body. A normal, common physiologic form of myoclonus is the sudden jerking movement observed while falling asleep. Pathologic myoclonus is most commonly seen in association with metabolic disorders, degenerative CNS diseases, or anoxic brain injury (Chap. 330). Although the distinction from other forms of myoclonus is imprecise, myoclonic seizures are considered to be true epileptic events because they are caused by cortical (versus subcortical or spinal) dysfunction. The EEG may show bilaterally synchronous spike-and-wave discharges synchronized with the myoclonus, although these can be obscured by movement artifact. Myoclonic seizures usually coexist with other forms of generalized seizures but are the predominant feature of juvenile myoclonic epilepsy (discussed below).
CURRENTLY UNCLASSIFIABLE SEIZURES
Not all seizure types can be designated as focal or generalized, and they should therefore be labeled as “unclassifiable” until additional evidence allows a valid classification. Epileptic spasms are such an example. These are characterized by a briefly sustained flexion or extension of predominantly proximal muscles, including truncal muscles. The EEG in these patients usually shows hypsarrhythmias, which consist of diffuse, giant slow waves with a chaotic background of irregular, multifocal spikes and sharp waves. During the clinical spasm, there is a marked suppression of the EEG background (the “electrodecremental response”). The electromyogram (EMG) also reveals a characteristic rhomboid pattern that may help distinguish spasms from brief tonic and myoclonic seizures. Epileptic spasms occur predominantly in infants and likely result from differences in neuronal function and connectivity in the immature versus mature CNS.
EPILEPSY SYNDROMES
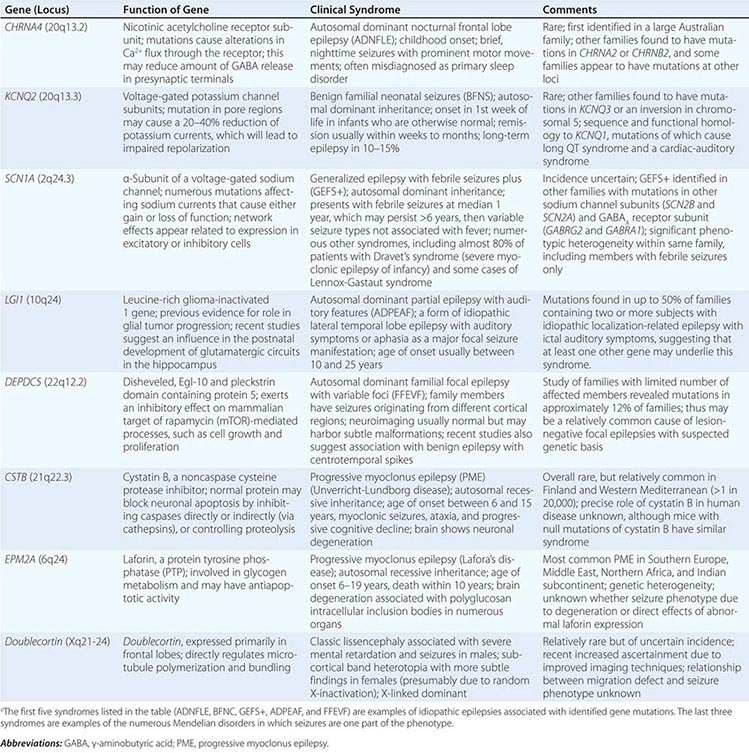
Epilepsy syndromes are disorders in which epilepsy is a predominant feature, and there is sufficient evidence (e.g., through clinical, EEG, radiologic, or genetic observations) to suggest a common underlying mechanism. Three important epilepsy syndromes are listed below; additional examples with a known genetic basis are shown in Table 445-2.
|
EXAMPLES OF GENES ASSOCIATED WITH EPILEPSY SYNDROMESa |
JUVENILE MYOCLONIC EPILEPSY
Juvenile myoclonic epilepsy (JME) is a generalized seizure disorder of unknown cause that appears in early adolescence and is usually characterized by bilateral myoclonic jerks that may be single or repetitive. The myoclonic seizures are most frequent in the morning after awakening and can be provoked by sleep deprivation. Consciousness is preserved unless the myoclonus is especially severe. Many patients also experience generalized tonic-clonic seizures, and up to one-third have absence seizures. Although complete remission is relatively uncommon, the seizures usually respond well to appropriate anticonvulsant medication. There is often a family history of epilepsy, and genetic linkage studies suggest a polygenic cause.
LENNOX-GASTAUT SYNDROME
Lennox-Gastaut syndrome occurs in children and is defined by the following triad: (1) multiple seizure types (usually including generalized tonic-clonic, atonic, and atypical absence seizures); (2) an EEG showing slow (<3 Hz) spike-and-wave discharges and a variety of other abnormalities; and (3) impaired cognitive function in most but not all cases. Lennox-Gastaut syndrome is associated with CNS disease or dysfunction from a variety of causes, including de novo mutations, developmental abnormalities, perinatal hypoxia/ischemia, trauma, infection, and other acquired lesions. The multifactorial nature of this syndrome suggests that it is a nonspecific response of the brain to diffuse neural injury. Unfortunately, many patients have a poor prognosis due to the underlying CNS disease and the physical and psychosocial consequences of severe, poorly controlled epilepsy.
MESIAL TEMPORAL LOBE EPILEPSY SYNDROME
Mesial temporal lobe epilepsy (MTLE) is the most common syndrome associated with focal seizures with dyscognitive features and is an example of an epilepsy syndrome with distinctive clinical, electroencephalographic, and pathologic features (Table 445-3). High-resolution magnetic resonance imaging (MRI) can detect the characteristic hippocampal sclerosis that appears to be essential in the pathophysiology of MTLE for many patients (Fig. 445-1). Recognition of this syndrome is especially important because it tends to be refractory to treatment with anticonvulsants but responds well to surgical intervention. Advances in the understanding of basic mechanisms of epilepsy have come through studies of experimental models of MTLE, discussed below.
|
CHARACTERISTICS OF THE MESIAL TEMPORAL LOBE EPILEPSY SYNDROME |
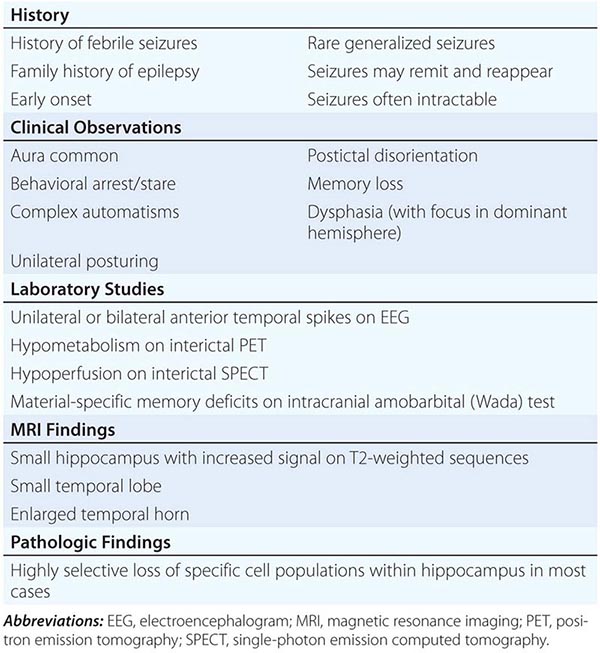
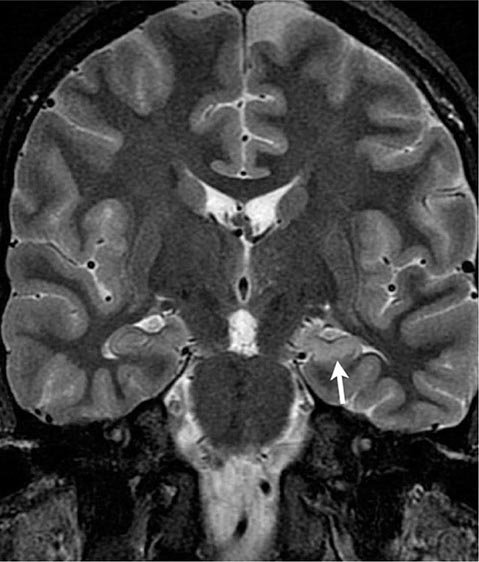
FIGURE 445-1 Mesial temporal lobe epilepsy. The electroencephalogram and seizure semiology were consistent with a left temporal lobe focus. This coronal high-resolution T2-weighted fast spin echo magnetic resonance image obtained at 3 tesla is at the level of the hippocampal bodies, and shows abnormal high signal intensity, blurring of internal laminar architecture, and reduced size of the left hippocampus (arrow) relative to the right. This triad of imaging findings is consistent with hippocampal sclerosis.
THE CAUSES OF SEIZURES AND EPILEPSY
Seizures are a result of a shift in the normal balance of excitation and inhibition within the CNS. Given the numerous properties that control neuronal excitability, it is not surprising that there are many different ways to perturb this normal balance, and therefore many different causes of both seizures and epilepsy. Three clinical observations emphasize how a variety of factors determine why certain conditions may cause seizures or epilepsy in a given patient.
1. The normal brain is capable of having a seizure under the appropriate circumstances, and there are differences between individuals in the susceptibility or threshold for seizures. For example, seizures may be induced by high fevers in children who are otherwise normal and who never develop other neurologic problems, including epilepsy. However, febrile seizures occur only in a relatively small proportion of children. This implies there are various underlying endogenous factors that influence the threshold for having a seizure. Some of these factors are genetic, as a family history of epilepsy has a clear influence on the likelihood of seizures occurring in otherwise normal individuals. Normal development also plays an important role, because the brain appears to have different seizure thresholds at different maturational stages.
2. There are a variety of conditions that have an extremely high likelihood of resulting in a chronic seizure disorder. One of the best examples of this is severe, penetrating head trauma, which is associated with up to a 45% risk of subsequent epilepsy. The high propensity for severe traumatic brain injury to lead to epilepsy suggests that the injury results in a long-lasting pathologic change in the CNS that transforms a presumably normal neural network into one that is abnormally hyperexcitable. This process is known as epileptogenesis, and the specific changes that result in a lowered seizure threshold can be considered epileptogenic factors. Other processes associated with epileptogenesis include stroke, infections, and abnormalities of CNS development. Likewise, the genetic abnormalities associated with epilepsy likely involve processes that trigger the appearance of specific sets of epileptogenic factors.
3. Seizures are episodic. Patients with epilepsy have seizures intermittently and, depending on the underlying cause, many patients are completely normal for months or even years between seizures. This implies there are important provocative or precipitating factors that induce seizures in patients with epilepsy. Similarly, precipitating factors are responsible for causing the single seizure in someone without epilepsy. Precipitants include those due to intrinsic physiologic processes such as psychological or physical stress, sleep deprivation, or hormonal changes associated with the menstrual cycle. They also include exogenous factors such as exposure to toxic substances and certain medications.
These observations emphasize the concept that the many causes of seizures and epilepsy result from a dynamic interplay between endogenous factors, epileptogenic factors, and precipitating factors. The potential role of each needs to be carefully considered when determining the appropriate management of a patient with seizures. For example, the identification of predisposing factors (e.g., family history of epilepsy) in a patient with febrile seizures may increase the necessity for closer follow-up and a more aggressive diagnostic evaluation. Finding an epileptogenic lesion may help in the estimation of seizure recurrence and duration of therapy. Finally, removal or modification of a precipitating factor may be an effective and safer method for preventing further seizures than the prophylactic use of anticonvulsant drugs.
CAUSES ACCORDING TO AGE
In practice, it is useful to consider the etiologies of seizures based on the age of the patient, because age is one of the most important factors determining both the incidence and the likely causes of seizures or epilepsy (Table 445-4). During the neonatal period and early infancy, potential causes include hypoxic-ischemic encephalopathy, trauma, CNS infection, congenital CNS abnormalities, and metabolic disorders. Babies born to mothers using neurotoxic drugs such as cocaine, heroin, or ethanol are susceptible to drug-withdrawal seizures in the first few days after delivery. Hypoglycemia and hypocalcemia, which can occur as secondary complications of perinatal injury, are also causes of seizures early after delivery. Seizures due to inborn errors of metabolism usually present once regular feeding begins, typically 2–3 days after birth. Pyridoxine (vitamin B6) deficiency, an important cause of neonatal seizures, can be effectively treated with pyridoxine replacement. The idiopathic or inherited forms of benign neonatal convulsions are also seen during this time period.
|
CAUSES OF SEIZURES |

The most common seizures arising in late infancy and early childhood are febrile seizures, which are seizures associated with fevers but without evidence of CNS infection or other defined causes. The overall prevalence is 3–5% and even higher in some parts of the world such as Asia. Patients often have a family history of febrile seizures or epilepsy. Febrile seizures usually occur between 3 months and 5 years of age and have a peak incidence between 18 and 24 months. The typical scenario is a child who has a generalized, tonic-clonic seizure during a febrile illness in the setting of a common childhood infection such as otitis media, respiratory infection, or gastroenteritis. The seizure is likely to occur during the rising phase of the temperature curve (i.e., during the first day) rather than well into the course of the illness. A simple febrile seizure is a single, isolated event, brief, and symmetric in appearance. Complex febrile seizures are characterized by repeated seizure activity, duration >15 minutes, or by focal features. Approximately one-third of patients with febrile seizures will have a recurrence, but <10% have three or more episodes. Recurrences are much more likely when the febrile seizure occurs in the first year of life. Simple febrile seizures are not associated with an increase in the risk of developing epilepsy, while complex febrile seizures have a risk of 2–5%; other risk factors include the presence of preexisting neurologic deficits and a family history of nonfebrile seizures.
Childhood marks the age at which many of the well-defined epilepsy syndromes present. Some children who are otherwise normal develop idiopathic, generalized tonic-clonic seizures without other features that fit into specific syndromes. Temporal lobe epilepsy usually presents in childhood and may be related to mesial temporal lobe sclerosis (as part of the MTLE syndrome) or other focal abnormalities such as cortical dysgenesis. Other types of focal seizures, including those that evolve into generalized seizures, may be the relatively late manifestation of a developmental disorder, an acquired lesion such as head trauma, CNS infection (especially viral encephalitis), or very rarely a CNS tumor.
The period of adolescence and early adulthood is one of transition during which the idiopathic or genetically based epilepsy syndromes, including JME and juvenile absence epilepsy, become less common, while epilepsies secondary to acquired CNS lesions begin to predominate. Seizures that arise in patients in this age range may be associated with head trauma, CNS infections (including parasitic infections such as cysticercosis), brain tumors, congenital CNS abnormalities, illicit drug use, or alcohol withdrawal. Autoantibodies directed against CNS antigens such as potassium channels or glutamate receptors are a newly recognized cause of epilepsy that also begins to appear in this age group (although cases of autoimmunity are being increasingly described in the pediatric population), including patients without an identifiable cancer. This etiology should be suspected when a previously normal individual presents with a particularly aggressive seizure pattern developing over weeks to months and characterized by increasingly frequent and prolonged seizures combined with cognitive decline (Chap. 122).
Head trauma is a common cause of epilepsy in adolescents and adults. The head injury can be caused by a variety of mechanisms, and the likelihood of developing epilepsy is strongly correlated with the severity of the injury. A patient with a penetrating head wound, depressed skull fracture, intracranial hemorrhage, or prolonged posttraumatic coma or amnesia has a 30–50% risk of developing epilepsy, whereas a patient with a closed head injury and cerebral contusion has a 5–25% risk. Recurrent seizures usually develop within 1 year after head trauma, although intervals of >10 years are well known. In controlled studies, mild head injury, defined as a concussion with amnesia or loss of consciousness of <30 min, was found to be associated with only a slightly increased likelihood of epilepsy. Nonetheless, most epileptologists know of patients who have focal seizures within hours or days of a mild head injury and subsequently develop chronic seizures of the same type; such cases may represent rare examples of chronic epilepsy resulting from mild head injury.
The causes of seizures in older adults include cerebrovascular disease, trauma (including subdural hematoma), CNS tumors, and degenerative diseases. Cerebrovascular disease may account for ~50% of new cases of epilepsy in patients older than age 65. Acute seizures (i.e., occurring at the time of the stroke) are seen more often with embolic rather than hemorrhagic or thrombotic stroke. Chronic seizures typically appear months to years after the initial event and are associated with all forms of stroke.
Metabolic disturbances such as electrolyte imbalance, hypo- or hyperglycemia, renal failure, and hepatic failure may cause seizures at any age. Similarly, endocrine disorders, hematologic disorders, vasculitides, and many other systemic diseases may cause seizures over a broad age range. A wide variety of medications and abused substances are known to precipitate seizures as well (Table 445-5).
|
DRUGS AND OTHER SUBSTANCES THAT CAN CAUSE SEIZURES |
aIn benzodiazepine-dependent patients.
BASIC MECHANISMS
MECHANISMS OF SEIZURE INITIATION AND PROPAGATION
Focal seizure activity can begin in a very discrete region of cortex and then slowly invade the surrounding regions. The hallmark of an established seizure is typically an electrographic “spike” due to intense near-simultaneous firing of a large number of local excitatory neurons, resulting in an apparent hypersynchronization of the excitatory bursts across a relatively large cortical region. The bursting activity in individual neurons (the “paroxysmal depolarization shift”) is caused by a relatively long-lasting depolarization of the neuronal membrane due to influx of extracellular calcium (Ca2+), which leads to the opening of voltage-dependent sodium (Na+) channels, influx of Na+, and generation of repetitive action potentials. This is followed by a hyperpolarizing afterpotential mediated by γ-aminobutyric acid (GABA) receptors or potassium (K+) channels, depending on the cell type. The synchronized bursts from a sufficient number of neurons result in a so-called spike discharge on the EEG.
The spreading seizure wavefront is slowed and ultimately halted by intact hyperpolarization and a “surround” inhibition created by feedforward activation of inhibitory neurons. With sufficient activation, there is a recruitment of surrounding neurons via a number of synaptic and nonsynaptic mechanisms, including: (1) an increase in extracellular K+, which blunts hyperpolarization and depolarizes neighboring neurons; (2) accumulation of Ca2+ in presynaptic terminals, leading to enhanced neurotransmitter release; (3) depolarization-induced activation of the N-methyl-D-aspartate (NMDA) subtype of the excitatory amino acid receptor, which causes additional Ca2+ influx and neuronal activation; and (4) ephaptic interactions related to changes in tissue osmolarity and cell swelling. The recruitment of a sufficient number of neurons leads to the propagation of excitatory currents into contiguous areas via local cortical connections and to more distant areas via long commissural pathways such as the corpus callosum.
Many factors control neuronal excitability, and thus there are many potential mechanisms for altering a neuron’s propensity to have bursting activity. Mechanisms intrinsic to the neuron include changes in the conductance of ion channels, response characteristics of membrane receptors, cytoplasmic buffering, second-messenger systems, and protein expression as determined by gene transcription, translation, and posttranslational modification. Mechanisms extrinsic to the neuron include changes in the amount or type of neurotransmitters present at the synapse, modulation of receptors by extracellular ions and other molecules, and temporal and spatial properties of synaptic and nonsynaptic input. Nonneural cells, such as astrocytes and oligodendrocytes, have an important role in many of these mechanisms as well.
Certain recognized causes of seizures are explained by these mechanisms. For example, accidental ingestion of domoic acid, which is an analogue of glutamate (the principal excitatory neurotransmitter in the brain), causes profound seizures via direct activation of excitatory amino acid receptors throughout the CNS. Penicillin, which can lower the seizure threshold in humans and is a potent convulsant in experimental models, reduces inhibition by antagonizing the effects of GABA at its receptor. The basic mechanisms of other precipitating factors of seizures such as sleep deprivation, fever, alcohol withdrawal, hypoxia, and infection, are not as well understood but presumably involve analogous perturbations in neuronal excitability. Similarly, the endogenous factors that determine an individual’s seizure threshold may relate to these properties as well.
Knowledge of the mechanisms responsible for initiation and propagation of most generalized seizures (including tonic-clonic, myoclonic, and atonic types) remains rudimentary and reflects the limited understanding of the connectivity of the brain at a systems level. Much more is understood about the origin of generalized spike-and-wave discharges in absence seizures. These appear to be related to oscillatory rhythms normally generated during sleep by circuits connecting the thalamus and cortex. This oscillatory behavior involves an interaction between GABAB receptors, T-type Ca2+ channels, and K+ channels located within the thalamus. Pharmacologic studies indicate that modulation of these receptors and channels can induce absence seizures, and there is good evidence that the genetic forms of absence epilepsy may be associated with mutations of components of this system.
MECHANISMS OF EPILEPTOGENESIS
Epileptogenesis refers to the transformation of a normal neuronal network into one that is chronically hyperexcitable. There is often a delay of months to years between an initial CNS injury such as trauma, stroke, or infection and the first seizure. The injury appears to initiate a process that gradually lowers the seizure threshold in the affected region until a spontaneous seizure occurs. In many genetic and idiopathic forms of epilepsy, epileptogenesis is presumably determined by developmentally regulated events.
Pathologic studies of the hippocampus from patients with temporal lobe epilepsy have led to the suggestion that some forms of epileptogenesis are related to structural changes in neuronal networks. For example, many patients with MTLE have a highly selective loss of neurons that may contribute to inhibition of the main excitatory neurons within the dentate gyrus. There is also evidence that, in response to the loss of neurons, there is reorganization or “sprouting” of surviving neurons in a way that affects the excitability of the network. Some of these changes can be seen in experimental models of prolonged electrical seizures or traumatic brain injury. Thus, an initial injury such as head injury may lead to a very focal, confined region of structural change that causes local hyperexcitability. The local hyperexcitability leads to further structural changes that evolve over time until the focal lesion produces clinically evident seizures. Similar models have provided strong evidence for long-term alterations in intrinsic, biochemical properties of cells within the network such as chronic changes in glutamate or GABA receptor function. Recent work has suggested that induction of inflammatory cascades may be a critical factor in these processes as well.
GENETIC CAUSES OF EPILEPSY
![]() The most important recent progress in epilepsy research has been the identification of genetic mutations associated with a variety of epilepsy syndromes (Table 445-2). Although most of the mutations identified to date cause rare forms of epilepsy, their discovery has led to extremely important conceptual advances. For example, it appears that many of the inherited, idiopathic epilepsies (i.e., the relatively “pure” forms of epilepsy in which seizures are the phenotypic abnormality and brain structure and function are otherwise normal) are due to mutations affecting ion channel function. These syndromes are therefore part of the larger group of channelopathies causing paroxysmal disorders such as cardiac arrhythmias, episodic ataxia, periodic weakness, and familial hemiplegic migraine. In contrast, gene mutations observed in symptomatic epilepsies (i.e., disorders in which other neurologic abnormalities such as cognitive impairment coexist with seizures) are proving to be associated with pathways influencing CNS development or neuronal homeostasis. De novo mutations may explain a significant proportion of these syndromes, especially those with onset in early childhood. A current challenge is to identify the multiple susceptibility genes that underlie the more common forms of idiopathic epilepsies. Recent studies suggest that ion channel mutations and copy number variants may contribute to causation in a subset of these patients.
The most important recent progress in epilepsy research has been the identification of genetic mutations associated with a variety of epilepsy syndromes (Table 445-2). Although most of the mutations identified to date cause rare forms of epilepsy, their discovery has led to extremely important conceptual advances. For example, it appears that many of the inherited, idiopathic epilepsies (i.e., the relatively “pure” forms of epilepsy in which seizures are the phenotypic abnormality and brain structure and function are otherwise normal) are due to mutations affecting ion channel function. These syndromes are therefore part of the larger group of channelopathies causing paroxysmal disorders such as cardiac arrhythmias, episodic ataxia, periodic weakness, and familial hemiplegic migraine. In contrast, gene mutations observed in symptomatic epilepsies (i.e., disorders in which other neurologic abnormalities such as cognitive impairment coexist with seizures) are proving to be associated with pathways influencing CNS development or neuronal homeostasis. De novo mutations may explain a significant proportion of these syndromes, especially those with onset in early childhood. A current challenge is to identify the multiple susceptibility genes that underlie the more common forms of idiopathic epilepsies. Recent studies suggest that ion channel mutations and copy number variants may contribute to causation in a subset of these patients.
MECHANISMS OF ACTION OF ANTIEPILEPTIC DRUGS
Antiepileptic drugs appear to act primarily by blocking the initiation or spread of seizures. This occurs through a variety of mechanisms that modify the activity of ion channels or neurotransmitters, and in most cases, the drugs have pleiotropic effects. The mechanisms include inhibition of Na+-dependent action potentials in a frequency-dependent manner (e.g., phenytoin, carbamazepine, lamotrigine, topiramate, zonisamide, lacosamide, rufinamide), inhibition of voltage-gated Ca2+ channels (phenytoin, gabapentin, pregabalin), facilitating the opening of potassium channels (ezogabine), attenuation of glutamate activity (lamotrigine, topiramate, felbamate), potentiation of GABA receptor function (benzodiazepines and barbiturates), increase in the availability of GABA (valproic acid, gabapentin, tiagabine), and modulation of release of synaptic vesicles (levetiracetam). The two most effective drugs for absence seizures, ethosuximide and valproic acid, probably act by inhibiting T-type Ca2+ channels in thalamic neurons.
In contrast to the relatively large number of antiepileptic drugs that can attenuate seizure activity, there are currently no drugs known to prevent the formation of a seizure focus following CNS injury. The eventual development of such “antiepileptogenic” drugs will provide an important means of preventing the emergence of epilepsy following injuries such as head trauma, stroke, and CNS infection.
APPROACH TO THE PATIENT:
Seizure
When a patient presents shortly after a seizure, the first priorities are attention to vital signs, respiratory and cardiovascular support, and treatment of seizures if they resume (see “Treatment: Seizures and Epilepsy”). Life-threatening conditions such as CNS infection, metabolic derangement, or drug toxicity must be recognized and managed appropriately.
When the patient is not acutely ill, the evaluation will initially focus on whether there is a history of earlier seizures (Fig. 445-2). If this is the first seizure, then the emphasis will be to: (1) establish whether the reported episode was a seizure rather than another paroxysmal event, (2) determine the cause of the seizure by identifying risk factors and precipitating events, and (3) decide whether anticonvulsant therapy is required in addition to treatment for any underlying illness.
FIGURE 445-2 Evaluation of the adult patient with a seizure. CBC, complete blood count; CNS, central nervous system; CT, computed tomography; EEG, electroencephalogram; MRI, magnetic resonance imaging.
In the patient with prior seizures or a known history of epilepsy, the evaluation is directed toward: (1) identification of the underlying cause and precipitating factors, and (2) determination of the adequacy of the patient’s current therapy.
HISTORY AND EXAMINATION
The first goal is to determine whether the event was truly a seizure. An in-depth history is essential, because in many cases the diagnosis of a seizure is based solely on clinical grounds—the examination and laboratory studies are often normal. Questions should focus on the symptoms before, during, and after the episode in order to differentiate a seizure from other paroxysmal events (see “Differential Diagnosis of Seizures” below). Seizures frequently occur out-of-hospital, and the patient may be unaware of the ictal and immediate postictal phases; thus, witnesses to the event should be interviewed carefully.
The history should also focus on risk factors and predisposing events. Clues for a predisposition to seizures include a history of febrile seizures, earlier auras or brief seizures not recognized as such, and a family history of seizures. Epileptogenic factors such as prior head trauma, stroke, tumor, or CNS infection should be identified. In children, a careful assessment of developmental milestones may provide evidence for underlying CNS disease. Precipitating factors such as sleep deprivation, systemic diseases, electrolyte or metabolic derangements, acute infection, drugs that lower the seizure threshold (Table 445-5), or alcohol or illicit drug use should also be identified.
The general physical examination includes a search for signs of infection or systemic illness. Careful examination of the skin may reveal signs of neurocutaneous disorders such as tuberous sclerosis or neurofibromatosis, or chronic liver or renal disease. A finding of organomegaly may indicate a metabolic storage disease, and limb asymmetry may provide a clue to brain injury early in development. Signs of head trauma and use of alcohol or illicit drugs should be sought. Auscultation of the heart and carotid arteries may identify an abnormality that predisposes to cerebrovascular disease.
All patients require a complete neurologic examination, with particular emphasis on eliciting signs of cerebral hemispheric disease (Chap. 437). Careful assessment of mental status (including memory, language function, and abstract thinking) may suggest lesions in the anterior frontal, parietal, or temporal lobes. Testing of visual fields will help screen for lesions in the optic pathways and occipital lobes. Screening tests of motor function such as pronator drift, deep tendon reflexes, gait, and coordination may suggest lesions in motor (frontal) cortex, and cortical sensory testing (e.g., double simultaneous stimulation) may detect lesions in the parietal cortex.
LABORATORY STUDIES
Routine blood studies are indicated to identify the more common metabolic causes of seizures such as abnormalities in electrolytes, glucose, calcium, or magnesium, and hepatic or renal disease. A screen for toxins in blood and urine should also be obtained from all patients in appropriate risk groups, especially when no clear precipitating factor has been identified. A lumbar puncture is indicated if there is any suspicion of meningitis or encephalitis, and it is mandatory in all patients infected with HIV, even in the absence of symptoms or signs suggesting infection. Testing for autoantibodies in the serum and cerebrospinal fluid (CSF) should be considered in patients presenting with a seemingly aggressive form of epilepsy associated with other abnormalities such as cognitive disturbances.
ELECTROPHYSIOLOGIC STUDIES
All patients who have a possible seizure disorder should be evaluated with an EEG as soon as possible. Details about the EEG are covered in Chap. 442e.
In the evaluation of a patient with suspected epilepsy, the presence of electrographic seizure activity during the clinically evident event (i.e., abnormal, repetitive, rhythmic activity having a discrete onset and termination) clearly establishes the diagnosis. The absence of electrographic seizure activity does not exclude a seizure disorder, however, because focal seizures may originate from a region of the cortex that cannot be detected by standard scalp electrodes. The EEG is always abnormal during generalized tonic-clonic seizures. Because seizures are typically infrequent and unpredictable, it is often not possible to obtain the EEG during a clinical event. Continuous monitoring for prolonged periods in video-EEG telemetry units for hospitalized patients or the use of portable equipment to record the EEG continuously for ≥24 h in ambulatory patients has made it easier to capture the electrophysiologic accompaniments of clinical events. In particular, video-EEG telemetry is now a routine approach for the accurate diagnosis of epilepsy in patients with poorly characterized events or seizures that are difficult to control.
The EEG may also be helpful in the interictal period by showing certain abnormalities that are highly supportive of the diagnosis of epilepsy. Such epileptiform activity consists of bursts of abnormal discharges containing spikes or sharp waves. The presence of epileptiform activity is not specific for epilepsy, but it has a much greater prevalence in patients with epilepsy than in normal individuals. However, even in an individual who is known to have epilepsy, the initial routine interictal EEG may be normal up to 60% of the time. Thus, the EEG cannot establish the diagnosis of epilepsy in many cases.
The EEG is also used for classifying seizure disorders and aiding in the selection of anticonvulsant medications. For example, episodic generalized spike-wave activity is usually seen in patients with typical absence epilepsy and may be seen with other generalized epilepsy syndromes. Focal interictal epileptiform discharges would support the diagnosis of a focal seizure disorder such as temporal lobe epilepsy or frontal lobe seizures, depending on the location of the discharges.
The routine scalp-recorded EEG may also be used to assess the prognosis of seizure disorders; in general, a normal EEG implies a better prognosis, whereas an abnormal background or profuse epileptiform activity suggests a poor outcome. Unfortunately, the EEG has not proved to be useful in predicting which patients with predisposing conditions such as head injury or brain tumor will go on to develop epilepsy, because in such circumstances epileptiform activity is commonly encountered regardless of whether seizures occur.
Magnetoencephalography (MEG) provides another way of looking noninvasively at cortical activity. Instead of measuring electrical activity of the brain, it measures the small magnetic fields that are generated by this activity. The source of epileptiform activity seen on MEG can be analyzed, and its source in the brain can be estimated using a variety of mathematical techniques. These source estimates can then be plotted on an anatomic image of the brain such as an MRI (discussed below), to generate a magnetic source image (MSI). MSI can be useful to localize potential seizure foci.
BRAIN IMAGING
Almost all patients with new-onset seizures should have a brain imaging study to determine whether there is an underlying structural abnormality that is responsible. The only potential exception to this rule is children who have an unambiguous history and examination suggestive of a benign, generalized seizure disorder such as absence epilepsy. MRI has been shown to be superior to computed tomography (CT) for the detection of cerebral lesions associated with epilepsy. In some cases, MRI will identify lesions such as tumors, vascular malformations, or other pathologies that need urgent therapy. The availability of newer MRI methods such as 3-tesla scanners, parallel imaging with multichannel head coils, three-dimensional structural imaging at submillimeter resolution, and widespread use of pulse sequences such as fluid-attenuated inversion recovery (FLAIR), has increased the sensitivity for detection of abnormalities of cortical architecture, including hippocampal atrophy associated with mesial temporal sclerosis, as well as abnormalities of cortical neuronal migration. In such cases, the findings may not lead to immediate therapy, but they do provide an explanation for the patient’s seizures and point to the need for chronic antiepileptic drug therapy or possible surgical resection.
In the patient with a suspected CNS infection or mass lesion, CT scanning should be performed emergently when MRI is not immediately available. Otherwise, it is usually appropriate to obtain an MRI study within a few days of the initial evaluation. Functional imaging procedures such as positron emission tomography (PET) and single-photon emission computed tomography (SPECT) are also used to evaluate certain patients with medically refractory seizures (discussed below).
DIFFERENTIAL DIAGNOSIS OF SEIZURES
Disorders that may mimic seizures are listed in Table 445-6. In most cases, seizures can be distinguished from other conditions by meticulous attention to the history and relevant laboratory studies. On occasion, additional studies such as video-EEG monitoring, sleep studies, tilt-table analysis, or cardiac electrophysiology, may be required to reach a correct diagnosis. Two of the more common nonepileptic syndromes in the differential diagnosis are detailed below.
|
DIFFERENTIAL DIAGNOSIS OF SEIZURES |
SYNCOPE
(See also Chap. 27) The diagnostic dilemma encountered most frequently is the distinction between a generalized seizure and syncope. Observations by the patient and bystanders that can help differentiate between the two are listed in Table 445-7. Characteristics of a seizure include the presence of an aura, cyanosis, unconsciousness, motor manifestations lasting >15 s, postictal disorientation, muscle soreness, and sleepiness. In contrast, a syncopal episode is more likely if the event was provoked by acute pain or anxiety or occurred immediately after arising from the lying or sitting position. Patients with syncope often describe a stereotyped transition from consciousness to unconsciousness that includes tiredness, sweating, nausea, and tunneling of vision, and they experience a relatively brief loss of consciousness. Headache or incontinence usually suggests a seizure but may on occasion also occur with syncope. A brief period (i.e., 1–10 s) of convulsive motor activity is frequently seen immediately at the onset of a syncopal episode, especially if the patient remains in an upright posture after fainting (e.g., in a dentist’s chair) and therefore has a sustained decrease in cerebral perfusion. Rarely, a syncopal episode can induce a full tonic-clonic seizure. In such cases, the evaluation must focus on both the cause of the syncopal event as well as the possibility that the patient has a propensity for recurrent seizures.
|
FEATURES THAT DISTINGUISH GENERALIZED TONIC-CLONIC SEIZURE FROM SYNCOPE |
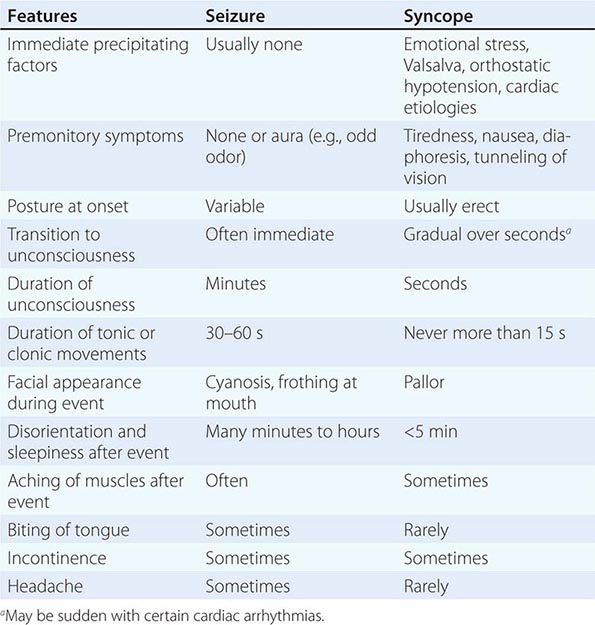
PSYCHOGENIC SEIZURES
Psychogenic seizures are nonepileptic behaviors that resemble seizures. They are often part of a conversion reaction precipitated by underlying psychological distress. Certain behaviors such as side-to-side turning of the head, asymmetric and large-amplitude shaking movements of the limbs, twitching of all four extremities without loss of consciousness, and pelvic thrusting are more commonly associated with psychogenic rather than epileptic seizures. Psychogenic seizures often last longer than epileptic seizures and may wax and wane over minutes to hours. However, the distinction is sometimes difficult on clinical grounds alone, and there are many examples of diagnostic errors made by experienced epileptologists. This is especially true for psychogenic seizures that resemble focal seizures with dyscognitive features, because the behavioral manifestations of focal seizures (especially of frontal lobe origin) can be extremely unusual, and in both cases, the routine surface EEG may be normal. Video-EEG monitoring is very useful when historic features are nondiagnostic. Generalized tonic-clonic seizures always produce marked EEG abnormalities during and after the seizure. For suspected focal seizures of temporal lobe origin, the use of additional electrodes beyond the standard scalp locations (e.g., sphenoidal electrodes) may be required to localize a seizure focus. Measurement of serum prolactin levels may also help to distinguish between organic and psychogenic seizures, because most generalized seizures and some focal seizures are accompanied by rises in serum prolactin (during the immediate 30-min postictal period), whereas psychogenic seizures are not. The diagnosis of psychogenic seizures does not exclude a concurrent diagnosis of epilepsy, because the two often coexist.
|
TREATMENT |
SEIZURES AND EPILEPSY |
Therapy for a patient with a seizure disorder is almost always multimodal and includes treatment of underlying conditions that cause or contribute to the seizures, avoidance of precipitating factors, suppression of recurrent seizures by prophylactic therapy with antiepileptic medications or surgery, and addressing a variety of psychological and social issues. Treatment plans must be individualized, given the many different types and causes of seizures as well as the differences in efficacy and toxicity of antiepileptic medications for each patient. In almost all cases, a neurologist with experience in the treatment of epilepsy should design and oversee implementation of the treatment strategy. Furthermore, patients with refractory epilepsy or those who require polypharmacy with antiepileptic drugs should remain under the regular care of a neurologist.
TREATMENT OF UNDERLYING CONDITIONS
If the sole cause of a seizure is a metabolic disturbance such as an abnormality of serum electrolytes or glucose, then treatment is aimed at reversing the metabolic problem and preventing its recurrence. Therapy with antiepileptic drugs is usually unnecessary unless the metabolic disorder cannot be corrected promptly and the patient is at risk of having further seizures. If the apparent cause of a seizure was a medication (e.g., theophylline) or illicit drug use (e.g., cocaine), then appropriate therapy is avoidance of the drug; there is usually no need for antiepileptic medications unless subsequent seizures occur in the absence of these precipitants.
Seizures caused by a structural CNS lesion such as a brain tumor, vascular malformation, or brain abscess may not recur after appropriate treatment of the underlying lesion. However, despite removal of the structural lesion, there is a risk that the seizure focus will remain in the surrounding tissue or develop de novo as a result of gliosis and other processes induced by surgery, radiation, or other therapies. Most patients are therefore maintained on an antiepileptic medication for at least 1 year, and an attempt is made to withdraw medications only if the patient has been completely seizure free. If seizures are refractory to medication, the patient may benefit from surgical removal of the epileptic brain region (see below).
AVOIDANCE OF PRECIPITATING FACTORS
Unfortunately, little is known about the specific factors that determine precisely when a seizure will occur in a patient with epilepsy. Some patients can identify particular situations that appear to lower their seizure threshold; these situations should be avoided. For example, a patient who has seizures in the setting of sleep deprivation should obviously be advised to maintain a normal sleep schedule. Many patients note an association between alcohol intake and seizures, and they should be encouraged to modify their drinking habits accordingly. There are also relatively rare cases of patients with seizures that are induced by highly specific stimuli such as a video game monitor, music, or an individual’s voice (“reflex epilepsy”). Because there is often an association between stress and seizures, stress reduction techniques such as physical exercise, meditation, or counseling may be helpful.
ANTIEPILEPTIC DRUG THERAPY
Antiepileptic drug therapy is the mainstay of treatment for most patients with epilepsy. The overall goal is to completely prevent seizures without causing any untoward side effects, preferably with a single medication and a dosing schedule that is easy for the patient to follow. Seizure classification is an important element in designing the treatment plan, because some antiepileptic drugs have different activities against various seizure types. However, there is considerable overlap between many antiepileptic drugs such that the choice of therapy is often determined more by the patient’s specific needs, especially his or her assessment of side effects.
When to Initiate Antiepileptic Drug Therapy Antiepileptic drug therapy should be started in any patient with recurrent seizures of unknown etiology or a known cause that cannot be reversed. Whether to initiate therapy in a patient with a single seizure is controversial. Patients with a single seizure due to an identified lesion such as a CNS tumor, infection, or trauma, in which there is strong evidence that the lesion is epileptogenic, should be treated. The risk of seizure recurrence in a patient with an apparently unprovoked or idiopathic seizure is uncertain, with estimates ranging from 31 to 71% in the first 12 months after the initial seizure. This uncertainty arises from differences in the underlying seizure types and etiologies in various published epidemiologic studies. Generally accepted risk factors associated with recurrent seizures include the following: (1) an abnormal neurologic examination, (2) seizures presenting as status epilepticus, (3) postictal Todd’s paralysis, (4) a strong family history of seizures, or (5) an abnormal EEG. Most patients with one or more of these risk factors should be treated. Issues such as employment or driving may influence the decision whether to start medications as well. For example, a patient with a single, idiopathic seizure whose job depends on driving may prefer taking antiepileptic drugs rather than risk a seizure recurrence and the potential loss of driving privileges.
Selection of Antiepileptic Drugs Antiepileptic drugs available in the United States are shown in Table 445-8, and the main pharmacologic characteristics of commonly used drugs are listed in Table 445-9. Worldwide, older medications such as phenytoin, valproic acid, carbamazepine, phenobarbital, and ethosuximide are generally used as first-line therapy for most seizure disorders because, overall, they are as effective as recently marketed drugs and significantly less expensive overall. Most of the new drugs that have become available in the past decade are used as add-on or alternative therapy, although many are now being used as first-line monotherapy.
|
SELECTION OF ANTIEPILEPTIC DRUGS |
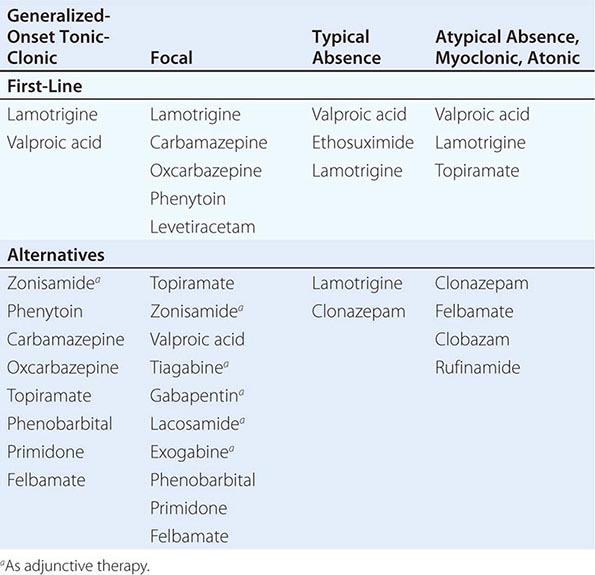
|
DOSAGE AND ADVERSE EFFECTS OF COMMONLY USED ANTIEPILEPTIC DRUGS |
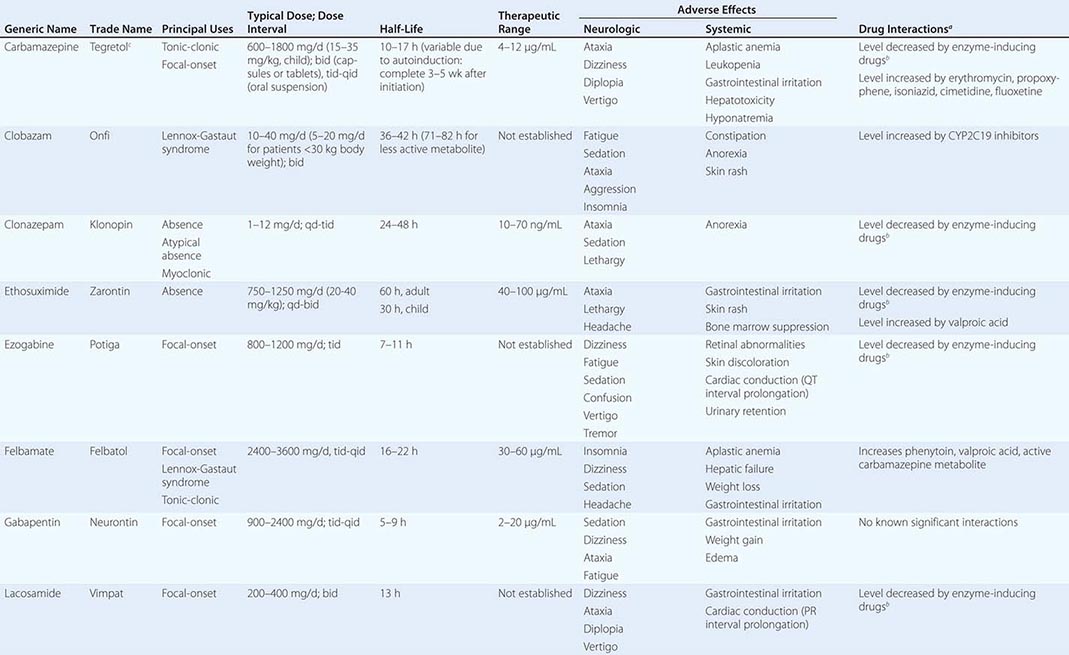
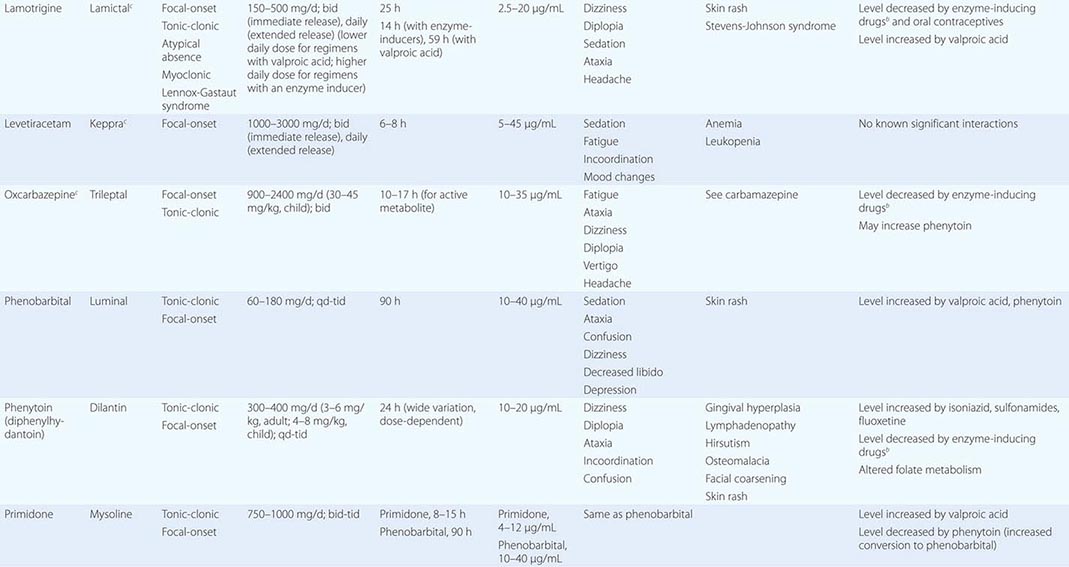
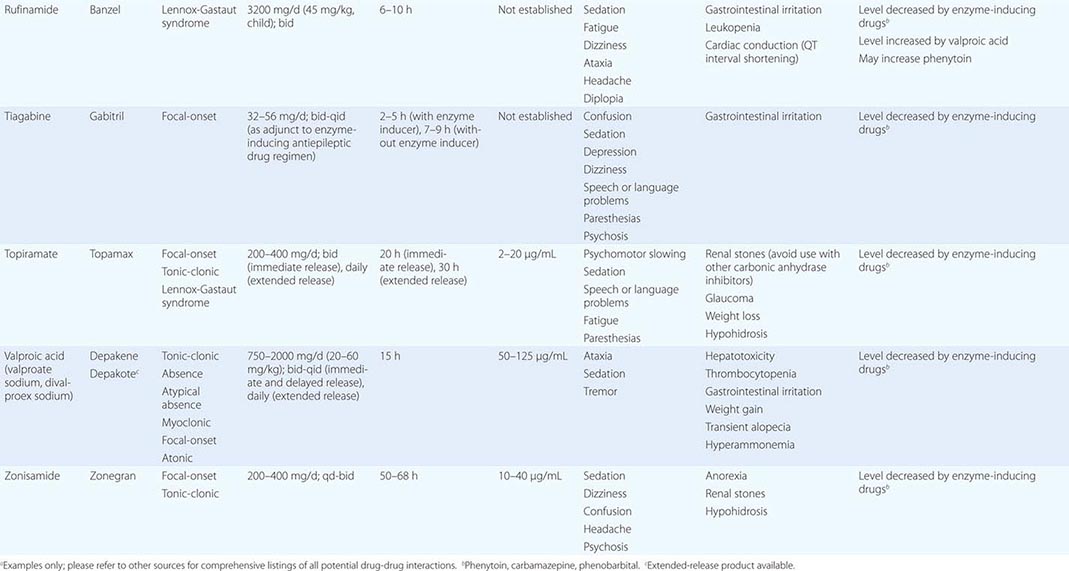
In addition to efficacy, factors influencing the choice of an initial medication include the convenience of dosing (e.g., once daily versus three or four times daily) and potential side effects. In this regard, a number of the newer drugs have the advantage of reduced drug-drug interactions and easier dosing. Almost all of the commonly used antiepileptic drugs can cause similar, dose-related side effects such as sedation, ataxia, and diplopia. Long-term use of some agents in adults, especially the elderly, can lead to osteoporosis. Close follow-up is required to ensure these side effects are promptly recognized and reversed. Most of the older drugs and some of the newer ones can also cause idiosyncratic toxicity such as rash, bone marrow suppression, or hepatotoxicity. Although rare, these side effects should be considered during drug selection, and patients must be instructed about symptoms or signs that should signal the need to alert their health care provider. For some drugs, laboratory tests (e.g., complete blood count and liver function tests) are recommended prior to the institution of therapy (to establish baseline values) and during initial dosing and titration of the agent. Importantly, studies have shown that Asian individuals carrying the human leukocyte antigen allele, HLA-B*1502, are at particularly high risk of developing serious skin reactions from carbamazepine and phenytoin. As a result, racial background and genotype are additional factors to consider in drug selection.
ANTIEPILEPTIC DRUG SELECTION FOR FOCAL SEIZURES Carbamazepine (or a related drug, oxcarbazepine), lamotrigine, phenytoin, and levetiracetam are currently the drugs of choice approved for the initial treatment of focal seizures, including those that evolve into generalized seizures. Overall they have very similar efficacy, but differences in pharmacokinetics and toxicity are the main determinants for use in a given patient. For example, an advantage of carbamazepine (which is also available in an extended-release form) is that its metabolism follows first-order pharmacokinetics, which allows for a linear relationship between drug dose, serum levels, and toxicity. Carbamazepine can cause leukopenia, aplastic anemia, or hepatotoxicity and would therefore be contraindicated in patients with predispositions to these problems. Oxcarbazepine has the advantage of being metabolized in a way that avoids an intermediate metabolite associated with some of the side effects of carbamazepine. Oxcarbazepine also has fewer drug interactions than carbamazepine. Lamotrigine tends to be well tolerated in terms of side effects. However, patients need to be particularly vigilant about the possibility of a skin rash during the initiation of therapy. This can be extremely severe and lead to Stevens-Johnson syndrome if unrecognized and if the medication is not discontinued immediately. This risk can be reduced by the use of low initial doses and slow titration. Lamotrigine must be started at lower initial doses when used as add-on therapy with valproic acid, because valproic acid inhibits lamotrigine metabolism and results in a substantially prolonged half-life. Phenytoin has a relatively long half-life and offers the advantage of once or twice daily dosing compared to two or three times daily dosing for many of the other drugs. However, phenytoin shows properties of nonlinear kinetics, such that small increases in phenytoin doses above a standard maintenance dose can precipitate marked side effects. This is one of the main causes of acute phenytoin toxicity. Long-term use of phenytoin is associated with untoward cosmetic effects (e.g., hirsutism, coarsening of facial features, gingival hypertrophy) and effects on bone metabolism. Due to these side effects, phenytoin is often avoided in young patients who are likely to require the drug for many years. Levetiracetam has the advantage of having no known drug-drug interactions, making it especially useful in the elderly and patients on other medications. However, a significant number of patients taking levetiracetam complain of irritability, anxiety, and other psychiatric symptoms. Topiramate can be used for both focal and generalized seizures. Similar to some of the other antiepileptic drugs, topiramate can cause significant psychomotor slowing and other cognitive problems. Additionally, it should not be used in patients at risk for the development of glaucoma or renal stones.
Valproic acid is an effective alternative for some patients with focal seizures, especially when the seizures generalize. Gastrointestinal side effects are fewer when using the delayed-release formulation (Depakote). Laboratory testing is required to monitor toxicity because valproic acid can rarely cause reversible bone marrow suppression and hepatotoxicity. This drug should generally be avoided in patients with preexisting bone marrow or liver disease. Irreversible, fatal hepatic failure appearing as an idiosyncratic rather than dose-related side effect is a relatively rare complication; its risk is highest in children <2 years old, especially those taking other antiepileptic drugs or with inborn errors of metabolism.
Zonisamide, tiagabine, gabapentin, lacosamide, and ezogabine are additional drugs currently used for the treatment of focal seizures with or without evolution into generalized seizures. Phenobarbital and other barbiturate compounds were commonly used in the past as first-line therapy for many forms of epilepsy. However, the barbiturates frequently cause sedation in adults, hyperactivity in children, and other more subtle cognitive changes; thus, their use should be limited to situations in which no other suitable treatment alternatives exist.
ANTIEPILEPTIC DRUG SELECTION FOR GENERALIZED SEIZURES Lamotrigine and valproic acid are currently considered the best initial choice for the treatment of primary generalized, tonic-clonic seizures. Topiramate, zonisamide, phenytoin, carbamazepine, and oxcarbazepine are suitable alternatives. Valproic acid is also particularly effective in absence, myoclonic, and atonic seizures. It is therefore the drug of choice in patients with generalized epilepsy syndromes having mixed seizure types. Importantly, carbamazepine, oxcarbazepine, and phenytoin can worsen certain types of generalized seizures, including absence, myoclonic, tonic, and atonic seizures. Ethosuximide is a particularly effective drug for the treatment of uncomplicated absence seizures, but it is not useful for tonic-clonic or focal seizures. Periodic monitoring of blood cell counts is required since ethosuximide rarely causes bone marrow suppression. Lamotrigine appears to be particularly effective in epilepsy syndromes with mixed, generalized seizure types such as JME and Lennox-Gastaut syndrome. Topiramate, zonisamide, and felbamate may have similar broad efficacy.
INITIATION AND MONITORING OF THERAPY
Because the response to any antiepileptic drug is unpredictable, patients should be carefully educated about the approach to therapy. The goal is to prevent seizures and minimize the side effects of treatment; determination of the optimal dose is often a matter of trial and error. This process may take months or longer if the baseline seizure frequency is low. Most antiepileptic drugs need to be introduced relatively slowly to minimize side effects. Patients should expect that minor side effects such as mild sedation, slight changes in cognition, or imbalance will typically resolve within a few days. Starting doses are usually the lowest value listed under the dosage column in Table 445-9. Subsequent increases should be made only after achieving a steady state with the previous dose (i.e., after an interval of five or more half-lives).
Monitoring of serum antiepileptic drug levels can be very useful for establishing the initial dosing schedule. However, the published therapeutic ranges of serum drug concentrations are only an approximate guide for determining the proper dose for a given patient. The key determinants are the clinical measures of seizure frequency and presence of side effects, not the laboratory values. Conventional assays of serum drug levels measure the total drug (i.e., both free and protein bound). However, it is the concentration of free drug that reflects extracellular levels in the brain and correlates best with efficacy. Thus, patients with decreased levels of serum proteins (e.g., decreased serum albumin due to impaired liver or renal function) may have an increased ratio of free to bound drug, yet the concentration of free drug may be adequate for seizure control. These patients may have a “subtherapeutic” drug level, but the dose should be changed only if seizures remain uncontrolled, not just to achieve a “therapeutic” level. It is also useful to monitor free drug levels in such patients. In practice, other than during the initiation or modification of therapy, monitoring of antiepileptic drug levels is most useful for documenting adherence.
If seizures continue despite gradual increases to the maximum tolerated dose and documented compliance, then it becomes necessary to switch to another antiepileptic drug. This is usually done by maintaining the patient on the first drug while a second drug is added. The dose of the second drug should be adjusted to decrease seizure frequency without causing toxicity. Once this is achieved, the first drug can be gradually withdrawn (usually over weeks unless there is significant toxicity). The dose of the second drug is then further optimized based on seizure response and side effects. Monotherapy should be the goal whenever possible.
WHEN TO DISCONTINUE THERAPY
Overall, about 70% of children and 60% of adults who have their seizures completely controlled with antiepileptic drugs can eventually discontinue therapy. The following patient profile yields the greatest chance of remaining seizure free after drug withdrawal: (1) complete medical control of seizures for 1–5 years; (2) single seizure type, either focal or generalized; (3) normal neurologic examination, including intelligence; and (4) normal EEG. The appropriate seizure-free interval is unknown and undoubtedly varies for different forms of epilepsy. However, it seems reasonable to attempt withdrawal of therapy after 2 years in a patient who meets all of the above criteria, is motivated to discontinue the medication, and clearly understands the potential risks and benefits. In most cases, it is preferable to reduce the dose of the drug gradually over 2–3 months. Most recurrences occur in the first 3 months after discontinuing therapy, and patients should be advised to avoid potentially dangerous situations such as driving or swimming during this period.
TREATMENT OF REFRACTORY EPILEPSY
Approximately one-third of patients with epilepsy do not respond to treatment with a single antiepileptic drug, and it becomes necessary to try a combination of drugs to control seizures. Patients who have focal epilepsy related to an underlying structural lesion or those with multiple seizure types and developmental delay are particularly likely to require multiple drugs. There are currently no clear guidelines for rational polypharmacy, although in theory a combination of drugs with different mechanisms of action may be most useful. In most cases, the initial combination therapy combines first-line drugs (i.e., carbamazepine, oxcarbazepine, lamotrigine, valproic acid, levetiracetam, and phenytoin). If these drugs are unsuccessful, then the addition of other drugs such as topiramate, zonisamide, lacosamide, or tiagabine is indicated. Patients with myoclonic seizures resistant to valproic acid may benefit from the addition of clonazepam or clobazam, and those with absence seizures may respond to a combination of valproic acid and ethosuximide. The same principles concerning the monitoring of therapeutic response, toxicity, and serum levels for monotherapy apply to polypharmacy, and potential drug interactions need to be recognized. If there is no improvement, a third drug can be added while the first two are maintained. If there is a response, the less effective or less well tolerated of the first two drugs should be gradually withdrawn.
SURGICAL TREATMENT OF REFRACTORY EPILEPSY
Approximately 20–30% of patients with epilepsy continue to have seizures despite efforts to find an effective combination of antiepileptic drugs. For some, surgery can be extremely effective in substantially reducing seizure frequency and even providing complete seizure control. Understanding the potential value of surgery is especially important when a patient’s seizures are not controlled with initial treatment, as such patients often do not respond to subsequent medication trials. Rather than submitting the patient to years of unsuccessful medical therapy and the psychosocial trauma and increased mortality associated with ongoing seizures, the patient should have an efficient but relatively brief attempt at medical therapy and then be referred for surgical evaluation.
The most common surgical procedure for patients with temporal lobe epilepsy involves resection of the anteromedial temporal lobe (temporal lobectomy) or a more limited removal of the underlying hippocampus and amygdala (amygdalohippocampectomy). Focal seizures arising from extratemporal regions may be abolished by a focal neocortical resection with precise removal of an identified lesion (lesionectomy). Localized neocortical resection without a clear lesion identified on MRI is also possible when other tests (e.g. MEG, PET, SPECT) implicate a focal cortical region as a seizure onset zone. When the cortical region cannot be removed, multiple subpial transection, which disrupts intracortical connections, is sometimes used to prevent seizure spread. Hemispherectomy or multilobar resection is useful for some patients with severe seizures due to hemispheric abnormalities such as hemimegalencephaly or other dysplastic abnormalities, and corpus callosotomy has been shown to be effective for disabling tonic or atonic seizures, usually when they are part of a mixed-seizure syndrome (e.g., Lennox-Gastaut syndrome).
Presurgical evaluation is designed to identify the functional and structural basis of the patient’s seizure disorder. Inpatient video-EEG monitoring is used to define the anatomic location of the seizure focus and to correlate the abnormal electrophysiologic activity with behavioral manifestations of the seizure. Routine scalp or scalp-sphenoidal recordings and a high-resolution MRI scan are usually sufficient for localization of the epileptogenic focus, especially when the findings are concordant. Functional imaging studies such as SPECT, PET, and MEG are adjunctive tests that may help to reveal or verify the localization of an apparent epileptogenic region. Once the presumed location of the seizure onset is identified, additional studies, including neuropsychological testing, the intracarotid amobarbital test (Wada test), and functional MRI may be used to assess language and memory localization and to determine the possible functional consequences of surgical removal of the epileptogenic region. In some cases, standard noninvasive evaluation is not sufficient to localize the seizure onset zone, and invasive electrophysiologic monitoring, such as implanted depth or subdural electrodes, is required for more definitive localization. The exact extent of the resection to be undertaken can also be determined by performing cortical mapping at the time of the surgical procedure, allowing for a tailored resection. This involves electrocorticographic recordings made with electrodes on the surface of the brain to identify the extent of epileptiform disturbances. If the region to be resected is within or near brain regions suspected of having sensorimotor or language function, electrical cortical stimulation mapping is performed on the awake patient to determine the function of cortical regions in question in order to avoid resection of so-called eloquent cortex and thereby minimize postsurgical deficits.
Advances in presurgical evaluation and microsurgical techniques have led to a steady increase in the success of epilepsy surgery. Clinically significant complications of surgery are <5%, and the use of functional mapping procedures has markedly reduced the neurologic sequelae due to removal or sectioning of brain tissue. For example, about 70% of patients treated with temporal lobectomy will become seizure free, and another 15–25% will have at least a 90% reduction in seizure frequency. Marked improvement is also usually seen in patients treated with hemispherectomy for catastrophic seizure disorders due to large hemispheric abnormalities. Postoperatively, patients generally need to remain on antiepileptic drug therapy, but the marked reduction of seizures following resective surgery can have a very beneficial effect on quality of life.
Not all medically refractory patients are suitable candidates for resective surgery. For example, some patients have seizures arising from more than one location, making the risk of ongoing seizures or potential harm from the surgery unacceptably high. Vagus nerve stimulation (VNS) has been used in some of these cases, although the results are limited and it is difficult to predict who will benefit. A new implantable device that can detect the onset of a seizure (in some instances before the seizure becomes clinically apparent) and deliver an electrical stimulation (Responsive NeuroStimulation) has recently been approved and may be of benefit in selected patients. Studies are currently evaluating the efficacy of stereotactic radiosurgery, laser thermoablation, and deep brain stimulation (DBS) as other options for surgical treatment of refractory epilepsy.
STATUS EPILEPTICUS
Status epilepticus refers to continuous seizures or repetitive, discrete seizures with impaired consciousness in the interictal period. Status epilepticus has numerous subtypes, including generalized convulsive status epilepticus (GCSE) (e.g., persistent, generalized electrographic seizures, coma, and tonic-clonic movements) and nonconvulsive status epilepticus (e.g., persistent absence seizures or focal seizures with confusion or partially impaired consciousness, and minimal motor abnormalities). The duration of seizure activity sufficient to meet the definition of status epilepticus has traditionally been specified as 15–30 min. However, a more practical definition is to consider status epilepticus as a situation in which the duration of seizures prompts the acute use of anticonvulsant therapy. For GCSE, this is typically when seizures last beyond 5 min.
GCSE is an emergency and must be treated immediately, because cardiorespiratory dysfunction, hyperthermia, and metabolic derangements can develop as a consequence of prolonged seizures, and these can lead to irreversible neuronal injury. Furthermore, CNS injury can occur even when the patient is paralyzed with neuromuscular blockade but continues to have electrographic seizures. The most common causes of GCSE are anticonvulsant withdrawal or noncompliance, metabolic disturbances, drug toxicity, CNS infection, CNS tumors, refractory epilepsy, and head trauma.
GCSE is obvious when the patient is having overt convulsions. However, after 30–45 min of uninterrupted seizures, the signs may become increasingly subtle. Patients may have mild clonic movements of only the fingers or fine, rapid movements of the eyes. There may be paroxysmal episodes of tachycardia, hypertension, and pupillary dilation. In such cases, the EEG may be the only method of establishing the diagnosis. Thus, if the patient stops having overt seizures, yet remains comatose, an EEG should be performed to rule out ongoing status epilepticus. This is obviously also essential when a patient with GCSE has been paralyzed with neuromuscular blockade in the process of protecting the airway.
The first steps in the management of a patient in GCSE are to attend to any acute cardiorespiratory problems or hyperthermia, perform a brief medical and neurologic examination, establish venous access, and send samples for laboratory studies to identify metabolic abnormalities. Anticonvulsant therapy should then begin without delay; a treatment approach is shown in Fig. 445-3.
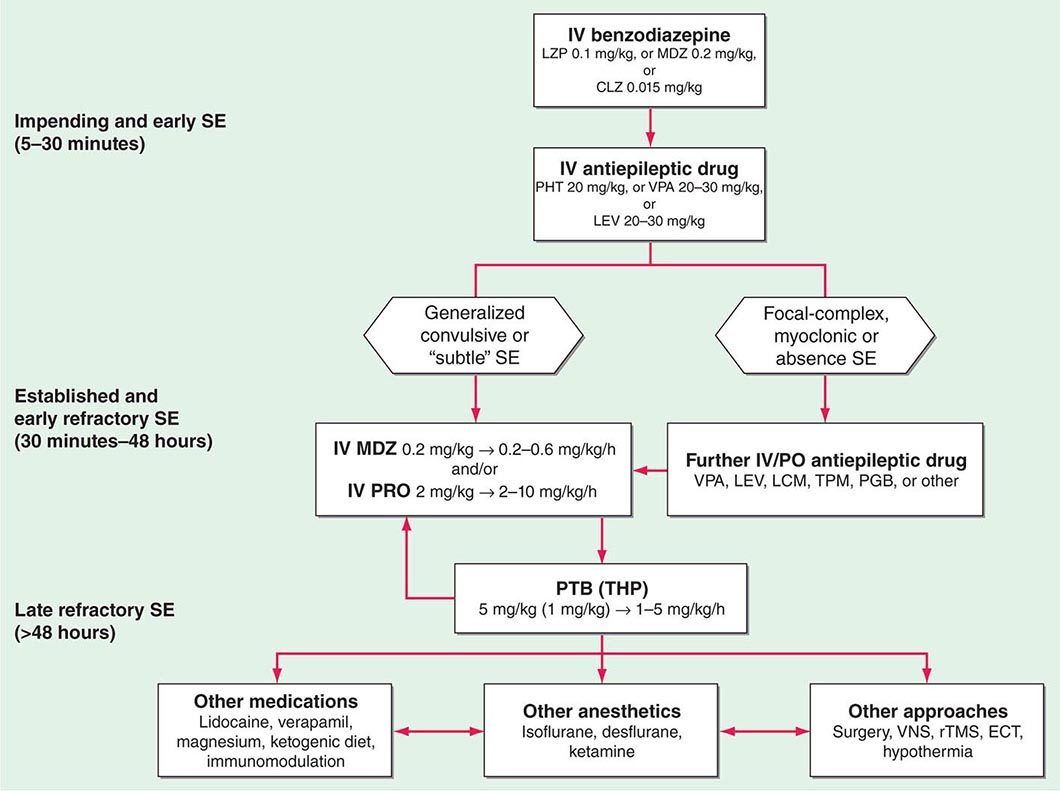
FIGURE 445-3 Pharmacologic treatment of generalized tonic-clonic status epilepticus (SE) in adults. CLZ, clonazepam; ECT, electroconvulsive therapy; LCM, lacosamide; LEV, levetiracetam; LZP, lorazepam; MDZ, midazolam; PGB, pregabalin; PHT, phenytoin or fos-phenytoin; PRO, propofol; PTB, pentobarbital; rTMS, repetitive transcranial magnetic stimulation; THP, thiopental; TPM, topiramate; VNS, vagus nerve stimulation; VPA, valproic acid. (From AO Rossetti, DH Lowenstein: Lancet Neurol 10:922, 2011.)
The treatment of nonconvulsive status epilepticus is thought to be less urgent than GCSE, because the ongoing seizures are not accompanied by the severe metabolic disturbances seen with GCSE. However, evidence suggests that nonconvulsive status epilepticus, especially that caused by ongoing, focal seizure activity, is associated with cellular injury in the region of the seizure focus; therefore this condition should be treated as promptly as possible using the general approach described for GCSE.
BEYOND SEIZURES: OTHER MANAGEMENT ISSUES
INTERICTAL BEHAVIOR
The adverse effects of epilepsy often go beyond clinical seizures, and the extent of these effects largely depends on the etiology of epilepsy, seizure frequency and severity, and side effects from antiepileptic therapy. Many epilepsy patients are completely normal between seizures and live highly successful and productive lives. In contrast, patients with seizures secondary to developmental abnormalities or acquired brain injury may have impaired cognitive function and other neurologic deficits. Frequent interictal EEG abnormalities are associated with subtle dysfunction of memory and attention. Patients with many seizures, especially those emanating from the temporal lobe, often note an impairment of short-term memory that may progress over time.
Patients with epilepsy are at risk of developing a variety of psychiatric problems, including depression, anxiety, and psychosis. This risk varies considerably depending on many factors, including the etiology, frequency, and severity of seizures and the patient’s age and previous personal or family history of psychiatric disorder. Depression occurs in ~20% of patients, and the incidence of suicide is higher in patients with epilepsy than in the general population. Depression should be treated through counseling or medication. The selective serotonin reuptake inhibitors (SSRIs) typically have minimal effect on seizures, whereas tricyclic antidepressants may lower the seizure threshold. Anxiety can be a seizure symptom, and anxious or psychotic behavior can occur during a postictal delirium. Postictal psychosis is a rare phenomenon that typically occurs after a period of increased seizure frequency. There is usually a brief lucid interval lasting up to a week, followed by days to weeks of agitated, psychotic behavior. The psychosis usually resolves spontaneously but frequently will require short-term treatment with antipsychotic or anxiolytic medications.
There is ongoing controversy as to whether some patients with epilepsy (especially temporal lobe epilepsy) have a stereotypical “interictal personality.” The predominant view is that atypical personality traits occur in diverse epilepsies (e.g., generalized and frontal lobe epilepsy) and may result from an underlying structural brain lesion, antiepileptic drug effects, and psychosocial factors related to suffering from a chronic disease, as well as the epilepsy itself.
MORTALITY OF EPILEPSY
Patients with epilepsy have a risk of death that is roughly two to three times greater than expected in a matched population without epilepsy. Most of the increased mortality is due to the underlying etiology of epilepsy (e.g., tumors or strokes in older adults). However, a significant number of patients die from accidents, status epilepticus, and a syndrome known as sudden unexpected death in epilepsy (SUDEP), which usually affects young people with convulsive seizures and tends to occur at night. The cause of SUDEP is unknown; it may result from brainstem-mediated effects of seizures on pulmonary, cardiac, and arousal functions. Recent studies suggest that, in some cases, a genetic mutation may be the cause of both epilepsy and a cardiac conduction defect that gives rise to sudden death.
PSYCHOSOCIAL ISSUES
There continues to be a cultural stigma about epilepsy, although it is slowly declining in societies with effective health education programs. Many patients with epilepsy harbor fears such as the fear of becoming mentally retarded or dying during a seizure. These issues need to be carefully addressed by educating the patient about epilepsy and by ensuring that family members, teachers, fellow employees, and other associates are equally well informed. A useful source of educational material is the Web site www.epilepsy.com.
EMPLOYMENT, DRIVING, AND OTHER ACTIVITIES
Many patients with epilepsy face difficulty in obtaining or maintaining employment, even when their seizures are well controlled. Federal and state legislation is designed to prevent employers from discriminating against patients with epilepsy, and patients should be encouraged to understand and claim their legal rights. Patients in these circumstances also benefit greatly from the assistance of health providers who act as strong patient advocates.
Loss of driving privileges is one of the most disruptive social consequences of epilepsy. Physicians should be very clear about local regulations concerning driving and epilepsy, because the laws vary considerably among states and countries. In all cases, it is the physician’s responsibility to warn patients of the danger imposed on themselves and others while driving if their seizures are uncontrolled (unless the seizures are not associated with impairment of consciousness or motor control). In general, most states allow patients to drive after a seizure-free interval (on or off medications) of between 3 months and 2 years.
Patients with incompletely controlled seizures must also contend with the risk of being in other situations where an impairment of consciousness or loss of motor control could lead to major injury or death. Thus, depending on the type and frequency of seizures, many patients need to be instructed to avoid working at heights or with machinery or to have someone close by for activities such as bathing and swimming.
SPECIAL ISSUES RELATED TO WOMEN AND EPILEPSY
CATAMENIAL EPILEPSY
Some women experience a marked increase in seizure frequency around the time of menses. This is believed to be mediated by either the effects of estrogen and progesterone on neuronal excitability or changes in antiepileptic drug levels due to altered protein binding or metabolism. Some patients may benefit from increases in antiepileptic drug dosages during menses. Natural progestins or intramuscular medroxyprogesterone may be of benefit to a subset of women.
PREGNANCY
Most women with epilepsy who become pregnant will have an uncomplicated gestation and deliver a normal baby. However, epilepsy poses some important risks to a pregnancy. Seizure frequency during pregnancy will remain unchanged in ~50% of women, increase in 30%, and decrease in 20%. Changes in seizure frequency are attributed to endocrine effects on the CNS, variations in antiepileptic drug pharmacokinetics (such as acceleration of hepatic drug metabolism or effects on plasma protein binding), and changes in medication compliance. It is useful to see patients at frequent intervals during pregnancy and monitor serum antiepileptic drug levels. Measurement of the unbound drug concentrations may be useful if there is an increase in seizure frequency or worsening of side effects of antiepileptic drugs.
The overall incidence of fetal abnormalities in children born to mothers with epilepsy is 5–6%, compared to 2–3% in healthy women. Part of the higher incidence is due to teratogenic effects of antiepileptic drugs, and the risk increases with the number of medications used (e.g., 10–20% risk of malformations with three drugs) and possibly with higher doses. A meta-analysis of published pregnancy registries and cohorts found that the most common malformations were defects in the cardiovascular and musculoskeletal system (1.4–1.8%). Valproic acid is strongly associated with an increased risk of adverse fetal outcomes (7–20%). Recent findings from a large pregnancy registry suggest that, other than topiramate, the newer antiepileptic drugs are far safer than valproic acid.
Because the potential harm of uncontrolled convulsive seizures on the mother and fetus is considered greater than the teratogenic effects of antiepileptic drugs, it is currently recommended that pregnant women be maintained on effective drug therapy. When possible, it seems prudent to have the patient on monotherapy at the lowest effective dose, especially during the first trimester. For some women, however, the type and frequency of their seizures may allow for them to safely wean off antiepileptic drugs prior to conception. Patients should also take folate (1–4 mg/d), because the antifolate effects of anticonvulsants are thought to play a role in the development of neural tube defects, although the benefits of this treatment remain unproved in this setting.
Enzyme-inducing drugs such as phenytoin, carbamazepine, oxcarbazepine, topiramate, phenobarbital, and primidone cause a transient and reversible deficiency of vitamin K–dependent clotting factors in ~50% of newborn infants. Although neonatal hemorrhage is uncommon, the mother should be treated with oral vitamin K (20 mg/d, phylloquinone) in the last 2 weeks of pregnancy, and the infant should receive intramuscular vitamin K (1 mg) at birth.
CONTRACEPTION
Special care should be taken when prescribing antiepileptic medications for women who are taking oral contraceptive agents. Drugs such as carbamazepine, phenytoin, phenobarbital, and topiramate can significantly decrease the efficacy of oral contraceptives via enzyme induction and other mechanisms. Patients should be advised to consider alternative forms of contraception, or their contraceptive medications should be modified to offset the effects of the antiepileptic medications.
BREAST-FEEDING
Antiepileptic medications are excreted into breast milk to a variable degree. The ratio of drug concentration in breast milk relative to serum ranges from ~5% (valproic acid) to 300% (levetiracetam). Given the overall benefits of breast-feeding and the lack of evidence for long-term harm to the infant by being exposed to antiepileptic drugs, mothers with epilepsy can be encouraged to breast-feed. This should be reconsidered, however, if there is any evidence of drug effects on the infant such as lethargy or poor feeding.
446 |
Cerebrovascular Diseases |
 Cerebrovascular diseases include some of the most common and devastating disorders: ischemic stroke and hemorrhagic stroke. Stroke is the second leading cause of death worldwide, causing 6.2 million deaths in 2011, and is double the rate of heart disease in China. Strokes cause ~200,000 deaths each year in the United States and are a major cause of disability. The incidence of cerebrovascular diseases increases with age, and the number of strokes is projected to increase as the elderly population grows, with a doubling in stroke deaths in the United States by 2030. A stroke, or cerebrovascular accident, is defined as an abrupt onset of a neurologic deficit that is attributable to a focal vascular cause. Thus, the definition of stroke is clinical, and laboratory studies including brain imaging are used to support the diagnosis. The clinical manifestations of stroke are highly variable because of the complex anatomy of the brain and its vasculature. Cerebral ischemia is caused by a reduction in blood flow that lasts longer than several seconds. Neurologic symptoms are manifest within seconds because neurons lack glycogen, so energy failure is rapid. If the cessation of flow lasts for more than a few minutes, infarction or death of brain tissue results. When blood flow is quickly restored, brain tissue can recover fully and the patient’s symptoms are only transient: this is called a transient ischemic attack (TIA). The definition of TIA requires that all neurologic signs and symptoms resolve within 24 h without evidence of brain infarction on brain imaging. Stroke has occurred if the neurologic signs and symptoms last for >24 h or brain infarction is demonstrated. A generalized reduction in cerebral blood flow due to systemic hypotension (e.g., cardiac arrhythmia, myocardial infarction, or hemorrhagic shock) usually produces syncope (Chap. 27). If low cerebral blood flow persists for a longer duration, then infarction in the border zones between the major cerebral artery distributions may develop. In more severe instances, global hypoxia-ischemia causes widespread brain injury; the constellation of cognitive sequelae that ensues is called hypoxic-ischemic encephalopathy (Chap. 330). Focal ischemia or infarction, conversely, is usually caused by thrombosis of the cerebral vessels themselves or by emboli from a proximal arterial source or the heart. Intracranial hemorrhage is caused by bleeding directly into or around the brain; it produces neurologic symptoms by producing a mass effect on neural structures, from the toxic effects of blood itself, or by increasing intracranial pressure.
Cerebrovascular diseases include some of the most common and devastating disorders: ischemic stroke and hemorrhagic stroke. Stroke is the second leading cause of death worldwide, causing 6.2 million deaths in 2011, and is double the rate of heart disease in China. Strokes cause ~200,000 deaths each year in the United States and are a major cause of disability. The incidence of cerebrovascular diseases increases with age, and the number of strokes is projected to increase as the elderly population grows, with a doubling in stroke deaths in the United States by 2030. A stroke, or cerebrovascular accident, is defined as an abrupt onset of a neurologic deficit that is attributable to a focal vascular cause. Thus, the definition of stroke is clinical, and laboratory studies including brain imaging are used to support the diagnosis. The clinical manifestations of stroke are highly variable because of the complex anatomy of the brain and its vasculature. Cerebral ischemia is caused by a reduction in blood flow that lasts longer than several seconds. Neurologic symptoms are manifest within seconds because neurons lack glycogen, so energy failure is rapid. If the cessation of flow lasts for more than a few minutes, infarction or death of brain tissue results. When blood flow is quickly restored, brain tissue can recover fully and the patient’s symptoms are only transient: this is called a transient ischemic attack (TIA). The definition of TIA requires that all neurologic signs and symptoms resolve within 24 h without evidence of brain infarction on brain imaging. Stroke has occurred if the neurologic signs and symptoms last for >24 h or brain infarction is demonstrated. A generalized reduction in cerebral blood flow due to systemic hypotension (e.g., cardiac arrhythmia, myocardial infarction, or hemorrhagic shock) usually produces syncope (Chap. 27). If low cerebral blood flow persists for a longer duration, then infarction in the border zones between the major cerebral artery distributions may develop. In more severe instances, global hypoxia-ischemia causes widespread brain injury; the constellation of cognitive sequelae that ensues is called hypoxic-ischemic encephalopathy (Chap. 330). Focal ischemia or infarction, conversely, is usually caused by thrombosis of the cerebral vessels themselves or by emboli from a proximal arterial source or the heart. Intracranial hemorrhage is caused by bleeding directly into or around the brain; it produces neurologic symptoms by producing a mass effect on neural structures, from the toxic effects of blood itself, or by increasing intracranial pressure.
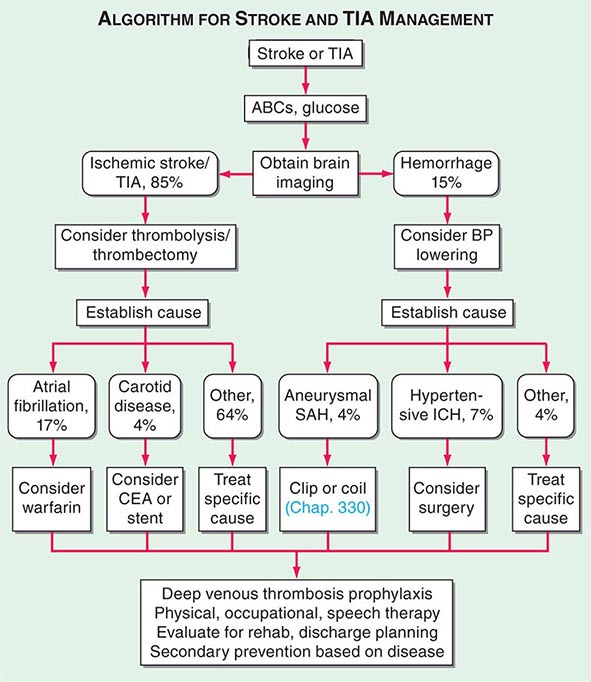
FIGURE 446-1 Medical management of stroke and TIA. Rounded boxes are diagnoses; rectangles are interventions. Numbers are percentages of stroke overall. ABCs, airway, breathing, circulation; BP, blood pressure; CEA, carotid endarterectomy; ICH, intracerebral hemorrhage; SAH, subarachnoid hemorrhage; TIA, transient ischemic attack.
ISCHEMIC STROKE
PATHOPHYSIOLOGY OF ISCHEMIC STROKE
Acute occlusion of an intracranial vessel causes reduction in blood flow to the brain region it supplies. The magnitude of flow reduction is a function of collateral blood flow, and this depends on individual vascular anatomy (which may be altered by disease), the site of occlusion, and systemic blood pressure. A decrease in cerebral blood flow to zero causes death of brain tissue within 4–10 min; values <16–18 mL/100 g tissue per minute cause infarction within an hour; and values <20 mL/100 g tissue per minute cause ischemia without infarction unless prolonged for several hours or days. If blood flow is restored to ischemic tissue before significant infarction develops, the patient may experience only transient symptoms, and the clinical syndrome is called a TIA. Another important concept is the ischemic penumbra, defined as the ischemic but reversibly dysfunctional tissue surrounding a core area of infarction. The penumbra can be imaged by perfusion-diffusion imaging using MRI or CT (see below and Figs. 446-15 and 446-16). The ischemic penumbra will eventually progress to infarction if no change in flow occurs, and hence saving the ischemic penumbra is the goal of revascularization therapies.
Focal cerebral infarction occurs via two distinct pathways (Fig. 446-2): (1) a necrotic pathway in which cellular cytoskeletal breakdown is rapid, due principally to energy failure of the cell; and (2) an apoptotic pathway in which cells become programmed to die. Ischemia produces necrosis by starving neurons of glucose and oxygen, which in turn results in failure of mitochondria to produce ATP. Without ATP, membrane ion pumps stop functioning and neurons depolarize, allowing intracellular calcium to rise. Cellular depolarization also causes glutamate release from synaptic terminals; excess extracellular glutamate produces neurotoxicity by activating postsynaptic glutamate receptors that increase neuronal calcium influx. Free radicals are produced by degradation of membrane lipids and mitochondrial dysfunction. Free radicals cause catalytic destruction of membranes and likely damage other vital functions of cells. Lesser degrees of ischemia, as are seen within the ischemic penumbra, favor apoptotic cellular death causing cells to die days to weeks later. Fever dramatically worsens brain injury during ischemia, as does hyperglycemia (glucose >11.1 mmol/L [200 mg/dL]), so it is reasonable to suppress fever and prevent hyperglycemia as much as possible. The value of induced mild hypothermia to improve stroke outcomes is the subject of continuing clinical research.
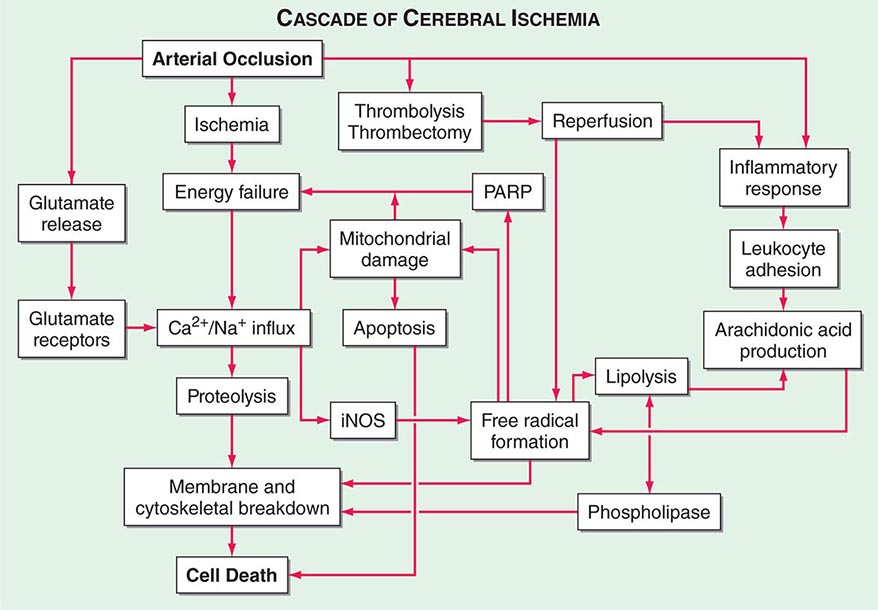
FIGURE 446-2 Major steps in the cascade of cerebral ischemia. See text for details. iNOS, inducible nitric oxide synthase; PARP, poly-A ribose polymerase.
|
TREATMENT |
ACUTE ISCHEMIC STROKE |
After the clinical diagnosis of stroke is made, an orderly process of evaluation and treatment should follow (Fig. 446-1). The first goal is to prevent or reverse brain injury. Attend to the patient’s airway, breathing, and circulation (ABCs), and treat hypoglycemia or hyperglycemia if identified. Perform an emergency noncontrast head CT scan to differentiate between ischemic stroke and hemorrhagic stroke; there are no reliable clinical findings that conclusively separate ischemia from hemorrhage, although a more depressed level of consciousness, higher initial blood pressure, or worsening of symptoms after onset favor hemorrhage, and a deficit that is maximal at onset, or remits, suggests ischemia. Treatments designed to reverse or lessen the amount of tissue infarction and improve clinical outcome fall within six categories: (1) medical support, (2) IV thrombolysis, (3) endovascular revascularization, (4) antithrombotic treatment, (5) neuroprotection, and (6) stroke centers and rehabilitation.
MEDICAL SUPPORT
When ischemic stroke occurs, the immediate goal is to optimize cerebral perfusion in the surrounding ischemic penumbra. Attention is also directed toward preventing the common complications of bedridden patients—infections (pneumonia, urinary, and skin) and deep venous thrombosis (DVT) with pulmonary embolism. Subcutaneous heparin (unfractionated and low-molecular-weight) is safe and can be used concomitantly. Use of pneumatic compression stockings is of proven benefit in reducing risk of DVT and is a safe alternative to heparin.
Because collateral blood flow within the ischemic brain may be blood pressure dependent, there is controversy about whether blood pressure should be lowered acutely. Blood pressure should be lowered if there is malignant hypertension (Chap. 298) or concomitant myocardial ischemia, or if blood pressure is >185/110 mmHg and thrombolytic therapy is anticipated. When faced with the competing demands of myocardium and brain, lowering the heart rate with a β1-adrenergic blocker (such as esmolol) can be a first step to decrease cardiac work and maintain blood pressure. Routine lowering of blood pressure has been found to worsen outcomes. Fever is detrimental and should be treated with antipyretics and surface cooling. Serum glucose should be monitored and kept at <10.0 mmol/L (180 mg/dL) using an insulin infusion if necessary.
Between 5 and 10% of patients develop enough cerebral edema to cause obtundation or brain herniation. Edema peaks on the second or third day but can cause mass effect for ~10 days. The larger the infarct, the greater the likelihood that clinically significant edema will develop. Water restriction and IV mannitol may be used to raise the serum osmolarity, but hypovolemia should be avoided because this may contribute to hypotension and worsening infarction. Combined analysis of three randomized European trials of hemicraniectomy (craniotomy and temporary removal of part of the skull) shows that hemicraniectomy markedly reduces mortality, and the clinical outcomes of survivors are acceptable. The size of the diffusion-weighted imaging volume of brain infarction during the acute stroke is a predictor of deterioration requiring hemicraniectomy.
Special vigilance is warranted for patients with cerebellar infarction. These strokes may mimic labyrinthitis because of prominent vertigo and vomiting; the presence of head or neck pain should alert the physician to consider cerebellar stroke from vertebral artery dissection. Even small amounts of cerebellar edema can acutely increase intracranial pressure (ICP) by obstructing cerebrospinal fluid (CSF) flow leading to hydrocephalus or by directly compressing the brainstem. The resulting brainstem compression can manifest as coma and respiratory arrest and require emergency surgical decompression. Prophylactic suboccipital decompression of large cerebellar infarcts before brainstem compression, although not tested rigorously in a clinical trial, is practiced at most stroke centers.
INTRAVENOUS THROMBOLYSIS
The National Institute of Neurological Disorders and Stroke (NINDS) rtPA Stroke Study showed a clear benefit for IV rtPA in selected patients with acute stroke. The NINDS study used IV rtPA (0.9 mg/kg to a 90-mg maximum; 10% as a bolus, then the remainder over 60 min) versus placebo in ischemic stroke within 3 h of onset. One-half of the patients were treated within 90 min. Symptomatic intracranial hemorrhage occurred in 6.4% of patients on rtPA and 0.6% on placebo. In the rTPA group, there was a significant 12% absolute increase in the number of patients with only minimal disability (32% on placebo and 44% on rtPA) and a nonsignificant 4% reduction in mortality (21% on placebo and 17% on rtPA). Thus, despite an increased incidence of symptomatic intracranial hemorrhage, treatment with IV rtPA within 3 h of the onset of ischemic stroke improved clinical outcome.
Three subsequent trials of IV rtPA did not confirm this benefit, perhaps because of the dose of rtPA used, the timing of its delivery, and small sample size. When data from all randomized IV rtPA trails were combined, however, efficacy was confirmed in the <3-h time window, and efficacy likely extended to 4.5 h and possibly to 6 h. Based on these combined results, the European Cooperative Acute Stroke Study (ECASS) III explored the safety and efficacy of rtPA in the 3- to 4.5-h time window. Unlike the NINDS study, patients older than 80 years of age and diabetic patients with a previous stroke were excluded. In this 821-patient randomized study, efficacy was again confirmed, although the treatment effect was less robust than in the 0- to 3-h time window. In the rtPA group, 52.4% of patients achieved a good outcome at 90 days, compared to 45.2% of the placebo group (odds ratio [OR] 1.34, p = .04). The symptomatic intracranial hemorrhage rate was 2.4% in the rtPA group and 0.2% in the placebo group (p = .008).
Based on these data, rtPA is approved in the 3- to 4.5-h window in Europe and Canada, but is still only approved for 0–3 h in the United States and Canada. Use of IV tPA is now considered a central component of primary stroke centers (see below). It represents the first treatment proven to improve clinical outcomes in ischemic stroke and is cost-effective and cost-saving. Advanced neuroimaging techniques (see neuroimaging section below) may help to select patients beyond the 4.5-h window who will benefit from thrombolysis, but this is currently investigational. The time of stroke onset is defined as the time the patient’s symptoms were witnessed to begin or the time the patient was last seen as normal. Patients who awaken with stroke have the onset defined as when they went to bed. Table 446-1 summarizes eligibility criteria and instructions for administration of IV rtPA.
|
ADMINISTRATION OF INTRAVENOUS RECOMBINANT TISSUE PLASMINOGEN ACTIVATOR (rtPA) FOR ACUTE ISCHEMIC STROKE (AIS)a |
ENDOVASCULAR REVASCULARIZATION
Ischemic stroke from large-vessel intracranial occlusion results in high rates of mortality and morbidity. Occlusions in such large vessels (middle cerebral artery [MCA], intracranial internal carotid artery, and the basilar artery) generally involve a large clot volume and often fail to open with IV rtPA alone. Therefore, there is growing interest in using thrombolytics via an intraarterial route to increase the concentration of drug at the clot and minimize systemic bleeding complications. The Prolyse in Acute Cerebral Thromboembolism (PROACT) II trial found benefit for intraarterial prourokinase in acute MCA occlusions up to the sixth hour following onset of stroke. Intraarterial treatment of basilar artery occlusions may also be beneficial for selected patients. Intraarterial administration of a thrombolytic agent for acute ischemic stroke (AIS) is not approved by the U.S. Food and Drug Administration (FDA); however, many stroke centers offer this treatment based on these data.
Endovascular mechanical thrombectomy has been studied as an alternative or adjunctive treatment of acute stroke in patients who are ineligible for, or have contraindications to, thrombolytics or in those who failed to achieve vascular recanalization with IV thrombolytics (see Fig. 446-15). The Mechanical Embolus Removal in Cerebral Ischemia (MERCI) and multi-MERCI single-arm trials found that an endovascular thrombectomy device restored patency of occluded intracranial vessels within 8 h of ischemic stroke symptoms compared with a historical control group. Recanalization of the target vessel occurred in 48–58% of treated patients and in 60–69% of patients after use of adjuvant endovascular methods, and successful recanalization at 90 days correlated well with favorable outcomes. Based on these nonrandomized data, the FDA approved this device as the first device for revascularization of occluded vessels in AIS even if the patient has been given rtPA and that therapy has failed. The Penumbra Pivotal Stroke trial tested another mechanical device that showed even higher rates of recanalization and led to FDA clearance of the tested device as well. More recently, two Stentriever devices (nondetachable stents) were shown to significantly improve vascular recanalization compared to the first approved MERCI device, approaching recanalization rates of 90% in most large intracranial vessels.
In 2013, three randomized endovascular trials with nonendovascular controls found no benefits to endovascular therapy. The largest was the Interventional Management of Stroke III trial that randomized 656 AIS patients within 3 h of onset to IV rtPA (0.9 mg/kg) alone versus IV rtPA (0.6 mg/kg) followed by endovascular adjuvant treatment with IA rtPA, or endovascular thrombectomy as soon as possible. Outcomes between these groups were not significantly different, and there were more complications (groin bleeding chiefly) in the endovascular group. The SYNTHESIS trial based in Italy randomized 363 patients to IV rtPA versus intraarterial rtPA for patients within 3 h of stroke onset. No differences were found between the groups at 90 days. These two relatively large trials indicate that endovascular therapy using principally intraarterial rtPA is not better than IV therapy, but many questions remain. Relatively few patients received mechanical clot retraction therapies, and those who did received what we now know were inferior devices. Trials assessing more efficacious thrombectomy devices are currently ongoing.
Because use of endovascular devices in combination with rtPA appears relatively safe, some centers continue to offer endovascular therapy. This applies to patients who are not eligible for IV rtPA (recent surgery, stroke following cardiac catheterization, etc.), and some continue to use thrombectomy because of perceived better outcomes in patients with more effective devices. Comprehensive stroke centers are now obtaining credentialing to offer this therapy in distinction to primary stroke centers that offer only IV rtPA.
ANTITHROMBOTIC TREATMENT
Platelet Inhibition Aspirin is the only antiplatelet agent that has been proven effective for the acute treatment of ischemic stroke; there are several antiplatelet agents proven for the secondary prevention of stroke (see below). Two large trials, the International Stroke Trial (IST) and the Chinese Acute Stroke Trial (CAST), found that the use of aspirin within 48 h of stroke onset reduced both stroke recurrence risk and mortality minimally. Among 19,435 patients in IST, those allocated to aspirin, 300 mg/d, had slightly fewer deaths within 14 days (9.0 vs 9.4%), significantly fewer recurrent ischemic strokes (2.8 vs 3.9%), no excess of hemorrhagic strokes (0.9 vs 0.8%), and a trend toward a reduction in death or dependence at 6 months (61.2 vs 63.5%). In CAST, 21,106 patients with ischemic stroke received 160 mg/d of aspirin or a placebo for up to 4 weeks. There were very small reductions in the aspirin group in early mortality (3.3 vs 3.9%), recurrent ischemic strokes (1.6 vs 2.1%), and dependency at discharge or death (30.5 vs 31.6%). These trials demonstrate that the use of aspirin in the treatment of AIS is safe and produces a small net benefit. For every 1000 acute strokes treated with aspirin, about 9 deaths or nonfatal stroke recurrences will be prevented in the first few weeks and ~13 fewer patients will be dead or dependent at 6 months.
Clopidogrel is being tested as a way to prevent stroke following TIA and minor ischemic stroke (see below).
Anticoagulation Numerous clinical trials have failed to demonstrate any benefit of anticoagulation in the primary treatment of atherothrombotic cerebral ischemia. Several trials have investigated antiplatelet versus anticoagulant medications given within 12–24 h of the initial event. The U.S. Trial of Organon 10172 in Acute Stroke Treatment (TOAST), an investigational low-molecular-weight heparin (LMWH), failed to show any benefit over aspirin. Use of SC unfractionated heparin versus aspirin was tested in IST. Heparin given SC afforded no additional benefit over aspirin and increased bleeding rates. Several trials of LMWHs have also shown no consistent benefit in AIS. Furthermore, trials generally have shown an excess risk of brain and systemic hemorrhage with acute anticoagulation. A recent meta-analysis of all forms of heparin found no benefit for acute stroke patients at high or low risk of thrombotic events. Therefore, trials do not support the use of heparin or other anticoagulants for patients with atherothrombotic stroke.
NEUROPROTECTION
Neuroprotection is the concept of providing a treatment that prolongs the brain’s tolerance to ischemia. Drugs that block the excitatory amino acid pathways have been shown to protect neurons and glia in animals, but despite multiple human trials, they have not yet been proven to be beneficial. Hypothermia is a powerful neuroprotective treatment in patients with cardiac arrest (Chap. 330) and is neuroprotective in animal models of stroke, but it has not been adequately studied in patients with ischemic stroke and is associated with an increase in pneumonia rates that could adversely impact stroke outcomes.
STROKE CENTERS AND REHABILITATION
Patient care in stroke units followed by rehabilitation services improves neurologic outcomes and reduces mortality. Use of clinical pathways and staff dedicated to the stroke patient can improve care. This includes use of standardized stroke order sets. Stroke teams that provide emergency 24-h evaluation of acute stroke patients for acute medical management and consideration of thrombolysis or endovascular treatments are essential components of primary and comprehensive stroke centers, respectively.
Proper rehabilitation of the stroke patient includes early physical, occupational, and speech therapy. It is directed toward educating the patient and family about the patient’s neurologic deficit, preventing the complications of immobility (e.g., pneumonia, DVT and pulmonary embolism, pressure sores of the skin, and muscle contractures), and providing encouragement and instruction in overcoming the deficit. Use of pneumatic compression stockings is of proven benefit in reducing risk of DVT and is a safe alternative to heparin. The goal of rehabilitation is to return the patient to home and to maximize recovery by providing a safe, progressive regimen suited to the individual patient. Additionally, the use of constrained movement therapy (immobilizing the unaffected side) has been shown to improve hemiparesis following stroke, even years after the stroke, suggesting that physical therapy can recruit unused neural pathways. Newer robotic therapies appear promising as well. The human nervous system is more adaptable than previously thought, and developing physical and pharmacologic strategies to enhance long-term neural recovery is an active area of research.
[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]
STEM CELLS AND TRANSPLANTATION
The nervous system is traditionally considered to be a nonmitotic organ, in particular with respect to neurons. These concepts have been challenged by the finding that neural progenitor or stem cells exist in the adult CNS that are capable of differentiation, migration over long distances, and extensive axonal arborization and synapse formation with appropriate targets. These capabilities also indicate that the repertoire of factors required for growth, survival, differentiation, and migration of these cells exists in the mature nervous system. In rodents, neural stem cells, defined as progenitor cells capable of differentiating into mature cells of neural or glial lineage, have been experimentally propagated from fetal CNS and neuroectodermal tissues and also from adult germinal matrix and ependyma regions. Human fetal CNS tissue is also capable of differentiation into cells with neuronal, astrocyte, and oligodendrocyte morphology when cultured in the presence of growth factors.
Once the repertoire of signals required for cell type specification is better understood, differentiation into specific neural or glial subpopulations can be directed in vitro; such cells could also be engineered to express therapeutic molecules. Another promising approach is to use growth factors, such as BDNF, to stimulate endogenous stem cells to proliferate and migrate to areas of neuronal damage.
A major advance has been the development of induced pluripotent stem cells. Using this technique, adult somatic cells such as skin fibroblasts are treated with four pluripotency factors (SOX2, KLF4, cMYC, and Oct4), and this generates induced pluripotent stem cells (iPSCs). These adult-derived stem cells sidestep the ethical issues of using stem cells derived from human embryos. The development of these cells has tremendous promise for both studying disease mechanisms and testing therapeutics. As yet there is no consensus on the best way to generate and differentiate the iPSCs; however, techniques to avoid using viral vectors and use of Cre-lox systems to remove reprogramming factors result in a better match of gene expression profiles with those of embryonic stem cells. Over the years, the field of directed differentiation has used three main strategies to specify neural lineages from human pluripotent stem cells. These strategies are embryoid body formation, coculture on neural-inducing feeders, and direct neural induction. Thus far, iPSCs have been made from patients with all of the major human neurodegenerative diseases, and studies using them are under way.
Although stem cells hold tremendous promise for the treatment of debilitating neurologic diseases, such as Parkinson’s disease and spinal cord injury, it should be emphasized that medical application is in its infancy. Major obstacles are the generation of position- and neurotransmitter-defined subtypes of neurons and their isolation as pure populations of the desired cells. This is crucial to avoid persistence of undifferentiated embryonic stem (ES) cells, which can generate tumors. The establishment of appropriate neural connections and afferent control is also critical. For instance, human ES motor neurons will need to be introduced at multiple segments in the neuraxis, and then their axons will need to regenerate from the spinal cord to distal musculature.
Experimental transplantation of human fetal dopaminergic neurons in patients with Parkinson’s disease has shown that these transplanted cells can survive within the host striatum; however, some patients developed disabling dyskinesias, and this approach is no longer in clinical development. The possibility that iPSCs will be used in Parkinson’s disease was strengthened by studies showing that they can be differentiated into dopaminergic neurons. The dopaminergic neurons were then shown to rescue the parkinsonian phenotype in a MPTP-induced primate model with excellent dopaminergic neuron survival function and lack of neural overgrowth. The correction of tau mutations in iPSC-derived neurons has been shown to reverse the toxic phenotype in dendrite retraction and cell death.
Another new use for iPSCs is to screen drugs as potential treatments for neurodegenerative and other diseases. The feasibility of this has been shown using iPSC-induced macrophages from patients with Gaucher’s disease, and verifying the efficacy of protein chaperones in these cells as a means of stabilizing the mutant glucocerebrosidase and increasing its activity and the duration of its effects. Other approaches are to attempt to reduce expression of proteins, such as amyloid, tau, and α-synuclein, implicated in the pathogenesis of neurodegenerative diseases. One difficulty has been that reprogramming cells to iPSCs resets their identity back to an embryonic age, which is a hurdle in modeling of late-onset diseases. One approach to this has been to express a fragment of the mutated gene, such as a portion of lamin A, which causes premature aging in progeria. This approach showed that dendrite degeneration and progressive loss of tyrosine hydroxylase expression, as well as enlarged mitochondria and Lewy body precursor inclusions, were induced in iPSC-derived dopaminergic neurons with progerin-induced aging.
Studies of transplantation for patients with Huntington’s disease have also reported encouraging, although very preliminary, results. OPCs transplanted into mice with a dysmyelinating disorder effectively migrated in the new environment, interacted with axons, and mediated myelination; such experiments raise hope that similar transplantation strategies may be feasible in human disorders of myelin such as MS. The promise of stem cells for treatment of both neurodegenerative diseases and neural injury is great, but development has been slowed by unresolved concerns over safety (including the theoretical risk of malignant transformation of transplanted cells), ethics (particularly with respect to use of fetal tissue), and efficacy.
In developing brain, the extracellular matrix provides stimulatory and inhibitory signals that promote neuronal migration, neurite outgrowth, and axonal extension. After neuronal damage, reexpression of inhibitory molecules such as chondroitin sulfate proteoglycans may prevent tissue regeneration. Chondroitinase degraded these inhibitory molecules and enhanced axonal regeneration and motor recovery in a rat model of spinal cord injury. Several myelin proteins, specifically Nogo, oligodendrocyte myelin glycoprotein (OMGP), and myelin-associated glycoprotein (MAG), may also interfere with axon regeneration. Sialidase, which cleaves one class of receptors for MAG, enhances axonal outgrowth. Antibodies against Nogo promote regeneration after experimental focal ischemia or spinal cord injury. Nogo, OMGP, and MAG all bind to the same neural receptor, the Nogo receptor, which mediates its inhibitory function via the p75 neurotrophin receptor signaling.
CELL DEATH: EXCITOTOXICITY AND APOPTOSIS
Excitotoxicity refers to neuronal cell death caused by activation of excitatory amino acid receptors (Fig. 444e-4). Compelling evidence for a role of excitotoxicity, especially in ischemic neuronal injury, is derived from experiments in animal models. Experimental models of stroke are associated with increased extracellular concentrations of the excitatory amino acid neurotransmitter glutamate, and neuronal damage is attenuated by denervation of glutamate-containing neurons or the administration of glutamate receptor antagonists. The distribution of cells sensitive to ischemia corresponds closely with that of N-methyl-D-aspartate (NMDA) receptors (except for cerebellar Purkinje cells, which are vulnerable to hypoxia-ischemia but lack NMDA receptors); and competitive and noncompetitive NMDA antagonists are effective in preventing focal ischemia. In global cerebral ischemia, non-NMDA receptors (kainic acid and α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate [AMPA]) are activated, and antagonists to these receptors are protective. Experimental brain damage induced by hypoglycemia is also attenuated by NMDA antagonists.
FIGURE 444e-4 Neurodegeneration caused by prions. A. In sporadic neurodegenerative diseases (NDs), wild-type (Wt) prions multiply through self-propagating cycles of posttranslational modification, during which the precursor protein (green circle) is converted into the prion form (red square), which generally is high in β-sheet content. Pathogenic prions are most toxic as oligomers and less toxic after polymerization into amyloid fibrils. The small polygons (blue) represent proteolytic cleavage products of the prion. Depending on the protein, the fibrils coalesce into Aβ amyloid plaques in Alzheimer’s disease (AD), neurofibrillary tangles in AD and Pick’s disease, or Lewy bodies in Parkinson’s disease and Lewy body dementia. Drug targets for the development of therapeutics include: (1) lowering the precursor protein, (2) inhibiting prion formation, and (3) enhancing prion clearance. B. Late-onset heritable neurodegeneration argues for two discrete events: The (i) first event is the synthesis of mutant precursor protein (green circle), and the (ii) second event is the age-dependent formation of mutant prions (red square). The highlighted yellow bar in the DNA structure represents mutation of a base pair within an exon, and the small yellow circles signify the corresponding mutant amino acid substitution. Green arrows represent a normal process; red arrows, a pathogenic process; and blue arrows, a process that is known to occur but unknown whether it is normal or pathogenic. (Micrographs prepared by Stephen J. DeArmond. Reprinted with permission from SB Prusiner: Biology and genetics of prions causing neurodegeneration. Annu Rev Genet 47:601, 2013.)
Excitotoxicity is not a single event but rather a cascade of cell injury. Excitotoxicity causes influx of calcium into cells, and much of the calcium is sequestered in mitochondria rather than in the cytoplasm. Increased cytoplasmic calcium causes metabolic dysfunction and free radical generation; activates protein kinases, phospholipases, nitric oxide synthase, proteases, and endonucleases; and inhibits protein synthesis. Activation of nitric oxide synthase generates nitric oxide (NO•), which can react with superoxide (O•2) to generate peroxynitrite (ONOO–), which may play a direct role in neuronal injury. Another critical pathway is activation of poly-ADP-ribose polymerase, which occurs in response to free radical–mediated DNA damage. Experimentally, mice with knockout mutations of neuronal nitric oxide synthase or poly-ADP-ribose polymerase, or those that overexpress superoxide dismutase, are resistant to focal ischemia.
Another aspect of excitotoxicity is that it has been demonstrated that stimulation of extrasynaptic NMDA receptors mediates cell death, where as stimulation of synaptic receptors is protective. This has been shown to play a role in excitotoxicity in transgenic mouse models of Huntington’s disease, in which using low-dose memantine to selectively block the extrasynaptic receptors is beneficial.
Although excitotoxicity is clearly implicated in the pathogenesis of cell death in stroke, to date treatment with NMDA antagonists has not proven to be clinically useful. One approach has been to use an inhibitor of the postsynaptic density-95 protein that uncouples NMDA receptors from neurotoxic pathways, including the generation of nitric oxide. This approach was effective in a primate stroke model and in a phase 2 clinical trial of stroke associated with endovascular repair of cerebral aneurysms. Transient receptor potentials (TRPs) are calcium channels that are activated by oxidative stress in parallel with excitotoxic signal pathways. In addition, glutamate-independent pathways of calcium influx via acid-sensing ion channels have been identified. These channels transport calcium in the setting of acidosis and substrate depletion, and pharmacologic blockade of these channels markedly attenuates stroke injury. These channels offer a potential new therapeutic target for stroke.
Apoptosis, or programmed cell death, plays an important role in both physiologic and pathologic conditions. During embryogenesis, apoptotic pathways operate to destroy neurons that fail to differentiate appropriately or reach their intended targets. There is mounting evidence for an increased rate of apoptotic cell death in a variety of acute and chronic neurologic diseases. Apoptosis is characterized by neuronal shrinkage, chromatin condensation, and DNA fragmentation, whereas necrotic cell death is associated with cytoplasmic and mitochondrial swelling followed by dissolution of the cell membrane. Apoptotic and necrotic cell death can coexist or be sequential events, depending on the severity of the initiating insult. Cellular energy reserves appear to have an important role in these two forms of cell death, with apoptosis favored under conditions in which ATP levels are preserved. Evidence of DNA fragmentation has been found in a number of degenerative neurologic disorders, including AD, Huntington’s disease, and ALS. The best characterized genetic neurologic disorder related to apoptosis is infantile spinal muscular atrophy (Werdnig-Hoffmann disease), in which two genes thought to be involved in the apoptosis pathways are causative.
Mitochondria are essential in controlling specific apoptosis pathways. The redistribution of cytochrome c, as well as apoptosis-inducing factor (AIF), from mitochondria during apoptosis leads to the activation of a cascade of intracellular proteases known as caspases. Caspase-independent apoptosis occurs after DNA damage, activation of poly-ADP-ribose polymerase, and translocation of AIF into the nucleus. Redistribution of cytochrome c is prevented by overproduction of the apoptotic protein BCL2 and is promoted by the proapoptotic protein BAX. These pathways may be triggered by activation of a large pore in the mitochondrial inner membrane known as the permeability transition pore, although in other circumstances, they occur independently. The permeability transition pore is made up of dimers of ATP synthase and is activated by cyclophilin D, leading to large calcium fluxes across the inner mitochondrial membrane. Certain forms of congenital muscular dystrophies are caused by mutations in collagen VI, which leads to increased activation of the permeability transition pore. Recent studies suggest that blocking the mitochondrial pore reduces both hypoglycemic and ischemic cell death. Mice deficient in cyclophilin D, a key protein involved in opening the permeability transition pore, are resistant to necrosis produced by focal cerebral ischemia.
PROTEIN AGGREGATION AND NEURODEGENERATION
The possibility that protein aggregation plays a role in the pathogenesis of neurodegenerative diseases is a major focus of current research. Protein aggregation is a major histopathologic hallmark of neurodegenerative diseases. Deposition of β-amyloid is strongly implicated in the pathogenesis of AD. Genetic mutations in familial AD cause increased production of β-amyloid with 42 amino acids, which has an increased propensity to aggregate, as compared to β-amyloid with 40 amino acids. Furthermore, mutations in the amyloid precursor protein (APP), which reduce the production of β-amyloid, protect against the development of AD and are associated with preserved cognition in the elderly. Mutations in genes encoding the MAPT lead to altered splicing of tau and the production of neurofibrillary tangles in frontotemporal dementia and progressive supranuclear palsy. Familial Parkinson’s disease is associated with mutations in leucine-rich repeat kinase 2 (LRRK2), α-synuclein, parkin, PINK1, and DJ-1. PINK1 is a mitochondrial kinase (see below), and DJ-1 is a protein involved in protection from oxidative stress. Parkin, which causes autosomal recessive early-onset Parkinson’s disease, is a ubiquitin ligase. The characteristic histopathologic feature of Parkinson’s disease is the Lewy body, an eosinophilic cytoplasmic inclusion that contains both neurofilaments and α-synuclein. Huntington’s disease and cerebellar degenerations are associated with expansions of polyglutamine repeats in proteins, which aggregate to produce neuronal intranuclear inclusions. Familial ALS is associated with superoxide dismutase mutations and cytoplasmic inclusions containing superoxide dismutase. An important finding was the discovery that ubiquinated inclusions observed in most cases of ALS and the most common form of frontotemporal dementia are composed of TAR DNA binding protein 43 (TDP-43). Subsequently, mutations in the TDP-43 gene, and in the fused in sarcoma gene (FUS), were found in familial ALS. Both of these proteins are involved in transcription regulation as well as RNA metabolism. In autosomal dominant neurohypophyseal diabetes insipidus, mutations in vasopressin result in abnormal protein processing, accumulation in the endoplasmic reticulum, and cell death.
Another key mechanism linked to cell death is mitochondrial dynamics, which refers to the processes involved in movement of mitochondria, as well as in mitochondrial fission and fusion, which play a critical role mitochondrial turnover and in replenishment of damaged mitochondria. Mitochondrial dysfunction is strongly linked to the pathogenesis of a number of neurodegenerative diseases such as Friedreich’s ataxia, which is caused by mutations in an iron-binding protein that plays an important role in transferring iron to iron-sulfur clusters in aconitase and complex I and II of the electron transport chain. Mitochondrial fission is dependent on the dynamin-related proteins (Drp1), which bind to its receptor Fis, whereas mitofuscins 1 and 2 (MF 1/2) and optic atrophy protein 1 (OPA1) are responsible for fusion of the outer and inner mitochondrial membrane, respectively. Mutations in MFN2 cause Charcot-Marie-Tooth neuropathy type 2A, and mutations in OPA1 cause autosomal dominant optic atrophy. Both β-amyloid and mutant huntingtin protein induce mitochondrial fragmentation and neuronal cell death associated with increased activity of Drp1. In addition, mutations in genes causing autosomal recessive Parkinson’s disease, parkin and PINK1, cause abnormal mitochondrial morphology and result in impairment of the ability of the cell to remove damaged mitochondria by autophagy.
One major scientific question is whether protein aggregates directly contribute to neuronal death or whether they are merely secondary bystanders. A current focus in all the neurodegenerative diseases is on small protein aggregates termed oligomers. These may be the toxic species of β-amyloid, α-synuclein, and proteins with expanded polyglutamines such as are associated with Huntington’s disease. Protein aggregates are usually ubiquinated, which targets them for degradation by the 26S component of the proteasome. An inability to degrade protein aggregates could lead to cellular dysfunction, impaired axonal transport, and cell death by apoptotic mechanisms.
Autophagy is the degradation of cystolic components in lysosomes. There is increasing evidence that autophagy plays an important role in degradation of protein aggregates in the neurodegenerative diseases, and it is impaired in AD, Parkinson’s disease, and Huntington’s disease. Autophagy is particularly important to the health of neurons, and failure of autophagy contributes to cell death. In Huntington’s disease, a failure of cargo recognition occurs, contributing to protein aggregates and cell death. Rapamycin, which induces autophagy, exerts beneficial therapeutic effects in transgenic mouse models of AD, Parkinson’s disease, and Huntington’s disease.
There is other evidence for lysosomal dysfunction and impaired autophagy in Parkinson’s disease. Mutations in glucocerebrosidase are associated with 5% of all Parkinson’s disease cases as well as 8–9% of patients with dementia with Lewy bodies. Therefore, this is the most important genetic cause of both disorders thus far identified. There appear to be reciprocal interactions between glucocerebrosidase and α-synuclein. It has been shown that glucocerebrosidase concentrations and enzymatic activity are reduced in the substantia nigra of sporadic Parkinson’s disease patients. Furthermore, α-synuclein is degraded by chaperone-mediated and macro autophagy. The degradation of α-synuclein has been shown to be impaired in transgenic mice deficient in glucocerebrosidase as well as in mice in which the enzyme has been inhibited. Furthermore, it is known that α-synuclein inhibits the activity of glucocerebrosidase. Therefore, there is bidirectional feedback between α-synuclein and glucocerebrosidase. An attractive therapeutic intervention could be to use protein chaperones to increase the activity and duration of action of glucocerebrosidase. This would also reduce α-synuclein levels and block the degeneration of dopaminergic neurons.
The retromer complex is a conserved membrane-associated protein complex that functions in the endosome-to-Golgi complex. The retromer complex contains a cargo selective complex consisting of VPS35, VPS26, and VPS29, along with a sorting nexin dimer. Recently, mutations in VPS35 were shown to be a cause of late-onset autosomal dominant Parkinson’s disease. The retromer also traffics APP away from endosomes, where it is cleaved to generate β-amyloid. Deficiencies of VPS35 and VPS26 were also identified in hippocampal brain tissue from AD. A new therapeutic approach to these diseases might therefore be to use chaperones to stabilize the retromer and reduce the generation of β-amyloid and α-synuclein.
The LRRK2 mutations were shown to have effects on clearance of Golgi-derived vesicles through the autophagy-lysosome system both in vitro and in vivo. LRRK2 mutations also are linked to elevated protein synthesis mediated by ribosomal protein s15 phosphorylation. Blocking this phosphorylation reduces LRRK2-mediated neurite loss and cell death in human dopamine and cortical neurons.
Interestingly, in experimental models of Huntington’s disease and cerebellar degeneration, protein aggregates are not well correlated with neuronal death and may be protective. A substantial body of evidence suggests that the mutant proteins with polyglutamine expansions in these diseases bind to transcription factors and that this contributes to disease pathogenesis. In Huntington’s disease, there is dysfunction of the transcriptional co-regulator, PGC-1α, a key regulator of mitochondrial biogenesis. There is evidence that impaired function of PGC-1α is also important in both Parkinson’s disease and AD, making it an attractive target for treatments. Agents that upregulate gene transcription are neuroprotective in animal models of these diseases. A number of compounds have been developed to block β-amyloid production and/or aggregation, and these agents are being studied in early clinical trials in humans. Another approach under investigation is immunotherapy with antibodies that bind β-amyloid, tau, or α-synuclein. These studies have shown efficacy in preventing the spread of amyloid, tau, and α-synuclein in animal studies, raising hopes that this could lead to effective therapies by blocking neuron-to-neuron propagation. Two large clinical trials of β-amyloid immunotherapy, however, did not show efficacy, although this therapeutic strategy is still being studied.
PRIONS AND NEURODEGENERATIVE DISEASES
As we have learned more about the etiology and pathogenesis of the neurodegenerative diseases, it has become clear that the histologic abnormalities that were once curiosities, in fact, are likely to reflect the etiologies. For example, the amyloid plaques in kuru and Creutzfeldt-Jakob disease (CJD) are filled with the PrPSc prions that have assembled into fibrils. The past three decades have witnessed an explosion of new knowledge about prions. For many years, kuru, CJD, and scrapie of sheep were thought to be caused by slow-acting viruses, but a large body of experimental evidence argues that the infectious pathogens causing these diseases are devoid of nucleic acid. Such pathogens are called prions, which are composed of host-encoded proteins that adopt alternative conformations (Chap. 453e). Prions are self-propagating by imposing their conformations on the normal, precursor protein; most prions are enriched for β-sheet and can assemble into amyloid fibrils.
Similar to the plaques in kuru and CJD that are composed of PrP prions, the amyloid plaques in AD are filled with Aβ prions that have polymerized into fibrils. This relationship between the neuropathologic findings and the etiologic prion was strengthened by the genetic linkage between familial CJD and mutations in the PrP gene, as well as (as noted above) between familial AD and mutations in the APP gene. Moreover, a mutation in the APP gene that prevents Aβ peptide formation was correlated with a decreased incidence of AD in Iceland.
The heritable neurodegenerative diseases offer an important insight into the pathogenesis of the more common, sporadic ones. Although the mutant proteins that cause these disorders are expressed in the brains of people early in life, the diseases do not occur for many decades. Many explanations for the late onset of familial neurodegenerative diseases have been offered, but none are supported by substantial experimental evidence. The late onset might be due to a second event in which a mutant protein, after its conversion into a prion, begins to accumulate at some rather advanced age (Fig. 444e-5). Such a formulation is also consistent with data showing that the protein quality-control mechanisms diminish in efficiency with age. Thus, the prion forms of both wild-type and mutant proteins are likely to be efficiently degraded in younger people but are less well handled in older individuals. This explanation is consistent with the view that neurodegenerative diseases are disorders of the aging nervous system.
FIGURE 444e-5 Video game training can enhance cognitive performance. A. An older participant engaging in NeuroRacer training (driving while responding to target signs), with (B) a screen shot of the experimental training session. C. NeuroRacer multitasking costs for target discrimination increased (i.e., a larger percentage decrease from single task performance when multitasking) in (i) a linear fashion across the lifespan and with (ii) costs before training 1 month after training and 6 months after training, showing a differential benefit of multitasking training compared to a no-contact control group and a single-task training group. D. Midline frontal theta activity obtained with electroencephalogram showed significantly enhanced activity only following multitasking training, mimicking the pattern of change in the behavioral data as well as performance improvements on untrained tests of working memory and sustained attention (not presented). For details, see JA Anguera et al: Nature 501:97, 2013.
A new classification for neurodegenerative diseases can be proposed based on not only the traditional phenotypic presentation and neuropathology, but also the prion etiology (Table 444e-4). Over the past decade, an expanding body of experimental data has accumulated implicating prions in each of these illnesses. In addition to kuru and CJD, Gerstmann-Sträussler-Scheinker disease (GSS) and fatal insomnia in humans are caused by PrPSc prions. In animals, PrPSc prions cause scrapie of sheep and goats, bovine spongiform encephalopathy (BSE), chronic wasting disease (CWD) of deer and elk, feline spongiform encephalopathy, and transmissible mink encephalopathy (TME). Similar to PrP, Aβ, tau, α-synuclein, superoxide dismutase 1 (SOD1), and possibly huntingtin all adopt alternative conformations that become self-propagating, and thus, each protein can become a prion and be transferred to synaptically connected neurons. Moreover, each of these prions causes a distinct constellation of neurodegenerative diseases.
|
A PRION-BASED CLASSIFICATION OF NEURODEGENERATIVE DISEASES |
Evidence for a prion etiology of AD comes from a series of transmission experiments initially performed in marmosets and more recently in transgenic (Tg) mice inoculated with a synthetic Aβ peptide folded into a prion. Studies with the tau protein have shown that it not only features in the pathogenesis of AD, but also causes such illnesses as the frontotemporal dementias including chronic traumatic encephalopathy, which has been reported in both contact sport athletes and military personnel who have suffered traumatic brain injuries. A series of incisive studies using cultured cells and Tg mice has demonstrated that tau can become a prion and multiply in the brain. In contrast to the Aβ and tau prions, a strain of α-synuclein prions found in the brains of patients who died of multiple system atrophy (MSA) killed the Tg mouse host ~90 days after intracerebral inoculation, whereas α-synuclein prions formed spontaneously in Tg mouse brains killed recipient mice in ~200 days.
For many years, the most frequently cited argument against prions was the existence of strains that produced distinct clinical presentations and different patterns of neuropathologic lesions. Some investigators argued that the biologic information carried in different prion strains could only be encoded within a nucleic acid. Subsequently, many studies demonstrated that strain-specified variation is enciphered in the conformation of PrPSc, but the molecular mechanisms responsible for the storage of biologic information remains enigmatic. The neuroanatomical patterns of prion deposition have been shown to be dependent on the particular strain of prion. Convincing evidence in support of this proposition has been accumulated for PrP, Aβ, tau, and α-synuclein prions.
Although the number of prions identified in mammals and in fungi continues to expand, the existence of prions in other phylogeny remains undetermined. Some mammalian prions perform vital functions and do not cause disease; such nonpathogenic prions include the cytoplasmic polyadenylation element binding (CPEB) protein, the mitochondrial antiviral-signaling (MAVS) protein, and T cell–restricted intracellular antigen 1 (TIA-1).
All mammalian prion proteins adopt a β-sheet-rich conformation and appear to readily oligomerize as this process becomes self-propagating. Control of the self-propagating state of benign mammalian prions is not well understood but is critical for the well-being of the host. In contrast, pathogenic mammalian prions appear to multiply exponentially, but the mechanisms by which they cause disease are poorly defined. We do not know if prions multiply as monomers or as oligomers; notably, the ionizing radiation target size of PrPSc prions seems to suggest it is a trimer. The oligomeric states of pathogenic mammalian prions are thought to be the toxic forms, and assembly into larger polymers, such as amyloid fibrils, seems to be a mechanism for minimizing toxicity.
To date, there is no medication that halts or even slows a human neurodegenerative disease. The development of drugs designed to inhibit the conversion of the normal precursor proteins into prions or to enhance the degradation of prions focuses on the initial step in prion accumulation. Although several drugs that cross the blood-brain barrier have been identified that prolong the lives of mice infected with scrapie prions, none have been identified that extend the lives of Tg mice that replicate human CJD prions. Despite doubling the length of incubation times in mice inoculated with scrapie prions, all of the mice eventually succumb to illness. Because all of the treated mice develop neurologic dysfunction at the same time, the mutation rate as judged by drug resistance is likely to approach 100%, which is much higher than mutation rates recorded for bacteria and viruses. Mutations in prions seem likely to represent conformational variants that are selected for in mammals where survival becomes limited by the fastest-replicating prions. The results of these studies make it likely that cocktails of drugs that attack a variety of prion conformers will be required for the development of effective therapeutics.
SYSTEMS NEUROSCIENCE
Systems neuroscience refers to study of the functions of neural circuits and how they relate to brain function, behavior, motor activity, and cognition. Brain imaging techniques, primarily functional magnetic resonance imaging (fMRI) and position emission tomography (PET), have made it possible to investigate, noninvasively and in awake individuals, cognitive processes such as perception, making judgments, paying attention, and thinking. This has allowed insights into how networks of neurons operate to produce behavior. Many of these studies at present are based on determining the connectivity of neural circuits and how they operate, and how this can be then modeled to produce improved understanding of physiologic processes. fMRI uses contrast mechanisms related to physiologic changes in tissue, and brain perfusion can be studied by observing the time course of changes in brain water signal as a bolus of injected paramagnetic gadolinium contrast moves through the brain. More recently, to study intrinsic contrast-related local changes in blood oxygenation with brain activity, blood-oxygen-level-dependent (BOLD) contrast has been used to provide a rapid noninvasive approach for functional assessment. These techniques have been reliably used in the field of both behavior and cognitive sciences. One example is the use of fMRI to demonstrate mirror neuron systems, imitative pathways activated when observing actions of others. Mirror neurons are thought to be important for social conditioning and for many forms of learning, and abnormalities in mirror neurons may underlie some autism disorders.
Both structural and functional connectivity methods show large-scale network dysfunction in AD and frontotemporal dementia. The networks targeted have been defined as the default network in AD and the salience network in frontotemporal dementia. The default network is characterized by an area of reduced glucose metabolism in the temporoparietal cortex, which precedes the onset of dementia and which is an area preferentially affected by amyloid deposition. These networks are now thought to be pathways accounting for the spread of abnormally templated proteins (prions; see above), including β-amyloid, tau, and α-synuclein.
Other examples of the use of fMRI include the study of memory, revealing that not only is hippocampal activity correlated with declarative memory consolidation, but it also involves activation in the ventral medial prefrontal cortex. Consolidation of memory over time results in decreased activity of the hippocampus and progressively stronger activation in the ventral medial prefrontal region associated with retrieval of consolidated memories. fMRI has also been used to identify sequences of brain activation involved in normal movements and alterations in their activation associated with both injury and recovery, to plan neurosurgical operations, and remarkably, to reconstruct actual visual images from the occipital cortex. Noninvasive brain-computer interfaces have extraordinary potential to advance the development of robotics and exoskeleton devices guided by brain activity for patients with a variety of nervous system afflictions. Diffusion tensor imaging is a recently developed MRI technique that can measure macroscopic axonal organization in nervous system tissues; it appears to be useful in assessing myelin and axonal injuries as well as brain development. Advances in understanding neural processing have led to the development of the ability to demonstrate that humans have online voluntary control of human temporal lobe neurons.
Multitasking capabilities, including attention to tasks when faced with distractions, decline as we age due to a decline in medial prefrontal cognitive control system. When faced with a multitasking challenge, video game training can improve cognitive control capabilities by augmenting prefrontal suppression of the default network and, as measured by electroencephalography and fMRI, result in improved performance that is sustained and, importantly, transfers to other cognitive tasks not associated with the training paradigm.
A significant recent advance in neuropathology has discovered that sodium dodecyl sulfate (SDS) detergent treatment can render the brain transparent (CLARITY), removing lipids while preserving most protein and structural elements and providing opportunities to identify brain structures and neural networks with unprecedented detail.
A therapeutic technology that has long-reaching implications for the development of novel interventions for neurologic, including behavioral, conditions has been the development of deep-brain stimulation as a highly effective therapeutic intervention for treating excessively firing neurons in the subthalamic nucleus of patients with Parkinson’s disease and the precingulate cortex in patients with depression.
BRAIN RESEARCH THROUGH ADVANCING INNOVATIVE NEUROTECHNOLOGIES (BRAIN) INITIATIVE
The BRAIN initiative, grand in scope, was launched in 2013 to speed development of advances to understand, treat, repair, and prevent common neurologic disorders that, in aggregate, affect more than 1 billion people worldwide. The initial goal of BRAIN is to bring together experts in neurobiology (including optogenetics), engineering, information technology, and other fields to develop novel visualization and electrophysiologic methods to better define and understand neural circuits and all the connections among individual neurons. The announcement of the BRAIN initiative followed just weeks after a similarly ambitious program, the Human Brain Project (HBP), was unveiled by the European Union. The HBP seeks to model individual neurons, neural circuits, and ultimately the entire brain using computer technologies. Its architects also envision layering clinical and biomarker data from large health care databases to identify biosignatures associated with human phenotypes, possibly leading to a fundamental reclassification of disease, a concept also proposed by others, including in a 2011 report of the National Academy of Sciences (Toward Precision Medicine: Building a Knowledge Network for Biomedical Research and a New Taxonomy of Disease). These two ambitious projects are expected to be complementary and over time will hopefully become increasingly integrated. Emerging discoveries are also certain to stimulate a range of ethical questions, many of which are not unique to the neurosciences but do come into sharpest focus in this area. These include possible risks to the privacy of personal information about our health, cognitive capabilities, or behavioral attributes; the sanctity of our private thoughts; as well as concerns regarding neuroenhancement technologies, including their potential military use. Both the BRAIN and HBP initiatives have put into place strong ethical components to ensure that these programs are carried out, from the outset and to the fullest extent possible, consistent with guiding ethical principles that include respect for individuals, public beneficence, justice and fairness, democratic deliberation, and transparency.
SECTION 2 |
DISEASES OF THE CENTRAL NERVOUS SYSTEM |
445 |
Seizures and Epilepsy |
A seizure (from the Latin sacire, “to take possession of”) is a paroxysmal event due to abnormal excessive or synchronous neuronal activity in the brain. Depending on the distribution of discharges, this abnormal brain activity can have various manifestations, ranging from dramatic convulsive activity to experiential phenomena not readily discernible by an observer. Although a variety of factors influence the incidence and prevalence of seizures, ~5–10% of the population will have at least one seizure, with the highest incidence occurring in early childhood and late adulthood.
The meaning of the term seizure needs to be carefully distinguished from that of epilepsy. Epilepsy describes a condition in which a person has recurrent seizures due to a chronic, underlying process. This definition implies that a person with a single seizure, or recurrent seizures due to correctable or avoidable circumstances, does not necessarily have epilepsy. Epilepsy refers to a clinical phenomenon rather than a single disease entity, because there are many forms and causes of epilepsy. However, among the many causes of epilepsy there are various epilepsy syndromes in which the clinical and pathologic characteristics are distinctive and suggest a specific underlying etiology.
Using the definition of epilepsy as two or more unprovoked seizures, the incidence of epilepsy is ~0.3–0.5% in different populations throughout the world, and the prevalence of epilepsy has been estimated at 5–30 persons per 1000.
CLASSIFICATION OF SEIZURES
Determining the type of seizure that has occurred is essential for focusing the diagnostic approach on particular etiologies, selecting the appropriate therapy, and providing potentially vital information regarding prognosis. The International League against Epilepsy (ILAE) Commission on Classification and Terminology, 2005–2009 has provided an updated approach to classification of seizures (Table 445-1). This system is based on the clinical features of seizures and associated electroencephalographic findings. Other potentially distinctive features such as etiology or cellular substrate are not considered in this classification system, although this will undoubtedly change in the future as more is learned about the pathophysiologic mechanisms that underlie specific seizure types.
|
CLASSIFICATION OF SEIZURES |
A fundamental principle is that seizures may be either focal or generalized. Focal seizures originate within networks limited to one cerebral hemisphere (note that the term partial seizures is no longer used). Generalized seizures arise within and rapidly engage networks distributed across both cerebral hemispheres. Focal seizures are usually associated with structural abnormalities of the brain. In contrast, generalized seizures may result from cellular, biochemical, or structural abnormalities that have a more widespread distribution. There are clear exceptions in both cases, however.
FOCAL SEIZURES
Focal seizures arise from a neuronal network either discretely localized within one cerebral hemisphere or more broadly distributed but still within the hemisphere. With the new classification system, the subcategories of “simple focal seizures” and “complex focal seizures” have been eliminated. Instead, depending on the presence of cognitive impairment, they can be described as focal seizures with or without dyscognitive features. Focal seizures can also evolve into generalized seizures. In the past this was referred to as focal seizures with secondary generalization, but the new system relies on specific descriptions of the type of generalized seizures that evolve from the focal seizure.
The routine interictal (i.e., between seizures) electroencephalogram (EEG) in patients with focal seizures is often normal or may show brief discharges termed epileptiform spikes, or sharp waves. Because focal seizures can arise from the medial temporal lobe or inferior frontal lobe (i.e., regions distant from the scalp), the EEG recorded during the seizure may be nonlocalizing. However, the seizure focus is often detected using sphenoidal or surgically placed intracranial electrodes.
Focal Seizures Without Dyscognitive Features Focal seizures can cause motor, sensory, autonomic, or psychic symptoms without impairment of cognition. For example, a patient having a focal motor seizure arising from the right primary motor cortex near the area controlling hand movement will note the onset of involuntary movements of the contralateral, left hand. These movements are typically clonic (i.e., repetitive, flexion/extension movements) at a frequency of ~2–3 Hz; pure tonic posturing may be seen as well. Since the cortical region controlling hand movement is immediately adjacent to the region for facial expression, the seizure may also cause abnormal movements of the face synchronous with the movements of the hand. The EEG recorded with scalp electrodes during the seizure (i.e., an ictal EEG) may show abnormal discharges in a very limited region over the appropriate area of cerebral cortex if the seizure focus involves the cerebral convexity. Seizure activity occurring within deeper brain structures is sometimes not detected by the standard EEG, however, and may require intracranial electrodes for its detection.
Three additional features of focal motor seizures are worth noting. First, in some patients, the abnormal motor movements may begin in a very restricted region such as the fingers and gradually progress (over seconds to minutes) to include a larger portion of the extremity. This phenomenon, described by Hughlings Jackson and known as a “Jacksonian march,” represents the spread of seizure activity over a progressively larger region of motor cortex. Second, patients may experience a localized paresis (Todd’s paralysis) for minutes to many hours in the involved region following the seizure. Third, in rare instances, the seizure may continue for hours or days. This condition, termed epilepsia partialis continua, is often refractory to medical therapy.
Focal seizures may also manifest as changes in somatic sensation (e.g., paresthesias), vision (flashing lights or formed hallucinations), equilibrium (sensation of falling or vertigo), or autonomic function (flushing, sweating, piloerection). Focal seizures arising from the temporal or frontal cortex may also cause alterations in hearing, olfaction, or higher cortical function (psychic symptoms). This includes the sensation of unusual, intense odors (e.g., burning rubber or kerosene) or sounds (crude or highly complex sounds), or an epigastric sensation that rises from the stomach or chest to the head. Some patients describe odd, internal feelings such as fear, a sense of impending change, detachment, depersonalization, déjá vu, or illusions that objects are growing smaller (micropsia) or larger (macropsia). These subjective, “internal” events that are not directly observable by someone else are referred to as auras.
Focal Seizures with Dyscognitive Features Focal seizures may also be accompanied by a transient impairment of the patient’s ability to maintain normal contact with the environment. The patient is unable to respond appropriately to visual or verbal commands during the seizure and has impaired recollection or awareness of the ictal phase. The seizures frequently begin with an aura (i.e., a focal seizure without cognitive disturbance) that is stereotypic for the patient. The start of the ictal phase is often a sudden behavioral arrest or motionless stare, which marks the onset of the period of impaired awareness. The behavioral arrest is usually accompanied by automatisms, which are involuntary, automatic behaviors that have a wide range of manifestations. Automatisms may consist of very basic behaviors such as chewing, lip smacking, swallowing, or “picking” movements of the hands, or more elaborate behaviors such as a display of emotion or running. The patient is typically confused following the seizure, and the transition to full recovery of consciousness may range from seconds up to an hour. Examination immediately following the seizure may show an anterograde amnesia or, in cases involving the dominant hemisphere, a postictal aphasia.
The range of potential clinical behaviors linked to focal seizures is so broad that extreme caution is advised before concluding that stereotypic episodes of bizarre or atypical behavior are not due to seizure activity. In such cases additional, detailed EEG studies may be helpful.
EVOLUTION OF FOCAL SEIZURES TO GENERALIZED SEIZURES
Focal seizures can spread to involve both cerebral hemispheres and produce a generalized seizure, usually of the tonic-clonic variety (discussed below). This evolution is observed frequently following focal seizures arising from a focus in the frontal lobe, but may also be associated with focal seizures occurring elsewhere in the brain. A focal seizure that evolves into a generalized seizure is often difficult to distinguish from a primary generalized-onset tonic-clonic seizure, because bystanders tend to emphasize the more dramatic, generalized convulsive phase of the seizure and overlook the more subtle, focal symptoms present at onset. In some cases, the focal onset of the seizure becomes apparent only when a careful history identifies a preceding aura. Often, however, the focal onset is not clinically evident and may be established only through careful EEG analysis. Nonetheless, distinguishing between these two entities is extremely important, because there may be substantial differences in the evaluation and treatment of epilepsies associated with focal versus generalized seizures.
GENERALIZED SEIZURES
Generalized seizures are thought to arise at some point in the brain but immediately and rapidly engage neuronal networks in both cerebral hemispheres. Several types of generalized seizures have features that place them in distinctive categories and facilitate clinical diagnosis.
Typical Absence Seizures Typical absence seizures are characterized by sudden, brief lapses of consciousness without loss of postural control. The seizure typically lasts for only seconds, consciousness returns as suddenly as it was lost, and there is no postictal confusion. Although the brief loss of consciousness may be clinically inapparent or the sole manifestation of the seizure discharge, absence seizures are usually accompanied by subtle, bilateral motor signs such as rapid blinking of the eyelids, chewing movements, or small-amplitude, clonic movements of the hands.
Typical absence seizures are associated with a group of genetically determined epilepsies with onset usually in childhood (ages 4–8 years) or early adolescence and are the main seizure type in 15–20% of children with epilepsy. The seizures can occur hundreds of times per day, but the child may be unaware of or unable to convey their existence. Because the clinical signs of the seizures are subtle, especially to parents who may not have had previous experience with seizures, it is not surprising that the first clue to absence epilepsy is often unexplained “daydreaming” and a decline in school performance recognized by a teacher.
The electrophysiologic hallmark of typical absence seizures is a generalized, symmetric, 3-Hz spike-and-wave discharge that begins and ends suddenly, superimposed on a normal EEG background. Periods of spike-and-wave discharges lasting more than a few seconds usually correlate with clinical signs, but the EEG often shows many more brief bursts of abnormal cortical activity than were suspected clinically. Hyperventilation tends to provoke these electrographic discharges and even the seizures themselves and is routinely used when recording the EEG.
Atypical Absence Seizures Atypical absence seizures have features that deviate both clinically and electrophysiologically from typical absence seizures. For example, the lapse of consciousness is usually of longer duration and less abrupt in onset and cessation, and the seizure is accompanied by more obvious motor signs that may include focal or lateralizing features. The EEG shows a generalized, slow spike-and-wave pattern with a frequency of ≤2.5 per second, as well as other abnormal activity. Atypical absence seizures are usually associated with diffuse or multifocal structural abnormalities of the brain and therefore may accompany other signs of neurologic dysfunction such as mental retardation. Furthermore, the seizures are less responsive to anticonvulsants compared to typical absence seizures.
Generalized, Tonic-Clonic Seizures Generalized-onset tonic-clonic seizures are the main seizure type in ~10% of all persons with epilepsy. They are also the most common seizure type resulting from metabolic derangements and are therefore frequently encountered in many different clinical settings. The seizure usually begins abruptly without warning, although some patients describe vague premonitory symptoms in the hours leading up to the seizure. This prodrome is distinct from the stereotypic auras associated with focal seizures that generalize. The initial phase of the seizure is usually tonic contraction of muscles throughout the body, accounting for a number of the classic features of the event. Tonic contraction of the muscles of expiration and the larynx at the onset will produce a loud moan or “ictal cry.” Respirations are impaired, secretions pool in the oropharynx, and cyanosis develops. Contraction of the jaw muscles may cause biting of the tongue. A marked enhancement of sympathetic tone leads to increases in heart rate, blood pressure, and pupillary size. After 10–20 s, the tonic phase of the seizure typically evolves into the clonic phase, produced by the superimposition of periods of muscle relaxation on the tonic muscle contraction. The periods of relaxation progressively increase until the end of the ictal phase, which usually lasts no more than 1 min. The postictal phase is characterized by unresponsiveness, muscular flaccidity, and excessive salivation that can cause stridorous breathing and partial airway obstruction. Bladder or bowel incontinence may occur at this point. Patients gradually regain consciousness over minutes to hours, and during this transition, there is typically a period of postictal confusion. Patients subsequently complain of headache, fatigue, and muscle ache that can last for many hours. The duration of impaired consciousness in the postictal phase can be extremely long (i.e., many hours) in patients with prolonged seizures or underlying central nervous system (CNS) diseases such as alcoholic cerebral atrophy.
[/not-level-membership-for-internal-medicine-category]
