155 Sedative-Hypnotic Agents
 Key Points
Key Points• Benzodiazepines account for the majority of overdoses with sedative-hypnotic drugs.
• Benzodiazepines can induce cardiovascular and pulmonary toxicity, but fatalities resulting from pure benzodiazepine overdoses are rare.
• Central nervous system depression is the primary symptom of sedative-hypnotic toxicity.
• Treatment should focus on supportive care, with particular attention to airway patency and respiratory function.
• Urinary alkalinization and multiple doses of activated charcoal can enhance the elimination of phenobarbital.
• Flumazenil is a reversal agent for benzodiazepine toxicity, but it should be used cautiously, to avoid the risk of seizures.
• Other possible causes of altered mental status should always be considered.
Epidemiology
Sedative-hypnotic agents are a heterogeneous group of agents that have tranquilizing (sedative) or sleep induction (hypnotic) properties. Grouped with antipsychotics, they comprise the fourth leading class of substances reported to poison centers, and they are the leading cause of reported fatalities.1 These drugs are widely used in clinical settings but are also used for suicide, illicit recreational activities, and facilitation of sexual assault (“date rape”). Several high-profile deaths have been attributed to sedative-hypnotic overdoses.
Benzodiazepines have largely supplanted older agents and have become the most widely used sedative-hypnotics in clinical settings. However, given their prevalence, benzodiazepines also account for the majority of sedative-hypnotic overdoses.1 Flunitrazepam, sometimes referred to as “roofies,”2 is a potent benzodiazepine that has been popularized as a street drug of abuse and has been implicated as a date-rape drug.3
Barbiturates were formerly the primary sedative-hypnotic agents used clinically. Currently, they are most often encountered as anticonvulsants, induction agents for anesthesia, and agents used for procedural sedation. Because the barbiturates have largely been replaced clinically by benzodiazepines due to safety concerns, their prevalence in overdoses has drastically decreased when compared with previous decades.1 The reported use of barbiturates among high school seniors experienced a slow but steady surge throughout the 1990s and reached a peak in 2005, only to experience a decline since then.4 Barbiturates accounted for only two single-substance deaths reported to poison centers in 2009.1
Gamma-hydroxybutyrate (GHB) was synthesized in 1960 as an anesthetic agent. Although it found limited use in this arena, GHB gained widespread acceptance in the body-building community in the 1990s as a purported anabolic agent. More recently, it has been used as a recreational drug for its euphoric and intoxicating effects.5 It has also been implicated in date rape because of its “knockout” and amnestic properties.
Several nonbenzodiazepine sedatives have been introduced for sleep induction. Examples include zolpidem (Ambien), zaleplon (Sonata), and eszopiclone (Lunesta). Cases of abuse and dependence have been reported, albeit with much less frequency compared with benzodiazepines.6 Nonbenzodiazepine sedatives have been implicated in cases of impaired driving.7
Chloral hydrate has been used as a sedative since the nineteenth century. In the early 1900s, chloral hydrate was used maliciously, added to alcoholic drinks consumed by unwary individuals to facilitate robberies. The drug-laced drink was referred to as a “Mickey Finn,” named after the owner of a Chicago bar who used these drinks to rob unsuspecting patrons.8 Currently, chloral hydrate is used primarily for procedural sedation.
Propofol is a short-acting sedative-hypnotic that has become widely used clinically for induction of anesthesia and procedural sedation. Despite its abuse potential, the literature is limited to case reports of toxicity from recreational use because of its limited availability to the general public.9 Most reported cases involve self-administration by medical personnel.10–12 Propofol is covered in greater detail elsewhere in this text.
Pathophysiology
Benzodiazepines
Benzodiazepines vary in onset and duration of action according to their lipid solubility and the presence or absence of active metabolites (Box 155.1). The more lipid soluble the agent is, the more rapidly it crosses the blood-brain barrier, thus yielding a faster onset of action. The duration of action depends largely on the elimination half-life of specific agents, which can range from hours to days. The duration of action is also affected by the metabolism of certain benzodiazepines because their active metabolites extend the duration of symptoms.
Barbiturates
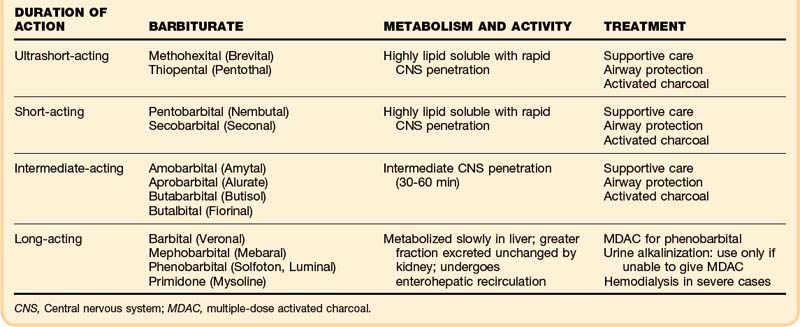
The barbiturates are often classified according to their therapeutic duration of action: ultrashort-acting, short-acting, intermediate-acting, and long-acting agents (Box 155.2). In overdoses, however, the duration of action varies with dose, rate of absorption, and rate of distribution and elimination. The ultrashort-acting and short-acting agents are highly lipid soluble and rapidly penetrate the CNS, so the onset of symptoms is also rapid. In addition, the ultrashort-acting barbiturates are more highly protein bound, have higher acid-dissociation constant (pKa), values, and have larger volumes of distribution. Long-acting agents such as phenobarbital are metabolized more slowly in the liver, with a greater fraction of unchanged drug excreted in the kidney. These factors help explain why enhanced renal elimination through alkalinization may be more effective with phenobarbital, which also has a lower pKa than the other barbiturates, thus making it more sensitive to alkalinization. In addition, phenobarbital undergoes enterohepatic recirculation, which makes repeated use of activated charcoal potentially advantageous.
The reticular activating system and the cerebellum appear to be the most susceptible to the depressant effects of barbiturates. Toxicity can lead to suppression of skeletal, smooth, and cardiac muscles, with resulting depressed myocardial contractility, bradycardia, vasodilation, and hypotension (Table 155.1).
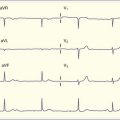
Gamma-Hydroxybutyrate
GHB is a metabolite of GABA that occurs naturally in the human brain.5 It is highly lipophilic and rapidly absorbed, and, unlike GABA, it readily crosses the blood-brain barrier. Presentation in a coma state and subsequent rapid recovery is characteristic of GHB overdose.
Chloral Hydrate
Although chloral hydrate has been used for more than a century to induce sedation, its mechanism of action is still largely unknown. It probably works through effects at the GABA receptor, similar to the other sedative-hypnotic agents discussed. Chloral hydrate can also induce cardiac dysrhythmias, most likely by increasing the sensitivity of the myocardium to catecholamines.13
Other Agents
Buspirone is a nonbenzodiazepine used to treat generalized anxiety. Its mechanism of action involves serotonin, rather than GABA. Although buspirone is one of the more commonly reported miscellaneous sedative-hypnotic exposures, significant toxicity from overdose is extremely rare.1 In one case report, overdose resulted in generalized seizure activity and subsequent full recovery.14
Presenting Signs and Symptoms
 Red Flags
Red Flags
Tips and Tricks
• Pear-like odor is associated with chloral hydrate.
• Chloral hydrate overdoses may manifest with cardiac toxicity as well as with central nervous system depression.
• Bullous lesions are sometimes associated with barbiturate overdoses.
• Consider rhabdomyolysis as a complication.
• A patient who appears brain dead after a severe sedative-hypnotic (barbiturate) overdose may not be brain dead.
• Use beta-blockers to treat chloral hydrate–induced cardiac dysrhythmias.
• Epinephrine and norepinephrine are relatively contraindicated in chloral hydrate overdose because the myocardium may have an increased sensitivity to these types of agents.
• The most common drug used to facilitate sexual assault is ethanol, not sedative-hypnotics.
• MDAC appears to be superior to urinary alkalinization in enhancing the elimination of phenobarbital, and performing both procedures concurrently has no apparent benefit.
Differential Diagnosis and Medical Decision Making
The differential diagnosis of sedative-hypnotic toxicity includes any condition or ingestion resulting in CNS depression. Many other substances are capable of producing profound CNS depression, including alcohol and opiates. However, care should be made not to miss other, nontoxicologic causes of CNS depression. Although sedative-hypnotic toxicity resolves with just supportive care, other mimickers of sedative-hypnotic overdose may need other acute interventions. Other diagnoses that should be considered include head trauma with intracranial hemorrhage, embolic and hemorrhagic stroke, electrolyte abnormalities, hypoglycemia, hyperglycemic crisis, hypoxemia, hypothyroidism, liver or renal failure, CNS infection, seizures, and significant alterations in temperature (Table 155.2).
Table 155.2 Differential Diagnosis of Sedative-Hypnotic Toxidromes and Priority Actions
| DIAGNOSTIC CONSIDERATION | PRIORITY ACTION(S) |
|---|---|
| Airway and respiratory status? |
MDAC, multiple-dose activated charcoal.
Treatment
Beta-blockers have been successfully used to treat cardiac dysrhythmias resulting from chloral hydrate toxicity because myocardial catecholamine sensitivity is believed to induce the dysrhythmia.15 Epinephrine and norepinephrine are relatively contraindicated because the myocardium may have increased sensitivity to these types of agents.
Repeat dosing of activated charcoal has been recommended for increased clearance of certain drugs, one of which is phenobarbital.16 This therapeutic procedure has been referred to as multiple-dose activated charcoal (MDAC). It is thought to be helpful for phenobarbital because this drug undergoes enterohepatic circulation and is excreted back into the gut, where activated charcoal present in the intestine may bind it before it is reabsorbed distally. Phenobarbital also has physical characteristics that allow it to diffuse from the blood into the intestinal lumen. With MDAC, activated charcoal avidly binds to the phenobarbital in the intestinal lumen, a process that creates a concentration gradient into the intestine and subsequently enhances the elimination of the phenobarbital.17,18 Although MDAC has been shown to increase clearance phenobarbital, it has not been shown to improve overall clinical outcomes.
Alkalinizing the urine with the intravenous administration of sodium bicarbonate can increase the elimination of phenobarbital. Urinary alkalinization with sodium bicarbonate to a pH of 7.5 to 8.0 can hasten the renal excretion of phenobarbital.19 Urinary alkalinization can be accomplished with an initial sodium bicarbonate bolus of 1 mEq/kg, followed by a continuous infusion. This infusion is made by adding 100 to 150 mEq of sodium bicarbonate to 850 mL of dextrose 5% in water and titrating it to maintain a urine pH of greater than 7.5 with an arterial pH less than 7.5. The rate must be assessed hourly to avoid excessive administration of fluid or bicarbonate, which can cause pulmonary or cerebral edema or electrolyte imbalance. Although expediting the elimination of phenobarbital from the body has theoretical benefit, no difference in clinical outcome has been shown. Alkalinization does not increase excretion of short- and medium-acting agents, which are more lipid soluble.
MDAC appears to be superior to urinary alkalinization for enhancing the elimination of phenobarbital.20 Performing both procedures concurrently appears to have no benefit.21 Urinary alkalinization may still be useful in a patient who cannot undergo MDAC.
In patients who are not responsive to standard therapeutic measures, or in patients with renal failure, hemodialysis may help eliminate long-acting barbiturates.22 These agents are less protein bound and less lipid soluble that the shorter-acting barbiturates, characteristics that enhance the role of hemodialysis. Fortunately, extracorporeal elimination is rarely indicated because most barbiturate overdoses resolve with supportive care alone.
Hemodialysis can enhance the elimination of chloral hydrate and its metabolites.23 However, supportive measures are generally effective. Hemodialysis may have a role if a patient with chloral hydrate toxicity is not responding to conservative therapy.
Flumazenil is a specific antagonist for benzodiazepines (Box 155.3). It competitively binds at the benzodiazepine receptor, displaces benzodiazepines from the site, and inhibits GABA potentiation. Flumazenil is lipid soluble and readily crosses the blood-brain barrier to exert its effects quickly. Typically, benzodiazepine-induced sedation is reversed within a couple of minutes.
In the setting of procedural sedation, flumazenil is an excellent rescue agent for inadvertent supratherapeutic administrations of a benzodiazepine agent.24 Flumazenil may also be helpful in the setting of an isolated known benzodiazepine overdose. Unfortunately, this situation rarely occurs clinically. The use of flumazenil in the setting of a multiple drug overdose that includes a benzodiazepine is less clear.
Overall, for an unknown overdose, the administration of flumazenil is not indicated.25 Flumazenil does not antagonize the CNS effects of alcohol, barbiturates, tricyclic antidepressants, or narcotics. Reports have noted precipitation of seizure activity in the setting of mixed overdose or benzodiazepine dependence.26 Because supportive therapy is usually effective in benzodiazepine overdose, the benefit of flumazenil may not outweigh the risks of administration when circumstances surrounding the toxic ingestion are unclear. Flumazenil use has been described in the setting of overdose of the newer, nonbenzodiazepine sedatives.27 However, supportive care is usually effective in these cases as well.
 Documentation
DocumentationParadoxical Reactions
Occasionally, patients who have been exposed to sedative-hypnotic agents experience a reaction that can be characterized by an increase in psychomotor activity. This has been most commonly described in benzodiazepines.28 The reactions can range from increased talkativeness and excessive movement to rage and hostility. As a whole, these reactions have been termed paradoxical because they seem counter to the sedative properties of these agents.
The mechanism of these paradoxical reactions is unclear, but some characteristics seem to increase the risk of these reactions. These characteristics include the younger and older age groups, as well as underlying psychiatric disorders. A genetic predisposition to these reactions may also exist.29
The use of flumazenil,30–32 haloperidol,33 and other agents has been described for the treatment of these reactions, with variable success. However, the mainstay of treatment should focus on supportive care. This attention to supportive care should be all that is required.
Chudnofsky CR. Safety and efficacy of flumazenil in reversing conscious sedation in the emergency department: Emergency Medicine Conscious Sedation Study Group. Acad Emerg Med. 1997;4:944–950.
Gueye PN, Hoffman JR, Taboulet P, et al. Empiric use of flumazenil in comatose patients: limited applicability of criteria to define low risk. Ann Emerg Med. 1996;27:730–735.
Hall R, Zisook S. Paradoxical reactions to benzodiazepines. Br J Clin Pharmacol. 1981;11(suppl 1):99S–104S.
Pond SM, Olson KR, Osterloh JD, Tong TG. Randomized study of the treatment of phenobarbital overdose with repeated doses of activated charcoal. JAMA. 1984;251:3104–3108.
1 Bronstein AC, Spyker DA, Cantilena LR, Jr., et al. 2008 annual report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 26th annual report. Clin Toxicol (Phila). 2009;47:911–1084.
2 Office of National Drug Control Policy. Street terms. http://www.expomed.com/drugtest/files/drugterms.pdf.
3 Waltzman ML. Flunitrazepam: a review of “roofies.”. Pediatr Emerg Care. 1999;15:59–60.
4 Johnston LD, O’Malley PM, Bachman JG, Schulenberg JE. Monitoring the Future national results on adolescent drug use: Overview of key findings, 2009. NIH publication no. 10-7583. Bethesda, MD: National Institute on Drug Abuse; 2010.
5 Gonzalez A, Nutt DJ. Gamma hydroxy butyrate abuse and dependency. J Psychopharmacol. 2005;19:195–204.
6 Cimolai N. Zopiclone: is it a pharmacologic agent for abuse? Can Fam Physician. 2007;53:2124–2129.
7 Gustavsen I, Al-Sammurraie M, Morland J, Bramness JG. Impairment related to blood drug concentrations of zopiclone and zolpidem compared to alcohol in apprehended drivers. Accid Anal Prev. 2009;41:462–466.
8 Baum CR. A century of Mickey Finn: but who was he? J Toxicol Clin Toxicol. 2000;38:683. author reply 685
9 Wilson C, Canning P, Caravati EM. The abuse potential of propofol. Clin Toxicol (Phila). 2010;48:165–170.
10 Kranioti EF, Mavroforou A, Mylonakis P, Michalodimitrakis M. Lethal self administration of propofol (Diprivan): a case report and review of the literature. Forensic Sci Int. 2007;167:56–58.
11 Roussin A, Mirepoix M, Lassabe G, et al. Death related to a recreational abuse of propofol at therapeutic dose range. Br J Anaesth. 2006;97:268.
12 Iwersen-Bergmann S, Rosner P, Kuhnau HC, et al. Death after excessive propofol abuse. Int J Legal Med. 2001;114:248–251.
13 Sing K, Erickson T, Amitai Y, Hryhorczuk D. Chloral hydrate toxicity from oral and intravenous administration. J Toxicol Clin Toxicol. 1996;34:101–106.
14 Catalano G, Catalano MC, Hanley PF. Seizures associated with buspirone overdose: case report and literature review. Clin Neuropharmacol. 1998;21:347–350.
15 Zahedi A, Grant MH, Wong DT. Successful treatment of chloral hydrate cardiac toxicity with propranolol. Am J Emerg Med. 1999;17:490–491.
16 Position statement and practice guidelines on the use of multi-dose activated charcoal in the treatment of acute poisoning: American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. J Toxicol Clin Toxicol. 1999;37:731–751.
17 Berg MJ, Berlinger WG, Goldberg MJ, et al. Acceleration of the body clearance of phenobarbital by oral activated charcoal. N Engl J Med. 1982;307:642–644.
18 Pond SM, Olson KR, Osterloh JD, Tong TG. Randomized study of the treatment of phenobarbital overdose with repeated doses of activated charcoal. JAMA. 1984;251:3104–3108.
19 Mawer GE, Lee HA. Value of forced diuresis in acute barbiturate poisoning. Br Med J. 1968;2:790–793.
20 Frenia ML, Schauben JL, Wears RL, et al. Multiple-dose activated charcoal compared to urinary alkalinization for the enhancement of phenobarbital elimination. J Toxicol Clin Toxicol. 1996;34:169–175.
21 Mohammed Ebid AH, Abdel-Rahman HM. Pharmacokinetics of phenobarbital during certain enhanced elimination modalities to evaluate their clinical efficacy in management of drug overdose. Ther Drug Monit. 2001;23:209–216.
22 Jacobs F, Brivet FG. Conventional haemodialysis significantly lowers toxic levels of phenobarbital. Nephrol Dial Transplant. 2004;19:1663–1664.
23 Ludwigs U, Divino Filho JC, Magnusson A, Berg A. Suicidal chloral hydrate poisoning. J Toxicol Clin Toxicol. 1996;34:97–99.
24 Chudnofsky CR. Safety and efficacy of flumazenil in reversing conscious sedation in the emergency department: Emergency Medicine Conscious Sedation Study Group. Acad Emerg Med. 1997;4:944–950.
25 Gueye PN, Hoffman JR, Taboulet P, et al. Empiric use of flumazenil in comatose patients: limited applicability of criteria to define low risk. Ann Emerg Med. 1996;27:730–735.
26 Spivey WH. Flumazenil and seizures: analysis of 43 cases. Clin Ther. 1992;14:292–305.
27 Yang CC, Deng JF. Utility of flumazenil in zopiclone overdose. Clin Toxicol (Phila). 2008;46:920–921.
28 Hall R, Zisook S. Paradoxical reactions to benzodiazepines. Br J Clin Pharmacol. 1981;11(suppl 1):99S–104S.
29 Paton C. Benzodiazepines and disinhibition: a review. Psychiatr Bull. 2002;26:460–462.
30 Rodrigo CR. Flumazenil reverses paradoxical reaction with midazolam. Anesth Prog. 1991;38:65–68.
31 Honan VJ. Paradoxical reaction to midazolam and control with flumazenil. Gastrointest Endosc. 1994;40:86–88.
32 Thurston TA, Williams CG, Foshee S. Reversal of a paradoxical reaction to midazolam with flumazenil. Anesth Analg. 1996;83:192.
33 Khan LC, Lustik SJ. Treatment of a paradoxical reaction to midazolam with haloperidol. Anesth Analg. 1997;85:213–215.