rhGH Safety and Efficacy Update
Growth hormone (GH) has been used to treat patients with GH deficiency (GHD) since 1960 when it was originally extracted from cadaveric pituitary glands [1]. In 1985, several cases of Creutzfeldt-Jakob disease were identified and attributed to pituitary-derived GH. Later that year, recombinant human GH (rhGH) was approved by the US Food and Drug Administration (FDA). The availability of an unlimited supply of rhGH improved access to therapy for children with GHD and the frequency of dosing increased from 3 times each week to 6 or 7 days each week [2]. The increased supply of rhGH allowed investigation into treatment of multiple conditions associated with short stature not associated with GHD leading to FDA approval for treatment of children with growth failure associated with chronic renal insufficiency (1993), Turner syndrome (TS; 1996), Prader-Willi Syndrome (PWS; 2000), small for gestational age (SGA) without adequate catch-up growth (2001), idiopathic short stature (ISS; 2003), short stature homeobox (SHOX) deficiency (2006), and Noonan syndrome (NS, 2007). In addition, rhGH therapy was FDA approved for acquired immune deficiency syndrome (AIDS) wasting (1996), adult GHD (1996), and short bowel syndrome (2003) (Table 1). Guidelines for the use of GH therapy have been reported previously by the American Association of Clinical Endocrinologists (AACE), Growth Hormone Research Society (GRS), and The Lawson Wilkins Pediatric Endocrinology Society (LWPES) [2,4].

GH registries have been an invaluable resource for determining the safety and efficacy of GH. These registries were originally mandated by the FDA in 1985 because rhGH was the first product approved in the United States derived from recombinant DNA technology. The FDA required Genentech to conduct a 5-year phase IV postmarketing safety study. However, the National Cooperative Growth Study (NCGS) was extended as a phase IV postmarketing safety and efficacy study until 2010 [5]. Other GH registries include GHMonitorSM (Serono) and the ongoing international registries American Norditropin Studies: Web Enabled Research (ANSWER; Novo Nordisk), Genetics and Neuroendocrinology of Short Stature International Study (GeNeSIS, Eli Lilly), and Pfizer International Growth Study (formerly Kabi International Growth Study [KIGS]) as well as national and pharmaceutical registries in many individual countries. Some countries continue to mandate safety and efficacy registries for children receiving GH therapy. The cumulative safety and efficacy data from these multiple studies involves more than 120,000 children treated for nearly half a million patient years. Numerous publications from these studies have educated the pediatric and adult endocrinology community about the safety and efficacy of GH therapy in children and adults.
GH registries have been, and continue to be, primarily conducted through entry of data relating to patient care without requiring adherence to specific practice guidelines. Thus, the data collected reflect the variation in clinical practice among the investigators who participate in the trial. Attempts have been made to use the registries to provide evidence to guide clinical practice [6] and to compare practice patterns with reported knowledge, attitudes, and beliefs [7]. However, one major weakness of these registries is the vast amount of data that are not entered by the participating sites. Although the study sites are compensated for the time required for data entry, because of the volume of data and work involved the number of fields required for payment were limited to high-priority items. Therefore, items not required for payment are less likely to be recorded. However, the sheer volume of these registries overcomes these limitations to some degree.
This article focuses on efficacy data for FDA-approved indications for promoting linear growth in children and available safety data related to rhGH treatment of all conditions in children. Effects that can occur after rhGH therapy is completed, or in individuals who continue rhGH therapy into adulthood, are also addressed.
Efficacy
Since rhGH became available, numerous clinical trials have been performed to show its efficacy in improving linear growth, quality of life, body composition, and bone mineral density [8–11]. Prospective, phase III clinical trials showing efficacy and safety have formed the basis of FDA approval for the numerous indications described earlier. Although small clinical trials have also shown efficacy in numerous other conditions associated with growth failure, including inflammatory bowel disease, chronic steroid use, cystic fibrosis, skeletal dysplasias, hypophosphatemic rickets, and juvenile rheumatoid arthritis, rhGH has not been FDA approved for these conditions. Phase IV postmarketing surveillance and efficacy studies (ie, registries) have also provided significant efficacy and safety information regarding FDA-approved indications and numerous other conditions treated with rhGH.
Pediatric GHD
rhGH was first approved for the treatment of childhood-onset GHD on October 18, 1985. The efficacy of GHD depends on several variables including cause, rhGH dose, and dosing schedule. Children with organic causes of GHD, including congenital panhypopituitarism, multiple pituitary hormone deficiency, and suprasellar tumors, tend to be more sensitive to rhGH and grow well in response to lower doses. However, the growth response in children with idiopathic GHD (IGHD) is more variable and may be predicted by the results of the GH stimulation tests [9]. GHD is diagnosed in children using auxologic features (short stature and/or poor growth velocity), radiologic investigations (bone age, magnetic resonance imaging of brain with focus on pituitary), and biochemical testing (insulin-like growth factor [IGF]-1, IGFBP-3, and GH stimulation testing). Peak GH responses of less than 10 ng/mL to two separate secretory stimuli have been considered the cutoff for abnormally low GH secretion and support a diagnosis of GHD. Because of the variability and lack of reproducibility of GH stimulation tests (GHSTs), a trial of GH therapy has been suggested as part of the diagnostic evaluation of a child who otherwise fits the criteria for GHD but has normal response to GHST. The recommended rhGH dose for children with GHD in the United States is 0.175 to 0.35 mg/kg/wk with 0.3 mg/kg/wk being used most commonly [2–4,6]. Because of the GH sensitivity of children with GHD, at some point in the dose curve, increasing the dosage does not translate into improvement in growth response (Fig. 1) [12]. Adjustment of rhGH dosing (0.4–0.7 mg/kg/wk) during puberty in an attempt to match the normal increase in GH secretion during puberty has also been shown to improve growth velocity during puberty [13]. A stepwise increase in rhGH dose with pubertal stages has been shown to improve near-adult height by 3.6 cm (or +0.49 mean height standard deviation score [SDS]) compared with age-matched historical controls [14].
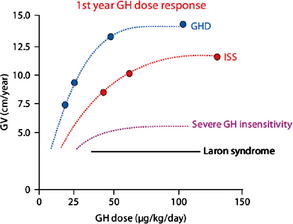
Fig. 1 First-year response to rhGH therapy.
(From Keni J, Cohen P, Keni J, et al. Optimizing growth hormone dosing in children with idiopathic short stature. Horm Res 2009;71 Suppl 1:71; with permission.)
The use of serum IGF-1 values to adjust rhGH dosing has been debated since the reliable assays for IGF-1 (formerly Somatomedin C) became available [15]. The impact of this treatment paradigm was shown in a clinical trial in which weight-based dosing of rhGH was compared with dosing of rhGH to target an IGF-1 value at the mean (0 SDS) or top of the normal range for age (+2 SDS) [16]. Children in the weight-based dosing arm of the study grew similarly to those titrated to an IGF-1 at 0 SDS. However, children whose rhGH doses were titrated to IGF-1 levels at +2 SDS grew significantly better than either other group. An important finding in this study was that the spectrum of rhGH doses that children required to achieve a particular target was highly variable. The weight-based arm was treated with 0.28 mg/kg/wk, whereas the 0 SDS target arm received an average of 0.23 mg/kg/wk and the GHD subjects in the +2 SDS target arm received an average of 0.46 mg/kg/wk. Children with GHD also showed better growth response to treatment in all 3 arms. In a 2006 survey of pediatric endocrinologists, 72% reported that they routinely monitor IGF-1 values in children receiving rhGH [7]. Demonstration of the use of IGF-based dosing in clinical practice was done by reviewing NCGS that which showed that 41% of children with GHD had IGF-1 values entered into the registry between 2000 and 2005 [7]. However, the long-term efficacy of IGF-based dosing has not been tested. In addition, the safety of the higher doses of rhGH required in some children to achieve a serum IGF-1 target of +2 SDS is unclear [17].
Short-term growth responses to rhGH in GHD have been reported in multiple articles [8–11]. The growth velocity (GV) seen in the first 12 months is characteristically the highest GV seen during therapy, with subsequent years remaining at more than the pretreatment values. In mathematical growth predication models, growth in children with the first-year growth response has been shown to be inversely correlated with age and height deficit, but positively correlated with body weight, birth weight, and GH dose [18]. A recent analysis of data from the NCGS provides expected growth responses to rhGH in prepubertal children for the first year of therapy (Fig. 2A–D). This analysis also shows that responsiveness to rhGH decreases with age [6].
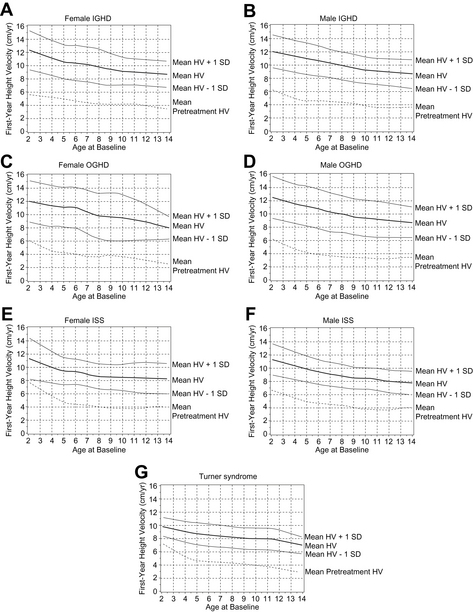
Fig. 2 First-year growth responses to daily rhGH. First-year growth responses to daily GH expressed as height velocity (HV) at age of treatment onset (x-axis) in naive, prepubertal girls and boys with IGHD (A, B), organic GHD (OGHD) (C, D), and ISS (E, F), and girls with TS (G). Data given for mean and mean ± 1 SD.
(From Bakker B, Frane J, Anhalt H, et al. Height velocity targets from the National Cooperative Growth Study for first-year growth hormone responses in short children. J Clin Endocrinol Metab 2008;93(2):355; with permission.)
Efficacy may also be termed lack of failure. However, growth parameters for failure to respond to rhGH therapy have not been well established. Criteria that have been used include a doubling of the pretreatment growth velocity, 2 cm/y faster than pretreatment GV, and 50% faster than pretreatment GV. It has been proposed that the children whose GV is greater than −1 SDS on the Bakker charts are showing an adequate response to therapy. The relationship of the early growth response to rhGH therapy has been shown to correlate with final adult height [19].
Determination of long-term growth response to rhGH using near-adult height has been done for 2 large GH registries [19,20]. In the NCGS, height improved in children with IGHD from −2.7 (± 0.7) SDS to −1.4 (± 1.0) SDS [20]. Enrollment height and GHST response were significant predictors for Δ height SDS in this cohort. In KIGS, median height SDS at the start of treatment were −2.4 (IGHD) and −2.9 (MPHD) for white boys and −2.6 (IGHD) and −3.4 (MPHD) for girls, respectively. With rhGH treatment, height improved to −0.8 (IGHD) and −0.7 (MPHD) for white boys and −1.0 (IGHD) and −1.1 (MPHD) for girls, respectively [19] The first-year increase in height SDS and prepubertal height gain was highly correlated with total height gain.
Chronic renal insufficiency before transplantation
Children with chronic renal insufficiency (CRI) frequently have growth failure that, if untreated, leads to adult short stature. Growth failure in CRI is multifactorial, with GH resistance playing a major role [21]. Before consideration of rhGH therapy, management of modifiable factors that may impair growth in CRI (eg, malnutrition, acidosis, metabolic bone disease, and/or the iatrogenic impact of therapies for CRI) needs to be optimized. rhGH therapy for children with growth failure (height <−1.88 SDS) in the setting of CRI before renal transplant was approved in 1993. GH treatment is recommended at a dosage of 0.35 mg/kg/wk, divided into 6 or 7 doses [2,22]. Treatment with rhGH is effective and has been shown to improve final adult height [22,24]. A recent analysis of data from the NCGS provides expected growth responses to rhGH in prepubertal children with CRI for the first year of therapy (Fig. 3) [25]. To maximize response to rhGH therapy in CRI, it is important that continued attention is paid to the management of modifiable factors that may impair growth. Long-term rhGH treatment of children with CRI showed improvement of height from −2.6 to −0.7 SDS after 5 years [26]. Final adult height was analyzed in a study of 38 children with CRI, showing that rhGH therapy led to improvement from a height of −3.1 ± 1.2 to −1.6 ± 1.2 SDS [23]. A group of 50 age-matched controls with CRI without growth failure at baseline did not receive rhGH therapy and had worsening of their height in the same time period, with a loss of 0.5 SDS [23]. Analysis of adult height in 49 patients with baseline height SDS between −2 and −3 revealed that 65% had an adult height of more than −2 SDS [27].
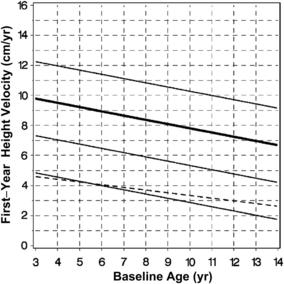
Fig. 3 First-year growth responses to daily rhGH in CRI. First-year growth response (fitted curves) expressed in HV for prepubertal children with chronic kidney disease during rhGH treatment compared with pretreatment HV for the same children.
(From Mahan JD, Warady BA, Frane J, et al. First-year response to rhGH therapy in children with CKD: a National Cooperative Growth Study report. Pediatr Nephrol 2010;25(6):1128; with permission.)
RhGH therapy following renal transplantation has been controversial and is not FDA approved despite multiple studies showing efficacy [28]. Despite a lack of FDA approval, analysis of the North American Pediatric Renal Transplant Cooperative Study (NAPRTCS) database has shown that the number of children receiving rhGH more than 5 years after transplantation is 9% of all patients and 14% of those whose height was less than −1.88 SDS 6 months after transplant [28]. In this population, there was a modest improvement in height of approximately +0.5 SDS following 5 years of rhGH use [28]. Analysis of NAPRTCS data shows that the final adult height in renal transplant recipients who received rhGH therapy (n = 71) compared with renal transplant recipients who did not receive rhGH therapy (n = 669) showed a height gain of +0.77 SDS (−1.83 ± 0.14 rhGH vs −2.60 ± 0.05, P<.001) [29].
Turner syndrome
Short stature is the most common clinical characteristic of TS. A significant proportion of the height deficit in girls with TS is caused by the presence of only a single copy of the SHOX gene (haploinsufficiency). The SHOX gene is a homeobox gene that regulates the growth of long bones. The SHOX gene is located in the pseudoautosomal region of both sex chromosomes and its actions are gene dose related [30]. Growth in children with TS is characterized by a slight intrauterine growth retardation (−1 SD), slow growth during infancy and childhood, and lack of pubertal growth spurt. More than half of girls with TS are at less than the fifth percentile by 2 years of age [31]. The average height of an adult woman with TS who has not received rhGH therapy is 143 cm, approximately 20 cm (8 inches) less than the average adult woman [32].
rhGH therapy was FDA approved in 1996 for treatment of short stature in TS, has been shown to significantly improve adult height in girls with TS, and may allow achievement of height in the normal adult female range [33–36]. Earlier diagnosis and initiation of GH treatment in children with TS should help patients to attain heights near the normal range at a younger age. Early initiation of rhGH therapy enables the children to start estrogen supplementation in a more age-appropriate manner [37]. According to the 2006 Turner Syndrome Consensus Study Group, puberty should not be delayed to promote statural growth because this approach undervalues the psychosocial importance of age-appropriate pubertal maturation [38]. Treatment with rhGH should commence when the child has experienced growth deceleration or is less than or equal to −2SD less than the target adult height based on midparental height. Earlier rhGH treatment of young patients with TS can abrogate the height loss compared with their peers [39]. If the rhGH is initiated later than 10 years, or in cases of extreme short stature, adjunct therapy with nonaromatizable androgens should be considered [40]. Therapy should be continued until final adult height is reached or the height achieved is acceptable [41]. Final height gains may be as high as 8 to 10 cm with or without delaying estrogen-replacement therapy [42–45]. A typical rhGH dose for treatment in children with TS is 0.35 mg/kg/wk [2,6] and is FDA approved up to 0.469 mg/kg/wk. Higher doses of rhGH may be associated with increased height gain [46], although the potential for adverse late effects has not been evaluated adequately. rhGH dose seems to be the most important predictor of height velocity in the first year of rhGH therapy, whereas, for height gain in years 2 to 4 of therapy, the velocity during the previous year is the most important predictor [46]. A recent analysis of data from the NCGS provides expected growth responses to rhGH in prepubertal children with TS for the first year of therapy (see Fig. 2G) [6].
The usefulness of monitoring IGF-1 levels during rhGH therapy in TS has not been well studied. In one study of 24 prepubertal children with TS, IGF-1 values were low at baseline, improved at 3 and 6 months after starting rhGH therapy, but returned to baseline at 12 months [47]. Although the IGF-1 response does not seem to have a clear relationship with rhGH dose [47] in girls with TS, rhGH-induced IGF-1 levels have been shown to correlate with the growth response [48]. In the largest randomized study of rhGH therapy for TS treated to near-final height, in which 232 children with TS received either 0.27 or 0.36 mg/kg/wk, baseline IGF-I concentrations were low normal and remained within 2 SDS of the mean during GH therapy in all but 2 subjects [34].
Short stature from prader-willi syndrome
RhGH treatment was FDA approved in 2000 for the treatment of growth failure in children with PWS. The FDA-approved dose is 0.24 mg/kg/wk with recommended rhGH dosing between 0.24 and 0.35 mg/kg/wk [4]. rhGH treatment in PWS has been proved to improve linear growth and increase muscle mass/body fat ratios [49,51]. Improved muscle tone and strength have also been clinically observed in children with PWS treated with rhGH [49,51]. GH therapy has also been shown to provide neurodevelopmental benefits in young children with PWS [51]. This finding suggests additional benefits of rhGH treatment of younger PWS children with growth failure and hypotonia. Near-adult height in PWS is also shown to be improved with rhGH therapy. In a study of 22 children with genetically-confirmed PWS treated with rhGH (0.30 mg/kg/wk) from age 6.9 to 18.1 years, the near-adult height for all children was within midparental height median −0.5 SDS (−1.4 to 0.7) and 0.9 SDS (0.1–1.9) for girls and boys, respectively [52]. Analysis of growth data in the Pfizer International Growth Study (KIGS) shows that two-thirds of children with PWS achieved a height within the normal range [52,53].
The impact of rhGH treatment on obesity in children with PWS is controversial, and stringent weight management techniques are necessary for all children with PWS regardless of rhGH treatment status. However, multiple studies with rhGH have shown overall improvement in body composition. KIGS data indicate that there is a decrease in body mass index (BMI) in the first year of GH therapy. BMI then tends to stabilize as a result of an increase in muscle mass and a decrease in fat mass associated with rhGH therapy [53]. Furthermore, at final adult height, body composition has been shown to be improved, but not normalized [52].
Small for gestational age
SGA (<−2 SD for birth weight and/or birth length) is defined at different gestational ages based on data from Usher and McLean [54]. The incidence of children born SGA in the United States has been estimated at 8.6% [55]. Eighty-five percent of children born SGA catch up to the normal curve by the age of 2 years, with most of them catching up by the age of 6 months [56,57]. Children who have not shown adequate catch-up growth by 2 years of age are unlikely to attain a normal adult height without rhGH therapy [56,58]. Children who are SGA have relative GH resistance, but rhGH therapy has been shown to improve final adult height and was FDA approved in 2001 [59,60]. Approved rhGH doses for treatment of SGA are up to 0.49 mg/kg/wk. Mathematic prediction models of the growth response of SGA children to rhGH show that rhGH dose is the most important predictor of the first-year growth response [61]. Age at start of therapy is negatively correlated with the first-year growth response [61]. Subsequent growth is dependent on the first-year response to rhGH therapy. Investigations comparing continuous and intermittent rhGH therapy in SGA children showed similar growth responses over time [59]. However, because of the degree of catch-down growth seen between therapy courses with intermittent therapy, continuous therapy has been adopted [62]. In a final height study of continuous rhGH therapy, 54 children started treatment with rhGH at 8.1 (±1.9) years at a dose of 0.23 or 0.46 mg/kg/wk. Final adult height was −1.1 (± 0.7) SDS in those who received 0.23 mg/kg/wk and −0.9 (±0.8) SDS in those who received 0.46 mg/kg/wk. rhGH treatment resulted in an adult height more than −2 SDS in 85% of the children after a mean rhGH treatment period of 7.8 (± 1.7) years [57].
Idiopathic short stature
Short stature is commonly defined as a height less than the third percentile (2 SD <the mean) compared with children of the same age and gender. Using this statistical definition, 1.8 million children (aged 5–14 years) in the United States have short stature at any given time [63]. The prevalence of short stature (<third percentile) and poor growth (GV<5 cm/y) has been predicted as 0.7% of children (420,000; 1:143) in a school-based study [64]. The rate of referral of children with short stature and poor growth has a major impact on the number of children prescribed GH. Estimates of the referral rate vary from 2.5% by records review (Adda Grimberg, personal communications, 2006) to 34% by hypothetical survey [65]. Assuming a 20% referral rate, 84,000 children could be expected to be evaluated at some point between the ages of 4 and 15 years. If 35% of children referred to a pediatric endocrinologist for short stature have a known pathologic condition that would respond to GH therapy [5,66], 65% of referrals (54,600 children) for short stature and poor growth to have no known cause for their growth failure. This value correlates well with data from the NCGS showing that 60.2% of children receiving GH therapy are in a combined category of IGHD and ISS [67].
By definition, ISS excludes known causes of short stature [68]. However, ISS most certainly includes a subset of children with GHD that have a normal response to GHST. The basis of the diagnosis can affect the interpretation of efficacy. For example, the cutoff for the GH peak is moved following GHST to diagnose GHD from less than 10 down to less than 7, less than 5, or less than 3, the spectrum of ISS and its response to rhGH therapy expands. Therefore, the ability to describe the cause of short stature in children with ISS is limited by the current diagnostic tools [69]. Many children with undiagnosed conditions that result in severe short stature respond to rhGH therapy [70,72].
The use of rhGH in ISS has been the focus of both ethical and financial controversies. Multiple natural history studies report that children with ISS have short stature as adults. Hintz and colleagues [70] showed that GH treatment in children with ISS resulted in an increase in final height of 5 cm compared with predicted height. A larger increase (9 cm) was seen when the final height of treated patients was compared with historical controls. In the only randomized, placebo-controlled trial of rhGH therapy for 68 children with ISS to final height, adult height was greater in the rhGH-treated group (−1.81 ± 0.11 SDS) than in the placebo-treated group (−2.32 ± 0.17 SDS) by 0.51 SDS (3.7 cm) [71]. In a meta-analysis of 40 studies of GH therapy in ISS, the investigators concluded that adult height was increased by 4 to 6 cm in those treated with GH [73]. However, they estimated the cost of treatment at $35,000 per inch. In May, 2003, the FDA approved the use of rhGH at a dose of up to 0.37 mg/kg/wk in children with ISS who are less than −2.25 height SDS and who are unlikely to catch up in height. A subsequent final height trial with higher dose and more frequent administration showed improved height outcome. This study compared the adult height of 50 patients with ISS given daily injections of GH at dosages of 0.24 mg/kg/wk, 0.24 mg/kg/wk for the first year and 0.37 mg/kg/wk thereafter, or 0.37 mg/kg/wk [72]. Mean height SDS increased by 1.55, 1.52, and 1.85, respectively. Final heights were 7.2 cm and 5.4 cm more than pretreatment predicted final heights for the 0.37 mg/kg/wk and 0.24 mg/kg/wk dose groups, respectively [72]. In addition, in those children who received 0.24 mg/kg/wk, 71% of final height SDS values were within the normal range and in those who received 0.37 mg/kg/wk, 94% of final height SDS values (all but 1) were within the normal range.
Determination of long-term growth response to rhGH for children with ISS using near-adult height has been done for 2 large GH registries [20,74]. In the NCGS, height improved in children with ISS from −2.8 (±0.6) SDS to −1.6 (±1.0) SDS [20]. The Δ height SDS for ISS was similar to that seen for IGHD. However, enrollment height and GHST response were not significant predictors of growth response in ISS. In the Pfizer International Growth Study (formerly KIGS), height improved in children with ISS from −2.5 to −1.4 SDS [74]. Age at onset of rhGH therapy and first-year responsiveness are the strongest predictors of long-term growth response in ISS.
The use of serum IGF-1 values to adjust rhGH dosing in ISS is also controversial. The impact of this treatment paradigm in ISS was shown in a clinical trial in which weight-based dosing of rhGH was compared with dosing of rhGH to target an IGF-1 value at the mean (0 SDS) or top of the normal range for age (+2 SDS) [16]. Children in the weight-based dosing arm of the study grew similarly to those titrated to an IGF-1 at 0 SDS. However, children whose rhGH doses were titrated to IGF-1 levels at +2 SDS grew significantly better than either other group. An important finding in this study was that the spectrum of rhGH doses that children required to achieve a particular target was highly variable. The weight-based arm was treated with 0.28 mg/kg/wk, whereas the 0 SDS target arm received an average of 0.23 mg/kg/wk and the ISS subjects in the +2 SDS target arm received a much higher average of 0.83 mg/kg/wk. This finding indicates that ISS represents partial GH insensitivity. Children with ISS also showed a poorer growth response to treatment in all 3 arms compared with children with GHD: Δ height SDS was 2.04 (±0.17) for GHD and 1.33 (±0.09) for ISS groups in those with IGF-1 values targeted at +2 SDS and 1.41 (±0.13) for children with GHD, and 0.84 (±0.07) for those with ISS with IGF-1 values targeted at 0 SDS [75]. This result indicates that ISS also represents a state of partial IGF-1-insensitivity. Demonstration of the use of IGF-based dosing in clinical practice was done by evaluation of IGF-1 data entered into NCGS, which showed that, when children with ISS were growing poorly, the dose was more likely to be changed if an IGF-1 value was obtained [7]. However, the long-term efficacy of IGF-based dosing in ISS has not been tested. In addition, the safety of the higher doses of rhGH required in some children to achieve a serum IGF-1 target of +2 SDS is unclear [17].
SHOX deficiency
SHOX is produced by the short stature homeobox gene in the pseudoautosomal regions on the distal ends of the X and Y chromosomes. Absence of 1 copy (haploinsufficiency) of the shox gene, caused by absence of X chromosome material, is a significant component of short stature in TS. The shox gene encodes a transcription factor that is important in regulating the growth of long bones. SHOX deficiency (SHOX-D), through mutation or deletion of the shox gene, is present in most individuals (∼70%) with Leri-Weill dyschondrosteosis (LWD) and in a small, but significant, number (2%–15%) of children with ISS [76,78].
There is a single published, randomized trial of rhGH in short prepubertal children with a molecularly proved SHOX gene defect [30]. In this multinational study, children with SHOX-D with both ISS and LWD phenotypes were randomized to treatment with rhGH. A comparison group of prepubertal girls with TS also received rhGH therapy. After 1 year, children with SHOX-D receiving rhGH therapy (n = 27, 12 boys; 0.35 mg/kg/wk; 8.7 ± 0.3 cm/y) showed a significant improvement in growth compared with untreated controls (n = 25, 12 boys; 5.2 ± 0.2 cm/y, P<.0001). The 1-year growth response was also comparable with a group of rhGH-treated children with TS (n = 26; 0.35 μg/kg/wk; 8.9 ± 0.4 cm/y, P = .592). After 2 years, rhGH-treated SHOX-deficient children had grown 5.9 cm more and gained +0.9 height SDS more than untreated controls (P<.001) [30]. rhGH therapy for SHOX-D was FDA approved in 2006 at a dose of 0.35 mg/kg/wk. A retrospective analysis of rhGH therapy showed similar height gain at final height in individuals with SHOX-D (n = 14, + 1.1± 0.2 SDS) to those with TS (n = 157, + 1.2 ± 0.1 SDS, P = .708) [79]. However, individuals with SHOX-D showed a trend toward shorter final heights than those with TS (149.0 ± 6.7 vs 151.4 ± 5.1 cm, P = .109) and had a lower final adult height SDS (−2.2 ± 0.8 vs −1.7 ± 0.8, P = .016) [79]. Further studies are needed to provide additional near-adult height efficacy data in this condition.
Noonan syndrome
NS, characterized by hypertelorism, down-slanting palpebral fissures, low-set posteriorly rotated ears, small chin, short neck, short stature, chest deformities, and heart defects (especially valvular pulmonic stenosis), was first described in 1963 and has an estimated incidence of 1 in 1000 to 1 in 2500 live births [80]. Mutations in multiple different genes involved in intracellular signaling (PTPN11, K-Ras, SOS1, Raf1, B-Raf, SHOC2, and N-Ras) have been found to cause NS in 61% of cases [80]. Short stature during childhood, caused by postnatal growth failure, is seen in more than 50% of individuals with NS. In untreated individuals with NS, mean adult height at age 19 years has been reported as 162.5 (±5.4) cm in men and 152.7 (±5.7) cm in girls [81]. However, because of the delayed puberty common to NS, untreated final adult height of boys with NS in another cohort was 167.4 cm [82]. The adult height of untreated North American individuals with NS is less than the third percentile in 54.5% of women (<151 cm) and 38% of men (<163.2 cm) [80]. There is a genotype-phenotype correlation, with individuals with NS caused by SOS1 mutations more likely to have normal stature and individuals with PTPN11 mutations more likely to have short stature [83,84]. Reports of GH secretion vary in the incidence of GHD in NS [85]. However, IGF-1 deficiency is a frequent finding in individuals with NS [80,85,86].
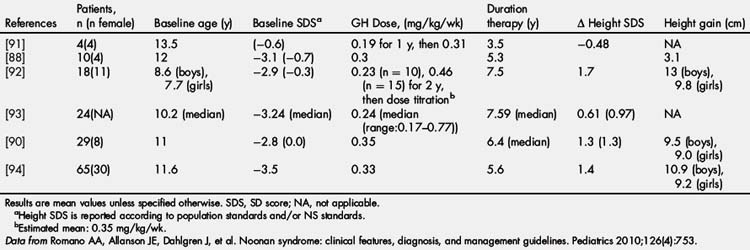
rhGH was FDA approved for the treatment of short stature in NS in 2007 at a dose of up to 0.462 mg/kg/wk. Early studies of rhGH therapy in NS showed only mildly improved height SDS and GV with therapy [87] or variable improvement with definite responders and nonresponders [88]. Short-term treatment studies showed poorer linear growth and IGF-1 generation to rhGH therapy in those with PTPN11 mutations [86,89]. However, long-term rhGH treatment studies have shown no difference in growth response between PTPN11-positive and PTPN11-negative individuals [90]. Long-term efficacy is shown in Table 2, with improved outcomes occurring with earlier initiation and longer duration of rhGH therapy [80]. Long-term height gains range from +1.3 to 1.7 SDS or 9.5 to 13 cm in boys and 9.0 to 9.8 cm in girls [80].
Safety
When considering a therapy for a medical condition, one of the guiding principles for physicians is Primum non nocere (First, do no harm). Therefore, when considering GH therapy for treatment of GHD and short stature, the medical, physical, psychological, and psychosocial impacts of the disease or condition need to be considered. The potential therapeutic benefit of rhGH therapy then needs to be balanced with the known and potential short-term and long-term side effects. This article defines the short-term side effects as those that can occur while receiving rhGH therapy during childhood, and the long-term side effects as those that occur after rhGH therapy has been discontinued or in those individuals who continue rhGH therapy into adulthood.
Short-term safety
GH registries have been an invaluable resource for determining the safety and efficacy of GH. Such postmarketing surveillance is necessary to accurately assess the safety profile of a drug because of the inability of clinical trials to identify rare side effects. In addition, spontaneous postmarketing adverse event reporting may be incomplete and does not allow for the determination of incidence rates for side effects. The cumulative safety data from the 2 largest studies (KIGS and NCGS) represent more than 120,000 children treated for nearly half a million patient years. Based on the most recent NCGS safety publication, the cumulative enrollment in the NCGS from December 1985 to January 1 2006 was 54,996 patients comprising 195,419 patient years of treatment with Genentech’s rhGH products [95]. Additional safety data were collected with continued enrollment in NCGS until December 2010 and data collection until June 2011 when the study was discontinued. Based on the most recent KIGS safety publication, the cumulative enrollment in the KIGS from January 1987 to August, 2008 was 58,603 patients comprising 197,173 patient years of treatment with Kabi, Pharmacia, and Pfizer’s rhGH products [96]. Additional safety and efficacy data continue to be collected with ongoing US and international enrollment in KIGS. GHMonitorSM enrolled 1335 subjects from January 2003 to August 2006 and is no longer collecting US data [97]. Safety and efficacy data also continue to be collected in the ongoing international registries ANSWER (5797 subjects enrolled from January 2002 to November 2009) [98] and GeNeSIS (estimated enrollment 23,500 from April 1999 to March 2010) [99].
The major safety questions that have been raised regarding rhGH therapy have been the risks of mortality and malignancy. The risk of mortality has focused particularly on sudden death in children with PWS, aortic dissection in children with TS, and cancer survivors. The risk of malignancy has focused particularly on children at increased risk for malignancy, including cancer survivors and those with genetic conditions predisposed to tumor formation.
Short-term risk of mortality
From the 54,996 patients enrolled in NCGS, there were 174 reported deaths, 19 (11%) of which were assessed as related to rhGH by the investigator, 4 were designated as not assessable, and no causality was provided for 21. Most of the 19 deaths assessed as related to rhGH (12 of 19) were caused by neoplasms (Table 3) [95]. The other 7 deaths assessed as related to rhGH were isolated cases as follows: cerebral edema in a patient with a glioma resection, and cerebral edema in a patient with a mitochondrial myopathy and hypertrophic cardiomyopathy; arrhythmia in a patient with 2 renal transplants with hypertrophic cardiomyopathy and coronary atherosclerosis at autopsy; diabetic hyperosmolar coma in a patient with craniopharyngioma who was receiving rhGH at the time the hyperosmolar coma developed; depression after stopping rhGH in a 21-year-old man with thalassemia major, with subsequent death from an overdose of street drugs; complications of neurofibromatosis in a patient with an optic glioma, an odontoma, ataxia, and facial swelling; and sudden death in a 4-year-old boy with PWS [95].
Mortality in prader-willi syndrome
The annual mortality in PWS has been described as 3% across all age groups and is increased particularly in relation to complications of obesity [100]. A review of all reported deaths in PWS conducted in 2007 identified 64 deaths in children up to age 19 years with PWS (42 boys) [101]. Twenty-eight (44%) of these children received rhGH treatment. Respiratory disorders were the most common cause of death (61%; 68% of rhGH treated, 56% of untreated). Seventy-five percent of deaths occurred in the first 9 months after the initiation of rhGH therapy. No significant differences were found between the groups in gender, prevalence of obesity, or sleep apnea. More deaths in the untreated group occurred in children less than 3 years of age (25 deaths, 22 untreated). However, deaths in those receiving rhGH were more evenly distributed by age.
It has been hypothesized that the causal connection between rhGH therapy and sudden death in PWS is the induction of tonsillar and adenoid hypertrophy. However, sleep studies have shown an improvement of sleep apnea, particularly central apnea, and 1 death has been reported in a child with a normal polysomnograph before and after initiation of rhGH therapy [102,103].
Increases in IGF-1 are associated with tonsillar hypertrophy. This finding suggests that it may be safer to start at low doses of rhGH and titrate the rhGH dose using IGF-1 values. However, there are no studies to support this practice. Tonsillectomy and adenoidectomy have also been proposed as a treatment of sleep apnea. However, this treatment may be dangerous in children with PWS and may not improve the sleep apnea [102].
Therefore, although polysomnography has been recommended before starting rhGH treatment, particularly in the presence of snoring or obesity, there is limited evidence that polysomnography is beneficial, because a normal result cannot guarantee 100% safety. It should be emphasized that PWS is a condition with an inherently high mortality risk. Children with PWS, particularly with obesity, sleep apnea, and respiratory illness, require careful clinical management whether or not they are receiving rhGH therapy. In those children with PWS receiving rhGH therapy, awareness of the risk for sudden death should always be present, but particularly during the first 9 months of treatment. Although a causal link between rhGH therapy, respiratory illness, and sudden death has not been established, discontinuation of rhGH therapy is recommended during respiratory illnesses. In addition, despite the potential metabolic and body composition benefits, rhGH therapy in severely obese children with PWS is contraindicated.
Mortality in turner syndrome
Aortic dissection, an often fatal event, occurs in 1% to 2% of the TS population and is usually preceded by dilatation of the aortic root and/or ascending aorta. Dissection occurs early in life, at a median age of 35 years (range 18–61 years) [104]. Longitudinal imaging every 5 to 10 years to assess aortic diameters is recommended, even in those with a normal initial cardiac study [105]. In the NCGS, 5 deaths from aortic dissection/rupture were reported among 5127 patients with TS [95].
Short-term risk of malignancy
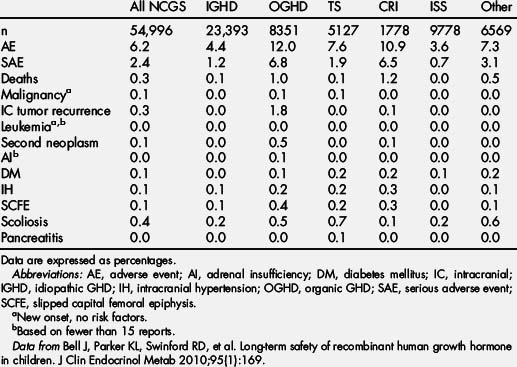
Since the inception of GH therapy, there has been ongoing concern about the potential for GH therapy to promote tumorigenesis either during therapy or in the period following therapy. Increased levels of IGF-I are associated with an increased risk of developing breast, prostate, and colon cancer in adults [106,108]. In addition, GH and IGF-1 signaling have become adjunctive targets in the treatment of certain forms of cancer [109]. Lessons from animal models have suggested that GH and IGF-1 play permissive rather than causative roles in tumorigenesis [110,111]. However, since the first report of leukemia in a child receiving rhGH in 1988 [112], the possibility of an increased risk of cancer in patients receiving rhGH therapy has been investigated. Because of evidence of a link between increased IGF-1 levels and certain forms of cancer [106,108], the concern about the potential of rhGH therapy as a child affecting the long-term risk of cancer after GH therapy resulted in long-term follow-up studies of GH-treated individuals. In one such study of childhood pituitary-derived GH-treated individuals, 2 cases of colorectal cancer were identified in a population in which 1 or less was expected [113]. In the Childhood Cancer Survivor Study, long-term follow-up of childhood cancer survivors has shown that there is an increased risk of second neoplasm developing in GH-treated children years after treatment (Fig. 4) [114]. In this study, the rate ratio of GH-treated survivors developing a second neoplasm was 2.15 (95% confidence interval [CI], 1.3–3.5; P<.002) compared with non–GH-treated survivors (Table 4) [114].
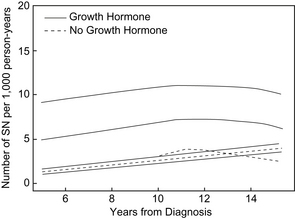
Fig. 4 Incidence of second neoplasms. Comparison of the number of second neoplasms estimated per 1000 person years for survivors who did and did not receive treatment with rhGH, plotted against time from diagnosis. The plot includes 95% confidence intervals.
(From Ergun-Longmire B, Mertens AC, Mitby P, et al. Growth hormone treatment and risk of second neoplasms in the childhood cancer survivor. J Clin Endocrinol Metab 2006;91(9):3496; with permission.)
Table 4 Risk factors for occurrence of second neoplasms
| Covariate | Risk ratio (95% CI) | P |
|---|---|---|
| Univariate model | ||
| Sex | — | <0.0001 |
| Female | 1.00 | — |
| Male | 0.53 (0.44–0.62) | — |
| Age at primary diagnosis (risk/y) | 1.08 (1.06–1.09) | <0.0001 |
| Alkylating agent | — | 0.0002 |
| No | 1.00 | — |
| Yes | 1.41 (1.18–1.69) | — |
| Radiation | — | <0.0001 |
| No | 1.00 | — |
| Yes | 3.00 (2.32–3.87) | — |
| Chemotherapy | — | 0.67 |
| No | 1.00 | — |
| Yes | 0.96 (0.78–1.17) | — |
| GH | — | 0.004 |
| No | 1.00 | — |
| Yes | 1.92 (1.22–2.99) | — |
| Multivariate model | — | — |
| Sex | — | <0.0001 |
| Female | 1.00 | — |
| Male | 0.52 (0.43–0.63) | — |
| Age at diagnosis (risk/y) | 1.07 (1.06–1.09) | <0.0001 |
| Alkylating agent | — | 0.004 |
| No | 1.00 | — |
| Yes | 1.30 (1.09–1.56) | — |
| Radiation | — | <0.0001 |
| No | 1.00 | — |
| Yes | 2.88 (2.20–3.78) | — |
| GH | — | 0.002 |
| No | 1.00 | — |
| Yes | 2.15 (1.33–3.47) | — |
Data from Ergun-Longmire B, Mertens AC, Mitby P, et al. Growth hormone treatment and risk of second neoplasms in the childhood cancer survivor. J Clin Endocrinol Metab 2006;91(9):3496.
This study included data from children who received pituitary-derived GH, rhGH, or both. Meningiomas were the most common second neoplasm (9/20). All individuals who developed meningiomas as second neoplasms had previously received radiation to the head or brain [114]. After adjusting for covariates, the relative risk of death for GH-treated patients compared with those not treated with GH was not statistically increased (1.20; 95% CI, 0.81–1.79; P = .36).
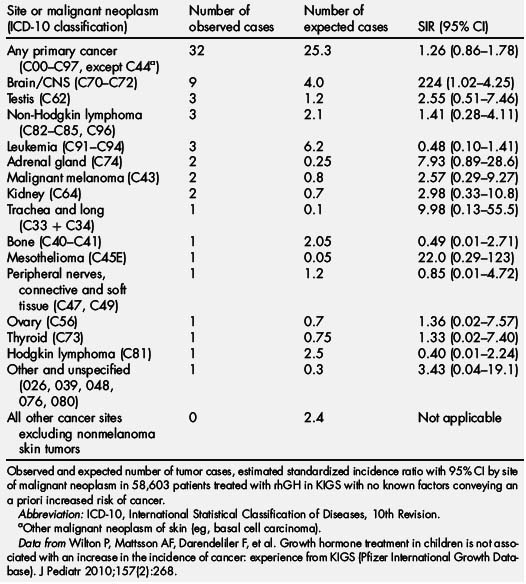
In the KIGS database, a total of 32 new malignant neoplasms were reported in 58,603 patients, versus the 25.3 expected (incidence, 16.4 per 100,000 patient years; standardized incidence ratio [SIR] 1.26; 95% CI, 0.86–1.78; Table 5) [96]. These 32 new malignant neoplasms were diagnosed at a mean age of 11.9 years (range, 5.0–17.6 years). The mean duration of rhGH therapy in KIGS before the diagnosis of cancer was 3.6 years (range, 0.08–9.70 years). Eight of 32 patients with a new cancer were treated with rhGH before enrollment in KIGS for a median of 0.7 years (range, 0.06–4.5 years). In 6 of 7626 patients observed in KIGS after rhGH was discontinued, a malignant neoplasm was reported for as long as 3.1 years after stopping GH. The mean dose of GH in patients in whom a neoplasm developed was 0.26 (±0.08) mg/kg/wk (range, 0.12–0.48). Serum IGF-I levels measured 1 to 12 months before the onset of a neoplasm were only available for 7 patients. Three patients had IGF-I SDS greater than 0 (range, 0.6–1.7), 4 patients had IGF SDS between −0.6 and −1.9, and none were greater than the normal range.
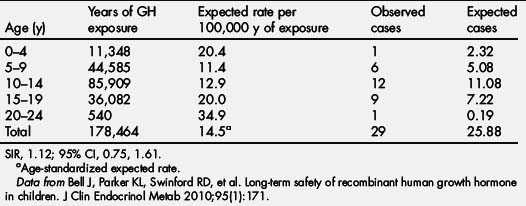
In the NCGS database, intracranial malignancies were reported in 243 patients, 44 of which were new-onset malignancies. Of the 44 new-onset intracranial malignancies, 15 had no predisposing factors. Extracranial malignancies, including leukemia, were reported in 87 patients, with 63 new-onset malignancies. Of the 63 new-onset tumors, 21 had no predisposing factors (Tables 6 and 7) [95]. Using the 29 children with new-onset tumors with confirmed NCGS enrollment, a comparison with the risk of new-onset tumor in the general pediatric population yielded an SIR that was not significantly increased (1.12; 95% CI 0.75–1.61; see Table 4 and Bell and colleagues [95]).
Table 6 New-onset malignancies in patients without risk factors: tumor types in 2 or more patients
| Malignancy | No. of patients (%) |
|---|---|
| Unspecified brain neoplasm | 5 (13.8) |
| Glioma | 4 (11.1) |
| Lymphoma | 4 (11.1) |
| Leukemia | 3 (8.3) |
| Thyroid carcinoma | 3 (8.3) |
| Astrocytoma | 2 (5.5) |
| Germ cell tumor | 2 (5.5) |
| Osteogenic sarcoma | 2 (5.5) |
| Soft tissue sarcoma | 2 (5.5) |
| Other | 9 (25.0) |
| Total | 36 (100) |
Data from Bell J, Parker KL, Swinford RD, et al. Long-term safety of recombinant human growth hormone in children. J Clin Endocrinol Metab 2010;95(1):171.
Based on these results, the large volume of data from the KIGS and NCGS databases provide no evidence that rhGH therapy in childhood growth disorders results in an increased risk of developing cancer relative to that expected in the normal population. However, surveillance for an extended time should continue to allow further assessment [96].
Other targeted safety events
Based on data collected during preclinical trials and postmarketing surveillance registries, the product insert for the various rhGH products includes several safety concerns. Many of these, including headaches and otitis media, are common in the general pediatric population and are minor concerns. Injection site lipoatrophy is a cosmetic concern that can be eliminated by using appropriate rotation of injection sites. Injection site rashes and allergic reactions are concerns with any injectable material and may be related to the diluent rather than the rhGH. Borderline hypothyroidism (primary or secondary) may be uncovered with rhGH therapy. Therefore, thyroid functions should be monitored during rhGH treatment. Fluid retention and edema are common side effects of rhGH therapy in adults, but are seen less in children and adolescents. Other targeted safety events including diabetes mellitus, intracranial hypertension, scoliosis, pancreatitis, adrenal insufficiency, and slipped capital femoral epiphysis are described later. In addition, concerns about rhGH therapy for short stature caused by CRI before or following renal transplantation are discussed.
Diabetes mellitus
Diabetes mellitus (DM) has been reported in 65 patients; 37 type 1 DM (T1DM), 20 type 2 DM (T2DM), and 8 unclassifiable [95]. The 33 T1DM cases, compared with that expected and adapted to an age-matched population, yielded an SIR of 0.90 (observed 33; expected 36.8). Based on the mechanism of action of rhGH, it would be unexpected that the incidence of T1DM would be increased. However, because of the relative insulin resistance caused by increased GH secretion or rhGH administration, it would be more likely for a child to develop T2DM while receiving rhGH therapy. If the 8 unclassifiable cases are considered T2DM, the incidence would be 14 per 100,000 patient years. However, it is difficult to compare this incidence with the general pediatric population because of the changing incidence of T2DM during the years of the NCGS database. It is also difficult to determine whether rhGH therapy causes a long-term increase in the risk of T2DM because the insulin resistance would be reduced once rhGH therapy was discontinued.
Intracranial hypertension
Although intracranial hypertension (IH), also known as pseudotumor cerebri, has been identified in children receiving rhGH therapy, no known mechanism has been identified. The annual incidence of IH in the general pediatric population is 0.9 per 100,000 [115]. IH was reported in 61 confirmed patients in the NCGS, with most events occurring within 6 months of starting rhGH [95]. Thus, the incidence of IH in children receiving rhGH therapy reported in the NCGS is 31 per 100,000 patient years [95]. IH was reported in 41 children with confirmed KIGS receiving rhGH therapy, with an incidence of 28 per 100,000 patient years [116].The highest incidence is seen in children with TS, organic GHD, PWS, and CRI [95,116]. Even though only 2 cases of IH have been seen in ISS, this is still higher than the general population [95]. It is unclear whether this is an ascertainment bias. However, IH in children receiving rhGH is generally resolved with discontinuation of rhGH therapy for a brief period followed by resumption of rhGH at a lower dose.
Scoliosis
rhGH therapy does not cause scoliosis. However, it can make it worse so that previously unrecognized scoliosis becomes evident or known scoliosis progresses. Scoliosis was reported in 238 patients in the NCGS of which 76 were preexisting. Scoliosis was more common in TS and PWS. Among the 64 patients with serious scoliosis, 37 reported surgical treatment. It is recommended that clinicians monitor regularly for the development or worsening of scoliosis. If scoliosis is present and/or worsening, involvement of a pediatric orthopedist is recommended. rhGH therapy in children with severe or worsening scoliosis should be monitored closely and should involve ongoing communication between the pediatric orthopedist and pediatric endocrinologist.
Pancreatitis
Pancreatitis has been reported in 10 children receiving rhGH therapy in the NCGS database and 1 individual 5 months after discontinuation of rhGH therapy. These data provide an incidence of 5.2 per 100,000 in rhGH-treated individuals, and yield an SIR of 1.44 and 95% CI of 0.70 to 2.67 [95]. The annual background incidence of pancreatitis is 3.6 per 100,000 in the pediatric age group [117]. These data fail to show a clearly significant relationship between rhGH therapy and pancreatitis.
Adrenal insufficiency
Concern about adrenal insufficiency (AI) in children with multiple pituitary hormone deficiencies has arisen because of the impact of GH on cortisol metabolism. The microsomal enzyme 11β-hydroxysteroid dehydrogenase type 1 (11βHSD-1) is required for conversion of cortisone to its active metabolite, cortisol. GH inhibits 11βHSD-1 activity [118]. Therefore, children with unrecognized central AI may develop an adrenal crisis caused by rhGH therapy. Alternatively, individuals with known central AI may require higher maintenance and stress doses of steroids when rhGH therapy is initiated [119]. Close dose monitoring and possible increase would be particularly needed in individuals receiving prednisone or cortisone acetate who require 11βHSD-1 activity for conversion to cortisol. In patients with organic GHD (OGHD) and idiopathic panhypopituitarism in the NCGS, 11 events consistent with acute AI, including 4 deaths, were recorded [95]. The time from onset of rhGH therapy to the event of AI ranged from within 24 hours of initiation to 7.4 years, with a mean of 2.5 years. Five nonfatal cases were assessed as serious by the investigator. Although rhGH may facilitate the occurrence of AI, patients with hypopituitarism are at lifelong risk for AI and sudden death whether or not they are receiving rhGH [120,121].
Slipped capital femoral epiphysis
The annual incidence of slipped capital femoral epiphysis (SCFE) in the general pediatric population is 2 to 13 per 100,000 [122,123]. The risk of SCFE is increased in obesity, hypogonadism, multiple pituitary hormone deficiency, and rapid growth during puberty. SCFE was reported in 93 confirmed patients in the NCGS receiving rhGH [95]. Thus the incidence of SCFE in children receiving rhGH therapy reported in the NCGS is 48 per 100,000 patient years [95]. SCFE was reported in 52 confirmed children in the KIGS receiving rhGH therapy, with an incidence of 73 per 100,000 patient years [116].The highest incidence is seen in children with TS, organic GHD, and CRI [95,116]. Wider growth plates and relative osteopenia seen in GHD, SHOX deficiency in TS, and metabolic bone disease in CRI may predispose the growth plates to slip during rapid growth [95,116].
Chronic renal insufficiency after transplantation
Primarily, there has been concern that rhGH therapy would increase the frequency of solid organ rejection. Data from the NAPRTCS showed no increase in rejection episodes in children receiving rhGH therapy after renal transplant compared with children not receiving rhGH. A potential increased risk of rejection was seen in patients receiving rhGH with a history of frequent previous rejection episodes [124,125]. However, the risk of rejection is also increased in children with a history of frequent previous rejection episodes who have not received rhGH. The use of rhGH before transplantation was associated with a borderline higher risk for posttransplant lymphoproliferative disorder (PTLD; odds ratio 1.88, 95% CI 1.00–3.55, P = .05). In contrast, use of rhGH during dialysis or after transplantation only was not associated with a higher risk for PTLD [126]. Because of these concerns, rhGH therapy in children following renal transplantation should be carefully considered by a multispecialty team including pediatric nephrologists, pediatric endocrinologists, and transplant surgeons.
Long-term safety
For the purposes of this article, long-term side effects are defined as those that occur after rhGH therapy has been discontinued or in those individuals who continue rhGH therapy into adulthood. In children with organic GHD and severe isolated GHD, rhGH therapy should be considered replacement therapy and, if replacement doses are not excessive, is not expected to have long-term negative side effects. However, subcutaneous doses given once daily are unlikely to match the natural profile of pulsatile GH secretion. If larger doses are given for OGHD or IGHD, or when treating conditions without GHD, then there may be more risk of long-term negative effects. The relative safety of rhGH during therapy, as documented in numerous publications and described earlier in this article ,makes long-term side effects less likely.
Measurement of IGF-1 and IGFBP-3 levels during therapy started as a measure of efficacy and compliance [15]. With the identification of a correlation between high IGF-1 levels and an increased risk of cancer in adults, monitoring IGF-1 and IGFBP-3 has become a safety practice [15]. However, it is unclear whether increased levels of IGF-1 or IGFBP-3 during childhood correlate with any adverse effects in adults. Studies showing an increased risk of cancer with high IGF-1 levels in adults show that the highest risk occurs in those with increased IGF-1 and low IGFBP-3 (Fig. 5) [15]. This finding may be caused by increased levels of free IGF-1, reduced IGF-independent actions of IGFBP-3, or both. Low levels of IGF-1 with high IGFBP-3 in adulthood are associated with an increased risk of cardiovascular disease [15].
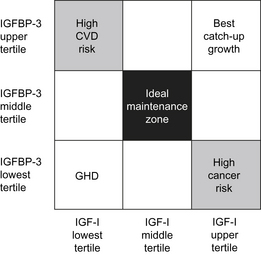
Fig. 5 The relationships between IGF-1, IGFBP-3, and rhGH treatment for GHD, catch-up growth, malignancy risk, and cardiovascular disease (CVD) risk shown in a two-dimensional grid.
(Data from Park P, Cohen P. The role of insulin-like growth factor I monitoring in growth hormone-treated children. Horm Res 2004;62 Suppl 1:61.)
Children treated with rhGH typically have low or low normal levels of IGF-1 before therapy. During rhGH therapy, IGF-1 and IGFBP-3 levels increase in tandem, a combination with a low cancer risk profile. The degree of increase of IGF-1 and IGFBP-3 during rhGH therapy is dependent on the rhGH dose, compliance, and GH-responsiveness. Following completion of rhGH therapy, IGF-1 and IGFBP-3 levels are likely to revert to pretreatment ranges with low IGF-1 and BP-3 that have a low cancer risk profile. It is unclear whether the temporary, although prolonged, increase of IGF-1 and IGFBP-3 levels during rhGH therapy has any long-term impact on the cancer risk or any other health risk factors. Although it has been proposed as a safety measure, it also remains unclear whether monitoring IGF-1 and IGFBP-3 levels, targeting IGF-1 and/or IGFBP-3 values to a certain range, or reducing rhGH doses if IGF-1 levels are high, has any impact on the long-term safety of rhGH therapy [16,17].
Although GH registries have been invaluable in the determination of safety and efficacy during rhGH therapy, once children discontinue therapy, they are not typically entered into the registries and frequently no longer see the pediatric endocrinologist. Therefore, long-term safety data in this population are sparse. There have been several studies that have attempted to investigate long-term effects of rhGH therapy in childhood. In one study, the risk of cancer was investigated in adults who received pituitary-derived GH between 1959 and 1985 [113]. There was no overall increase in the incidence of cancer in this study. However, this study raised concerns about an increased risk of colorectal cancer (SIR 7.9; 1.0–28.7, P = .05) and Hodgkin lymphoma (SIR 2.3; 0.3–8.5, P = .42) occurring in individuals with IGHD. Two cases of each type of these cancers were identified in this population. One of the cases of colorectal cancer occurred in an individual with an unclear history of possible polyposis coli. If that case is excluded, neither tumor had a significantly increased incidence compared with the normal population.
Because of the unanswered questions regarding long-term safety of rhGH therapy, further long-term follow-up studies are necessary. These studies could be prospective cohort studies or cross-sectional studies. One such effort is the ongoing retrospective observational study entitled Safety and Appropriateness of Growth Hormone Treatments in Europe (SAGhE). On December 10 2010, the French drug agency Agence Francaise de Securite Sanitaire des Produits de Santé and the European Medicines Agency (EMA) issued press releases based on preliminary mortality data from the French portion of this study, Santé Adulte GH Enfant (FrSAGhE) regarding potential increased mortality in adults previously treated with rhGH [127,128]. According to the reports, the FrSAGhE study evaluated data from about 7000 patients who had received rhGH during childhood, initiated between 1985 and 1996, for treatment of IGHD (∼75% of the cohort) or ISS/SGA. They reported an increased risk of all-cause mortality (93 deaths) compared with the general French population (70 deaths estimated), with an abnormally high death rate caused by cerebrovascular complications and bone tumors. The risk was also reported to be associated with higher doses of rhGH.
The Committee for Medicinal Products (CHMP) for Human Use of the EMA reviewed the available data and determined “that there is no need for regulatory action based on the presented data.” On December 16 2010, the CHMP of the EMA issued the following statement reporting their plans for further review of the safety of rhGH: “While this review is ongoing, the CHMP confirms that there is no immediate concern. However, prescribers are reminded to strictly follow the indications and the approved doses. The maximum recommended dose of 50 μg/kg weight/day for somatropin-containing medicines should not be exceeded.” [129] The FDA issued a similar report on December 22 2010 [130]. The EMA and FDA reported that they would review all available information on the potential risk of rhGH therapy and communicate any new recommendations once the review was complete. The FDA issued the following statement: “At this time, FDA believes the benefits of recombinant GH continue to outweigh its potential risks” [130].
These preliminary results regarding potential mortality related to rhGH therapy emphasize the need for ongoing long-term studies of adults previously treated with rhGH. It is also necessary to ensure that appropriate reference or control populations are evaluated to determine whether the risk is related to rhGH therapy or a predisposition related to the underlying condition (IGHD, ISS, or SGA). It is unclear whether there is a selection bias for individuals participating in such a study that may influence the risk of reporting adverse events. Studies of cancer survivors have shown that the increased risk of second neoplasms compared with the reference population was reduced simply by extension of the follow-up period [114]. This argues that long-term studies are necessary to determine the risk of long-term side effects related to rhGH therapy. The CHMP of the EMA has emphasized the need for the European SAGhE study to continue. Similar studies should be considered in the United States where the prescribed rhGH doses are typically higher than in Europe. However, tracking individuals previously treated with rhGH over time may be difficult.
Overall, the rhGH short-term safety profile is reassuring, with the fewest adverse events occurring in children with ISS. It is unclear whether rhGH treatment during childhood has any long-term impact on morbidity and mortality. However, because short stature has low morbidity and mortality, it would not be acceptable to expose children to even a small increase in the long-term risk of a serious adverse event related to rhGH treatment for the relative improvement in growth. Therefore, the pediatric endocrinology community, rhGH manufacturers, and research funding and regulatory agencies need to work together to design studies that will identify the long-term risk of rhGH therapy.
Future directions
This article focuses on currently available data regarding safety and efficacy of rhGH therapy. The use of rhIGF-1, approved in 2005 for severe primary IGF-1 deficiency, for treatment of children with less severe growth issues as a single agent or in combination with rhGH therapy has been the subject of clinical trials [131,132]. The safety data for rhIGF-1 alone, or in combination with rhGH, for this population is limited to small clinical trials. The short-term and long-term safety of rhIGF-1 is currently being studied in a postmarketing surveillance registry [133]. Long-term safety after rhIGF-1 therapy also needs to be monitored in the future.
Long-acting rhGH was previously available as Nutropin Depot. The safety profile of this medication was similar to that of daily rhGH [134]. However, the medication was only commercially available for 4.5 years and no long-term studies have been reported. Efficacy of Nutropin Depot was diminished compared with daily rhGH [134]. Several forms of long-acting rhGH are currently in development and may soon be commercially available [135]. If these long-acting rhGH preparations are approved, it will be important to establish their short-term and long-term safety and efficacy.
References
[1] P.C. Sizonenko, P.E. Clayton, P. Cohen, et al. Diagnosis and management of growth hormone deficiency in childhood and adolescence. Part 1: diagnosis of growth hormone deficiency. Growth Horm IGF Res. 2001;11(3):137-165.
[2] H. Gharib, D.M. Cook, P.H. Saenger, et al. American Association of Clinical Endocrinologists medical guidelines for clinical practice for growth hormone use in adults and children–2003 update. Endocr Pract. 2003;9(1):64-76.
[3] Growth Hormone Research Society. Consensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: summary statement of the GH Research Society. GH Research Society. [see comment]. J Clin Endocrinol Metab. 2000;85(11):3990-3993.
[4] T.A. Wilson, S.R. Rose, P. Cohen, et al. Update of guidelines for the use of growth hormone in children: the Lawson Wilkins Pediatric Endocrinology Society Drug and Therapeutics Committee. J Pediatr. 2003;143(4):415-421.
[5] G.P. August, B.M. Lippe, S.L. Blethen, et al. Growth hormone treatment in the United States: demographic and diagnostic features of 2331 children. J Pediatr. 1990;116(6):899-903.
[6] B. Bakker, J. Frane, H. Anhalt, et al. Height velocity targets from the National Cooperative Growth Study for first-year growth hormone responses in short children. J Clin Endocrinol Metab. 2008;93(2):352-357.
[7] B.S. Miller, D.I. Shulman, A. Shillington, et al. Consensus and discordance in the management of growth hormone-treated patients: results of a knowledge, attitudes, beliefs, and practices survey. Int J Pediatr Endocrinol. 2010;2010:891571.
[8] M.B. Ranke, A. Lindberg, K.I. Board, et al. Observed and predicted growth responses in prepubertal children with growth disorders: guidance of growth hormone treatment by empirical variables. J Clin Endocrinol Metab. 2010;95(3):1229-1237.
[9] M.B. Ranke, A. Lindberg, P. Chatelain, et al. Derivation and validation of a mathematical model for predicting the response to exogenous recombinant human growth hormone (GH) in prepubertal children with idiopathic GH deficiency. KIGS International Board. Kabi Pharmacia International Growth Study. J Clin Endocrinol Metab. 1999;84(4):1174-1183.
[10] M.B. Ranke, A. Lindberg, M.B. Ranke, et al. Predicting growth in response to growth hormone treatment. Growth Horm IGF Res. 2009;19(1):1-11.
[11] T. Tanaka, P. Cohen, P.E. Clayton, et al. Diagnosis and management of growth hormone deficiency in childhood and adolescence–part 2: growth hormone treatment in growth hormone deficient children. Growth Horm IGF Res. 2002;12(5):323-341.
[12] J. Keni, P. Cohen, J. Keni, et al. Optimizing growth hormone dosing in children with idiopathic short stature. Horm Res. 2009;71(Suppl 1):70-74.
[13] N. Mauras, K.M. Attie, E.O. Reiter, et al. High dose recombinant human growth hormone (GH) treatment of GH-deficient patients in puberty increases near-final height: a randomized, multicenter trial. Genentech, Inc, Cooperative Study Group. J Clin Endocrinol Metab. 2000;85(10):3653-3660.
[14] L. Riddick, C. Alter, D.A. Davis, et al. A stepwise increase in recombinant human growth hormone dosing during puberty achieves improved pubertal growth: a National Cooperative Growth Study report. J Pediatr Endocrinol. 2009;22(7):623-628.
[15] P. Park, P. Cohen. The role of insulin-like growth factor I monitoring in growth hormone-treated children. Horm Res. 2004;62(Suppl 1):59-65.
[16] P. Cohen, A.D. Rogol, C.P. Howard, et al. Insulin growth factor-based dosing of growth hormone therapy in children: a randomized, controlled study. J Clin Endocrinol Metab. 2007;92(7):2480-2486.
[17] J. Baron. Editorial: growth hormone therapy in childhood: titration versus weight-based dosing? J Clin Endocrinol Metab. 2007;92(7):2436-2438.
[18] M.B. Ranke, A. Lindberg, D.D. Martin, et al. The mathematical model for total pubertal growth in idiopathic growth hormone (GH) deficiency suggests a moderate role of GH dose. J Clin Endocrinol Metab. 2003;88(10):4748-4753.
[19] E.O. Reiter, D.A. Price, P. Wilton, et al. Effect of growth hormone (GH) treatment on the near-final height of 1258 patients with idiopathic GH deficiency: analysis of a large international database. J Clin Endocrinol Metab. 2006;91(6):2047-2054.
[20] J.P. Frindik, S.F. Kemp, J.J. Hunold, et al. Near adult heights after growth hormone treatment in patients with idiopathic short stature or idiopathic growth hormone deficiency. J Pediatr Endocrinol. 2003;16(Suppl 3):607-612.
[21] B. Tonshoff, D. Kiepe, S. Ciarmatori. Growth hormone/insulin-like growth factor system in children with chronic renal failure. Pediatr Nephrol. 2005;20(3):279-289.
[22] R.N. Fine, E.C. Kohaut, D. Brown, et al. Growth after recombinant human growth hormone treatment in children with chronic renal failure: report of a multicenter randomized double-blind placebo-controlled study. Genentech Cooperative Study Group. J Pediatr. 1994;124(3):374-382.
[23] D. Haffner, F. Schaefer, R. Nissel, et al. Effect of growth hormone treatment on the adult height of children with chronic renal failure. German Study Group for Growth Hormone Treatment in Chronic Renal Failure. N Engl J Med. 2000;343(13):923-930.
[24] V.H. Koch, B.M. Lippe, P.A. Nelson, et al. Accelerated growth after recombinant human growth hormone treatment of children with chronic renal failure. J Pediatr. 1989;115(3):365-371.
[25] J.D. Mahan, B.A. Warady, J. Frane, et al. First-year response to rhGH therapy in children with CKD: a National Cooperative Growth Study report. Pediatr Nephrol. 2010;25(6):1125-1130.
[26] R.N. Fine, E. Kohaut, D. Brown, et al. Long-term treatment of growth retarded children with chronic renal insufficiency, with recombinant human growth hormone. Kidney Int. 1996;49(3):781-785.
[27] E. Berard, J.L. Andre, G. Guest, et al. Long-term results of rhGH treatment in children with renal failure: experience of the French Society of Pediatric Nephrology. Pediatr Nephrol. 2008;23(11):2031-2038.
[28] R.N. Fine, K. Martz, D. Stablein, et al. What have 20 years of data from the North American Pediatric Renal Transplant Cooperative Study taught us about growth following renal transplantation in infants, children, and adolescents with end-stage renal disease? Pediatr Nephrol. 2010;25(4):739-746.
[29] R.N. Fine, D. Stablein, R.N. Fine, et al. Long-term use of recombinant human growth hormone in pediatric allograft recipients: a report of the NAPRTCS Transplant Registry. Pediatr Nephrol. 2005;20(3):404-408.
[30] W.F. Blum, B.J. Crowe, C.A. Quigley, et al. Growth hormone is effective in treatment of short stature associated with short stature homeobox-containing gene deficiency: two-year results of a randomized, controlled, multicenter trial. J Clin Endocrinol Metab. 2007;92(1):219-228.
[31] M.L. Davenport, N. Punyasavatsut, P.W. Stewart, et al. Growth failure in early life: an important manifestation of Turner syndrome. Horm Res. 2002;57(5–6):157-164.
[32] B. Lippe. Turner syndrome. Endocrinol Metab Clin North Am. 1991;20(1):121-152.
[33] J.C. Carel, L. Mathivon, C. Gendrel, et al. Near normalization of final height with adapted doses of growth hormone in Turner’s syndrome. J Clin Endocrinol Metab. 1998;83(5):1462-1466.
[34] C.A. Quigley, B.J. Crowe, D.G. Anglin, et al. Growth hormone and low dose estrogen in Turner syndrome: results of a United States multi-center trial to near-final height. J Clin Endocrinol Metab. 2002;87(5):2033-2041.
[35] R.G. Rosenfeld, K.M. Attie, J. Frane, et al. Growth hormone therapy of Turner’s syndrome: beneficial effect on adult height. J Pediatr. 1998;132(2):319-324.
[36] D.K. Stephure, Canadian Growth Hormone Advisory Committee. Impact of growth hormone supplementation on adult height in Turner syndrome: results of the Canadian randomized controlled trial. J Clin Endocrinol Metab. 2005;90(6):3360-3366.
[37] E.O. Reiter, S.L. Blethen, J. Baptista, et al. Early initiation of growth hormone treatment allows age-appropriate estrogen use in Turner’s syndrome. J Clin Endocrinol Metab. 2001;86(5):1936-1941.
[38] C.A. Bondy, Turner Syndrome Study Group. Care of girls and women with Turner syndrome: a guideline of the Turner Syndrome Study Group. J Clin Endocrinol Metab. 2007;92(1):10-25.
[39] M.L. Davenport, B.J. Crowe, S.H. Travers, et al. Growth hormone treatment of early growth failure in toddlers with Turner syndrome: a randomized, controlled, multicenter trial. J Clin Endocrinol Metab. 2007;92(9):3406-3416.
[40] R.G. Rosenfeld, R.L. Hintz, A.J. Johanson, et al. Methionyl human growth hormone and oxandrolone in Turner syndrome: preliminary results of a prospective randomized trial. J Pediatr. 1986;109(6):936-943.
[41] J.L. Frias, M.L. Davenport, Committee on Genetics and Section on Endocrinology. Health supervision for children with Turner syndrome. Pediatrics. 2003;111(3):692-702.
[42] S.D. Chernausek, K.M. Attie, J.F. Cara, et al. Growth hormone therapy of Turner syndrome: the impact of age of estrogen replacement on final height. Genentech, Inc, Collaborative Study Group. J Clin Endocrinol Metab. 2000;85(7):2439-2445.
[43] L. Plotnick, K.M. Attie, S.L. Blethen, et al. Growth hormone treatment of girls with Turner syndrome: the National Cooperative Growth Study experience. Pediatrics. 1998;102(2 Pt 3):479-481.
[44] P. Saenger. Growth-promoting strategies in Turner’s syndrome. J Clin Endocrinol Metab. 1999;84(12):4345-4348.
[45] Y.K. van Pareren, S.M. de Muinck Keizer-Schrama, T. Stijnen, et al. Final height in girls with Turner syndrome after long-term growth hormone treatment in three dosages and low dose estrogens. J Clin Endocrinol Metab. 2003;88(3):1119-1125.
[46] M.B. Ranke, A. Lindberg, P. Chatelain, et al. Prediction of long-term response to recombinant human growth hormone in Turner syndrome: development and validation of mathematical models. KIGS International Board. Kabi International Growth Study. J Clin Endocrinol Metab. 2000;85(11):4212-4218.
[47] F. Darendeliler, Z. Aycan, E. Cetinkaya, et al. Effects of growth hormone on growth, insulin resistance and related hormones (ghrelin, leptin and adiponectin) in Turner syndrome. Horm Res. 2007;68(1):1-7.
[48] P.F. Collett-Solberg, A.N. Pessoa de Queiroz, M.E. Cardoso, et al. The correlation of the IGF-I, IGFBP-3, and ALS generation test to height velocity after 6 months of recombinant growth hormone therapy in girls with Turner syndrome. Growth Horm IGF Res. 2006;16(4):240-246.
[49] A.L. Carrel, S.E. Myers, B.Y. Whitman, et al. Long-term growth hormone therapy changes the natural history of body composition and motor function in children with Prader-Willi syndrome. J Clin Endocrinol Metab. 2010;95(3):1131-1136.
[50] A.L. Carrel, V. Moerchen, S.E. Myers, et al. Growth hormone improves mobility and body composition in infants and toddlers with Prader-Willi syndrome. J Pediatr. 2004;145(6):744-749.
[51] S.E. Myers, B.Y. Whitman, A.L. Carrel, et al. Two years of growth hormone therapy in young children with Prader-Willi syndrome: physical and neurodevelopmental benefits. Am J Med Genet A. 2007;143(5):443-448.
[52] A.C. Lindgren, A. Lindberg. Growth hormone treatment completely normalizes adult height and improves body composition in Prader-Willi syndrome: experience from KIGS (Pfizer International Growth Database). Horm Res. 2008;70(3):182-187.
[53] M. Tauber, W. Cutfield, M. Tauber, et al. KIGS highlights: growth hormone treatment in Prader-Willi syndrome. Horm Res. 2007;68(Suppl 5):48-50.
[54] R. Usher, F. McLean. Intrauterine growth of live-born Caucasian infants at sea level: standards obtained from measurements in 7 dimensions of infants born between 25 and 44 weeks of gestation. J Pediatr. 1969;74(6):901-910.
[55] M.L. Hediger, M.D. Overpeck, K.R. Maurer, et al. Growth of infants and young children born small or large for gestational age: findings from the Third National Health and Nutrition Examination Survey. Arch Pediatr Adolesc Med. 1998;152(12):1225-1231.
[56] J.P. Karlberg, K. Albertsson-Wikland, E.Y. Kwan, et al. The timing of early postnatal catch-up growth in normal, full-term infants born short for gestational age. Horm Res. 1997;1:17-24.
[57] Y. Van Pareren, P. Mulder, M. Houdijk, et al. Adult height after long-term, continuous growth hormone (GH) treatment in short children born small for gestational age: results of a randomized, double-blind, dose-response GH trial. J Clin Endocrinol Metab. 2003;88(8):3584-3590.
[58] K. Albertsson-Wikland, J. Karlberg. Postnatal growth of children born small for gestational age. Acta Paediatr Suppl. 1997;423:193-195.
[59] F. de Zegher, K. Albertsson-Wikland, H.A. Wollmann, et al. Growth hormone treatment of short children born small for gestational age: growth responses with continuous and discontinuous regimens over 6 years. J Clin Endocrinol Metab. 2000;85(8):2816-2821.
[60] M. Ranke, P. Wilton, editors. Growth hormone therapy in KIGS: 10 years’ experience. Heidelberg (Germany): Barth Verlag, 1999.
[61] M.B. Ranke, A. Lindberg, C.T. Cowell, et al. Prediction of response to growth hormone treatment in short children born small for gestational age: analysis of data from KIGS (Pharmacia International Growth Database). J Clin Endocrinol Metab. 2003;88(1):125-131.
[62] A. Fjellestad-Paulsen, D. Simon, P. Czernichow, et al. Short children born small for gestational age and treated with growth hormone for three years have an important catch-down five years after discontinuation of treatment. J Clin Endocrinol Metab. 2004;89(3):1234-1239.
[63] US Census Bureau. Profile of general demographic characteristics. Geographic area: United States. US Census 2000. Table DP-1.
[64] R. Lindsay, M. Feldkamp, D. Harris, et al. Utah growth study: growth standards and the prevalence of growth hormone deficiency. J Pediatr. 1994;125(1):29-35.
[65] B.S. Finkelstein, J.B. Silvers, U. Marrero, et al. Insurance coverage, physician recommendations, and access to emerging treatments: growth hormone therapy for childhood short stature. [see comment]. JAMA. 1998;279(9):663-668.
[66] F. Lifshitz, C. Cervantes. Short stature. In: F. Lifschitz, editor. Pediatric endocrinology. 3rd edition. New York: Marcel Dekker; 1996:1-18.
[67] D.M. Wilson, J. Frane. A brief review of the use and utility of growth hormone stimulation testing in the NCGS: do we need to do provocative GH testing? Growth Horm IGF Res. 2005;15(Suppl A):S21-S25.
[68] P. Cohen, A.D. Rogol, C.L. Deal, et al. Consensus statement on the diagnosis and treatment of children with idiopathic short stature: a summary of the Growth Hormone Research Society, the Lawson Wilkins Pediatric Endocrine Society, and the European Society for Paediatric Endocrinology Workshop. J Clin Endocrinol Metab. 2008;93(11):4210-4217.
[69] B.S. Miller, D. Zimmerman. Idiopathic short stature in children. Pediatr Ann. 2004;33(3):177-181.
[70] R.L. Hintz, K.M. Attie, J. Baptista, et al. Effect of growth hormone treatment on adult height of children with idiopathic short stature. Genentech Collaborative Group. [see comment]. N Engl J Med. 1999;340(7):502-507.
[71] E.W. Leschek, S.R. Rose, J.A. Yanovski, et al. Effect of growth hormone treatment on adult height in peripubertal children with idiopathic short stature: a randomized, double-blind, placebo-controlled trial. [see comment]. J Clin Endocrinol Metab. 2004;89(7):3140-3148.
[72] J.M. Wit, L.T. Rekers-Mombarg, G.B. Cutler, et al. Growth hormone (GH) treatment to final height in children with idiopathic short stature: evidence for a dose effect. [see comment]. J Pediatr. 2005;146(1):45-53.
[73] B.S. Finkelstein, T.F. Imperiale, T. Speroff, et al. Effect of growth hormone therapy on height in children with idiopathic short stature: a meta-analysis. Arch Pediatr Adolesc Med. 2002;156(3):230-240.
[74] M.B. Ranke, A. Lindberg, D.A. Price, et al. Age at growth hormone therapy start and first-year responsiveness to growth hormone are major determinants of height outcome in idiopathic short stature. Horm Res. 2007;68(2):53-62.
[75] P. Cohen, J. Germak, A.D. Rogol, et al. Variable degree of growth hormone (GH) and insulin-like growth factor (IGF) sensitivity in children with idiopathic short stature compared with GH-deficient patients: evidence from an IGF-based dosing study of short children. J Clin Endocrinol Metab. 2010;95(5):2089-2098.
[76] G. Binder, M.B. Ranke, D.D. Martin, et al. Auxology is a valuable instrument for the clinical diagnosis of SHOX haploinsufficiency in school-age children with unexplained short stature. J Clin Endocrinol Metab. 2003;88(10):4891-4896.
[77] C. Huber, M. Rosilio, A. Munnich, et al. High incidence of SHOX anomalies in individuals with short stature. J Med Genet. 2006;43(9):735-739.
[78] G.A. Rappold, M. Fukami, B. Niesler, et al. Deletions of the homeobox gene SHOX (short stature homeobox) are an important cause of growth failure in children with short stature. J Clin Endocrinol Metab. 2002;87(3):1402-1406.
[79] W.F. Blum, D. Cao, V. Hesse, et al. Height gains in response to growth hormone treatment to final height are similar in patients with SHOX deficiency and Turner syndrome. Horm Res. 2009;71(3):167-172.
[80] A.A. Romano, J.E. Allanson, J. Dahlgren, et al. Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics. 2010;126(4):746-759.
[81] M.B. Ranke, P. Heidemann, C. Knupfer, et al. Noonan syndrome: growth and clinical manifestations in 144 cases. Eur J Pediatr. 1988;148(3):220-227.
[82] A.C. Shaw, K. Kalidas, A.H. Crosby, et al. The natural history of Noonan syndrome: a long-term follow-up study. Arch Dis Child. 2007;92(2):128-132.
[83] M. Zenker, G. Buheitel, R. Rauch, et al. Genotype-phenotype correlations in Noonan syndrome. J Pediatr. 2004;144(3):368-374.
[84] J.M. Ko, J.M. Kim, G.H. Kim, et al. PTPN11, SOS1, KRAS, and RAF1 gene analysis, and genotype-phenotype correlation in Korean patients with Noonan syndrome. J Hum Genet. 2008;53(11–12):999-1006.
[85] C. Noordam, I. van der Burgt, C.G. Sweep, et al. Growth hormone (GH) secretion in children with Noonan syndrome: frequently abnormal without consequences for growth or response to GH treatment. Clin Endocrinol. 2001;54(1):53-59.
[86] J.M. Limal, B. Parfait, S. Cabrol, et al. Noonan syndrome: relationships between genotype, growth, and growth factors. J Clin Endocrinol Metab. 2006;91(1):300-306.
[87] C.E. MacFarlane, D.C. Brown, L.B. Johnston, et al. Growth hormone therapy and growth in children with Noonan’s syndrome: results of 3 years’ follow-up. J Clin Endocrinol Metab. 2001;86(5):1953-1956.
[88] J.M. Kirk, P.R. Betts, G.E. Butler, et al. Short stature in Noonan syndrome: response to growth hormone therapy. Arch Dis Child. 2001;84(5):440-443.
[89] L.V. Ferreira, S.A. Souza, I.J. Arnhold, et al. PTPN11 (protein tyrosine phosphatase, nonreceptor type 11) mutations and response to growth hormone therapy in children with Noonan syndrome. J Clin Endocrinol Metab. 2005;90(9):5156-5160.
[90] C. Noordam, P.G. Peer, I. Francois, et al. Long-term GH treatment improves adult height in children with Noonan syndrome with and without mutations in protein tyrosine phosphatase, non-receptor-type 11. Eur J Endocrinol. 2008;159(3):203-208.
[91] G. Municchi, A.M. Pasquino, I. Pucarelli, et al. Growth hormone treatment in Noonan syndrome: report of four cases who reached final height. Horm Res. 1995;44(4):164-167.
[92] D. Osio, J. Dahlgren, K.A. Wikland, et al. Improved final height with long-term growth hormone treatment in Noonan syndrome. Acta Paediatr. 2005;94(9):1232-1237.
[93] R. Raaijmakers, C. Noordam, G. Karagiannis, et al. Response to growth hormone treatment and final height in Noonan syndrome in a large cohort of patients in the KIGS database. J Pediatr Endocrinol. 2008;21(3):267-273.
[94] A.A. Romano, K. Dana, B. Bakker, et al. Growth response, near-adult height, and patterns of growth and puberty in patients with Noonan syndrome treated with growth hormone. J Clin Endocrinol Metab. 2009;94(7):2338-2344.
[95] J. Bell, K.L. Parker, R.D. Swinford, et al. Long-term safety of recombinant human growth hormone in children. J Clin Endocrinol Metab. 2010;95(1):167-177.
[96] P. Wilton, A.F. Mattsson, F. Darendeliler, et al. Growth hormone treatment in children is not associated with an increase in the incidence of cancer: experience from KIGS (Pfizer International Growth Database). J Pediatr. 2010;157(2):265-270.
[97] P. Pitukcheewanont, P. Desrosiers, J. Steelman, et al. Issues and trends in pediatric growth hormone therapy–an update from the GHMonitor observational registry. Pediatr Endocrinol Rev. 2008;5(Suppl 2):702-707.
[98] J. Ross, P.A. Lee, R. Gut, et al. Factors influencing the one- and two-year growth response in children treated with growth hormone: analysis from an observational study. Int J Pediatr Endocrinol. 2010;2010:494656.
[99] ClinicalTrials.gov. Observational study of somatropin treatment in children (GeNeSIS). Available at: http://clinicaltrials.gov/ct2/show/NCT01088412, 2011. Published 2010. Updated 3/16/10. Accessed May 3, 2011
[100] J.E. Whittington, A.J. Holland, T. Webb, et al. Population prevalence and estimated birth incidence and mortality rate for people with Prader-Willi syndrome in one UK Health Region. J Med Genet. 2001;38(11):792-798.
[101] M. Tauber, G. Diene, C. Molinas, et al. Review of 64 cases of death in children with Prader-Willi syndrome (PWS). Am J Med Genet A. 2008;146(7):881-887.
[102] D.A. Festen, A.W. de Weerd, R.A. van den Bossche, et al. Sleep-related breathing disorders in prepubertal children with Prader-Willi syndrome and effects of growth hormone treatment. J Clin Endocrinol Metab. 2006;91(12):4911-4915.
[103] J. Miller, J. Silverstein, J. Shuster, et al. Short-term effects of growth hormone on sleep abnormalities in Prader-Willi syndrome. J Clin Endocrinol Metab. 2006;91(2):413-417.
[104] C.H. Gravholt, K. Landin-Wilhelmsen, K. Stochholm, et al. Clinical and epidemiological description of aortic dissection in Turner’s syndrome. Cardiol Young. 2006;16(5):430-436.
[105] M.L. Davenport, M.L. Davenport. Approach to the patient with Turner syndrome. J Clin Endocrinol Metab. 2010;95(4):1487-1495.
[106] J.M. Chan, M.J. Stampfer, E. Giovannucci, et al. Plasma insulin-like growth factor-I and prostate cancer risk: a prospective study. Science. 1998;279(5350):563-566.
[107] E. Giovannucci, M. Pollak, E.A. Platz, et al. Insulin-like growth factor I (IGF-I), IGF-binding protein-3 and the risk of colorectal adenoma and cancer in the Nurses’ Health Study. Growth Horm IGF Res. 2000;10(Suppl A):S30-S31.
[108] S.E. Hankinson, W.C. Willett, G.A. Colditz, et al. Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. Lancet. 1998;351(9113):1393-1396.
[109] D. Sachdev, D. Yee, D. Sachdev, et al. Disrupting insulin-like growth factor signaling as a potential cancer therapy. Mol Cancer Ther. 2007;6(1):1-12.
[110] J.K. Perry, B.S. Emerald, H.C. Mertani, et al. The oncogenic potential of growth hormone. Growth Horm IGF Res. 2006;16(5–6):277-289.
[111] S. Yakar, D. Leroith, P. Brodt, et al. The role of the growth hormone/insulin-like growth factor axis in tumor growth and progression: lessons from animal models. Cytokine Growth Factor Rev. 2005;16(4–5):407-420.
[112] S. Watanabe, Y. Tsunematsu, J. Fujimoto, et al. Leukaemia in patients treated with growth hormone. Lancet. 1988;1(8595):1159-1160.
[113] A.J. Swerdlow, C.D. Higgins, P. Adlard, et al. Risk of cancer in patients treated with human pituitary growth hormone in the UK, 1959–85: a cohort study. Lancet. 2002;360(9329):273-277.
[114] B. Ergun-Longmire, A.C. Mertens, P. Mitby, et al. Growth hormone treatment and risk of second neoplasms in the childhood cancer survivor. J Clin Endocrinol Metab. 2006;91(9):3494-3498.
[115] K. Gordon. Pediatric pseudotumor cerebri: descriptive epidemiology. Can J Neurol Sci. 1997;24(3):219-221.
[116] F. Darendeliler, G. Karagiannis, P. Wilton, et al. Headache, idiopathic intracranial hypertension and slipped capital femoral epiphysis during growth hormone treatment: a safety update from the KIGS database. Horm Res. 2007;68(Suppl 5):41-47.
[117] A. Nydegger, R.G. Heine, R. Ranuh, et al. Changing incidence of acute pancreatitis: 10-year experience at the Royal Children’s Hospital, Melbourne. J Gastroenterol Hepatol. 2007;22(8):1313-1316.
[118] S.V. Gelding, N.F. Taylor, P.J. Wood, et al. The effect of growth hormone replacement therapy on cortisol-cortisone interconversion in hypopituitary adults: evidence for growth hormone modulation of extrarenal 11 beta-hydroxysteroid dehydrogenase activity. Clin Endocrinol. 1998;48(2):153-162.
[119] C. Giavoli, R. Libe, S. Corbetta, et al. Effect of recombinant human growth hormone (GH) replacement on the hypothalamic-pituitary-adrenal axis in adult GH-deficient patients. J Clin Endocrinol Metab. 2004;89(11):5397-5401.
[120] J.L. Mills, L.B. Schonberger, D.K. Wysowski, et al. Long-term mortality in the United States cohort of pituitary-derived growth hormone recipients. J Pediatr. 2004;144(4):430-436.
[121] D.I. Shulman, M.R. Palmert, S.F. Kemp, et al. Adrenal insufficiency: still a cause of morbidity and death in childhood. Pediatrics. 2007;119(2):e484-e494.
[122] B. Henrikson. The incidence of slipped capital femoral epiphysis. Acta Orthop Scand. 1969;40(3):365-372.
[123] J.L. Kelsey, K.J. Keggi, W.O. Southwick. The incidence and distribution of slipped capital femoral epiphysis in Connecticut and Southwestern United States. J Bone Joint Surg Am. 1970;52(6):1203-1216.
[124] R.N. Fine, D. Stablein, A.H. Cohen, et al. Recombinant human growth hormone post-renal transplantation in children: a randomized controlled study of the NAPRTCS. Kidney Int. 2002;62(2):688-696.
[125] B. Tonshoff, D. Haffner, O. Mehls, et al. Efficacy and safety of growth hormone treatment in short children with renal allografts: three year experience. Members of the German Study Group for Growth Hormone Treatment in Children with Renal Allografts. Kidney Int. 1993;44(1):199-207.
[126] V.R. Dharnidharka, L.I. Talley, K.L. Martz, et al. Recombinant growth hormone use pretransplant and risk for post-transplant lymphoproliferative disease–a report of the NAPRTCS. Pediatr Transplant. 2008;12(6):689-695.
[127] European Medicines Agency. European Medicines Agency to review the safety of somatropin-containing medicines. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Press_release/2010/12/WC500099699.pdf Published 2010. Updated 12/10/2010. Accessed May 3, 2011
[128] AFSSAPS. Hormone de Croissance synthétique (somatropine recombinante): premiers résultats de l’étude épidémiologique sur la tolérance à long terme – communiqué. Available at: http://www.afssaps.fr/Infos-de-securite/Communiques-Points-presse/Hormone-de-Croissance-synthetique-somatropine-recombinante-Premiers-resultats-de-l-etude-epidemiologique-sur-la-tolerance-a-long-terme-Communique Published 2010. Updated 12/10/2010. Accessed May 3, 2011
[129] European Medicines Agency. Update on somatropin-containing medicines. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Press_release/2010/12/WC500099936.pdf Published 2010. Updated 12/16/2010. Accessed May 3, 2011
[130] U.S. Food & Drug Administration. Recombinant human growth hormone (somatropin): ongoing safety review – possible increased risk of death. Available at: http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm237969.htm Published 2010. Updated 12/22/2010. Accessed May 3, 2011
[131] ClinicalTrials.gov. rhGH and rhIGF-1 combination therapy in children with short stature associated with IGF-1 deficiency. Available at: http://clinicaltrials.gov/ct2/show/NCT00572156 Published 2007. Updated 9/14/10. Accessed May 3, 2011
[132] L.K. Midyett, A.D. Rogol, Q.L. Van Meter, et al. Recombinant insulin-like growth factor (IGF)-I treatment in short children with low IGF-I levels: first-year results from a randomized clinical trial. J Clin Endocrinol Metab. 2010;95(2):611-619.
[133] ClinicalTrials.gov. IGFD Registry: a patient registry for monitoring long-term safety and efficacy of increlex. Available at:http://clinicaltrials.gov/ct2/show/NCT00747604 Published 2008. Updated 1/26/2011. Accessed May 3, 2011.
[134] B.L. Silverman, S.L. Blethen, E.O. Reiter, et al. A long-acting human growth hormone (Nutropin Depot): efficacy and safety following two years of treatment in children with growth hormone deficiency. J Pediatr Endocrinol. 2002;15(Suppl 2):715-722.
[135] U. Fuhr, D. Tuculanu, A. Berghout, et al. Bioequivalence between novel ready-to-use liquid formulations of the recombinant human GH Omnitrope and the original lyophilized formulations for reconstitution of Omnitrope and Genotropin. Eur J Endocrinol. 2010;162(6):1051-1058.