[level-membership-for-basic-science-category]
CHAPTER 42 S Phase and DNA Replication
Accurate replication of DNA, which is crucial for cellular propagation and survival, occurs during the S phase (DNA synthesis phase) of the cell cycle. This chapter begins with a brief primer on the events of replication and then discusses its regulation. Next, the chapter covers the proteins that bind origins of replication and ensure that each region of DNA is replicated once and only once per cell cycle. It closes by discussing how the structure of the nucleus influences replication.
DNA Replication: A Primer
In the basic reaction of DNA replication, the 3′ hydroxyl at the end of the growing DNA strand makes a nucleophilic attack on the α-phosphate of the incoming nucleoside triphosphate to form a phosphodiester bond. This incorporates the nucleotide into the growing chain and releases pyrophosphate (Fig. 42-1). Subsequent hydrolysis of the pyrophosphate provides the driving force for the reaction. This reaction requires the presence of a template strand of DNA that specifies, through base pairing, which of the four nucleoside triphosphates is added to the growing complementary strand.
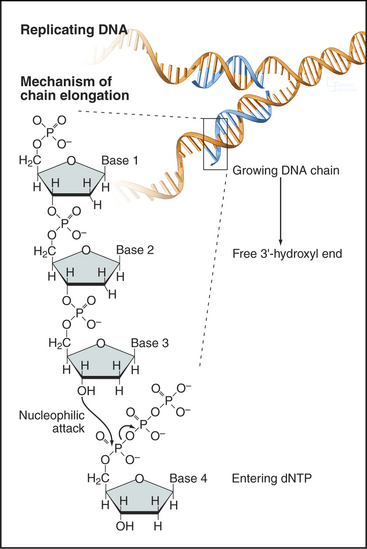
Before discussing DNA replication and its regulation, an introduction to some terminology describing the geometry of replicating DNA is required. The exact site on the chromosomal DNA where replication begins is termed the origin of bidirectional replication. As the termbidirectional implies, two sets of DNA replication machinery head off in opposite directions from the origin. Each set of replication machinery, together with the DNA that it is replicating, is called a replication fork because at the site of replication, one parental DNA molecule splits into two (Fig. 42-2). It is not known whether replication forks move along the DNA like trains along a track or whether the fork sits at a stationary site (referred to as a replication factory) through which the DNA is “reeled in” as it is replicated.
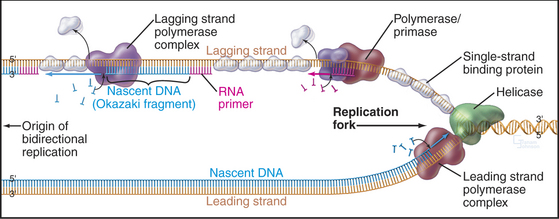
The bidirectional nature of DNA replication causes a fundamental problem, as DNA synthesis invariably proceeds in a 5′ to 3′ direction. Replication of the so-called leading strand poses no problems. This is the strand along which the fork moves in a 3′ to 4′ direction, so the newly synthesized DNA is laid down smoothly in a 5′ to 3′ direction (Fig. 42-2). However, the other template strand faces in the opposite direction, apparently requiring DNA polymerase to synthesize DNA in the wrong direction as the replication fork progresses away from the origin (i.e., adding nucleotides in a 3′ to 4′ direction). No DNA polymerase with this polarity has been found. Instead, this lagging strand replicates in a series of short segments. Every time the DNA strands have been peeled apart (unwound) by 250 nucleotides or so, a polymerase/primase complex (see Fig. 42-11) initiates DNA synthesis on the lagging strand, with the polymerase running back toward the replication origin in a 5′ to 3′ direction. Locally, synthesis on the lagging strand proceeds in a direction opposite to the overall direction of fork movement. Synthesis of each lagging strand fragment stops when DNA polymerase runs into the 5′ end of the previous fragment. Thus, the lagging strand is copied in a highly discontinuous fashion into short fragments known as Okazaki fragments (named after their discoverer [Fig. 42-2]). Fig. 42-11 describes the enzymes and events at the replication fork in greater detail.
Origins of Replication
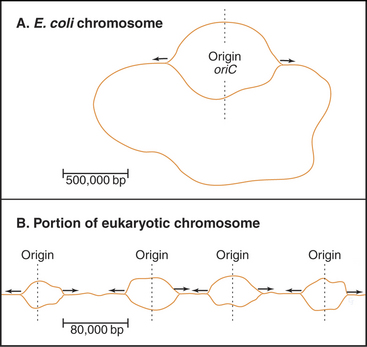
Bacteria such as Escherichia coli replicate their circular chromosomes using two replication forks starting from a single origin of replication (Fig. 42-3A), but eukaryotes must use multiple origins of replication to duplicate their large genomes during a relatively short S phase, which can be limited to as little as a few minutes in some early embryos. These numerous origins are distributed along the chromosome: up to 400 in budding yeast and about 60,000 in human cells. These origins are positioned so that all of the DNA is replicated in the available time, and to be on the safe side, more origins are prepared than are actually needed.
The existence of multiple origins creates a potential hazard: If any origin were used more or less than once per cell cycle, genes would be duplicated or lost. How is the “firing” of all of these origins orchestrated so that each is used once and only once per S phase? Cells manage this problem by a mechanism termed licensing, which ensures that each origin is used once and only once per S phase. Each origin is licensed to replicate once and only once per cell cycle. Replication of the origin removes the license, which cannot normally be renewed until the cell has completely traversed the cycle and has passed through mitosis.
A unit of chromosomal DNA whose replication is initiated at a single origin is termed a replicon. The origin is defined genetically as a replicator element. The classic replicon is the E. coli chromosome (which is 4 × 106 base pairs [bp] in size); this has a single replicator site called oriC (Fig. 42-3). An initiator protein (product of the E. coli DnaA gene [Fig. 42-12]) binds to this origin and either directly or indirectly promotes melting of the DNA duplex, giving the replication machinery access to two single strands of DNA. Other factors bind to the initiator, and their concerted action produces a wave of DNA replication proceeding outward in both directions along the DNA (a replication “bubble”) at about 750 to 1250 bases per second.
An average human chromosome contains about 150 × 106 bp of DNA. Because the replication machinery in mammals moves only about 20 to 100 bases per second (probably reflecting the fact that the DNA is packaged into chromatin [see Chapter 13]), it would take up to 2000 hours to replicate this length of DNA from a single origin. In most human cells, the duration of the S phase is about eight hours. This means that at least 25 to 125 origins of replication would be required to replicate an average chromosome in the allotted time. In fact, origins of replication are much more closely spaced than this. It has been estimated that mammalian origins of replication are spaced about 100,000 to 150,000 bp apart. Thus, approximately 60,000 origins of replication participate in replication of the entire human genome.
Replication Origins in S. Cerevisiae
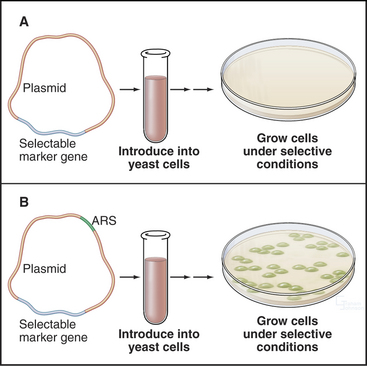
About 400 origins of replication participate in replicating the budding yeast genome. A major breakthrough in understanding DNA replication in S. cerevisiae was the identification of short (100 to 150 bp) segments of DNA that act as replication origins in vivo when cloned into a yeast plasmid (circular DNA molecule). These autonomously replicating sequences (or ARS elements) allow yeast plasmids to replicate in parallel with the cellular chromosomes (Fig. 42-4). ARS elements are often, although not always, bona fide replication origins in their native chromosomal context. Replication always initiates within ARS elements, but not all ARS ele-ments act as origins of DNA replication in every cell cycle.
Budding yeast ARS elements share a common DNA sequence motif called the ARS core consensus sequence: 5′-(A/T)TTTAT(A/G)TTT(A/T)-3′ (Fig. 42-5). Single base mutations at several locations within this sequence completely inactivate ARS activity. Other, less well-conserved DNA sequences also contribute to the activity of the ARS as a replication origin. One of these, termed B1, together with the ARS core, forms the binding site for a complex of six proteins (five of which are AAA ATPases) termed the origin recognition complex (ORC [see later section]). The DNA unwinding element is thought to be another short sequence (B2) located a bit further along the DNA. DNA synthesis begins at an origin of bidirectional replication midway between the ORC binding site and the DNA unwinding element.
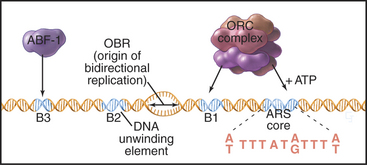
ORC was identified by its ability to bind the 11-bp ARS core sequence (Fig. 42-5). This binding has two noteworthy features. First, it requires adenosine triphosphate (ATP), which remains associated with the ORC complex. Second, in yeast, the ORC complex remains bound to the origins of replication across the entire cell cycle. Thus, something other than the presence of ORC must be responsible for regulating the periodic activation of origins in the S phase (see Fig. 42-14). In metazoans, ORC behavior is more complex; the largest subunit, Orc1, cycles on and off the DNA in a cell-cycle-regulated manner.
ARS elements typically contain binding sites for other sequence-specific DNA binding proteins, such as transcription factors. For example, a transcription factor called ARS-binding factor 1 (ABF-1) binds to the B3 sequence within the ARS1 element (Fig. 42-5). Deletion of the ABF-1 binding site only slightly reduces the ability of ARS1 to act as a replication origin in vivo. Furthermore, substitution of DNA binding sequences for other transcription factors within the B3 sequence has little effect on replication efficiency.
In addition to their role in DNA replication, several ORC components also seem to regulate heterochromatin formation and transcription (see Chapters 13 and 15). This cross talk between the machinery used for transcription and DNA replication may explain why regions of chromosomes with actively transcribed genes typically replicate early in the S phase (see the discussion that follows). The Orc6 subunit also functions in mitosis at kinetochores and during cytokinesis. Its detailed role in those processes is not known.
Replication Origins in Mammalian Cells
At present, two types of mammalian replication origins are known. The first is exemplified by the origin of replication adjacent to the lamin B2 gene (Fig. 42-6A). This origin “fires” within the first several minutes of the S phase, and a variety of methods have succeeded in mapping it to a stretch of less than 500 bp. Within this region, a single origin of bidirectional replication appears to be used. Thus, the lamin B2 origin of replication appears to be analogous to the well-characterized budding yeast origins.
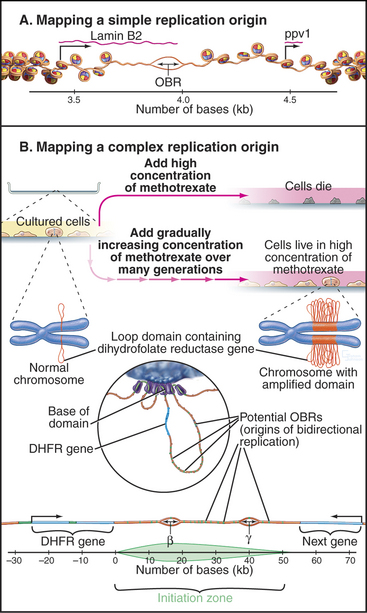
The second is exemplified by the widely studied replication origin lying just downstream of the hamster gene for dihydrofolate reductase, an enzyme that is essential for biosynthesis of thymidine. This origin is accessible to experimental study because it is possible to select for cells with this chromosomal region amplified as hundreds or even thousands of copies (Fig. 42-6B). By looking for the first regions of the amplified DNA to replicate, the origin of replication was initially located within a region of about 55,000 bp. It now appears that DNA replication can initiate with low efficiency at roughly 20 sites distributed throughout this broad zone. Two of these sites are used with relatively higher efficiency, accounting for about 20% of all initiation in the region. These sites, termed Ori-β and Ori-γ (Fig. 42-6), each encompass about 0.5 to 2 kb of DNA.
Assembly of the Prereplication Complex
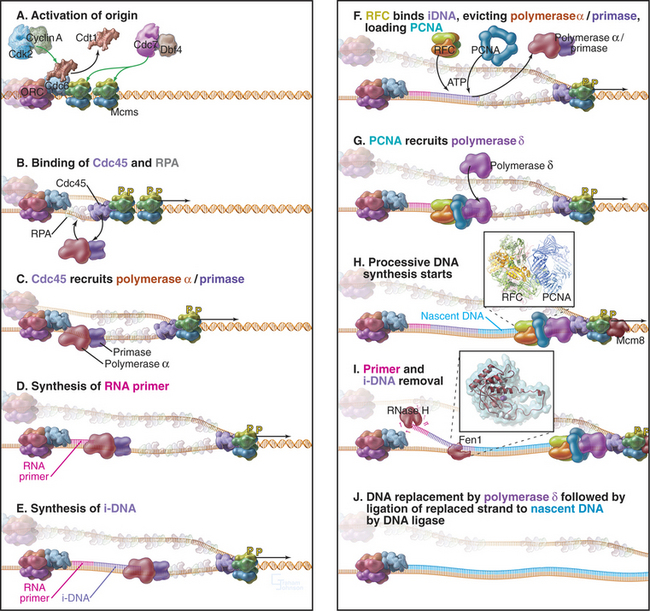
Recall that yeast ORC is stably bound to replication origins throughout the cell cycle. However, ORC is not the trigger for DNA replication. Rather, it acts as a “landing pad” for assembly of a prereplication complex of other proteins that initiates DNA replication. During late anaphase or very early G1 phase, several proteins, including Cdc6p and Cdt1, bind to the ORC complex at origins of replication (Table 42-1). ORC-Cdc6p-Cdt1 then recruits a complex of Mcm proteins to the origin and loads it onto the DNA. This prereplication com-plex of ORC, Cdc6p, CDT1, and minichromosome maintenance (Mcm) proteins (Fig. 42-7) assembles at each replication origin before the onset of the S phase.
Table 42-1 BIOCHEMICAL ACTIVITIES REQUIRED FOR REPLICATION OF DNA IN EUKARYOTES
| Activity | Name of Protein |
|---|---|
| Origin recognition | ORC (origin recognition complex; five of six subunits are AAA ATPases) |
| Pre-replication complex | Cdc6 (recruits Mcm 2–7) |
| Cdt1 (recruits Mcm 2–7) | |
| Mcm 10 (stimulates Cdc45 and polymerase α binding) | |
| Origin activation | Cdk2-cyclin A |
| Cdc7p-Dbf4p | |
| Cdc45p (recruits RPA and polymerases. Needed for elongation of growing chain) | |
| GINS complex (needed for polymerase binding and elongation of growing chain) | |
| DNA unwinding (helicase) | Mcm 2–7 proteins (precede other fork components) |
| Mcm 8 (controversial, may be elongation helicase) | |
| Stabilization of single-stranded DNA | RPA (binds single-stranded DNA) |
| Polymerase/primase | DNA polymerase α (no editing function) |
| Replicative polymerases | DNA polymerase δ |
| DNA polymerase ε (both have 3′–5′ exonuclease editing capability) | |
| Processivity factor | PCNA (ring-shaped clamp that slides along the DNA. Keeps polymerases δ and ε attached to the template strand so that they make longer chains; coordination of cell cycle control and replication; role in repair) |
| PCNA loader | RF-C (Binds primer: template junction. AAA ATPase. Loading factor for PCNA, important for polymerase switch) |
| Closing Factors | |
| Removal of RNA primer | Fen1 5′ 3′ exonuclease |
| RNase H | |
| Ligation of discontinuous DNA fragments | DNA ligase I |
| Releasing superhelical tension | DNA topoisomerase I |
| Disentangling daughter strands | DNA topoisomerase II |
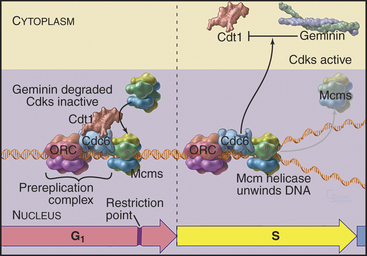
Figure 42-7 COMPONENTS OF THE PREREPLICATION COMPLEX AND THEIR FATE AFTER THE INITIATION OF DNA REPLICATION.
Mcm proteins were identified in a screen for genes of budding yeast that are required for the stability of small artificial chromosomes. Six of these Mcm genes encode a structurally related group of proteins, termed Mcm 2–7, that are required for DNA replication. Mcm 2–7 proteins form a hexameric complex that is thought to be shaped like a doughnut. Somehow, Cdc6p–Cdt1 uses ATP hydrolysis to thread DNA through the central hole of the Mcm doughnut. Although the function of the Mcm 2–7 complex is not known for certain, the predominant view is that it is a DNA helicase, an enzyme that uses ATP hydrolysis to separate DNA strands (see Fig. 42-11). It is currently thought that Mcm 2–7 binding to the prereplication complex is the key point of regulation at which origins are “licensed” so that they replicate only once per cell cycle.
In mammals, licensing occurs in several stages, all before passage of the restriction point (see Chapter 41). During telophase, Cdc6, Cdt1, and Mcm 2–7 bind to origins all across the chromosomes. Later, in the early G1 phase, these licensed origins are somehow processed to select the subset of origins that will fire in the subsequent S phase. A third step establishes the relative temporal order in which origins will fire.
At least three mechanisms regulate licensing. The first involves negative regulation of Cdc6p activity by Cdks, which inhibit Cdc6p–Cdt1 from loading Mcm proteins onto DNA. At the exit from mitosis, destruction of cyclins and synthesis of inhibitory proteins inactivates Cdks, creating a window of time between anaphase and the restriction point for licensing replication origins (see Fig. 40-18). Once mammalian cells pass the restriction point, the levels of Cdk2–cyclin E, and subsequently, Cdk2–cyclin A rise again (see Fig. 41-11) preventing the reassembly of prereplication complexes until after the next mitosis. In yeasts, the single Cdk that is complexed with B-type cyclins inhibits prereplication complex reassembly. Experimental inactivation of Cdk1 during the G2 phase in the fission yeast Schizosaccharomyces pombe demonstrated the importance of kinase activation: Cells lacking Cdk1 activity assembled prereplication complexes on already replicated DNA and then carried out further rounds of “illegal” DNA replication without division.
In vertebrates a protein called geminin is a critical regulator of origin “licensing.” Geminin binds to Cdt1 and prevents it from loading Mcm proteins onto DNA. The anaphase-promoting complex/cyclosome (APC/C) (see Fig. 40-16) degrades or inactivates geminin, keeping its concentration very low from anaphase through late G1 when prereplication complexes assemble. Accumulation of geminin starting in the S phase prevents the assembly of new prereplication complexes until after the next mitosis. Yeasts lack geminin, but in vertebrates, regulation of geminin and Cdt1 levels by proteolysis appears to be the primary method of controlling origin licensing.
A third way to regulate origin “licensing” involves sequestering molecules that are required to assemble the prereplication complex in the cytoplasm following the onset of the S phase. This was first suggested by studies of DNA replication in Xenopus egg extracts (see Fig. 40-8) in which nuclei replicate their DNA once and only once unless their nuclear envelopes are perforated, in which case the DNA replicates again. In living cells, this regulation by the nuclear envelope appears to be significant only in yeasts, in which factors that are excluded from the nucleus after replication include Mcm proteins.
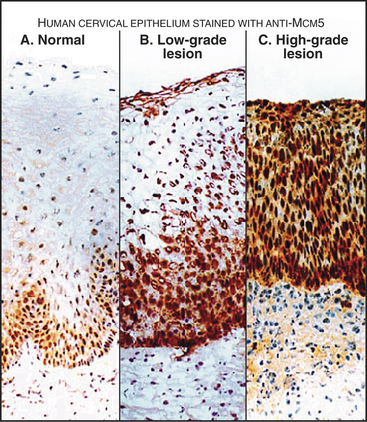
Components of the prereplication complex are absent from differentiated (G0) cells. In fact, detection of these proteins with antibodies in cells from cervical smears is currently being developed as a sensitive method for the early detection of cancer cells (Fig. 42-8).
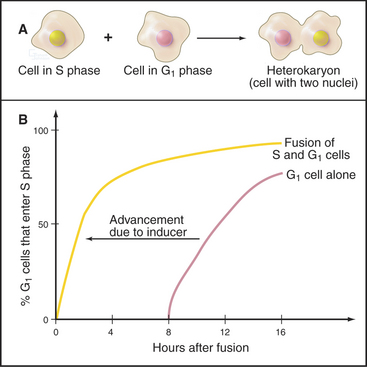
Signals That Start Replication
A classic experiment (Fig. 42-9) demonstrated that (1) a cytoplasmic inducer triggers the transition into the S phase and (2) this inducer triggers DNA replication in a G1 nucleus but not in a G2 nucleus. The inducer is very likely a combination of protein kinases, including Cdk-cyclin pairs, as well as a specialized kinase, Cdc7p-Dbf4p. In mammals, Cdk2–cyclin E, whose activity is maximal at the G1/S transition (Fig. 42-10), phosphorylates Rb, thereby opening the restriction point “gate” and allowing the E2F/DP dimer to function as a transcription factor and stimulate the transcription of genes involved in DNA replication (see Chapter 41). In addition to cyclin E itself, genes targeted by E2F include cyclin A, Cdc25A, enzymes required for synthesis of DNA precursors (dihydrofolate reductase, thymidine kinase, and thymidylate synthase), origin-binding proteins Cdc6p, Orc1, Cdt1 and its inhibitor geminin, and two components of the replication machinery (DNA polymerase a and proliferating cell nuclear antigen [PCNA]; see Fig. 42-11).
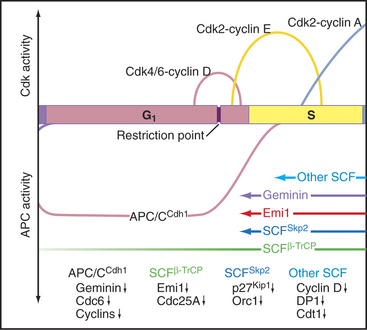
In the S phase, the Cdk inhibitor p27Kip1 is a target for the SCFSkp2 ubiquitin ligase complex, which marks it for destruction by proteasomes (see Chapter 40). SCF gets its name from three of its components: Skp2, cullin, and F-box proteins (see Fig. 40-17). Skp2, which is short for “S-phase kinase-associated protein,” got its name because it was first identified in a complex with Cdk2–cyclin A. This kinase targets proteins for recognition by SCF, which recognizes and ubiquitinates its substrates only after they have been phosphorylated at certain key positions. E2F/DP and cyclin E are also degraded when cells enter the S phase, and this is apparently triggered by Cdk2–cyclin A (Fig. 42-10).
At the onset of DNA replication, each origin of replication has bound to it the ORC complex, Cdc6p-Cdt1, and multiple hexameric Mcm complexes. (See Table 42-1 for a description of the major activities involved in DNA replication. See Box 42-1 for an introduction to DNA replication in E. coli.)
BOX 42-1 DNA Replication in Escherichia Coli
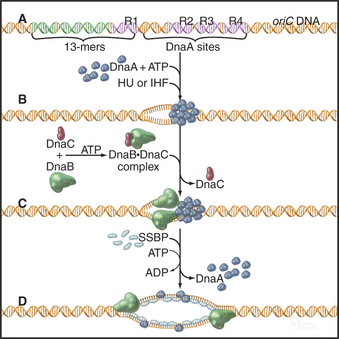
Initiation: E. coli chromosomal DNA replication initiates within a 245-bp region, termed oriC. This region contains four 9-bp binding sites for the E. coli initiator protein, DnaA. Nearby are three repeats of a 13-bp A: T-rich sequence. oriC also contains specific binding sites for two small histone-like proteins called HU and IHF. Replication is initiated with the cooperative binding of 10 to 20 DnaA monomers to their specific binding sites (Fig. 42-12). To be active, these monomers must each have bound ATP. Binding of DnaA permits unwinding of the DNA at the 13-bp repeats, in a reaction that requires the histone-like proteins. Next, DnaC binds to DnaB and escorts it to the unwound DNA. DnaB is the key helicase that will drive DNA replication by unwinding the double helix, but it binds DNA poorly on its own in the absence of its DnaC escort. Once DnaB has docked onto the DNA, DnaC is released, and the helicase can then start to unwind the DNA, provided that ATP, SSB, and DNA gyrase are present. SSB is a single-stranded DNA binding protein that stabilizes the unwound DNA, and DNA gyrase is a topoisomerase (see Chapter 13) that removes the twist that is generated when the two strands of the double helix are separated.
Mechanism of DNA Synthesis
Locally, DNA replication appears to start when the Cdk2-cyclin A and Cdc7p-Dbf4p kinases activate the prereplication complex. Key phosphorylated proteins include Mcm 2–7 and Cdc6 (Fig. 42-11A). Phosphorylation triggers a change in the binding of Cdc6p and Cdt1 to the DNA. Cdc6p remains bound to the chromatin throughout the S phase, while Cdt1 is released and degraded. Activation recruits to the origin a protein called Cdc45p together with a single-strand DNA-binding protein, RPA (Fig. 42-11B). Several other proteins also bind at this time, but their detailed functions are still being elucidated (Table 42-1).
Cdc45p appears to associate with the Mcm proteins and promote the binding of RPA, forming a complex that somehow activates the Mcm helicase. Cdc45p and RPA then recruit DNA polymerase to the origin (Fig. 42-11C). As the helicase starts to separate the DNA strands, moving outward in both directions from the origin of bidirectional replication, RPA stabilizes the separated strands, ensuring that they do not base-pair with one another again. Recent results suggest that the Mcm 8 protein may take over as the helicase once the replication fork has moved away from the replication origin (Fig. 42-11H), but this remains under investigation.
The separated DNA strands are ready for replication, but DNA synthesis always involves addition of an incoming nucleoside triphosphate to a free 3′ OH group at the terminus of a preexisting nascent polynucleotide (Fig. 42-1). In the absence of a nascent DNA chain, how does DNA polymerase get started? This problem is solved by a DNA-dependent RNA polymerase called a primase, which, like other RNA polymerases, can initiate synthesis de novo without the need for a 3′ OH group. In eukaryotes, all DNA chains are started by a complex of DNA polymerase a and a primase subunit, collectively known as Pol α/Primase. Primase synthesizes an RNA chain of about 10 nucleotides to which DNA polymerase a adds another 20 to 30 nucleotides of so-called initiator DNA (iDNA) (Fig. 42-11D–E). These initiating reactions are potentially hazardous, because DNA polymerase a lacks proofreading ability. Any errors in matching up an incoming base would create a mutation. Given the huge number of initiation events that are required to replicate an entire genome, this potential for errors is not acceptable. Therefore, the RNA primer and most or all of the initiator DNA laid down by Pol α/Primase are subsequently replaced.
Once Pol α/Primase has done its job, two further essential factors act. A pentameric protein complex called replication factor C (RFC) binds the 3′ end of the initiator DNA. RFC uses energy from ATP hydrolysis to load the trimeric protein PCNA onto the DNA (see Fig. 42-11F–G). The PCNA trimer is doughnut-shaped, and when the DNA is inserted into its central hole, it is topologically locked onto the DNA. RFC binding and PCNA loading displace Pol α/Primase from the DNA, and PCNA then recruits DNA polymerases δ and ε to the DNA. Moving along with the sliding platform of PCNA, these polymerases then process along the DNA, synthesizing DNA continuously on the leading strand (see Fig. 42-11H). On the lagging strand, they synthesize about 250 bp of DNA until they run into the next Okazaki fragment. Cdc45p may be a scaffolding factor that holds the Mcm hexamer and the replicative DNA polymerases together as the fork moves.
The final steps of DNA replication are removal of the RNA primer (and probably initiator DNA) and ligation of adjacent stretches of newly synthesized DNA. Removal of the primer can be accomplished in two ways (Fig. 42-11I). On one hand, an RNA exonuclease called RNase H can chew in from the 5′ end of the primer. However, this enzyme cannot remove the last ribonucleotide that is joined to initiator DNA. That requires a second nuclease, called Fen1. Alternatively, Fen1 can do the whole job itself if it gets help from a helicase. In this case, the helicase peels the RNA (and possibly the initiator DNA) away from the template, creating a sort of flap. Fen1 then cleaves at the junction where the flap is anchored to the DNA template, removing the oligomer of unwanted nucleotides in one step.
Following removal of initiator RNA, the Pol d/PCNA complex extends the upstream nascent chain until it runs into the 5′ end created by Fen1. DNA ligase I then joins the two stretches of DNA together (Fig. 42-11J).
Higher-Order Organization of DNA Replication in the Nucleus
A wide variety of experimental evidence revealed that the unit of replication in eukaryotic chromosomes is not the individual replicon but rather a replicon cluster. Evidence for this higher-order organization of DNA replication within the nucleus was first obtained by fiber autoradiography. Cells were fed radioactive precursors for DNA synthesis and then examined by electron microscopy. (For an explanation of this technique, see Fig. 40-3.) The spatial distribution of DNA replication during the S phase is more readily observed by using BrdU, a nucleotide base analog that is incorporated into DNA by the replication machinery in place of thymidine (this is called by its more correct name of Br-dUTP in Fig. 42-13). Incorporation of BrdU into DNA makes the newly synthesized daughter DNA strand heavier, allowing its separation from the parental DNA by centrifugation on a cesium chloride density gradient (see Chapter 6). In addition, specific antibodies that recognize DNA containing either BrdU or the related reagents IdU and CldU can be used to localize the changing patterns of DNA synthesis as cells traverse the S phase. More recently, analogs have been developed in which the Br in Fig. 42-13A has been replaced by a fluorescent group. This allows the newly replicated DNA to be observed directly in living cells.
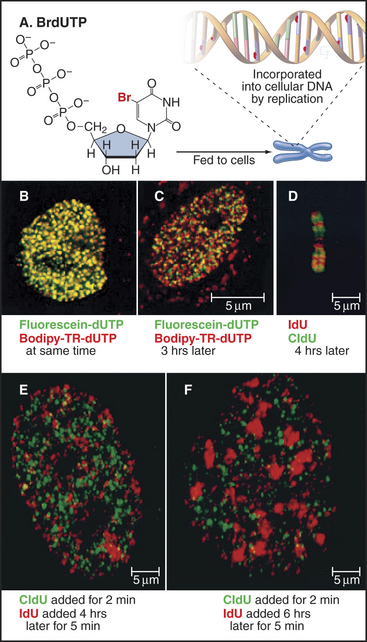
These methods reveal up to 1000 sites of active replication, called replication foci, at any one time during the S phase in a mammalian cell nucleus (Fig. 42-13B–C and E–F). Given that each of these replication foci is active for only about one hour out of the eight- to ten-hour S phase, a cell will replicate DNA at about 10,000 of these foci. Given roughly 60,000 origins in a mammalian cell, each replication focus represents five or six replication origins that are activated coordinately. These replication foci may be associated with the nuclear matrix or nucleoskeleton (see Chapter 13).
Temporal Control of Replication during the S Phase
The term S phase gives the impression that all DNA replicates more or less synchronously, but this is far from true. At any given time during the S phase, only 10% to 15% of the replicons actively synthesize DNA. Some replicate earlier, others later. It is important to note that this pattern of replication is not random; some origins consistently replicate early in the S phase, whereas others consistently replicate late in the S phase. Overall, the human genome can be subdivided into at least 1000 “zones,” each of which replicates at a characteristic time during the S phase. The organization of replication zones corresponds roughly to the organization of chromosomes into banding patterns: early-replicating regions typically correspond to gene-rich R bands, whereas late-replicating regions typically correspond to gene-poor G-bands (Fig. 42-13D; compare with Fig. 13-14). A similar division of chromosomes into early- and late-replicating regions also holds true for budding yeast, although many fewer replication origins are involved.
BrdU labeling experiments show that the basic unit of chromosomal DNA replication is a cluster of roughly five replication origins that fire coordinately. What must now be superimposed on this view of the replicating chromosome is a second level of regulation: the time at which each replicon cluster fires during the S phase. This can be seen clearly by synchronizing cells at the beginning of the S phase, releasing them from cell-cycle arrest, and then exposing them to BrdU at various times thereafter. This experiment reveals very distinctive patterns of DNA synthesis occurring at different times during the S phase (Fig. 42-13B–C and E–F). Early on, euchromatin replicates throughout the nucleus. Later, replicating regions appear concentrated around nucleoli and other areas of more condensed chromatin. Toward the end of the S phase, replication is largely concentrated in blocks of heterochromatin. These observations show that DNA replication occurs throughout the nucleus, wherever DNA is located. DNA does not move to a small number of discrete sites to be replicated (as was previously thought).
The most striking aspect of these patterns of DNA synthesis is their reproducibility from one cell cycle to the next. For example, regions of DNA labeled early in the S phase overlap little or not at all with DNA labeled three hours later (Fig. 42-13C and E). However, DNA labeled at corresponding points of the S phase in two successive cell cycles superimposes almost entirely. Thus, the chromosomal substructure that gives rise to replication foci is stable from one cell cycle to the next. This strongly suggests that particular regions of chromosomes are organized into reproducible structural domains and that each domain has a particular “window” during the S phase during which it replicates. This is significant. In one study, chromosomal regions that replicated at the wrong time during the S phase as a result of a mutation in an ORC subunit had a defective condensed structure in the next mitosis.
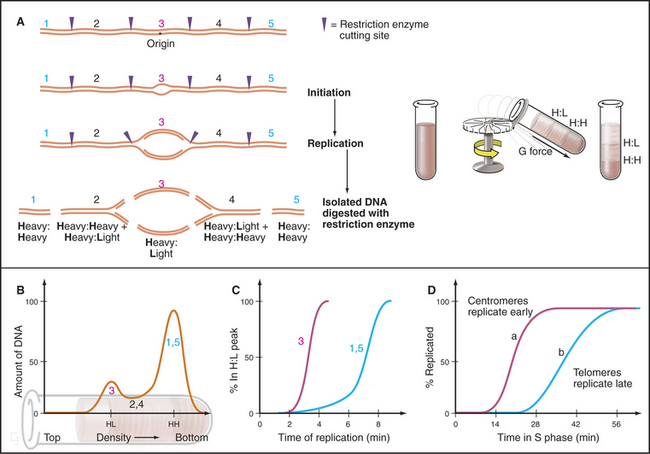
The timing of replication of particular replication origins has been studied most carefully in budding yeast. First, a procedure was developed whereby all cells in a population could be induced to enter the S phase synchronously. Next, the shift in the density of the DNA following BrdU incorporation was used to distinguish between DNA that had replicated and DNA that had not (Fig. 42-14). It then became relatively simple to take DNA probes from different regions of the chromosome and determine when each replicated (changed its density) during the S phase. This protocol demonstrated that each ARS element replicates at a characteristic time during the S phase.
The Intra-S Checkpoint
A powerful group of three checkpoints, which we here collectively call the intra-S checkpoint, monitors the process of DNA replication and stops it if DNA breaks or stalled replication forks are detected (Fig. 42-15). A third aspect of these checkpoints is to delay the onset of mitosis until the replication of the genome is complete.
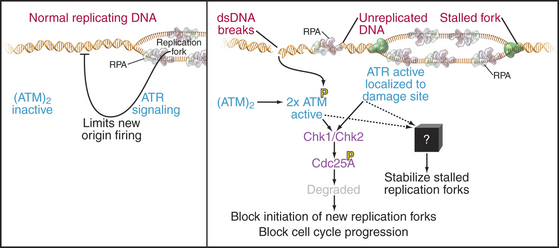
These checkpoints have similarities and differences compared to other DNA damage checkpoints. For example, unlike the G1 DNA damage checkpoint, p53-mediated transcription is not required. However, as is the case in the G1 and G2 phases, if DNA breaks are detected, the kinases ATM and ATR and their downstream effectors phosphorylate Cdc25A, triggering its rapid destruction mediated by SCFb-TrCP (see Fig. 41-12). The resulting inactivation of Cdks during the S phase prevents Cdc45p from loading onto prereplication complexes and blocks the initiation of new replication forks. This prevents replication forks from running across DNA breaks, which could lead to chromosome breaks and the loss of genetic material, with lethal consequences for the cell.
The intra-S checkpoint also detects stalled replication forks. Why would a replication fork stall? This could happen if, for example, the fork encounters a damaged DNA base or bases that it cannot “read.” Stopping the fork gives time for the DNA repair machinery to detect and repair the damage (see Box 43-1). Stalled forks activate the ATR kinase, leading to Cdc25A inactivation as described earlier and the cessation of new fork initiation. In addition, through an unknown mechanism, the intra-S checkpoint also has a mechanism to protect existing forks from disassembly. This is important because replication forks contain unwound and nicked DNA molecules that could be turned into breaks if the structure disassembled.
ATR kinase is activated by binding single-stranded DNA associated with RPA. As this is normally present at every replication fork during DNA replication, ATR signaling appears to be an intrinsic aspect of the replication process. It has been proposed that ATR normally limits excessive firing of replication origins by keeping the concentration of Cdc25A low and coordinates replication with other cell-cycle events. For example, Chk2, a kinase activated by ATM and ATR (Fig. 42-15), is required for the dependence of late origin firing on completion of early replication. This role during the normal S phase could explain why ATR is essential for the life of the cell.
Synthesis of the Histone Proteins
Synthesis of histones during the S phase is tightly coupled to ongoing DNA replication. If replication is blocked either by addition of drugs or by temperature-sensitive mutants, histone synthesis declines abruptly shortly thereafter. This link between histone synthesis and DNA replication appears to involve at least three components (Fig. 42-16).
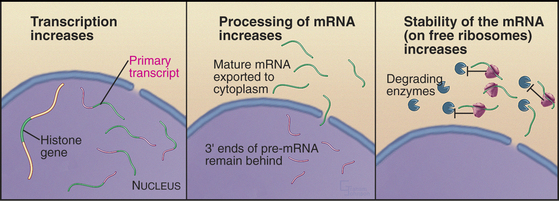
Second, theprocessing of histone mRNAs increases sixfold to 10-fold as cells enter the S phase. Histone mRNAs are not polyadenylated, and the primary transcripts are considerably longer than the mature forms. Processing of the 3′ end of histone pre-mRNAs involves the U7 snRNP (see Chapter 16), a portion of which recognizes histone mRNA and base-pairs with it during processing. Cell-cycle-dependent regulation of processing appears to involve changes in the accessibility of the necessary portion of U7 snRNA. This region is inaccessible in G0 cells but becomes accessible when cells that have reentered the cycle begin the S phase. The mechanism for this change in RNA conformation is not known.
As discussed in Chapter 13, specialized variant forms of histones are synthesized and inserted into the chromatin outside of the S phase. These histones are encoded by mRNAs with introns and normal poly(A) tails and are therefore not processed by the specific S phase–associated pathway (see Chapter 16). Their insertion into chromatin is typically correlated with RNA transcription rather than DNA replication.
Other Events of the S Phase
Although the bulk of attention on the S phase focuses on the duplication of the chromosomes, at least one other essential function required for stability of the genome also occurs at this time. This is duplication of the centrosomes, which will go on at the next mitosis to set up the poles of the mitotic spindle that are responsible for accurate partitioning of the replicated chromosomes. (See Chapter 34 for a discussion of centrosome duplication.)
With the completion of DNA replication and duplication of the centrosomes, the cell is ready to divide. As the levels of Cdk activity rise toward the threshold that is sufficient to trigger mitotic entry and other factors necessary for mitosis accumulate, the cell continues to screen the integrity of the DNA to ensure that the genome has been replicated correctly and that no harmful DNA damage has occurred in the interim. These checks, together with other ongoing preparations for mitosis, are the principal events of the G2 phase (see Chapter 43).
Baker TA, Wickner SH. Genetics and enzymology of DNA replication in Escherichia coli. Annu Rev Genet. 1992;26:447-477.
Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nat Rev Mol Cell Biol. 2004;5:792-804.
Bell SP, Dutta A. DNA replication in eukaryotic cells. Annu Rev Biochem. 2002;71:333-374.
Diffley JF. Regulation of early events in chromosome replication. Curr Biol. 2004;14:R778-R786.
Gilbert DM. In search of the holy replicator. Nat Rev Mol Cell Biol. 2004;5:848-855.
Hübscher U, Maga G, Spadari S. Eukaryotic DNA polymerases. Annu Rev Biochem. 2002;71:133-163.
Jónsson ZO, Hübscher U. Proliferating cell nuclear antigen: More than a clamp for DNA polymerases. BioEssays. 1997;19:967-975.
Kearsey SE, Cotterill S. Enigmatic variations: Divergent modes of regulating eukaryotic DNA replication. Mol Cell. 2003;12:1067-1075.
Kearsey SE, Maiorano D, Holmes EC, Todorov IT. The role of MCM proteins in the cell cycle control of genome duplication. BioEssays. 1996;18:183-190.
Reed SI. Ratchets and clocks: The cell cycle, ubiquitylation and protein turnover. Nat Rev Mol Cell Biol. 2003;4:855-864.
Stillman B. Cell cycle control of DNA replication. Science. 1996;274:1659-1664.
Waga S, Stillman B. The DNA replication fork in eukaryotic cells. Annu Rev Biochem. 1998;67:721-751.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
CHAPTER 42 S Phase and DNA Replication
Accurate replication of DNA, which is crucial for cellular propagation and survival, occurs during the S phase (DNA synthesis phase) of the cell cycle. This chapter begins with a brief primer on the events of replication and then discusses its regulation. Next, the chapter covers the proteins that bind origins of replication and ensure that each region of DNA is replicated once and only once per cell cycle. It closes by discussing how the structure of the nucleus influences replication.
DNA Replication: A Primer
In the basic reaction of DNA replication, the 3′ hydroxyl at the end of the growing DNA strand makes a nucleophilic attack on the α-phosphate of the incoming nucleoside triphosphate to form a phosphodiester bond. This incorporates the nucleotide into the growing chain and releases pyrophosphate (Fig. 42-1). Subsequent hydrolysis of the pyrophosphate provides the driving force for the reaction. This reaction requires the presence of a template strand of DNA that specifies, through base pairing, which of the four nucleoside triphosphates is added to the growing complementary strand.
Before discussing DNA replication and its regulation, an introduction to some terminology describing the geometry of replicating DNA is required. The exact site on the chromosomal DNA where replication begins is termed the origin of bidirectional replication. As the termbidirectional implies, two sets of DNA replication machinery head off in opposite directions from the origin. Each set of replication machinery, together with the DNA that it is replicating, is called a replication fork because at the site of replication, one parental DNA molecule splits into two (Fig. 42-2). It is not known whether replication forks move along the DNA like trains along a track or whether the fork sits at a stationary site (referred to as a replication factory) through which the DNA is “reeled in” as it is replicated.
The bidirectional nature of DNA replication causes a fundamental problem, as DNA synthesis invariably proceeds in a 5′ to 3′ direction. Replication of the so-called leading strand poses no problems. This is the strand along which the fork moves in a 3′ to 4′ direction, so the newly synthesized DNA is laid down smoothly in a 5′ to 3′ direction (Fig. 42-2). However, the other template strand faces in the opposite direction, apparently requiring DNA polymerase to synthesize DNA in the wrong direction as the replication fork progresses away from the origin (i.e., adding nucleotides in a 3′ to 4′ direction). No DNA polymerase with this polarity has been found. Instead, this lagging strand replicates in a series of short segments. Every time the DNA strands have been peeled apart (unwound) by 250 nucleotides or so, a polymerase/primase complex (see Fig. 42-11) initiates DNA synthesis on the lagging strand, with the polymerase running back toward the replication origin in a 5′ to 3′ direction. Locally, synthesis on the lagging strand proceeds in a direction opposite to the overall direction of fork movement. Synthesis of each lagging strand fragment stops when DNA polymerase runs into the 5′ end of the previous fragment. Thus, the lagging strand is copied in a highly discontinuous fashion into short fragments known as Okazaki fragments (named after their discoverer [Fig. 42-2]). Fig. 42-11 describes the enzymes and events at the replication fork in greater detail.
Origins of Replication
Bacteria such as Escherichia coli replicate their circular chromosomes using two replication forks starting from a single origin of replication (Fig. 42-3A), but eukaryotes must use multiple origins of replication to duplicate their large genomes during a relatively short S phase, which can be limited to as little as a few minutes in some early embryos. These numerous origins are distributed along the chromosome: up to 400 in budding yeast and about 60,000 in human cells. These origins are positioned so that all of the DNA is replicated in the available time, and to be on the safe side, more origins are prepared than are actually needed.
The existence of multiple origins creates a potential hazard: If any origin were used more or less than once per cell cycle, genes would be duplicated or lost. How is the “firing” of all of these origins orchestrated so that each is used once and only once per S phase? Cells manage this problem by a mechanism termed licensing, which ensures that each origin is used once and only once per S phase. Each origin is licensed to replicate once and only once per cell cycle. Replication of the origin removes the license, which cannot normally be renewed until the cell has completely traversed the cycle and has passed through mitosis.
A unit of chromosomal DNA whose replication is initiated at a single origin is termed a replicon. The origin is defined genetically as a replicator element. The classic replicon is the E. coli chromosome (which is 4 × 106 base pairs [bp] in size); this has a single replicator site called oriC (Fig. 42-3). An initiator protein (product of the E. coli DnaA gene [Fig. 42-12]) binds to this origin and either directly or indirectly promotes melting of the DNA duplex, giving the replication machinery access to two single strands of DNA. Other factors bind to the initiator, and their concerted action produces a wave of DNA replication proceeding outward in both directions along the DNA (a replication “bubble”) at about 750 to 1250 bases per second.
An average human chromosome contains about 150 × 106 bp of DNA. Because the replication machinery in mammals moves only about 20 to 100 bases per second (probably reflecting the fact that the DNA is packaged into chromatin [see Chapter 13]), it would take up to 2000 hours to replicate this length of DNA from a single origin. In most human cells, the duration of the S phase is about eight hours. This means that at least 25 to 125 origins of replication would be required to replicate an average chromosome in the allotted time. In fact, origins of replication are much more closely spaced than this. It has been estimated that mammalian origins of replication are spaced about 100,000 to 150,000 bp apart. Thus, approximately 60,000 origins of replication participate in replication of the entire human genome.
Replication Origins in S. Cerevisiae
About 400 origins of replication participate in replicating the budding yeast genome. A major breakthrough in understanding DNA replication in S. cerevisiae was the identification of short (100 to 150 bp) segments of DNA that act as replication origins in vivo when cloned into a yeast plasmid (circular DNA molecule). These autonomously replicating sequences (or ARS elements) allow yeast plasmids to replicate in parallel with the cellular chromosomes (Fig. 42-4). ARS elements are often, although not always, bona fide replication origins in their native chromosomal context. Replication always initiates within ARS elements, but not all ARS ele-ments act as origins of DNA replication in every cell cycle.
Budding yeast ARS elements share a common DNA sequence motif called the ARS core consensus sequence: 5′-(A/T)TTTAT(A/G)TTT(A/T)-3′ (Fig. 42-5). Single base mutations at several locations within this sequence completely inactivate ARS activity. Other, less well-conserved DNA sequences also contribute to the activity of the ARS as a replication origin. One of these, termed B1, together with the ARS core, forms the binding site for a complex of six proteins (five of which are AAA ATPases) termed the origin recognition complex (ORC [see later section]). The DNA unwinding element is thought to be another short sequence (B2) located a bit further along the DNA. DNA synthesis begins at an origin of bidirectional replication midway between the ORC binding site and the DNA unwinding element.
ORC was identified by its ability to bind the 11-bp ARS core sequence (Fig. 42-5). This binding has two noteworthy features. First, it requires adenosine triphosphate (ATP), which remains associated with the ORC complex. Second, in yeast, the ORC complex remains bound to the origins of replication across the entire cell cycle. Thus, something other than the presence of ORC must be responsible for regulating the periodic activation of origins in the S phase (see Fig. 42-14). In metazoans, ORC behavior is more complex; the largest subunit, Orc1, cycles on and off the DNA in a cell-cycle-regulated manner.
ARS elements typically contain binding sites for other sequence-specific DNA binding proteins, such as transcription factors. For example, a transcription factor called ARS-binding factor 1 (ABF-1) binds to the B3 sequence within the ARS1 element (Fig. 42-5). Deletion of the ABF-1 binding site only slightly reduces the ability of ARS1 to act as a replication origin in vivo. Furthermore, substitution of DNA binding sequences for other transcription factors within the B3 sequence has little effect on replication efficiency.
In addition to their role in DNA replication, several ORC components also seem to regulate heterochromatin formation and transcription (see Chapters 13 and 15). This cross talk between the machinery used for transcription and DNA replication may explain why regions of chromosomes with actively transcribed genes typically replicate early in the S phase (see the discussion that follows). The Orc6 subunit also functions in mitosis at kinetochores and during cytokinesis. Its detailed role in those processes is not known.
Replication Origins in Mammalian Cells
At present, two types of mammalian replication origins are known. The first is exemplified by the origin of replication adjacent to the lamin B2 gene (Fig. 42-6A). This origin “fires” within the first several minutes of the S phase, and a variety of methods have succeeded in mapping it to a stretch of less than 500 bp. Within this region, a single origin of bidirectional replication appears to be used. Thus, the lamin B2 origin of replication appears to be analogous to the well-characterized budding yeast origins.
The second is exemplified by the widely studied replication origin lying just downstream of the hamster gene for dihydrofolate reductase, an enzyme that is essential for biosynthesis of thymidine. This origin is accessible to experimental study because it is possible to select for cells with this chromosomal region amplified as hundreds or even thousands of copies (Fig. 42-6B). By looking for the first regions of the amplified DNA to replicate, the origin of replication was initially located within a region of about 55,000 bp. It now appears that DNA replication can initiate with low efficiency at roughly 20 sites distributed throughout this broad zone. Two of these sites are used with relatively higher efficiency, accounting for about 20% of all initiation in the region. These sites, termed Ori-β and Ori-γ (Fig. 42-6), each encompass about 0.5 to 2 kb of DNA.
[/not-level-membership-for-basic-science-category]
