[level-membership-for-neonatal-and-perinatal-medicine-category]
21 Right Atrial Isomerism
I. CASE
A. Fetal echocardiography findings
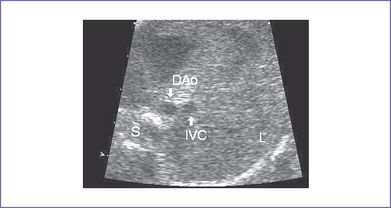
1. Imaging of the inferior vena cava (IVC) and aorta shows the two vessels running together on the left side of the spine (right atrial isomerism [RAI]) (Fig. 21-1).
2. The liver is midline, and the stomach is on the right and posterior.
3. The heart is in the left chest with a left aortic arch, a normal cardiac axis, and a normal size (cardiothoracic ratio = 0.34). The heart rate is 154 bpm.
4. The four-chamber view shows significant disproportion, with an unbalanced atrioventricular (AV) canal. There is a significantly large right-sided morphologic right ventricle (RV).
5. There is mild holosystolic common AV valve regurgitation.
6. The outflow assessment reveals normally related great arteries but with asymmetry (aorta–to–pulmonary artery diameter ratio = 1.1:0.6), and both arise from the RV.
7. No forward flow can be demonstrated by color or pulsed Doppler through the pulmonary outflow and main pulmonary artery. There are small confluent branch pulmonary arteries.
8. The aortic annulus is increased in size, and there is normal blood velocity by Doppler through it (1.2 m/s).
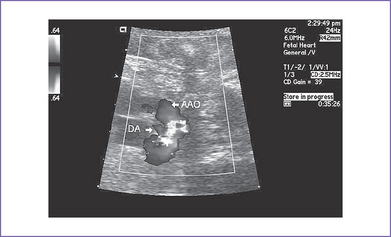
9. The ductal arch is tortuous and small and has retrograde flow (filling the pulmonary artery) (Fig. 21-2).
10. There is a large atrial septal defect (ASD) creating a common atrium, with a strand of muscle extending from anterior to posterior.
11. The pulmonary venous connection is to a posterior confluence and then to a descending vein coursing to the liver, where it is obstructed (abnormal venous flow pattern) before draining into the IVC.
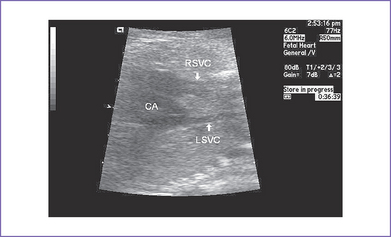
12. There are bilateral superior venae cavae (SVC) to the respective atria and no coronary sinus (Fig. 21-3).
13. The RV Tei index (myocardial performance index) is normal, with good ventricular function and no signs of heart failure.
D. Fetal management and counseling
a. The risk from amniocentesis is probably higher than the risk of abnormal karyotype in isomerism.
b. Aneuploidy is extremely rare and reportable in atrial isomerism.
2. Follow-up includes serial antenatal studies at 4- to 6-week intervals to monitor:
a. Heart size and ventricular function.
b. Evidence of hydrops fetalis, including pericardial effusion, ascites, and other features.
c. Progressive common AV valve regurgitation.
d. Size of the LV (when it is unbalanced at such an early age, it will remain so to delivery, and therefore reassessment of LV size is of no real value).
e. Size of the pulmonary arteries.
f. Pulmonary vein growth and flow patterns that could suggest progressive obstruction.
E. Delivery
1. In RAI with complex congenital heart disease, delivery should be at term or as close to term as possible in a tertiary care center given the risk of pulmonary stenosis and atresia and pulmonary vein obstruction.
2. With this ductus-dependent lesion, early initiation of prostaglandin E1 (PGE1) is mandatory. However, if obstruction of the pulmonary veins coexists, then increasing pulmonary perfusion can cause pulmonary edema.
F. Neonatal managment
a. PGE1 infusion should be started to keep the ductus open to increase pulmonary blood flow and raise arterial oxygen saturation to greater than 70%. If saturation does not increase, then repair of a total anomalous pulmonary venous connection (TAPVC) may be indicated.
b. Administration of oxygen can increase oxygen saturation by decreasing pulmonary vascular resistance (PVR) and by increasing blood flow.
c. Intubation and mechanical ventilation for progressive cyanosis might be needed.
d. With hypotension, volume and inotropic support are indicated to improve ventricular function.
e. Antibiotics: Asplenic patients should receive amoxicillin orally once per day at dose of 20 mg/kg body weight.
G. Follow-up
1. Subacute bacterial endocarditis (SBE) prophylaxis must be taken for life.
2. If the patient has residual hemodynamic abnormalities, anticongestive medications and some restriction of activities may be required.
3. The patient must be examined at least yearly with 12-lead electrocardiogram (ECG) and echocardiogram. Attention is paid to the patency of the pulmonary venous confluence–to–atrium connection.
4. Reoperation or stenting of the site of the previous confluence-to-atrium surgical connection may be necessary. However, it is not likely to be successful for long-term single-ventricle palliation.
5. If there is any history of unexplained palpitations or abnormal heart rhythm, a metabolic stress test with 24-hour Holter monitoring is recommended.
H. Risk of recurrence
1. If the mother is an obligate carrier of ZIC3 mutation (i.e., she is physically normal), the recurrence risk is 25% for having an affected son because this is an X-linked recessive single-gene disorder.
2. For every son there is a 50% chance of being affected.
3. ZIC3 mutation is an X-linked condition that can be associated with either right or left atrial isomerism (LAI).
4. Some families demonstrate autosomal recessive inheritance in which 25% of all children might be affected.
I. Outcome of this case
1. Labor was induced at 38 weeks.
2. The baby was born with good Apgar scores of 8 at 1 minute and 9 at 5 minutes and good weight.
3. Severe cyanosis was noted at birth. Pulse oximeter reading was 57%.
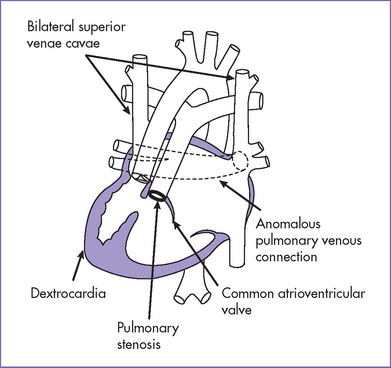
4. Postnatal echocardiography confirmed the diagnosis of right isomerism, common inlet right ventricle, transposed great arteries with double outlet and pulmonary stenosis, TAPVC obstructed below the diaphragm, and left- and right-sided SVCs (Fig. 21-4).
5. Chest x-ray showed a small heart and increased pulmonary vascular markings.
6. The baby was mechanically ventilated for progressive cyanosis, respiratory distress, and increased oxygen requirements. He was started on epinephrine and milrinone infusion for decreased cardiac function.
7. There was a common atrium and a patent ductus arteriosus. PGE1 infusion was started.
8. Abdominal ultrasound confirmed kidney abnormalities, a midline liver, and no spleen. No splenic artery could be identified.
9. High-penetration chest x-ray showed bilateral right bronchi.
10. The baby had an immediate repair of the TAPVC with direct anastomosis of the confluence to the left-sided atrium as well as a palliative aortopulmonary shunt.
11. The baby was placed on prophylactic amoxicillin for asplenia and infection prophylaxis.
12. Howell–Jolly bodies (pitted red blood cells) were noted on the complete blood count.
13. The postoperative period was complicated by pulmonary hypertension, seizures, wound infection, and accelerated junctional rhythm. Stenosis of the right pulmonary veins at the entrance to the back of the left-sided atrium was diagnosed at 1 month of age.
14. At 2 months of life the baby could not be weaned off assisted ventilation and died of multisystem organ failure.
II. YOUR HANDY REFERENCE
A. Prevalence
1. Syndromes involving abnormalities of lateralization of the viscera are commonly associated with structural cardiac abnormalities.
2. The incidence of this group of abnormalities is difficult to establish because the entity has been classified by so many names, and the condition may be underrecognized, particularly LAI, where there may be insignificant heart disease.
3. Visceral heterotaxy with congenital absence of the spleen (RAI) and complex cardiac malformation was reported in 2.8% of patients with congenital heart disease in the cardiac registry of Children’s Hospital in Boston.
B. Outcome
1. High surgical mortality has been reported in patients with RAI and complex heart disease, particularly in those presenting early in the neonatal period.
2. In a series reported by Sadiq and colleagues (1996), the mortality was 88% in patients requiring surgery in the first months of life, falling to 22% in patients requiring surgery after the first months of life.
3. Of 94 cases reported by Hashmi and colleagues (1998), the mortality rate in patients requiring surgery in the neonatal period was 75%, as against 51% in those requiring surgery later.
4. The usual indication for early surgery is most often to relieve obstructed anomalous pulmonary venous drainage or to augment inadequate pulmonary blood flow, or both. The best chance for survival is when the pulmonary veins are not anomalous or are anomalous without obstruction and there is only mild pulmonary outflow obstruction.
C. Associated syndromes and extracardiac anomalies
1. Heterotaxy is not associated with increased risk of chromosomal abnormalities.
a. Paradoxically, it is suggested that abnormal visceral situs is strongly predictive of normal karyotype.
b. Rarely, there have been associations of microdeletion of chromosome 22 and rarer forms of translocations.
2. Maternal diabetes is considered a risk factor for atrial isomerism.
3. In RAI, the lungs are bilaterally bilobed, no spleen is visualized, and the stomach can be on either side of the abdomen.
4. Bowel obstruction can evolve, at least in part due to malrotation of the gut.
D. Clues to fetal sonographic diagnosis
1. Discordant stomach and apex are common.
2. The IVC (which is intact) and aorta lie on same side of spine.
3. The IVC lies directly anterior to the aorta in the upper abdomen.
4. RAI is usually, if not always, associated with complex intracardiac anomalies such as atrioventricular septal defect (particularly unbalanced forms) with double outlet right ventricle (DORV), and is usually associated with pulmonary stenosis or pulmonary atresia.
5. RAI is often associated with anomalous pulmonary venous connection. At times, however, the pulmonary veins connect with a confluence that then connects to the back wall of the atrium.
6. RAI is often associated with bilateral SVC, which can also complicate the outcome of such infants, particularly at bidirectional Glenn surgery.
E. Immediate postnatal management for patients without prenatal diagnosis
1. Check pulse oximeter. An acceptable reading is greater than 92%; if it is lower, do a hyperoxia test.
2. Check the four-limb blood pressure.
a. If there is any discrepancy between upper and lower limb blood pressures (>20 mm Hg), suspect aortic coarctation.
b. This test is of value only if the ductus arteriosus is closed.
c. Absence of discrepancy between upper and lower limb blood pressure does not rule out coarctation if the ductus arteriosus is still open.
3. Check the chest x-ray for cardiac apex and midline liver and bronchi as well as evidence of pulmonary edema.
4. The ECG shows clues in cases of AV canal (left axis deviation) with qR pattern in lead aVL.
a. A qR pattern in lead V1 is always associated with complex congenital heart disease.
b. The ECG might show an abnormal p-wave axis (due to the presence of two sinus nodes), and serial ECGs might show a shifting p-wave axis.
5. Transfer the patient to the neonatal or pediatric (cardiac if available) intensive care unit (ICU) for further assessment and management including stat echocardiogram.
F. Pathophysiology
Asplenia syndrome (RAI, Ivemark’s syndrome) is associated with absence of the spleen (a left-sided organ) and bilateral right-sidedness (see Fig. 21-4).
1. Noncardiac malformations (Box 21-1).
a. Bilateral three-lobed right lungs with bilateral epitracheal bronchi.
c. Malrotation of the intestine and various other gastrointestinal malformations.
2. Cardiac malformations: In general, asplenia syndrome has more-severe cardiac malformations than polysplenia syndrome does.
a. The IVC is intact in all patients, but it may be left-sided (35%). Azygous continuation is almost never seen. Hepatic veins might join the IVC or some might join the atrium separately from the IVC (partial hepatic venous connection).
b. Total anomalous pulmonary venous return (TAPVR) with extracardiac connection (75%) often occurs with pulmonary venous obstruction. The pulmonary veins can return through a supracardiac or supradiaphragmatic connection, such as to the innominate vein or SVC, either directly or to a descending infradiaphragmatic vertical vein, where they might connect with the hepatic veins near the ductus venosus or reach the heart through the portal venous system.
c. Bilateral right atria (bilateral sinus node), primum ASD, and an additional upper interatrial communication are seen in the majority. This type of anatomy has been called a common atrium or an atrial strand.
d. Most patients have a single AV valve (90%) with single ventricle (50%) or with two ventricles (50%).
e. DORV with transposed or malposed arrangement of the great arteries is seen in 70%. The pulmonary valve can be stenotic (40%) or atretic (40%).
f. The P axis is normal or left atrial: +90-degree to +180-degree quadrant.
g. There is usually complete mixing of systemic and pulmonary venous blood in the heart because of multiple cardiovascular malformations. When pulmonary blood flow is reduced, as in RAI with asplenia syndrome, severe cyanosis results.
III. TAKE-HOME MESSAGE
Hashmi A, Abu-Sulaiman R, McCrindle BW, et al. Management and outcomes of right atrial isomerism: A 26-year experience. J Am Coll Cardiol. 1998;31(5):1120-1126.
Ho SY, Cook A, Anderson RH, et al. Isomerism of the atrial appendages in the fetus. Pediatr Pathol. 1991;11(4):589-608.
Huhta JC, Smallhorn JF, Macartney FJ. Two-dimensional echocardiographic diagnosis of situs. Br Heart J. 1982;48(2):97-108.
Machado MV, Crawford DC, Anderson RH, Allan LD. Atrioventricular septal defect in prenatal life. Br Heart J. 1988;59(3):352-355.
Machado MV, Tynan MJ, Curry PV, Allan LD. Fetal complete heart block. Br Heart J. 1988;60(6):512-515.
Rubino M, Van Praagh S, Kadoba K, et al. Systemic and pulmonary venous connections in visceral heterotaxy with asplenia: Diagnostic and surgical considerations based on seventy-two autopsied cases. J Thorac Cardiovasc Surg. 1995;110(3):641-650.
Sadiq M, Stümper O, De Giovanni JV, et al. Management and outcome of infants and children with right atrial isomerism. Heart. 1996;75(3):314-319.
[/level-membership-for-neonatal-and-perinatal-medicine-category][not-level-membership-for-neonatal-and-perinatal-medicine-category]
21 Right Atrial Isomerism
I. CASE
A. Fetal echocardiography findings
1. Imaging of the inferior vena cava (IVC) and aorta shows the two vessels running together on the left side of the spine (right atrial isomerism [RAI]) (Fig. 21-1).
2. The liver is midline, and the stomach is on the right and posterior.
3. The heart is in the left chest with a left aortic arch, a normal cardiac axis, and a normal size (cardiothoracic ratio = 0.34). The heart rate is 154 bpm.
4. The four-chamber view shows significant disproportion, with an unbalanced atrioventricular (AV) canal. There is a significantly large right-sided morphologic right ventricle (RV).
5. There is mild holosystolic common AV valve regurgitation.
6. The outflow assessment reveals normally related great arteries but with asymmetry (aorta–to–pulmonary artery diameter ratio = 1.1:0.6), and both arise from the RV.
7. No forward flow can be demonstrated by color or pulsed Doppler through the pulmonary outflow and main pulmonary artery. There are small confluent branch pulmonary arteries.
8. The aortic annulus is increased in size, and there is normal blood velocity by Doppler through it (1.2 m/s).
9. The ductal arch is tortuous and small and has retrograde flow (filling the pulmonary artery) (Fig. 21-2).
10. There is a large atrial septal defect (ASD) creating a common atrium, with a strand of muscle extending from anterior to posterior.
11. The pulmonary venous connection is to a posterior confluence and then to a descending vein coursing to the liver, where it is obstructed (abnormal venous flow pattern) before draining into the IVC.
12. There are bilateral superior venae cavae (SVC) to the respective atria and no coronary sinus (Fig. 21-3).
13. The RV Tei index (myocardial performance index) is normal, with good ventricular function and no signs of heart failure.
D. Fetal management and counseling
a. The risk from amniocentesis is probably higher than the risk of abnormal karyotype in isomerism.
b. Aneuploidy is extremely rare and reportable in atrial isomerism.
2. Follow-up includes serial antenatal studies at 4- to 6-week intervals to monitor:
a. Heart size and ventricular function.
b. Evidence of hydrops fetalis, including pericardial effusion, ascites, and other features.
c. Progressive common AV valve regurgitation.
d. Size of the LV (when it is unbalanced at such an early age, it will remain so to delivery, and therefore reassessment of LV size is of no real value).
e. Size of the pulmonary arteries.
f. Pulmonary vein growth and flow patterns that could suggest progressive obstruction.
E. Delivery
1. In RAI with complex congenital heart disease, delivery should be at term or as close to term as possible in a tertiary care center given the risk of pulmonary stenosis and atresia and pulmonary vein obstruction.
2. With this ductus-dependent lesion, early initiation of prostaglandin E1 (PGE1) is mandatory. However, if obstruction of the pulmonary veins coexists, then increasing pulmonary perfusion can cause pulmonary edema.
F. Neonatal managment
a. PGE1 infusion should be started to keep the ductus open to increase pulmonary blood flow and raise arterial oxygen saturation to greater than 70%. If saturation does not increase, then repair of a total anomalous pulmonary venous connection (TAPVC) may be indicated.
b. Administration of oxygen can increase oxygen saturation by decreasing pulmonary vascular resistance (PVR) and by increasing blood flow.
c. Intubation and mechanical ventilation for progressive cyanosis might be needed.
d. With hypotension, volume and inotropic support are indicated to improve ventricular function.
e. Antibiotics: Asplenic patients should receive amoxicillin orally once per day at dose of 20 mg/kg body weight.
[/not-level-membership-for-neonatal-and-perinatal-medicine-category]
