Chapter 80 Rheumatic Disease
An approach to the assessment of the patient with possible rheumatic disease
Common ocular presentations of rheumatic disease
Keratoconjunctivitis sicca and other corneal presentations
Keratoconjunctivitis sicca (KCS, or “dry eye syndrome”) is a common ocular manifestation of a number of rheumatic diseases including rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), scleroderma, and relapsing polychondritis. Symptoms vary from slight irritation and burning in mild disease to severe pain and blurred vision arising from increasing corneal involvement. Clinical examination using slit-lamp biomicroscopy reveals a small or absent tear meniscus with a tear film break-up time of less than 10 seconds. Corneal abnormalities, which may be highlighted with fluorescein drops and a cobalt blue light, include punctate epitheliopathy, mucus filaments, strands, and plaques. Additional staining with Rose Bengal or Lissamine Green drops reveals a characteristic interpalpebral pattern, with greatest staining nasal and temporal to the corneal limbus. Tear production, as measured by Schirmer’s test, is reduced. Wetting of the test strip by less than 5 mm after 5 minutes in the un-anesthetized eye indicates severe tear deficiency. It should be noted that the correlation of dry eye symptoms with observed disease is poor. Many more patients report “dry eyes” than have visible disease, and many asymptomatic patients do have some degree of keratoconjunctivitis sicca.1
Other less common corneal presentations of rheumatic disease include the sight-threatening peripheral ulcerative keratitis (PUK). The etiology is uncertain but it has been suggested that immune complex deposition at the corneal limbus results in an obliterative vasculitis and stromal melt. It is most commonly associated with RA or systemic vasculitis, in particular granulomatosis with polyangiitis (previously known as Wegener’s granulomatosis).2,3 Clinical features include variable pain and redness and reduced vision, uni/bilateral peripheral corneal ulceration with epithelial defect and stromal thinning, associated limbal inflammation, and scleritis.
Scleritis/episcleritis
Scleritis
Scleritis is associated with systemic disease in around 40–50% of patients, of which most are cases of a rheumatic disease, such as RA, granulomatosis with polyangiitis, relapsing polychondritis, SLE, sarcoidosis, polyarteritis nodosa, inflammatory bowel disease, psoriatic arthritis, ankylosing spondylitis, and gout.4 It is commonest in middle-aged women. Scleritis is bilateral in 50% of cases, but both eyes may not be affected at the same time.5 The pain (constant/deep/boring) can be so severe that it may wake the patient at night. The eye has an intense red/dark red appearance. The globe may be very tender to touch. A bluish hue implies scleral thinning from previous active scleritis due to the underlying blue/black uveal tissue showing through the translucent sclera. Scleral thinning can eventually result in high degrees of astigmatism. The degree of redness and scleral thinning is more easily seen under room light or in daylight than by the slit lamp. Topical phenylephrine 2.5% causes blanching of the more superficial episcleral vessels but does not change the engorgement of deeper scleral vessels and can often help differentiate between scleritis and episcleritis. The most severe type is necrotizing anterior scleritis with inflammation. Apart from the severe pain and redness, there may be tearing and photophobia. White avascular areas surrounded by injected edematous sclera are present that may lead to scleral necrosis. An associated anterior uveitis suggests advanced disease. Complications of scleritis include peripheral ulcerative keratitis, acute stromal keratitis, sclerosing keratitis, uveitis, cataract, astigmatism, glaucoma, and globe perforation.
Episcleritis
This common condition is a benign, recurrent inflammation of the episclera. Being superficial it is easily distinguished from deeper scleral inflammation, in that it is less painful and the involved vessels blanch on instillation of topical phenylephrine 2.5%. It is more common in young women, often self-limiting, and may require little or no treatment. It is not usually associated with any systemic disease, although around 10% may have an underlying rheumatic disease.6
Uveitis
Acute anterior uveitis
In acute anterior uveitis (AAU) patients typically present with pain, photophobia, redness, and blurred vision. Examination findings are of anterior segment inflammation including circumlimbal injection, keratic precipitates (especially inferior), anterior chamber (AC) flare, cells, and fibrin (fibrin is a key feature in HLA-B27-associated uveitis). A hypopyon is suggestive of HLA-B27-associated disease, Behçet disease, or severe intraocular infection.7 Posterior synechiae are common in both idiopathic and HLA-B27-associated AAU and every effort should be made to break them at time of presentation. Vitreous cells may be seen as “spill-over” inflammation, but vitritis is not a dominant feature. Occasionally cystoid macular edema (CME) may be seen (especially in HLA-B27 disease), but this is more commonly a feature of intermediate, posterior or pan-uveitis. It is estimated that up to one-third of patients with AAU have ankylosing spondylitis (AS).8 Treatment is with intensive topical corticosteroid and mydriatic. If severe, subconjunctival corticosteroid and mydriatic, oral corticosteroid or even intravenous corticosteroid may be given. Recurrent disease (especially if frequent or severe) may be an indication for maintenance systemic treatment.
Therapeutic considerations
Many patients will require systemic treatment, such as corticosteroids, immunosuppressants (methotrexate, azathioprine, mycophenolate mofetil, cyclosporine), and biologics (anti-TNF, rituximab – anti-CD20) for their rheumatic and ophthalmic disease. Where possible this should be undertaken in conjunction with a rheumatologist who has an expertise in this type of therapy. Ophthalmologists should only prescribe these drugs if they have a detailed knowledge of their action, route of administration, side-effects, and how to monitor for them, as the treatments themselves have potentially serious complications. Most patients with sight-threatening disease are likely to be prescribed oral corticosteroid, and these have numerous, well-recognized side-effects, including glucocorticoid-induced bone disease.9 Fortunately, in patients with ocular inflammation the commonly used immunosuppressants do not appear to increase overall or cancer mortality.10
Disease-specific section
Rheumatoid arthritis
Epidemiology
RA is the commonest of the inflammatory arthritides, with an incidence of around 3 in 10 000 per annum and a prevalence of 1% among adults in industrialized nations.11–13 Epidemiological risk factors include age (increases with age), female gender (3–5 times greater risk than male),13 and smoking.14
Articular and systemic disease
Typical features of the arthritis of RA are its symmetrical small joint distribution and its deforming nature, giving rise to the classic appearance of ulnar deviation (“rheumatoid hands”). All synovial joints may be affected, including the metacarpophalangeal joints (MCP), proximal interphalangeal joints (PIP), the interphalangeal joint of the thumb, metatarsophalangeal joints, wrist, elbows, hip joints, knees, and the atlanto-axial joint.15 Degenerative changes of the atlanto-axial joint may make intubation hazardous and should be considered when planning anesthesia. In contrast to noninflammatory degenerative arthritis such as osteoarthritis, patients commonly complain of morning stiffness that improves with exercise.15
Extra-articular features are common and affect many systems.16 One-quarter of patients with RA develop solid lesions within the subcutaneous tissues of extensor surfaces known as rheumatoid nodules. Cardiac complications includes accelerated atherosclerotic coronary artery disease, pericarditis, heart block or valvular dysfunction.17 Respiratory complications include pleural effusions, nodules of the pleura or lung tissue, and interstitial fibrosis; a severe variant of interstitial fibrosis associated with RA and coal-miners’ pneumoconiosis is known as Caplan syndrome.18 Other systemic complications include renal amyloidosis and a chronic anemia and/or leucopenia. The combination of RA, splenomegaly, and leucopenia is known as Felty syndrome.19 Vasculitis is a rare but important complication of RA which ranges in severity from nail-fold infarcts to severe life-threatening systemic vasculitis.20
In addition to clinical assessment, the diagnosis may be supported by systemic investigations such as the measurement of inflammatory markers, rheumatoid factor, and anticitrullinated protein antibodies (ACPA) (Box 80.1).21
Box 80.1
The 2010 American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) classification criteria for rheumatoid arthritis21
Classification criteria for RA
| Score | |
| A. Joint involvement | |
| 1 large joint | 0 |
| 2–10 large joints | 1 |
| 1–3 small joints (with or without involvement of large joints) | 2 |
| 4–10 small joints (with or without involvement of large joints) | 3 |
| >10 joints (at least 1 small joint) | 5 |
| B. Serology (at least 1 test result is needed for classification) | |
| Negative RF and negative ACPA | 0 |
| Low-positive RF or low-positive ACPA | 2 |
| High-positive RF or high-positive ACPA | 3 |
| C. Acute-phase reactants (at least 1 test result is needed for classification) | |
| Normal CRP and normal ESR | 0 |
| Abnormal CRP or abnormal ESR | 1 |
| D. Duration of symptoms | |
| <6 weeks | 0 |
| ≥6 weeks | 1 |
(Adapted from Aletaha D, Neogi T, Silman AJ, et al. 2010 Rheumatoid Arthritis Classification Criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010;62:2569–81.)
Ocular disease
Scleritis occurs in 1–6% of patients with RA, and in up to 14% of patients with rheumatoid vasculitis.2,4,5 Scleritis in the context of RA may present with severe pain and may be diffuse or nodular, anterior or posterior, and necrotizing or non-necrotizing in pattern.6 Of greatest concern is scleromalacia perforans, in which scleral destruction is not typically painful and not associated with visible signs of inflammation.2,6 Episcleritis may also be seen in the context of RA.6,22
Corneal complications of RA are wide-ranging with varying risk of perforation. Marginal keratitis without apparent inflammation may result in peripheral thinning giving rise to the appearance of “contact lens cornea”. More significant are peripheral ulcerative keratitis and keratolysis (“corneal melt”) both of which have a high risk of perforation.2 Necrotizing keratitis or scleritis in the context of RA are associated with increased mortality.23
Posterior segment lesions in RA include posterior scleritis and rarely a retinal vasculitis. Retinal vasculitis is probably underdiagnosed. In one study of 60 patients with RA the rate of retinal vasculitis was 18% even in the absence of clinical features of retinal vasculitis.24 This study is supported by a number of case reports describing typical retinal vasculitis associated with fluorescein angiographic evidence of leakage and a single case of retinal exudation in the context of scleritis that the authors attribute to vasculitis.25,26
Treatment
Treatment of systemic disease
The goals of treatment in RA are to alleviate symptoms and to prevent tissue destruction (usually of joints) and loss of function. Permanent damage to the joints may occur early in the disease and thus best practice is now to treat early and aggressively. The 2008 recommendations of the American College of Rheumatology (ACR) advise a hierarchical approach based on disease activity (low, moderate or high) and duration of disease (<6 months, 6–24 months, and 24 months).27 In addition to appropriate anti-inflammatory pharmacological interventions (such as NSAIDs, intra-articular and oral corticosteroids), disease-modifying antirheumatic drugs (DMARDs) such as methotrexate and lefluonimide are recommended first-line and may be used in combination according to activity and duration of disease. The ACR recommendations for use of biologics (such as anti-TNF therapies) include high disease activity, poor prognostic features (such as the presence of rheumatoid nodules, secondary Sjögren syndrome, rheumatoid vasculitis, longer duration of disease, and failure of DMARDs).27 Anti-TNF therapy may also be used with methotrexate. Other biologic therapies include rituximab (anti-CD20) and abatacept (fusion protein of CTLA-4 and IgG).28
Treatment of ocular disease
Mild superficial ocular disease (such as mild keratoconjunctivitis sicca or episcleritis) may be adequately controlled with topical therapies such as artificial tear substitutes.29 Non-necrotizing anterior scleritis may be managed with oral NSAIDs, but necrotizing disease frequently requires systemic corticosteroid. Uncontrolled ocular inflammation warrants escalation of systemic treatment that should be coordinated with a rheumatologist. In addition, sight-threatening inflammation such as necrotizing scleritis or corneal melt requires urgent rescue therapy such as pulsed intravenous methylprednisolone.2 In our practice we administer up to three pulses of 500–1000 mg methylprednisolone on consecutive days that is usually followed by commencing or increasing a course of oral corticosteroid (in addition to DMARD/biologic therapy).
Seronegative spondyloarthropathies
General considerations
Spondyloarthropathy is a term used to describe a group of interrelated inflammatory arthropathies affecting the synovium and extra-articular sites (Box 80.2).30 The spondylarthropathies include the following conditions: ankylosing spondylitis, reactive arthritis, inflammatory bowel disease-related arthritis, juvenile spondyloarthropathies, and psoriatic arthritis. Clinical manifestations include inflammatory back pain, enthesitis (inflammation of the entheses, where tendons or ligaments insert into the bone), dactylitis (inflammation of an entire digit), uveitis, and usually an asymmetrical arthritis that affects lower limbs. There is a strong association with the class I MHC molecule HLA-B27. Based on a systematic review, which included nearly 30 000 patients, the mean prevalence of uveitis in spondylarthropathies has been estimated at 33% overall, with acute anterior uveitis being the most common type seen.31
Box 80.2
The European Spondyloarthropathy Study Group (ESSG) spondyloarthropathy classification criteria
(Reproduced with permission from Dougados M, van der Linden S, Juhlin R, et al. The European Spondylarthropathy Study Group preliminary criteria for the classification of spondylarthropathy. Arthritis Rheum 1991;34:1218–27.)
Epidemiology
Spondyloarthropathies occur particularly in individuals who are positive for HLA-B27 but additional environmental factors are also thought to play a role. It can be difficult to differentiate these disorders, because their clinical features may overlap and undifferentiated forms of spondyloarthropathy are well recognized.32 Spondyloarthropathies as a whole have a prevalence of 0.5–1.9% of the population.
Ankylosing spondylitis
Epidemiology
AS is the commonest of the spondyloarthropathies. Its prevalence varies between 0.1 and 0.4% of the population, depending on the frequency of HLA-B27 in that population. This results in significant geographic variation with extremely low rates of AS in South Africa, low rates in Japan, higher rates in Germany compared to other European countries and very high rates in the natives of Eurasia and the North American circumpolar/sub-Arctic areas.33 AS is commoner in males, with a male: female ratio of 2–3 : 1, although it has been suggested that it may be underdiagnosed in females due to them having milder disease.34
Articular and systemic disease
The hallmark of AS is inflammatory back pain presenting with buttock pain, early morning stiffness (minimum 30 minutes), relieved by exercise and NSAIDs, worse with rest, and night pain.35 The shoulders and hips are regarded as axial joints and are affected in up to 50% of patients.36 In AS an asymmetrical oligoarthritis is uncommon, but may be a predictor of more severe disease if it presents early in the disease course.36
Enthesitis is a characteristic feature of AS and may occur at any enthesis, but is most commonly seen in the foot at the insertion of the Achilles tendon and of the plantar fascia onto the calcaneus. The classic cardiac abnormalities in AS are aortitis, aortic regurgitation, and conduction abnormalities, which are seen in up to 9% of long-term follow-up patients.37
Ocular disease
The commonest ocular complication of AS is recurrent AAU. It is almost always unilateral but may affect both eyes sequentially (described as “flip-flop” pattern). Rarely the anterior uveitis may become persistent. The presentation is of typical AAU but inflammation is classically more severe (often with fibrin in the anterior chamber) and recurrences more frequent than in idiopathic AAU.38–40 Hyopyon may also be present in severe cases. AAU may lead to sight loss via recurrent or persistent CME, secondary glaucoma, and cataract.38–40 Treatment-related ocular complications of AS include cataract and elevated intraocular pressure (secondary to corticosteroid usage).
Treatment
Treatment of systemic disease
The goal of treatment in AS is to restore and maintain posture and movement to as near normal as possible, which is achieved through lifelong physical therapy, and medical and surgical treatment. NSAIDs (non-steroidal anti-inflammatory drugs) are the basis of treating AS. They reduce pain and stiffness within 48–72 hours in several studies; a good response may also be used to support the diagnosis.41,42 Unlike many inflammatory rheumatic diseases, systemic corticosteroids do not have a major part in the treatment of AS although peripheral arthritis often responds to steroid treatment.41
DMARDs (disease-modifying antirheumatic drugs), such as sulfasalazine and methotrexate, are effective for the peripheral manifestations of AS, but there is limited efficacy for treating axial manifestations.43,44 Anti-TNF agents, such as etanercept, infliximab, and adalimumab are recommended as treatment options for patients with AS if disease is not sufficiently controlled after treatment with two or more DMARDs.
Treatment of ocular disease
The mainstay of treatment for AS-associated AAU is intensive topical corticosteroids and a mydriatic (as for idiopathic AAU), but it should be noted that subconjunctival treatment and oral corticosteroids are more commonly required to adequately control inflammation in HLA-B27-associated versus idiopathic AAU. Anti-TNF agents, such as infliximab, etanercept, and adalimumab, as part of studies for the treatment of the underlying AS can also reduce the frequency of AAU recurrences.45,46
Reactive arthritis (previously known as Reiter syndrome)
Epidemiology
There are a few population-based studies which have estimated the incidence to be between 10 and 30 per 100 000 per annum.47 HLA-B27 is positive in 60–80% of patients with reactive arthritis. The most commonly associated infections are urogenital (Chlamydia trachomatis) or gastrointestinal (Yersinia, Salmonella, Shigella and Campylobacter).48–50
Articular and systemic disease
Characteristically patients will have an asymmetric oligoarthritis of the large lower limb joints, but the upper limbs and small joints (usually PIP joints rather than MCP joints) are affected. Other features include inflammatory lower back pain similar to AS, dactylitis, enthesitis, erythema nodosum, and keratoderma blenorrhagica (pustular skin lesions on the soles of feet), indistinguishable from pustular psoriasis.48
Ocular disease
The commonest ocular complications of ReA are anterior segment disease. Conjunctivitis forms part of the classical triad of ReA, but is usually only seen at first presentation and early in the disease . Of more consequence is recurrent AAU that may occur in up to 50% of patients, although in only up to 20% patients at the initial attack.40,48 As described for other forms of AAU, these attacks may be associated with spillover vitreous cells and CME, and very rarely optic disc edema.
Rarely ReA may be associated with panuveitis or multifocal choroiditis;51,52 the rarity of such cases is underlined by many series of ReA patients in which no cases of panuveitis or posterior uveitis were identified.48,53
Treatment
Treatment of systemic disease
The underlying infection should be treated as appropriate.54 Acute arthritis can be treated with NSAIDs and intra-articular corticosteroids. DMARDs such as sulphasalazine or methotrexate should be considered in patients with a prolonged disease course.54
Inflammatory bowel disease
Epidemiology
The incidence of peripheral arthritis is reported to be between 5% and 10% in ulcerative colitis and 10% and 20% in Crohn’s disease, respectively.55 Men and women are affected equally. Spondylitis occurs in 1–26% of patients with IBD and males are more often affected than females. In addition, the prevalence of AS in IBD (1–6%) is higher than in the general population.55
Articular and systemic disease
IBD-related arthritis is often a clinical diagnosis as radiology is often normal with no joint erosion or deformity. Two distinct types of arthritis have been described: type 1 (pauciarticular) and type 2 (polyarticular).55,56
Ocular disease
Ophthalmic complications are estimated to occur in 3.5–12% of patients with IBD, occurring more commonly earlier in the disease.57 The commonest ocular complications of IBD are uveitis, episcleritis, and scleritis.
Uveitis is usually of recurrent AAU type, occurring in around 5% of IBD patients, but in up to 50% of IBD patients who are also positive for HLA-B27.58 Less commonly, a chronic bilateral anterior uveitis may be seen, which has a female gender preponderance.59 Scleritis is a well-recognized feature of IBD and has been reported to parallel the activity of the bowel disease. Scleritis is usually anterior but may be posterior; it may be non-necrotizing or necrotizing, leading to a risk of scleromalacia perforans.60 Retinal artery occlusions and ischemic optic neuropathy are reported and may reflect the prothrombotic tendency seen in some patients with IBD. Other reported associations with IBD include keratitis, retinal vasculitis, posterior uveitis, cystoid macular edema, optic neuritis, neuroretinitis, Brown’s syndrome and orbital myositis.60 A recent community survey also suggested that there is a high prevalence of “dry eye” (up to 42%), which was associated with 5-aminosalicylate use, although it was not clear if this was causative or a surrogate marker of disease activity.61 Interestingly, a family history of IBD has been proposed as an independent risk factor for the development of idiopathic ocular inflammation, including uveitis.62
Treatment
Treatment of systemic disease
Treatment depends on the severity of symptoms. Patients with mild oligoarthritis usually respond to relative rest, physiotherapy, and intra-articular corticosteroid injections.55,56 Most of the patients respond to NSAIDs because they control the symptoms and joint and enthesis inflammation, but they do not stop joint destruction, and they may have significant side-effects including exacerbation of IBD and produce small intestine and colon ulcers. Hence, they are recommended for patients with mild exacerbations, to control symptoms in arthritis flares, but their use must be limited to the minimal effective dose and time.55,56
Type 1 arthritis is related to disease activity and therefore therapy of the underlying IBD is the treatment of choice. Treatment of type 2 IBD arthritis and axial arthropathies generally requires long-term treatment with a DMARD, such as sulfasalazine or methotrexate. In addition treatment with systemic on intra-articular corticosteroids can be used.55 A number of IBD patients will also be using anti-TNF agents to control their bowel symptoms.
Treatment of ocular disease
IBD-associated AAU is treated as for idiopathic AAU with intensive topical corticosteroids and a mydriatic; it should be noted that the more chronic form of anterior uveitis may require more prolonged treatment.63 The treatment of scleritis will depend on the pattern and severity of disease seen, but will often require immunosuppression.63
Psoriatic arthritis
General considerations
Psoriatic arthritis (PsA) is the combination of an inflammatory arthritis (peripheral arthritis and/or sacroiliitis or spondylitis), psoriasis, and the absence of serological tests for rheumatoid factor.30 In 2006, however, the CASPAR (ClASsification Criteria for Psoriatic ARthritis) was developed (Box 80.3). This has been demonstrated to have high sensitivity and specificity for the diagnosis of PsA (see below).30,64
Box 80.3
The CASPAR criteria for psoriatic arthritis
(Adapted with permission from Taylor W, Gladman D, Helliwell P, et al. Classification criteria for psoriatic arthritis: development of new criteria from a large international study. Arthritis Rheum 2006;54:2665–73.)
Articular and systemic disease
PsA can cause a variety of articular symptoms, varying from an isolated monoarthritis to an extensive destructive arthritis. Articular involvement can be divided into five subtypes: DIP (distal interphalangeal) joint involvement, mono/oligoarticular, symmetrical polyarthritis, arthritis mutilans, and spondylarthropathy. It is important that patients with longstanding PsA have cervical spine X-rays before a general anesthetic, as there can be a clinically silent erosive/inflammatory arthritis causing atlantoaxial or subaxial instability, as in RA.67 Dactylitis or “sausage digit” occurs in 30–40% of patients with PsA. In addition 20–40% of patients have symptomatic enthesitis generally affecting the foot at the insertion of the Achilles tendon and of the plantar fascia onto the calcaneus.64,68 There appears no correlation between the severity of skin psoriasis and joint involvement, but skin symptoms do tend to precede joint symptoms. Psoriasis can be assessed using a variety of tools including: PASI (Psoriasis Area and Severity Index), health assessment questionnaires, and Psoriatic Arthritis Response Criteria (PsARC).
Ocular disease
Ophthalmic complications are estimated to occur in 10% of patients with psoriasis,69 and 31% of patients with PsA.70 The most common presentations are conjunctivitis (up to 20%) or uveitis (7%).70 Paiva and coworkers compared the nature of uveitis in PsA to that seen with a previous cohort of spondylarthropathy patients. Interestingly this suggested that PsA-associated uveitis was more likely to be insidious in onset (19% versus 3%), bilateral (38% versus 7%), chronic in duration (31% versus 6%) or posterior (44% versus 17%).71 Episcleritis, scleritis, keratoconjunctivitis sicca, and keratitis are also reported.69,70
Treatment
Treatment of systemic disease
NSAIDs are normally used for musculoskeletal symptoms, based on the evidence from other rheumatic diseases.72 Intra-articular corticosteroid can be used, but oral corticosteroids should be used cautiously as they can be associated with a “post-steroid psoriasis flare.” Methotrexate can be used for both skin psoriasis and PsA, but care must be taken as the risk of hepatotoxicity seems to be higher in patients with psoriasis, possibly due to a higher tendency of non-alcoholic steatohepatitis (NASH). Sulfasalazine can be used for articular symptoms; there is no benefit for the skin psoriasis. Leflunomide has demonstrated to be effective at treating PsA.72 Cyclosporine can achieve rapid improvement of the skin lesions of psoriasis, but there is little evidence regarding its effectiveness in PsA. There are concerns regarding possibly causing hypertension and renal insufficiency, so the dose needs to be kept as low as possible and careful monitoring is indicated as these reversible events are reversible if picked up soon after onset.72
Anti-TNF agents (etanercept/infliximab/adalimumab) are generally reserved for severe disease. They have been shown to be effective at treating peripheral arthritis, psoriasis, enthesitis, and dactylitis.73 Further work is currently being undertaken to look at other possible biologic targets including: alefacept, which is a fully human fusion protein that blocks interaction between LFA-3 on the antigen-presenting cell; rituximab, an anti-CD20 agent; and ustekinumab, an IL-12/IL-23 inhibitor.73
Treatment of ocular disease
PsA-associated AAU is treated as for idiopathic AAU with intensive topical corticosteroids and a mydriatic, but more persistent anterior uveitis may require prolonged treatment. Cataract, which may be associated with chronic intraocular inflammation, corticosteroid usage, and possibly P-UVA treatment,74 requires surgical treatment with appropriate immunosuppressive perioperative care.
Juvenile idiopathic arthritis
Epidemiology
The reported incidence of JIA varies between 0.8 to 23 per 100 000 per annum, with a prevalence rate between 7 and 400 per 100 000 children. There are reported differences between different ethnic groups: JIA is more frequent in children of European descent than in children of African, Asian or East Indian origin.75
Articular and systemic disease
JIA is classified using the ILAR (International League of Associations of Rheumatologists) classification (Box 80.4). The main clinical feature of JIA is defined as: “swelling within a joint or limitation in range of movement with joint pain or tenderness, which persists for a minimum of 6 weeks, observed by a physician and which is not due to primary mechanical disorders or to other identifiable causes.”76
Box 80.4
The International League of Associations of Rheumatologists (ILAR) classification of juvenile idiopathic arthritis
Psoriatic arthritis
Definition: Arthritis and psoriasis, or arthritis and at least two of the following:
Enthesitis-related arthritis
1. The presence of or a history of sacroiliac joint tenderness and/or inflammatory lumbosacral pain
2. The presence of HLA-B27 antigen
3. Onset of arthritis in a male over 6 years of age
4. Acute (symptomatic) anterior uveitis
5. History of ankylosing spondylitis, enthesitis-related arthritis, sacroiliitis with inflammatory bowel disease, Reiter syndrome, or acute anterior uveitis in a first-degree relative
Exclusions
a. Psoriasis or a history of psoriasis in the patient or first-degree relative
b. Arthritis in an HLA-B27-positive male beginning after the 6th birthday
c. Ankylosing spondylitis, enthesitis-related arthritis, sacroiliitis with inflammatory bowel disease, Reiter syndrome, or acute anterior uveitis, or a history of one of these disorders in a first-degree relative
d. The presence of IgM rheumatoid factor on at least two occasions at least 3 months apart
(Reproduced with permission from Petty RE, Southwood TR, Manners P, et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol 2004;31:390–2.)
Ocular disease
The major ocular manifestation of JIA is uveitis. Uveitis is present in around 10% of JIA patients at presentation but may occur in up to one-third of patients at some point during their disease although the exact estimate depends on the type of population sampled.77–81 Inflammation is typically a chronic anterior uveitis with a white eye, which is usually bilateral (70%) but may initially present with unilateral disease. Recurrent acute anterior uveitis is less commonly seen and when it does occur is usually in the context of HLA-B27.
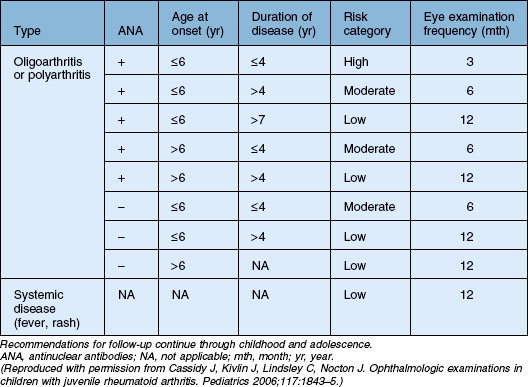
The risk of developing uveitis in association with JIA has been stratified according to age of onset, type of arthritis, and the presence of ANA, with uveitis occurring in up to half of the highest-risk group (oligoarticular – persistent and extended, ANA-positive disease). Interestingly, a recent study has suggested that age and ANA status may be less useful in predicting risk in boys (versus girls).82 It should be noted that the gold standard for determining ANA is via immunofluorescence on HEp-2 cells; ELISA-determined ANA was not found to be predictive.83 Antihistone antibodies are also associated with risk of uveitis but are less widely used in clinical practice.83 Since the chronic anterior uveitis has minimal if any of the symptoms commonly associated with inflammation, screening is recommended. The recommendations of the American Academy of Pediatrics are summarized in Table 80.1.84
Table 80.1 American Academy of Pediatrics guidelines on frequency of ophthalmologic examination in patients with juvenile idiopathic arthritis
JIA-associated uveitis is more commonly seen in girls,81 but male gender is a risk factor for worse disease, with significantly higher rates of CME by 5 years of follow-up (50% versus 4%) and need for cataract surgery (59% versus 32%). Other reported predictors of worse outcome are uveitis present at time of diagnosis85,86 and elevated laser flare values.87
The key sight-threatening complications of JIA-associated uveitis are band keratopathy (60%), cataract (40%), glaucoma (10–25%), and CME (10%). Posterior synechiae are present in most cases.77,88,89 Less commonly, vitritis and a peripheral retinal vasculitis are reported.77,80,88,89
Treatment
Treatment of systemic disease
The management of JIA is based on a combination of medical treatment, physical and occupational therapy, and surgical management. NSAIDs can be used for all types of mild JIA, to treat pain and stiffness. Oral corticosteroids must be used minimally in children because of the effects on bone and growth, the main indications are severe fever, serositis and macrophage activation syndrome. Intra-articular corticosteroids can be used, and encouraging results have been reported in children with monoarthritis.90 Methotrexate is used for patients with polyarthritis, other DMARDs that have been shown to be effective include sulfasalazine and leflunomide. Etanercept can be used in patients who fail to respond to methotrexate therapy and has been demonstrated to be effective both in the short and long term management of JIA.91–93 Infliximab, was not shown to be superior to placebo in polyarticular JIA.94 Adalimumab is licensed for use in JIA, and was found to be effective in children with polyarticular JIA.95 Abatacept is an alternative biological agent which is a selective T-cell co-stimulator inhibitor. In randomized controlled trials (RCTs) abatacept was superior to placebo in children with polyarticular arthritis, including those who were anti-TNF treatment failures.96,97 There is some concern regarding the potential increased risk of cancers in children using anti-TNF agents.98 Further studies are being undertaken to investigate the potential future role of other biologics, including rituximab, IL(interleukin)-1 and IL-6 antagonists.
Treatment of ocular disease
Systemic immunosuppression is usually required to control both the systemic disease and its ocular manifestations, although some children may only require topical corticosteroid and a mydriatic. What constitutes a “safe” level of topical corticosteroid usage in children is controversial. In a recent retrospective study by Thorne and coworkers topical corticosteroid use was associated with development of cataract and this was independent of active uveitis or presence of posterior synechiae.99 Importantly, there appeared to be no significant increase of cataract when chronic administration was no more than twice daily.99 The need for frequent topical corticosteroid to control the uveitis usually requires the introduction of methotrexate often given by the subcutaneous route weekly rather than orally. If methotrexate cannot adequately control the inflammation then it requires the addition of an anti-TNF agent; adalimumab appears to be the drug of choice, with effective control of the uveitis in 16/18 children in one study.100 There are some reports of uveitis occurring in association with etanercept.101
Cataract surgery is challenging and requires very careful preparation, perioperative care and intensive postoperative management. It is vital that the patient’s carers are fully aware of the importance of adherence to prescribed therapy and follow-up visits. Traditionally cataract removal for these patients was by pars plana vitrectomy/lensectomy followed by aphakia, but it is now more common to use intraocular lenses at the time of surgery.102–104 Postoperative posterior synechiae and intraocular lens deposits are common, but overall visual improvement is encouraging, with a recent series of 17 eyes reporting an improvement in visual acuity of 2 lines or more in all patients with no increase of CME, glaucoma or hypotony,103 but it should be noted that other studies do report significant rates of secondary glaucoma and CME.104
Systemic lupus erythematosus
General considerations
Systemic lupus erythematosus (SLE) is a multisystem autoimmune disease predominantly affecting women of childbearing age. The classification criteria for SLE are summarized in Box 80.5.105
Box 80.5
The 1997 updated American College of Rheumatology criteria for systemic lupus erythematosus
Fixed erythema, flat or raised, over the malar eminences, tending to spare the nasolabial folds
Skin rash as a result of unusual reaction to sunlight, by patient history or physician observation
Oral or nasopharyngeal ulceration, usually painless, observed by physician
Involving 2 or more peripheral joints, characterized by tenderness, swelling, or effusion
At least one of the following:
At least one of the following:
(Modified from Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1997;40:1725.)
Epidemiology
SLE has a prevalence of around 28 cases per 100 000,106 predominantly affecting women of child-bearing age (15–45 years); the female: male ratio peak is 12 : 1. SLE is more common in non-Caucasians, in whom it is both more severe and earlier in onset.107
Articular and systemic disease
SLE is a systemic disease that can cause constitutional or organ-specific symptoms. The skin, mucous membranes, joints, kidney, brain, serous membranes, lung, heart, and occasionally the gastrointestinal tract may all be involved.105,106 Arthritis in SLE can divided into a deforming and nondeforming arthropathy. Constitutional symptoms consist of fever, malaise, fatigue, weight loss, lymphadenopathy, and anorexia.106 There are multiple cutaneous manifestations of lupus: commonly these include photosensitivity (>50% of patients), butterfly/malar rash, painful/painless oral ulcers, diffuse alopecia, and livedo reticularis.106
Renal disease is one of the most serious SLE manifestations and a prognostic indicator of disease severity. Renal biopsy is used to determine the class of nephropathy as classified by the International Society of Nephrology (ISN)/Renal Pathology Society (RPS) guidelines.108
Neuropsychiatric SLE (NPSLE) is a major diagnostic and treatment problem. The ACR has provided classification criteria, describing central and peripheral types of neurological involvement that may be found in lupus patients.106
Pulmonary features of SLE include pleurisy, pneumonitis, pulmonary hemorrhage, pulmonary embolism, pulmonary hypertension, and diaphragmatic weakness causing shrinking lungs. Pericarditis is the most common cardiological manifestation; others include myocarditis, endocarditis, accelerated atherosclerosis, and, rarely, pericardial tamponade.106
Abdominal pain, nausea, vomiting, and diarrhea occur in up to 50% of SLE patients. Gastrointestinal involvement includes mesenteric vasculitis (high risk of death), aseptic peritonitis (with or without ascites), subacute bowel obstruction, hepatitis, sclerosing cholangitis, protein-losing enteropathy, pancreatitis, and ascites.109
Cytopenias, including anemia, leucopenia or thrombocytopenia, are commonly associated with SLE; they may be immune-mediated or due to other factors, e.g. menstrual losses. Antiphospholipid antibodies and lupus anticoagulant are found in about 30–40% of patients, associated with venous and arterial thrombosis, recurrent fetal loss, pre-eclampsia, headache, and epilepsy.106
A firm diagnosis of lupus is made based on appropriate clinical findings and the measurement of at least one antibody. There are a number of antibodies associated with SLE, the most common being ANA (antinuclear antibody). Other associated autoantibodies include anti-dsDNA (double stranded DNA) in approximately 60% of patients, the highly specific anti-Sm antibody (Smith proteins) in 10–30% of patients, and anti-RNP (ribonucleoprotein) also in 10–30% of patients.105,106
Ocular disease
Ophthalmic complications are common in SLE, affecting up to one-third of patients. The commonest complication is keratoconjunctivitis sicca but sight-threatening posterior segment and neuro-ophthalmic disease is also seen (reviewed by Sivaraj and coworkers).110
Common ocular surface diseases related to SLE include KCS (25% of SLE patients).111,112 Peripheral ulcerative keratitis is a rare but serious complication requiring urgent immunosuppression.2
Episcleritis is observed in 1–2% and scleritis in 1% of patients with SLE.6,113 Scleritis may be anterior or posterior, necrotizing or non-necrotizing, and may indicate activity of the underlying disease.
Occasionally SLE may cause orbital inflammation and present with acute proptosis, lid edema, conjunctival injection and chemosis, reduced ocular motility and elevated intraocular pressure, with myositis and panniculitis also reported.114,115 Orbital inflammation may also be associated with posterior scleritis.
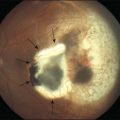
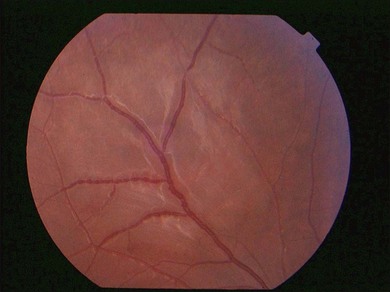
Lupus retinopathy was first described by Bergmeister in 1929. Its prevalence is estimated at around 10%, although this is variable depending on the population studied.116 The classic clinical picture is of cotton-wool spots, retinal hemorrhages, and vascular abnormalities (arterial narrowing with capillary dilation, and venous dilation and tortuosity) (Figs 80.1–80.3). Additional features may include retinal edema, hard exudates, and microaneurysms. Retinopathy is usually bilateral but may be asymmetric.117
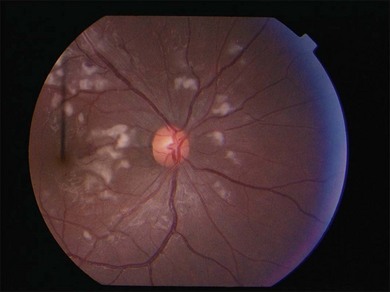
Fig. 80.1 Acute lupus retinopathy with cotton-wool spots, arterial narrowing, venous dilation, and tortuosity.
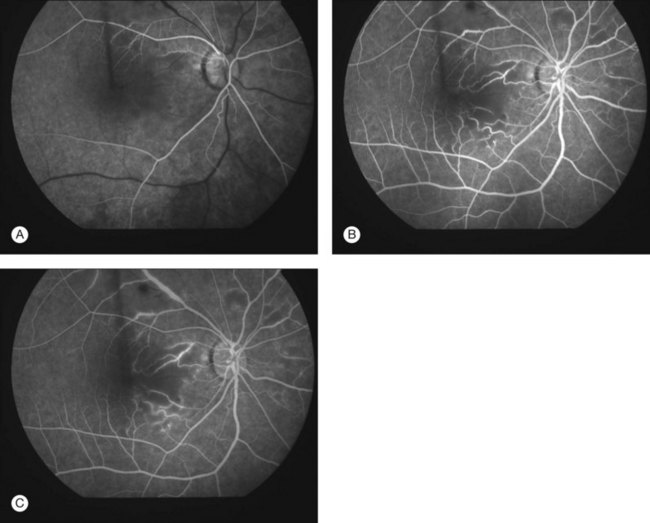
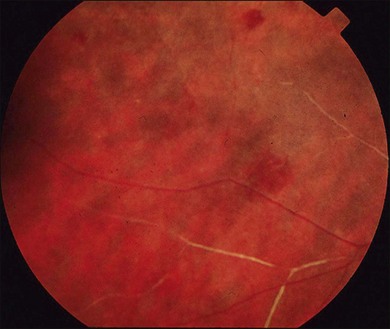
Severe vaso-occlusive retinopathy is much less common but potentially devastating. Whereas mild retinopathy may be picked up as an incidental finding, vaso-occlusive retinopathy usually presents with visual symptoms such as reduced acuity, visual field loss, and distortion. In addition it is strongly associated with the life-threatening complication of CNS lupus.118 The clinical appearance is of widespread arteriolar occlusion and capillary nonperfusion. Subsequently neovascularization is common (up to 72% cases) (Figs 80.4, 80.5),118 and may be complicated by vitreous hemorrhage (up to 63%), retinal traction, and detachment (up to 27%).118 The occurrence of vaso-occlusive disease is strongly associated with the presence of antiphospholipid antibodies (up to fourfold increased risk).119 Primary antiphospholipid syndrome (i.e. antiphospholipid antibodies in the absence of SLE or any other systemic disease) may be associated with a similar severe vaso-occlusive retinopathy but with cotton-wool spots being less common. The severe vaso-occlusive retinopathy is sometimes described as a retinal vasculitis, but this is not generally supported by histological examination.120
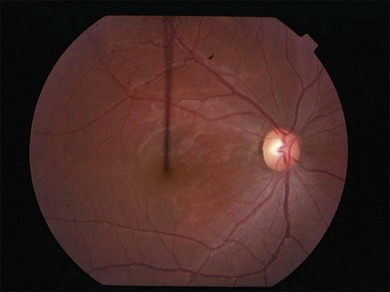
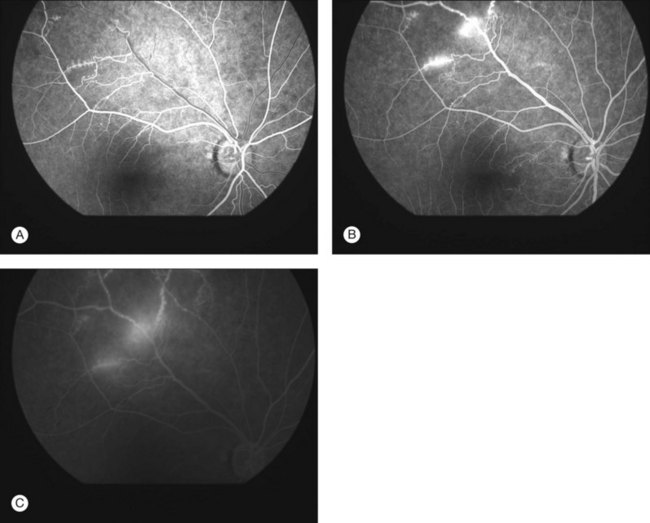
Occlusion of the larger retinal vessels may occur and again is associated with the presence of antiphospholipid antibodies. Manifestations include branch retinal arteriolar or central retinal artery occlusion (B/CRAO) (Fig. 80.6), branch or central retinal vein occlusion (B/CRVO), and combined retinal arterial and venous occlusions.118,120
Other retinal manifestations of SLE include an unusual bilateral pigmentary retinopathy that may resemble retinitis pigmentosa but is proposed to be ischemic in origin.121 Coexisting systemic hypertension may result in features of hypertensive retinopathy. Choroidal involvement in SLE (lupus choroidopathy) may also cause significant visual morbidity. Choroidopathy results in single or multifocal serous detachments of the retina and retinal pigment epithelium (RPE), which may mimic idiopathic central serous chorioretinopathy (CSC).122 The degree of visual symptoms depends on the anatomical location of the detachment(s). These serous detachments may become extensive over time but conversely may reverse with control of the underlying systemic disease.122 Fluorescein angiography (and indocyanine green angiography) is helpful as it not only demonstrates typical CSC-like leakage from the choroid into the subretinal and sub-RPE spaces but also demonstrates the degree of choroidal ischemia.
Optic nerve disease is uncommon (about 1% patients with SLE). The spectrum of disease described includes anterior and posterior ischemic optic neuropathy, and acute optic neuritis (which is also thought to be ischemic in origin).123 Bilateral optic disc swelling in SLE may arise due to either idiopathic intracranial hypertension or accelerated systemic hypertension, both of which are more common in SLE.
Other neuro-ophthalmic complications of SLE include ocular motility abnormalities (causes include brain stem infarcts, cranial neuropathies, tenosynovitis, myositis and Miller–Fisher syndrome), nystagmus, ptosis, and migraine.110
Finally, it should be noted that patients with SLE are immunosuppressed (both due to disease and treatment) and may present with severe intraocular infections. Retinal necrosis due to herpes simplex virus, varicella zoster virus, and cytomegalovirus have been reported. Other ocular infections include tuberculous choroidal abscess, and nocardia endophthalmitis.124,125
Treatment
Treatment of systemic disease
Rituximab is a monoclonal antibody against the B-lymphocyte marker CD20 expressed on B cells, it has been used in SLE patients since 2002, and observational studies have suggested that rituximab is effective in treating active SLE refractory to standard immunosuppressant.126 Recent data have demonstrated that repeated treatment with rituximab is effective in treating refractory SLE and has a favorable safety profile.127 Lightstone and coworkers are currently investigating the possibility of corticosteroid avoidance regimes.128
Treatment of ocular disease
In SLE, control of the systemic disease often improves the ophthalmic disease. The presence of severe ophthalmic disease should prompt the rheumatologist to look for evidence of systemic activity, and warrants escalation of systemic therapy. Additional local and regional treatments may also be indicated depending on the type of ocular complication. For example, KCS may benefit from a range of treatments, including tear replacement therapy (preservative-free preparations preferred), punctal occlusion, lid hygiene, topical corticosteroids or cyclosporine, and environmental measures.110
Mild anterior segment inflammation may respond to topical corticosteroids (keratitis or anterior uveitis), or topical NSAIDs (episcleritis). In more severe anterior segment inflammation, such as scleritis or disease affecting the posterior segment or orbit, systemic treatment is required. Non-necrotizing scleritis may respond to oral NSAIDs, but most severe inflammatory disease will require high-dose systemic corticosteroid often in combination with the immunosuppressive agents listed above. Significant retinal vascular occlusions associated with antiphospholipid antibodies may be treated with warfarin or low-dose acetylsalicylic acid (in addition to immunosuppression). Retinal neovascularization usually requires panretinal photocoagulation. Persistent vitreous hemorrhage or tractional retinal detachment may require vitreoretinal surgery.110,118
Sjögren syndrome
Epidemiology
Sjögren syndrome predominantly affects females in the fourth to fifth decade of life. The female: male ratio is 9 : 1. In a population-based study in Minnesota, incidence of Sjögren syndrome was estimated to be 3.9 per 100 000 per year.129
Articular and systemic disease
Sjögren syndrome is clinically characterized by sicca symptoms: dry eyes and mouth due to failure of the salivary and mucosal glands. Sjögren syndrome may be primary or secondary to a pre-existing disorder such as SLE, rheumatoid arthritis, systemic sclerosis, vasculitis, autoimmune thyroid disease or primary biliary cirrhosis.130,131
The primary syndrome is associated with hypergammaglobulinemia with very high total IgG levels and strongly positive antinuclear antibody, rheumatoid factor, and anti-Ro and anti-La antibody levels. The extraglandular manifestations include arthralgia, Raynaud phenomenon, peripheral neuropathy, myositis, liver and interstitial nephritis or renal tubular acidosis. Immune complex deposition resulting from ongoing B-cell hyperactivity is associated with increased morbidity and lymphoma risk.130
Ocular disease
The cardinal ocular sign of Sjögren syndrome is keratoconjunctivitis sicca (KCS), which may range from mild irritation in its early stage to severe tear deficiency with ocular surface inflammation and damage resulting in severe visual loss.1 Assessment of tear production, tear stability, and careful examination of the ocular surface are key.1 Posterior segment disease is rare. Rosenbaum and Bennett described a series of eight patients with Sjögren syndrome and uveitis, reporting that in all cases the disease was bilateral and chronic; in their report they describe anterior and posterior disease (but no chorioretinitis) with posterior synechiae, cataract, and pars plana exudation being common.132
Treatment
Treatment of systemic disease
Sjögren syndrome is a chronic disease with a wide clinical spectrum, making it necessary for regular follow-up. Treatment of sicca symptoms is essential, and includes general measures such as avoidance of dry atmospheres, humidification of rooms, and chewing sugarless chewing gum.131 Hydroxychloroquine can be effective for treating the subgroup of Sjögren sufferers who have inflammatory myalgias and arthralgias. Anti-TNF-α agents have not shown clinical efficacy, and larger controlled trials are needed to establish the efficacy of rituximab.131,133
Treatment of ocular disease
The treatment of KCS is predominantly with frequent use of preservative-free tear substitutes, with a range of viscosities to suit the patient, their visual needs, and even the time of day. RCTs have shown topical 0.05% cyclosporine to be beneficial for patients with moderate to severe dry eye disease.134,135 In the presence of associated inflammation of the ocular surface, topical glucocorticoids may be required.29
Familial juvenile systemic granulomatosis (Blau syndrome)
General considerations
Familial juvenile systemic granulomatosis (Blau syndrome or Jabs syndrome) is a rare autosomal dominant disorder, associated with mutations in the NOD2/CARD 15 gene.136–139 It was described by Blau in 1990, as a triad of polyarthritis, iritis, and granulomatous papulosquamous rash. There may also be an environmental role, with one series finding Mycobacterium avium ss. paratuberculosis DNA to be present in Blau syndrome tissue in all cases (n = 5).140
Epidemiology
Little is known about the epidemiology of familial juvenile systemic granulomatosis. It is thought to occur worldwide, with equal gender and race distribution.138 In a study looking at the experience of two centers (four families), all affected members carried a NOD2/CARD 15 mutation while it was absent in all the unaffected members.138 The disease is autosomal dominant in nature, with an observed element of “anticipation” (worsening of symptoms in succeeding generations).138
Articular and systemic disease
Familial juvenile systemic granulomatosis presents with a polyarticular arthritis, associated with synovial and tenosynovial cysts, resulting in swelling of the affected joints and tendons.138 Campylodactyly (multidigit contracture of the interphalangeal joints) can occur secondary to inflammation.138
Familial juvenile systemic granulomatosis can be distinguished from childhood sarcoidosis by the absence of pulmonary involvement.138,141 The rash is described as papulo-erythematous, and usually affects the trunk and extremities.138 In addition, familial juvenile systemic granulomatosis can be associated with a large vessel vasculitis, and cranial nerve palsies;138 Crohn’s disease is reported to occur in 30% of patients.142
Ocular disease
The cardinal ocular sign of familial juvenile systemic granulomatosis is a chronic anterior uveitis or panuveitis with multifocal choroiditis.143 Complications of uveitis are common including cataract, glaucoma, band keratopathy, and CME. Also reported are subepithelial corneal infiltrates, optic disc edema, ischemic optic neuropathy, and retinal vasculopathy.143,144
Treatment
Treatment of systemic disease
Treatment for familial juvenile systemic granulomatosis is largely empirical. Prednisone can be used (dose will be dependent on the severity of disease).138,139 Immunosuppressive agents, such as methotrexate and azathioprine, have been used with little effect.138,139 There are also isolated reports of benefit from infliximab and anakinra (IL-1 receptor antagonist) in refractory cases.138,139
Scleroderma
Epidemiology
The precise estimate for the incidence and prevalence of scleroderma are unknown. This is likely due to a combination of true variation over different populations and differences in case ascertainment and disease classification.145 Reported prevalence estimates in North America have varied from 13.8 cases per 100 000 from 1950 to 1979 to 28.6 cases per 100 000 in 1985.146 A Canadian study estimated the prevalence in Quebec in 2003 to be 44.3 cases per 100 000.145,147 Scleroderma is more common in women, with a female : male ratio of 4–6 : 1. Multiple studies have demonstrated increased incidence and severity of scleroderma in people of African descent.
Articular and systemic disease
Cutaneous manifestations may initially present with inflammation, edema, and reduced sweat and oil production. The characteristic cutaneous features include scleroderma (symmetrical skin thickening proximal to MCP joints), Raynaud phenomenon, Barnett’s sign (vertical striation on neck extension), telangiectasia, calcinosis, “mauskopf” facies (caused by tightening of skin and a reduced oral aperture. Gastrointestinal effects include esophageal dysmotility, gastro-esophageal reflux disease (GORD), and intestinal dysmotility.148
Ocular disease
Ocular involvement is common, particularly of the eyelids and anterior segment. Lid involvement occurs in up to two-thirds of patients, resulting in progressive skin tightness, blepharophimosis, and occasionally lagophthalmos. Small lid telangiectasia occurs in up to 21% of patients.149,150 Ocular surface disease is also very common, with KCS affecting up to 79% patients. KCS may occur as part of secondary Sjögren syndrome. It should be noted when interpreting IOP measurements in scleroderma that central corneal thickness increases during the first eight years of disease and may affect IOP readings.151,152
Although retinopathy may be seen in patients with scleroderma it is usually in the context of secondary hypertension, and is of a clinical appearance typical of hypertensive retinopathy (cotton-wool spots, exudation, neuroretinal edema, hemorrhages). Milder retinal changes may also occur in normotensive patients with scleroderma, as shown by Ushiyama et al. where 34% of normotensive scleroderma patients (versus 8% controls) had retinal findings such as hard exudates and vascular tortuosity.116 Other reported retinal features include combined CRVO and CRAO, bilateral CRVO, BRVO and parafoveal telangiectasia. Interestingly, fundus fluorescein angiography (FFA) studies suggest that abnormality of the choroidal vasculature occurs in around one-third of patients, with hyperfluorescence in the late phase corresponding with areas of hypopigmentation.153 Other reported ocular complications include cranial nerve palsies, Brown syndrome, and ophthalmoplegia.154
Treatment of systemic disease
Scleroderma is a heterogeneous condition, with multiple degrees of severity, so optimal management involves an individual patient approach: assessing current organ involvement, assessing potential problems, and aggressive treatment of more serious organ involvement.155 Optimal management of blood pressure is a key aspect for managing these patients. Corticosteroids are commonly used at low doses for inflammatory arthritis, but there is no convincing RCT evidence. Cyclophosphamide has been demonstrated in two recent RCTs to be beneficial in scleroderma-associated lung fibrosis and skin disease. Methotrexate has been demonstrated to be effective for skin manifestations in early disease. Mycophenolate mofetil has been demonstrated to be effective for skin, lung, and survival in open-label trials, but work is being undertaken to assess this further.156 At present there is some evidence that anti-TNF agents may improve inflammatory arthritis, disability, and possibly skin manifestations.157 In patients with severe diffuse cutaneous scleroderma autologous hematopoietic stem cell transplantation results in sustained improvement of skin thickening and stabilization of organ function. There are two large RCTs currently under way in Europe and the United States to investigate this further.158
Polymyositis and dermatomyositis
Epidemiology
Several classification criteria have been proposed, the most frequent are the Bohan and Peter criteria (Box 80.6).159 The precise incidence of myositis is unknown, but is estimated to be between 2 and 10 new cases per million persons at risk per year.160,161 The reported female: male incidence ratio is 2.5 : 1.161 Similar to SLE and systemic sclerosis, there is a higher incidence in people of African descent, with a younger age of onset.
Box 80.6
The Bohan and Peter criteria for the diagnosis of polymyositis and dermatomyositis
(Modified from Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med 1975;292:344–7.)
Myositis can be associated with other autoimmune conditions, particularly scleroderma and mixed connective tissue disease, and occasionally in SLE, RA, and Sjögren syndrome.162 There is an increased risk of malignancy in both polymyositis and dermatomyositis. The greatest risk is in dermatomyositis (standardized incidence ratio (SIR): 3.0) which is associated with ovarian, lung, pancreatic, stomach, colorectal and non-Hodgkin lymphoma. Polymyositis is associated with a significant but lower increased risk (SIR: 1.2–1.5), of malignancy particularly non-Hodgkin lymphoma, and lung and bladder cancer.163
Articular and systemic disease
Myositis often presents as a subtle, progressive, painless symmetrical weakness over 3–6 months, affecting proximal more than distal muscles.161 Dermatomyositis is characteristically associated with a heliotrope rash (purplish rash over the periorbital area) and Gottron papules (scaly or erythematous papules and plaques over bony prominences, particularly elbows and knees). Subcutaneous calcinosis (nodules or plaques of calcification over the elbows, forearms, knuckles, axillae, or buttocks), occurs particularly in juvenile dermatomyositis, but occasionally also in adult cases.161
Arthralgias and synovitis of small or large joints may occur in patients with myositis, even without an associated connective tissue disease. A deforming arthropathy of the proximal and distal interphalangeal joints occurs typically in patients with inflammatory myopathy and antisynthetase antibodies.161,162 Interstitial lung disease is also more likely to develop in DM or PM patients with anti-Jo-1 or other antisynthetase antibodies. Subclinical cardiac involvement including myocarditis, pericarditis, arrhythmias, and congestive cardiac failure have been reported. Gastrointestinal tract musculature involvement may occur, causing dysphonia, dysphagia, pseudo-obstruction or malabsorption.161,162
Ocular disease
The classic heliotrope eyelid eruption of dermatomyositis is a familiar periocular sign of disease. Actual ocular involvement with myositis of the extraocular muscles is rare.164 A retinopathy with cotton-wool spots is described in both these conditions, most commonly seen in children and usually (but not exclusively) in the context of systemic vasculitis. Retinopathy is usually mild, but in its severe retinal vasculitis form may lead to permanent visual loss.165,166 Conjunctivitis, anterior uveitis, and episcleritis are also reported.166,167
Treatment
Treatment of systemic disease
There are no large RCTs exploring the treatment of myositis, so treatment is based on case series, open-label trials, and small RCTs. General measures for treating are rehabilitation, avoidance of aspiration, and sun protection. Primary initial therapy is oral corticosteroid.162,168 Initially patients should be treated with prednisone 1 mg/kg, daily for 4–6 weeks before tapering the dose, but in more severe disease IV methylprednisolone up to 1 g/day for three consecutive days is recommended. Immunosuppressant therapy at an early stage may be required to facilitate corticosteroid reduction and side-effects from corticosteroids, and the first choice agents are methotrexate and azathioprine. In severe cases, in particular those associated with vasculitis or interstitial lung disease, cyclophosphamide has been recommended.162,168 Intravenous immunoglobulin has been proposed in patients with rapidly progressing disease, and alternative agents include cyclosporine, mycophenolate mofetil, possibly rituximab (ongoing phase II clinical trials), and anti-TNF agents (although studies have found an increased risk of disease flare).169
Relapsing polychondritis
Epidemiology
Relapsing polychondritis has an estimated incidence rate of 3.5 per million per annum; the peak onset is between 40 and 60 years. Equal frequency has been reported in both genders and all racial groups. Over 30% of cases are associated with existing autoimmune and hematological conditions, including RA, SLE, Sjögren syndrome, AS, lymphoma, and IBD.170
Articular and systemic disease
Relapsing polychondritis is a multisystem disease that can affect the cartilaginous structures in the eyes, ears, nose, laryngotracheobronchial, and costal cartilages (Table 80.2). It causes inflammation of hyaline cartilage with a predilection for ear cartilage.171 Involvement of the parasternal joints, including the sternoclavicular, costochondral, and manubriosternal articulations, is typical for this condition. Peripheral joint disease is reported in 70% of patients, and is usually nonerosive and asymmetric.171
| Organ involvement | Clinical manifestation |
|---|---|
| Ear | External inflammation, loss of hearing, tinnitus, vertigo |
| Eye | Episcleritis, scleritis, ulcerative keratitis, uveitis, proptosis |
| Nose | Crusting, rhinorrhea, epistaxis, saddle nose |
| Large airways | Hoarseness, aphonia, wheezing, inspiratory stridor, nonproductive cough, dyspnea |
| Joints | Parasternal joints, peripheral joints (mono- or oligoarticular) |
| Heart | Aortic and mitral valvular disease |
| Skin | Aphthous ulcers, purpura, papules, nodules or ulcerations |
(Reproduced with permission from Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev 2010;9:540–6.)
Ocular disease
Ophthalmic disease occurs in around half of patients with relapsing polychondritis. In a series of 112 patients, Isaak and coworkers reported that 19% patients had ocular symptoms at the onset of disease, with 51% developing ocular symptoms during the course of disease. Episcleritis (39%) and scleritis (14%) are common.172 Anterior uveitis was reported as just 9% in the Isaak series but has been reported to be as prevalent as 30% in other series.173 Other anterior segment findings include KCS and peripheral ulcerative keratitis.174 The commonest posterior segment presentation is posterior scleritis, which may be severe and be associated with serous retinal detachments and frank proptosis.175–177 Retinopathy consisting of cotton-wool spots and intraretinal hemorrhages is reported to occur in up to 9% of patients. Other posterior segment features are branch or central retinal vein occlusions and ischemic optic neuropathy.172 Cranial neuropathies may also be seen.
Treatment
Treatment of systemic disease
First-line therapy for relapsing polychondritis is corticosteroid, initially started at 1 mg/kg prednisone daily; if necessary three pulses IV methylprednisolone can be administered. Immunosuppressives should be used for severe disease causing organ compromise or where corticosteroids have not provided a satisfactory response within a few weeks. The choice of immunosuppressant is empirical; the most commonly used agents are cyclophosphamide, azathioprine, cyclosporine, and methotrexate.170 There are case reports reporting success with anti-TNF agents (etanercept/infliximab/adalimumab), anakinra, and rituximab.178
Primary systemic vasculitis
The vasculitides are a heterogeneous group of diseases involving inflammation of blood vessels with subsequent tissue destruction and/or organ damage. The vasculitides can be considered to be primary or secondary (commonly associated with another connective tissue disease or infection). They are predominantly arterial in nature, though capillaries and less commonly veins are involved. Local tissue disruption is caused by inflammatory cell infiltrate in the vessel wall and subsequent tissue ischemia from vessel occlusion. The primary systemic vasculitides are an uncommon group of diseases (combined annual incidence >100 new cases per million).179 Classification is usually based on the predominantly affected vessel (Box 80.7). Recently the term ANCA-associated vasculitis (AAV) has been used to describe those small vessel conditions where there are similar immunopathological tissue mechanism: granulomatosis with polyangiitis (GPA, Wegener’s granulomatosis), microscopic polyangiitis (MPA), and Churg–Strauss syndrome (CSS).180,181
Box 80.7
Classification of vasculitides according to the Chapel Hill consensus
Secondary vasculitides
Recently Watts et al. have suggested a possible fourth category, no predominant vessel size, to describe Behçet disease, primary central nervous system (CNS) vasculitis and Cogan syndrome.180
(Modified from Jennette JC, Falk RJ, Andrassy K, et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum 1994;37:187–92.)
Progression and prognosis of primary systemic necrotizing vasculitis
Classification of the vasculitides is most often based on the size of vessel involved (Box 80.7).182 The initial and predominant inflammatory process is granulomatous in some cases, with the vasculitic phase of the illness only presenting later. This is typical of GPA, where constitutional and upper respiratory symptoms may be present for several years prior to diagnosis, thus delaying the commencement of appropriate therapy and adding to the morbidity and mortality of the condition.
Though the vasculitides are characteristically relapsing diseases, the frequency of relapse depends on the specific underlying diagnosis. In polyarteritis nodosa (PAN) the risk is low, which contrasts with ANCA-associated vasculitides where relapse is as high as 50%.183 Prior to the introduction of effective therapy, the 5-year survival of systemic necrotizing vasculitis (SNV) was only 15%; corticosteroids improved this figure to 48%, while the combination corticosteroids and cyclophosphamide (CP) gave a significant improvement, with 5-year survival reaching 80%. However, this improved survival came at a cost with recurrent flares of disease activity leading to the accumulation of organ damage, with appreciable morbidity also related to drug toxicity.
Aims of therapy
The aim of therapy in SNV must be to suppress disease activity so that organ damage is limited.
Clinical tools to assess disease activity and damage are used to aid in assessment and management of these complex diseases. These scoring systems have predictive value for severe disease where patients are at higher risk of mortality, thus supporting a more aggressive approach to therapy. The Birmingham Vasculitis Activity Score (BVAS) provides a weighted numerical score based on the specific organ involved and the severity of that involvement. A high score reflects either critical organ involvement or multisystem disease and predicts a higher mortality.184 The Vasculitis Damage Index (VDI) is a cumulative score where items of organ damage must be present for a period of at least 3 months and be attributable to effects of the disease, its therapy or other undefined causes. A high VDI score identifies a subgroup of patients with more severe or fatal disease.185
Induction stage
Cyclophosphamide in combination with corticosteroids are the drugs of choice for remission induction. Continuous oral cyclophosphamide (2 mg/kg/day) in conjunction with oral prednisone (1 mg/kg reducing to 10 mg daily by 3 months) induces remission in most by 3 months.186 Remission induction takes longer in some patients, increasing the risk of drug-related toxicity. A safer and equally effective approach is to use intermittent pulses of intravenous cyclophosphamide.187 The pulse interval is an important factor and a suggested induction regimen has been provided (Table 80.3).
| Drug doses | Methylprednisolone 10 mg/kg plus Cyclophosphamide 15 mg/kg |
| Dose interval | 0, 2, 4, 7, 10, 13 weeks Switch after six pulses to consolidation phase with monthly infusions ×3. If in remission, maintenance treatment with methotrexate or azathioprine can be commenced |
| Dose reductions | Age (>70 yr), renal impairment, infection, neutropenia |
| Toxicity | Nausea, alopecia, neutropenia, infertility, hemorrhagic cystitis |
Treatment of relapse
Fewer items of damage accumulate after relapse than at first presentation,185 but for major relapses a short course of cyclophosphamide (six pulses) with an early transfer to maintenance methotrexate, azathioprine or cyclosporine is one approach. Patients with recurrent relapses may be exposed to several courses of cyclophosphamide, increasing the risk of drug-related toxicity (bladder cancer, infertility, myelodysplasia). Methotrexate may be used as an alternative to cyclophosphamide for less severe relapse.
Alternative approaches to therapy
A number of alternative therapies have been trialed. A short course of higher-dose pulsed cyclophosphamide may induce an early remission, but has an increased risk of neutropenia and infection. Autologous stem cell transplantation after intensive immunosuppression has been successfully carried out in a few patients with severe unremitting disease. Anti-T cell monoclonal antibodies (Campath-1H and anti-CD4) have produced dramatic responses in some patients. Anti-TNF agents have not shown any benefit over standard therapy,188 although several case reports suggest a benefit in some cases of treatment-resistant vasculitis. More promising results have been shown using the B-cell depleting anti-CD20 antibody rituximab.189
Large vessel vasculitides
Giant cell arteritis
General considerations
Giant cell arteritis (GCA) or temporal arteritis is a vasculitis that typically effects elderly patients, and is highly corticosteroid-responsive. Symptoms tend to begin insidiously, most commonly headaches, scalp tenderness, myalgias, fever, anorexia, and weight loss.190 In some cases, however, disease can present abruptly with a major complication such as loss of vision.191
Epidemiology
GCA is predominantly a disease of the elderly. It rarely affects those under 50 years, with a mean age of presentation of 70–75 years. There is a 2 : 1 female:male ratio. It is estimated to affect approximately 220 patients per million per year.180,192,193
Articular and systemic disease
GCA targets branches of the external carotid artery; patient symptoms include headaches, scalp tenderness, jaw and tongue claudication. There is an increased risk of transient ischemic attack (TIA) or stroke, as a result of arteritis of the vertebral and basilar arteries. Systemic features of GCA, such as fever, malaise, fatigue, weakness, anorexia, weight loss, and depression are present in 40–50% of patients. The arteritic process can involve other large vessels and subclavian or brachial arteries presenting with upper limb claudication or an aortitis (thoracic > abdominal) is well recognized in 10–20% of patients. There is thought to be an association with polymyalgia rheumatica.194
Ocular disease
Many patients present with temporal headache but no visual loss, but anterior ischemic optic neuropathy (AION) due to arteritis of the short posterior ciliary arteries is the major complication of GCA, and typically presents as acute painless loss of vision. The diagnosis is straightforward when seen with devastating loss of vision (often perception of light), a relative afferent pupillary defect, and typical optic disc edema in the context of systemic features typical of the disease. With time optic atrophy ensues with complete loss of vision. Nevertheless in some patients it is difficult to distinguish between an arteritic and nonarteritic cause of AION. Failure to differentiate these two can be catastrophic as involvement of the second eye in arteritic AION ranges from 10% if treated to 95% if untreated. A detailed ocular and general history is essential and examination reveals a tender, thickened, nonpulsatile superficial temporal artery. Characteristically there is an elevated erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP). A definitive diagnosis is made on temporal artery biopsy (TAB) but a positive biopsy is not always required to make a diagnosis (American College of Rheumatology classification criteria for GCA – Box 80.8).195 Nevertheless all patients with a clinical and/or laboratory diagnosis, or where the diagnosis is in doubt should have therapy instigated and a TAB performed. A TAB is normally performed within 48–72 hours of commencing corticosteroid therapy. A recent study has shown that on 459 cases of biopsy-proven GCA the odds of a positive biopsy were 1.5 times greater with an ESR of 47–107 mm/hr, 5.3 times greater with a CRP >2.45 mg/dl, and 4.2 times greater with platelets >400 /µL.196
Box 80.8
The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis
1. Age ≥50 years at disease onset
2. New onset of localized headache
3. Temporal artery tenderness or decreased pulse
5. Arterial biopsy with necrotizing arteritis with a predominance of mononuclear cell infiltrates or granulomatous process with multinucleate giant cells
(Reproduced with permission from Hunder GG, Bloch DA, Michel BA, et al. The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum 1990;33:1122–8.)
More problematic are those presenting with posterior ischemic optic neuropathy (with an apparently normal optic disc in the acute phase) and who may not report typical features; in the series by Hayreh, around 10% of the IONs seen were posterior ischemic optic neuropathy (PION).191 The estimates of visual loss from GCA vary widely from 13% to 70%, with lower estimates being seen in a recent series that may reflect improved diagnosis and earlier treatment. The visual loss is usually severe (<20/200), and visual recovery is uncommon despite appropriate therapy.197 Visual field loss may be complete, altitudinal or occasionally an arcuate (Bjerrum-type) scotoma. Amaurosis fugax may be a warning of impending ION or other serious ischemic pathology. An ischemic retinopathy with cotton-wool spots (and sometimes retinal hemorrhages) may be seen and can precede optic nerve involvement.198,199 Other ophthalmic complications of biopsy-proven GCA include cilioretinal artery occlusion (CRAO), and ocular ischemic syndrome occurring in about 14%, 20%, and 1% respectively of patients. Occasionally there is extraocular muscle dysfunction with symptoms of transient or permanent diplopia.191
Treatment
Corticosteroids are the mainstay of treatment, with almost all patients who present to a rheumatologist responding to 40–60 mg oral prednisone. Those patients presenting to an ophthalmologist with or without vision loss should have three IV pulses of methylprednisolone 1 g for 3 days, then oral prednisone 1 mg/kg/day. Corticosteroid taper can occur after 4 weeks with resolution of symptoms and a fall in inflammatory markers.200 It is reasonable to aim for a dose of 15 mg by 3 months of therapy. In addition, low-dose aspirin (to reduce the risk of arterial thrombus and visual loss) and bone protection (bisphosphonates and calcium and vitamin D supplementation) with or without a proton pump inhibitor could be considered.200 On average 18 months of corticosteroid treatment is required, but up to 40% may need long-term therapy due to relapsing disease. A corticosteroid sparing agent may need to be introduced, such as methotrexate, azathioprine, and leflunomide. The role of biological agents in treatment is not currently established. There is no evidence that they are beneficial when used at disease onset but may be helpful in relapsing disease to minimize corticosteroid dose and thus side-effects. Recently encouraging results have been demonstrated with the biologics infliximab, etanercept, rituximab, and tociluzimab.201–203
Takayasu’s arteritis
Epidemiology
Takayasu’s arteritis is very rare, with an annual incidence of 2.6 per million in North America and 1.2 per million in Japan. It is classically described in women of childbearing age of Southeast Asian, South African, and Latin American background204–206 (see Box 80.9207).
Box 80.9
The American College of Rheumatology criteria for the classification of Takayasu arteritis
1. Age at disease onset < 40 years
2. Claudication of extremities
3. Decreased brachial artery pulse
(Reproduced with permission from Arend WP, Michel BA, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Takayasu arteritis. Arthritis Rheum 1990;33:1129–34.)
Articular and systemic disease
At the time of diagnosis, 10–20% of patients are clinically asymptomatic and the disease is diagnosed incidentally on vascular examination. The most common findings on examination are hypertension, bruits, diminished or absent pulses, and asymmetric blood pressure readings in the extremities. Of symptomatic patients, 80–90% can present with either systemic or vascular symptoms, or a combination of both. Systemic features include fever, malaise, weight loss, arthralgia, and night sweats.204–206
Vascular symptoms are more common and are as a result of active vasculitis or vascular damage from previous inflammation resulting in stenosis or aneurysm formation. Involvement of the carotid and vertebral arteries can lead to central nervous system involvement. Clinically patients may be asymptomatic, or have symptoms including transient ischemic attacks, stroke, dizziness, syncope, headache or visual changes. Mesenteric involvement is common. The most common cardiac manifestation is aortic regurgitation. Cutaneous manifestations such as erythema nodosum and pyroderma granulosum have been observed in 3–28% of patients.204–206
Ocular disease
Ophthalmic disease occurs in around one-third of patients with Takayasu arteritis. The most common ocular complication is hypertensive retinopathy (16–30%), followed by Takayasu retinopathy (13–15%), and ocular ischemic syndrome (around 4%).208,209 Takayasu retinopathy may progress from small vessel dilation (stage 1), to microaneurysms (stage 2), arteriovenous anastomoses (stage 3), and finally additional complications, such as retinal neovascularization and vitreous hemorrhage (stage 4).208 Other recognized complications include AION, PION, and neovascular glaucoma. The presence of ischemic ocular complications is associated with non-recordable right upper limb blood pressure.209
Treatment
Corticosteroids are the mainstay of treatment. Initially it is recommended that prednisone is started at 1 mg/kg/day (maximum 60 mg/day), for a month then tapered gradually.210,211 If patients have severe disease or refractory disease, steroid-sparing agents need to be considered.
Corticosteroid-sparing agents that can be considered are azathioprine (2 mg/kg/day), and methotrexate (20–25 mg/week).210,212 Other agents with limited data available include cyclophosphamide, mycophenolate mofetil, leflunomide and minocycline, and the biological agents infliximab, tocliziumab, and abatacept.210,212
Non-medical interventions such as angioplasty, stent insertion, arterial reconstruction, and bypass grafting may also be required.210,211
Medium vessel vasculitides
Polyarteritis nodosa
Epidemiology
PAN is a very uncommon disease, being estimated to occur at less than 1 per million per year (United Kingdom data). The major environmental risk factor is hepatitis B; other associated viruses are HIV and parvovirus B19.193
Articular and systemic disease
PAN has a wide spectrum of disease, ranging from very mild limited disease to life threatening organ involvement. Arthralgia or arthritis may be present in up to 50% of patients. An intermittent, asymmetrical, predominantly lower limb, nondeforming arthritis may occur in up to 20% of cases. Myalgias may occur in up to 50% of patients.213,214
Cutaneous features are seen in 25–60%: these include infarctions, ulcerations, livedo reticularis, subcutaneous nodules, and ischemic changes of distal digits.215 Peripheral neuropathy or mononeuritis multiplex may be seen. CNS involvement with headaches, seizures, cranial nerve dysfunction, stroke, and cerebral hemorrhage are less common. Renal involvement can result in hypertension as a result of renal artery stenosis or renal impairment/failure due to multiple microaneurysms and infarcts as a consequence of vascular occlusion. Abdominal pain, which can occur in up to 70% of patients as a result of gastrointestinal involvement, can be severe, with bowel ischemia occurring.213,214
Ocular disease
Ophthalmic disease occurs in around 10–20% of patients with PAN, and may reflect direct ocular involvement or be secondary to its systemic effects (notably hypertension from renal disease). Retinal disease includes retinal vasculitis and hypertensive retinopathy. Other ophthalmic complications include peripheral ulcerative keratitis, episcleritis, scleritis (both anterior and posterior), serous retinal detachments, ischemic optic neuropathy, cranial neuropathies, and cerebral disease resulting in visual field defects.216–218
Treatment
Pulsed IV cyclophosphamide and corticosteroids has been found to achieve better disease control and sustained remission compared to corticosteroids alone, but the long-term survival remains unchanged.219,220 Patients with hepatitis-associated PAN are recommended to have high-dose corticosteroid therapy tapered over 2 weeks followed by antiviral agents, and plasma exchange.219,220
Kawasaki disease
Epidemiology
The highest incidence rate is in Japan – 216 cases per 100 000 per year in children under 5 years of age – with a peak incidence at between 9 and 11 months of age.221 There are possible familial and infectious links.193
Articular and systemic disease
Kawasaki disease is usually defined as a persistent fever for a minimum of 5 days with a minimum of four out of the following: (1) polymorphous rash; (2) bilateral conjunctival injection without exudates; (3) oropharyngeal involvement, including red fissured lips, strawberry tongue, and red pharynx without exudates; (4) peripheral changes including edema of extremities, sole erythema, and periungual desquamation; and (5) cervical lymphadenopathy. Arthritis of the small joints is common, and is associated with a bluish discoloration over the PIPs. Axial arthropathy and effusions can also occur, but the articular symptoms will usually resolve after the acute phase.222,223
The most serious manifestation is that of the cardiovascular system, which can result in aneurysms (especially the coronary arteries), myocarditis, and congestive cardiac failure. Other clinical manifestations include gastrointestinal manifestations, urethritis, facial nerve palsy (rare), and sensorineural hearing loss.222,223
Ocular disease
Bilateral conjunctival injection without purulent discharge is one of the hallmarks of Kawasaki disease. Additionally, bilateral anterior uveitis is common in the first week of illness.224 Rare ocular manifestations include optic neuritis and ophthalmic artery obstruction.225
Treatment
Kawasaki disease is treated with a single IV dose of immunoglobulin and high-dose aspirin. The aspirin dose can be reduced once the fever finishes. If patients fail to respond to the first dose of IV immunoglobulin, treatment options include a repeat infusion, infliximab, IV methylprednisolone, and plasmapheresis.226–229
Small vessel vasculitides
Granulomatosis with polyangiitis (Wegener’s granulomatosis)
Epidemiology
GPA has a slight male predominance, which is more marked in Caucasians. GPA is a rare disease, with a prevalence ranging from 3 per 100 000 in the United States to up to 16 per 100 000 in Northern Europe.230,231 GPA predominantly affects older individuals, but 15% of cases have been reported in childhood.232 GPA is highly associated with PR3 (proteinase 3) ANCA.233
Articular and systemic disease
Presenting symptoms in up to 75% of patients will be of upper respiratory tract disease (nasal, sinus, tracheal and/or ear abnormalities). The “saddle nose deformity” results from nasal bridge collapse, and other nasal manifestations include nasal pain, stuffiness, crusts, epistaxis, mucosal erosion, and septal perforation. A characteristic feature of mouth involvement is “strawberry gums,” an intense gingivitis that responds to systemic treatment. A serious complication is subglottic stenosis secondary to tracheal inflammation and scarring, as this may require a tracheostomy.3 Nonspecific systemic features may occur, particularly during active disease, which include fever, malaise, arthralgias, anorexia, and weight loss.3
Ocular disease
Ophthalmic disease occurs in around 50% of patients in this condition and is an important cause of morbidity and blindness, with vision loss occurring in around 8%.3 Although orbital disease is the most common ocular complication of the disease, almost any part of the ophthalmic system may be affected.
Orbital disease occurs in up to 20% of patients with GPA. It may present with acute or subacute proptosis, and may be associated with ocular motility disturbance (resulting in diplopia), and optic nerve compression or infiltration potentially leading to blindness. Severe proptosis may also lead to sight-threatening exposure keratopathy. It should be noted that although proptosis is common in these patients, chronic orbital inflammation may lead to orbital socket contraction with enophthalmos, restrictive ophthalmopathy, and chronic pain.234
Important nonorbital presentations of GPA include scleritis (7–10% of patients), peripheral ulcerative keratitis, and an ulcerative conjunctivitis with a chronic cicatrizing course.235 The scleritis may be anterior or posterior; necrotizing scleritis is often associated with corneal disease (marginal corneal ulcer/PUK). Uveitis is uncommon (around 3%). Anterior, posterior and pan-uveitis have all been described in this context and may be isolated or associated with scleritis.236,237 Retinal vasculitis may occur and ranges in severity from cotton-wool spots to severe vaso-occlusive disease, with neovascularization and related consequences. Neuro-ophthalmic consequences most commonly occur secondary to orbital involvement, but may also arise due to vasculitis causing ischemic optic neuropathy. In a study of 59 patients with ANCA-associated vasculitis and ocular inflammation, 75% had scleritis, and with time these patients had a 2.75-fold higher mortality than other patients with inflammatory eye disease.238
Treatment
Like other vasculitides, the suggested treatment to induce remission is pulsed IV cyclophosphamide with oral steroids.219,239 Corticosteroid-sparing agents that can be substituted for cyclophosphamide once remission has been achieved are methotrexate or azathioprine. If disease is limited then methotrexate in combination with oral corticosteroid can be used rather than cyclophosphamide.219 Biologics are now increasingly used in the management, initially infliximab and more recently rituximab.240 Rituximab has been used in clinical trials to induce remission in GPA, with favorable results compared to cyclophosphamide, and in sight-threatening ocular and orbital disease.189,241 Treatment is primarily directed against the systemic disease, although it should be noted that the severity of ophthalmic involvement may be an important factor in escalating therapy.
Microscopic polyangiitis
Epidemiology
MPA has an incidence of 5 per million per year, with a peak age of onset of between 65 and 75 years; it is more common in men.231
Articular and systemic disease
Renal involvement is very common, affecting up to 90% of patients. Rapidly progressive glomerulonephritis may occur, resulting in acute renal failure necessitating renal dialysis. Varying degrees of pulmonary involvement can occur; these can range from mild dyspnea to life-threatening pulmonary hemorrhage. Involvement of both the central and peripheral nervous system is described, and includes peripheral neuropathy, mononeuritis multiplex, cerebral hemorrhage/infarction, seizures or headache. Other systemic manifestations include cardiac, gastrointestinal, otorhinolaryngeal, and venous thromboembolism.215,242,243
Ocular disease
Ophthalmic disease is rare in MPA, with one series noting ocular involvement in only 1 of 85 patients with MPA.242 In small case series and isolated reports the following ophthalmic complications have been noted: scleritis, anterior uveitis with hypopyon, retinal vasculitis (ranging from cotton-wool spots to neovascularization and vitreous hemorrhage) and peripheral nonulcerative keratitis.244,245
Treatment
Recommended treatment for severe MPA is oral corticosteroid with pulsed IV cyclophosphamide to induce remission.219,239 In milder active disease (no threatened vital organ disease or damage), methotrexate or azathioprine and oral corticosteroid can be used, but there is a higher risk of relapse.239 If patients have failed to achieve remission and have persistent low activity, IV immunoglobulin can be used to achieve remission.219 There is sparse data in the literature regarding the use of biologics, mainly infliximab and rituximab.246
Churg–Strauss syndrome
Epidemiology
CSS is estimated to have a prevalence of 10 to 15 per million. The mean age of diagnosis is 55 years. It has equal gender incidence.247
Articular and systemic disease
Churg–Strauss syndrome can affect virtually any organ system in the body. Systemic symptoms include fever, weight loss, arthralgias, and, rarely, arthritis.247
Abdominal pain is the most common gastrointestinal symptom. Cardiac involvement includes eosinophilic endomyocarditis, coronary vasculitis, valvular heart disease, congestive heart failure, hypertension, and pericarditis. Skin lesions are common and typically include nonthrombocytopenic palpable purpura, with erythematous, maculopapular, or pustular lesions being reported. Renal involvement is fairly typical, but unlike other necrotizing vasculitides such as GPA or microscopic polyangiitis, renal failure is rare.247–249
Ocular disease
Ophthalmic disease may arise from two processes: vasculitis and granuloma formation. Clinical presentations include conjunctival nodules,250 peripheral ulcerative keratitis, episcleritis, scleritis, uveitis (rare), retinal vasculitis,251 retinal artery occlusion, ischemic optic neuropathy, cranial neuropathies252 and orbital disease (presenting with an orbital inflammatory syndrome).251
Treatment
As with other ANCA-positive vasculitides, remission induction is normally achieved with oral corticosteroids and cyclophosphamide or with a DMARD such as methotrexate or azathioprine in less severe disease.219,239 Rituximab is currently undergoing clinical trials, but there have been promising results published.253
Ocular complications of rheumatological therapies
Corticosteroids
Most treatment-related visual morbidity is associated with corticosteroid treatment. The association between posterior subcapsular cataract and exogenous corticosteroids is well established.254 Cataract surgery in patients with rheumatic diseases is generally successful, although there must be strict control of any intraocular inflammation prior to surgery and in the postoperative phase. Prognosis will be worse if there is visually significant posterior segment disease. Increased intraocular pressure due to exogenous corticosteroids may occur in up to 30% of the normal population, with 5% experiencing an increase of more than 15 mmHg (reviewed by Clark255). Corticosteroid-induced ocular hypertension must be monitored and treated where there is a risk of progression to secondary glaucoma.
Antimalarials
The aminoquinolones chloroquine and hydroxychloroquine have been widely used in the treatment of SLE. These drugs can cause a reversible, visually insignificant keratopathy (cornea verticillata) and, more importantly, an irreversible sight-threatening maculopathy. Clinical progression is of loss of the foveal reflex followed by a fine granular appearance of the macula and finally a “bull’s eye” maculopathy presenting as a central scotoma. Endstage disease includes generalized atrophy, peripheral pigmentation, arteriolar attenuation, and optic atrophy.256
Retinopathy is rare with hydroxychloroquine when used at currently recommended doses (<6.5 mg/kg/d), but increases markedly towards 1% after 5–7 years of usage or a cumulative dose of 1000 g of hydroxychloroquine.257,258 The risk with chloroquine is thought to be significantly greater, with an increased risk at over 460 g chloroquine. In both cases, risk increases with increasing dose, increasing duration, and reduced renal function. The American Academy of Ophthalmology (AAO) 2011 guidelines recommend screening for all patients at baseline or within first year of use, and to start annual screening after 5 years of use (or earlier if additional risk factors).258 Assessments should include dilated retinal examinations, and white 10–2 automated visual field testing which should be interpreted with a low threshold for abnormality and with retesting if abnormalities are seen. Additionally, they recommend that one or more of the following should be performed where available: spectral domain optical coherence tomography, multifocal electroretinography or fundus autofluorescence.258 High-risk patients include those on a dose of >6.5 mg/kg, with a duration of >5 years, obese patients, those with renal or hepatic disease, those with pre-existing retinal disease or those over the age of 60 years.258 Amsler grid testing is no longer recommended. Although fundus examinations are advised for documentation, the aim is to detect changes before visible maculopathy. The AAO authors stress the importance of counseling patients regarding the risk of toxicity and the rationale for screening.
1 The definition and classification of dry eye disease: report of the Definition and Classification Subcommittee of the International Dry Eye WorkShop (2007). Ocul Surf. 2007;5:75–92.
2 Galor A, Thorne JE. Scleritis and peripheral ulcerative keratitis. Rheum Dis Clin North Am. 2007;33:835–854. vii
3 Tarabishy AB, Schulte M, Papaliodis GN, et al. Wegener’s granulomatosis: clinical manifestations, differential diagnosis, and management of ocular and systemic disease. Surv Ophthalmol. 2010;55:429–444.
4 Akpek EK, Thorne JE, Qazi FA, et al. Evaluation of patients with scleritis for systemic disease. Ophthalmology. 2004;111:501–516.
5 Galor A, Thorne JE, Jabs DA. Rheumatic disease and scleritis. Ophthalmology. 2007;114:1232.
6 Watson PG, Hayreh SS. Scleritis and episcleritis. Br J Ophthalmol. 1976;60:163–191.
7 Zaidi AA, Ying GS, Daniel E, et al. Hypopyon in patients with uveitis. Ophthalmology. 2010;117:366–372.
8 Rothova A, van Veenedaal WG, Linssen A, et al. Clinical features of acute anterior uveitis. Am J Ophthalmol. 1987;103:137–145.
9 Weinstein RS. Clinical practice. Glucocorticoid-induced bone disease. N Engl J Med. 2011;365:62–70.
10 Kempen JH, Daniel E, Dunn JP, et al. Overall and cancer related mortality among patients with ocular inflammation treated with immunosuppressive drugs: retrospective cohort study. BMJ. 2009;339:b2480.
11 Symmons D, Turner G, Webb R, et al. The prevalence of rheumatoid arthritis in the United Kingdom: new estimates for a new century. Rheumatology (Oxford). 2002;41:793–800.
12 Gabriel SE, Crowson CS, O’Fallon WM. The epidemiology of rheumatoid arthritis in Rochester, Minnesota, 1955–1985. Arthritis Rheum. 1999;42:415–420.
13 Gabriel SE. The epidemiology of rheumatoid arthritis. Rheum Dis Clin North Am. 2001;27:269–281.
14 Sugiyama D, Nishimura K, Tamaki K, et al. Impact of smoking as a risk factor for developing rheumatoid arthritis: a meta-analysis of observational studies. Ann Rheum Dis. 2010;69:70–81.
15 Majithia V, Geraci SA. Rheumatoid arthritis: diagnosis and management. Am J Med. 2007;120:936–939.
16 Turesson C, O’Fallon WM, Crowson CS, et al. Extra-articular disease manifestations in rheumatoid arthritis: incidence trends and risk factors over 46 years. Ann Rheum Dis. 2003;62:722–727.
17 Avina-Zubieta JA, Choi HK, Sadatsafavi M, et al. Risk of cardiovascular mortality in patients with rheumatoid arthritis: a meta-analysis of observational studies. Arthritis Rheum. 2008;59:1690–1697.
18 Schreiber J, Koschel D, Kekow J, et al. Rheumatoid pneumoconiosis (Caplan’s syndrome). Eur J Intern Med. 2010;21:168–172.
19 Balint GP, Balint PV. Felty’s syndrome. Best Pract Res Clin Rheumatol. 2004;18:631–645.
20 Bartels CM, Bridges AJ. Rheumatoid vasculitis: vanishing menace or target for new treatments? Curr Rheumatol Rep. 2010;12:414–419.
21 Aletaha D, Neogi T, Silman AJ, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010;62:2569–2581.
22 Akpek EK, Uy HS, Christen W, et al. Severity of episcleritis and systemic disease association. Ophthalmology. 1999;106:729–731.
23 Foster CS, Forstot SL, Wilson LA. Mortality rate in rheumatoid arthritis patients developing necrotizing scleritis or peripheral ulcerative keratitis. Effects of systemic immunosuppression. Ophthalmology. 1984;91:1253–1263.
24 Giordano N, D’Ettorre M, Biasi G, et al. Retinal vasculitis in rheumatoid arthritis: an angiographic study. Clin Exp Rheumatol. 1990;8:121–125.
25 Martin MF, Scott DG, Gilbert C, et al. Retinal vasculitis in rheumatoid arthritis. Br Med J Clin Res Ed. 1981;282:1745–1746.
26 Matsuo T, Koyama T, Morimoto N, et al. Retinal vasculitis as a complication of rheumatoid arthritis. Ophthalmologica. 1990;201:196–200.
27 Saag KG, Teng GG, Patkar NM, et al. American College of Rheumatology 2008 recommendations for the use of nonbiologic and biologic disease-modifying antirheumatic drugs in rheumatoid arthritis. Arthritis Rheum. 2008;59:762–784.
28 Maxwell L, Singh JA. Abatacept for rheumatoid arthritis. Cochrane Database Syst Rev. 2009. CD007277
29 Management and therapy of dry eye disease: report of the Management and Therapy Subcommittee of the International Dry Eye WorkShop (2007). Ocul Surf. 2007;5:163–178.
30 Dougados M, van der Linden S, Juhlin R, et al. The European Spondylarthropathy Study Group preliminary criteria for the classification of spondylarthropathy. Arthritis Rheum. 1991;34:1218–1227.
31 Zeboulon N, Dougados M, Gossec L. Prevalence and characteristics of uveitis in the spondyloarthropathies: a systematic literature review. Ann Rheum Dis. 2008;67:955–959.
32 Rudwaleit M, Taylor WJ. Classification criteria for psoriatic arthritis and ankylosing spondylitis/axial spondyloarthritis. Best Pract Res Clin Rheumatol. 2010;24:589–604.
33 Gabriel SE, Michaud K. Epidemiological studies in incidence, prevalence, mortality, and comorbidity of the rheumatic diseases. Arthritis Res Ther. 2009;11:229.
34 Lee W, Reveille JD, Davis JC, Jr., et al. Are there gender differences in severity of ankylosing spondylitis? Results from the PSOAS cohort. Ann Rheum Dis. 2007;66:633–638.
35 van der Heijde D, Lie E, Kvien TK, et al. ASDAS, a highly discriminatory ASAS-endorsed disease activity score in patients with ankylosing spondylitis. Ann Rheum Dis. 2009;68:1811–1818.
36 Zochling J, van der Heijde D, Burgos-Vargas R, et al. ASAS/EULAR recommendations for the management of ankylosing spondylitis. Ann Rheum Dis. 2006;65:442–452.
37 Amor B, Dougados M, Listrat V, et al. Are classification criteria for spondylarthropathy useful as diagnostic criteria? Rev Rhum Engl Ed. 1995;62:10–15.
38 Rosenbaum JT. Acute anterior uveitis and spondyloarthropathies. Rheum Dis Clin North Am. 1992;18:143–151.
39 Linssen A, Dekker-Saeys AJ, Dijkstra PF, et al. The use of HLA-B27 as a diagnostic and prognostic aid in acute anterior uveitis AAU in The Netherlands. Doc Ophthalmol. 1986;64:217–223.
40 Tay-Kearney ML, Schwam BL, Lowder C, et al. Clinical features and associated systemic diseases of HLA-B27 uveitis. Am J Ophthalmol. 1996;121:47–56.
41 Dougados M, Dijkmans B, Khan M, et al. Conventional treatments for ankylosing spondylitis. Ann Rheum Dis. 2002;61(Suppl 3):iii40–iii50.
42 van der Heijde D, Baraf HS, Ramos-Remus C, et al. Evaluation of the efficacy of etoricoxib in ankylosing spondylitis: results of a fifty-two-week, randomized, controlled study. Arthritis Rheum. 2005;52:1205–1215.
43 Clegg DO, Reda DJ, Abdellatif M. Comparison of sulfasalazine and placebo for the treatment of axial and peripheral articular manifestations of the seronegative spondylarthropathies: a Department of Veterans Affairs cooperative study. Arthritis Rheum. 1999;42:2325–2329.
44 Roychowdhury B, Bintley-Bagot S, Bulgen DY, et al. Is methotrexate effective in ankylosing spondylitis? Rheumatology (Oxford). 2002;41:1330–1332.
45 Rudwaleit M, Rodevand E, Holck P, et al. Adalimumab effectively reduces the rate of anterior uveitis flares in patients with active ankylosing spondylitis: results of a prospective open-label study. Ann Rheum Dis. 2009;68:696–701.
46 Braun J, Baraliakos X, Listing J, Sieper J. Decreased incidence of anterior uveitis in patients with ankylosing spondylitis treated with the anti-tumor necrosis factor agents infliximab and etanercept. Arthritis Rheum. 2005;52:2447–2451.
47 Soderlin MK, Kautiainen H, Puolakkainen M, et al. Infections preceding early arthritis in southern Sweden: a prospective population-based study. J Rheumatol. 2003;30:459–464.
48 Lee DA, Barker SM, Su WP, et al. The clinical diagnosis of Reiter’s syndrome. Ophthalmic and nonophthalmic aspects. Ophthalmology. 1986;93:350–356.
49 Saari KM, Kauranen O. Ocular inflammation in Reiter’s syndrome associated with Campylobacter jejuni enteritis. Am J Ophthalmol. 1980;90:572–573.
50 Saari KM, Vilppula A, Lassus A, et al. Ocular inflammation in Reiter’s disease after Salmonella enteritis. Am J Ophthalmol. 1980;90:63–68.
51 Needham AD, Harding SP, Carey P. Bilateral multifocal choroiditis in Reiter syndrome. Arch Ophthalmol. 1997;115:684–685.
52 Conway RM, Graham SL, Lassere M. Incomplete Reiter’s syndrome with focal involvement of the posterior segment. Aust N Z J Ophthalmol. 1995;23:63–66.
53 Rosenbaum JT. Characterization of uveitis associated with spondyloarthritis. J Rheumatol. 1989;16:792–796.
54 Leirisalo-Repo M. Reactive arthritis. Scand J Rheumatol. 2005;34:251–259.
55 Rothfuss KS, Stange EF, Herrlinger KR. Extraintestinal manifestations and complications in inflammatory bowel diseases. World J Gastroenterol. 2006;12:4819–4831.
56 Rodriguez-Reyna TS, Martinez-Reyes C, Yamamoto-Furusho JK. Rheumatic manifestations of inflammatory bowel disease. World J Gastroenterol. 2009;15:5517–5524.
57 Hopkins DJ, Horan E, Burton IL, et al. Ocular disorders in a series of 332 patients with Crohn’s disease. Br J Ophthalmol. 1974;58:732–737.
58 Greenstein AJ, Janowitz HD, Sachar DB. The extra-intestinal complications of Crohn’s disease and ulcerative colitis: a study of 700 patients. Medicine (Baltimore). 1976;55:401–412.
59 Lyons JL, Rosenbaum JT. Uveitis associated with inflammatory bowel disease compared with uveitis associated with spondyloarthropathy. Arch Ophthalmol. 1997;115:61–64.
60 Ghanchi FD, Rembacken BJ. Inflammatory bowel disease and the eye. Surv Ophthalmol. 2003;48:663–676.
61 Cury DB, Moss AC. Ocular manifestations in a community-based cohort of patients with inflammatory bowel disease. Inflamm Bowel Dis. 2010;16:1393–1396.
62 Lin P, Tessler HH, Goldstein DA. Family history of inflammatory bowel disease in patients with idiopathic ocular inflammation. Am J Ophthalmol. 2006;141:1097–1104.
63 Soukiasian SH, Foster CS, Raizman MB. Treatment strategies for scleritis and uveitis associated with inflammatory bowel disease. Am J Ophthalmol. 1994;118:601–611.
64 Taylor W, Gladman D, Helliwell P, et al. Classification criteria for psoriatic arthritis: development of new criteria from a large international study. Arthritis Rheum. 2006;54:2665–2673.
65 Shbeeb M, Uramoto KM, Gibson LE, et al. The epidemiology of psoriatic arthritis in Olmsted County, Minnesota, USA, 1982–1991. J Rheumatol. 2000;27:1247–1250.
66 Ibrahim G, Waxman R, Helliwell PS. The prevalence of psoriatic arthritis in people with psoriasis. Arthritis Rheum. 2009;61:1373–1378.
67 Jenkinson T, Armas J, Evison G, et al. The cervical spine in psoriatic arthritis: a clinical and radiological study. Br J Rheumatol. 1994;33:255–259.
68 van Romunde LK, Cats A, Hermans J, et al. Psoriasis and arthritis. II. A cross-sectional comparative study of patients with “psoriatic arthritis” and seronegative and seropositive polyarthritis: clinical aspects. Rheumatol Int. 1984;4:61–65.
69 Rehal B, Modjtahedi BS, Morse LS, et al. Ocular psoriasis. J Am Acad Dermatol. 2011;65:1202–1212.
70 Lambert JR, Wright V. Eye inflammation in psoriatic arthritis. Ann Rheum Dis. 1976;35:354–356.
71 Paiva ES, Macaluso DC, Edwards A, et al. Characterisation of uveitis in patients with psoriatic arthritis. Ann Rheum Dis. 2000;59:67–70.
72 Nash P, Clegg DO. Psoriatic arthritis therapy: NSAIDs and traditional DMARDs. Ann Rheum Dis. 2005;64(Suppl 2):ii74–ii77.
73 Mease PJ. Psoriatic arthritis: update on pathophysiology, assessment and management. Ann Rheum Dis. 2011;70(Suppl 1):i77–i84.
74 Current status of oral PUVA therapy for psoriasis. Eye protection revisions. J Am Acad Dermatol. 1982;6:851–855.
75 Saurenmann RK, Rose JB, Tyrrell P, et al. Epidemiology of juvenile idiopathic arthritis in a multiethnic cohort: ethnicity as a risk factor. Arthritis Rheum. 2007;56:1974–1984.
76 Petty RE, Southwood TR, Manners P, et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol. 2004;31:390–392.
77 BenEzra D, Cohen E, Behar-Cohen F. Uveitis and juvenile idiopathic arthritis: a cohort study. Clin Ophthalmol. 2007;1:513–518.
78 Berk AT, Kocak N, Unsal E. Uveitis in juvenile arthritis. Ocul Immunol Inflamm. 2001;9:243–251.
79 Boone MI, Moore TL, Cruz OA. Screening for uveitis in juvenile rheumatoid arthritis. J Pediatr Ophthalmol Strabismus. 1998;35:41–43.
80 Chylack LT, Jr. The ocular manifestations of juvenile rheumatoid arthritis. Arthritis Rheum. 1977;20:217–223.
81 Saurenmann RK, Levin AV, Feldman BM, et al. Prevalence, risk factors, and outcome of uveitis in juvenile idiopathic arthritis: a long-term followup study. Arthritis Rheum. 2007;56:647–657.
82 Saurenmann RK, Levin AV, Feldman BM, et al. Risk factors for development of uveitis differ between girls and boys with juvenile idiopathic arthritis. Arthritis Rheum. 2010;62:1824–1828.
83 Nordal EB, Songstad NT, Berntson L, et al. Biomarkers of chronic uveitis in juvenile idiopathic arthritis: predictive value of antihistone antibodies and antinuclear antibodies. J Rheumatol. 2009;36:1737–1743.
84 Cassidy J, Kivlin J, Lindsley C, et al. Ophthalmologic examinations in children with juvenile rheumatoid arthritis. Pediatrics. 2006;117:1843–1845.
85 Ayuso VK, Ten Cate HA, van der Does P, et al. Male gender and poor visual outcome in uveitis associated with juvenile idiopathic arthritis. Am J Ophthalmol. 2010;149:987–993.
86 Kalinina AV, Ten Cate HA, van der Does P, et al. Male gender as a risk factor for complications in uveitis associated with juvenile idiopathic arthritis. Am J Ophthalmol. 2010;149:994–999.
87 Christoph T, Carsten H, Martin R, et al. Elevated laser flare values correlate with complicated course of anterior uveitis in patients with juvenile idiopathic arthritis. Acta Ophthalmol. 2011;896:e521–e527.
88 Key SN, III., Kimura SJ. Iridocyclitis associated with juvenile rheumatoid arthritis. Am J Ophthalmol. 1975;80:425–429.
89 Rosenberg AM, Oen KG. The relationship between ocular and articular disease activity in children with juvenile rheumatoid arthritis and associated uveitis. Arthritis Rheum. 1986;29:797–800.
90 Beukelman T, Guevara JP, Albert DA. Optimal treatment of knee monarthritis in juvenile idiopathic arthritis: a decision analysis. Arthritis Rheum. 2008;59:1580–1588.
91 Prakken B, Albani S, Martini A. Juvenile idiopathic arthritis. Lancet. 2011;377:2138–2149.
92 Giannini EH, Ilowite NT, Lovell DJ, et al. Long-term safety and effectiveness of etanercept in children with selected categories of juvenile idiopathic arthritis. Arthritis Rheum. 2009;60:2794–2804.
93 Lovell DJ, Giannini EH, Reiff A, et al. Etanercept in children with polyarticular juvenile rheumatoid arthritis. Pediatric Rheumatology Collaborative Study Group. N Engl J Med. 2000;342:763–769.
94 Ruperto N, Lovell DJ, Cuttica R, et al. A randomized, placebo-controlled trial of infliximab plus methotrexate for the treatment of polyarticular-course juvenile rheumatoid arthritis. Arthritis Rheum. 2007;56:3096–3106.
95 Lovell DJ, Ruperto N, Goodman S, et al. Adalimumab with or without methotrexate in juvenile rheumatoid arthritis. N Engl J Med. 2008;359:810–820.
96 Ruperto N, Lovell DJ, Quartier P, et al. Long-term safety and efficacy of abatacept in children with juvenile idiopathic arthritis. Arthritis Rheum. 2010;62:1792–1802.
97 Ruperto N, Lovell DJ, Quartier P, et al. Abatacept in children with juvenile idiopathic arthritis: a randomised, double-blind, placebo-controlled withdrawal trial. Lancet. 2008;372:383–391.
98 Diak P, Siegel J, La GL, et al. Tumor necrosis factor alpha blockers and malignancy in children: forty-eight cases reported to the Food and Drug Administration. Arthritis Rheum. 2010;62:2517–2524.
99 Thorne JE, Woreta FA, Dunn JP, et al. Risk of cataract development among children with juvenile idiopathic arthritis-related uveitis treated with topical corticosteroids. Ophthalmology. 2010;117:1436–1441.
100 Biester S, Deuter C, Michels H, et al. Adalimumab in the therapy of uveitis in childhood. Br J Ophthalmol. 2007;91:319–324.
101 Lim LL, Fraunfelder FW, Rosenbaum JT. Do tumor necrosis factor inhibitors cause uveitis? A registry-based study. Arthritis Rheum. 2007;56:3248–3252.
102 Quinones K, Cervantes-Castaneda RA, Hynes AY, et al. Outcomes of cataract surgery in children with chronic uveitis. J Cataract Refract Surg. 2009;35:725–731.
103 Grajewski RS, Zurek-Imhoff B, Roesel M, et al. Favourable outcome after cataract surgery with IOL implantation in uveitis associated with juvenile idiopathic arthritis. Acta Ophthalmol. ePub 11 February, 2011. doi: 10.1111/j.1755–3768.2011.02110.x
104 Kotaniemi K, Penttila H. Intraocular lens implantation in patients with juvenile idiopathic arthritis-associated uveitis. Ophthalmic Res. 2006;38:318–323.
105 Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725.
106 Smith PP, Gordon C. Systemic lupus erythematosus: clinical presentations. Autoimmun Rev. 2010;10:43–45.
107 Alarcon GS, Friedman AW, Straaton KV, et al. Systemic lupus erythematosus in three ethnic groups: III. A comparison of characteristics early in the natural history of the LUMINA cohort. LUpus in MInority populations: NAture vs Nurture. Lupus. 1999;8:197–209.
108 Weening JJ, D’Agati VD, Schwartz MM, et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. Kidney Int. 2004;65:521–530.
109 Sultan SM, Ioannou Y, Isenberg DA. A review of gastrointestinal manifestations of systemic lupus erythematosus. Rheumatology (Oxford). 1999;38:917–932.
110 Sivaraj RR, Durrani OM, Denniston AK, et al. Ocular manifestations of systemic lupus erythematosus. Rheumatology (Oxford). 2007;46:1757–1762.
111 Jensen JL, Bergem HO, Gilboe IM, et al. Oral and ocular sicca symptoms and findings are prevalent in systemic lupus erythematosus. J Oral Pathol Med. 1999;28:317–322.
112 Read RW. Clinical mini-review: systemic lupus erythematosus and the eye. Ocul Immunol Inflamm. 2004;12:87–99.
113 Lachmann SM, Hazleman BL, Watson PG. Scleritis and associated disease. Br Med J. 1978;1:88–90.
114 Stavrou P, Murray PI, Batta K, et al. Acute ocular ischaemia and orbital inflammation associated with systemic lupus erythematosus. Br J Ophthalmol. 2002;86:474–475.
115 Grimson BS, Simons KB. Orbital inflammation, myositis, and systemic lupus erythematosus. Arch Ophthalmol. 1983;101:736–738.
116 Ushiyama O, Ushiyama K, Koarada S, et al. Retinal disease in patients with systemic lupus erythematosus. Ann Rheum Dis. 2000;59:705–708.
117 Coppeto JR. Retinopathy and systemic lupus erythematosus. Arch Ophthalmol. 1984;102:1748–1749.
118 Jabs DA, Fine SL, Hochberg MC, et al. Severe retinal vaso-occlusive disease in systemic lupus erythematous. Arch Ophthalmol. 1986;104:558–563.
119 Asherson RA, Merry P, Acheson JF, et al. Antiphospholipid antibodies: a risk factor for occlusive ocular vascular disease in systemic lupus erythematosus and the “primary” antiphospholipid syndrome. Ann Rheum Dis. 1989;48:358–361.
120 Graham EM, Spalton DJ, Barnard RO, et al. Cerebral and retinal vascular changes in systemic lupus erythematosus. Ophthalmology. 1985;92:444–448.
121 Sekimoto M, Hayasaka S, Noda S, et al. Pseudoretinitis pigmentosa in patients with systemic lupus erythematosus. Ann Ophthalmol. 1993;25:264–266.
122 Jabs DA, Hanneken AM, Schachat AP, et al. Choroidopathy in systemic lupus erythematosus. Arch Ophthalmol. 1988;106:230–234.
123 Jabs DA, Miller NR, Newman SA, et al. Optic neuropathy in systemic lupus erythematosus. Arch Ophthalmol. 1986;104:564–568.
124 Wang JC, Chuah GC, Yap EY. Tuberculous choroidal granulomas in a patient with systemic lupus erythematosus. A case report. Int Ophthalmol. 2001;24:107–109.
125 Ishibashi Y, Watanabe R, Hommura S, et al. Endogenous Nocardia asteroides endophthalmitis in a patient with systemic lupus erythematosus. Br J Ophthalmol. 1990;74:433–436.
126 Favas C, Isenberg DA. B-cell-depletion therapy in SLE – what are the current prospects for its acceptance? Nat Rev Rheumatol. 2009;5:711–716.
127 Turner-Stokes T, Lu TY, Ehrenstein MR, et al. The efficacy of repeated treatment with B-cell depletion therapy in systemic lupus erythematosus: an evaluation. Rheumatology (Oxford). 2011.
128 Lightstone L. Lupus nephritis: where are we now? Curr Opin Rheumatol. 2010;22:252–256.
129 Pillemer SR, Matteson EL, Jacobsson LT, et al. Incidence of physician-diagnosed primary Sjögren syndrome in residents of Olmsted County, Minnesota. Mayo Clin Proc. 2001;76:593–599.
130 Fox RI. Sjögren’s syndrome. Lancet. 2005;366:321–331.
131 Mavragani CP, Moutsopoulos NM, Moutsopoulos HM. The management of Sjögren’s syndrome. Nat Clin Pract Rheumatol. 2006;2:252–261.
132 Rosenbaum JT, Bennett RM. Chronic anterior and posterior uveitis and primary Sjogren’s syndrome. Am J Ophthalmol. 1987;104:346–352.
133 Ramos-Casals M, Tzioufas AG, Stone JH, et al. Treatment of primary Sjögren syndrome: a systematic review. JAMA. 2010;304:452–460.
134 Sall K, Stevenson OD, Mundorf TK, et al. Two multicenter, randomized studies of the efficacy and safety of cyclosporine ophthalmic emulsion in moderate to severe dry eye disease. CsA Phase 3 Study Group. Ophthalmology. 2000;107:631–639.
135 Barber LD, Pflugfelder SC, Tauber J, et al. Phase III safety evaluation of cyclosporine 0.1% ophthalmic emulsion administered twice daily to dry eye disease patients for up to 3 years. Ophthalmology. 2005;112:1790–1794.
136 Rothova A. Ocular involvement in sarcoidosis. Br J Ophthalmol. 2000;84:110–116.
137 Fretzayas A, Moustaki M, Vougiouka O. The puzzling clinical spectrum and course of juvenile sarcoidosis. World J Pediatr. 2011;7:103–110.
138 Becker ML, Rose CD. Blau syndrome and related genetic disorders causing childhood arthritis. Curr Rheumatol Rep. 2005;7:427–433.
139 Punzi L, Furlan A, Podswiadek M, et al. Clinical and genetic aspects of Blau syndrome: a 25–year follow-up of one family and a literature review. Autoimmun Rev. 2009;8:228–232.
140 Dow CT, Ellingson JL. Detection of Mycobacterium avium ss. paratuberculosis in Blau syndrome tissues. Autoimmune Dis. 2011;2011:127692.
141 James DG. A comparison of Blau’s syndrome and sarcoidosis. Sarcoidosis. 1994;11:100–101.
142 Geha RS, Notarangelo LD, Casanova JL, et al. Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. J Allergy Clin Immunol. 2007;120:776–794.
143 Latkany PA, Jabs DA, Smith JR, et al. Multifocal choroiditis in patients with familial juvenile systemic granulomatosis. Am J Ophthalmol. 2002;134:897–904.
144 Latkany P. Blau syndrome. Ophthalmology. 2004;111:853–854.
145 Bernatsky S, Joseph L, Pineau CA, et al. Scleroderma prevalence: demographic variations in a population-based sample. Arthritis Rheum. 2009;61:400–404.
146 Mayes MD, Lacey JV, Jr., Beebe-Dimmer J, et al. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. 2003;48:2246–2255.
147 Chifflot H, Fautrel B, Sordet C, et al. Incidence and prevalence of systemic sclerosis: a systematic literature review. Semin Arthritis Rheum. 2008;37:223–235.
148 Ebert EC. Esophageal disease in progressive systemic sclerosis. Curr Treat Options Gastroenterol. 2008;11:64–69.
149 West RH, Barnett AJ. Ocular involvement in scleroderma. Br J Ophthalmol. 1979;63:845–847.
150 Horan EC. Ophthalmic manifestations of progressive systemic sclerosis. Br J Ophthalmol. 1969;53:388–392.
151 Emre S, Kayikcioglu O, Ates H, et al. Corneal hysteresis, corneal resistance factor, and intraocular pressure measurement in patients with scleroderma using the Reichert ocular response analyzer. Cornea. 2010;29:628–631.
152 Serup J, Serup L. Increased central cornea thickness in localized scleroderma morphoea. Metab Pediatr Syst Ophthalmol. 1985;8:11–14.
153 Serup L, Serup J, Hagdrup H. Fundus fluorescein angiography in generalized scleroderma. Ophthalmic Res. 1987;19:303–308.
154 Tailor R, Gupta A, Herrick A, et al. Ocular manifestations of scleroderma. Surv Ophthalmol. 2009;54:292–304.
155 Kowal-Bielecka O, Landewe R, Avouac J, et al. EULAR recommendations for the treatment of systemic sclerosis: a report from the EULAR Scleroderma Trials and Research group EUSTAR. Ann Rheum Dis. 2009;68:620–628.
156 Le EN, Wigley FM, Shah AA, et al. Long-term experience of mycophenolate mofetil for treatment of diffuse cutaneous systemic sclerosis. Ann Rheum Dis. 2011;70:1104–1107.
157 Phumethum V, Jamal S, Johnson SR. Biologic therapy for systemic sclerosis: a systematic review. J Rheumatol. 2011;38:289–296.
158 Vonk MC, Marjanovic Z, van den Hoogen FH, et al. Long-term follow-up results after autologous haematopoietic stem cell transplantation for severe systemic sclerosis. Ann Rheum Dis. 2008;67:98–104.
159 Bohan A, Peter JB. Polymyositis and dermatomyositis first of two parts. N Engl J Med. 1975;292:344–347.
160 Patrick M, Buchbinder R, Jolley D, et al. Incidence of inflammatory myopathies in Victoria, Australia, and evidence of spatial clustering. J Rheumatol. 1999;26:1094–1100.
161 Mastaglia FL, Phillips BA. Idiopathic inflammatory myopathies: epidemiology, classification, and diagnostic criteria. Rheum Dis Clin North Am. 2002;28:723–741.
162 Mastaglia FL, Garlepp MJ, Phillips BA, et al. Inflammatory myopathies: clinical, diagnostic and therapeutic aspects. Muscle Nerve. 2003;27:407–425.
163 Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96–100.
164 Susac JO, Garcia-Mullin R, Glaser JS. Ophthalmoplegia in dermatomyositis. Neurology. 1973;23:305–310.
165 Yeo LM, Swaby DS, Situnayake RD, et al. Irreversible visual loss in dermatomyositis. Br J Rheumatol. 1995;34:1179–1181.
166 Cohen BH, Sedwick LA, Burde RM. Retinopathy of dermatomyositis. J Clin Neuroophthalmol. 1985;5:177–179.
167 Backhouse O, Griffiths B, Henderson T, et al. Ophthalmic manifestations of dermatomyositis. Ann Rheum Dis. 1998;57:447–449.
168 McHugh N. Disease management dermatomyositis/polymyositis. Rheumatology (Oxford). 2011;50:12–14.
169 Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362:971–982.
170 Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev. 2010;9:540–546.
171 Balsa A, Expinosa A, Cuesta M, et al. Joint symptoms in relapsing polychondritis. Clin Exp Rheumatol. 1995;13:425–430.
172 Isaak BL, Liesegang TJ, Michet CJ, Jr. Ocular and systemic findings in relapsing polychondritis. Ophthalmology. 1986;93:681–689.
173 Matas BR. Iridocyclitis associated with relapsing polychondritis. Arch Ophthalmol. 1970;84:474–476.
174 Messmer EM, Foster CS. Vasculitic peripheral ulcerative keratitis. Surv Ophthalmol. 1999;43:379–396.
175 Magargal LE, Donoso LA, Goldberg RE, et al. Ocular manifestations of relapsing polychondritis. Retina. 1981;1:96–99.
176 Anderson B, Sr. Ocular lesions in relapsing polychondritis and other rheumatoid syndromes. The Edward Jackson memorial lecture. Am J Ophthalmol. 1967;64:35–50.
177 McKay DA, Watson PG, Lyne AJ. Relapsing polychondritis and eye disease. Br J Ophthalmol. 1974;58:600–605.
178 McCarthy EM, Cunnane G. Treatment of relapsing polychondritis in the era of biological agents. Rheumatol Int. 2010;30:827–828.
179 Basu N, Watts R, Bajema I, et al. EULAR points to consider in the development of classification and diagnostic criteria in systemic vasculitis. Ann Rheum Dis. 2010;69:1744–1750.
180 Watts RA, Suppiah R, Merkel PA, et al. Systemic vasculitis – is it time to reclassify? Rheumatology (Oxford). 2011;50:643–645.
181 Falk RJ, Gross WL, Guillevin L, et al. Granulomatosis with polyangiitis Wegener’s: an alternative name for Wegener’s granulomatosis. Arthritis Rheum. 2011;63:863–864.
182 Jennette JC, Falk RJ, Andrassy K, et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum. 1994;37:187–192.
183 Gordon M, Luqmani RA, Adu D, et al. Relapses in patients with a systemic vasculitis. Q J Med. 1993;86:779–789.
184 Luqmani RA, Bacon PA, Moots RJ, et al. Birmingham Vasculitis Activity Score (BVAS) in systemic necrotizing vasculitis. QJM. 1994;87:671–678.
185 Exley AR, Carruthers DM, Luqmani RA, et al. Damage occurs early in systemic vasculitis and is an index of outcome. QJM. 1997;90:391–399.
186 Langford CA, Talar-Williams C, Barron KS, et al. Use of a cyclophosphamide-induction methotrexate-maintenance regimen for the treatment of Wegener’s granulomatosis: extended follow-up and rate of relapse. Am J Med. 2003;114:463–469.
187 Adu D, Pall A, Luqmani RA, et al. Controlled trial of pulse versus continuous prednisolone and cyclophosphamide in the treatment of systemic vasculitis. QJM. 1997;90:401–409.
188 Morgan MD, Drayson MT, Savage CO, et al. Addition of infliximab to standard therapy for ANCA-associated vasculitis. Nephron Clin Pract. 2011;117:c89–c97.
189 Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363:221–232.
190 Hunder GG. Clinical features of GCA/PMR. Clin Exp Rheumatol. 2000;18:S6–S8.
191 Hayreh SS, Podhajsky PA, Zimmerman B. Ocular manifestations of giant cell arteritis. Am J Ophthalmol. 1998;125:509–520.
192 Direskeneli H, Aydin SZ, Kermani, et al. Development of outcome measures for large-vessel vasculitis for use in clinical trials: opportunities, challenges, and research agenda. J Rheumatol. 2011;38:1471–1479.
193 Watts RA, Scott DG. Epidemiology of the vasculitides. Semin Respir Crit Care Med. 2004;25:455–464.
194 Cantini F, Niccoli L, Storri L, et al. Are polymyalgia rheumatica and giant cell arteritis the same disease? Semin Arthritis Rheum. 2004;33:294–301.
195 Hunder GG, Bloch DA, Michel BA, et al. The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum. 1990;33:1122–1128.
196 Walvick MD, Walvick MP. Giant cell arteritis: laboratory predictors of a positive temporal artery biopsy. Ophthalmology. 2011;118:1201–1204.
197 Danesh-Meyer H, Savino PJ, Gamble GG. Poor prognosis of visual outcome after visual loss from giant cell arteritis. Ophthalmology. 2005;112:1098–1103.
198 Melberg NS, Grand MG, Dieckert JP, et al. Cotton-wool spots and the early diagnosis of giant cell arteritis. Ophthalmology. 1995;102:1611–1614.
199 Johnson MC, Lee AG. Giant cell arteritis presenting with cotton wool spots. Semin Ophthalmol. 2008;23:141–142.
200 Ghosh P, Borg FA, Dasgupta B. Current understanding and management of giant cell arteritis and polymyalgia rheumatica. Expert Rev Clin Immunol. 2010;6:913–928.
201 Hoffman GS, Cid MC, Rendt-Zagar KE, et al. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med. 2007;146:621–630.
202 Martinez-Taboada VM, Rodriguez-Valverde V, Carreno L, et al. A double-blind placebo controlled trial of etanercept in patients with giant cell arteritis and corticosteroid side effects. Ann Rheum Dis. 2008;67:625–630.
203 Bhatia A, Ell PJ, Edwards JC. Anti-CD20 monoclonal antibody (rituximab) as an adjunct in the treatment of giant cell arteritis. Ann Rheum Dis. 2005;64:1099–1100.
204 Cong XL, Dai SM, Feng X, et al. Takayasu’s arteritis: clinical features and outcomes of 125 patients in China. Clin Rheumatol. 2010;29:973–981.
205 Hall S, Barr W, Lie JT, et al. Takayasu arteritis. A study of 32 North American patients. Medicine (Baltimore). 1985;64:89–99.
206 Nasu T. Takayasu’s truncoarteritis in Japan. A statistical observation of 76 autopsy cases. Pathol Microbiol (Basel). 1975;43:140–146.
207 Arend WP, Michel BA, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Takayasu arteritis. Arthritis Rheum. 1990;33:1129–1134.
208 Chun YS, Park SJ, Park IK, et al. The clinical and ocular manifestations of Takayasu arteritis. Retina. 2001;21:132–140.
209 Peter J, David S, Danda D, et al. Ocular manifestations of Takayasu arteritis: a cross-sectional study. Retina. 2011;31:1170–1178.
210 Mukhtyar C, Guillevin L, Cid MC, et al. EULAR recommendations for the management of large vessel vasculitis. Ann Rheum Dis. 2009;68:318–323.
211 Park MC, Lee SW, Park YB, et al. Clinical characteristics and outcomes of Takayasu’s arteritis: analysis of 108 patients using standardized criteria for diagnosis, activity assessment, and angiographic classification. Scand J Rheumatol. 2005;34:284–292.
212 Valsakumar AK, Valappil UC, Jorapur V, et al. Role of immunosuppressive therapy on clinical, immunological, and angiographic outcome in active Takayasu’s arteritis. J Rheumatol. 2003;30:1793–1798.
213 Pettigrew HD, Teuber SS, Gershwin ME. Polyarteritis nodosa. Compr Ther. 2007;33:144–149.
214 Dillon MJ, Eleftheriou D, Brogan PA. Medium-size-vessel vasculitis. Pediatr Nephrol. 2010;25:1641–1652.
215 Kluger N, Pagnoux C, Guillevin L, et al. Comparison of cutaneous manifestations in systemic polyarteritis nodosa and microscopic polyangiitis. Br J Dermatol. 2008;159:615–620.
216 Blodi FC, Sullivan PB. Involvement of the eyes in periarteritis nodosa. Trans Am Acad Ophthalmol Otolaryngol. 1959;63:161–165.
217 Moore JG, Sevel D. Corneo-scleral ulceration in periarteritis nodosa. Br J Ophthalmol. 1966;50:651–655.
218 Yamamoto S, Takeuchi S. Episcleritis as the primary clinical manifestation in a patient with polyarteritis nodosa. Jpn J Ophthalmol. 2000;44:151–153.
219 Mukhtyar C, Guillevin L, Cid MC, et al. EULAR recommendations for the management of primary small and medium vessel vasculitis. Ann Rheum Dis. 2009;68:310–317.
220 Gayraud M, Guillevin L, le TP, et al. Long-term followup of polyarteritis nodosa, microscopic polyangiitis, and Churg–Strauss syndrome: analysis of four prospective trials including 278 patients. Arthritis Rheum. 2001;44:666–675.
221 Nakamura Y, Yashiro M, Uehara R, et al. Epidemiologic features of Kawasaki disease in Japan: results of the 2007–2008 nationwide survey. J Epidemiol. 2010;20:302–307.
222 Gedalia A, Cuchacovich R. Systemic vasculitis in childhood. Curr Rheumatol Rep. 2009;11:402–409.
223 Gedalia A. Kawasaki disease: an update. Curr Rheumatol Rep. 2002;4:25–29.
224 Ohno S, Miyajima T, Higuchi M, et al. Ocular manifestations of Kawasaki’s disease (mucocutaneous lymph node syndrome). Am J Ophthalmol. 1982;93:713–717.
225 Farvardin M, Kashef S, Aleyasin S, et al. Sudden unilateral blindness in a girl with Kawasaki disease. J Pediatr Ophthalmol Strabismus. 2007;44:303–304.
226 Ogata S, Bando Y, Kimura S, et al. The strategy of immune globulin resistant Kawasaki disease: a comparative study of additional immune globulin and steroid pulse therapy. J Cardiol. 2009;53:15–19.
227 Imagawa T, Mori M, Miyamae T, et al. Plasma exchange for refractory Kawasaki disease. Eur J Pediatr. 2004;163:263–264.
228 Sundel RP, Burns JC, Baker A, et al. Gamma globulin re-treatment in Kawasaki disease. J Pediatr. 1993;123:657–659.
229 Sundel RP, Baker AL, Fulton DR, et al. Corticosteroids in the initial treatment of Kawasaki disease: report of a randomized trial. J Pediatr. 2003;142:611–616.
230 Schilder AM. Wegener’s granulomatosis vasculitis and granuloma. Autoimmun Rev. 2010;9:483–487.
231 Ntatsaki E, Watts RA, Scott DG. Epidemiology of ANCA-associated vasculitis. Rheum Dis Clin North Am. 2010;36:447–461.
232 Mahr AD, Neogi T, Merkel PA. Epidemiology of Wegener’s granulomatosis: lessons from descriptive studies and analyses of genetic and environmental risk determinants. Clin Exp Rheumatol. 2006;24:S82–S91.
233 Rao NV, Wehner NG, Marshall BC, et al. Characterization of proteinase-3 PR-3, a neutrophil serine proteinase. Structural and functional properties. J Biol Chem. 1991;266:9540–9548.
234 Talar-Williams C, Sneller MC, Langford CA, et al. Orbital socket contracture: a complication of inflammatory orbital disease in patients with Wegener’s granulomatosis. Br J Ophthalmol. 2005;89:493–497.
235 Robinson MR, Lee SS, Sneller MC, et al. Tarsal-conjunctival disease associated with Wegener’s granulomatosis. Ophthalmology. 2003;110:1770–1780.
236 Tanihara H, Nakayama Y, Honda Y. Wegener’s granulomatosis with rapidly progressive retinitis and anterior uveitis. Acta Ophthalmol (Copenh). 1993;71:853–855.
237 Huong du LT, Tran TH, Piette JC. Granulomatous uveitis revealing Wegener’s granulomatosis. J Rheumatol. 2006;33:1209–1210.
238 Watkins AS, Kempen JH, Choi D, et al. Ocular disease in patients with ANCA-positive vasculitis. J Ocul Biol Dis Infor. 2009;3:12–19.
239 Lapraik C, Watts R, Bacon P, et al. BSR and BHPR guidelines for the management of adults with ANCA associated vasculitis. Rheumatology (Oxford). 2007;46:1615–1616.
240 Booth A, Harper L, Hammad T, et al. Prospective study of TNFalpha blockade with infliximab in anti-neutrophil cytoplasmic antibody-associated systemic vasculitis. J Am Soc Nephrol. 2004;15:717–721.
241 Taylor SR, Salama AD, Joshi L, et al. Rituximab is effective in the treatment of refractory ophthalmic Wegener’s granulomatosis. Arthritis Rheum. 2009;60:1540–1547.
242 Guillevin L, Durand-Gasselin B, Cevallos R, et al. Microscopic polyangiitis: clinical and laboratory findings in eighty-five patients. Arthritis Rheum. 1999;42:421–430.
243 Lhote F, Cohen P, Genereau T, et al. Microscopic polyangiitis: clinical aspects and treatment. Ann Med Interne (Paris). 1996;147:165–177.
244 Hara A, Ohta S, Takata M, et al. Microscopic polyangiitis with ocular manifestations as the initial presenting sign. Am J Med Sci. 2007;334:308–310.
245 Gallagher MJ, Ooi KG, Thomas M, et al. ANCA associated pauci-immune retinal vasculitis. Br J Ophthalmol. 2005;89:608–611.
246 Walsh M, Jayne D. Rituximab in the treatment of anti-neutrophil cytoplasm antibody associated vasculitis and systemic lupus erythematosus: past, present and future. Kidney Int. 2007;72:676–682.
247 Noth I, Strek ME, Leff AR. Churg–Strauss syndrome. Lancet. 2003;361:587–594.
248 Guillevin L, Cohen P, Gayraud M, et al. Churg–Strauss syndrome. Clinical study and long-term follow-up of 96 patients. Medicine (Baltimore). 1999;78:26–37.
249 Masi AT, Hunder GG, Lie JT, et al. The American College of Rheumatology 1990 criteria for the classification of Churg–Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum. 1990;33:1094–1100.
250 Margolis R, Kosmorsky GS, Lowder CY, et al. Conjunctival involvement in Churg–Strauss syndrome. Ocul Immunol Inflamm. 2007;15:113–115.
251 Takanashi T, Uchida S, Arita M, et al. Orbital inflammatory pseudotumor and ischemic vasculitis in Churg–Strauss syndrome: report of two cases and review of the literature. Ophthalmology. 2001;108:1129–1133.
252 Weinstein JM, Chui H, Lane S, et al. Churg–Strauss syndrome (allergic granulomatous angiitis). Neuro-ophthalmologic manifestations. Arch Ophthalmol. 1983;101:1217–1220.
253 Pepper RJ, Fabre MA, Pavesio C, et al. Rituximab is effective in the treatment of refractory Churg–Strauss syndrome and is associated with diminished T-cell interleukin-5 production. Rheumatology (Oxford). 2008;47:1104–1105.
254 West SK, Valmadrid CT. Epidemiology of risk factors for age-related cataract. Surv Ophthalmol. 1995;39:323–334.
255 Clark AF, Wordinger RJ. The role of steroids in outflow resistance. Exp Eye Res. 2009;88:752–759.
256 Hobbs HE, Sorsby A, Freedman A. Retinopathy following chloroquine therapy. Lancet. 1959;2:478–480.
257 Lee AG. Hydroxychloroquine screening. Br J Ophthalmol. 2005;89:521–522.
258 Marmor MF, Kellner U, Lai TY, et al. Revised recommendations on screening for chloroquine and hydroxychloroquine retinopathy. Ophthalmology. 2011;118:415–422.