[level-membership-for-opthalmology-category]10
Retina and Choroid
Branch Retinal Artery Occlusion
Central Retinal Artery Occlusion
Central / Hemiretinal Vein Occlusion
Coats’ Disease / Leber’s Miliary Aneurysms
Familial Exudative Vitreoretinopathy and Norrie’s Disease
Retinopathies Associated with Blood Abnormalities
Acquired Retinal Arterial Macroaneurysm
Age-Related Macular Degeneration
Retinal Angiomatous Proliferation
Polypoidal Choroidal Vasculopathy
Myopic Degeneration / Pathologic Myopia
Central Serous Chorioretinopathy
Vitreomacular Adhesion and Traction
Epiretinal Membrane / Macular Pucker
Peripheral Retinal Degenerations
Proliferative Vitreoretinopathy
Intermediate Uveitis / Pars Planitis
Posterior Uveitis: White Dot Syndromes
Posterior Uveitis: Other Inflammatory Disorders
Posterior Uveitis: Evaluation / Management
Hereditary Chorioretinal Dystrophies
Hereditary Macular Dystrophies
Trauma
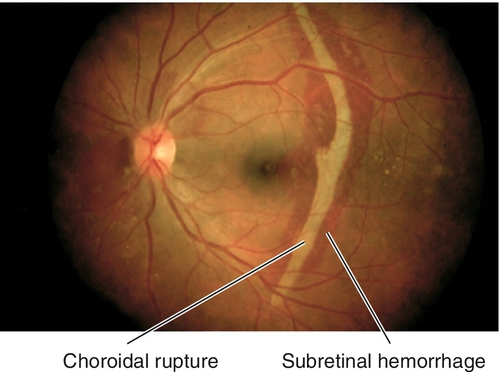
Choroidal Rupture
Tear in choroid, Bruch’s membrane, and retinal pigment epithelium (RPE) is usually seen after blunt trauma. Acutely, the rupture site may be obscured by hemorrhage; scars form over 3–4 weeks with RPE hyperplasia at the margin of the rupture site. Anterior ruptures are usually parallel to the ora serrata; posterior ruptures are usually crescent-shaped and concentric to the optic nerve. Patients may have decreased vision if commotio retinae or subretinal hemorrhage is present, or if the rupture is located in the macula; increased risk of developing a choroidal neovascular membrane (CNV) during the healing process (months to years after trauma). Good prognosis if the macula is not involved, but poor if the fovea is involved.
• No treatment recommended, unless CNV occurs.
• Laser photocoagulation of juxtafoveal and extrafoveal CNV; consider anti-VEGF agent for subfoveal CNV (experimental).
• Monitor for CNV with Amsler grid.
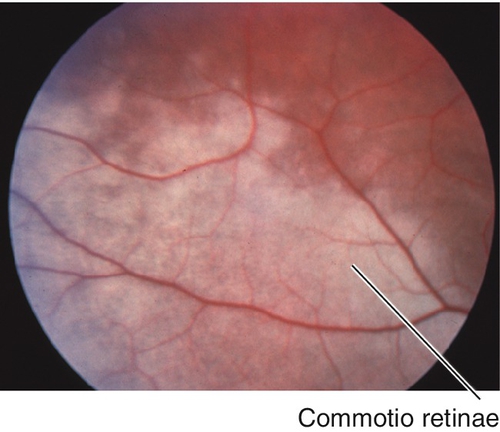
Commotio Retinae (Berlin’s Edema)
Gray-white discoloration of the outer retina due to photoreceptor outer segment disruption following blunt eye trauma; can affect any area of the retina and may be accompanied by hemorrhages or choroidal rupture. There is no intercellular edema; whitening is due to intracellular edema and disorganization of outer retinal layers. It is termed Berlin’s edema if involving the macula, and commotion retinae in all other areas. Can cause acute decrease in vision if located within the macula, which resolves as the retinal discoloration disappears; may cause permanent loss of vision if the fovea is damaged, but usually resolves without sequelae. Visual acuity does not always correlate with the degree of retinal whitening seen on exam. Occasionally, a macular hole can form in the area of commotio with variable prognosis.
• Fluorescein angiogram: Early blocked fluorescence in the areas of commotio retinae.
• No treatment recommended.
Purtscher’s Retinopathy
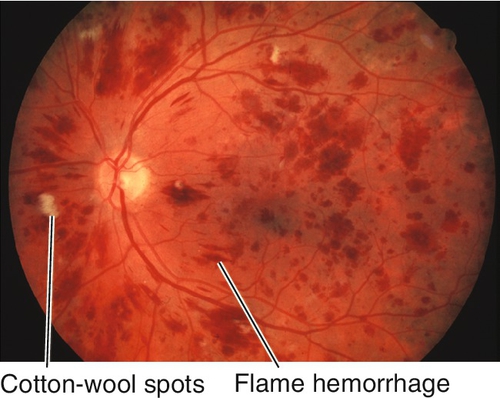
Multiple patches of retinal whitening, large cotton-wool spots, and hemorrhages that surround the optic disc following multiple long-bone fractures with fat emboli or severe compressive injuries to the chest or head. May have optic disc edema and a relative afferent pupillary defect (RAPD). Usually resolves over weeks to months.
In the absence of trauma, a Purtscher’s-like retinopathy may be associated with acute pancreatitis, collagen–vascular disease, leukemia, dermatomyositis, and amniotic fluid embolus.
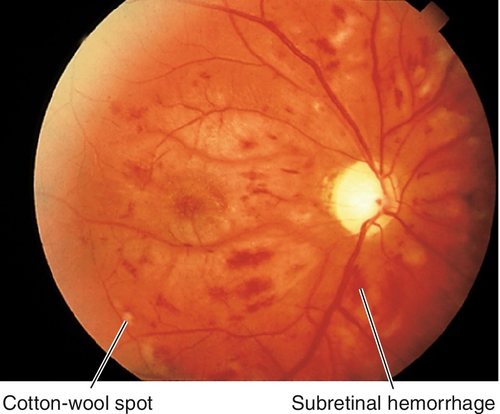
Figure 10-4 Multiple patches of retinal whitening, cotton-wool spots, and intraretinal hemorrhages secondary to Purtscher’s retinopathy.
• Fluorescein angiogram: Leakage from retinal vasculature with late venous staining.
• No treatment recommended.
Traumatic Retinal Holes
Full-thickness tear in the retina, often horseshoe shaped; usually occurs along the vitreous base, posterior border of lattice degeneration, or at cystic retinal tufts (areas with strong vitreoretinal adhesions). As most patients are young, the formed vitreous tamponades the tear and prevents a retinal detachment. Associated with pigmented vitreous cells (“tobacco-dust”, also known as Schaffer’s sign), vitreous hemorrhages, operculum (often located over the retinal hole), and posterior vitreous detachment. Patients usually report photopsias and floaters that shift with eye movement. Liquefied vitreous can pass through the tear into the subretinal space, causing retinal detachment even months to years after the tear forms; chronic tears have a ring of pigment around the retinal hole.
Giant Retinal Tear
Traumatic retinal hole measuring > 90° in circumferential extent or > 3 clock hours.
Avulsion of Vitreous Base
Separation of vitreous base from ora serrata that is pathognomonic for trauma.
Oral Tear
Tear at the ora serrata due to split of vitreous that has a fish-mouth appearance.
Preoral Tear
Tear at anterior border of vitreous base most often occurs superotemporally.
Retinal Dialysis
Most common form after trauma; circumferential separation of the retina at the ora serrata, usually in superotemporal (22%) or inferotemporal (31%) quadrant. Risk of retinal detachment increases over time with 10% at initial examination and 80% by 2 years.
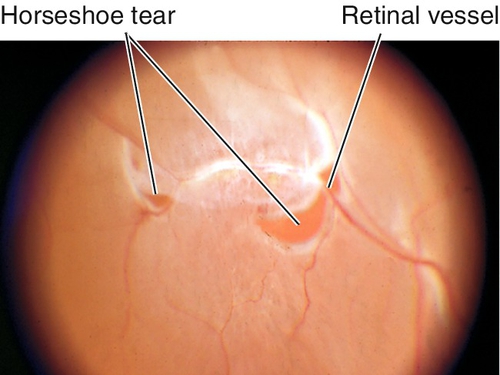
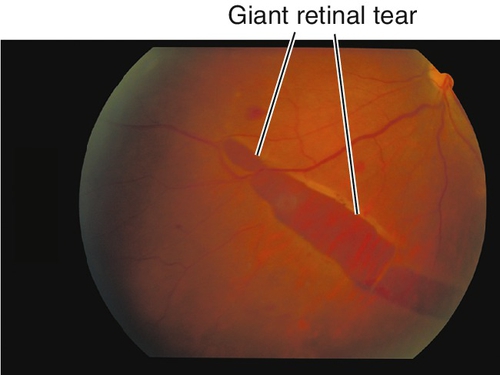
• Retinal surgery required if rhegmatogenous retinal detachment, retinal dialysis, avulsion of the vitreous base, or giant retinal tear exists; should be performed by a retina specialist.
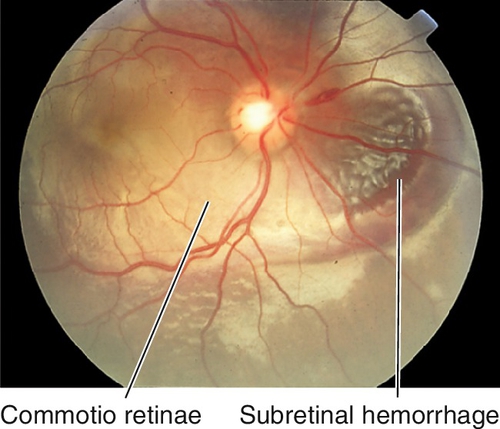
Chorioretinitis Sclopeteria
Trauma to retina and choroid caused by transmitted shock waves from high-velocity projectile that causes choroidal rupture, retinal hemorrhages, and commotio retinae. Vitreous hemorrhage is common. Lesions heal with white fibrous scar and RPE changes. Low risk of retinal detachment in young patients with a formed vitreous; however, the appearance can simulate retinal detachment in these patients.
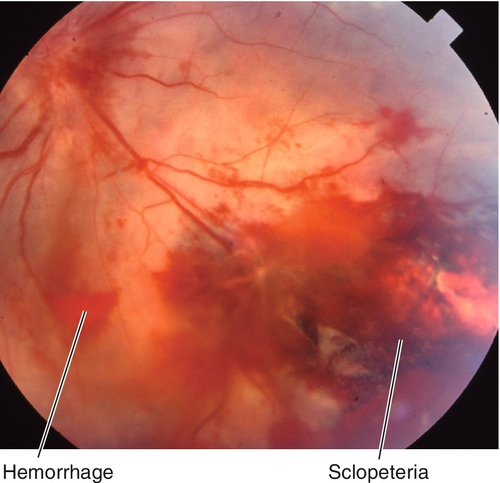
Figure 10-7 Chorioretinitis sclopeteria with subretinal hemorrhage and commotio retinae (same patient as Figure 1-6).
Hemorrhages
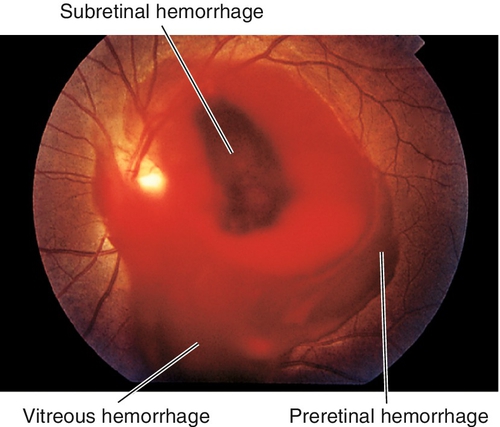
Preretinal Hemorrhage
Hemorrhage located between the retina and posterior vitreous face (subhyaloid) or under the internal limiting membrane of the retina (sub-ILM). Often amorphous or boat-shaped, with flat upper border and curved lower border, which obscures the underlying retina. Caused by trauma, retinal neovascularization (diabetic retinopathy, radiation retinopathy, breakthrough bleeding from a choroidal neovascular membrane), hypertensive retinopathy, Valsalva retinopathy, retinal artery macroaneurysm, posterior vitreous detachment, shaken-baby syndrome, or retinal breaks, and less frequently by vascular occlusion, retinopathy of blood disorders, or leukemia.
Intraretinal Hemorrhage
Bilateral intraretinal hemorrhages are associated with systemic disorders (e.g., diabetes mellitus and hypertension); unilateral intraretinal hemorrhages generally occur in venous occlusive diseases or ocular ischemic syndrome.
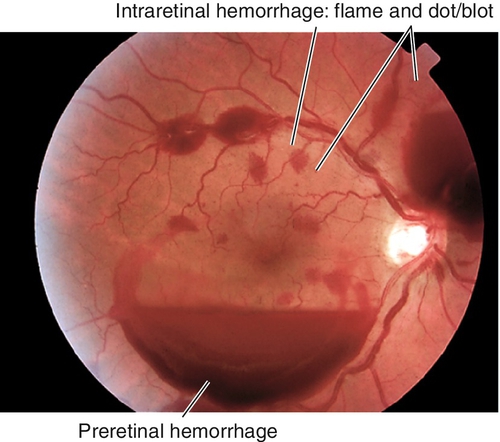
Flame-Shaped Hemorrhage
Located in the superficial retina oriented with the nerve fiber layer; feathery borders. Usually occurs in hypertensive retinopathy and vein occlusion; may be peripapillary in glaucoma, especially in normal-tension glaucoma (splinter hemorrhage) and disc edema.
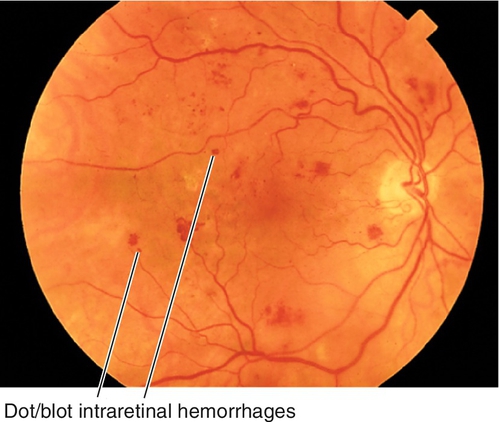
Dot / Blot Hemorrhage
Located in the outer plexiform layer, confined by the anteroposterior orientation of the photoreceptor, bipolar, and Müller’s cells; round dots or larger blots. Usually occurs in diabetic retinopathy.
Roth Spot
Hemorrhage with white center that represents an embolus with lymphocytic infiltration. Classically associated with subacute bacterial endocarditis (occurs in 1–5% of such patients); also occurs in leukemia, severe anemia, sickle cell disease, collagen vascular diseases, diabetes mellitus, multiple myeloma, and acquired immunodeficiency syndrome (AIDS) (see Figures 10-40, 10-43).
Subretinal Hemorrhage
Amorphous hemorrhage located under the neurosensory retina or RPE; appears dark and is deep to the retinal vessels. Associated with trauma, subretinal and choroidal neovascular membranes, and macroaneurysms (see Figure 10-71).
All three types of hemorrhages may occur together in several disorders including age-related macular degeneration (AMD), acquired retinal arterial macroaneurysm, Eales’ disease, and capillary hemangioma.
Cotton-Wool Spot
Asymptomatic, yellow-white, fluffy lesions in the superficial retina (see Figure 10-4). Nonspecific finding due to multiple etiologies including: retinal ischemia (retinal vascular occlusions, severe anemia, ocular ischemic syndrome), emboli (Purtcher’s retinopathy [white blood cell emboli], intravenous drug abuse [talc], cardiac/carotid emboli, deep venous emboli), infections (acquired immunodeficiency syndrome, Rocky Mountain spotted fever, cat-scratch fever [Bartonella henselae], leptospirosis, onchocerciasis, bacteremia, fungemia), collagen vascular diseases (systemic lupus erythematosus, dermatomyositis, polyarteritis nordosa, scleroderma, giant cell arteritis), drugs (interferon, chemotherapeutic agents), neoplasms (lymphoma, leukemia, metastatic carcinoma, multiple myeloma), retinal traction (epiretinal membrane), trauma (nerve fiber layer laceration, long-bone fractures, severe chest compression [white blood cell emboli]), systemic diseases (acute pancreatitis, hypertension, diabetes mellitus, high-altitude retinopathy), and radiation. Appears as thickening of the nerve fiber layer on OCT. Thought to develop secondary to obstruction of a retinal arteriole with resultant ischemia leading to blockage of axoplasmic flow within the nerve fiber layer.
• Treat underlying etiology (identified in 95% of cases).
Branch Retinal Artery Occlusion
Definition
Disruption of the vascular perfusion in a branch of the central retinal artery, leading to focal retinal ischemia.
Etiology
Mainly due to embolism from cholesterol (Hollenhorst’s plaques), calcifications (heart valves), platelet–fibrin plugs (ulcerated atheromatous plaques due to arteriosclerosis); rarely due to leukoemboli (vasculitis, Purtcher’s retinopathy), fat emboli (long-bone fractures), amniotic fluid emboli, tumor emboli (atrial myxoma), or septic emboli (heart valve vegetations in bacterial endocarditis or IV drug abuse). The site of the obstruction is usually at the bifurcation of retinal arteries. May result from vasospasm (migraine), compression, or coagulopathies.
Epidemiology
Usually occurs in elderly patients (seventh decade); associated with hypertension (67%), carotid occlusive disease (25%), diabetes mellitus (33%), and cardiac valvular disease (25%). CRAO is more common (57%) than BRAO (38%) or cilioretinal artery occlusion (5%) (in 32% of eyes, a cilioretinal artery is present).
Symptoms
Sudden, unilateral, painless, partial loss of vision, with a visual field defect corresponding to the location of the occlusion. May have history of amaurosis fugax (fleeting episodes of visual loss), prior cerebrovascular accident (CVA), or transient ischemic attacks (TIAs).
Signs
Visual field defect with normal or decreased visual acuity; focal, wedge-shaped area of retinal whitening within the distribution of a branch arteriole; 90% involve temporal retinal vessels; emboli (visible in 62% of cases) or Hollenhorst’s plaques may be visible at retinal vessel bifurcations. Retinal whitening resolves over several weeks and visual acuity can improve. In chronic stages, arterial attenuation with sector nerve fiber layer loss may be seen; artery-to-artery collaterals may form and are pathognomonic.
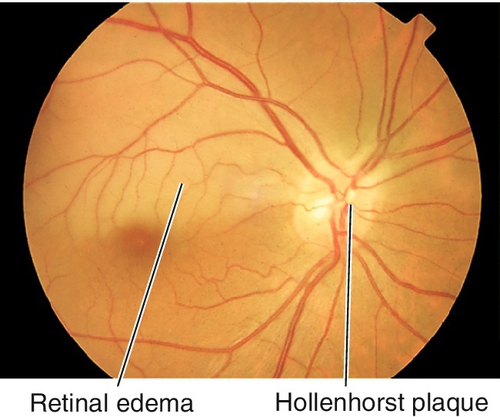
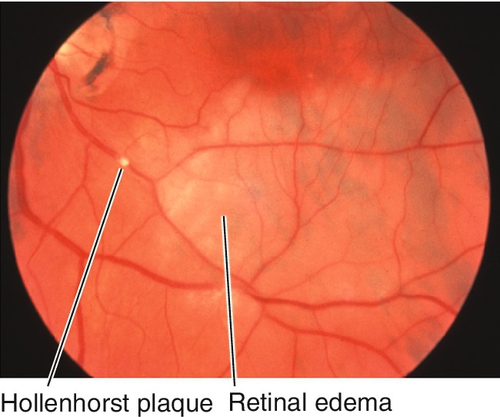
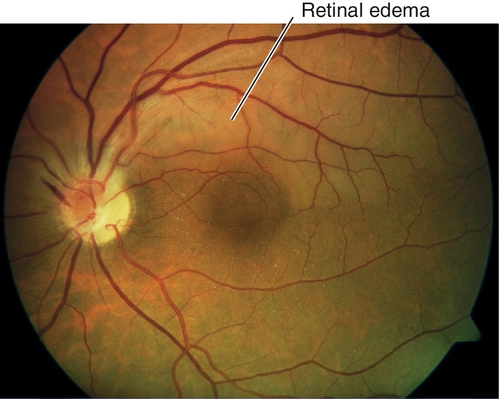
Figure 10-13 Superior branch artery occlusion demonstrating retinal edema.
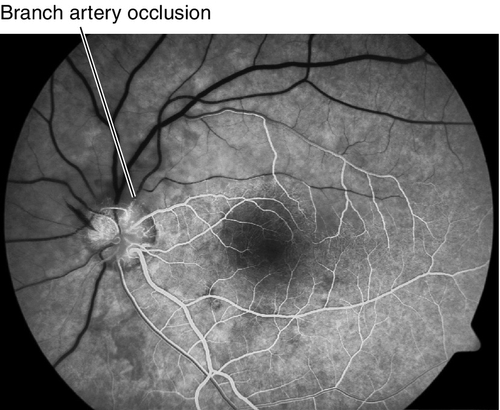
Figure 10-14 Fluorescein angiogram of same patient as Figure 10-13 demonstrating no filling of superior retinal vessels and delayed filling of affected veins.
Differential Diagnosis
Commotio retinae, branch retinal vein occlusion, CRAO with cilioretinal artery sparing, combined artery and vein occlusion.
Evaluation
• Check blood pressure.
• Lab tests: Fasting blood glucose (FBS), glycosylated hemoglobin, and complete blood count (CBC) with differential. Consider platelets, prothrombin time/partial thromboplastin time (PT / PTT), protein C, protein S, factor V Leiden mutation, antithrombin III, homocysteine level, antinuclear antibody (ANA), rheumatoid factor (RF), sickle cell disease, antiphospholipid antibody, serum protein electrophoresis, hemoglobin electrophoresis, Venereal Disease Research Laboratory (VDRL) test, and fluorescent treponemal antibody absorption (FTA-ABS) test in patients < 50 years of age. In patients > 50 years old, check erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) to rule out arteritic ischemic optic neuropathy due to giant cell arteritis. If positive and / or if the patient’s history and exam are consistent, start giant cell arteritis treatment immediately (see Chapter 11). If the BRAO is accompanied by optic nerve edema and / or retinitis, consider serologic testing for infectious etiologies such as Bartonella, Lyme, and toxoplasmosis.
• Fluorescein angiogram: Delayed or absent retinal arterial filling in a branch of the central retinal artery; delayed arteriovenous transit time; capillary nonperfusion in wedge-shaped area supplied by the branch artery; staining of occlusion site and vessel wall in late views. When occlusion dissolutes, retinal blood flow is usually restored.
• Optical coherence tomography (OCT): Thickened and hyperreflective inner retinal layers during acute occlusion that corresponds to intracellular edema. Reflectivity of outer retina is blocked. Later, the retina is thinned with atrophy of the inner retina.
• Consider B-scan ultrasonography or orbital computed tomography (CT) scan to rule out a compressive lesion if the history suggests this etiology.
• Medical consultation for complete cardiovascular evaluation including baseline electrocardiogram, echocardiogram (may require transesophageal echocardiogram to rule out valvular disease), and carotid Doppler ultrasonography.
• In patients < 50 years of age, a hypercoagulability evaluation should be considered.
Prognosis
Retinal pallor fades and circulation is restored over several weeks. Good if fovea is spared; 80% have ≥ 20 / 40 vision, but most have some degree of permanent visual field loss; 10% risk in fellow eye.
Central Retinal Artery Occlusion
Definition
Disruption of the vascular perfusion in the central retinal artery (CRAO) leading to global retinal ischemia.
Etiology
Due to emboli (only visible in 20–40% of cases) or thrombus at the level of the lamina cribosa; other etiologies are the same as for BRAO including temporal arteritis, leukoemboli in collagen vascular diseases, fat emboli, trauma (through compression, spasm, or direct vessel damage), hypercoagulation disorders, syphilis, sickle cell disease, amniotic fluid emboli, mitral valve prolapse, particles (talc) from IV drug abuse, and compressive lesions; associated with optic disc drusen, papilledema, prepapillary arterial loops, and primary open-angle glaucoma.
Epidemiology
Usually occurs in elderly patients; associated with hypertension (67%), carotid occlusive disease (25%), diabetes mellitus (33%), and cardiac valvular disease (25%). CRAO is more common (57%) than BRAO (38%) or cilioretinal artery occlusion (5%) (in 32% of eyes, a cilioretinal artery is present); rarely bilateral.
Symptoms
Sudden, unilateral, painless, profound loss of vision; may have history of amaurosis fugax (fleeting episodes of visual loss), prior CVA, or TIAs.
Signs
Decreased visual acuity in the count fingers (CF) to light perception (LP) range; RAPD may be present; diffuse retinal whitening and arteriole constriction with segmentation (boxcaring) of blood flow; visible emboli (20–40%) rarely occur in central retinal artery; cherry-red spot in the macula (thin fovea allows visualization of the underlying choroidal circulation). In ciliary retinal artery sparing CRAO (25%), a small wedge-shaped area of perfused retina may be present temporal to the optic disc (10% spare the foveola, in which case visual acuity improves to 20 / 50 or better in 80%). Note: Ophthalmic artery obstruction usually does not produce a cherry-red spot owing to underlying choroidal ischemia.
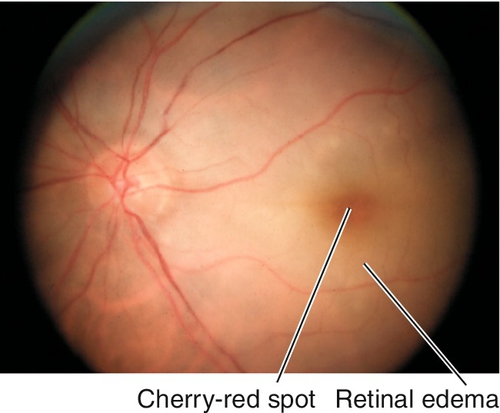
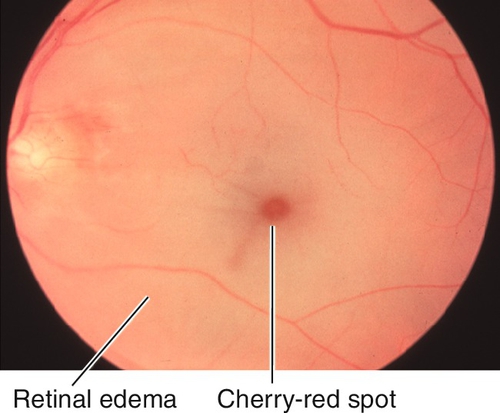
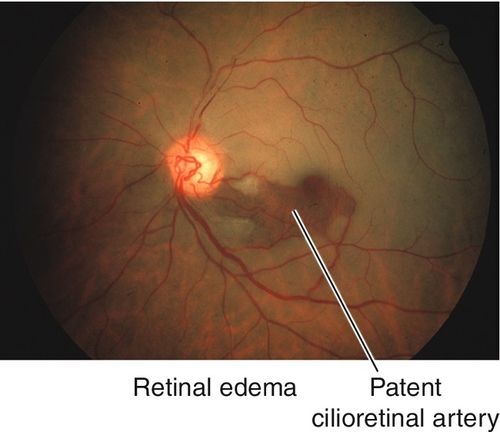
Figure 10-17 Cilioretinal artery sparing central retinal artery occlusion with patent cilioretinal artery allowing perfusion (thus no edema) in a small section of the macula.
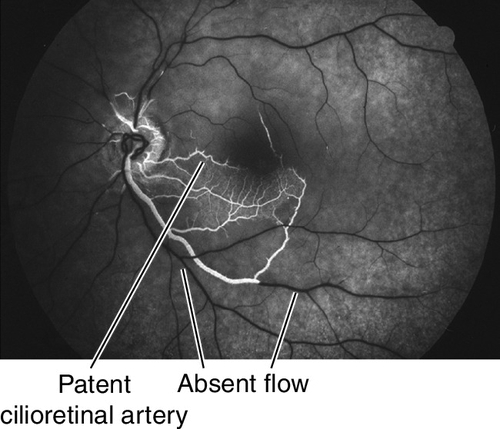
Figure 10-18 Fluorescein angiogram of same patient in Figure 10-17 demonstrating no filling of retinal vessels except in cilioretinal artery and surrounding branches.
Differential Diagnosis
Ophthalmic artery occlusion, commotio retinae, cherry-red spot due to inherited metabolic or lysosomal storage diseases, methanol toxicity.
Evaluation
• Check blood pressure.
• Lab tests: Fasting blood glucose (FBS), glycosylated hemoglobin, and complete blood count (CBC) with differential. Consider platelets, prothrombin time/partial thromboplastin time (PT / PTT), protein C, Protein S, factor V Leiden mutation, antithrombin III, homocysteine level, antinuclear antibody (ANA), rheumatoid factor (RF), sickle cell disease, antiphospholipid antibody, serum protein electrophoresis, hemoglobin electrophoresis, Venereal Disease Research Laboratory (VDRL) test, and fluorescent treponemal antibody absorption (FTA-ABS) test in patients < 50 years of age. In patients > 50 years old, check erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) to rule out arteritic ischemic optic neuropathy due to giant cell arteritis. If positive and /or if the patient’s history and exam are consistent, start giant cell arteritis treatment immediately (see Chapter 11). If the CRAO is accompanied by optic nerve edema and /or retinitis consider serologic testing for infectious etiologies such as Bartonella, Lyme disease, and toxoplasmosis.
• Fluorescein angiogram: Delayed retinal arterial filling and arteriovenous transit time with normal choroidal filling and perfusion of optic nerve from ciliary branches; prolonged arteriovenous circulation times; extensive capillary nonperfusion.
• Optical coherence tomography: Thickened and hyperreflective inner retinal layers during acute occlusion that corresponds to intracellular edema. Reflectivity of outer retina is blocked. Later, the retina is thinned with atrophy of the inner retina.
• Electrophysiologic testing: ERG (reduced b-wave amplitude, normal a-wave).
• Consider B-scan ultrasonography or orbital CT scan to rule out compressive lesion if history suggests compression.
• Medical consultation for complete cardiovascular evaluation including electrocardiogram, echocardiogram (may require transesophageal echocardiogram to rule out valvular disease), and carotid Doppler ultrasound.
Management
• Treat immediately before starting workup (if patient presents within 24 hours of visual loss), but best hope is to treat within 90 minutes.
• Digital ocular massage to try to dislodge emboli.
• Systemic acetazolamide (Diamox 500 mg IV or po).
• Topical ocular hypotensive drops: β-blocker (timolol 0.5% 1 gtt q15min × 2, repeat as necessary).
• Anterior chamber paracentesis (immediately lowers IOP to 0 mmHg): This procedure is easily performed at the slit lamp after prepping the eye with topical anesthetic, broad-spectrum antibiotic, and povidone-iodine. A lid speculum is placed, the eye is grasped with forceps at the nasal limbus to prevent movement and provide counter-traction, and either a disposable microsurgical knife (15° or MVR blade) or else a 30-gauge  inch (13 mm) needle on a 1 mL syringe without the plunger, is inserted parallel to the iris through the peripheral cornea at the temporal limbus. If necessary, gentle pressure can be applied to the posterior lip of the paracentesis site so that aqueous can be released in a controlled fashion. Treat with a topical broad-spectrum antibiotic (gatifloxacin [Zymaxid] or moxifloxacin [Vigamox] qid for 3 days).
inch (13 mm) needle on a 1 mL syringe without the plunger, is inserted parallel to the iris through the peripheral cornea at the temporal limbus. If necessary, gentle pressure can be applied to the posterior lip of the paracentesis site so that aqueous can be released in a controlled fashion. Treat with a topical broad-spectrum antibiotic (gatifloxacin [Zymaxid] or moxifloxacin [Vigamox] qid for 3 days).
• Consider admission to hospital for carbogen treatment (95% oxygen–5% carbon dioxide for 10 minutes q2h for 24–48 hours) to attempt to increase oxygenation and induce vasodilation.
• Unproven treatments include hyperbaric oxygen, antifibrinolytic drugs, retrobulbar vasodilators, sublingual nitroglycerine, and Nd : YAG laser to dislodge the emboli.
• If arteritic anterior ischemic optic neuropathy (see Chapter 11) is suspected: Systemic steroids (methylprednisolone 1 g IV qd in divided doses for 3 days, then prednisone at least 1 mg / kg po qd for at least a month with a very slow taper; decrease by no more than 2.5 mg /wk). Most patients will require a year of high-dose steroid treatment.
Prognosis
Retinal pallor fades and circulation is restored over several weeks. Poor prognosis; most have persistent severe visual loss with constricted retinal arterioles and optic atrophy. Rubeosis (20%) and disc /retinal neovascularization (2–3%) can rarely occur. Presence of visible embolus associated with increased mortality; most common cause of mortality is myocardial infarction.
Ophthalmic Artery Occlusion
Definition
Obstruction at the level of the ophthalmic artery that affects both the retinal and choroidal circulation leading to ischemia more severe than CRAO.
Etiology
Usually due to emboli or thrombus, but can be caused by any of the etiologies listed for CRAO.
Epidemiology
Usually occurs in elderly patients; associated with hypertension (67%), carotid occlusive disease (25%), diabetes mellitus (33%), and cardiac valvular disease (25%).
Symptoms
Sudden, unilateral, painless, profound loss of vision up to the level of light perception or even no light perception.
Signs
Marked constriction of the retinal vessels, marked retinal edema often without a cherry red spot (although it may be present); may have RAPD; later, optic atrophy, retinal vascular sclerosis, and diffuse pigmentary changes.
Differential Diagnosis
Central retinal artery occlusion, commotio retinae, cherry-red spot due to inherited metabolic or lysosomal storage diseases, methanol toxicity.
Evaluation
• Check blood pressure.
• Lab tests: Fasting blood glucose (FBS), glycosylated hemoglobin, and complete blood count (CBC) with differential. Consider platelets, prothrombin time/partial thromboplastin time (PT / PTT), protein C, Protein S, factor V Leiden mutation, antithrombin III, homocysteine level, antinuclear antibody (ANA), rheumatoid factor (RF), sickle cell disease, antiphospholipid antibody, serum protein electrophoresis, hemoglobin electrophoresis, Venereal Disease Research Laboratory (VDRL) test, and fluorescent treponemal antibody absorption (FTA-ABS) test in patients < 50 years of age. In patients > 50 years old, check erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) to rule out arteritic ischemic optic neuropathy due to giant cell arteritis. If positive and / or if the patient’s history and exam are consistent, start giant cell arteritis treatment immediately (see Chapter 11).
• Fluorescein angiogram: Delayed or absent choroidal and retinal vascular filling, extensive capillary nonperfusion.
• Electrophysiologic testing: ERG (reduced or absent a and b wave amplitudes).
• Medical consultation for complete cardiovascular evaluation including electrocardiogram, echocardiogram (may require transthoracic echocardiogram to rule out valvular disease), and carotid Doppler ultrasound.
Prognosis
Severe visual loss is usually permanent.
Branch Retinal Vein Occlusion
Definition
Occlusion of a branch retinal vein (BRVO). Two types:
Nonischemic (64%)
< 5 disc areas of capillary nonperfusion on fluorescein angiogram.
Ischemic
≥ 5 disc areas of capillary nonperfusion on fluorescein angiogram.
Etiology
Usually caused by a thrombus at arteriovenous crossings where a thickened artery compresses the underlying venous wall due to a common vascular sheath; associated with hypertension, coronary artery disease, diabetes mellitus, and peripheral vascular disease; rarely associated with hypercoagulable states (e.g., macroglobulinemia, cryoglobulinemia), hyperviscosity states (polycythemia vera, Waldenström’s macroglobulinemia), systemic lupus erythematosus, syphilis, sarcoid, homocystinuria, malignancies (e.g., multiple myeloma, polycythemia vera, leukemia), optic nerve drusen, and external compression. In younger patients, associated with oral contraceptive pills, collagen vascular disease, AIDS, protein S /protein C /antithrombin III deficiency, factor XII (Hageman factor) deficiency, antiphospholipid antibody syndrome, or activated protein-C resistance (factor V Leiden PCR assay).
Epidemiology
Usually occurs in elderly patients, 60–70 years old; associated with hypertension (50–70%), cardiovascular disease, diabetes mellitus, increased body mass index, and open-angle glaucoma; slight male and hyperopic predilection. Second most common vascular disease after diabetic retinopathy.
Symptoms
Sudden, unilateral, painless, visual field loss. Patients may have normal vision, especially when macula is not involved.
Signs
Quadrantic visual field defect; dilated, tortuous retinal veins with superficial, retinal hemorrhages, and cotton-wool spots in a wedge-shaped area radiating from an arteriovenous crossing (usually arterial over-crossing where an arteriole and venule share a common vascular sheath). More common superotemporally (60%) than inferotemporally (40%; rare nasally since usually asymptomatic). The closer the obstruction is to the optic disc, the greater the area of retina involved and the more serious the complications. Microaneurysms or macroaneurysms, macular edema (50%), epiretinal membranes (20%), retinal and /or iris /angle neovascularization (very rare), and vitreous hemorrhage may develop; neovascular glaucoma is rare.
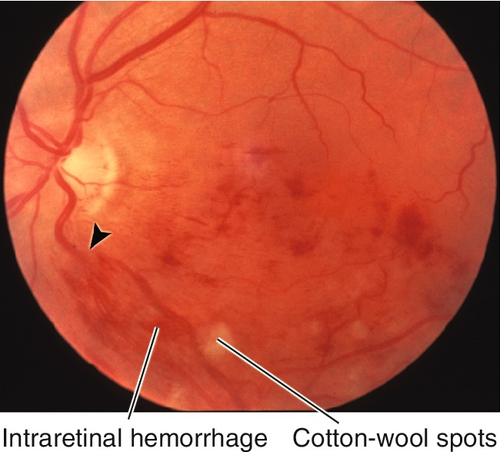
Figure 10-19 Inferior branch retinal vein occlusion demonstrating wedge-shaped area of intraretinal hemorrhages and cotton-wool spots.
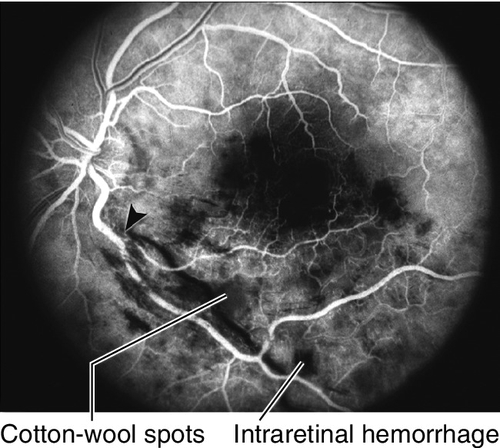
Figure 10-20 Fluorescein angiogram of same patient as Figure 10-19 demonstrating lack of perfusion in inferior retinal vein with blocking defects from the intraretinal hemorrhages. Site of occlusion is shown with an arrowhead.
Differential Diagnosis
Venous stasis retinopathy, ocular ischemic syndrome, hypertensive retinopathy, leukemic retinopathy, retinopathy of anemia, diabetic retinopathy, papilledema, papillophlebitis (in young patients).
Evaluation
• Check visual fields.
• Check blood pressure.
• Lab tests: Fasting blood glucose, glycosylated hemoglobin; consider CBC with differential, platelets, PT / PTT, ANA, RF, angiotensin converting enzyme (ACE), ESR, serum protein electrophoresis, lipid profile, hemoglobin electrophoresis (in African Americans), VDRL, and FTA-ABS depending on clinical situation. In a patient < 40 years old and in whom a hypercoagulable state is being considered: check human immunodeficiency virus (HIV) status, functional protein S assay, functional protein C assay, functional antithrombin III assay (type II heparin-binding mutation), antiphospholipid antibody titer, lupus anticoagulant, anticardiolipin antibody titer (IgG and IgM), homocysteine level (if elevated test for folate, B12, and creatinine), factor XII (Hageman factor) levels, and activated protein C resistance (factor V Leiden mutation PCR assay); if these tests are normal and clinical suspicion for a hypercoagulable state still exists: add plasminogen antigen assay, heparin cofactor II assay, thrombin time, reptilase time, and fibrinogen functional assay.
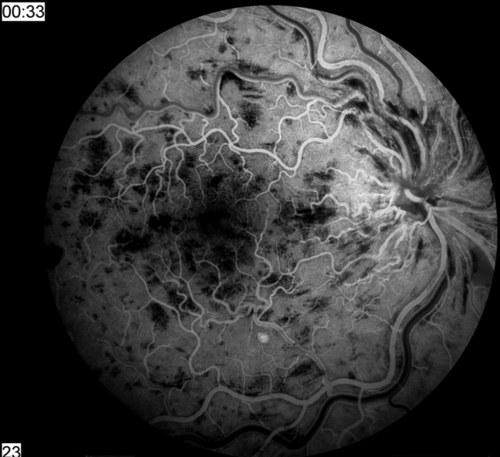
• Fluorescein angiogram: Delayed retinal venous filling in a branch of the central retinal vein, increased transit time in affected venous distribution, blocked fluorescence in areas of retinal hemorrhages, and capillary nonperfusion (ischemic defined as ≥ 5 disc areas of capillary nonperfusion) in the area supplied by the involved retinal vein. Retinal edema with cystic changes is not present acutely, but appears later. Wide-field angiography is being used increasingly to visualize peripheral nonperfusion.
• Optical coherence tomography: Monitor for cystic macular edema and intraretinal swelling. Useful to monitor treatment response.
• Medical consultation for complete cardiovascular evaluation.
Prognosis
Central / Hemiretinal Vein Occlusion
Definition
Occlusion of the central retinal vein (CRVO); hemiretinal occlusion (HRVO) occurs when the superior and inferior retinal drainage does not merge into a central retinal vein (20%) and is occluded (more like CRVO than BRVO). Two types:
Nonischemic / Perfused (67%)
< 10 disc areas of capillary nonperfusion on fluorescein angiogram.
Ischemic / Nonperfused
≥ 10 disc areas of capillary nonperfusion on fluorescein angiogram.
Etiology
Usually caused by a thrombus in the area of the lamina cribosa; associated with hypertension (60%), coronary artery disease, diabetes mellitus, peripheral vascular disease, and primary open-angle glaucoma (40%); rarely associated with hypercoagulable states (e.g., macroglobulinemia, cryoglobulinemia), hyperviscosity states especially in bilateral cases (polycythemia vera, Waldenström’s macroglobulinemia), systemic lupus erythematosus, syphilis, sarcoid, homocystinuria, malignancies (e.g., multiple myeloma, polycythemia vera, leukemia), optic nerve drusen, and external compression. In younger patients, associated with oral contraceptive pills, collagen vascular disease, acquired immunodeficiency syndrome (AIDS), protein S /protein C /antithrombin III deficiency, factor XII (Hageman factor) deficiency, antiphospholipid antibody syndrome, or activated protein C resistance (factor V Leiden polymerase chain reaction [PCR] assay).
Epidemiology
Usually occurs in elderly patients (90% are > 50 years old); slight male predilection. Ischemic disease is more common in older patients and those with cardiovascular disease. Younger patients can get inflammatory condition termed papillophlebitis or benign retinal vasculitis with benign clinical course.
Symptoms
Sudden, unilateral, loss of vision or less frequently history of transient obscuration of vision with complete recovery. Some report pain and present initially with neovascularization of the iris and neovascular glaucoma following a loss of vision 3 months earlier (“90-day glaucoma”). Patients may have normal vision if perfused, especially when the macula is not involved.
Signs
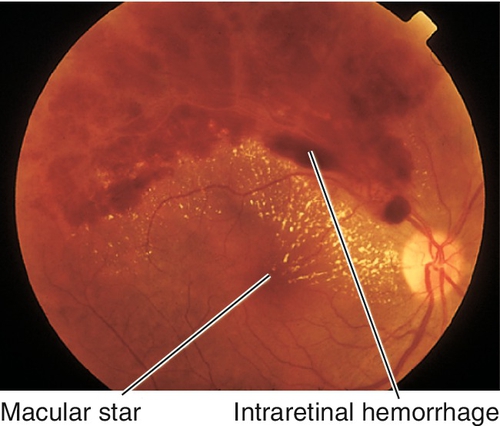
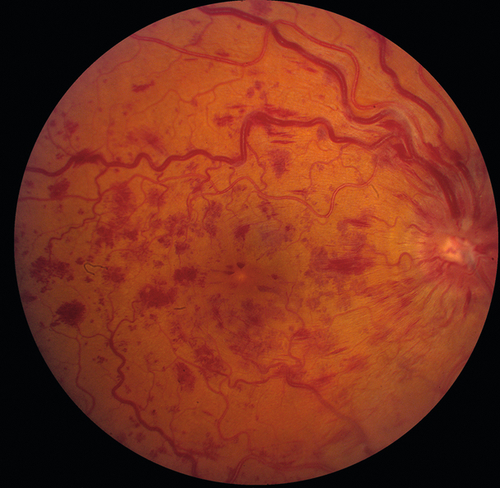
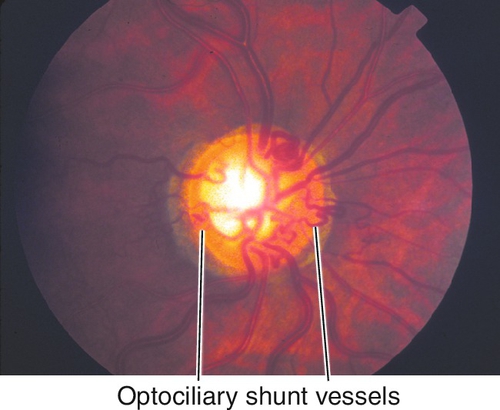
Decreased visual acuity ranging from 20 / 20 to hand motion (HM) with most worse than 20 / 200 (vision worse in ischemic type; usually > 20/ 200 in nonischemic); dilated, tortuous retinal veins with superficial, retinal hemorrhages, and cotton-wool spots in all four quadrants extending to periphery; optic disc hyperemia, disc edema, and macular edema common; RAPD (degree of defect correlates with amount of ischemia). Nonischemic disease rarely produces neovascularization; ischemic disease can produce rubeosis (20% in CRVO, rare in BRVO), disc/retinal neovascularization (border of perfused/nonperfused retina), neovascular glaucoma, and vitreous hemorrhages. Collateral optociliary shunt vessels between retinal and ciliary circulations (50%) occur late. Impending CRVO may have absence of spontaneous venous pulsations (but this can also occur in normal individuals). Transient patchy ischemic retinal whitening may occur early in nonischemic CRVO.
Differential Diagnosis
Venous stasis retinopathy, ocular ischemic syndrome, hypertensive retinopathy, leukemic retinopathy, retinopathy of anemia, diabetic retinopathy, radiation retinopathy, and papilledema.
Evaluation
• Check blood pressure.
• Lab tests: Fasting blood glucose, glycosylated hemoglobin; consider CBC with differential, platelets, PT / PTT, ANA, RF, ACE, ESR, serum protein electrophoresis, lipid profile, hemoglobin electrophoresis (in African American), VDRL, and FTA-ABS depending on clinical situation. In a patient < 40 years old and in whom a hypercoagulable state is being considered: check human immunodeficiency virus (HIV) status, functional protein S assay, functional protein C assay, functional antithrombin III assay (type II heparin-binding mutation), antiphospholipid antibody titer, lupus anticoagulant, anticardiolipin antibody titer (IgG and IgM), homocysteine level (if elevated test for folate, B12, and creatinine), factor XII (Hageman factor) levels, and activated protein C resistance (factor V Leiden mutation PCR assay); if these tests are normal and clinical suspicion for a hypercoagulable state still exists: add plasminogen antigen assay, heparin cofactor II assay, thrombin time, reptilase time, and fibrinogen functional assay.
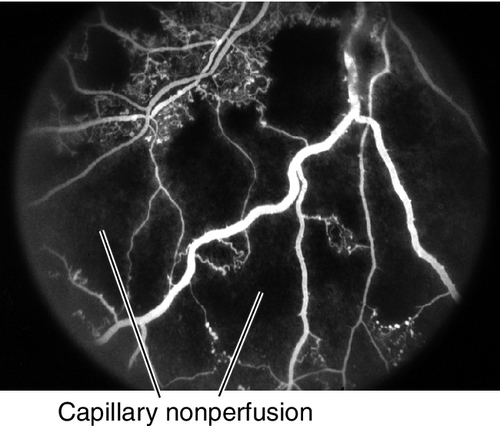
• Fluorescein angiogram: Delayed retinal venous filling, increased transit time (> 20 seconds increases risk of rubeosis), extensive capillary nonperfusion (ischemic defined in CVOS as ≥ 10 disc areas of capillary nonperfusion), staining of vascular walls, and blocking defects due to retinal hemorrhages. Retinal edema with cystic changes that are not present acutely, but appear later. Wide-field angiography is being used increasingly to visualize peripheral nonperfusion.
• Optical coherence tomography: monitor for cystic macular edema and intraretinal swelling. Useful to monitor treatment response.
• Electrophysiologic testing: ERG (reduced b wave amplitude [< 60% of normal more likely ischemic], reduced b : a-wave ratio [< 1 associated with increased risk of ischemia and neovascularization], prolonged b-wave implicit time).
• Medical consultation for complete cardiovascular evaluation.
Prognosis
Clinical course is variable; evaluate monthly for first 6 months. Nonischemic type has better prognosis (10% will completely resolve). Risk of neovascularization depends on amount of ischemia (CVOS conclusion); 16% of nonischemic patients progress to ischemic disease; 60% of ischemic patients develop neovascularization and 33% develop neovascular glaucoma.
Venous Stasis Retinopathy
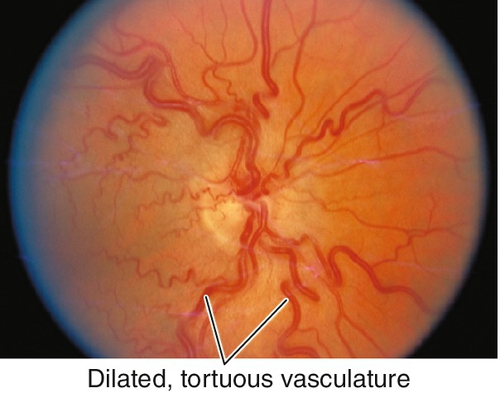
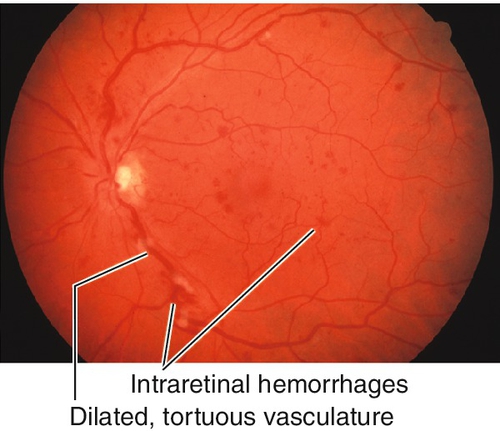
Milder form of nonischemic central retinal vein occlusion (CRVO) representing patients with better perfusion. Dot/blot/flame hemorrhages, dilated/tortuous vasculature, and microaneurysms occur, usually bilateral; more benign course. Associated with hyperviscosity syndromes including polycythemia vera, multiple myeloma, and Waldenström’s macroglobulinemia.
Ocular Ischemic Syndrome
Definition
Widespread ischemia of both the anterior and posterior segments of one eye due to ipsilateral carotid occlusive disease (less frequently obstruction of the ipsilateral ophthalmic artery), carotid dissection, or arteritis (rare).
Etiology
Due to a 90% or greater occlusion of the ipsilateral carotid artery or rarely ophthalmic artery.
Epidemiology
Usually occurs in patients aged 50–70 years old (mean = 65 years); 80% unilateral; male predilection (2 : 1). Associated with atherosclerosis, ischemic heart disease (50%), hypertension (67%), diabetes mellitus (50%), previous stroke (25%), and peripheral arterial disease (20%); rarely due to inflammatory conditions including giant cell arteritis. Blood flow to the eye is relatively unaffected until carotid obstruction exceeds 70%; ocular ischemic syndrome usually does not occur until it reaches 90% (decreasing CRA perfusion by 50%); 50% of patients have complete ipsilateral carotid artery obstruction.
Symptoms
Gradual loss of vision (90%) over days to weeks with accompanying dull eye pain/headache (40%) or “ocular angina”; patients may also report amaurosis fugax (10%) or a delayed recovery of vision after exposure to bright light due to impaired photoreceptor regeneration. May occur suddenly in 12% of cases where a cherry-red spot is also present.
Signs
Gradual or sudden decreased visual acuity ranging from 20 / 20 to NLP; retinal arterial narrowing and venous dilatation without tortuousity, retinal hemorrhages (80% midperipheral), microaneurysms, macular edema, cotton-wool spots, disc/retinal neovascularization (37%), and spontaneous pulsations of the retinal arteries; anterior segment signs including episcleral injection, corneal edema, anterior chamber cells and flare (keratic precipitates are absent and flare is often disproportionate to the amount of cell present), iris atrophy, chronic conjunctivitis, and rubeosis (66%) are common. Intraocular pressure may be elevated, but may also be normal even with 360° synechia. Light digital pressure on the globe through the eyelid often produces arterial pulsations (does not occur in other diseases in differential) and can shut down perfusion of the central retinal artery.
Differential Diagnosis
Nonischemic CRVO, venous stasis retinopathy, diabetic retinopathy, hypertensive retinopathy, aortic arch disease, parafoveal telangiectasis, radiation retinopathy, Takayasu’s disease.
Evaluation
• Check blood pressure.
• Fluorescein angiogram: Delayed arteriovenous transit time (> 11 seconds) in 95%; delayed or patchy choroidal filling (> 5 seconds) in 60%, arterial vascular staining in 85%.
• Electrophysiologic testing: ERG (reduced or absent a-wave and b-wave amplitudes).
• Medical consultation for complete cardiovascular evaluation including duplex and carotid Doppler ultrasound scans (≥ 90% obstruction of the ipsilateral internal or common carotid arteries). Carotid angiography is usually not needed except in cases where ultrasound is equivocal.
Prognosis
Poor prognosis; 5-year mortality rate is 40% mainly owing to cardiovascular disease. Sixty percent of patients have count fingers or worse vision at 1 year follow-up; only 25% have better than 20 / 50 vision. When rubeosis is present, 90% will be count fingers or worse within 1 year. One-third of patients have improved vision after carotid endarterectomy, one-third remain unchanged, and one-third worsen despite surgery.
Retinopathy of Prematurity
Definition
Abnormal retinal vasculature development in premature infants, especially after supplemental oxygen therapy.
Epidemiology
Usually bilateral; associated risk factors include premature birth (< 32 weeks’ gestation), low birth weight (< 750 g: 90% develop ROP and 16% develop threshold disease; 1000–1250 g: 45% develop ROP and 2% develop threshold disease), supplemental oxygen therapy (> 50 days), and a complicated hospital course.
Symptoms
Asymptomatic; later may have decreased vision.
Signs
Shallow anterior chamber, corneal edema, iris atrophy, poor pupillary dilation, posterior synechiae, ectropion uveae, leukocoria, vitreous hemorrhage, retinal detachment, and retrolental fibroplasia; may have strabismus.
International classification of ROP describes the retinal changes in five stages:
Stage 1
Thin, circumferential, flat, white, demarcation line develops between posterior vascularized and peripheral avascular retina (beyond line).
Stage 2
Demarcation line becomes elevated and organized into a pink-white ridge, no fibrovascular growth visible.
Stage 3
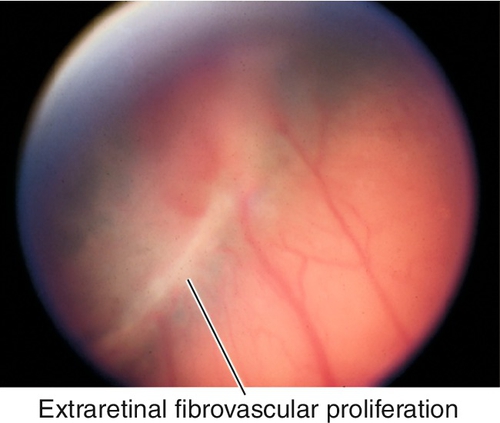
Extraretinal fibrovascular proliferation from surface of the ridge.
Stage 4
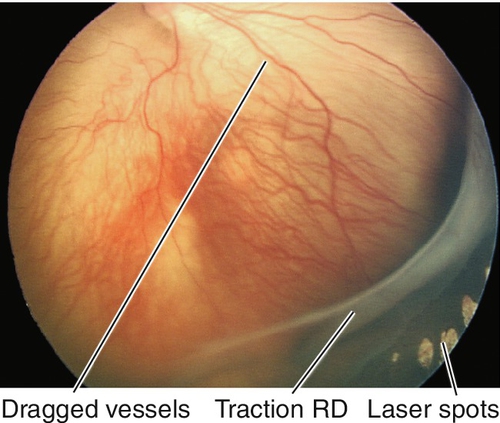
Dragging of vessels, and subtotal traction retinal detachment (4A is macula attached, 4B involves the macula).
Stage 5
Total retinal detachment (almost always funnel detachment).
International classification of ROP also describes the extent of retina involved by number of clock hours and location by zone (centered on optic disc, not the fovea because retinal vessels emanate from disc):
Zone 1
Inner zone (posterior pole) corresponding to the area enclosed by a circle around the optic disc with radius equal to twice the distance from the disc to the macula (diameter of 60°).
Zone 2
The area between zone 1 and a circle centered on the optic disc and tangent to the nasal ora serrata.
Zone 3
Remaining temporal crescent of retina (last area to become vascularized).
Finally, international classification of ROP defines “plus” disease:
“Plus” Disease
At least two quadrants (usually 6 or more clock hours) of shunted blood causing vascular engorgement in the posterior pole with tortuous arteries, dilated veins, pupillary rigidity due to iris vascular engorgement, and vitreous haze.
Differential Diagnosis
Coats’ disease, Eales’ disease, familial exudative vitreoretinopathy, sickle cell retinopathy, juvenile retinoschisis, persistent hyperplastic primary vitreous, incontinentia pigmenti (Bloch–Sulzberger syndrome), and other causes of leukocoria (see Chapter 7).
Evaluation
• The first exam should be either prior to discharge from the hospital, 4 weeks chronological age, or by 31 weeks postgestational age, whichever is later.
• Complete ophthalmic history with attention to birth history and birth weight.
• Complete eye exam with attention to iris, lens, and ophthalmoscopy (retinal vasculature and retinal periphery with scleral depression).
• Cycloplegic refraction as many develop refractive errors especially myopia.
• Pediatric consultation.
Prognosis
Depends on the amount and stage of ROP; 80–90% will spontaneously regress; may develop amblyopia, macular dragging, strabismus; stage 5 disease carries a poor prognosis (functional success in only 3%); may develop high myopia, glaucoma, cataracts, keratoconus, band keratopathy, and retinal detachment.
Coats’ Disease / Leber’s Miliary Aneurysms
Unilateral (80–95%), idiopathic, progressive, developmental retinal vascular abnormality (telangiectatic and aneurysmal vessels with a predilection for the macula); usually occurs in young males (10 : 1) < 20 years old (two-thirds present before age 10). Retinal microaneurysms, retinal telangiectasia, lipid exudation, “light-bulb” vascular dilatations, capillary nonperfusion and occasionally neovascularization, exudative retinal detachments, and subretinal cholesterol crystals occur primarily in the temporal quadrants, especially on fluorescein angiogram where microaneurysm leakage is common. May present with poor vision, strabismus, or leukocoria. Spectrum of disease from milder form in older patients with equal sex predilection and often bilateral (Leber’s miliary aneurysms) to severe form with localized exudative retinal detachments and yellowish subretinal masses, and is included in the differential diagnosis of leukocoria (Coats’ disease). Clinical course varies but generally progressive. Rarely associated with systemic disorders including Alport’s disease, fascioscapulohumeral dystrophy, muscular dystrophy, tuberous sclerosis, Turner’s syndrome, and Senior–Loken syndrome. On histopathologic examination there is loss of vascular endothelium and pericytes with subsequent mural disorganization. Classified into five stages:
Stage 1
Telangectasia only
Stage 2
Exudation (a = extrafoveal, b = subfoveal)
Stage 3
Exudative retinal detachment (a = subtotal, b = total)
Stage 4
RD with glaucoma
Stage 5
End-stage disease
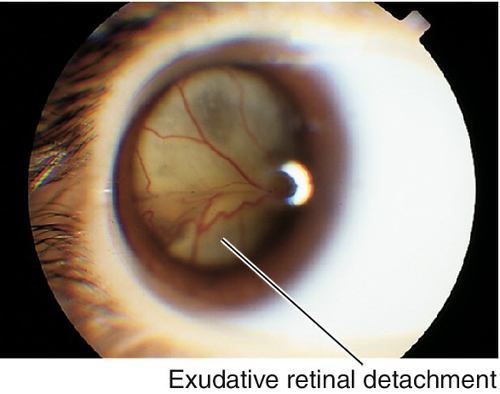
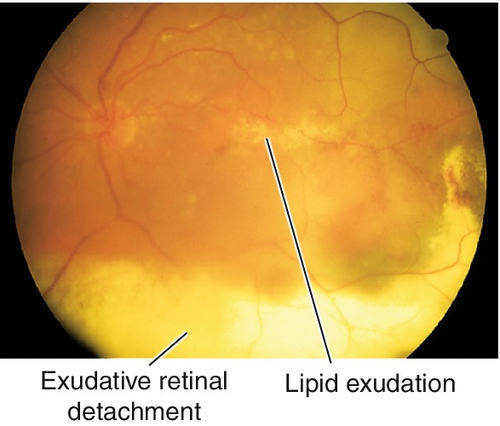
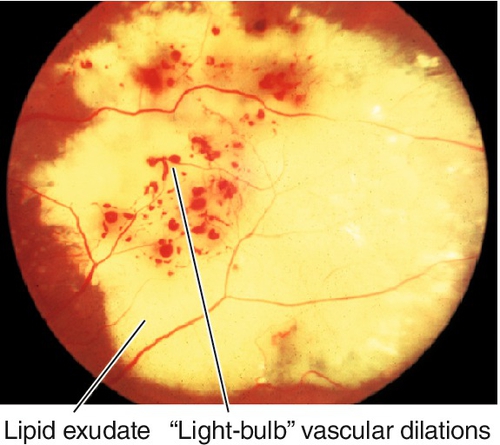
Figure 10-32 Leber’s miliary aneurysms demonstrating dilated arterioles with terminal “light-bulbs.”
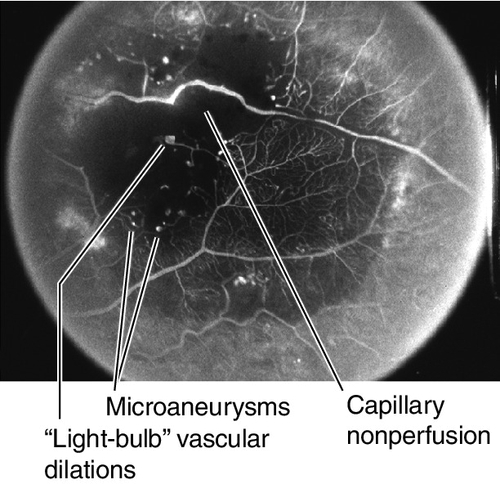
Figure 10-33 Fluorescein angiogram of same patient as Figure 10-32 demonstrating capillary nonperfusion, microaneurysms, and “light-bulb” vascular dilations.
• Treatment: Scatter laser photocoagulation to posterior or cryotherapy to anterior areas of abnormal vasculature, telangiectasia, and areas of nonperfusion when symptomatic. May require multiple treatment sessions. Goal is to ablate areas of vascular leakage and to allow resorption of exudate.
Familial Exudative Vitreoretinopathy and Norrie’s Disease (X-Linked Recessive)
(See Hereditary Vitreoretinal Degenerations section below.)
Incontinentia Pigmenti (X-Linked Dominant)
Ocular, CNS, dermatologic, and dental findings including skin blisters, retinal neovascularization, vitreous hemorrhage, and traction retinal detachment. Associated with mutation in the NEMO gene located on chromosome Xq28.
• Fluorescein angiogram: Shows perpheral nonperfusion; wide-angle angiography is especially useful.
• Treatment: Scatter laser photocoagulation to ischemic retina when neovascularization develops. Consider vitrectomy when traction retinal detachment or nonclearing vitreous hemorrhage is present should be performed by a retina specialist.
Eales’ Disease
Bilateral, idiopathic, peripheral obliterative vasculopathy that occurs in healthy, young adults aged 20–30 years old, with male predilection. Patients usually notice floaters and decreased vision and have areas of perivascular sheathing, vitreous cells, peripheral retinal nonperfusion, microaneurysms, intraretinal hemorrhages, white sclerotic ghost vessels, disc/iris/retinal neovascularization, and vitreous hemorrhages. Fibrovascular proliferation may lead to tractional retinal detachments. May have signs of ocular inflammation with keratic precipitates, anterior chamber cells and flare, and cystoid macular edema; variable prognosis. Eales’ disease is a diagnosis of exclusion; must rule out other causes of inflammation or neovascularization including BRVO, diabetic retinopathy, sickle cell retinopathy, multiple sclerosis, sarcoidosis, tuberculosis, SLE, and other collagen–vascular diseases.
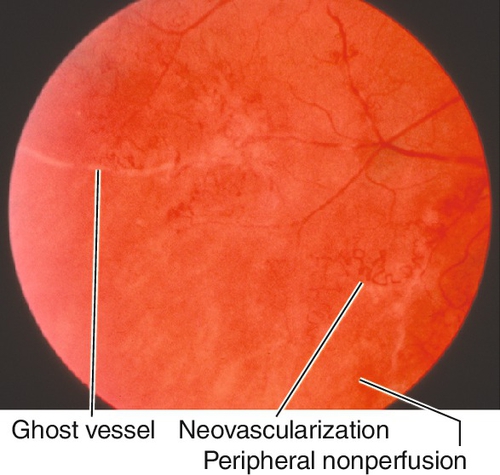
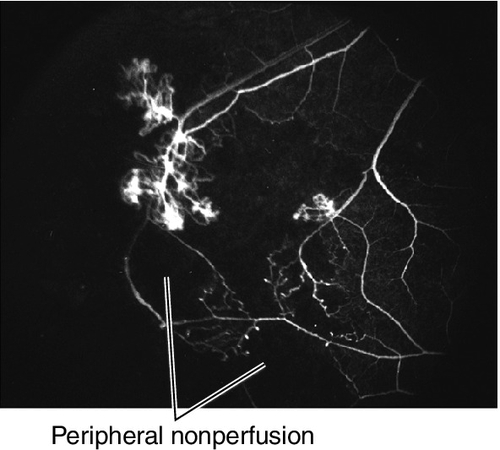
• Treatment: Scatter laser photocoagulation to nonperfused retina when neovascularization develops. If vitreous hemorrhage obscures view of retina, peripheral cryotherapy can be applied to ablate peripheral avascular retina.
• Consider periocular or systemic steroids for inflammatory component.
Macular Telangiectasia (Idiopathic Juxtafoveal / Perifoveal Telangiectasia)
Group of retinal vascular disorders with abnormal perifoveal capillaries confined to the juxtafoveal region (1–199 μm from center of fovea). Several forms:
Type 1A (Unilateral Congenital Parafoveal Telangiectasia)
Occurs in men in the fourth to fifth decades. Yellow exudate at outer edge of telangiectasis usually temporal to the fovea and 1–2 disc diameters in area; decreased vision ranging from 20 / 25 to 20 / 40 from macular edema and exudate. May represent mild presentation of Coats’ disease in an adult.
• Optical coherence tomography: Characteristic outer retinal hyporeflective cavities that do not correspond to leakage on FA. May eventually lead to atrophy.
• Treatment: Consider focal laser photocoagulation to leaking, nonsubfoveal vessels.
Type 1B (Unilateral Idiopathic Parafoveal Telangiectasia)
Occurs in middle-aged men. Minimal exudate usually confined to 1 clock hour at the edge of the foveal avascular zone; usually asymptomatic with vision better than 20/25.
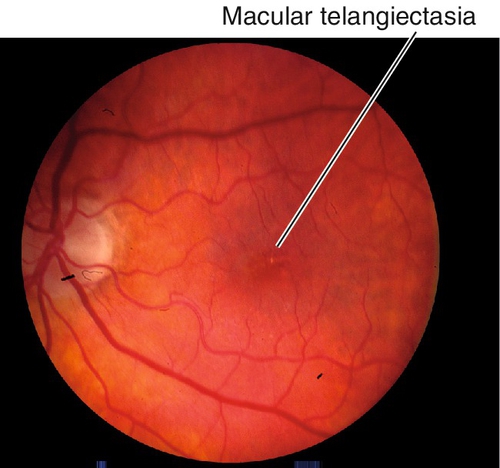
Figure 10-36 Macular telangiectasia type 1b with mild retinal pigment epithelium changes at edge of fovea.
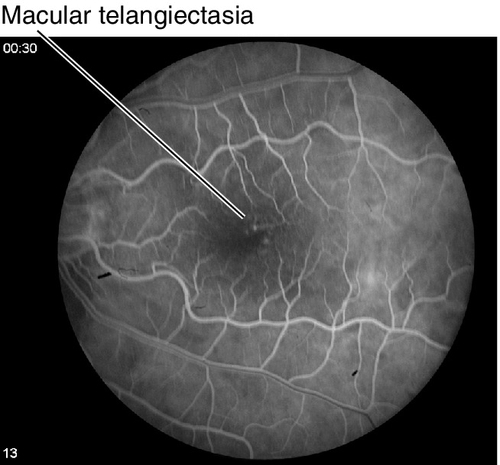
Figure 10-37 Fluorescein angiogram of same patient as Figure 10-36, demonstrating hyperfluorescent leakage from telangiectatic vessels.
• Optical coherence tomography: Characteristic outer retinal hyporeflective cavities that do not correspond to leakage on FA. May eventually lead to atrophy.
• No treatment recommended.
Type 2 (Bilateral Acquired Parafoveal Telangiectasia)
Onset of symptoms in the fifth to sixth decades with equal sex distribution. Symmetric, bilateral, right-angle venules within 1 disc diameter of the central fovea; usually found temporal to the fovea but may surround the fovea; mild blurring of central vision early, slowly progressive loss of central vision over years; blunting or grayish discoloration of the foveal reflex, right-angle retinal venules, and characteristic stellate retinal pigment epithelial hyperplasia/atrophy; leakage from telangiectatic vessels, but no exudates; associated with CNV, hemorrhagic macular detachments, and retinochoroidal anastomosis. May be caused by chronic venous stasis in the macula from unknown reasons.
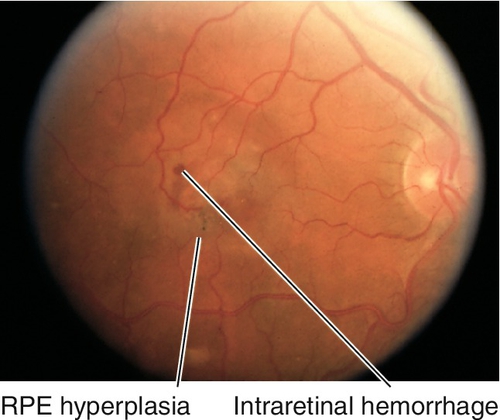
Figure 10-38 Macular telangiectasia type 2 with abnormal foveal reflex, intraretinal hemorrhages and retinal pigment epithelium changes.
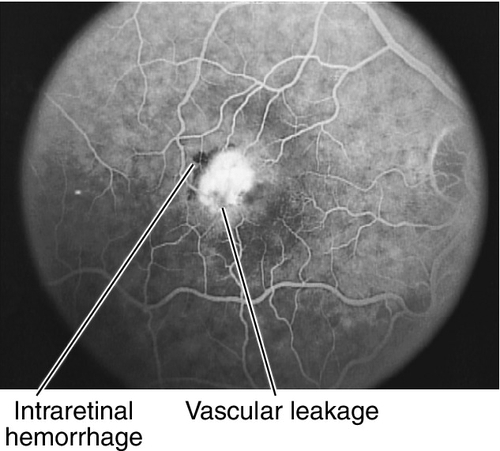
Figure 10-39 Fluorescein angiogram of patient shown in Figure 10-38, demonstrating hyperfluorescent leakage from telangiectatic vessels and blockage from the hemorrhages.
• Optical coherence tomography: Characteristic outer retinal hyporeflective cavities that do not correspond to leakage on FA. May eventually lead to atrophy.
• No treatment recommended unless CNV develops because focal laser photocoagulation to leaking, nonsubfoveal vessels and anti-VEGF injections do not prevent visual loss.
• Consider focal laser photocoagulation of juxtafoveal and extrafoveal CNV, and intravitreal anti-VEGF agents such as 1.25 mg bevacizumab (Avastin) for subfoveal CNV (experimental).
Type 3 (Bilateral Perifoveal Telangiectasis with Capillary Obliteration)
Rare form; occurs in adults in the fifth decade; no sex predilection. Slowly progressive loss of vision due to the marked aneurysmal dilatation and obliteration of the perifoveal telangiectatic capillary network; no leakage from telangiectasis; associated with optic nerve pallor, hyperactive deep tendon reflexes, and other central nervous system symptoms.
• No treatment recommended unless CNV develops.
• Consider focal laser photocoagulation of juxtafoveal and extrafoveal CNV, and intravitreal anti-VEGF agents such as 1.25 mg bevacizumab (Avastin) for subfoveal CNV (experimental).
• Neurology consultation to rule out central nervous system disease.
Retinopathies Associated with Blood Abnormalities
Retinopathy of Anemia
Superficial, flame-shaped, intraretinal hemorrhages, cotton-wool spots, and rarely exudates, retinal edema, and vitreous hemorrhage in patients with anemia (hemoglobin < 8 g / 100 mL). Retinopathy is worse when associated with thrombocytopenia. Roth spots are found in pernicious anemia and aplastic anemia.
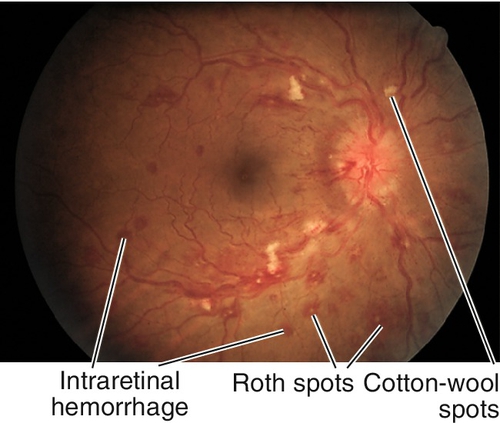
Figure 10-40 Retinopathy of anemia demonstrating intraretinal hemorrhages, cotton-wool spots, and Roth spots.
• Resolves with treatment of anemia.
• Medical or hematology consultation.
Leukemic Retinopathy
Ocular involvement in leukemia is common (80%). Patients are usually asymptomatic. Characterized by superficial, flame-shaped, intraretinal (24%), preretinal, and vitreous hemorrhages (2%), microaneurysms, Roth spots (11%), cotton-wool spots (16%), dilated/tortuous vessels, perivascular sheathing, and disc edema; rarely direct leukemic infiltrates (3%). Direct choroidal involvement appears with choroidal infiltrates, choroidal thickening, and an overlying serous retinal detachment. “Sea fan”-shaped retinal neovascularization can occur late. Retinopathy is due to the associated anemia, thrombocytopenia, and hyperviscosity. Opportunistic infections are also found in patients with leukemia, but are not considered part of leukemic retinopathy.
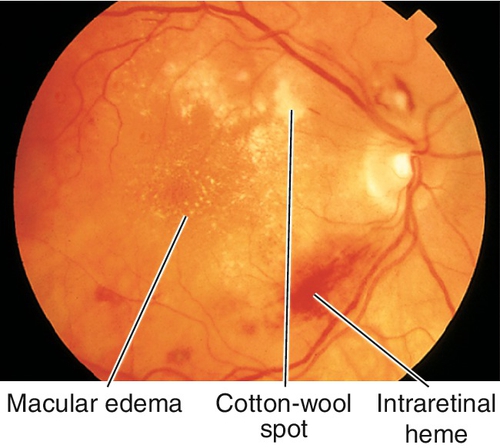
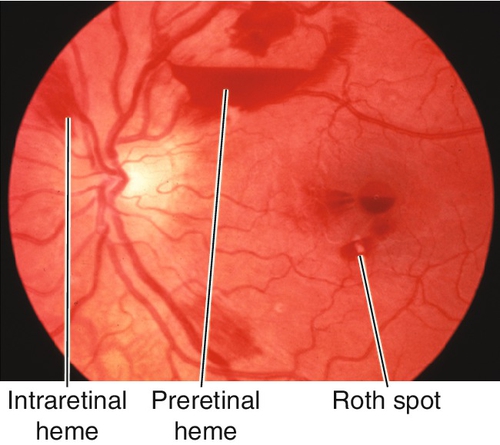
Figure 10-43 Leukemic retinopathy with intraretinal and preretinal hemorrhages, cotton-wool spots, and Roth spots.
• Lab tests: CBC, platelets, bone marrow biopsy.
• Resolves with treatment of underlying hematologic abnormality.
• Treat direct leukemic infiltrates with systemic chemotherapy to control the underlying problem and/or ocular radiation therapy if systemic therapy fails; should be performed by an experienced tumor specialist.
• Medical or oncology consultation.
Sickle Cell Retinopathy
Nonproliferative and proliferative vascular changes due to the sickling hemoglobinopathies; results from mutations in hemoglobin (Hb) where the valine is substituted for glutamate at the 6th position in the polypeptide chain (linked to chromosome 11p15) altering Hb conformation and deformability in erythrocytes. This leads to poor flow through capillaries. Proliferative changes (response to retinal ischemia) are more common with Hb SC (most severe) and Hb SThal variants; Hb SS is associated with angioid streaks; Hb AS and Hb AC mutations rarely cause ocular manifestations. Patients are usually asymptomatic, but may have decreased vision, visual field loss, floaters, photopsias, scotomas, and dyschromatopsia; more common in people of African and Mediterranean descent. Retinopathy follows an orderly progression:
Stage I
Background (nonproliferative) stage with venous tortuosity, “salmon patch” hemorrhages (pink intraretinal hemorrhages), iridescent spots (schisis cavity with refractile elements), cotton-wool spots, hairpin vascular loops, macular infarction, angioid streaks, black “sunburst” chorioretinal scars, comma-shaped conjunctival and optic nerve head vessels, and peripheral arteriole occlusions.
Stage II
Arteriovenous (AV) anastomosis stage with peripheral “silver-wire” vessels and shunt vessels between arterioles and medium-sized veins at border of perfused and nonperfused retina.
Stage III
Neovascular (proliferative) stage with sea-fan peripheral neovascularization (spontaneously regresses in 60% of cases due to autoinfarction); sea-fans grow along retinal surface in a circumferential pattern and have a predilection for superotemporal quadrant (develop approximately 18 months after formation of AV anastamosis).
Stage IV
Vitreous hemorrhage stage with vitreous traction bands contracting around the sea-fans, causing vitreous hemorrhages (most common in SC variant, 21–23%; SS, 2–3%).
Stage V
Retinal detachment stage with tractional/rhegmatogenous retinal detachments from contraction of the vitreous traction bands.
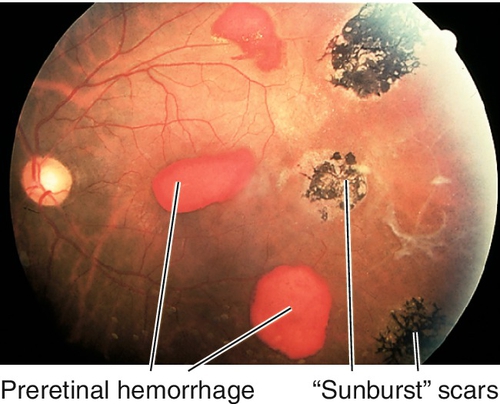
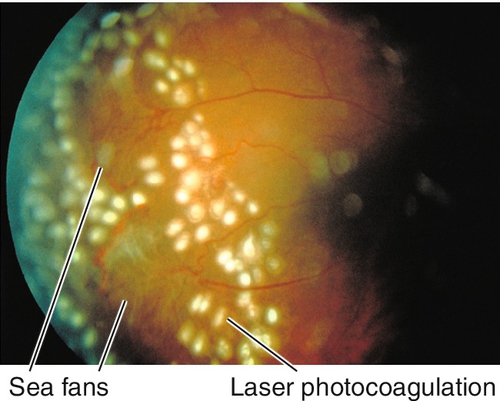
Figure 10-45 Proliferative sickle cell retinopathy demonstrating sea-fans following laser treatment.
• Fluorescein angiogram: Capillary nonperfusion near hairpin loops, enlarged foveal avascular zone, peripheral nonperfusion, arteriovenous anastomosis, and sea-fan neovascularization. Wide-field angiography is especially useful to evaluate for peripheral nonperfusion.
• When active peripheral neovascularization develops, scatter laser photocoagulation (500 μm spots) to nonperfused retina.
• If neovascularization persists, then complete panretinal photocoagulation and consider adding direct laser photocoagulation to neovascularization or feeder vessels (increases risk of complications including vitreous hemorrhage).
• The use of triple freeze–thaw cryotherapy for peripheral neovascularization is controversial; should be performed by a retina specialist.
• Retinal surgery for traction retinal detachment and nonclearing, vitreous hemorrhage (> 6 months); should be performed by a retina specialist. Consider exchange transfusion preoperatively (controversial); avoid scleral buckling to prevent ocular ischemia.
• Medical or hematology consultation.
Diabetic Retinopathy
Definition
Retinal vascular complication of diabetes mellitus; classified into nonproliferative diabetic retinopathy (NPDR) and proliferative diabetic retinopathy (PDR).
Epidemiology
Leading cause of blindness in US population aged 20–64 years old.
Insulin-Dependent Diabetes (Type I)
Juvenile onset, usually occurs before 30 years of age; most patients are free of retinopathy during first 5 years after diagnosis; 95% of patients with insulin-dependent diabetes mellitus (IDDM) get DR after 15 years; 72% will develop PDR and 42% will develop clinically significant macular edema (CSME); severity worsens with increasing duration of diabetes mellitus.
Non-Insulin-Dependent Diabetes (Type II)
Adult onset, usually diagnosed after 30 years of age; more common form (90%) with optimal control without insulin; DR commonly exists at the time of diagnosis (60%) in non-insulin-dependent diabetes mellitus (NIDDM) with 3% having PDR or CSME at diagnosis of diabetes; 30% will have retinopathy in 5 years and 80% in 15 years. Risk of DR increases with hypertension, chronic hyperglycemia, renal disease, hyperlipidemia, and pregnancy.
Symptoms
Asymptomatic, may have decreased or fluctuating vision. Advanced retinopathy can lead to complete blindness.
Signs
Nonproliferative Diabetic Retinopathy
Grading of NPDR (see Box 10-1) and risk of progression to PDR depend on the amount and location of hard and soft exudates, intraretinal hemorrhages, microaneurysms (MA), venous beading and loops, and intraretinal microvascular abnormalities (IRMA). Cotton-wool spots, dot and blot hemorrhages, posterior subcapsular cataracts, and induced myopia/hyperopia (from lens swelling due to high blood sugar) are common; may have macular edema, which can be clinically significant (CSME); usually bilateral.
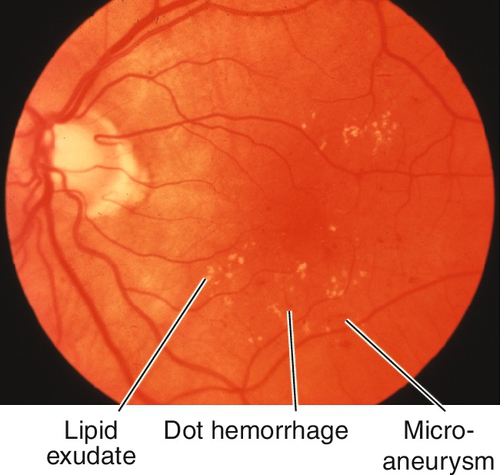
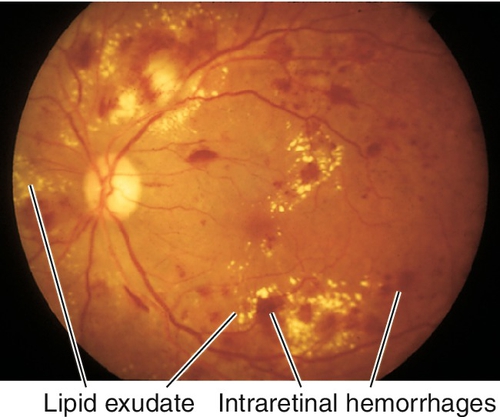
Figure 10-46 Moderate nonproliferative diabetic retinopathy with intraretinal hemorrhages, microaneurysms, and lipid exudate.
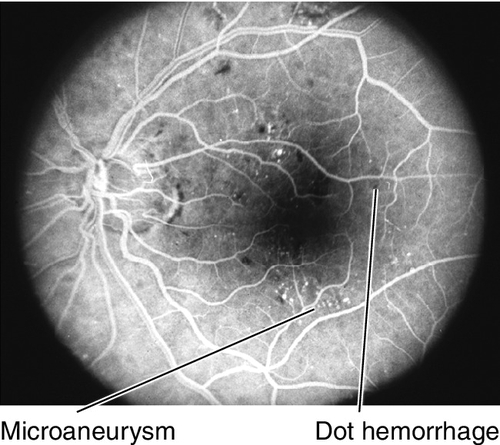
Figure 10-47 Fluorescein angiogram of same patient as Figure 10-46 demonstrating tiny blocking defects from the intraretinal hemorrhages and spots of hyperfluorescence due to microaneurysms.
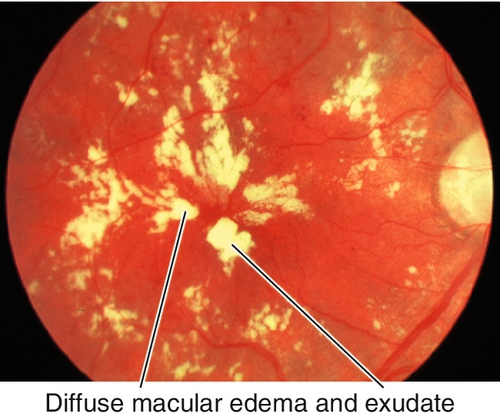
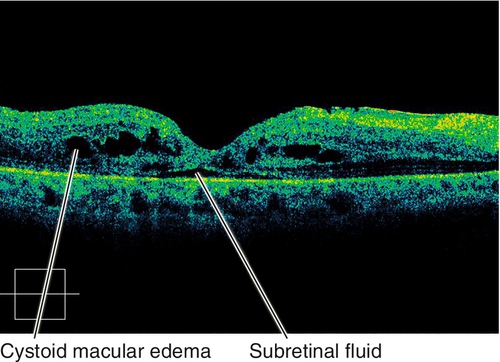
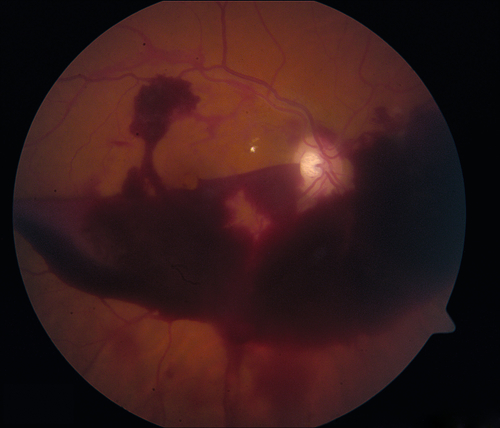
Proliferative Diabetic Retinopathy
Findings of NPDR often present in addition to neovascularization of the disc (NVD) or elsewhere in the retina (NVE), preretinal and vitreous hemorrhages, fibrovascular proliferation on posterior vitreous surface or extending into the vitreous cavity, and tractional retinal detachments; may develop neovascularization of the iris (NVI) and subsequent neovascular glaucoma (NVG). Usually asymmetric, but eventually bilateral.
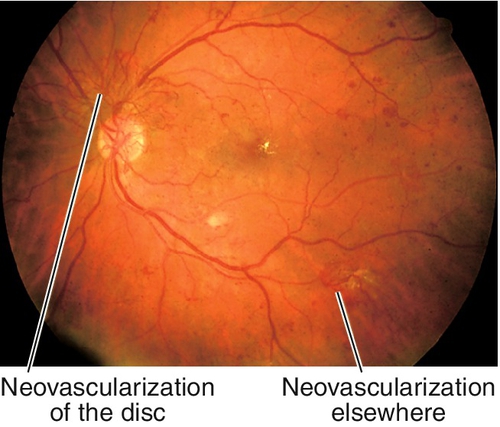
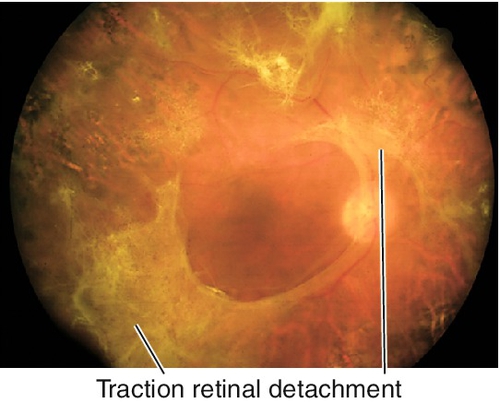
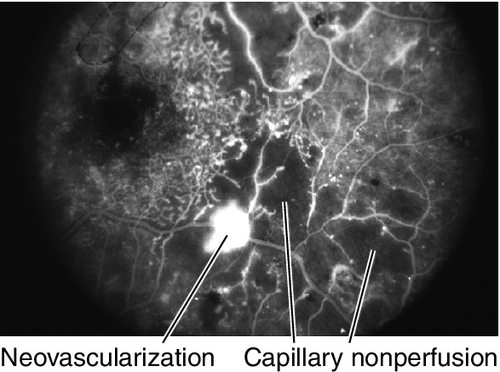
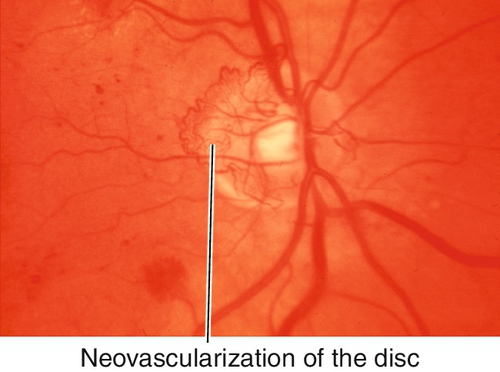
Differential Diagnosis
Hypertensive retinopathy, CRVO, BRVO, ocular ischemic syndrome, radiation retinopathy, retinopathy associated with blood disorders, Eales’ disease, hypertensive retinopathy.
Evaluation
NIDDM Type II: Examine at diagnosis of diabetes mellitus, then annually if no retinopathy is detected.
During pregnancy: Examine before pregnancy, each trimester, and 3–6 months post partum.
• Lab tests: Fasting blood glucose, hemoglobin A1C, blood urea nitrogen (BUN), and creatinine.
• B-scan ultrasonography to rule out tractional retinal detachment in eyes when dense vitreous hemorrhage obscures view of fundus.
• Fluorescein angiogram: Capillary nonperfusion, microaneurysms, macular edema, and disc/retinal neovascularization. Wide-field angiography is helpful to evaluate peripheral nonperfusion and to find early neovascularization.
• Optical coherence tomography: Increased retinal thickness, cysts, and subretinal fluid in cases of macular edema; can highlight the presence of posterior hyaloidal traction and traction macular detachment.
• Medical consultation with attention to blood pressure, cardiovascular system, renal status, weight, and glycemic control.
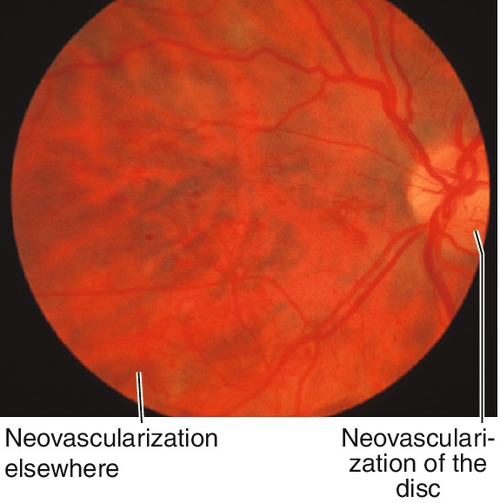
Figure 10-56 Proliferative diabetic retinopathy before laser treatment.
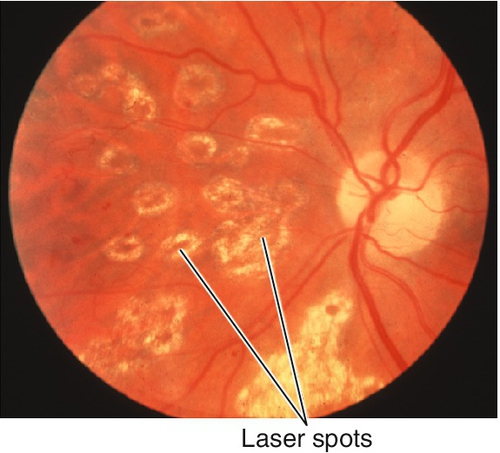
Figure 10-57 Same patient as Figure 10-56 demonstrating quiescent proliferative diabetic retinopathy following pan-retinal photocoagulation. Note absence of neovascularization.
Prognosis
Early treatment allows better control. Good for NPDR without CSME. After adequate treatment, diabetic retinopathy often becomes quiescent for extended periods of time. Focal laser photocoagulation improves vision in 17% of cases (ETDRS conclusion). Complications include cataracts (often posterior subcapsular) and neovascular glaucoma.
Hypertensive Retinopathy
Definition
Retinal vascular changes secondary to chronic or acutely (malignant) elevated systemic blood pressure.
Epidemiology
Hypertension defined as blood pressure > 140 / 90 mmHg; 60 million Americans over 18 years of age have hypertension; more prevalent in African Americans.
Symptoms
Asymptomatic; rarely, decreased vision.
Signs
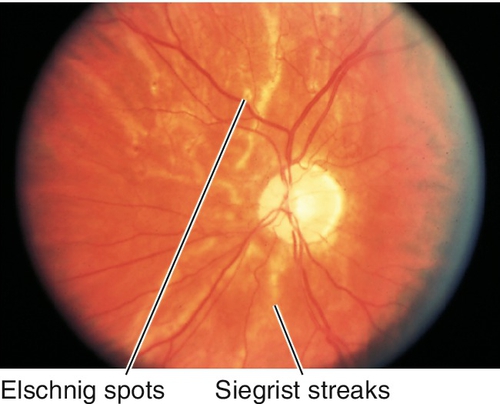
Retinal arteriole narrowing/straightening, copper- or silver-wire arteriole changes (arteriolosclerosis), arteriovenous crossing changes (nicking), cotton-wool spots, microaneurysms, flame hemorrhages, hard exudates (may be in a circinate or macular star pattern), Elschnig spots (yellow [early] or hyperpigmented [late] patches of retinal pigment epithelium overlying infarcted choriocapillaris lobules), Siegrist streaks (linear hyperpigmented areas over choroidal vessels), arterial macroaneurysms, and disc hyperemia or edema with dilated tortuous vessels (in malignant hypertension).
Fundus findings are graded/classified as follows:
Keith Wagener Barker grades
Grade 2: Grade 1 + arteriovenous crossing changes (nicking)
Grade 3: Grade 2 + cotton wool spots and flame hemorrhages
Grade 4: Grade 3 + swelling of the optic disc (optic disc edema).
Proposed classification scheme
2. Mild: Focal or generalized arteriolar narrowing, AV nicking, silver/copper wiring
3. Moderate: Hemorrhages, microaneurysms, cotton-wool spots, hard exudates
4. Malignant: Moderate plus optic disc swelling or severely elevated blood pressure.
Differential Diagnosis
Diabetic retinopathy, radiation retinopathy, vein occlusion, leukemic retinopathy, retinopathy of anemia, collagen vascular disease, ocular ischemia syndrome, neuroretinitis, anterior ischemic optic neuropathy, papilledema.
Evaluation
• Check blood pressure.
• Fluorescein angiogram: Retinal arteriole narrowing/straightening, microaneurysms, capillary nonperfusion, and macular edema.
• Medical consultation with attention to cardiovascular and cerebrovascular systems.
Prognosis
Usually good.
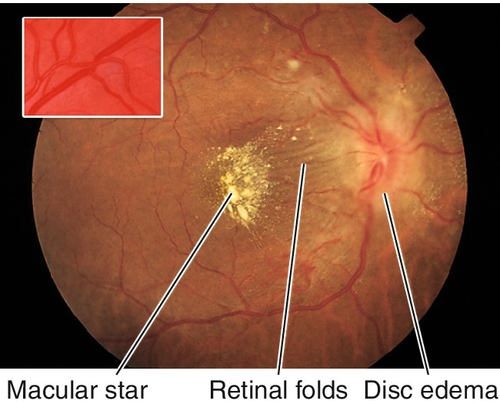
Toxemia of Pregnancy
Severe hypertension, proteinuria, edema (pre-eclampsia), and seizures (eclampsia) occur in 2–5% of obstetric patients in the third trimester. Patients have decreased vision, photopsias, and floaters usually just before or after delivery. Signs include focal arteriolar narrowing, cotton-wool spots, retinal hemorrhages, hard exudates, Elschnig spots (RPE changes from choroidal infarction), bullous exudative retinal detachments, neovascularization, and disc edema (all due to hypertension-related changes).
• Usually resolves without sequelae after treating hypertension and delivery.
• Emergent obstetrics consultation if presenting to ophthalmologist.
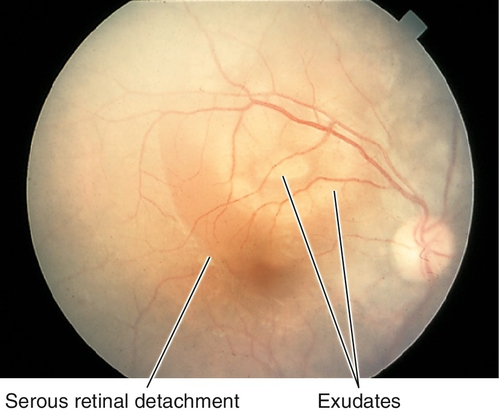
Acquired Retinal Arterial Macroaneurysm
Focal dilatation of retinal artery (> 100 μm) often at bifurcation or crossing site; more common in women > 60 years old with hypertension (50–70%) or atherosclerosis. Usually asymptomatic, unilateral, and solitary; may cause sudden loss of vision from vitreous hemorrhage; macroaneurysms nasal to the optic disc are less likely to cause symptoms. Subretinal, intraretinal, preretinal, or vitreous hemorrhages (multilevel hemorrhages) from rupture of aneurysm, and surrounding circinate exudates are common. May spontaneously sclerose forming a Z-shaped kink at old aneurysm site.
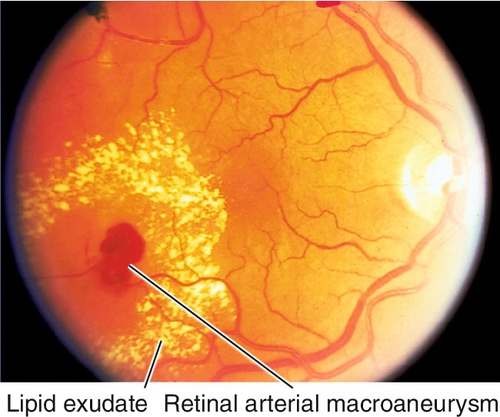
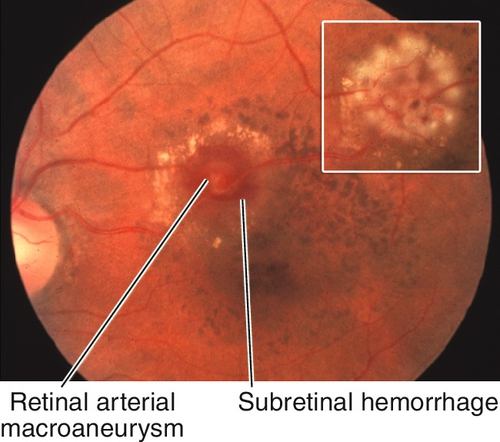
• Indocyanine green angiogram: Uniform, focal filling of the macroaneurysm; it is very useful to identify RAM in the presence of intra- and preretinal hemorrhage.
• Most require no treatment, especially in the absence of loss of vision.
• Low-intensity, longer-duration, argon green or yellow laser photocoagulation to microvascular changes around leaking aneurysm if decreased acuity is present (direct treatment controversial because it may cause a vitreous hemorrhage, distal ischemia, or a branch retinal artery occlusion).
• Consider pars plana vitrectomy with surgical evacuation of subretinal hemorrhage (with or without injection of subretinal tissue plasminogen activator) in cases of massive, subfoveal hemorrhage < 10 days old (experimental).
• Medical consultation for hypertension.
Radiation Retinopathy
Definition
Alteration in retinal vascular permeability after receiving local ionizing radiation usually from external beam radiotherapy or plaque brachytherapy.
Etiology
Endothelial cell DNA damage secondary to the radiation leading to progressive cell death and damage to the retinal blood vessels.
Epidemiology
Usually requires > 30–35 Gy (3000–3500 rads) total radiation dose; appears 0.5–2 years after ionizing radiation; diabetics and patients receiving chemotherapy have a lower threshold.
Symptoms
Often asymptomatic until retinopathy involves macula; decreased vision.
Signs
Microaneurysms, telangiectasia, cotton-wool spots, hard exudates, retinal hemorrhages, macular edema, vascular sheathing, disc edema, retinal/disc/iris neovascularization; may have cataract, dry eye disease, lid abnormalities.
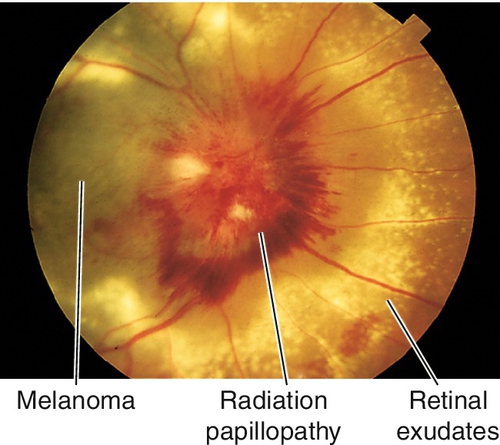
Differential Diagnosis
Diabetic retinopathy, sickle cell retinopathy, hypertensive retinopathy, retinal vascular occlusion, retinopathy of anemia/thrombocytopenia, and leukemic retinopathy.
Evaluation
• Complete eye exam with attention to tonometry, gonioscopy, iris, lens, noncontact biomicroscopic or contact lens fundus exam, and ophthalmoscopy.
• Fluorescein angiogram: Capillary nonperfusion, macular edema, and neovascularization may be present.
• Optical coherence tomography: Intraretinal fluid, cstic spaces, and subretinal fluid; can monitor for treatment response.
Prognosis
Fair; complications include cataract, macular edema/ischemia, optic atrophy, vitreous hemorrhage, and neovascular glaucoma. Two-thirds of patients maintain vision better than 20/200.
Age-Related Macular Degeneration
Definition
Progressive degenerative disease of the retinal pigment epithelium, Bruch’s membrane, and choriocapillaris. Generally classified into two types: (1) nonexudative or “dry” AMD (85%) and (2) exudative or “wet” AMD characterized by CNV and eventually disciform scarring (15%).
Epidemiology
Leading cause of blindness in US population aged > 50 years old, as well as the most common cause of blindness in the Western world; 6.4% of patients 65–74 years old and 19.7% of patients > 75 years old had signs of AMD in the Framingham Eye Study; more prevalent in Caucasians. Risk factors include increasing age (> 75 years old), positive family history, cigarette smoking, hyperopia, light iris color, hypertension, hypercholesterolemia, female gender, and presence of cardiovascular disease; nutritional factors and light toxicity also play a role in pathogenesis. Associated with variants of genes encoding the alternative complement pathway including Y402H single-nucleotide polymorphism (SNP) of complement factor H (CFH) on chromosome 1q31, ARMS2 / HTRA1 on chromosome 10q and LOC387715 on chromosome 10q, tissue inhibitor of metalloproteinase 3 (TIMP3), LIUPC, complement factor B and C2 on chromosome 6p21, complement factor I, and C3. Homozygotes (6 ×) and heterozygotes (2.5 ×) for CFH mutations are more likely to develop AMD. Their risk is even greater if they smoke (odds ratio 34 vs 7.6 in nonsmokers), have elevated ESR, and / or have elevated C-reactive protein.
Nonexudative (Dry) Macular Degeneration
Symptoms
Initially asymptomatic or may have decreased vision, metamorphopsia early. Advanced atrophic form (see Box 10-2) may have central or pericentral scotoma.
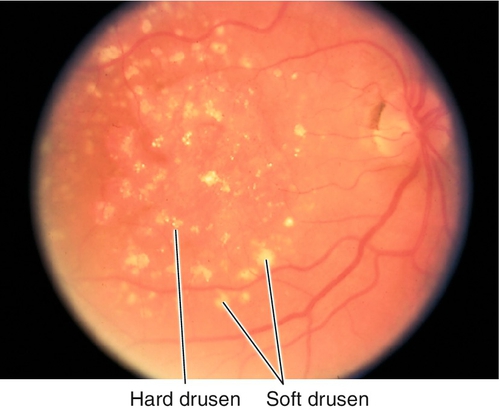
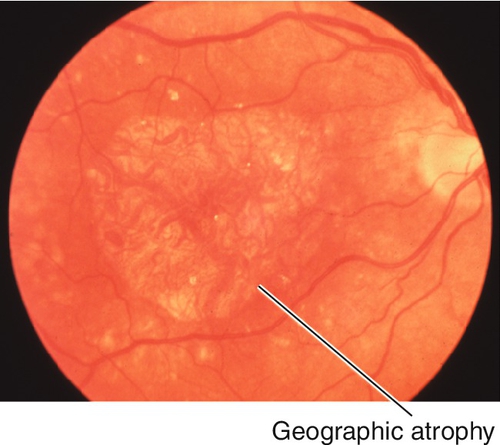
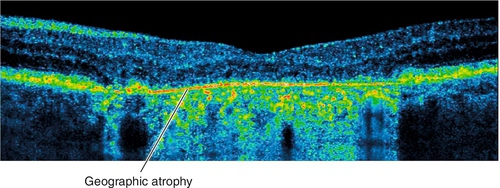
Figure 10-67 Advanced atrophic, nonexudative, age-related macular degeneration demonstrating subfoveal geographic atrophy.
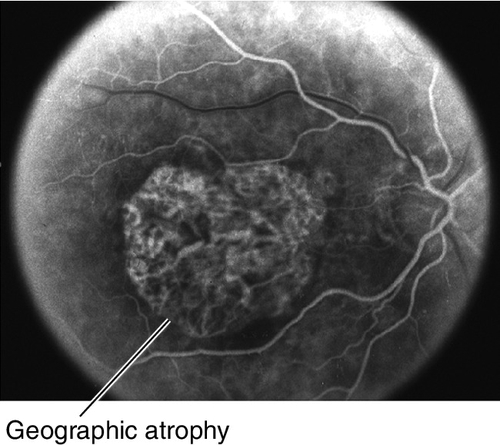
Figure 10-68 Fluorescein angiogram of same patient as Figure 10-67 demonstrating well-defined window defect corresponding to the area of geographic atrophy.
Signs
Normal or decreased visual acuity; abnormal Amsler grid (central/paracentral scotomas or metamorphopsia); small hard drusen, larger soft drusen, geographic atrophy (GA) of the retinal pigment epithelium (RPE), RPE clumping, and blunted foveal reflex.
Differential Diagnosis
Dominant drusen, pattern dystrophy, Best’s disease, Stargardt’s disease, cone dystrophy, and drug toxicity.
Evaluation
• Fluorescein angiogram: Window defects from GA and punctate hyperfluorescent staining of drusen (no late leakage).
• Fundus autofluoresence: To evaluate areas of geographic atropy that appear dark. Hyperautofluorescent areas on the edges of GA are likely to portend GA enlargement.
• Optical coherence tomography: Areas of drusen and GA can be quantified on OCT. Also useful to rule out wet AMD.
Prognosis
Usually good unless central GA or exudative AMD develops. Severe visual loss (defined as loss of > 6 lines) occurs in 12% of nonexudative cases; presence of large soft drusen and focal RPE hyperpigmentation increases risk of developing exudative form (MPS conclusion). Risk of advanced AMD over 5 years varies depending on category: Category 1 and 2 (1.8%), Category 3 (18%), Category 4 (43%) (AREDS conclusion).
Exudative (Wet) Macular Degeneration
Symptoms
Metamorphopsia, central scotoma, rapid visual loss.
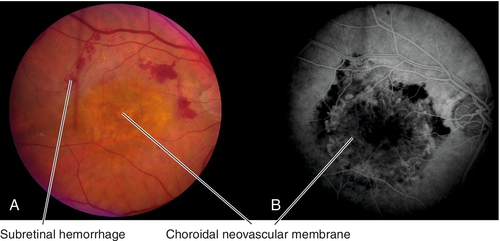
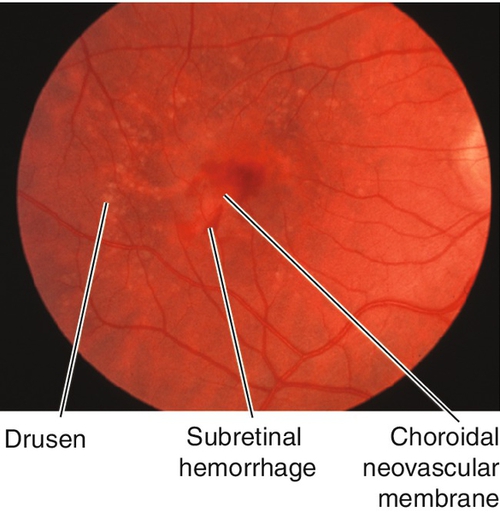
Figure 10-71 Exudative age-related macular degeneration demonstrating subretinal hemorrhage from choroidal neovascular membrane.
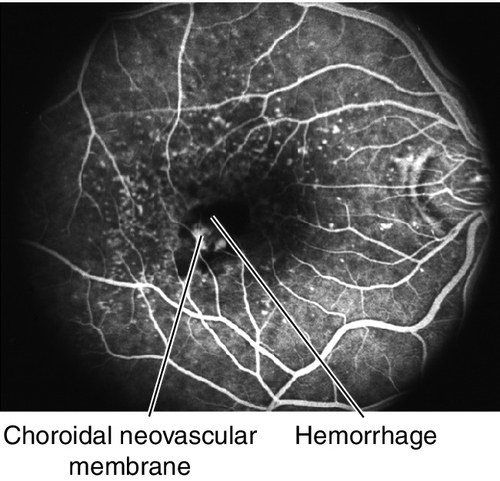
Figure 10-72 Fluorescein angiogram of same patient as Figure 10-71 demonstrating leakage from the CNV and blocking from the surrounding subretinal blood.
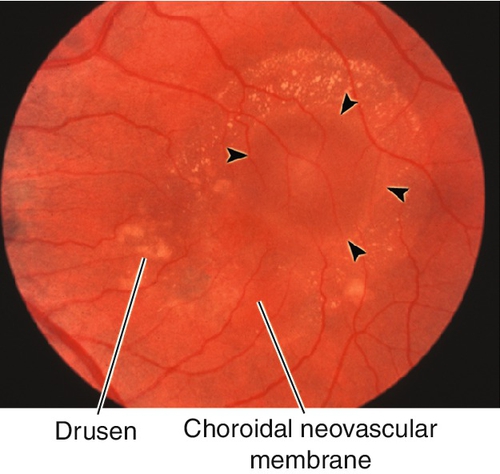
Figure 10-73 Exudative age-related macular degeneration drusen, pigmentary changes, and an occult choroidal neovascular membrane with associated serous pigment epithelial detachment (arrowheads).
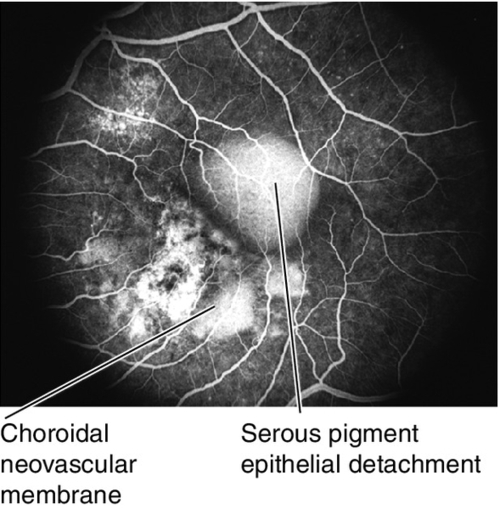
Figure 10-74 Fluorescein angiogram of same patient as Figure 10-73 demonstrating hyperfluorescent staining of pigmentary changes and drusen, leakage from the CNV and pooling of fluorescein dye within the serous pigment epithelial detachment.
Signs
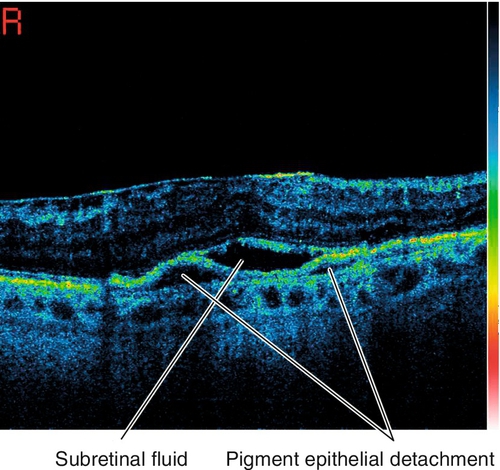
CNV, lipid exudates, subretinal or intraretinal hemorrhage/fluid, pigment epithelial detachment (PED), and retinal pigment epithelial tears; may have late fibrovascular disciform scars.
Differential Diagnosis
Dominant drusen, pattern dystrophy, Best’s disease, central serous retinopathy, Stargardt’s disease, cone dystrophy, drug toxicity, and choroidal neovascularization from other causes, including presumed ocular histoplasmosis syndrome, angioid streaks, myopic degeneration, traumatic choroidal rupture, retinal dystrophies, inflammatory choroidopathies, and optic nerve drusen.
Evaluation
• Fluorescein angiogram: Two forms of leakage from CNV: (1) classic leakage, defined as lacy, network of bright fluorescence during early choroidal filling views that increases in fluorescence throughout the angiogram and leaks beyond its borders in late views; (2) occult leakage, defined as stippled nonhomogeneous hyperfluorescence at the level of the RPE (best seen on stereoscopic views) that persists through to late views, but the leakage is not as bright as classic lesions (type 1 or fibrovascular PED), or late leakage of undetermined origin (type 2), where the early views show no apparent leakage, but as the angiogram progresses there is hyperfluorescent stippling at the level of the RPE in late views.
• Indocyanine green angiogram: Useful when the CNV is poorly demarcated or obscured by hemorrhage on fluorescein angiogram, or if fibrovascular pigment epithelial detachment is present (to identify areas of focal neovascularization or polypoidal choroidal vasculopathy); focal hotspots likely represent retinal angiomatous proliferation (see below); CNV also appears as plaque of late hyperfluorescence. In general, ICGA should be performed when there is lack of response to anti-VEGF therapy to rule out PCV and other masquerade syndromes.
• Optical coherence tomography: Increased retinal thickness, intraretinal fluid, cystoid spaces, subretinal fluid, pigment epithelial detachment, drusen, drusenoid PED, and/or CNV may all be seen on scans. Also useful to determine whether the CNV is type 1 (below the RPE) or type 2 (above the RPE). Usually thinned choroid on enhanced depth imaging.
Prognosis
Long-term prognosis is not known. CNV may recur or persist after treatment; the risk of the fellow eye developing CNV is 4–12% annually.
Retinal Angiomatous Proliferation
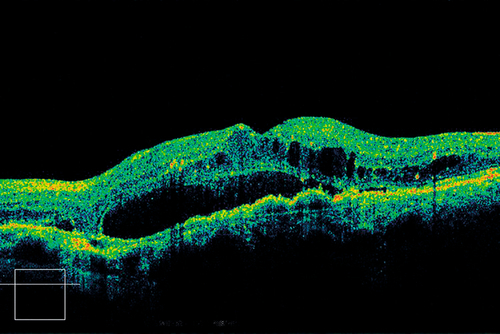
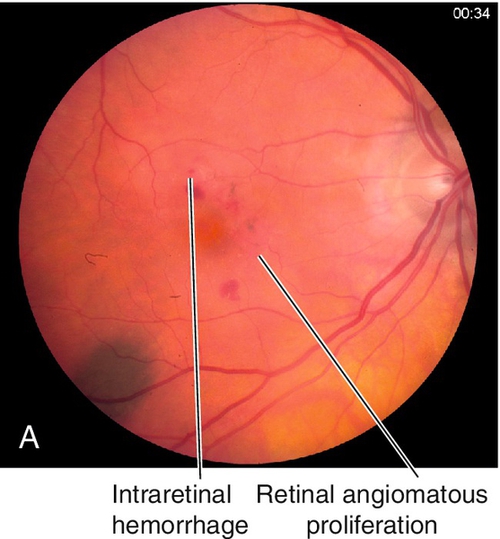
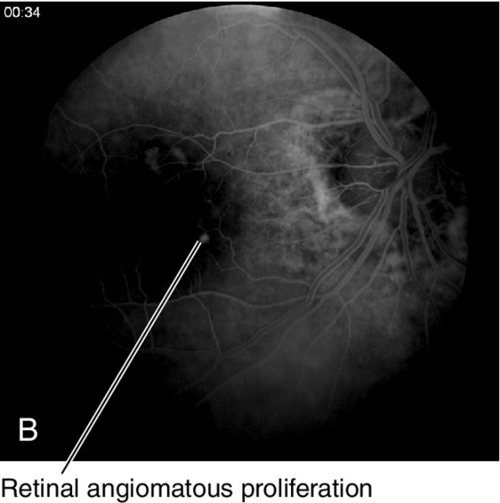
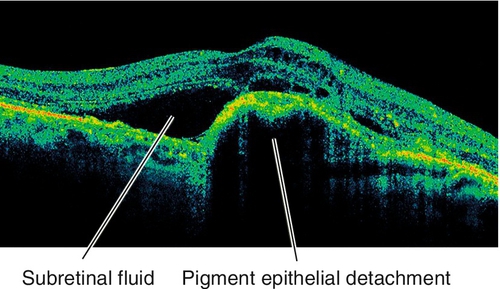
Type 3 CNV (intraretinal neovascularization) in which neovascularization forms a retinal choroidal anastomosis as the retinal vessels grow into the subretinal space; it is considered a subset of AMD. Angiomatous proliferation within the retina is the earliest finding, which manifests as focal intraretinal hemorrhages at the site of the neovascularization with associated pigment epithelial detachment (PED). The lesions are associated with intraretinal and subretinal hemorrhage and exudates. Generally, RAP lesions are more difficult to treat than other types of CNV.
• Indocyanine green angiogram: Ideal for visualizing the focal area of intense hyperfluorescence (hot spot) of a RAP lesion within the hypofluorescent PED. As the RAP lesion anastomoses with the choroidal circulation it may become indistinguishable from an occult CNV.
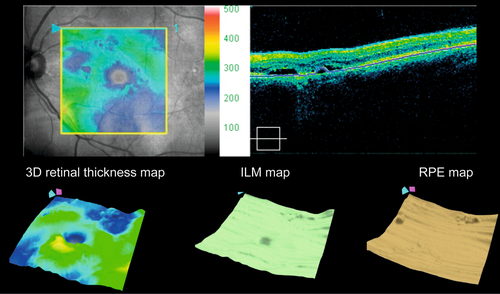
• Optical coherence tomography: PED is present and often the retinal choroidal anastomosis can be visualized.
• Treat RAP lesions with PDT and anti-VEGF agents such as intravitreal 0.5 mg ranibizumab [Lucentis], 2.0 mg aflibercept [Eylea] or 1.25 mg bevacizumab [Avastin] like AMD (see above).
• Extrafoveal RAP lesions can be treated with focal laser photocoagulation.
Polypoidal Choroidal Vasculopathy
Subretinal, orange-red nodules with polyps seen on ICGA. Variant of type 1 choroidal neovascularization (location below RPE); controversial if this is a subset of AMD or separate disease. Often unilateral presentation, but also bilateral disease consisting of orange-red nodular elevations of the RPE (notched PED) and neurosensory retina, often with subretinal hemorrhage (may be massive), retinal pigment epithelial atrophy, and, in late stages, subretinal fibrosis. More common in African American and Asian patients; in Asians, it is more common in males, macular in location and bilateral; in African American and Caucasian patients it is more common in females, unilateral, and peripapillary in location. Occurs in 4–10% of Caucasians diagnosed with wet AMD. Patients are younger than AMD patients. Risk factors include smoking, hypertension, and diabetes. Genetic factors associated with PCV are similar to AMD and include ARMS2, Y402H, and I62V on CFH, HTRA1, and C2. Differential diagnosis includes any disease that can produce occult or minimally classic CNV; usually occurs in patients aged 50–65 years old so a CNV diagnosis in these populations should make one consider PCV. Better prognosis and slower course than typical exudative AMD with loss of one to three lines over 2 years; may spontaneously regress.
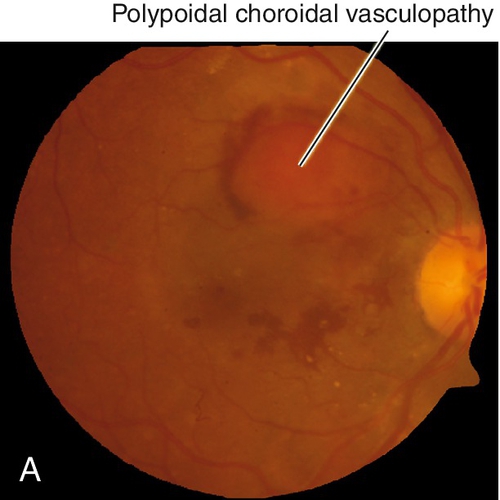
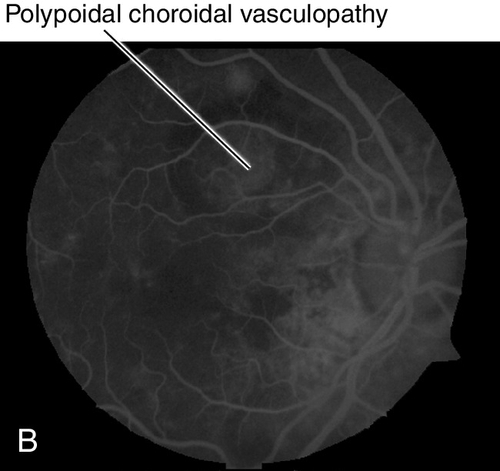
Figure 10-80 Polypoidal choroidal vasculopathy demonstrating the multiple, orange, serosanguinous pigment epithelial detachments as seen on (A) clinical photo, and (B) fluorescein angiogram.
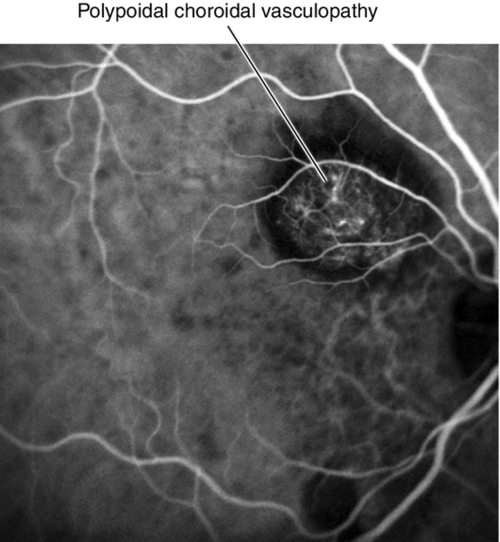
Figure 10-81 Indocyanine green angiogram of same patient as Figure 10-80 illustrating the polypoidal choroidal lesions.
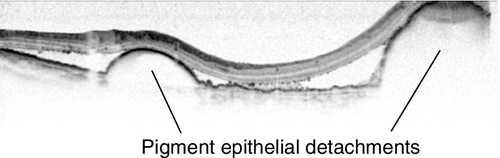
• Indocyanine green angiogram: Delineates the single or multiple, grape-like, hyperfluorescent polypoidal lesion(s) early with or without an associated branching vascular network (BVN) that appear within the first 5 minutes of ICGA that measure 100–500 μm in width. The vascular abnormalities hyperfluoresce centrally early with a surrounding hypofluorescent halo surrounding the lesions. If an orange-red subretinal nodule corresponds to the hyperfluorescence, this is pathognomonic. With dynamic ICGA, pulsatile filling of the hyperfluorescent nodules may be seen. In the late phase, the lesion core may become hypofluorescent because of washout producing a ring-like appearance to the polyp. The vessels are not located in the choroid. In general, ICGA should be performed for the diagnosis of PCV when routine ophthalmoscopic examination indicates a serosanguineous maculopathy with one of the following features: clinically visible orange-red subretinal nodules, spontaneous massive subretinal hemorrhage, or a notched or hemorrhagic pigment epithelium detachment (PED).
• Optical coherence tomography: RPE detachment; may see “string of pearls” of hyperreflective material underneath RPE detachment. In some cuts, may be able to see ring of hyperreflectance under RPE and above Bruch’s membrance that corresponds to polyp. Usually associated with thickened choroid on enhanced depth imaging.
• Observation in cases without foveal hemorrhage, exudative changes, or signs of symptomatic activity defined as either: a drop in vision of ≥ 5 letters, subretinal/intraretinal fluid, PED, subretinal hemorrhage, or FA leakage.
• Full or reduced fluence verteporfin (Visudyne) photodynamic therapy (PDT) alone or in combination with anti-VEGF agents such as intravitreal 0.5 mg ranibizumab (Lucentis), 1.25 mg bevacizumab (Avastin), 2.0 mg aflibercept (Eylea) has shown benefit (EVEREST 1 study result).
• Can treat the entire lesion including the polyps with focal laser photocoagulation or PDT for extrafoveal lesions.
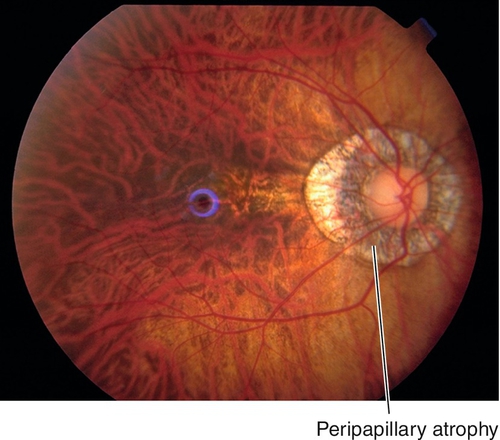
Myopic Degeneration / Pathologic Myopia
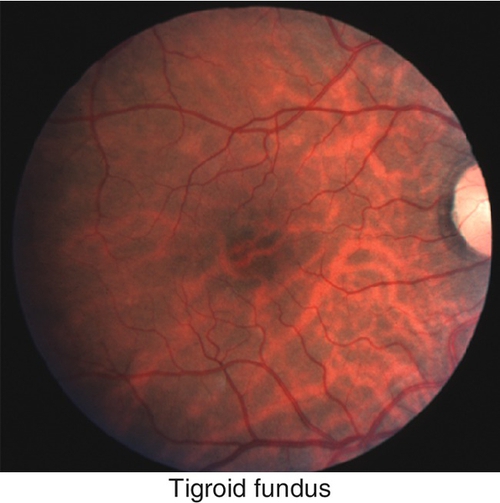
Progressive retinal degeneration that occurs in high myopia (≥ − 6.00 diopters, axial length > 26.5 mm) and pathologic myopia (≥ − 8.00 diopters, axial length > 32.5 mm); incidence of 2% in US population. Findings include scleral thinning, posterior staphyloma, lacquer cracks (irregular, yellow streaks), peripapillary, atrophic temporal crescent, tilted optic disc, Fuchs’ spots (dark spots due to RPE hyperplasia in macula), “tigroid” fundus due to thinning of RPE allowing visualization of larger choroidal vessels, subretinal hemorrhage (especially near lacquer cracks) and chorioretinal atrophy; increased incidence of posterior vitreous detachment, premature cataract formation, glaucoma, lattice degeneration, giant retinal tears, retinal detachments, macular hole, and CNV. Visual field defects may be present.
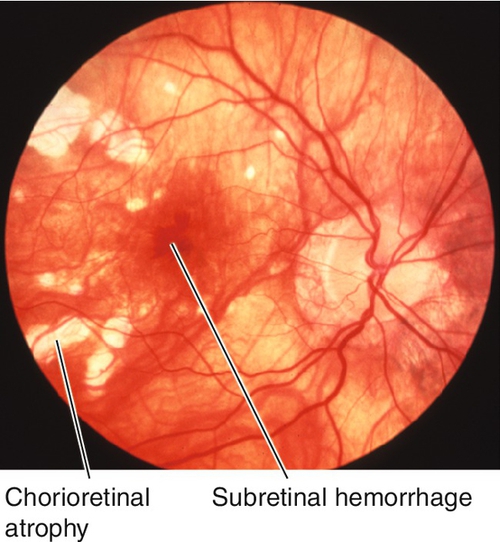
Figure 10-83 Myopic degeneration with peripapillary and chorioretinal atrophy.
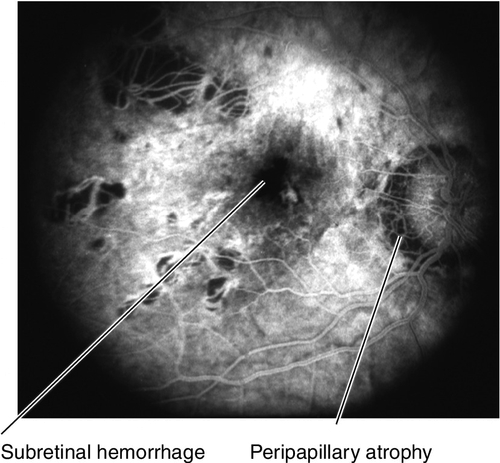
Figure 10-84 Fluorescein angiogram of same patient as Figure 10-83 demonstrating blocking defect from subretinal hemorrhage and window defects from chorioretinal and peripapillary atrophy.
• Genetics: Mapped to chromosomes 18p11.31 and 12q21-q23.
• Fluorescein angiogram: To evaluate for CNV if suspected clinically. Atrophic areas appear as window defects, lacquer cracks are hyperfluorescent linear areas that stain in late views.
• Correct any refractive error; contact lenses help reduce image minification and prismatic effect of glasses.
• Recommend polycarbonate safety glasses for sports (increased risk of choroidal rupture with minor trauma).
• Follow for signs of complications (CNV, retinal detachment, retinal breaks, macular holes, glaucoma, and cataracts).
• Treat CNV with focal laser photocoagulation per MPS guidelines in extrafoveal lesions (see Age-Related Macular Degeneration section), photodynamic therapy or anti-VEGF agents for juxtafoveal (since laser scar enlargement [“scar creep”] is common in pathologic myopia after laser treatment) and subfoveal lesions (Verteporfin in Photodynamic Therapy Pathologic Myopia Study – VIP-PM conclusion); 1.25 mg bevacizumab (Avastin), 0.5 mg ranibizumab (Lucentis), and 2 mg aflibercept (Eylea) (MYRROR Study result) have shown benefit.
• Treat retinal detachment and macular holes with vitreoretinal surgery performed by a retina specialist.
Angioid Streaks
Definition
Full-thickness breaks in calcified, thickened Bruch’s membrane with disruption of overlying RPE.
Etiology
Idiopathic or associated with systemic diseases (50% of cases) including pseudoxanthoma elasticum (PXE, 60%; redundant skin folds in the neck, gastrointestinal bleeding, hypertension), Paget’s disease (8%; extraskeletal calcification, osteoarthritis, deafness, vertigo, increased serum alkaline phosphatase and urine calcium levels), senile elastosis, calcinosis, abetalipoproteinemia, sickle cell disease (5%), thalassemia, hereditary spherocytosis, and Ehlers–Danlos syndrome (blue sclera, hyperextendable joints, elastic skin); also associated with optic disc drusen, acromegaly, lead poisoning, Marfan’s syndrome, and retinitis pigmentosa.
Symptoms
Usually asymptomatic; may have decreased vision, metamorphopsia if choroidal neovascular membrane develops.
Signs
Normal or decreased visual acuity; linear, irregular, deep, dark red-brown streaks radiating from the optic disc in a spoke-like pattern; often have “peau d’orange” retinal pigmentation, peripheral salmon spots, “histo-like” scars, and pigmentation around the streaks; may have subretinal hemorrhage/fluid, retinal pigment epithelial detachments, macular degeneration, and central/paracentral scotomas if CNV develops.
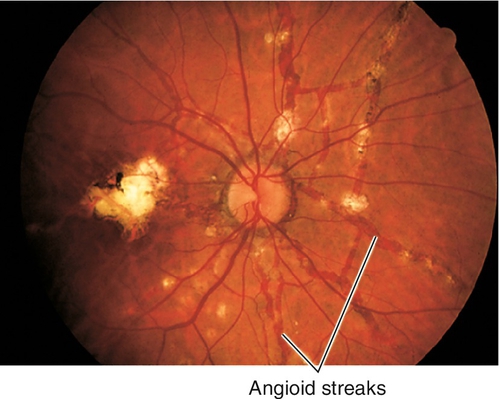
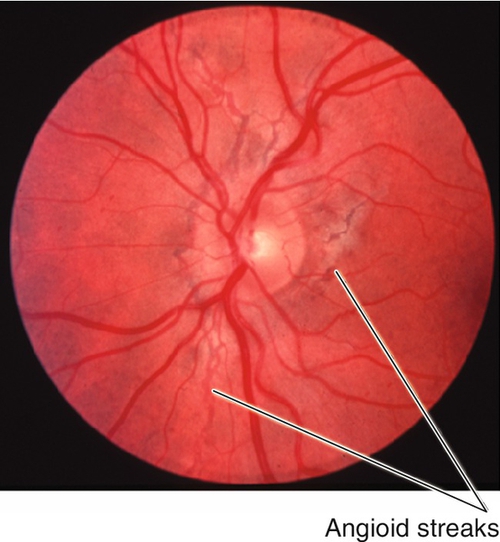
Figure 10-88 Angioid streaks radiating from the optic nerve.
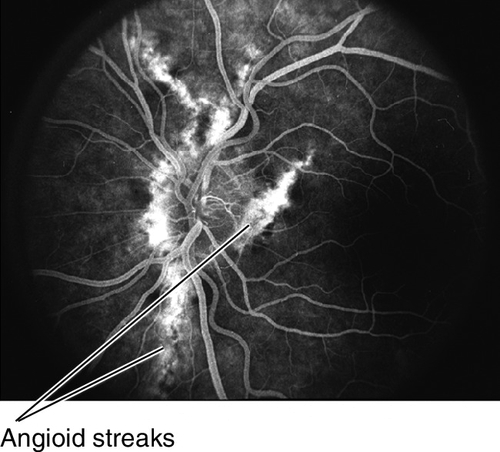
Figure 10-89 Fluorescein angiogram of same patient as shown in Figure 10-88, demonstrating hyperfluorescent window defects corresponding to the angioid streaks.
Differential Diagnosis
Age-related macular degeneration, lacquer cracks, myopic degeneration, choroidal rupture, choroidal folds, hypertensive retinopathy (Siegrist streaks), ophthalmic artery occlusion.
Evaluation
• Check Amsler grid to rule out CNV.
• Lab tests: Sickle cell prep, hemoglobin electrophoresis (sickle cell disease), serum alkaline phosphatase, serum lead levels, urine calcium, stool guaiac, skin biopsy.
• Fluorescein angiogram: To evaluate for CNV if suspected clinically. Usually occurs along the track of an angioid streak and has granular pattern of hyperfluorescence.
• Medical consultation to rule out systemic diseases including skin biopsy and radiographs.
Prognosis
Good unless CNV develops (high recurrence rates).
Central Serous Chorioretinopathy
Definition
Idiopathic leakage of fluid from the choroid into the subretinal space (94%), under the RPE (3%), or both (3%), presumably due to RPE or choroidal dysfunction.
Epidemiology
Usually occurs in males (10:1) aged 20–50 years old; in women, it tends to occur at a slightly older age. Usually unilateral, but can be bilateral; more common in Caucasians, Hispanics, and Asians; rare in African Americans. Associated with type-A personality, stress, hypochondriasis; also associated with pregnancy, steroid use, hypertension, Cushing’s syndrome, systemic lupus erythematosus, and organ transplantation.
Symptoms
Decreased vision, micropsia, metamorphopsia, central scotoma, and mild dyschromatopsia; may be asymptomatic.
Signs
Normal or decreased visual acuity ranging from 20 / 20 to 20 / 200 (visual acuity improves with pinhole or plus lenses); induced hyperopia, abnormal Amsler grid (central / paracentral scotomas or metamorphopsia); single or multiple, round- or oval-shaped shallow, serous retinal detachment or pigment epithelial detachment with deep-yellow spots at the level of the retinal pigment epithelium; areas of retinal pigment epithelium atrophy may occur at sites of previous episodes. Subretinal fibrin suggests active leakage. Rarely associated with type 1 CNV and subretinal fluid.
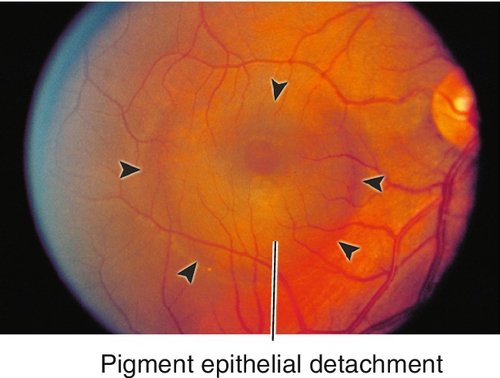
Figure 10-90 Idiopathic central serous retinopathy with large serous retinal detachment.
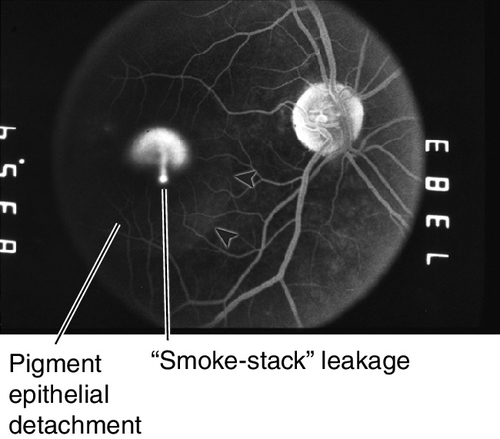
Figure 10-91 Fluorescein angiogram of same patient as shown in Figure 10-90, demonstrating classic smoke-stack appearance.
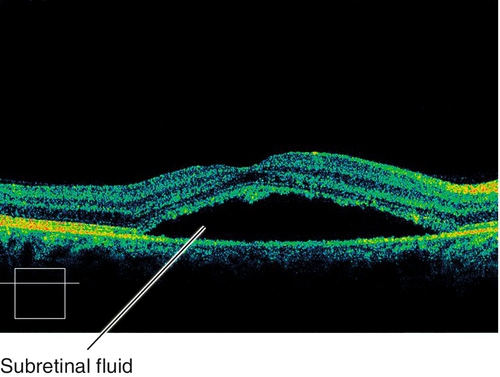
Differential Diagnosis
Age-related macular degeneration (especially in patients > 50 years old), Vogt–Koyanagi–Harada syndrome or other inflammatory choroidal disorders, uveal effusion syndrome, toxemia of pregnancy, optic nerve pit, choroidal tumors, vitelliform macular detachment, pigment epithelial detachment from other causes including PCV and CNV.
Evaluation
• Fluorescein angiogram: Focal dot of hyperfluorescence early that leaks in a characteristic smoke-stack pattern (10%) or gradually pools into a pigment epithelial detachment (90%); more than one site may be present simultaneously (30%); often punctate window defects are seen in other areas in both eyes; recurrent leakage sites are often close to original sites.
• Indocyanine green angiogram: Choroidal hyperpermeability.
• Optical coherence tomography: Enhanced depth imaging of the choroid shows a thickened choroid in affected eye and often in the fellow eye; subretinal fluid and often sub-RPE fluid visible. Useful to follow patients for progression/regression.
• Fundus autofluorescence: Characteristic teardrop-shaped hyperautofluorescence pattern extending from the site of leakage downward. May have other hyperautofluorescent areas from previous episodes.
Prognosis
Good; 94% regain ≥ 20 / 30 acuity; 95% of pigment epithelial detachments resolve spontaneously in 3–4 months, acuity improves over 21 months; recurrences common (45%) and usually occur within a year. Recovery of visual acuity is faster following laser treatment but recovery of contrast sensitivity is prolonged and may ultimately be reduced; 5% develop PCV or CNV. Prognosis is worse for patients with recurrent disease, multiple areas of detachment, or chronic course.
Cystoid Macular Edema
Definition
Accumulation of extracellular fluid in the macular region with characteristic cystoid spaces in the outer plexiform layer.
Etiology
Postoperative (especially in older patients and if the posterior capsule is violated with vitreous loss; CME following cataract surgery is called Irvine–Gass syndrome with peak incidence 4–6 weeks after surgery), post laser treatment (neodymium : yttrium–aluminum– garnet [Nd : YAG] laser capsulotomy, especially if performed within 3 months of cataract surgery), uveitis, diabetic retinopathy, macular or retinal telangiectasia, retinal vein occlusions, retinal vasculitis, epiretinal membrane, hereditary retinal dystrophies (dominant CME, retinitis pigmentosa), medications (epinephrine in aphakic patients, dipivefrin, and prostaglandin analogues), hypertensive retinopathy, exudative AMD, occult rhegmatogenous retinal detachment, intraocular tumors, collagen vascular diseases, hypotony, and chronic inflammation.
Symptoms
Decreased or washed-out vision.
Signs
Decreased visual acuity, loss of foveal reflex, thickened fovea, foveal folds, intraretinal cystoid spaces, lipid exudates; may have signs of uveitis or surgical complications including open posterior capsule, vitreous to the wound, peaked pupil, or iris incarceration in wound.
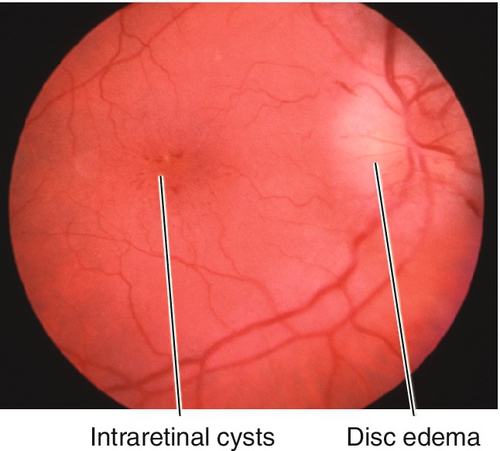
Figure 10-93 Cystoid macular edema with decreased foveal reflex, cystic changes in fovea, and intraretinal hemorrhages.
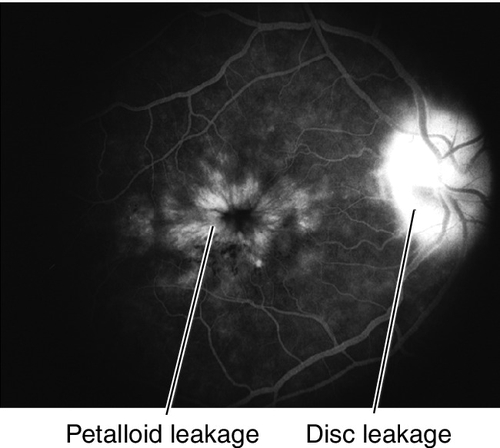
Figure 10-94 Fluorescein angiogram of same patient as shown in Figure 10-93 demonstrating characteristic petalloid appearance with optic nerve leakage.
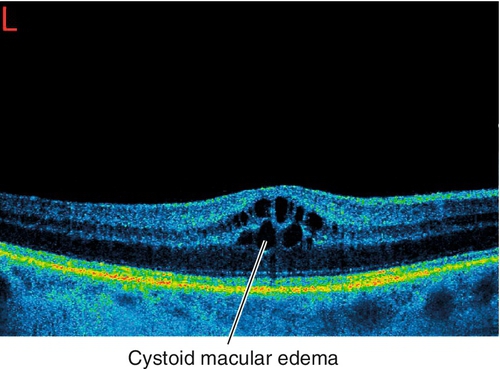
Differential Diagnosis
Macular hole (stage 1), foveal retinoschisis, central serous retinopathy, choroidal neovascular membrane, pseudocystoid macular edema (no leakage on fluorescein angiography) such as x-linked retinoschisis, Goldmann–Favre disease, and nicotinic acid maculopathy.
Evaluation
• Fluorescein angiogram: Early, perifoveal, punctate hyperfluorescence and characteristic late leakage in a petalloid pattern. Late leakage of optic nerve head seen with Irvine–Gass syndrome. Note: No leakage occurs in pseudocystoid macular edema from juvenile retinoschisis, nicotinic acid (niacin) maculopathy, Goldmann–Favre disease, and some forms of retinitis pigmentosa.
• Optical coherence tomography: Increased retinal thickness with round, cystoid spaces and loss of normal foveal contour with or without subsensory fluid.
Prognosis
Usually good; spontaneous resolution in weeks to months (postsurgical); poorer for chronic CME (> 6 months), may develop macular hole.
Macular Hole
Definition
Retinal hole in the fovea.
Etiology
Idiopathic; other risk factors are cystoid macular edema, vitreomacular traction, trauma, post surgery, myopia, post laser treatment and post inflammatory.
Epidemiology
Senile (idiopathic) macular holes (83%) usually occur in women (3 : 1) aged 60–80 years old; traumatic holes rare (5%); 25–30% are bilateral.
Symptoms
Decreased vision, metamorphopsia, and less commonly central scotoma.
Signs
Decreased visual acuity ranging from 20 / 40 in stage 1 to 20 / 100 to HM in stages 3 / 4; retinal detachments rare except in high myopes. Fundus findings were classified into five stages by Gass:
Stage 0
Vitreomacular adhesion or traction in fellow eye of patient with full-thickness macular hole in other eye.
Stage 1
Premacular hole (impending hole) with foveal detachment, absent foveal reflex, macular cyst (1A = yellow foveal spot, 100–200 μm in diameter, 1B = yellow ring, 200–350 μm in diameter); in OCT classification scheme.
Stage 2
Early, small, full-thickness hole either centrally within the ring or eccentrically at the ring’s margin. Seventy-five percent will progress to stage 3 or 4 holes.
Stage 3
Full-thickness hole (≥ 300 μm) with yellow deposits at level of retinal pigment epithelium (Klein’s tags), operculum, cuff of subretinal fluid, cystoid macular edema, and positive Watzke–Allen sign (subjective interruption of slit beam on biomicroscopy).
Stage 4
Stage 3 and posterior vitreous detachment (PVD).
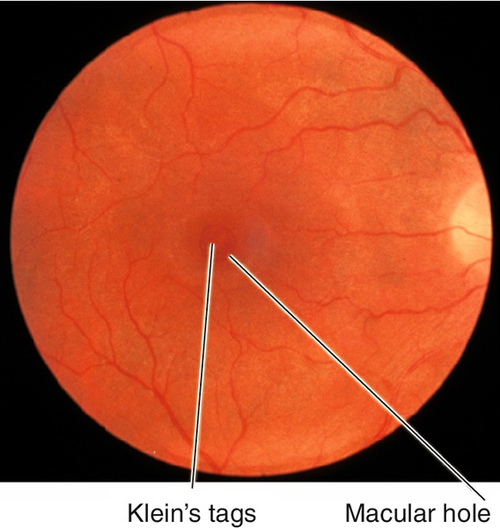
Figure 10-96 Macular hole with multiple yellow spots (Klein’s tags) at the base of the hole.
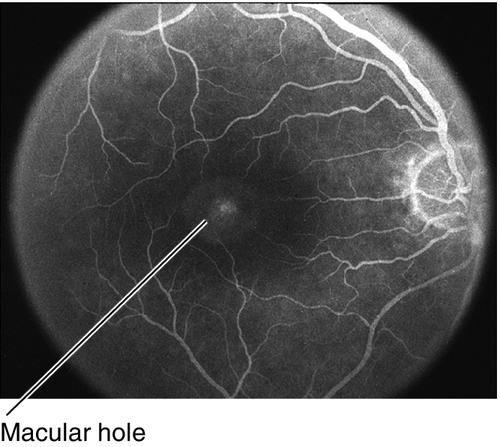
Figure 10-97 Fluorescein angiogram of same patient in Figure 10-96 demonstrating early hyperfluorescence of the hole that does not leak in late views.
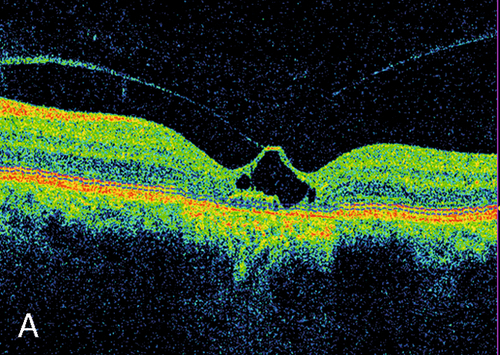
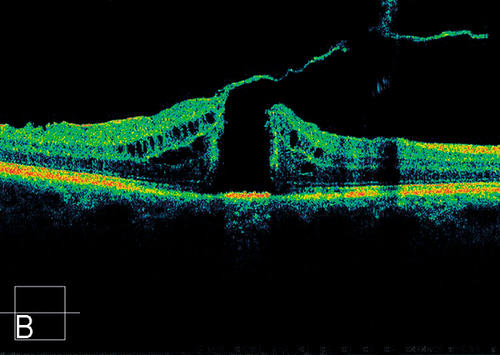
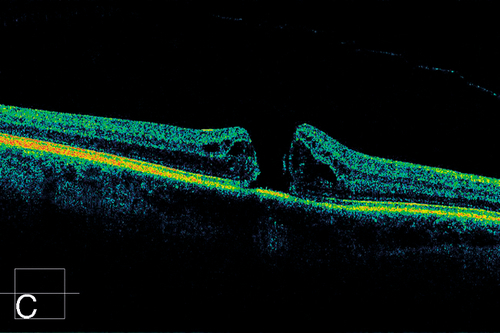
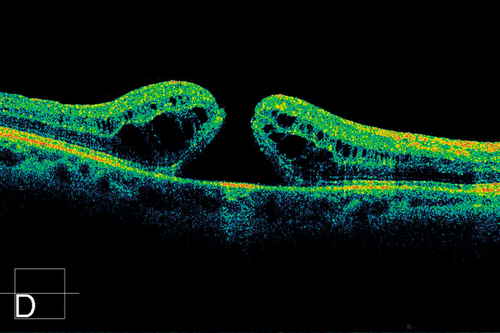
Also can be classified by OCT findings with subclassification based on size of smallest retinal aperature, presence/absence of traction, and presence/absence of other conditions (e.g., trauma, high myopia):
Medium: 250–400 μm
Large: > 400 μm.
Differential Diagnosis
Epiretinal membrane with pseudohole, solar retinopathy, central serous chorioretinopathy, vitreomacular traction syndrome, cystoid macular edema, solitary druse, and lamellar holes can appear clinically like MH, but are easily differentiated by OCT.
Evaluation
• Fluorescein angiogram: Hyperfluorescent window defect in the central fovea.
• Optical coherence tomography: Full-thickness defect in retina with or without traction on edges of hole; can differentiate lamellar holes and cysts from true macular holes; useful for determining treatment options and surgical planning.
Prognosis
Good for recent-onset holes; surgery has successful anatomic results in 60–95% depending on duration, of which 73% have improved acuity; preoperative visual acuity is inversely correlated with the absolute amount of visual improvement; poor for holes > 1 year’s duration.
Vitreomacular Adhesion and Traction
Vitreomacular traction (VMT) is defined as complete or partial adhesion of the vitreous cortex to the macular surface due to anomalous posterior vitreous detachment. Usually not symptomatic and can be observed until symptoms develop.
• Symptomatic vitreomacular traction can be treated with intravitreal injection of 2.5 mg / mL ocriplasmin (Jetrea) (MIVI-TRUST Study result). Good candidates would have one or more of the following features: no epiretinal membrane, adhesion < 1500 μm in width, age < 65 years, and phakic.
• In severe cases, vitrectomy and membrane peel can be performed by a retina specialist.
Epiretinal Membrane / Macular Pucker
Definition
Cellular proliferation along the internal limiting membrane and retinal surface; contraction of this membrane causes the retinal surface to become wrinkled (pucker/cellophane maculopathy).
Etiology
Risk factors include prior retinal surgery, intraocular inflammation, retinal vascular occlusion, sickle cell retinopathy, vitreous hemorrhage, trauma, macular holes, intraocular tumors such as angiomas and hamartomas, telangiectasis, retinal arterial macroaneurysms, retinitis pigmentosa, laser photocoagulation, PVD, retinal break, and cryotherapy; often idiopathic.
Epidemiology
Incidence increases with increasing age; it occurs in 2% of population > 50 years old and in 20% > 75 years old; 20–30% are bilateral, although often asymmetric. Slight female predilection (3 : 2); diabetes has been found to be associated with idiophathic ERMs.
Symptoms
Asymptomatic with normal or near-normal vision; mild distortion or blurred vision; less commonly macropsia, central photopsia, or monocular diplopia if macular pucker exists.
Signs
Normal or decreased visual acuity; abnormal Amsler grid; thin, translucent membrane appears as mild sheen (cellophane) along macula; may have dragged or tortuous vessels, retinal striae, pseudoholes, foveal ectopia, and cystoid macular edema. Occasionally multiple punctate hemorrhages occur in the inner retina.
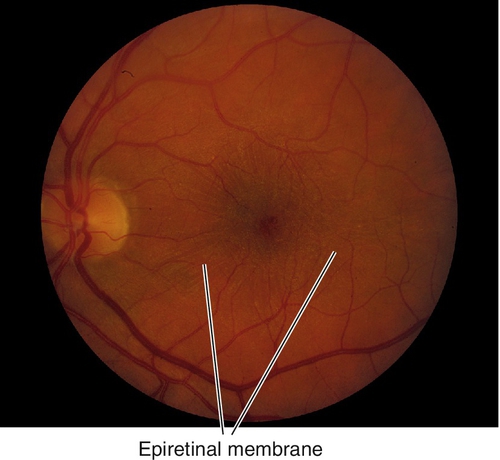
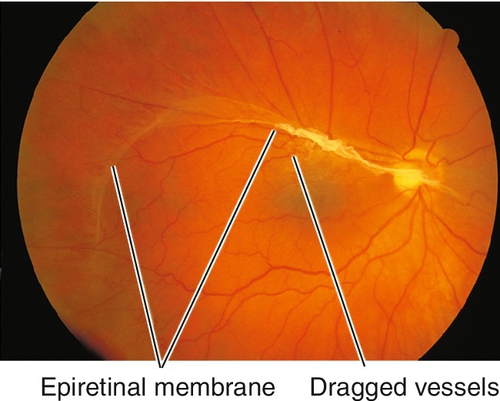
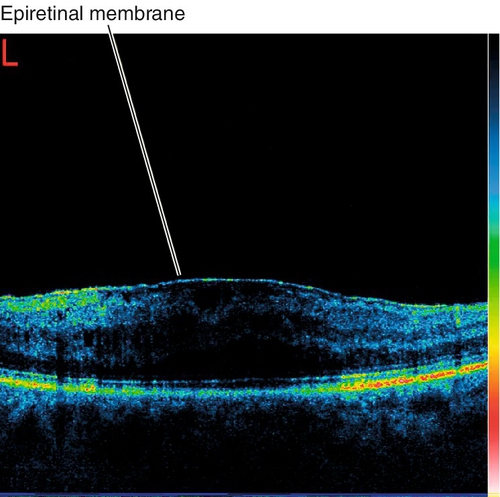
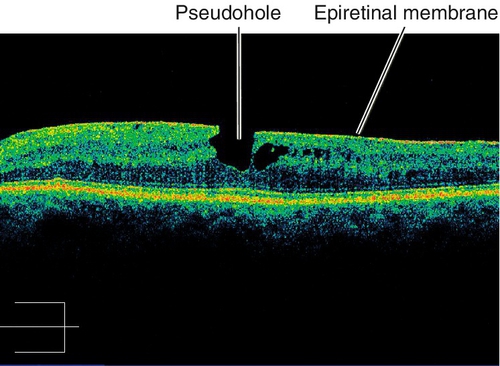
Differential Diagnosis
Traction retinal detachment from diabetic retinopathy, sickle cell retinopathy, or radiation retinopathy; choroidal folds.
Evaluation
• Optical coherence tomography to evaluate retinal thickening, hole status, and traction.
Prognosis
Good; 75% of patients have improvement in symptoms and acuity after surgery.
Myelinated Nerve Fibers
Abnormal myelination of ganglion cell axons anterior to the lamina cribosa; appears as yellow-white patches with feathery borders in the superficial retina (nerve fiber layer). Typically unilateral (80%) and occurs adjacent to the optic nerve, but can be located anywhere in the posterior pole. Obscures underlying retinal vasculature and can be confused with cotton-wool spots, astrocytic hamartomas, commotio retinae, or rarely retinal artery occlusion if extensive. Patients are usually asymptomatic, but scotomas corresponding to the areas of myelination can be demonstrated on visual fields; slight male predilection.
• Consider visual fields.
Solar / Photic Retinopathy
Bilateral decreased vision ranging from 20 / 40 to 20 / 100, metamorphopsia, photophobia, dyschromatopsia, after-images, scotomas, headaches, and orbital pain 1–4 hours after unprotected, long-term sun gazing. Retinal damage ranges from no changes to a yellow spot with surrounding pigmentary changes in the foveolar region in the early stages. Late changes include lamellar holes or depressions in the fovea. Vision can improve over 3–6 months, with residual scotomas and metamorphopsia. Similar problems may occur from unprotected viewing of lasers, welding arcs, and extended exposure to operating microscope lights (unilateral).
Toxic (Drug) Maculopathies
Aminoglycosides (Gentamicin / Tobramicin / Amikacin)
Aminoglycosides may be toxic when delivered into the eye by any technique including subconjunctival injection without apparent scleral perforation, diffusion through cataract wound from subconjunctival injection, or when used with a collagen corneal shield. Gentamicin (Garamycin) demonstrates more toxicity than amikacin (Amikin) or tobramycin (Nebcin). Toxicity, due to occlusion of the retinal capillaries by granulocytes, has occurred at doses as low as 0.1 mg of gentamicin or 0.2 mg of amikacin. It leads to acute, severe, permanent visual loss. Retinal toxic reaction with marked retinal whitening (especially in macula), arteriolar attenuation, venous beading, and widespread retinal hemorrhages; optic atrophy and pigmentary changes occur later. Poor visual prognosis.
• No effective treatment.
Canthaxanthine (Orobronze)
The carotenoid pigment canthaxanthine is prescribed for photosensitivity disorders and vitiligo. Toxicity produces characteristic refractile yellow spots in a wreath-like pattern around the fovea (gold-dust retinopathy). Usually asymptomatic or causes mild metamorphopsia and decreased vision while this oral tanning agent is being taken. Occurs with cumulative doses > 35 g.
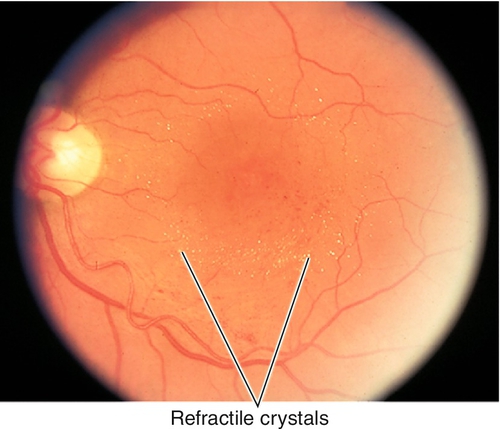
• Check visual fields (central 10°).
• Decrease or discontinue the medication if toxicity develops.
Chloroquine (Aralen) / Hydroxychloroquine (Plaquenil)
Quinolines were first used as an antimalarial agent in World War II and now are used to treat rheumatologic disorders such as systemic lupus erythematosis, rheumatoid arthritis, and for short-term pulse treatment for graft-versus-host disease, as well as amoebiasis. Toxicity produces central/paracentral scotomas, blurry vision, nyctalopia, photopsias, dyschromatopsia, photophobia, and, in late stages, constriction of visual fields, loss of color vision, decreased vision, and absolute scotomas. Early retinal changes include loss of foveal reflex and abnormal macular pigmentation (reversible); “bull’s eye” maculopathy (not reversible), peripheral bone spicules, vasculature attenuation, and disc pallor appear later; late stages can appear similar to end-stage retinitis pigmentosa. May also develop eyelash whitening and whorl-like subepithelial corneal deposits (cornea verticillata, vortex keratopathy). Doses > 3.5 mg / kg / day or 300 g total (chloroquine), and > 6.5 mg / kg / day of ideal body weight or 700 g total (hydroxychloroquine) may produce the maculopathy; total daily dose seems more critical than total accumulative dose; in patients with renal insufficiency, lower doses are required. Ideal body weight for men is calculated as 110 pounds (50 kg) for 5 feet (1.52 m) tall, plus 5 pounds (2.27 kg) for each inch (2.54 cm) in height over 5 feet, for women it is calculated as 100 pounds (45 kg) for 5 feet tall, plus 5 pounds for each inch in height over 5 feet. Quinolines are stored to a greater degree in lean body tissues than in fat; dosages based on actual, rather than ideal, body weight will lead to overdoses in obese patients; toxicity often progresses after medications are discontinued because the drug concentrates in the eye. Hydroxychloroquine appears safer since it does not readily cross the blood–retinal barrier (toxicity rarely occurs with use < 7 years).
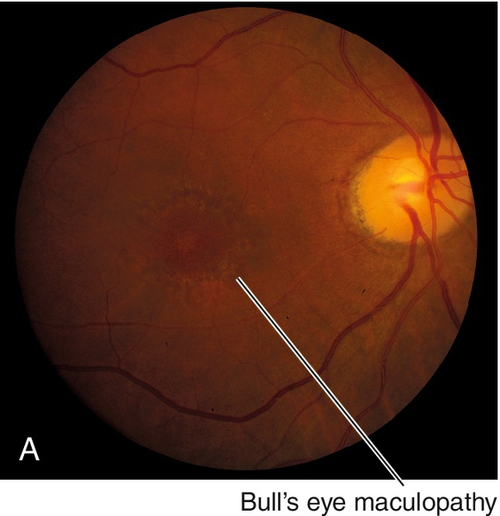
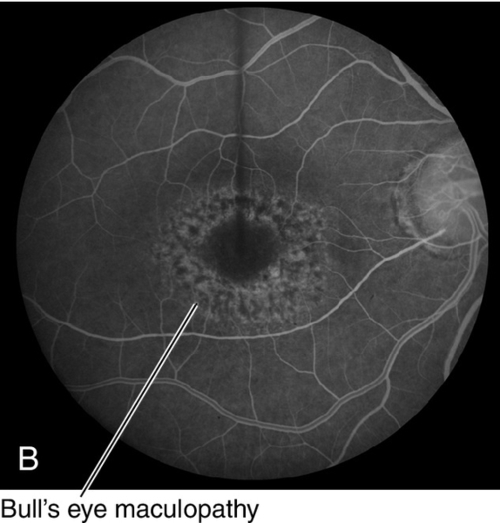
Figure 10-108 Bull’s eye maculopathy due to Plaquenil toxicity as seem on (A) clinical photo, and (B) fluorescein angiogram demonstrating same pattern with a circular window defect.
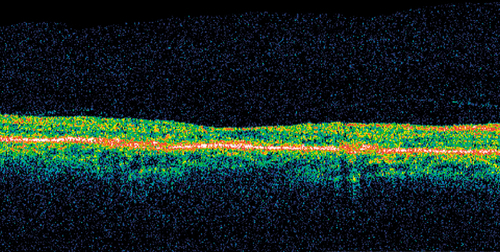
Figure 10-110 OCT of same patient as Figure 10-108 demonstrating thinning of the retina in the area of the bull’s eye.
• Check visual acuity, visual field (central 10° with white test object), and if available one or more of the following: spectral domain optical coherence tomography (flying saucer sign), multifocal electroretinogram, or fundus autofluoresence at baseline and every 6 months (chloroquine) or 12 months (hydroxychloroquine) after 5 years of use while patient is taking medications; patients with drug use > 5 years with high-fat-level body habitus, renal or liver disease, and age > 60 years old, especially if frail or extremely thin, are at higher risk of developing toxicity and should be checked more frequently.
• Low-risk patients (defined as nonobese individuals under age 60 years old, using less than 3 mg/ kg /day of chloroquine or 6.5 mg / kg /day of hydroxychloroquine for fewer than 5 years, and without concomitant renal, hepatic, or retinal disease) require no additional screening evaluations.
• Decrease or discontinue the medication if toxicity develops.
Chlorpromazine (Thorazine)
Patients have pigment deposition in eyelids, cornea, lens, and retina with toxic doses > 1200–1400 mg /day for at least 1 year.
• Decrease or discontinue the medication if toxicity develops.
Deferoxamine (Desferal)
Chelator of iron and aluminum that is prescribed for patients undergoing multiple blood transfusions. Toxicity causes decreased vision, nyctalopia, and visual field loss. The most common initial finding is a subtle gray macular discoloration, although a bull’s eye lesion may develop; a generalized pigmentary disturbance develops over weeks, which may persist despite drug discontinuation. Toxicity may occur after a single dose.
• Decrease or discontinue the medication if toxicity develops.
Interferon α
Interferon-α antiviral agents used to treat hepatitis cause vascular occlusion due to presumed immune-complex deposition. Toxicity causes cotton-wool spots, intraretinal hemorrhages, cystoid macular edema, capillary nonperfusion, and rarely vascular occlusion.
• Decrease or discontinue the medication if toxicity develops.
Methoxyflurane (Penthrane)
This inhaled anesthetic that is rarely used today may cause irreversible renal failure, partly through calcium oxalate crystalline deposition and retinal toxicity with yellow-white crystalline deposits in the posterior pole and along the arterioles. Methoxyflurane is metabolized to oxalate, which binds calcium to form insoluble calcium oxalate salts that are permanent.
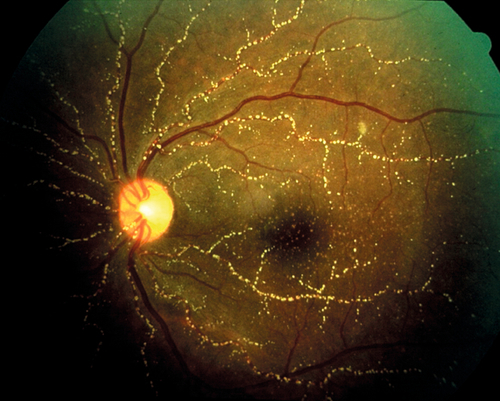
Niacin
Used to treat hypercholesterolemia. May produce decreased vision and metamorphopsia due to pseudocystoid macular edema (nicotinic acid maculopathy) caused by intracellular edema of Müller’s cells.
• Fluorescein angiogram: Early, perifoveal, punctate hyperfluorescence as in CME but no leakage.
• Optical coherence tomography: Cystoid spaces.
• Decrease or discontinue the medication if toxicity develops.
Quinine (Quinamm)
A quinolin, used to treat benign muscle cramps, that acutely causes retinal edema with venous engorgement and a cherry-red spot progressing to RPE mottling, retinal vascular attenuation, and optic atrophy; although the end stage resembles a vascular occlusion, the toxic effects appear to concentrate within the neurosensory retina. Toxicity causes generalized neurologic symptoms and blurred vision, visual field loss, and photophobia; acute overdose (single dose > 4 g) may cause permanent blindness.
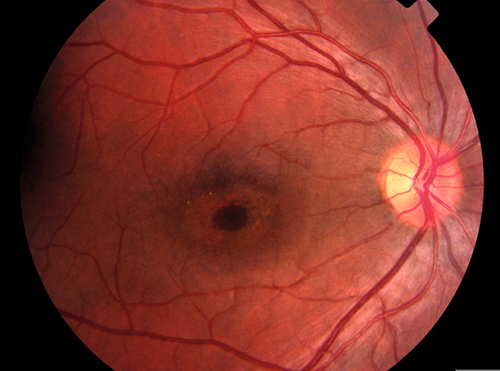
Sildenafil (Viagra)
This selective phosphodiesterase 5 (PDE-5) inhibitor commonly prescribed for erectile dysfunction, demonstrates cross-activity with the PDE-6 receptors in the photoreceptor layer. Produces reversible changes in color perception including a blue or blue-green tint or central haze of vision (may be pink or yellow); changes in light perception including darker colors appearing darker, increased perception of brightness, and flashing lights within 15–30 minutes of ingesting drug that peak within 1–2 hours; may also have photophobia and conjunctival hyperemia; resolves within 1 hour at doses < 50 mg, 2 hours with 100 mg, and 4–6 hours for 200 mg. The drug modifies the transduction cascade in photoreceptors (blocks PDE-5 10 × more than PDE-6 leading to interference in cGMP); occurs in 3% of patients taking a dose of 25–50 mg, 11% of those taking 100-mg dose, and in 40–50% of those taking > 100 mg; incidence is the same for all ages. No permanent visual effects have been reported; long-term effects are not known. Use with extreme caution in patients with retinitis pigmentosa (RP) and congenital stationary night blindness. There have been some reports of ischemic optic neuropathy, although no true association or causal relationship has been determined.
• Decrease or discontinue the medication if toxicity develops.
• No effective treatment of ischemic optic neuropathy.
Tadalafil (Cialis) / Vardenafil (Levitra)
Similar to sildenafil; there have been some reports of ischemic optic neuropathy, although no true association or causal relationship has been determined. The FDA has advised patients to discontinue the use of these medications if they experience sudden or decreased vision loss in one or both eyes.
• No effective treatment of ischemic optic neuropathy.
Talc
Magnesium silicate (talc) has no medicinal value, but serves as a vehicle for several oral medications, including methylphenidate (Ritalin) and methadone. Refractive yellow deposits near or in arterioles occur in IV drug abusers; similar findings occur in IV drug abusers injecting suspensions of crushed methylphenidate (Ritalin) tablets. Talc particles smaller than an erythrocyte will clear the pulmonary capillary network and enter the arterial system. Repeated intravenous injection appears to induce shunt formation, allowing larger particles access to the ophthalmic artery.
Tamoxifen (Nolvadex)
Used to treat metastatic breast adenocarcinoma. Produces refractile yellow-white crystals scattered throughout the posterior pole in a donut-shaped pattern, mild cystoid macular edema, and retinal pigmentary changes later; may develop whorl-like, white, subepithelial corneal deposits. Usually asymptomatic, but may cause mild decreases in vision and dyschromatopsia. Occurs with doses > 30 mg /day; at the initial higher dosage levels crystals often occur, but can resolve with a lowered dose.
• Fluorescein angiogram: Characteristic petalloid leakage from CME.
• Optical coherence tomography: Cystoid spaces from CME.
• Decrease or discontinue the medication if toxicity develops.
Thioridazine (Mellaril)
Phenothiazine, introduced in 1952 for the treatment of psychoses, may produce nyctalopia, decreased vision, ring/paracentral scotomas, and brown discoloration of vision. Pigment granularity/clumping in the midperiphery appears first (reversible), then progresses and coalesces into large areas of pigmentation (salt-and-pepper pigment retinopathy) or chorioretinal atrophy with short-term, high-dose use. A variant, termed nummular retinopathy, with chorioretinal atrophy posterior to the equator occurs with chronic use. Late stages can appear similar to end-stage retinitis pigmentosa or tapetoretinal degeneration with arteriolar attenuation, optic atrophy, and widespread pigmentary disturbances. Doses > 800 mg /day (300 mg recommended) can produce retinopathy; total daily dose seems more critical than total accumulative dose; may progress after medication is withdrawn because the drug is stored in the eye.
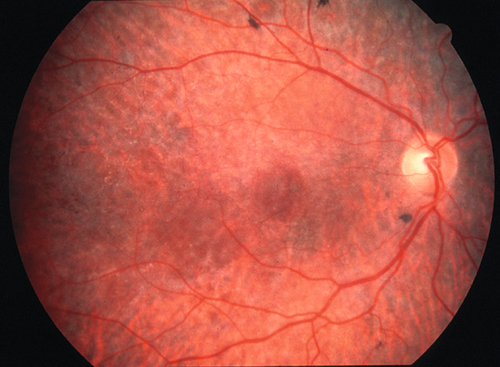
• Check vision, color vision and visual fields every 6 months while on medication.
• Fluorescein angiogram: Salt-and-pepper pattern of hypofluorescent spots and hyperfluorescent window defects; nummular pattern produces large areas of RPE loss.
• Optical coherence tomography: Inner retinal striae may be visible. All changes are reversible with cessation of the drug.
• Electrophysiologic testing: Electroretinogram (ERG) (normal early; reduced amplitude and abnormal dark adaptation later).
• Decrease or discontinue the medication if toxicity develops.
Topiramate
Oral anticonvulsant used for the treatment of seizures, prophylaxis for migraines and off-label in the treatment of bipolar disorder as well as second-line therapy for idiopathic intracranial hypertension for patients intolerant of acetazolamide. May produce induced myopia, bilateral angle-closure glaucoma, and retinal striae caused by vitreomacular traction. It is postulated that uveal effusion or ciliary edema leads to forward displacement of the lens–iris diaphragm and thickening of the lens by relaxation of the zonules. Laser iridotomy is not useful in correcting the angle closure as the mechanism of angle closure is not pupillary block.
• Optical coherence tomography: Inner retinal striae may be visible.
• All changes are reversible with cessation of the drug.
Lipid Storage Diseases
Sphingolipid storage diseases cause accumulation of ceramide in liposomes, especially in retinal ganglion cells, giving a characteristic cherry-red spot in the macula.
Farber’s Disease (Glycolipid) (Autosomal Recessive [AR])
Mild cherry-red spot, failure to thrive, subcutaneous nodules, hoarse cry, progressive arthropathy, and early mortality by 6–18 years of age.
Mucolipidosis (Mucopolysaccharidoses) (AR)
Cherry-red spot, nystagmus, myoclonus, corneal clouding, optic atrophy, cataracts, Hurler-like facies, hepatosplenomegaly, and failure to thrive.
Niemann–Pick Disease (Ceramide Phosphatidyl Choline) (AR)
Prominent cherry-red spot, corneal stromal opacities, splenomegaly, bone marrow foam cells, and hyperlipidemia.
Sandhoff’s Disease (Gangliosidosis Type II) (AR)
Prominent cherry-red spot and optic atrophy with associated lipid-storage problems in the kidney, liver, pancreas, and other gastrointestinal organs.
Tay–Sachs Disease (Gangliosidosis Type I) (AR)
Prominent cherry-red spot, blindness, deafness, convulsions; mainly occurs in Ashkenazic Jewish children.
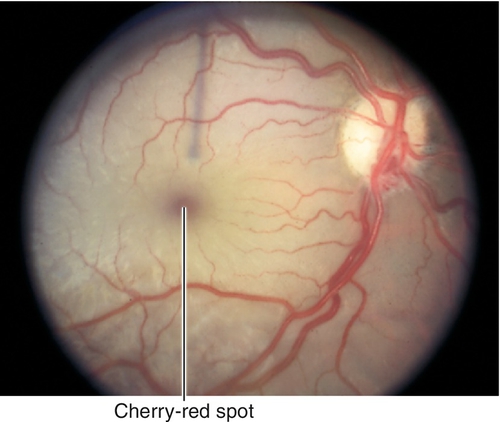
Peripheral Retinal Degenerations
Lattice Degeneration
Occurs in 7–10% of general population; more common in myopes; 33–50% are bilateral. Oval, circumferential area of retinal thinning and overlying vitreous liquefaction are found anterior to the equator; appears as criss-crossing, white lines (sclerotic vessels) with variable overlying retinal pigmentation that clusters in the inferior and superior peripheral retina. Atrophic holes (25%) are common; retinal tears can occur with posterior vitreous separation pulling on the atrophic, thinned retina; increased risk of retinal detachment.
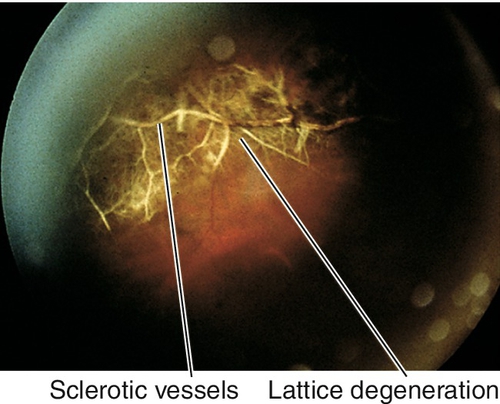
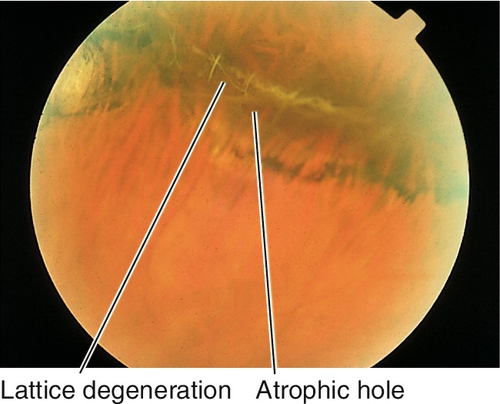
• Symptomatic lesions (photopsias/floaters) should receive prophylactic treatment with either cryopexy or two to three rows of laser photocoagulation around lattice degeneration and holes.
Pavingstone (Cobblestone) Degeneration
Occurs in 22–27% of general population; 33% bilateral. Appears as round, discrete, yellow-white spots ½ to 2 disc diameters in size with darkly pigmented borders found anterior to the equator adjacent to ora; corresponds to areas of thinned outer retina with loss of choriocapillaris and retinal pigment epithelium; usually found inferiorly; normal vitreous over lesions. May protect against retinal detachment due to adherence of thinned retina and choroid; increased incidence with age and myopia.
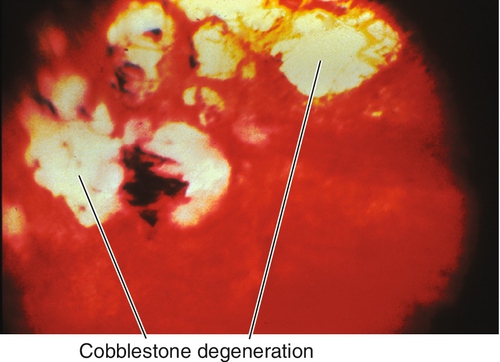
Peripheral Cystoid Degeneration
Clusters of tiny intraretinal cysts (Blessig–Iwanoff cysts) in the outer plexiform layer just posterior to ora serrata; the bubble-like cysts can coalesce and progress to typical degenerative retinoschisis; no increased risk of retinal detachment.
Snail Track Degeneration
Chains of fine, white dots that occur circumferentially in the peripheral retina; it is associated with myopia. Atrophic holes may develop in the areas of degeneration, increasing the risk of retinal detachment.
• Symptomatic lesions (photopsias/floaters) should receive prophylactic treatment with either cryopexy or two to three rows of laser photocoagulation around tears or holes.
Retinoschisis
Definition
Splitting of the retina. Two types:
Acquired
Senile, degenerative process with splitting between the inner nuclear and outer plexiform layers.
Juvenile
Congenital process with splitting of the nerve fiber layer.
Epidemiology
Acquired
More common; occurs in 4–7% of general population especially in patients > 40 years old; 50–75% bilateral, often symmetric; also associated with hyperopia.
Juvenile (X-linked recessive)
Onset in first decade; may be present at birth. Mapped to XLRS1 / Retinoschisin gene on chromosome Xp22 that codes proteins necessary for cell–cell adhesion; rarely autosomal; 98% bilateral.
Symptoms
Acquired
Usually asymptomatic and nonprogressive; may have visual field defect with sharp borders.
Juvenile
Decreased vision (often due to vitreous hemorrhage), or may be asymptomatic.
Signs
Acquired
Bilateral, smooth, convex, elevated schisis cavity usually in inferotemporal quadrant (70%); height of elevation constant even with change in head position; white dots (Gunn’s dots), “snowflakes” or “frosting” and sheathed retinal vessels (sclerotic in periphery) occur in the elevated inner retinal layer; outer layer breaks are common, large, well-delineated, have rolled margins, and appear pock-marked on scleral depression; inner layer breaks, vitreous hemorrhage, and rhegmatogenous retinal detachments are rare; intact outer retinal layer whitens with scleral depression; cystoid degeneration at the ora serrata; absolute scotoma.
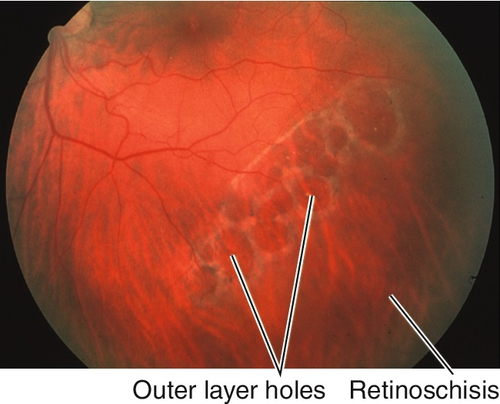
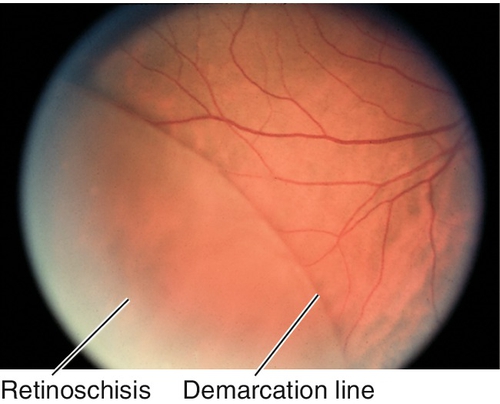
Juvenile
Slowly progressive decreased visual acuity ranging from 20 / 25 to 20 / 80; nystagmus and strabismus often occur; foveal schisis with fine, radiating folds from fovea (occurs in 100% of cases), spoke-like foveal cysts, pigment mottling, and microcystic foveal elevation (looks like cystoid macular edema but does not stain on fluorescein angiogram) are common; may have vitreous hemorrhage, vitreous veils, retinal vessels bridging inner and outer layers; peripheral retinoschisis (50%) with peripheral pigmentation and loss of retinal vessels, especially in inferotemporal quadrant, is often found.
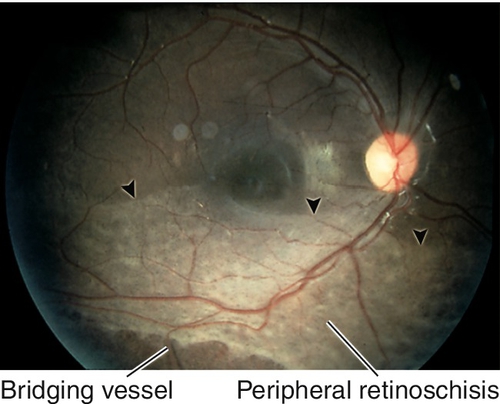
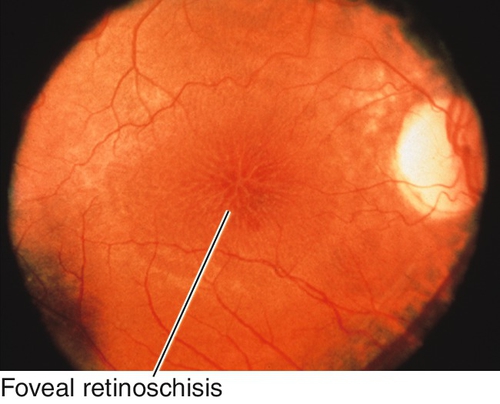
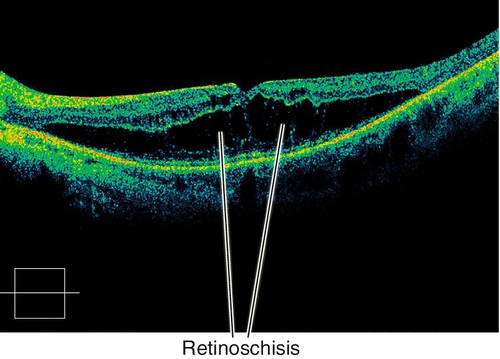
Differential Diagnosis
Retinal detachment, Goldmann–Favre disease, hereditary macular disease. Differentiating features from retinal detachment include no underlying RPE degeneration, blanching of RPE with laser treatment (no blanching with RD), no tobacco dust, and absolute scotoma (relative scotoma with RD).
Evaluation
• Color vision: Initial tritan defect followed by deutan–tritan defect (less severe than for cone–rod dystrophy).
• Visual fields: Absolute scotomas corresponding to areas of schisis.
• Fluorescein angiogram: Macular cysts in foveal schisis do not leak fluorescein.
• Optical coherence tomography: Macular cysts occur in foveal schisis; can also differentiate retinoschisis from retinal detachment.
• Electrophysiologic testing (in juvenile cases): Electroretinogram (ERG) (select decrease in b-wave amplitude, normal a-wave; Schubert–Bornsheim tracing or electronegative ERG), electro-oculogram (EOG) (normal in mild cases to subnormal in advanced cases), and dark adaptation (normal to subnormal).
Prognosis
Good; usually stationary for years.
Retinal Detachment
Separation of the neurosensory retina from the retinal pigment epithelium. Three types:
Rhegmatogenous Retinal Detachment
Definition
From Greek rhegma = rent; retinal detachment due to full-thickness retinal break (tear/hole/dialysis) that allows vitreous fluid access to subretinal space.
Etiology
Lattice degeneration (30%), posterior vitreous detachment (especially with vitreous hemorrhage), myopia, trauma (5–10%), and previous ocular surgery (especially with vitreous loss) increase risk of rhegmatogenous retinal detachments; retinal dialysis and giant retinal tears (> 3 clock hours in extent) more common after trauma.
Symptoms
Acute onset of photopsias, floaters (“shade” or “cobwebs”), shadow or curtain across visual field, decreased vision; may be asymptomatic.
Signs
Undulating, mobile, convex retina with corrugated folds; clear subretinal fluid that does not shift with body position; retinal break usually seen; may have “tobacco-dust” (Shafer’s sign: pigment cells in the vitreous), vitreous hemorrhage, or operculum; usually lower intraocular pressure in the affected eye and may have RAPD; chronic rhegmatogenous retinal detachments (RRD) may have pigmented demarcation lines, intraretinal cysts, fixed folds, or subretinal precipitates. Configuration of detachment helps localize retinal break:
Superotemporal/nasal detachment: Break within 1–1.5 clock hours of highest border.
Superior detachment that straddles 12 o’clock: Break between 11 and 1 o’clock.
Inferior detachment with one higher side: Break within 1–1.5 clock hours of highest border.
Inferior detachment equally high on either side: Break between 5 and 7 o’clock.
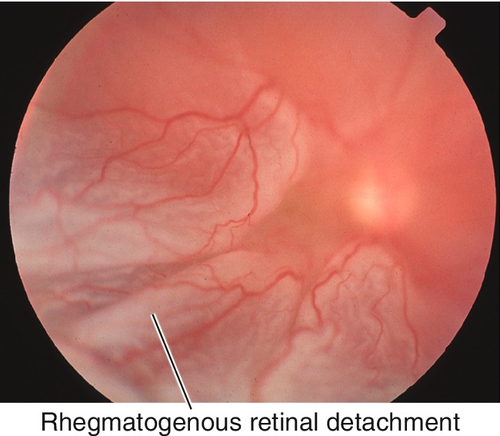
Figure 10-123 Rhegmatogenous retinal detachment demonstrating corrugated folds.
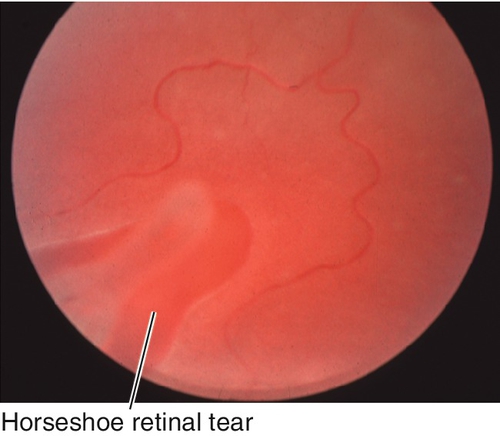
Figure 10-124 Same patient as Figure 10-123 demonstrating peripheral horseshoe tear that caused the rhegmatogenous retinal detachment.
Differential Diagnosis
Retinoschisis, choroidal detachment.
Evaluation
• B-scan ultrasonography: If unable to visualize the fundus; smooth, convex, freely mobile retina appears as highly reflective echo in the vitreous cavity that is attached at the optic nerve head and ora serrata; retinal tears can be visualized in the periphery.
Prognosis
Variable (depends on underlying etiology); 5–10% of rhegmatogenous retinal detachment repairs develop proliferative vitreoretinopathy (PVR).
Serous (Exudative) Retinal Detachment
Definition
Nonrhegmatogenous retinal detachment (not secondary to a retinal break) due to subretinal transudation of fluid from tumor, inflammatory process, vascular lesions, or degenerative lesions.
Etiology
Vogt–Koyanagi–Harada syndrome, Harada’s disease, idiopathic uveal effusion syndrome, choroidal tumors, central serous retinopathy, posterior scleritis, hypertensive retinopathy, Coats’ disease, optic nerve pit, retinal coloboma, and toxemia of pregnancy.
Symptoms
Usually asymptomatic until serous retinal detachment involves macula; may have acute onset of photopsias, floaters (“shade” or “cobwebs”), shadow across visual field, or decreased vision.
Signs
Smooth, serous elevation of retina; subretinal fluid shifts with changing head position; there is no retinal break by definition; mild RAPD may be observed.
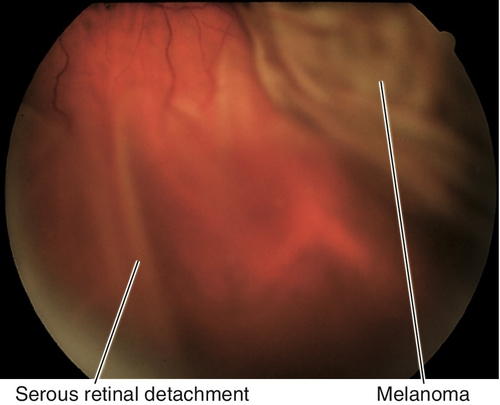
Differential Diagnosis
Retinoschisis, choroidal detachment, rhegmatogenous retinal detachment.
Evaluation
• B-scan ultrasonography: If unable to visualize the fundus, smooth, convex, freely mobile echoes that shifts with changing head position; retina appears as highly reflective echo in the vitreous cavity that is attached at the optic nerve head and ora serrata.
Prognosis
Variable (depends on underlying etiology).
Traction Retinal Detachment
Definition
Nonrhegmatogenous retinal detachment (not secondary to a retinal break) due to fibrovascular or fibrotic proliferation and subsequent contraction pulling retina up.
Etiology
Diabetic retinopathy, sickle cell retinopathy, retinopathy of prematurity, proliferative vitreoretinopathy, toxocariasis, and familial exudative vitreoretinopathy.
Symptoms
May be asymptomatic if traction retinal detachment does not involve macula; acute onset of photopsias, floaters (“shade” or “cobwebs”), shadow across visual field, or decreased vision.
Signs
Smooth, concave, usually localized, does not extend to the ora serrata; usually with fibrovascular proliferation; may have pseudoholes or true holes in a combined traction–rhegmatogenous detachment that progresses more rapidly than traction retinal detachment (TRD) alone; if a retinal tear develops, detachment may become convex.
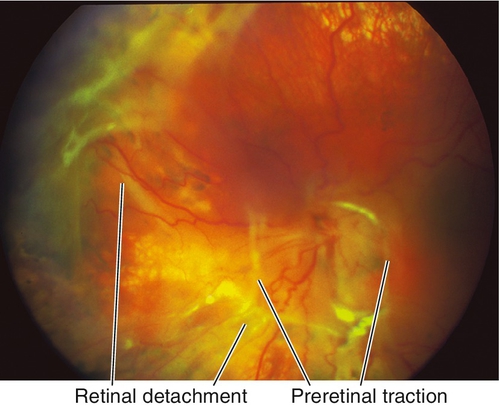
Differential Diagnosis
Retinoschisis, choroidal detachment, rhegmatogenous retinal detachment.
Evaluation
• B-scan ultrasonography: If unable to visualize the fundus; usually has tented appearance with vitreous adhesions; retina appears as highly reflective echo in the vitreous cavity that is attached at the optic nerve head and ora serrata.
Prognosis
Variable (depends on underlying etiology).
Choroidal Detachment
Smooth, bullous, orange-brown elevation of retina and choroid; usually extends 360° around the periphery in a lobular configuration; the ora serrata is visible without scleral depression. There are two forms:
Choroidal Effusion / Serous
Often asymptomatic with decreased intraocular pressure; may have shallow anterior chamber. Associated with acute ocular hypotony, postsurgical (excessive filtration through filtering bleb, wound leak, cyclodialysis cleft, postscleral buckling surgery), posterior scleritis, Vogt–Koyanagi–Harada syndrome, trauma (open globe), intraocular tumors, or uveal effusion syndrome. It transilluminates.
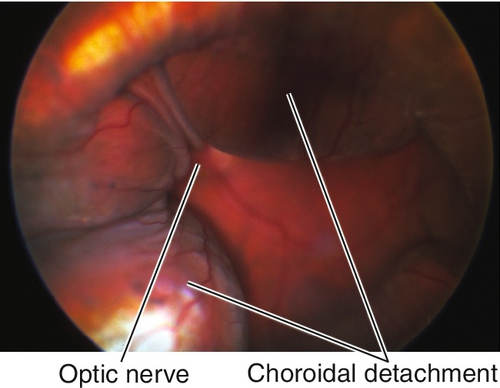
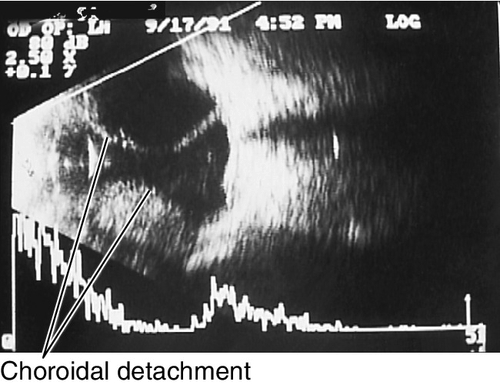
Choroidal Hemorrhage
Causes pain (often severe), decreased vision, red eye, intraocular inflammation, and increased intraocular pressure. Classically occurs acutely during anterior segment surgery, but may be delayed up to 1–7 days after surgery or trauma especially in patients with hypertension or taking anticoagulants. It does not transilluminate.
• Treat intraoperative choroidal hemorrhage with immediate closure of surgical wound and, if massive hemorrhage, perform sclerotomies to allow drainage of blood, and to close surgical wound; total intraoperative drainage is usually not possible.
• Topical cycloplegic (atropine 1% bid) and steroid (prednisolone acetate 1% qid); systemic steroids have been reported to have variable effect.
• May require treatment of increased intraocular pressure (see Primary Open-Angle Glaucoma section in Chapter 11).
• Consider surgical drainage when appositional or “kissing” (temporal and nasal choroid touch), severe intraocular pressure elevation despite maximal medical treatment, or corneal decompensation; visual results in appositional choroidal hemorrhage are very poor.
• Treat underlying condition.
Chorioretinal Folds
Definition
Folds of the choroid and retina.
Etiology
Compression of the sclera produces a series of folds in the inner choroid, Bruch’s membrane, RPE, and retina. This may be idiopathic or occur secondarily due to tumors (choroidal, orbital), hypotony, inflammation (posterior scleritis, idiopathic orbital inflammation, thyroid-related ophthalmopathy, and autoimmune disorders), choroidal neovascular membranes, papilledema, and extraocular hardware (scleral buckle, radiotherapy plaque, orbital implants for fractures).
Symptoms
Asymptomatic or may have metamorphopsia or decreased vision if folds involve fovea.
Signs
Normal or decreased visual acuity; may have true or induced hyperopia and abnormal Amsler grid (metamorphopsia); chorioretinal folds appear as curvilinear, parallel, or circular oriented alternating light and dark bands, usually in the posterior pole or temporal fundus; the crest of the fold is pale and broad, while the trough between the folds is darker and narrower; idiopathic folds are usually bilateral and symmetric, while unilateral folds are more common with tumors and external lesions. May have signs of the underlying etiology (i.e., scleral injection, wound leak, proptosis, choroidal lesion, optic disc swelling).
Differential Diagnosis
Retinal folds, which are usually due to epiretinal membranes (thinner, subtler, irregular folds that do not appear on fluorescein angiography), optic disc swelling (Paton’s lines), rhegmatogenous or traction retinal detachments, ROP, toxocariasis, and congenital.
Evaluation
• Fluorescein angiogram: Characteristic alternating bands of hyper- and hypofluorescence that correspond to the peaks (where the RPE is stretched) and troughs (where the RPE is compressed) of the folds, respectively.
• Optical coherence tomography: Scans perpendicular to the direction of the folds show the hollows and bulges of the folds.
• Consider lab tests (CBC, RF, ANA, C-ANCA) for suspected autoimmune disease.
• Consider B-scan ultrasonography for posterior scleritis and to rule out a tumor.
• Consider orbital CT scan for suspected retrobulbar mass, idiopathic orbital inflammation, and thyroid-related ophthalmopathy.
• May require medical consultation depending on the etiology.
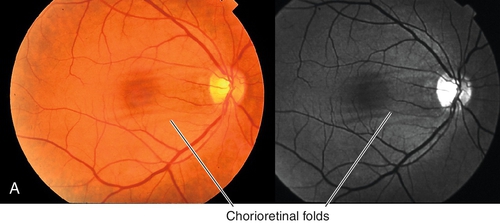
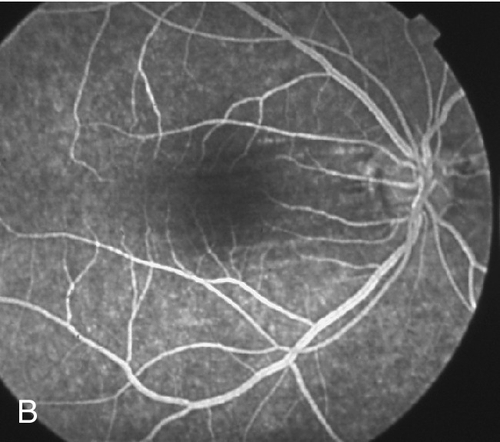
Prognosis
Depends on underlying etiology.
Chorioretinal Coloboma
Defect in retina, retinal pigment epithelium, and choroid due to incomplete closure of the embryonic fissure; usually located inferonasally. Variable sizes, may involve macula; appears as yellow-white lesion with pigmented margins. Associated with other ocular colobomata; increased risk of retinal detachment and CNV at margin of coloboma.
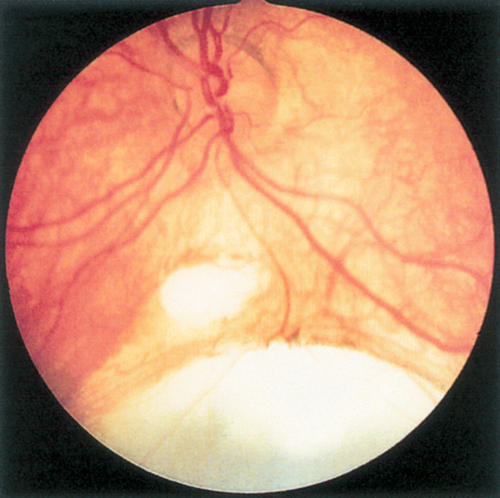
Proliferative Vitreoretinopathy
Fibrotic membranes composed of retinal pigment epithelial, glial, and inflammatory cells that form after retinal detachment or retinal surgery (8–10%); the membranes contract and pull on the retinal surface (6–8 weeks after surgery); may be preretinal or subretinal; primary cause of redetachment after successful retinal detachment surgery. Risk factors include previous retinal surgery, vitreous hemorrhage, choroidal detachment, giant retinal tears, multiple retinal breaks, penetrating trauma, excessive cryotherapy, and failure to reattach the retina at primary surgery. Final anatomic reattachment rate is 72–96%, variable visual prognosis (14–37% achieve > 20 / 100 vision).
Intermediate Uveitis / Pars Planitis
Definition
Intermediate uveitis is an inflammation primarily limited to the vitreous cavity that usually involves the pars plana and ciliary body of unknown etiology. Pars planitis is a form of intermediate uveitis, classically with vitritis, pars plana exudate, and peripheral retinal vasculitis of unknown etiology.
Epidemiology
Occurs in children and young adults; average age 23–28 years old; 75–90% are bilateral; associated with multiple sclerosis (up to 15%) and sarcoidosis. No sexual predilection; rare in African Americans and Asians. Represents roughly 5–8% of all uveitis cases with an incidence between 2 and 5 : 100,000.
Symptoms
Decreased vision, floaters; no red eye, pain, or photophobia.
Signs
Decreased visual acuity, fibrovascular exudates especially along the inferior pars plana (“snow balls or snow bank”), extensive vitreous cells (100%), vitreous cellular aggregates (“snowballs”) inferiorly, posterior vitreous detachment, vasculitis (10–32%), periphlebitis, cystoid macular edema (50–85%), and papillitis (3–20%); minimal anterior segment findings including mild anterior chamber cells and flare, fine keratic precipitate, posterior synechia, and endotheliitis; may develop neovascularization and vitreous hemorrhage in the pars plana exudate.
Differential Diagnosis
Sarcoidosis, multiple sclerosis, tuberculosis, toxocariasis, Lyme disease, Behçet’s disease, masquerade syndromes (especially lymphoma), syphilis, cat-scratch disease, leptospirosis, Whipple’s disease, HTLV-1-associated uveitis, posterior uveitis, amyloidosis, familial exudative vitreoretinopathy, Irvine–Gass syndrome (cystoid macular edema after cataract extraction), toxoplasmosis, candidiasis, fungal endophthalmitis, Eales’ disease, Vogt–Koyanagi–Harada (VKH) syndrome, Fuchs heterochromic iridocyclitis, and retinoblastoma.
Evaluation
• Lab tests: Are used to rule out other causes from differentital diagnosis although HLA-DR2 sometimes associated. ACE, chest radiographs, and serum lysozyme (sarcoidosis), CBC (masquerade syndromes), VDRL, FTA-ABS, Lyme titers, toxocariasis and toxoplasmosis IgG and IgM serology (infection).
• Fluorescein angiogram: Petalloid leakage from cystoid macular edema occurs in late views.
• CT scan of the chest to rule out mediastinal lymphadenopathy.
• Consider brain MRI or lumbar puncture to rule out multiple sclerosis if high level of suspicion.
Prognosis
Fifty-one percent of patients will achieve 20 / 30 vision; 10–20% may have self-limited disease; 40–60% will have a smoldering, chronic course with episodic exacerbations and remissions. Macular edema generally determines visual outcome. Zero tolerance for inflammation and aggressive treatment of active inflammation is a key factor in determining a good outcome.
Neuroretinitis (Leber’s Idiopathic Stellate Neuroretinitis)
Definition
Optic disc edema and macular star formation with no other systemic abnormalities.
Etiology
Due to pleomorphic Gram-negative bacillus Bartonella henselae (formerly known as Rochalimaea); associated with cat-scratch disease.
Symptoms
Mild, unilateral decreased vision, rarely pain with eye movement; may have viral prodrome (52%) with fever, malaise, lymphadenopathy, upper respiratory, gastrointestinal, or urinary tract infection.
Signs
Decreased visual acuity, visual field defects (cecocentral/central scotomas), RAPD, optic disc edema with macular star, peripapillary exudative retinal detachment, vitreous cells, rare anterior chamber cells and flare, yellow-white lesions at level of retinal pigment epithelium.
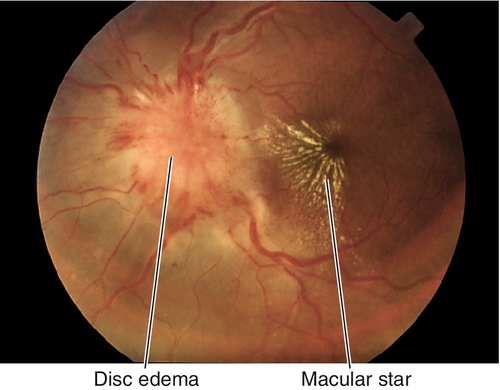
Differential Diagnosis
Hypertensive retinopathy, diabetic retinopathy, anterior ischemic optic neuropathy (AION), retinal vein occlusion, syphilis, diffuse unilateral subacute neuroretinitis (DUSN), acute macular neuroretinopathy, viral retinitis, sarcoidosis, toxocariasis, toxoplasmosis, tuberculosis, papilledema.
Evaluation
• Check blood pressure.
• Lab tests: VDRL, FTA-ABS, PPD and controls, indirect fluorescent antibody test for Bartonella henselae (Rochalimaea).
• Fluorescein angiogram: Leakage from optic disc capillaries, no perifoveal leakage.
Prognosis
Good; 67% regain ≥ 20 / 20 vision, and 97% > 20 / 40 vision; usually spontaneous recovery; disc edema resolves over 8–12 weeks, macular star over 6–12 months; optic atrophy may develop.
Posterior Uveitis: Infections
Acute Retinal Necrosis
Fulminant retinitis/vitritis due to the herpes zoster virus (HZV), herpes simplex virus (HSV), or rarely cytomegalovirus (CMV). Occurs in healthy, as well as immunocompromised, patients; male predilection (2 : 1 over females). Patients have pain, decreased vision, and floaters after a recent herpes simplex or zoster infection. Starts with small, well-demarcated, areas of retinal necrosis outside the vascular arcades that spread rapidly and circumferentially into large, confluent areas of white, retinal necrosis with retinal vascular occlusions and small satellite lesions; 36% bilateral (BARN); associated with granulomatous anterior uveitis and retinal vasculitis. In the cicatricial phase (1–3 months later), retinal detachments (50–75%) with multiple holes and giant tears are common; poor visual prognosis (only 30% achieve > 20 / 200 vision).
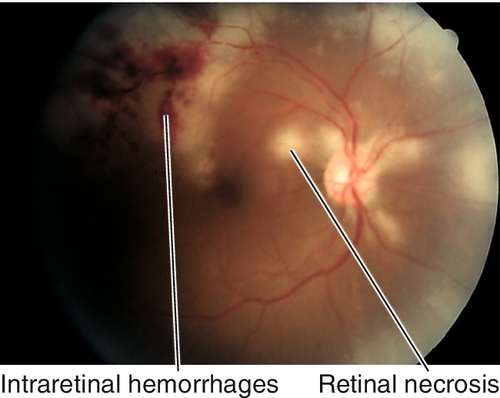
Figure 10-134 Acute retinal necrosis (ARN) demonstrating hemorrhage and yellow-white patches of necrosis.
[/level-membership-for-opthalmology-category][not-level-membership-for-opthalmology-category]10
Retina and Choroid
Branch Retinal Artery Occlusion
Central Retinal Artery Occlusion
Central / Hemiretinal Vein Occlusion
Coats’ Disease / Leber’s Miliary Aneurysms
Familial Exudative Vitreoretinopathy and Norrie’s Disease
Retinopathies Associated with Blood Abnormalities
Acquired Retinal Arterial Macroaneurysm
Age-Related Macular Degeneration
Retinal Angiomatous Proliferation
Polypoidal Choroidal Vasculopathy
Myopic Degeneration / Pathologic Myopia
Central Serous Chorioretinopathy
Vitreomacular Adhesion and Traction
Epiretinal Membrane / Macular Pucker
Peripheral Retinal Degenerations
Proliferative Vitreoretinopathy
Intermediate Uveitis / Pars Planitis
Posterior Uveitis: White Dot Syndromes
Posterior Uveitis: Other Inflammatory Disorders
Posterior Uveitis: Evaluation / Management
Hereditary Chorioretinal Dystrophies
Hereditary Macular Dystrophies
Trauma
Choroidal Rupture
Tear in choroid, Bruch’s membrane, and retinal pigment epithelium (RPE) is usually seen after blunt trauma. Acutely, the rupture site may be obscured by hemorrhage; scars form over 3–4 weeks with RPE hyperplasia at the margin of the rupture site. Anterior ruptures are usually parallel to the ora serrata; posterior ruptures are usually crescent-shaped and concentric to the optic nerve. Patients may have decreased vision if commotio retinae or subretinal hemorrhage is present, or if the rupture is located in the macula; increased risk of developing a choroidal neovascular membrane (CNV) during the healing process (months to years after trauma). Good prognosis if the macula is not involved, but poor if the fovea is involved.
• No treatment recommended, unless CNV occurs.
• Laser photocoagulation of juxtafoveal and extrafoveal CNV; consider anti-VEGF agent for subfoveal CNV (experimental).
• Monitor for CNV with Amsler grid.
Commotio Retinae (Berlin’s Edema)
Gray-white discoloration of the outer retina due to photoreceptor outer segment disruption following blunt eye trauma; can affect any area of the retina and may be accompanied by hemorrhages or choroidal rupture. There is no intercellular edema; whitening is due to intracellular edema and disorganization of outer retinal layers. It is termed Berlin’s edema if involving the macula, and commotion retinae in all other areas. Can cause acute decrease in vision if located within the macula, which resolves as the retinal discoloration disappears; may cause permanent loss of vision if the fovea is damaged, but usually resolves without sequelae. Visual acuity does not always correlate with the degree of retinal whitening seen on exam. Occasionally, a macular hole can form in the area of commotio with variable prognosis.
• Fluorescein angiogram: Early blocked fluorescence in the areas of commotio retinae.
• No treatment recommended.
Purtscher’s Retinopathy
Multiple patches of retinal whitening, large cotton-wool spots, and hemorrhages that surround the optic disc following multiple long-bone fractures with fat emboli or severe compressive injuries to the chest or head. May have optic disc edema and a relative afferent pupillary defect (RAPD). Usually resolves over weeks to months.
In the absence of trauma, a Purtscher’s-like retinopathy may be associated with acute pancreatitis, collagen–vascular disease, leukemia, dermatomyositis, and amniotic fluid embolus.
Figure 10-4 Multiple patches of retinal whitening, cotton-wool spots, and intraretinal hemorrhages secondary to Purtscher’s retinopathy.
• Fluorescein angiogram: Leakage from retinal vasculature with late venous staining.
• No treatment recommended.
Traumatic Retinal Holes
Full-thickness tear in the retina, often horseshoe shaped; usually occurs along the vitreous base, posterior border of lattice degeneration, or at cystic retinal tufts (areas with strong vitreoretinal adhesions). As most patients are young, the formed vitreous tamponades the tear and prevents a retinal detachment. Associated with pigmented vitreous cells (“tobacco-dust”, also known as Schaffer’s sign), vitreous hemorrhages, operculum (often located over the retinal hole), and posterior vitreous detachment. Patients usually report photopsias and floaters that shift with eye movement. Liquefied vitreous can pass through the tear into the subretinal space, causing retinal detachment even months to years after the tear forms; chronic tears have a ring of pigment around the retinal hole.
Giant Retinal Tear
Traumatic retinal hole measuring > 90° in circumferential extent or > 3 clock hours.
Avulsion of Vitreous Base
Separation of vitreous base from ora serrata that is pathognomonic for trauma.
Oral Tear
Tear at the ora serrata due to split of vitreous that has a fish-mouth appearance.
Preoral Tear
Tear at anterior border of vitreous base most often occurs superotemporally.
Retinal Dialysis
Most common form after trauma; circumferential separation of the retina at the ora serrata, usually in superotemporal (22%) or inferotemporal (31%) quadrant. Risk of retinal detachment increases over time with 10% at initial examination and 80% by 2 years.
• Retinal surgery required if rhegmatogenous retinal detachment, retinal dialysis, avulsion of the vitreous base, or giant retinal tear exists; should be performed by a retina specialist.
Chorioretinitis Sclopeteria
Trauma to retina and choroid caused by transmitted shock waves from high-velocity projectile that causes choroidal rupture, retinal hemorrhages, and commotio retinae. Vitreous hemorrhage is common. Lesions heal with white fibrous scar and RPE changes. Low risk of retinal detachment in young patients with a formed vitreous; however, the appearance can simulate retinal detachment in these patients.
Figure 10-7 Chorioretinitis sclopeteria with subretinal hemorrhage and commotio retinae (same patient as Figure 1-6).
Hemorrhages
Preretinal Hemorrhage
Hemorrhage located between the retina and posterior vitreous face (subhyaloid) or under the internal limiting membrane of the retina (sub-ILM). Often amorphous or boat-shaped, with flat upper border and curved lower border, which obscures the underlying retina. Caused by trauma, retinal neovascularization (diabetic retinopathy, radiation retinopathy, breakthrough bleeding from a choroidal neovascular membrane), hypertensive retinopathy, Valsalva retinopathy, retinal artery macroaneurysm, posterior vitreous detachment, shaken-baby syndrome, or retinal breaks, and less frequently by vascular occlusion, retinopathy of blood disorders, or leukemia.
Intraretinal Hemorrhage
Bilateral intraretinal hemorrhages are associated with systemic disorders (e.g., diabetes mellitus and hypertension); unilateral intraretinal hemorrhages generally occur in venous occlusive diseases or ocular ischemic syndrome.
Flame-Shaped Hemorrhage
Located in the superficial retina oriented with the nerve fiber layer; feathery borders. Usually occurs in hypertensive retinopathy and vein occlusion; may be peripapillary in glaucoma, especially in normal-tension glaucoma (splinter hemorrhage) and disc edema.
Dot / Blot Hemorrhage
Located in the outer plexiform layer, confined by the anteroposterior orientation of the photoreceptor, bipolar, and Müller’s cells; round dots or larger blots. Usually occurs in diabetic retinopathy.
Roth Spot
Hemorrhage with white center that represents an embolus with lymphocytic infiltration. Classically associated with subacute bacterial endocarditis (occurs in 1–5% of such patients); also occurs in leukemia, severe anemia, sickle cell disease, collagen vascular diseases, diabetes mellitus, multiple myeloma, and acquired immunodeficiency syndrome (AIDS) (see Figures 10-40, 10-43).
Subretinal Hemorrhage
Amorphous hemorrhage located under the neurosensory retina or RPE; appears dark and is deep to the retinal vessels. Associated with trauma, subretinal and choroidal neovascular membranes, and macroaneurysms (see Figure 10-71).
All three types of hemorrhages may occur together in several disorders including age-related macular degeneration (AMD), acquired retinal arterial macroaneurysm, Eales’ disease, and capillary hemangioma.
Cotton-Wool Spot
Asymptomatic, yellow-white, fluffy lesions in the superficial retina (see Figure 10-4). Nonspecific finding due to multiple etiologies including: retinal ischemia (retinal vascular occlusions, severe anemia, ocular ischemic syndrome), emboli (Purtcher’s retinopathy [white blood cell emboli], intravenous drug abuse [talc], cardiac/carotid emboli, deep venous emboli), infections (acquired immunodeficiency syndrome, Rocky Mountain spotted fever, cat-scratch fever [Bartonella henselae], leptospirosis, onchocerciasis, bacteremia, fungemia), collagen vascular diseases (systemic lupus erythematosus, dermatomyositis, polyarteritis nordosa, scleroderma, giant cell arteritis), drugs (interferon, chemotherapeutic agents), neoplasms (lymphoma, leukemia, metastatic carcinoma, multiple myeloma), retinal traction (epiretinal membrane), trauma (nerve fiber layer laceration, long-bone fractures, severe chest compression [white blood cell emboli]), systemic diseases (acute pancreatitis, hypertension, diabetes mellitus, high-altitude retinopathy), and radiation. Appears as thickening of the nerve fiber layer on OCT. Thought to develop secondary to obstruction of a retinal arteriole with resultant ischemia leading to blockage of axoplasmic flow within the nerve fiber layer.
• Treat underlying etiology (identified in 95% of cases).
Branch Retinal Artery Occlusion
Definition
Disruption of the vascular perfusion in a branch of the central retinal artery, leading to focal retinal ischemia.
Etiology
Mainly due to embolism from cholesterol (Hollenhorst’s plaques), calcifications (heart valves), platelet–fibrin plugs (ulcerated atheromatous plaques due to arteriosclerosis); rarely due to leukoemboli (vasculitis, Purtcher’s retinopathy), fat emboli (long-bone fractures), amniotic fluid emboli, tumor emboli (atrial myxoma), or septic emboli (heart valve vegetations in bacterial endocarditis or IV drug abuse). The site of the obstruction is usually at the bifurcation of retinal arteries. May result from vasospasm (migraine), compression, or coagulopathies.
Epidemiology
Usually occurs in elderly patients (seventh decade); associated with hypertension (67%), carotid occlusive disease (25%), diabetes mellitus (33%), and cardiac valvular disease (25%). CRAO is more common (57%) than BRAO (38%) or cilioretinal artery occlusion (5%) (in 32% of eyes, a cilioretinal artery is present).
Symptoms
Sudden, unilateral, painless, partial loss of vision, with a visual field defect corresponding to the location of the occlusion. May have history of amaurosis fugax (fleeting episodes of visual loss), prior cerebrovascular accident (CVA), or transient ischemic attacks (TIAs).
Signs
Visual field defect with normal or decreased visual acuity; focal, wedge-shaped area of retinal whitening within the distribution of a branch arteriole; 90% involve temporal retinal vessels; emboli (visible in 62% of cases) or Hollenhorst’s plaques may be visible at retinal vessel bifurcations. Retinal whitening resolves over several weeks and visual acuity can improve. In chronic stages, arterial attenuation with sector nerve fiber layer loss may be seen; artery-to-artery collaterals may form and are pathognomonic.
Figure 10-13 Superior branch artery occlusion demonstrating retinal edema.
Figure 10-14 Fluorescein angiogram of same patient as Figure 10-13 demonstrating no filling of superior retinal vessels and delayed filling of affected veins.
Differential Diagnosis
Commotio retinae, branch retinal vein occlusion, CRAO with cilioretinal artery sparing, combined artery and vein occlusion.
Evaluation
• Check blood pressure.
• Lab tests: Fasting blood glucose (FBS), glycosylated hemoglobin, and complete blood count (CBC) with differential. Consider platelets, prothrombin time/partial thromboplastin time (PT / PTT), protein C, protein S, factor V Leiden mutation, antithrombin III, homocysteine level, antinuclear antibody (ANA), rheumatoid factor (RF), sickle cell disease, antiphospholipid antibody, serum protein electrophoresis, hemoglobin electrophoresis, Venereal Disease Research Laboratory (VDRL) test, and fluorescent treponemal antibody absorption (FTA-ABS) test in patients < 50 years of age. In patients > 50 years old, check erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) to rule out arteritic ischemic optic neuropathy due to giant cell arteritis. If positive and / or if the patient’s history and exam are consistent, start giant cell arteritis treatment immediately (see Chapter 11). If the BRAO is accompanied by optic nerve edema and / or retinitis, consider serologic testing for infectious etiologies such as Bartonella, Lyme, and toxoplasmosis.
• Fluorescein angiogram: Delayed or absent retinal arterial filling in a branch of the central retinal artery; delayed arteriovenous transit time; capillary nonperfusion in wedge-shaped area supplied by the branch artery; staining of occlusion site and vessel wall in late views. When occlusion dissolutes, retinal blood flow is usually restored.
• Optical coherence tomography (OCT): Thickened and hyperreflective inner retinal layers during acute occlusion that corresponds to intracellular edema. Reflectivity of outer retina is blocked. Later, the retina is thinned with atrophy of the inner retina.
• Consider B-scan ultrasonography or orbital computed tomography (CT) scan to rule out a compressive lesion if the history suggests this etiology.
• Medical consultation for complete cardiovascular evaluation including baseline electrocardiogram, echocardiogram (may require transesophageal echocardiogram to rule out valvular disease), and carotid Doppler ultrasonography.
• In patients < 50 years of age, a hypercoagulability evaluation should be considered.
Prognosis
Retinal pallor fades and circulation is restored over several weeks. Good if fovea is spared; 80% have ≥ 20 / 40 vision, but most have some degree of permanent visual field loss; 10% risk in fellow eye.
Central Retinal Artery Occlusion
Definition
Disruption of the vascular perfusion in the central retinal artery (CRAO) leading to global retinal ischemia.
Etiology
Due to emboli (only visible in 20–40% of cases) or thrombus at the level of the lamina cribosa; other etiologies are the same as for BRAO including temporal arteritis, leukoemboli in collagen vascular diseases, fat emboli, trauma (through compression, spasm, or direct vessel damage), hypercoagulation disorders, syphilis, sickle cell disease, amniotic fluid emboli, mitral valve prolapse, particles (talc) from IV drug abuse, and compressive lesions; associated with optic disc drusen, papilledema, prepapillary arterial loops, and primary open-angle glaucoma.
Epidemiology
Usually occurs in elderly patients; associated with hypertension (67%), carotid occlusive disease (25%), diabetes mellitus (33%), and cardiac valvular disease (25%). CRAO is more common (57%) than BRAO (38%) or cilioretinal artery occlusion (5%) (in 32% of eyes, a cilioretinal artery is present); rarely bilateral.
Symptoms
Sudden, unilateral, painless, profound loss of vision; may have history of amaurosis fugax (fleeting episodes of visual loss), prior CVA, or TIAs.
Signs
Decreased visual acuity in the count fingers (CF) to light perception (LP) range; RAPD may be present; diffuse retinal whitening and arteriole constriction with segmentation (boxcaring) of blood flow; visible emboli (20–40%) rarely occur in central retinal artery; cherry-red spot in the macula (thin fovea allows visualization of the underlying choroidal circulation). In ciliary retinal artery sparing CRAO (25%), a small wedge-shaped area of perfused retina may be present temporal to the optic disc (10% spare the foveola, in which case visual acuity improves to 20 / 50 or better in 80%). Note: Ophthalmic artery obstruction usually does not produce a cherry-red spot owing to underlying choroidal ischemia.
Figure 10-17 Cilioretinal artery sparing central retinal artery occlusion with patent cilioretinal artery allowing perfusion (thus no edema) in a small section of the macula.
Figure 10-18 Fluorescein angiogram of same patient in Figure 10-17 demonstrating no filling of retinal vessels except in cilioretinal artery and surrounding branches.
Differential Diagnosis
Ophthalmic artery occlusion, commotio retinae, cherry-red spot due to inherited metabolic or lysosomal storage diseases, methanol toxicity.
Evaluation
• Check blood pressure.
• Lab tests: Fasting blood glucose (FBS), glycosylated hemoglobin, and complete blood count (CBC) with differential. Consider platelets, prothrombin time/partial thromboplastin time (PT / PTT), protein C, Protein S, factor V Leiden mutation, antithrombin III, homocysteine level, antinuclear antibody (ANA), rheumatoid factor (RF), sickle cell disease, antiphospholipid antibody, serum protein electrophoresis, hemoglobin electrophoresis, Venereal Disease Research Laboratory (VDRL) test, and fluorescent treponemal antibody absorption (FTA-ABS) test in patients < 50 years of age. In patients > 50 years old, check erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) to rule out arteritic ischemic optic neuropathy due to giant cell arteritis. If positive and /or if the patient’s history and exam are consistent, start giant cell arteritis treatment immediately (see Chapter 11). If the CRAO is accompanied by optic nerve edema and /or retinitis consider serologic testing for infectious etiologies such as Bartonella, Lyme disease, and toxoplasmosis.
• Fluorescein angiogram: Delayed retinal arterial filling and arteriovenous transit time with normal choroidal filling and perfusion of optic nerve from ciliary branches; prolonged arteriovenous circulation times; extensive capillary nonperfusion.
• Optical coherence tomography: Thickened and hyperreflective inner retinal layers during acute occlusion that corresponds to intracellular edema. Reflectivity of outer retina is blocked. Later, the retina is thinned with atrophy of the inner retina.
• Electrophysiologic testing: ERG (reduced b-wave amplitude, normal a-wave).
• Consider B-scan ultrasonography or orbital CT scan to rule out compressive lesion if history suggests compression.
• Medical consultation for complete cardiovascular evaluation including electrocardiogram, echocardiogram (may require transesophageal echocardiogram to rule out valvular disease), and carotid Doppler ultrasound.
Management
• Treat immediately before starting workup (if patient presents within 24 hours of visual loss), but best hope is to treat within 90 minutes.
• Digital ocular massage to try to dislodge emboli.
• Systemic acetazolamide (Diamox 500 mg IV or po).
• Topical ocular hypotensive drops: β-blocker (timolol 0.5% 1 gtt q15min × 2, repeat as necessary).
• Anterior chamber paracentesis (immediately lowers IOP to 0 mmHg): This procedure is easily performed at the slit lamp after prepping the eye with topical anesthetic, broad-spectrum antibiotic, and povidone-iodine. A lid speculum is placed, the eye is grasped with forceps at the nasal limbus to prevent movement and provide counter-traction, and either a disposable microsurgical knife (15° or MVR blade) or else a 30-gauge inch (13 mm) needle on a 1 mL syringe without the plunger, is inserted parallel to the iris through the peripheral cornea at the temporal limbus. If necessary, gentle pressure can be applied to the posterior lip of the paracentesis site so that aqueous can be released in a controlled fashion. Treat with a topical broad-spectrum antibiotic (gatifloxacin [Zymaxid] or moxifloxacin [Vigamox] qid for 3 days).
• Consider admission to hospital for carbogen treatment (95% oxygen–5% carbon dioxide for 10 minutes q2h for 24–48 hours) to attempt to increase oxygenation and induce vasodilation.
• Unproven treatments include hyperbaric oxygen, antifibrinolytic drugs, retrobulbar vasodilators, sublingual nitroglycerine, and Nd : YAG laser to dislodge the emboli.
• If arteritic anterior ischemic optic neuropathy (see Chapter 11) is suspected: Systemic steroids (methylprednisolone 1 g IV qd in divided doses for 3 days, then prednisone at least 1 mg / kg po qd for at least a month with a very slow taper; decrease by no more than 2.5 mg /wk). Most patients will require a year of high-dose steroid treatment.
Prognosis
Retinal pallor fades and circulation is restored over several weeks. Poor prognosis; most have persistent severe visual loss with constricted retinal arterioles and optic atrophy. Rubeosis (20%) and disc /retinal neovascularization (2–3%) can rarely occur. Presence of visible embolus associated with increased mortality; most common cause of mortality is myocardial infarction.
Ophthalmic Artery Occlusion
Definition
Obstruction at the level of the ophthalmic artery that affects both the retinal and choroidal circulation leading to ischemia more severe than CRAO.
Etiology
Usually due to emboli or thrombus, but can be caused by any of the etiologies listed for CRAO.
Epidemiology
Usually occurs in elderly patients; associated with hypertension (67%), carotid occlusive disease (25%), diabetes mellitus (33%), and cardiac valvular disease (25%).
Symptoms
Sudden, unilateral, painless, profound loss of vision up to the level of light perception or even no light perception.
Signs
Marked constriction of the retinal vessels, marked retinal edema often without a cherry red spot (although it may be present); may have RAPD; later, optic atrophy, retinal vascular sclerosis, and diffuse pigmentary changes.
Differential Diagnosis
Central retinal artery occlusion, commotio retinae, cherry-red spot due to inherited metabolic or lysosomal storage diseases, methanol toxicity.
Evaluation
• Check blood pressure.
• Lab tests: Fasting blood glucose (FBS), glycosylated hemoglobin, and complete blood count (CBC) with differential. Consider platelets, prothrombin time/partial thromboplastin time (PT / PTT), protein C, Protein S, factor V Leiden mutation, antithrombin III, homocysteine level, antinuclear antibody (ANA), rheumatoid factor (RF), sickle cell disease, antiphospholipid antibody, serum protein electrophoresis, hemoglobin electrophoresis, Venereal Disease Research Laboratory (VDRL) test, and fluorescent treponemal antibody absorption (FTA-ABS) test in patients < 50 years of age. In patients > 50 years old, check erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) to rule out arteritic ischemic optic neuropathy due to giant cell arteritis. If positive and / or if the patient’s history and exam are consistent, start giant cell arteritis treatment immediately (see Chapter 11).
• Fluorescein angiogram: Delayed or absent choroidal and retinal vascular filling, extensive capillary nonperfusion.
• Electrophysiologic testing: ERG (reduced or absent a and b wave amplitudes).
• Medical consultation for complete cardiovascular evaluation including electrocardiogram, echocardiogram (may require transthoracic echocardiogram to rule out valvular disease), and carotid Doppler ultrasound.
Prognosis
Severe visual loss is usually permanent.
Branch Retinal Vein Occlusion
Definition
Occlusion of a branch retinal vein (BRVO). Two types:
Nonischemic (64%)
< 5 disc areas of capillary nonperfusion on fluorescein angiogram.
Ischemic
≥ 5 disc areas of capillary nonperfusion on fluorescein angiogram.
Etiology
Usually caused by a thrombus at arteriovenous crossings where a thickened artery compresses the underlying venous wall due to a common vascular sheath; associated with hypertension, coronary artery disease, diabetes mellitus, and peripheral vascular disease; rarely associated with hypercoagulable states (e.g., macroglobulinemia, cryoglobulinemia), hyperviscosity states (polycythemia vera, Waldenström’s macroglobulinemia), systemic lupus erythematosus, syphilis, sarcoid, homocystinuria, malignancies (e.g., multiple myeloma, polycythemia vera, leukemia), optic nerve drusen, and external compression. In younger patients, associated with oral contraceptive pills, collagen vascular disease, AIDS, protein S /protein C /antithrombin III deficiency, factor XII (Hageman factor) deficiency, antiphospholipid antibody syndrome, or activated protein-C resistance (factor V Leiden PCR assay).
Epidemiology
Usually occurs in elderly patients, 60–70 years old; associated with hypertension (50–70%), cardiovascular disease, diabetes mellitus, increased body mass index, and open-angle glaucoma; slight male and hyperopic predilection. Second most common vascular disease after diabetic retinopathy.
Symptoms
Sudden, unilateral, painless, visual field loss. Patients may have normal vision, especially when macula is not involved.
Signs
Quadrantic visual field defect; dilated, tortuous retinal veins with superficial, retinal hemorrhages, and cotton-wool spots in a wedge-shaped area radiating from an arteriovenous crossing (usually arterial over-crossing where an arteriole and venule share a common vascular sheath). More common superotemporally (60%) than inferotemporally (40%; rare nasally since usually asymptomatic). The closer the obstruction is to the optic disc, the greater the area of retina involved and the more serious the complications. Microaneurysms or macroaneurysms, macular edema (50%), epiretinal membranes (20%), retinal and /or iris /angle neovascularization (very rare), and vitreous hemorrhage may develop; neovascular glaucoma is rare.
Figure 10-19 Inferior branch retinal vein occlusion demonstrating wedge-shaped area of intraretinal hemorrhages and cotton-wool spots.
Figure 10-20 Fluorescein angiogram of same patient as Figure 10-19 demonstrating lack of perfusion in inferior retinal vein with blocking defects from the intraretinal hemorrhages. Site of occlusion is shown with an arrowhead.
Differential Diagnosis
Venous stasis retinopathy, ocular ischemic syndrome, hypertensive retinopathy, leukemic retinopathy, retinopathy of anemia, diabetic retinopathy, papilledema, papillophlebitis (in young patients).
Evaluation
• Check visual fields.
• Check blood pressure.
• Lab tests: Fasting blood glucose, glycosylated hemoglobin; consider CBC with differential, platelets, PT / PTT, ANA, RF, angiotensin converting enzyme (ACE), ESR, serum protein electrophoresis, lipid profile, hemoglobin electrophoresis (in African Americans), VDRL, and FTA-ABS depending on clinical situation. In a patient < 40 years old and in whom a hypercoagulable state is being considered: check human immunodeficiency virus (HIV) status, functional protein S assay, functional protein C assay, functional antithrombin III assay (type II heparin-binding mutation), antiphospholipid antibody titer, lupus anticoagulant, anticardiolipin antibody titer (IgG and IgM), homocysteine level (if elevated test for folate, B12, and creatinine), factor XII (Hageman factor) levels, and activated protein C resistance (factor V Leiden mutation PCR assay); if these tests are normal and clinical suspicion for a hypercoagulable state still exists: add plasminogen antigen assay, heparin cofactor II assay, thrombin time, reptilase time, and fibrinogen functional assay.
• Fluorescein angiogram: Delayed retinal venous filling in a branch of the central retinal vein, increased transit time in affected venous distribution, blocked fluorescence in areas of retinal hemorrhages, and capillary nonperfusion (ischemic defined as ≥ 5 disc areas of capillary nonperfusion) in the area supplied by the involved retinal vein. Retinal edema with cystic changes is not present acutely, but appears later. Wide-field angiography is being used increasingly to visualize peripheral nonperfusion.
• Optical coherence tomography: Monitor for cystic macular edema and intraretinal swelling. Useful to monitor treatment response.
• Medical consultation for complete cardiovascular evaluation.
Prognosis
Central / Hemiretinal Vein Occlusion
Definition
Occlusion of the central retinal vein (CRVO); hemiretinal occlusion (HRVO) occurs when the superior and inferior retinal drainage does not merge into a central retinal vein (20%) and is occluded (more like CRVO than BRVO). Two types:
Nonischemic / Perfused (67%)
< 10 disc areas of capillary nonperfusion on fluorescein angiogram.
Ischemic / Nonperfused
≥ 10 disc areas of capillary nonperfusion on fluorescein angiogram.
Etiology
Usually caused by a thrombus in the area of the lamina cribosa; associated with hypertension (60%), coronary artery disease, diabetes mellitus, peripheral vascular disease, and primary open-angle glaucoma (40%); rarely associated with hypercoagulable states (e.g., macroglobulinemia, cryoglobulinemia), hyperviscosity states especially in bilateral cases (polycythemia vera, Waldenström’s macroglobulinemia), systemic lupus erythematosus, syphilis, sarcoid, homocystinuria, malignancies (e.g., multiple myeloma, polycythemia vera, leukemia), optic nerve drusen, and external compression. In younger patients, associated with oral contraceptive pills, collagen vascular disease, acquired immunodeficiency syndrome (AIDS), protein S /protein C /antithrombin III deficiency, factor XII (Hageman factor) deficiency, antiphospholipid antibody syndrome, or activated protein C resistance (factor V Leiden polymerase chain reaction [PCR] assay).
Epidemiology
Usually occurs in elderly patients (90% are > 50 years old); slight male predilection. Ischemic disease is more common in older patients and those with cardiovascular disease. Younger patients can get inflammatory condition termed papillophlebitis or benign retinal vasculitis with benign clinical course.
Symptoms
Sudden, unilateral, loss of vision or less frequently history of transient obscuration of vision with complete recovery. Some report pain and present initially with neovascularization of the iris and neovascular glaucoma following a loss of vision 3 months earlier (“90-day glaucoma”). Patients may have normal vision if perfused, especially when the macula is not involved.
Signs
Decreased visual acuity ranging from 20 / 20 to hand motion (HM) with most worse than 20 / 200 (vision worse in ischemic type; usually > 20/ 200 in nonischemic); dilated, tortuous retinal veins with superficial, retinal hemorrhages, and cotton-wool spots in all four quadrants extending to periphery; optic disc hyperemia, disc edema, and macular edema common; RAPD (degree of defect correlates with amount of ischemia). Nonischemic disease rarely produces neovascularization; ischemic disease can produce rubeosis (20% in CRVO, rare in BRVO), disc/retinal neovascularization (border of perfused/nonperfused retina), neovascular glaucoma, and vitreous hemorrhages. Collateral optociliary shunt vessels between retinal and ciliary circulations (50%) occur late. Impending CRVO may have absence of spontaneous venous pulsations (but this can also occur in normal individuals). Transient patchy ischemic retinal whitening may occur early in nonischemic CRVO.
Differential Diagnosis
Venous stasis retinopathy, ocular ischemic syndrome, hypertensive retinopathy, leukemic retinopathy, retinopathy of anemia, diabetic retinopathy, radiation retinopathy, and papilledema.
Evaluation
• Check blood pressure.
• Lab tests: Fasting blood glucose, glycosylated hemoglobin; consider CBC with differential, platelets, PT / PTT, ANA, RF, ACE, ESR, serum protein electrophoresis, lipid profile, hemoglobin electrophoresis (in African American), VDRL, and FTA-ABS depending on clinical situation. In a patient < 40 years old and in whom a hypercoagulable state is being considered: check human immunodeficiency virus (HIV) status, functional protein S assay, functional protein C assay, functional antithrombin III assay (type II heparin-binding mutation), antiphospholipid antibody titer, lupus anticoagulant, anticardiolipin antibody titer (IgG and IgM), homocysteine level (if elevated test for folate, B12, and creatinine), factor XII (Hageman factor) levels, and activated protein C resistance (factor V Leiden mutation PCR assay); if these tests are normal and clinical suspicion for a hypercoagulable state still exists: add plasminogen antigen assay, heparin cofactor II assay, thrombin time, reptilase time, and fibrinogen functional assay.
• Fluorescein angiogram: Delayed retinal venous filling, increased transit time (> 20 seconds increases risk of rubeosis), extensive capillary nonperfusion (ischemic defined in CVOS as ≥ 10 disc areas of capillary nonperfusion), staining of vascular walls, and blocking defects due to retinal hemorrhages. Retinal edema with cystic changes that are not present acutely, but appear later. Wide-field angiography is being used increasingly to visualize peripheral nonperfusion.
• Optical coherence tomography: monitor for cystic macular edema and intraretinal swelling. Useful to monitor treatment response.
• Electrophysiologic testing: ERG (reduced b wave amplitude [< 60% of normal more likely ischemic], reduced b : a-wave ratio [< 1 associated with increased risk of ischemia and neovascularization], prolonged b-wave implicit time).
• Medical consultation for complete cardiovascular evaluation.
Prognosis
Clinical course is variable; evaluate monthly for first 6 months. Nonischemic type has better prognosis (10% will completely resolve). Risk of neovascularization depends on amount of ischemia (CVOS conclusion); 16% of nonischemic patients progress to ischemic disease; 60% of ischemic patients develop neovascularization and 33% develop neovascular glaucoma.
Venous Stasis Retinopathy
Milder form of nonischemic central retinal vein occlusion (CRVO) representing patients with better perfusion. Dot/blot/flame hemorrhages, dilated/tortuous vasculature, and microaneurysms occur, usually bilateral; more benign course. Associated with hyperviscosity syndromes including polycythemia vera, multiple myeloma, and Waldenström’s macroglobulinemia.
Ocular Ischemic Syndrome
Definition
Widespread ischemia of both the anterior and posterior segments of one eye due to ipsilateral carotid occlusive disease (less frequently obstruction of the ipsilateral ophthalmic artery), carotid dissection, or arteritis (rare).
Etiology
Due to a 90% or greater occlusion of the ipsilateral carotid artery or rarely ophthalmic artery.
Epidemiology
Usually occurs in patients aged 50–70 years old (mean = 65 years); 80% unilateral; male predilection (2 : 1). Associated with atherosclerosis, ischemic heart disease (50%), hypertension (67%), diabetes mellitus (50%), previous stroke (25%), and peripheral arterial disease (20%); rarely due to inflammatory conditions including giant cell arteritis. Blood flow to the eye is relatively unaffected until carotid obstruction exceeds 70%; ocular ischemic syndrome usually does not occur until it reaches 90% (decreasing CRA perfusion by 50%); 50% of patients have complete ipsilateral carotid artery obstruction.
Symptoms
Gradual loss of vision (90%) over days to weeks with accompanying dull eye pain/headache (40%) or “ocular angina”; patients may also report amaurosis fugax (10%) or a delayed recovery of vision after exposure to bright light due to impaired photoreceptor regeneration. May occur suddenly in 12% of cases where a cherry-red spot is also present.
Signs
Gradual or sudden decreased visual acuity ranging from 20 / 20 to NLP; retinal arterial narrowing and venous dilatation without tortuousity, retinal hemorrhages (80% midperipheral), microaneurysms, macular edema, cotton-wool spots, disc/retinal neovascularization (37%), and spontaneous pulsations of the retinal arteries; anterior segment signs including episcleral injection, corneal edema, anterior chamber cells and flare (keratic precipitates are absent and flare is often disproportionate to the amount of cell present), iris atrophy, chronic conjunctivitis, and rubeosis (66%) are common. Intraocular pressure may be elevated, but may also be normal even with 360° synechia. Light digital pressure on the globe through the eyelid often produces arterial pulsations (does not occur in other diseases in differential) and can shut down perfusion of the central retinal artery.
Differential Diagnosis
Nonischemic CRVO, venous stasis retinopathy, diabetic retinopathy, hypertensive retinopathy, aortic arch disease, parafoveal telangiectasis, radiation retinopathy, Takayasu’s disease.
Evaluation
• Check blood pressure.
• Fluorescein angiogram: Delayed arteriovenous transit time (> 11 seconds) in 95%; delayed or patchy choroidal filling (> 5 seconds) in 60%, arterial vascular staining in 85%.
• Electrophysiologic testing: ERG (reduced or absent a-wave and b-wave amplitudes).
• Medical consultation for complete cardiovascular evaluation including duplex and carotid Doppler ultrasound scans (≥ 90% obstruction of the ipsilateral internal or common carotid arteries). Carotid angiography is usually not needed except in cases where ultrasound is equivocal.
Prognosis
Poor prognosis; 5-year mortality rate is 40% mainly owing to cardiovascular disease. Sixty percent of patients have count fingers or worse vision at 1 year follow-up; only 25% have better than 20 / 50 vision. When rubeosis is present, 90% will be count fingers or worse within 1 year. One-third of patients have improved vision after carotid endarterectomy, one-third remain unchanged, and one-third worsen despite surgery.
Retinopathy of Prematurity
Definition
Abnormal retinal vasculature development in premature infants, especially after supplemental oxygen therapy.
Epidemiology
Usually bilateral; associated risk factors include premature birth (< 32 weeks’ gestation), low birth weight (< 750 g: 90% develop ROP and 16% develop threshold disease; 1000–1250 g: 45% develop ROP and 2% develop threshold disease), supplemental oxygen therapy (> 50 days), and a complicated hospital course.
Symptoms
Asymptomatic; later may have decreased vision.
Signs
Shallow anterior chamber, corneal edema, iris atrophy, poor pupillary dilation, posterior synechiae, ectropion uveae, leukocoria, vitreous hemorrhage, retinal detachment, and retrolental fibroplasia; may have strabismus.
International classification of ROP describes the retinal changes in five stages:
Stage 1
Thin, circumferential, flat, white, demarcation line develops between posterior vascularized and peripheral avascular retina (beyond line).
Stage 2
Demarcation line becomes elevated and organized into a pink-white ridge, no fibrovascular growth visible.
Stage 3
Extraretinal fibrovascular proliferation from surface of the ridge.
Stage 4
Dragging of vessels, and subtotal traction retinal detachment (4A is macula attached, 4B involves the macula).
Stage 5
Total retinal detachment (almost always funnel detachment).
International classification of ROP also describes the extent of retina involved by number of clock hours and location by zone (centered on optic disc, not the fovea because retinal vessels emanate from disc):
Zone 1
Inner zone (posterior pole) corresponding to the area enclosed by a circle around the optic disc with radius equal to twice the distance from the disc to the macula (diameter of 60°).
Zone 2
The area between zone 1 and a circle centered on the optic disc and tangent to the nasal ora serrata.
Zone 3
Remaining temporal crescent of retina (last area to become vascularized).
Finally, international classification of ROP defines “plus” disease:
“Plus” Disease
At least two quadrants (usually 6 or more clock hours) of shunted blood causing vascular engorgement in the posterior pole with tortuous arteries, dilated veins, pupillary rigidity due to iris vascular engorgement, and vitreous haze.
Differential Diagnosis
Coats’ disease, Eales’ disease, familial exudative vitreoretinopathy, sickle cell retinopathy, juvenile retinoschisis, persistent hyperplastic primary vitreous, incontinentia pigmenti (Bloch–Sulzberger syndrome), and other causes of leukocoria (see Chapter 7).
Evaluation
• The first exam should be either prior to discharge from the hospital, 4 weeks chronological age, or by 31 weeks postgestational age, whichever is later.
• Complete ophthalmic history with attention to birth history and birth weight.
• Complete eye exam with attention to iris, lens, and ophthalmoscopy (retinal vasculature and retinal periphery with scleral depression).
• Cycloplegic refraction as many develop refractive errors especially myopia.
• Pediatric consultation.
Prognosis
Depends on the amount and stage of ROP; 80–90% will spontaneously regress; may develop amblyopia, macular dragging, strabismus; stage 5 disease carries a poor prognosis (functional success in only 3%); may develop high myopia, glaucoma, cataracts, keratoconus, band keratopathy, and retinal detachment.
Coats’ Disease / Leber’s Miliary Aneurysms
Unilateral (80–95%), idiopathic, progressive, developmental retinal vascular abnormality (telangiectatic and aneurysmal vessels with a predilection for the macula); usually occurs in young males (10 : 1) < 20 years old (two-thirds present before age 10). Retinal microaneurysms, retinal telangiectasia, lipid exudation, “light-bulb” vascular dilatations, capillary nonperfusion and occasionally neovascularization, exudative retinal detachments, and subretinal cholesterol crystals occur primarily in the temporal quadrants, especially on fluorescein angiogram where microaneurysm leakage is common. May present with poor vision, strabismus, or leukocoria. Spectrum of disease from milder form in older patients with equal sex predilection and often bilateral (Leber’s miliary aneurysms) to severe form with localized exudative retinal detachments and yellowish subretinal masses, and is included in the differential diagnosis of leukocoria (Coats’ disease). Clinical course varies but generally progressive. Rarely associated with systemic disorders including Alport’s disease, fascioscapulohumeral dystrophy, muscular dystrophy, tuberous sclerosis, Turner’s syndrome, and Senior–Loken syndrome. On histopathologic examination there is loss of vascular endothelium and pericytes with subsequent mural disorganization. Classified into five stages:
Stage 1
Telangectasia only
Stage 2
Exudation (a = extrafoveal, b = subfoveal)
Stage 3
Exudative retinal detachment (a = subtotal, b = total)
Stage 4
RD with glaucoma
Stage 5
End-stage disease
Figure 10-32 Leber’s miliary aneurysms demonstrating dilated arterioles with terminal “light-bulbs.”
Figure 10-33 Fluorescein angiogram of same patient as Figure 10-32 demonstrating capillary nonperfusion, microaneurysms, and “light-bulb” vascular dilations.
• Treatment: Scatter laser photocoagulation to posterior or cryotherapy to anterior areas of abnormal vasculature, telangiectasia, and areas of nonperfusion when symptomatic. May require multiple treatment sessions. Goal is to ablate areas of vascular leakage and to allow resorption of exudate.
Familial Exudative Vitreoretinopathy and Norrie’s Disease (X-Linked Recessive)
(See Hereditary Vitreoretinal Degenerations section below.)
Incontinentia Pigmenti (X-Linked Dominant)
Ocular, CNS, dermatologic, and dental findings including skin blisters, retinal neovascularization, vitreous hemorrhage, and traction retinal detachment. Associated with mutation in the NEMO gene located on chromosome Xq28.
• Fluorescein angiogram: Shows perpheral nonperfusion; wide-angle angiography is especially useful.
• Treatment: Scatter laser photocoagulation to ischemic retina when neovascularization develops. Consider vitrectomy when traction retinal detachment or nonclearing vitreous hemorrhage is present should be performed by a retina specialist.
Eales’ Disease
Bilateral, idiopathic, peripheral obliterative vasculopathy that occurs in healthy, young adults aged 20–30 years old, with male predilection. Patients usually notice floaters and decreased vision and have areas of perivascular sheathing, vitreous cells, peripheral retinal nonperfusion, microaneurysms, intraretinal hemorrhages, white sclerotic ghost vessels, disc/iris/retinal neovascularization, and vitreous hemorrhages. Fibrovascular proliferation may lead to tractional retinal detachments. May have signs of ocular inflammation with keratic precipitates, anterior chamber cells and flare, and cystoid macular edema; variable prognosis. Eales’ disease is a diagnosis of exclusion; must rule out other causes of inflammation or neovascularization including BRVO, diabetic retinopathy, sickle cell retinopathy, multiple sclerosis, sarcoidosis, tuberculosis, SLE, and other collagen–vascular diseases.
• Treatment: Scatter laser photocoagulation to nonperfused retina when neovascularization develops. If vitreous hemorrhage obscures view of retina, peripheral cryotherapy can be applied to ablate peripheral avascular retina.
• Consider periocular or systemic steroids for inflammatory component.
Macular Telangiectasia (Idiopathic Juxtafoveal / Perifoveal Telangiectasia)
Group of retinal vascular disorders with abnormal perifoveal capillaries confined to the juxtafoveal region (1–199 μm from center of fovea). Several forms:
Type 1A (Unilateral Congenital Parafoveal Telangiectasia)
Occurs in men in the fourth to fifth decades. Yellow exudate at outer edge of telangiectasis usually temporal to the fovea and 1–2 disc diameters in area; decreased vision ranging from 20 / 25 to 20 / 40 from macular edema and exudate. May represent mild presentation of Coats’ disease in an adult.
• Optical coherence tomography: Characteristic outer retinal hyporeflective cavities that do not correspond to leakage on FA. May eventually lead to atrophy.
•
[/not-level-membership-for-opthalmology-category]
