[level-membership-for-opthalmology-category]8
Lens
Congenital Anomalies
Definition
Variety of developmental lens abnormalities:
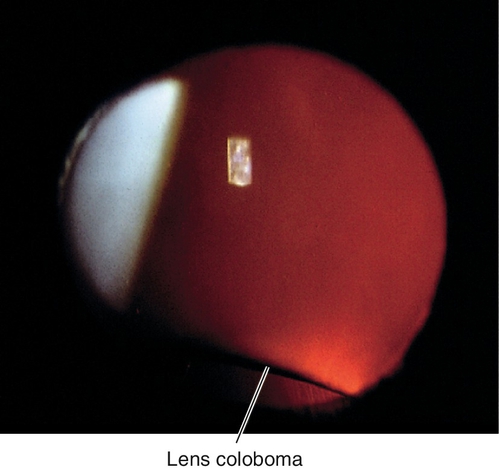
Coloboma
Focal inferior lens flattening due to ciliary body coloboma with absence of zonular support (not a true coloboma); other ocular colobomata (iris) usually exist. Ciliary body tumors may cause a secondary lens “coloboma.”
Lenticonus
Cone-shaped lens due to bulging from a thin lens capsule; either anteriorly or posteriorly, rarely in both directions.
Anterior
Usually males; bilateral; may be associated with Alport’s syndrome (see Congenital Cataract section below).
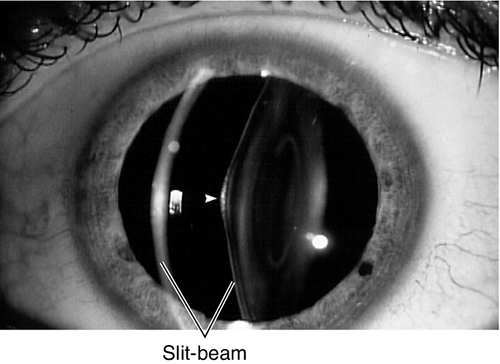
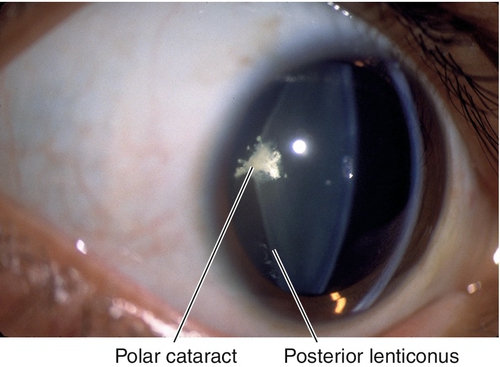
Posterior
More common; slight female predilection; may have associated cortical lens opacities, may be associated with Lowe’s syndrome (see Congenital Cataract section below).
Lentiglobus
Globe-shaped lens caused by bulging from thin lens capsule; rare.
Microspherophakia
Small spherical lens; may be an isolated anomaly or part of a syndrome (i.e., dominant spherophakia, Weill–Marchesani syndrome, Lowe’s syndrome, Alport’s syndrome, Peter’s anomaly, rubella).
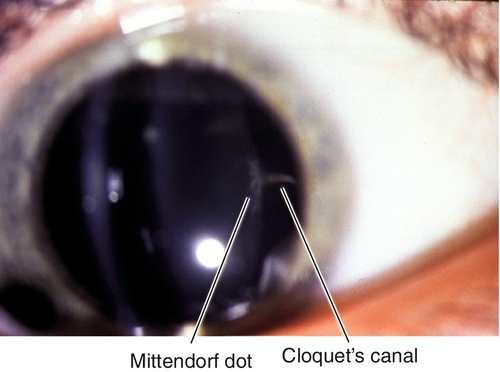
Mittendorf Dot
A small white spot on the posterior lens capsule that represents a remnant of the posterior tunica vasculosa lentis where the former hyaloid artery attached.
Symptoms
Asymptomatic (Mittendorf dot, coloboma); may have decreased vision (lenticonus, lentiglobus, and microspherophakia), diplopia, or symptoms of angle-closure glaucoma (microspherophakia).
Signs
Normal or decreased visual acuity; may have amblyopia, strabismus, nystagmus, myopia, and an “oil-droplet” fundus reflex on retroillumination in lenticonus and lentiglobus; may have dislocated lens and increased intraocular pressure in microspherophakia.
Differential Diagnosis
See above.
Evaluation
Prognosis
Usually good; poorer if amblyopia exists.
Congenital Cataract
Definition
Congenital opacity of the crystalline lens usually categorized by location or etiology:
Capsular
Opacity of the lens capsule, usually anteriorly.
Lamellar or Zonular
Central, circumscribed opacity surrounding the nucleus; “sand dollar” appearance.

Lenticular or Nuclear
Opacity of the lens nucleus.
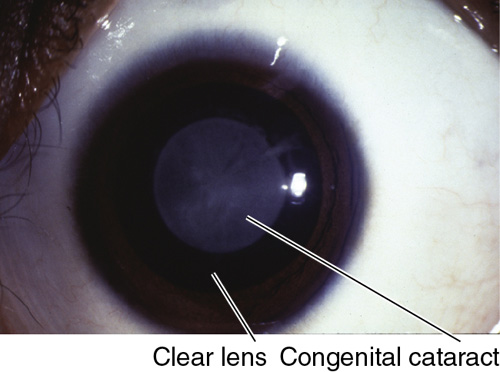
Figure 8-6 Congenital nuclear cataract with central white discoid appearance.
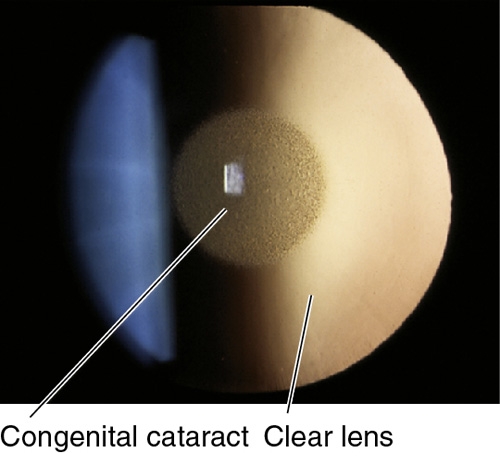
Figure 8-7 Same patient as Figure 8-6 demonstrating congenital nuclear cataract as viewed with retroillumination.
Polar
Central opacity located near the lens capsule, anteriorly or posteriorly.
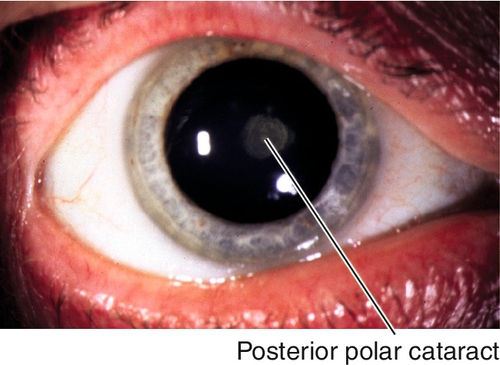
Figure 8-8 Congenital posterior polar cataract.
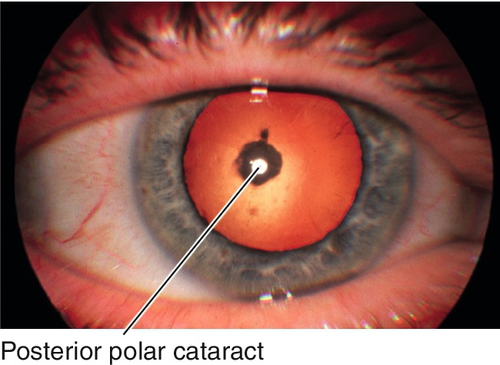
Figure 8-9 Same patient as Figure 8-8 demonstrating congenital polar cataract in retroillumination.
Sutural
Opacity of the Y-shaped sutures in the center of the lens.
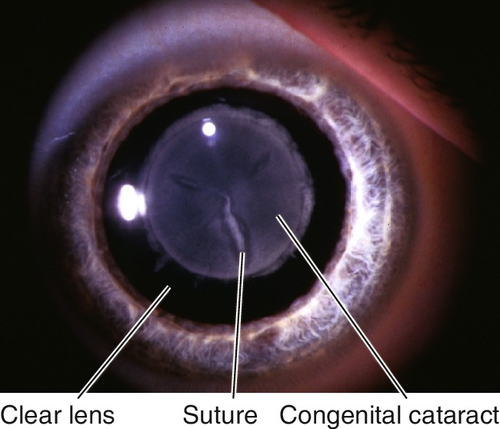
Figure 8-10 Congenital cataract with prominent suture lines.
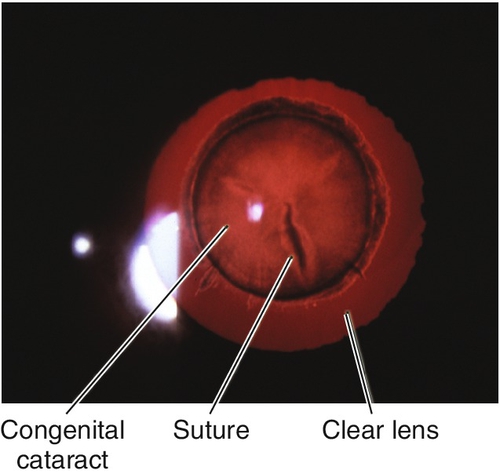
Figure 8-11 Same patient as Figure 8-10 demonstrating congenital sutural cataract in retroillumination.
Etiology
Hereditary or Syndromes
Without chromosomal abnormalities
Autosomal dominant (AD), autosomal recessive (AR), X-linked.
With chromosomal abnormalities
Down’s syndrome (snowflake cataracts), Turner’s syndrome, and others.
Other syndromes
Craniofacial, central nervous system, skin.
Intrauterine Infections
Congenital rubella syndrome
Cataracts, glaucoma, microcornea, microphthalmos, iris hypoplasia, and retinopathy with characteristic fine, granular, salt-and-pepper appearance (most common finding). Other complications include prematurity, mental retardation, neurosensory deafness, congenital heart disease, growth retardation, hepatosplenomegaly, interstitial pneumonitis, and encephalitis.
Congenital varicella syndrome
Cataracts, chorioretinitis, optic nerve atrophy or hypoplasia, nystagmus, and Horner’s syndrome. Systemic findings include hemiparesis, bulbar palsies, dermatomal cicatricial skin lesions, developmental delay, and learning difficulties.
Metabolic
Galactosemia
Bilateral, oil-droplet cataracts from accumulation of galactose metabolites (galactitol) due to hereditary enzymatic deficiency; usually galactose 1-phosphate uridyltransferase, also galactokinase. Associated with mental retardation, hepatosplenomegaly, cirrhosis, malnutrition, and failure to thrive. Mapped to chromosomes 1p (GALE gene), 9p (GALT gene) and 17q (GALK1 gene).
Lowe’s oculocerebrorenal syndrome (X-linked)
Small discoid lens, posterior lenticonus, and glaucoma. Systemic findings include acidosis, aminoaciduria, renal rickets, hypotonia, mental retardation. Female carriers have posterior, white, punctate cortical opacities and subcapsular, plaque-like opacities. Mapped to chromosome Xq25 (OCRL gene).
Alport’s syndrome (AD)
Basement membrane disease associated with acute hemorrhagic nephropathy, deafness, anterior lenticonus, anterior polar or cortical cataracts, and albipunctatus-like spots in the fundus. Mapped to chromosome Xq22 (COL4A5 gene [80% of cases], also COL4A3 and COL4A4 genes).
Other metabolic diseases
Hypoglycemia, hypocalcemia (diffuse lamellar punctate opacities), Fabry’s disease (spoke-like cataracts in 25%), mannosidosis (posterior spoke-like opacities).
Ocular Disorders
Persistent hyperplastic primary vitreous, Peter’s anomaly, Leber’s congenital amaurosis, retinopathy of prematurity, aniridia, posterior lenticonus, tumors.
Other
Birth trauma, idiopathic, and maternal drug ingestion.
Epidemiology
Congenital cataracts occur in approximately 1 of 2000 live births. Roughly one-third are isolated, one-third are familial (usually dominant), and one-third are associated with a syndrome; most unilateral cases are not metabolic or genetic.
Symptoms
Variable decreased vision; may notice white pupil or eye turn.
Signs
Decreased visual acuity, leukocoria, amblyopia; may have strabismus (usually with unilateral cataract), nystagmus (usually does not appear until 2–3 months of age; rarely when cataracts develop after age 6 months), amblyopia; may have other ocular or systemic findings if syndrome exists.
Differential Diagnosis
Evaluation
• Complete eye exam with attention to cycloplegic refraction, retinoscopy, tonometry, gonioscopy, lens (size and density of the opacity as viewed with retroillumination), and ophthalmoscopy.
• May require examination under anesthesia.
• Keratometry and biometry when intraocular lens (IOL) implantation is anticipated.
• Lab tests: TORCH titers (toxoplasmosis, other infections [syphilis], rubella, cytomegalovirus, and herpes simplex), fasting blood sugar (hypoglycemia), urine-reducing substances after milk feeding (galactosemia), calcium (hypocalcemia), and urine amino acids (Lowe’s syndrome).
• B-scan ultrasonography if unable to visualize the fundus (can perform through the lids of a crying child).
• Pediatric consultation.
Prognosis
Depends on age and duration of visually significant cataract prior to surgery; poor if amblyopia exists.
Acquired Cataract
Definition
Lenticular opacity usually categorized by location or etiology:
Cortical Degeneration
Caused by swelling and liquefaction of the cortical fiber cells. Various types:
Spokes and vacuoles
Asymmetrically located, radial, linear opacities and punctate dots.
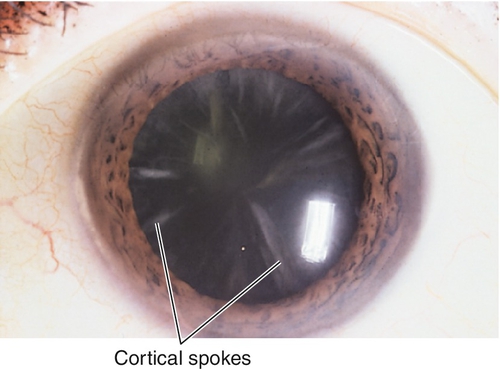
Mature cataract
Completely opacified cortex causing the lens to appear white; no red reflex visible from fundus.

Morgagnian cataract
Mature cataract with dense nucleus displaced inferiorly in completely liquefied, white cortex.
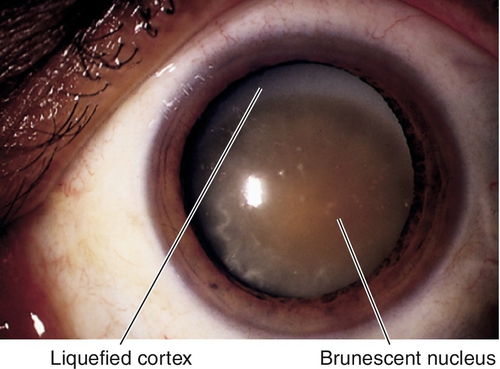
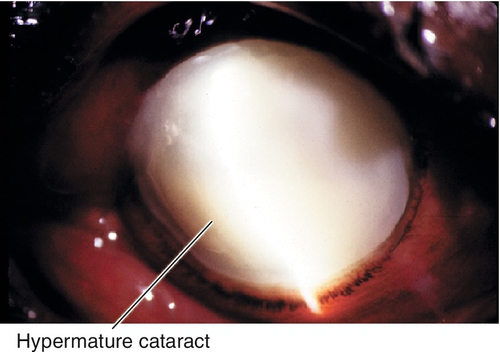
Hypermature cataract
After morgagnian cataract formation, the lens shrinks, the capsule wrinkles, and calcium deposits can form; proteins may leak into the anterior chamber causing phacolytic glaucoma.
Nuclear Sclerosis
Centrally located lens discoloration / opalescence (yellow-green or brown [brunescent]); caused by deterioration of the central lens fiber cells.
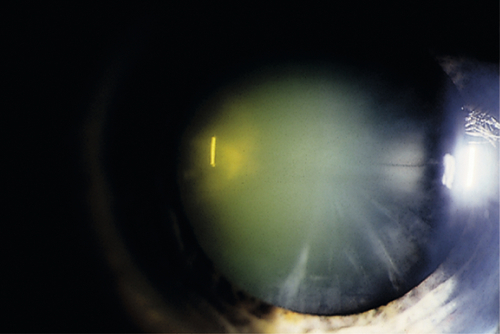
Subcapsular Cataract
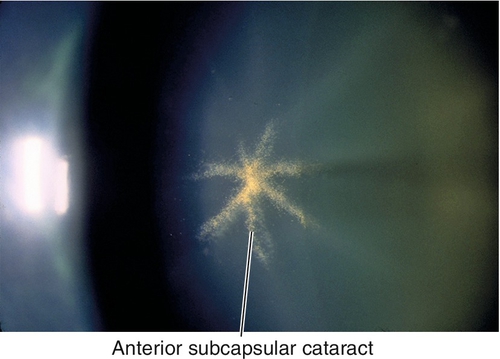
Anterior subcapsular
Central fibrous plaque caused by metaplasia of the central zone lens epithelial cells beneath the anterior lens capsule. Medications can cause anterior subcapsular stellate changes; acute angle-closure attacks can cause anterior subcapsular opacities (glaukomflecken) due to lens epithelial necrosis.
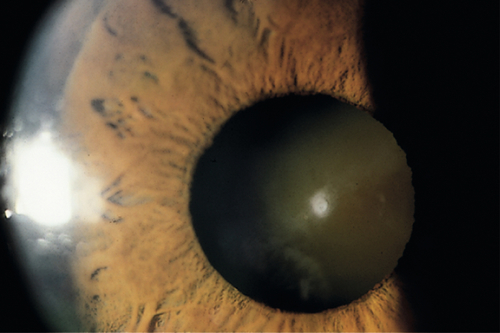
Posterior subcapsular
Asymmetric granular opacities with a frosted-glass appearance at the posterior surface of the lens; caused by posterior migration of epithelial cells and formation of bladder (Wedl) cells.
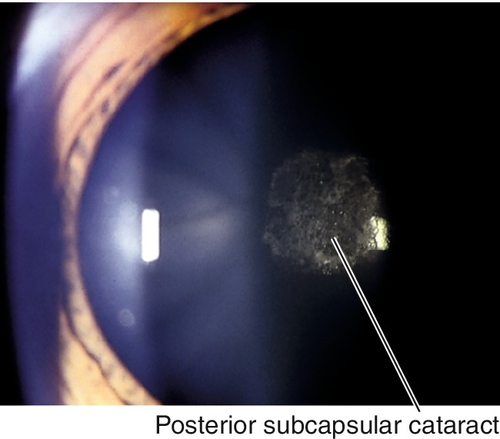
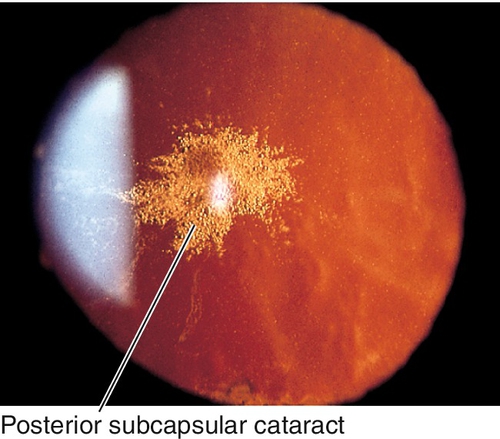
Etiology
Senile
Most common; due to age-related lens changes (cortical, nuclear, and /or subcapsular).
Systemic Disease
Many different systemic diseases can cause cataracts, including:
Diabetes mellitus
“Sugar” cataracts are cortical or posterior subcapsular opacities that occur earlier in diabetic patients than in age-matched controls, progress rapidly, and are related to poor glucose control more than duration of disease.
Hypocalcemia
Lens opacities are usually small white dots but can aggregate into larger flakes.
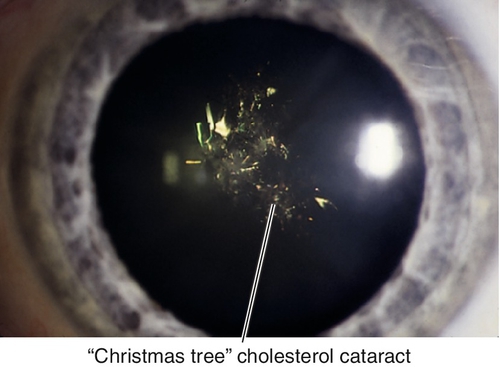
Myotonic dystrophy
Central, polychromatic, iridescent, cortical crystals (Christmas tree cataract); may develop a posterior subcapsular cataract later (see Chronic Progressive External Ophthalmoplegia section in Chapter 2).
Wilson’s disease
Sunflower cataract due to copper deposition (chalcosis lentis); green-brown surface opacity in the central lens with short stellate processes rather than the full flower petal pattern that occurs in chalcosis (copper foreign body).
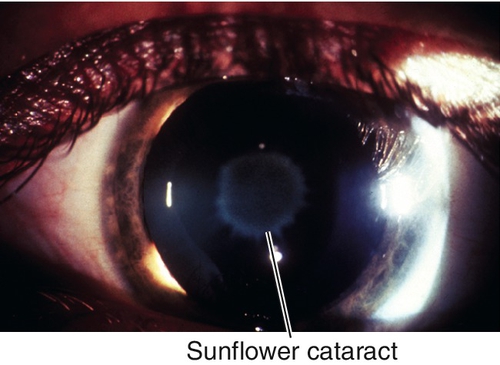
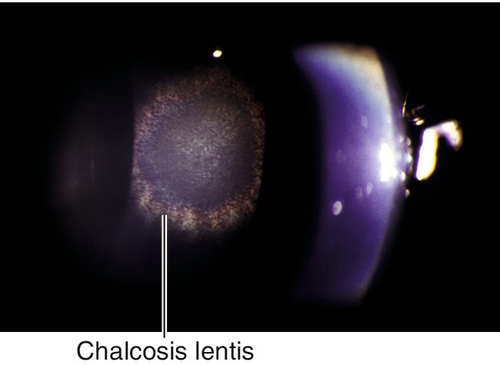
Figure 8-21 Sunflower cataract (chalcosis lentis) with green-brown central opacities in the lens (same patient as Figure 5-90 with Kayser–Fleischer ring).
Others
Fabry’s disease, atopic dermatitis (anterior subcapsular shield-like plaque), NF-2 (posterior subcapsular cataracts), and ectodermal dysplasia.
Other Eye Diseases
Uveitis, angle-closure glaucoma (glaukomflecken), retinal detachment, myopia, intraocular tumors, retinitis pigmentosa (posterior subcapsular cataracts), Refsum’s disease (posterior subcapsular cataracts), Stickler’s syndrome (cortical cataracts), phthisis bulbi.
Toxic
Steroids (posterior subcapsular cataracts), miotic agents, phenothiazines, amiodarone, busulfan; ionizing (X-rays, gamma rays, and neutrons), infrared, ultraviolet, microwave, and shortwave radiation; electricity, and chemicals.
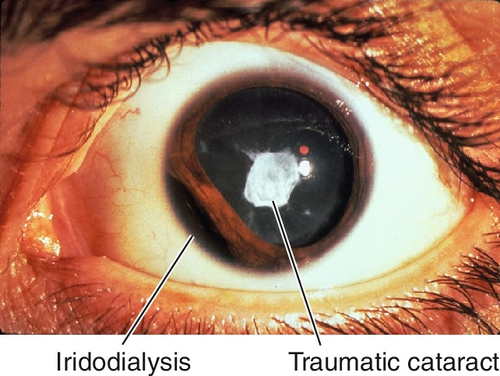
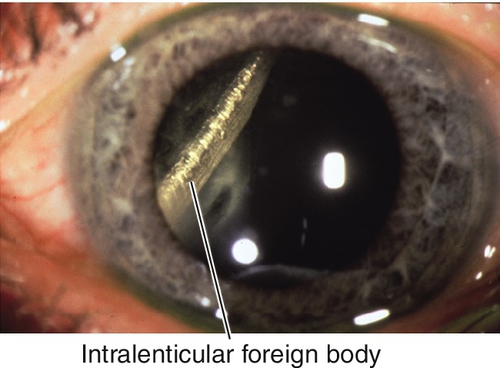
Trauma
Blunt or penetrating; intraocular foreign bodies (iron, copper); and postoperative (i.e., pars plana vitrectomy, trabeculectomy).
Epidemiology
Senile cataracts represent senescent lens changes related, in part, to ultraviolet B radiation. In the Framingham Eye Study, the prevalence of senile cataracts was 42% in adults 52–64 years old, 73% in 65–74-year-olds, and 91% in 75–85-year-olds. African-American men and women were more likely to have cataracts in every age category. Cataracts are the leading cause of blindness worldwide.
Symptoms
Painless, progressive loss of vision, decreased contrast and color sensitivity, glare, starbursts; rarely monocular diplopia.
Signs
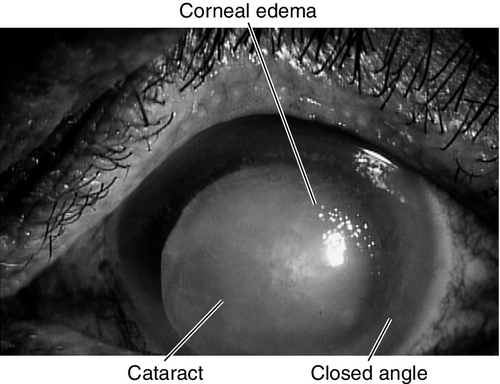
Decreased visual acuity (distance vision usually affected more than near vision in nuclear sclerosis, and near vision affected more than distance vision in posterior subcapsular), decreased contrast sensitivity, focal or diffuse lens opacification (yellow, green, brown, or white; often best appreciated with retroillumination), change in refractive error (often a myopic shift); intumescent cataracts may cause the iris to bow forward and lead to secondary angle-closure (see Chapter 6); hypermature cataracts may leak lens proteins and cause phacolytic glaucoma (see Lens-Induced Glaucoma section).
Differential Diagnosis
See above; senile cataract is a diagnosis of exclusion, must rule out secondary causes.
Evaluation
• Complete eye exam with attention to visual acuity, refraction, contrast sensitivity, cornea, gonioscopy, lens, and ophthalmoscopy.
• Consider brightness acuity tester (BAT) and potential acuity meter (PAM) testing (the latter is used to estimate visual potential, especially when posterior segment pathology exists).
• B-scan ultrasonography if unable to visualize the fundus.
• Keratometry and biometry to calculate the IOL implant power before cataract surgery; also consider specular microscopy and pachymetry if cornea guttata or corneal edema exists.
Prognosis
Very good; success rate for routine cataract surgery is > 96%; increased risk of complications for posterior polar and traumatic cataracts, pseudoexfoliation syndrome (see below), ectopia lentis, small pupil, intraoperative floppy iris syndrome (IFIS, caused by α1-adrenergic antagonist drugs [i.e., Flomax]).
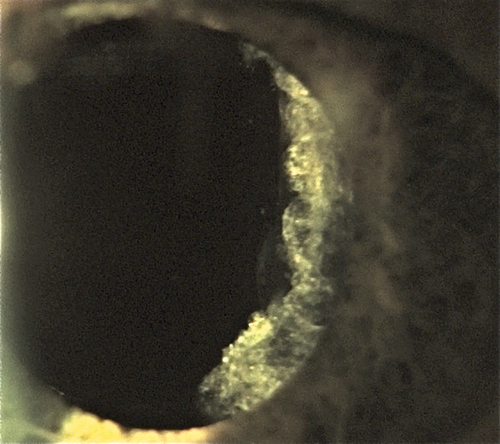
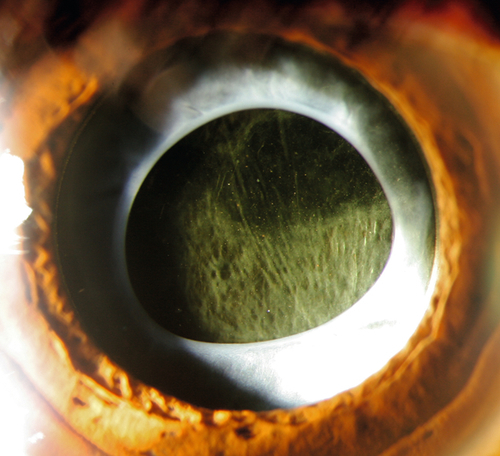
Posterior Capsular Opacification
Definition
Clouding of the posterior lens capsule after extracapsular cataract extraction.
Epidemiology
After cataract extraction, up to 50% of adult patients may develop posterior capsule opacification; increased incidence in children and patients with uveitis (approaches 100%). IOL material (acrylic) and design (square edge) have reduced the incidence of posterior capsular opacification to less than 10%.
Etiology
Epithelial cell proliferation (Elschnig pearls) and fibrosis of the capsule.
Symptoms
Asymptomatic or may have decreased vision and glare depending on severity and location with respect to the visual axis.
Signs
Posterior capsule opacification; graded on a 1 to 4 scale according to density; may appear as haze, striae, Elschnig pearls, or any combination.
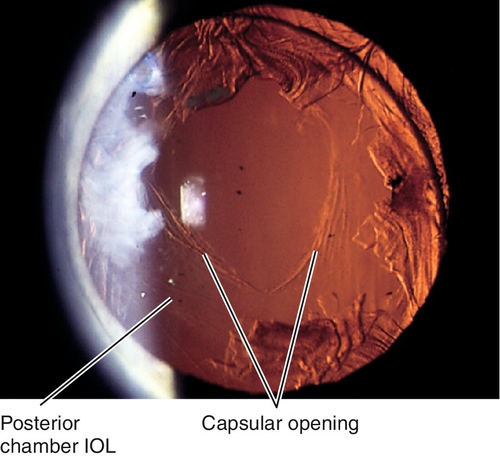
Evaluation
Prognosis
Very good; complications of Nd : YAG capsulotomy are rare but include increased intraocular pressure, IOL damage or dislocation, corneal burn, retinal detachment, cystoid macular edema, and hyphema.
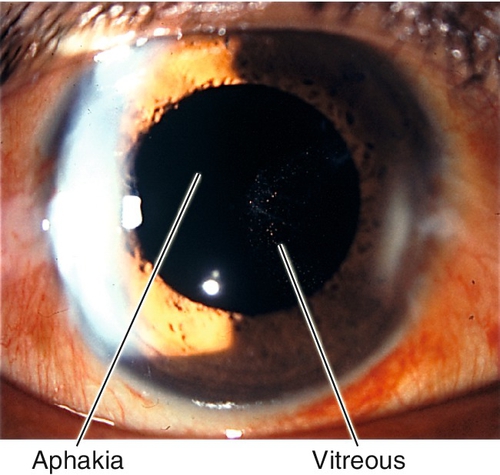
Aphakia
Definition
Absence of crystalline lens; usually secondary to surgery, rarely traumatic (total dislocation of crystalline lens [see Figure 8-39]), or very rarely congenital.
Symptoms
Loss of accommodation and decreased uncorrected vision.
Signs
Decreased uncorrected visual acuity (usually very high hyperopia), no lens, iridodonesis; may have a visible surgical wound, peripheral iridectomy, vitreous in the anterior chamber, complications from surgery (bullous keratopathy, increased intraocular pressure, iritis, posterior capsule opacification, cystoid macular edema), or evidence of ocular trauma (see appropriate sections).
Evaluation
• Complete ophthalmic history with attention to previous ocular surgery or trauma.
• Complete eye exam with attention to refraction, cornea, tonometry, anterior chamber, gonioscopy, iris, lens, and ophthalmoscopy.
• Consider specular microscopy and pachymetry if cornea guttata or corneal edema exists.
Prognosis
Usually good; increased risk of retinal detachment, especially for high myopes and if posterior capsule is not intact.
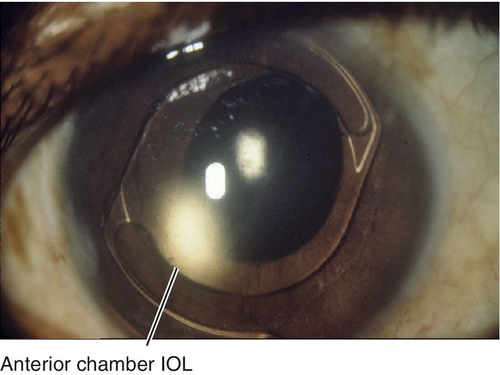
Pseudophakia
Definition
Presence of IOL implant after crystalline lens has been removed; may be inserted primarily or secondarily. There are numerous types of IOLs including: anterior or posterior chamber; rigid or foldable; 1- or 3-piece; loop or plate haptic; round or square edge; polymethylmethacrylate, acrylic, silicone, hydrogel, or collamer; monofocal, multifocal, accommodating, toric, and aspheric.
Symptoms
Asymptomatic; may have decreased vision, loss of accommodation, edge glare, monocular diplopia or polyopia, or induced ametropia with decentered or tilted IOL.
Signs
IOL implant (may be in anterior chamber, iris plane, capsular bag, or ciliary sulcus with or without suture fixation to iris or sclera); may have a visible surgical wound, peripheral iridectomy, complications from surgery (bullous keratopathy, iris capture, decentered IOL, increased intraocular pressure, iritis, hyphema, opacified posterior capsule, cystoid macular edema).


Evaluation
Prognosis
Usually good; increased risk of retinal detachment, especially for high myopes and if posterior capsule is not intact.
Exfoliation
Definition
True exfoliation is delamination, or schisis, of the anterior lens capsule into sheet-like lamellae.
Etiology
Infrared and thermal radiation; also senile form.
Epidemiology
Rare; classically occurs in glass blowers.
Symptoms
Asymptomatic.
Signs
Splitting of anterior lens capsule, appears as scrolls; may have posterior subcapsular cataract.
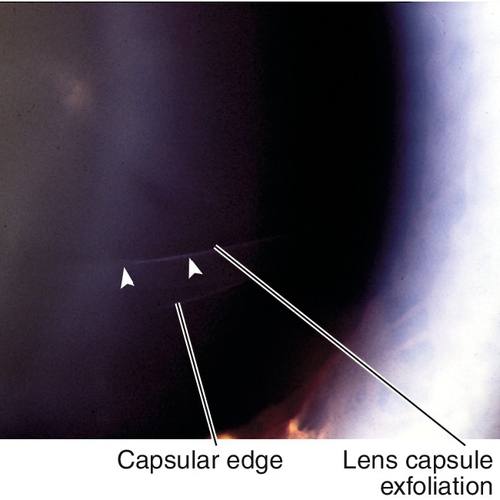
Figure 8-32 True lens exfoliation demonstrating scrolling of split anterior lens capsule (arrowheads).
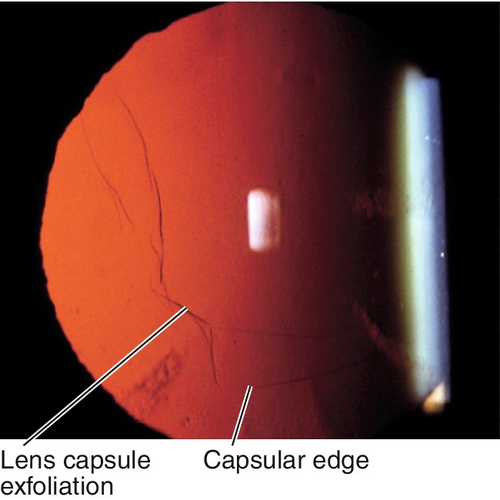
Figure 8-33 Same patient as Figure 8-32, demonstrating appearance of lens capsule exfoliation as viewed with retroillumination.
Differential Diagnosis
Pseudoexfoliation syndrome.
Evaluation
• Complete ophthalmic history with attention to infrared and thermal radiation exposure.
• Complete eye exam with attention to lens.
Prognosis
Good.
Pseudoexfoliation Syndrome
Definition
Pseudoexfoliation is a generalized disorder of elastin formation that results in the abnormal accumulation of small, gray-white fibrillar aggregates (resembling amyloid) on the lens capsule, iris, anterior segment structures, and systemically (may involve the skin, heart, and lungs).
Epidemiology
Usually asymmetric, bilateral more often than unilateral. Occurs in all racial groups, common in Scandinavians, South African Blacks, Navaho Indians, and Australian Aborigines; almost absent in Eskimos. Age-related, rare in individuals under 50 years old, incidence increases after age 60 years (4–6% in patients over 60 years old). Up to 60% develop ocular hypertension or glaucoma (see below); in the United States, 20% have elevated intraocular pressure at initial examination, and 15% develop it within 10 years. Mapped to chromosome 15q24 (LOXL1 gene).
Symptoms
Asymptomatic.
Signs
Loss of pupillary ruff, iris transillumination defects, pigment deposits on the iris, trabecular meshwork, and anterior to Schwalbe’s line (Sampaolesi’s line); target pattern of exfoliative material on lens capsule (central disc and peripheral ring with intervening clear area); white exfoliation material is also seen on zonules, anterior hyaloid, iris, and pupillary margin; shallow anterior chamber due to forward displacement of the lens–iris diaphragm; may have phacodonesis, cataract (40%), or signs of glaucoma with increased intraocular pressure, optic nerve cupping, nerve fiber layer defects, and visual field defects.
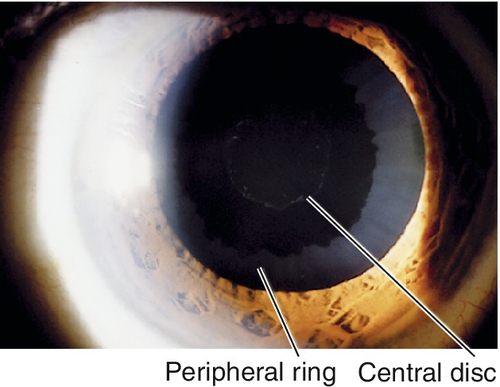
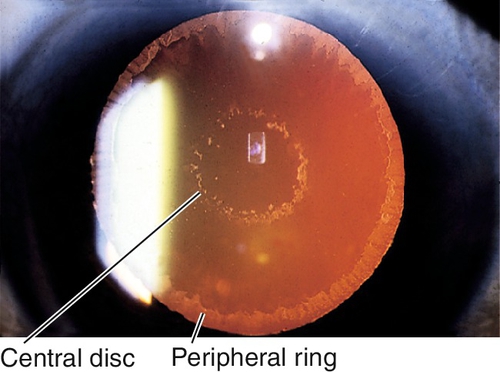
Figure 8-35 Central disc and peripheral ring of exfoliative material as seen with retroillumination.
Evaluation
• Check visual fields in patients with elevated intraocular pressure or optic nerve cupping to rule out glaucoma.
Prognosis
Good; poorer if pseudoexfoliation glaucoma develops; increased incidence of complications at cataract surgery due to weak zonules and increased lens mobility.
Pseudoexfoliation Glaucoma
Definition
A form of secondary open-angle glaucoma associated with pseudoexfoliation syndrome.
Epidemiology
Most common cause of secondary open-angle glaucoma; affects 2% of US population over 50 years old. Up to 60% with pseudoexfoliation syndrome develop ocular hypertension or glaucoma; 50–60% bilateral, often asymmetric; age-related (rare in individuals under 50 years old, increases after age 60 years). Mapped to chromosome 15q24 (LOXL1 gene).
Mechanism
(2) Abnormally weak lamina cribrosa (composed of elastin) that renders the optic nerve more susceptible to elevated IOP.
Symptoms
Asymptomatic; may have decreased vision or constricted visual fields in late stages.
Signs
Normal or decreased visual acuity, increased intraocular pressure (can be very high and asymmetric); similar ocular signs as in pseudoexfoliation syndrome (see above), optic nerve cupping, nerve fiber layer defects, and visual field defects.
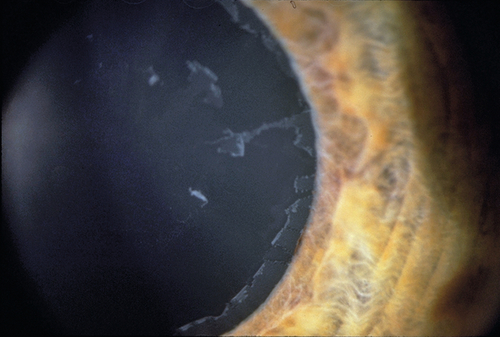
Differential Diagnosis
Primary open-angle glaucoma, other forms of secondary open-angle glaucoma.
Evaluation
• Check visual fields.
• Stereo optic nerve photos are useful for comparison at subsequent evaluations.
Prognosis
Poorer than primary open-angle glaucoma; increased incidence of angle-closure; lens removal has no effect on progression of disease; increased incidence of complications at cataract surgery due to weak zonules (composed of elastin) and increased lens mobility.
Lens-Induced Glaucoma
Definition
Secondary glaucoma due to lens-induced abnormalities.
Etiology/ mechanism
Lens Particle
Retained cortex or nucleus after cataract surgery or penetrating trauma causes inflammatory reaction and obstructs trabecular meshwork; more anterior segment inflammation than phacolytic.
Phacolytic
Lens proteins from hypermature cataract leak through intact capsule and are ingested by macrophages; can occur with intact, dislocated lens; lens proteins and macrophages obstruct trabecular meshwork.
Phacomorphic
Enlarged, cataractous lens pushes the iris forward, causing secondary angle-closure (see Chapter 6).
Symptoms
Decreased vision, pain, photophobia, red eye; may have halos around lights and other symptoms of angle-closure glaucoma (see Chapter 6); may have constricted visual fields.
Signs
Decreased visual acuity, increased intraocular pressure, ciliary injection, anterior chamber cells and flare, peripheral anterior synechiae, cataract or residual lens material, signs of recent surgery or trauma including surgical wounds, sutures, and signs of an open globe (see Chapter 4); may have optic nerve cupping, nerve fiber layer defects, and visual field defects.
Differential Diagnosis
See above; other forms of secondary glaucoma, uveitis, endophthalmitis.
Evaluation
• B-scan ultrasonography if unable to visualize the fundus.
• Check visual fields.
Prognosis
Good if definitive treatment is performed early and pressure control is achieved.
Dislocated Lens (Ectopia Lentis)
Definition
Congenital, developmental, or acquired displacement of the crystalline lens; may be incomplete (subluxation) or complete (luxation) dislocation of the lens into the anterior chamber or vitreous.
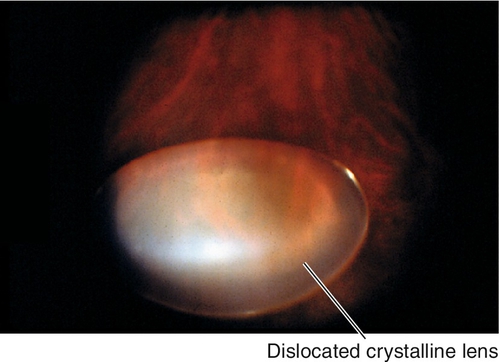
Figure 8-39 Dislocated crystalline lens resting on the retina.
Etiology
Ectopia Lentis et Pupillae (AR)
Associated with oval or slit-like pupils; pupil displacement is in opposite direction of the lens displacement.
Homocystinuria (AR)
Enzymatic disorder of methionine metabolism with elevated levels of homocystine and methionine; direction of lens displacement is typically down and in. Not present at birth; progressive thereafter, with more than 90% having dislocated lenses by the third decade. Patients develop seizures, osteoporosis, mental retardation, and thromboembolism.
Hyperlysinemia
Inability to metabolize lysine; lens subluxation, muscular hypotony, and mental retardation.
Marfan’s Syndrome (AD)
Usually bilateral; occurs in about two-thirds of Marfan’s patients due to defective zonules; direction of lens displacement is typically up and out. Other signs include marfanoid habitus with disproportionate growth of extremities, arachnodactyly, joint laxity, pectus deformities, scoliosis, and increasing dilation of the ascending aorta with aortic insufficiency (may cause death). Mapped to chromosome 15q (FBN1 [fibrillin-1] gene).
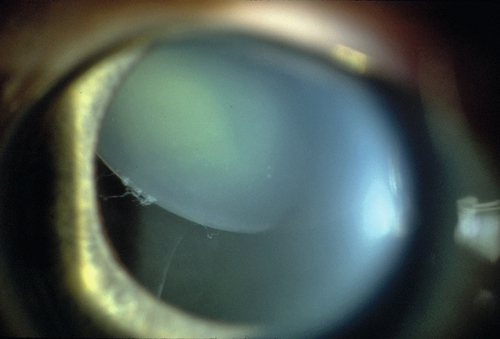
Microspherophakia
Small spheric lens; occurs as an isolated anomaly or as part of a syndrome (i.e., dominant spherophakia, Weill–Marchesani syndrome, Lowe’s syndrome, Alport’s syndrome, Peter’s anomaly, rubella); direction of lens displacement is often inferiorly or anteriorly.
Simple Ectopia Lentis (AD)
Often present at birth; lens is small and spheric (microphakic and spherophakic); direction of lens displacement is typically up and out.
Sulfite Oxidase Deficiency (AR)
Error of sulfur metabolism with ectopia lentis, seizures, and mental retardation.
Other
Aniridia, Ehlers–Danlos syndrome, trauma, syphilis, pseudoexfoliation syndrome, megalocornea.
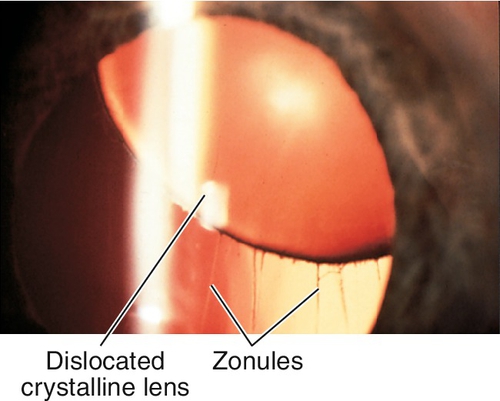
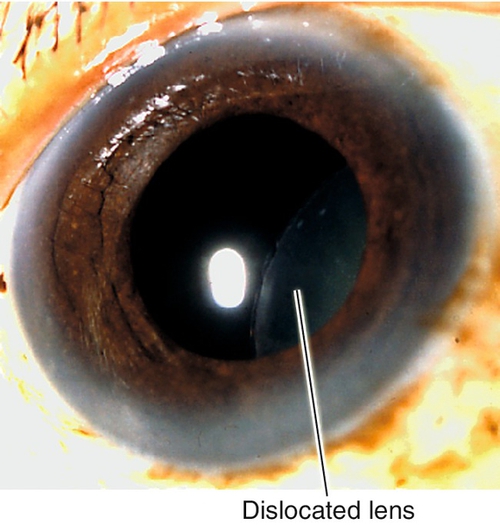
Epidemiology
Most common cause of lens subluxation or luxation is trauma (up to 50%); associated with cataract and rhegmatogenous retinal detachment. Most frequent cause of heritable lens dislocation is Marfan’s syndrome.
Symptoms
Asymptomatic; may have decreased vision, diplopia, symptoms of angle-closure glaucoma (see Chapter 6).
Signs
Normal or decreased visual acuity, subluxated or luxated lens, phacodonesis, iridodonesis; may have increased intraocular pressure, anterior chamber cells and flare, vitreous in the anterior chamber, iris transillumination defects, angle abnormalities, and other signs of ocular trauma.
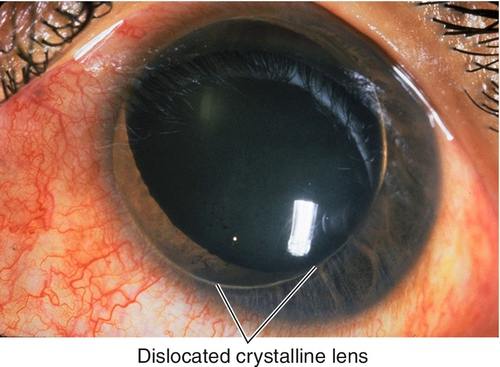
Differential Diagnosis
See above.
Evaluation
• Consider B-scan ultrasonography if unable to visualize the fundus.
• Lab tests: Venereal Disease Research Laboratory (VDRL) test, fluorescent Treponema antibody absorption (FTA-ABS) test, and lumbar puncture if syphilis suspected.
• Medical consultation for systemic diseases.
Prognosis
Depends on etiology.
[/level-membership-for-opthalmology-category][not-level-membership-for-opthalmology-category]8
Lens
Congenital Anomalies
Definition
Variety of developmental lens abnormalities:
Coloboma
Focal inferior lens flattening due to ciliary body coloboma with absence of zonular support (not a true coloboma); other ocular colobomata (iris) usually exist. Ciliary body tumors may cause a secondary lens “coloboma.”
Lenticonus
Cone-shaped lens due to bulging from a thin lens capsule; either anteriorly or posteriorly, rarely in both directions.
Anterior
Usually males; bilateral; may be associated with Alport’s syndrome (see Congenital Cataract section below).
Posterior
More common; slight female predilection; may have associated cortical lens opacities, may be associated with Lowe’s syndrome (see Congenital Cataract section below).
Lentiglobus
Globe-shaped lens caused by bulging from thin lens capsule; rare.
Microspherophakia
Small spherical lens; may be an isolated anomaly or part of a syndrome (i.e., dominant spherophakia, Weill–Marchesani syndrome, Lowe’s syndrome, Alport’s syndrome, Peter’s anomaly, rubella).
Mittendorf Dot
A small white spot on the posterior lens capsule that represents a remnant of the posterior tunica vasculosa lentis where the former hyaloid artery attached.
Symptoms
Asymptomatic (Mittendorf dot, coloboma); may have decreased vision (lenticonus, lentiglobus, and microspherophakia), diplopia, or symptoms of angle-closure glaucoma (microspherophakia).
Signs
Normal or decreased visual acuity; may have amblyopia, strabismus, nystagmus, myopia, and an “oil-droplet” fundus reflex on retroillumination in lenticonus and lentiglobus; may have dislocated lens and increased intraocular pressure in microspherophakia.
Differential Diagnosis
See above.
Evaluation
Prognosis
Usually good; poorer if amblyopia exists.
Congenital Cataract
Definition
Congenital opacity of the crystalline lens usually categorized by location or etiology:
Capsular
Opacity of the lens capsule, usually anteriorly.
Lamellar or Zonular
Central, circumscribed opacity surrounding the nucleus; “sand dollar” appearance.
Lenticular or Nuclear
Opacity of the lens nucleus.
Figure 8-6 Congenital nuclear cataract with central white discoid appearance.
Figure 8-7 Same patient as Figure 8-6 demonstrating congenital nuclear cataract as viewed with retroillumination.
Polar
Central opacity located near the lens capsule, anteriorly or posteriorly.
Figure 8-8 Congenital posterior polar cataract.
Figure 8-9 Same patient as Figure 8-8 demonstrating congenital polar cataract in retroillumination.
Sutural
Opacity of the Y-shaped sutures in the center of the lens.
Figure 8-10 Congenital cataract with prominent suture lines.
Figure 8-11 Same patient as Figure 8-10 demonstrating congenital sutural cataract in retroillumination.
Etiology
Hereditary or Syndromes
Without chromosomal abnormalities
Autosomal dominant (AD), autosomal recessive (AR), X-linked.
With chromosomal abnormalities
Down’s syndrome (snowflake cataracts), Turner’s syndrome, and others.
Other syndromes
Craniofacial, central nervous system, skin.
Intrauterine Infections
Congenital rubella syndrome
Cataracts, glaucoma, microcornea, microphthalmos, iris hypoplasia, and retinopathy with characteristic fine, granular, salt-and-pepper appearance (most common finding). Other complications include prematurity, mental retardation, neurosensory deafness, congenital heart disease, growth retardation, hepatosplenomegaly, interstitial pneumonitis, and encephalitis.
Congenital varicella syndrome
Cataracts, chorioretinitis, optic nerve atrophy or hypoplasia, nystagmus, and Horner’s syndrome. Systemic findings include hemiparesis, bulbar palsies, dermatomal cicatricial skin lesions, developmental delay, and learning difficulties.
Metabolic
Galactosemia
Bilateral, oil-droplet cataracts from accumulation of galactose metabolites (galactitol) due to hereditary enzymatic deficiency; usually galactose 1-phosphate uridyltransferase, also galactokinase. Associated with mental retardation, hepatosplenomegaly, cirrhosis, malnutrition, and failure to thrive. Mapped to chromosomes 1p (GALE gene), 9p (GALT gene) and 17q (GALK1 gene).
Lowe’s oculocerebrorenal syndrome (X-linked)
Small discoid lens, posterior lenticonus, and glaucoma. Systemic findings include acidosis, aminoaciduria, renal rickets, hypotonia, mental retardation. Female carriers have posterior, white, punctate cortical opacities and subcapsular, plaque-like opacities. Mapped to chromosome Xq25 (OCRL gene).
Alport’s syndrome (AD)
Basement membrane disease associated with acute hemorrhagic nephropathy, deafness, anterior lenticonus, anterior polar or cortical cataracts, and albipunctatus-like spots in the fundus. Mapped to chromosome Xq22 (COL4A5 gene [80% of cases], also COL4A3 and COL4A4 genes).
Other metabolic diseases
Hypoglycemia, hypocalcemia (diffuse lamellar punctate opacities), Fabry’s disease (spoke-like cataracts in 25%), mannosidosis (posterior spoke-like opacities).
Ocular Disorders
Persistent hyperplastic primary vitreous, Peter’s anomaly, Leber’s congenital amaurosis, retinopathy of prematurity, aniridia, posterior lenticonus, tumors.
Other
Birth trauma, idiopathic, and maternal drug ingestion.
Epidemiology
Congenital cataracts occur in approximately 1 of 2000 live births. Roughly one-third are isolated, one-third are familial (usually dominant), and one-third are associated with a syndrome; most unilateral cases are not metabolic or genetic.
Symptoms
Variable decreased vision; may notice white pupil or eye turn.
Signs
Decreased visual acuity, leukocoria, amblyopia; may have strabismus (usually with unilateral cataract), nystagmus (usually does not appear until 2–3 months of age; rarely when cataracts develop after age 6 months), amblyopia; may have other ocular or systemic findings if syndrome exists.
Differential Diagnosis
Evaluation
• Complete eye exam with attention to cycloplegic refraction, retinoscopy, tonometry, gonioscopy, lens (size and density of the opacity as viewed with retroillumination), and ophthalmoscopy.
• May require examination under anesthesia.
• Keratometry and biometry when intraocular lens (IOL) implantation is anticipated.
• Lab tests: TORCH titers (toxoplasmosis, other infections [syphilis], rubella, cytomegalovirus, and herpes simplex), fasting blood sugar (hypoglycemia), urine-reducing substances after milk feeding (galactosemia), calcium (hypocalcemia), and urine amino acids (Lowe’s syndrome).
• B-scan ultrasonography if unable to visualize the fundus (can perform through the lids of a crying child).
• Pediatric consultation.
Prognosis
Depends on age and duration of visually significant cataract prior to surgery; poor if amblyopia exists.
Acquired Cataract
Definition
Lenticular opacity usually categorized by location or etiology:
Cortical Degeneration
Caused by swelling and liquefaction of the cortical fiber cells. Various types:
Spokes and vacuoles
Asymmetrically located, radial, linear opacities and punctate dots.
Mature cataract
Completely opacified cortex causing the lens to appear white; no red reflex visible from fundus.
Morgagnian cataract
Mature cataract with dense nucleus displaced inferiorly in completely liquefied, white cortex.
Hypermature cataract
After morgagnian cataract formation, the lens shrinks, the capsule wrinkles, and calcium deposits can form; proteins may leak into the anterior chamber causing phacolytic glaucoma.
Nuclear Sclerosis
Centrally located lens discoloration / opalescence (yellow-green or brown [brunescent]); caused by deterioration of the central lens fiber cells.
Subcapsular Cataract
Anterior subcapsular
Central fibrous plaque caused by metaplasia of the central zone lens epithelial cells beneath the anterior lens capsule. Medications can cause anterior subcapsular stellate changes; acute angle-closure attacks can cause anterior subcapsular opacities (glaukomflecken) due to lens epithelial necrosis.
Posterior subcapsular
[/not-level-membership-for-opthalmology-category]
