[level-membership-for-neurosurgery-category]
CHAPTER 342 Rehabilitation of Patients with Traumatic Brain Injury
Accidents happen. Brain injury.
Unsolicited. Unanticipated. Unwelcome.
In the blink of an eye, everything changes.
—www.biama.org (Brain Injury Association of Massachusetts)
Definitions and Epidemiology
This excludes lacerations or contusions of the face, eye, ear or scalp, or fractures of the facial bones without other criteria listed above; as well as birth trauma, primary anoxic, inflammatory, infectious, toxic or metabolic encephalopathies which are not complications of head trauma, neoplasms or ischemic/hemorrhagic stroke without associated trauma.1
Using data collected from 1999-2001, the U.S. Centers for Disease Control and Prevention estimated that the annual incidence of TBI was approximately 1.4 million people per year.2 This number has remained relatively steady, as the most recent data from 2003 estimated an incidence of 1,565,000 with an overall mortality rate of 17.5/100,000 people. TBI accounts for an estimated 1.1 million emergency department visits, 235,000 hospitalizations, and 50,000 deaths in the United States per year.3 Morbidity rates remain high and are estimated at about 5.3 million Americans; in other words, about 2% of the population needs long-term help to perform activities of daily living because of TBI.3 Despite being quite impressive, these numbers likely underestimate the current impact of TBI on the American population for two reasons: (1) a new influx of returning veterans from Iraq and Afghanistan, in which incidence of TBI likely approaches 22%, if not higher4; and (2) likely underreporting of mild brain injury (concussion). For example, though sports-related concussion has been estimated at an annual incidence of 1.6 to 3.8 million,5 only 200,000 such patients are treated in emergency departments per year.3
Patients between the ages of 15 and 44 years make up about 45% of cases, with males making up about 60% of the population. Most recent data from 2003 still suggest that falls remain the most common cause of injury (32%), with motor vehicle accidents as the second most common cause (19.2%). Because patients are often injured in the prime of their lives, the socioeconomic impact can be devastating. Direct and indirect costs of TBI in the United States were estimated to be $60 billion in 2000.6 Thus despite advances in treatment of TBI, it remains a significant cause of morbidity and mortality in the United States.
Prognosis in TBI
| Category | Description |
|---|---|
| 1. Death | Death |
| 2. Persistent vegetative state | Prolonged unconsciousness with no verbalization or following of commands. Absent awareness of self and environment. Patient may open eyes and have reflexive actions. Sleep-wake cycles are present. |
| 3. Severe disability | Patient unable to be independent for any 24-hour period due to physical or mental disability |
| 4. Moderate disability | Patient can travel by public transportation and work in a sheltered environment. There may be residual deficits that do not interfere with independent daily life. |
| 5. Good recovery | Return to normal life with minor or no residual deficits. |
Some important predictive factors are listed below:
The best predictor of outcomes after TBI is the length of posttraumatic amnesia (PTA). The longer the duration of PTA, the worse outcomes generally are. Clinically, resolution of PTA occurs when patients are able to incorporate daily events into working memory. Patients with PTA of less than 24 hours generally have a quick and full recovery with few exceptions, and patients with PTA of more than 4 weeks generally have permanent deficits. However, more stringent “threshold” criteria have been used in previous textbooks to preserve hope for caregivers. These criteria state that severe disability (on GOS) is unlikely with PTA of less than 2 months, and good recovery (on GOS) is unlikely when PTA is more than 3 months.7 It has been shown that 21% patients with PTA between 1 and 2 months did return to work within a year, whereas only 9% of patients with PTA of more olthan 70 days were able to return to work within a year.8
Length of coma, defined as time from injury to follow commands, can be an important milestone as well. Generally, severe disability (GOS) is likely when length of coma is more than 2 weeks. Good recovery (GOS) is unlikely when length of coma is more than 4 weeks. For more information, please see the section on Disorders of Consciousness.
The best Glasgow Coma Scale (GCS) score within 24 hours correlates with outcomes, with lower scores associated with worse outcomes, and with the best motor response subset as the most accurate predictor of outcomes. Unfortunately, there are no “threshold” criteria because patients with severe injury can have good recovery. Indeed, recent reviews of GCS in outcomes of TBI suggest that other measures, such as age and pupillary response, improve its predictive capacity.9
Neuroimaging has become increasingly important in predicting outcome. It has been suggested that an adequate threshold marker is the presence of bilateral brainstem lesions; good recovery (GOS) is unlikely when bilateral brainstem lesions are present on an early MRI. CT scan findings of subarachnoid hemorrhage, midline shift, extradural hematoma, bilateral injury, or transventricular injury correlate with worse outcomes, though a full range of outcomes is still possible with these injuries.7
Research continues in this field to delineate other possible markers of prognosis and outcome. Biomarkers such as S100, genetic markers such as APOE 4 status, the ratio of total brain volume to third ventricle size, T2 gradient echo MRI lesions, PET scanning, diffusion tensor imaging, magnetic resonance spectroscopy, and somatosensory evoked potential responses are currently under study and may provide important prognostic information in the future.10
Given the uncertainty surrounding various predictors, several authors are currently trying to mathematically model outcomes of TBI using predictive variables.11–13 One such model, based on a large population study, is available at http://www.tbi-impact.org/. It must be noted that these models are still undergoing refinement, though they may also provide important prognostic information in the future.
Mechanisms of Recovery
Introduction
Much basic science research has been done on the mechanisms of neural recovery, particularly in stroke literature. Exact comparisons may be difficult for TBI because the nature of the injury is often diffuse but it seems likely that the basic mechanisms of recovery are similar. Trauma disrupts neuronal connections including the connectivity and organization of networks mediating executive functions and sensory and motor responses.14 In addition to the focal damage of TBI, dysfunction can occur in areas anatomically removed, likely because of diaschisis involving tissue hypometabolism, neurovascular uncoupling, and aberrant neurotransmission.15 In addition to local repair, neuroplasticity is thought to be a crucial feature of recovery from TBI with research from as far back as the 1960s demonstrating how brain structure and chemistry can be affected by environmental manipulation.16
Overview
Recovery of neurological function is dynamic and multifaceted but often limited partly because of the inability for dramatic anatomic reorganization in the brain following lesions. Three distinct phases of recovery have been described. The first involved activation of cell repair and reversal of diaschisis; the second, plasticity of existing neuronal pathways to produce a functional change; the third, neuroanatomic plasticity to produce nascent connections. Where damage is focal, recovery primarily engages ipsilateral brain tissue, but with more severe TBI it is likely that contralateral brain regions are also recruited.16 The recovery process begins immediately and extends over the course of many months to years.
Therapeutic Targets
One area of interest is growth promoters and inhibitors. When administered within days of the insult, functional recovery is promoted by growth factors through stimulation of neuronal plasticity. Several agents are currently in clinical trials and are diverse in nature, including erythropoietin, granulocyte colony stimulating factor, statin drugs, and phosphodiesterase-5 inhibitors.15 By contrast, growth inhibitors impair recovery with particular insights being gained into the effects of growth cone extension. Myelin-associated proteins, including Nogo-A, oligodendrocyte myelin glycoprotein, and myelin-associated glycoprotein (MAG), activate the inhibitory Nogo-66 receptor to prevent axonal sprouting.17 Intravenous or intrathecal delivery of antibodies or peptides aimed at either the Nogo protein or receptor dramatically enhance functional recovery after neural injury in animal models.
Finally, therapy may in the future focus on brain activation with early studies showing that amphetamine and possibly L-dopa enhanced recovery of function in animal models. The suggestion was that adrenergic and cholinergic neurotransmitter systems are critical to the recovery process, but human trials have had limited results.18 Brain tissue can also be activated by physical and environmental means, for example, repetitive physical activity or sensory experiences. As part of environmental stimulation, social interaction is felt to be important in that it induces multiple biologic effects on the brain.
While many questions remain about the mechanisms of recovery in TBI, the current sophisticated molecular tools available for researchers continue to yield insights and hopefully will lead to promising new therapies.15
Disorders of Consciousness
Up to 15% of patients with severe TBI are unable to respond consistently to commands at 4 weeks after injury, and the majority of these will have impairments of consciousness.19 There has been much confusion surrounding the different states of consciousness. By definition:
Vegetative state (VS)—arousable neurobehavioral unresponsiveness. Patients have periods of eye opening, either spontaneously or in response to external stimuli; subcortical responses to external stimuli (including posturing, tachycardia, diaphoresis with pain); return of autonomic functions (sleep-wake cycle, normalization of respiratory and digestive functions); roving eye movements without concomitant visual tracking ability.20
Minimally conscious state (MCS)—a condition of severely altered consciousness in which the patient demonstrates minimal but definite behavioral evidence of self- or environmental awareness. The Aspen workgroup proposed that one or more of the following should be present: (1) following simple commands; (2) manipulation of objects; (3) gestural or verbal yes/no responses; (4) intelligible verbalization; or (5) stereotypical movements that occur in a meaningful relationship to the eliciting stimulus and are not attributable to reflexive activity. These responses must be reproducible or occur on a sustained basis.21
Amantadine, bromocriptine, amphetamine, and methylphenidate and other neurostimulants have been proposed to help with arousal and vigilance.22 There has been a case report of a patient in MCS who regained full consciousness after treatment with amantadine who declined when the medication was withdrawn.10 However, Cochrane Reviews on monoaminergic agonists and excitatory amino acid inhibitors have not found good randomized trials to review.23,24 Similarly, another recent Cochrane Review determined that there have not been any good studies to support the use of sensory stimulation in MCS or VS patients.25 Unfortunately, most research surrounding treatment of disorders of consciousness in brain injury is poor, involving case reports or small numbers of patients without controls. More research is needed to further clarify treatment algorithms in the future.
Motor Recovery
As with everything in brain injury, motor recovery is highly individualized and varies from person to person. Though it seems that the majority of motor recovery happens in the first 6 months after injury, it is possible for patients to continue gaining function after that. Swaine and coworkers studied a small population of patients with severe TBI (GCS ≤ 8) for 6 weeks after injury, and demonstrated that, while the percentage of patients with basic primitive reflexes (asymmetric tonic neck reflex, flexor withdrawal) stayed relatively stable, equilibrium reactions, protective reactions, sitting reactions, and simple motor skills slowly increased over time. The sequence of recovery—reactions in sitting appeared before those in kneeling and before those in standing. Recovery of equilibrium reactions tended to occur before development of protective reactions. Similarly, with motor skills, rolling ability recovered before ability to sit unsupported, followed by the ability to stand, then walk independently, then climb stairs, then hop on one foot, then the ability to run. Surprisingly, some simple motor skills (rolling, sitting, even walking unsupported) seemed to recover faster than equilibrium. Also surprisingly, patients did not necessarily lose primitive reflexes before recovering simple motor tasks. Of note, in this case series, 60% of patients by 6 weeks could sit independently and 56% of patients could walk without assistance.26 Watson and associates have suggested a path-tree analysis instead of a simple linear progression of motor skills and demonstrated that while there is an underlying pattern of recovery, there are many routes that a patient can follow.27
Motor Trephine Syndrome
Following craniectomy, patients may suffer delayed or insidious headache, fatigue, and mood disturbance. A few may have delayed contralateral monoparesis that is reversible with cranioplasty—the motor trephine syndrome. In one retrospective review, up to 26% of patients with decompressive craniectomy developed delayed contralateral upper extremity weakness that improved with cranioplasty, with full recovery 1 month after cranioplasty. Weakness developed an average of 4.5 months later, was predominantly distal upper extremity 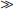 proximal, causing difficulty with grip and extension of metacarpals, and was slowly progressive. There was no sensory or language impairment. These patients more often had blossoming of cerebral contusions after craniectomy and cerebrospinal fluid (CSF) flow disturbances (hygromas and hydrocephalus), and had a longer time to cranioplasty than patients without motor trephine syndrome. Mechanistically, it has been proposed that motor trephine syndrome is caused by disturbances of CSF flow, leading to edema in brain parenchyma. Cranioplasty allows egress of CSF, which resolves brain edema and restores function.28
proximal, causing difficulty with grip and extension of metacarpals, and was slowly progressive. There was no sensory or language impairment. These patients more often had blossoming of cerebral contusions after craniectomy and cerebrospinal fluid (CSF) flow disturbances (hygromas and hydrocephalus), and had a longer time to cranioplasty than patients without motor trephine syndrome. Mechanistically, it has been proposed that motor trephine syndrome is caused by disturbances of CSF flow, leading to edema in brain parenchyma. Cranioplasty allows egress of CSF, which resolves brain edema and restores function.28
Cognitive Dysfunction in Traumatic Brain Injury
Cognitive complaints are common after TBI, and often are due to a combination of multiple factors. The initial injury surely can result in cognitive impairments; however, medical issues, medications, sleep disturbance, emotional and psychiatric issues often complicate management. Studies have shown that there are significant deficits in attention, processing speed, executive functioning, and memory compared with controls, even up to 10 years postinjury.29 Cognitive recovery after injury improves with younger age and better outcomes correlate with higher premorbid IQs.30 Recovery follows an asymptotic trajectory, with the majority of recovery occurring in the first 6 months. Different domains of cognition recover at different rates; for example, the domains of memory and manual dexterity improved faster compared to executive function and word knowledge.31
Treatment of Cognitive Dysfunction
Overall, there have been very few class I studies to guide treatment of cognitive dysfunction in brain injury.32 However, some possible targets and treatments are outlined below (Table 342-1).
Methylphenidate: Recently, a Cochrane review concluded that there was not enough evidence to warrant using a monoaminergic agonist to improve recovery after TBI because none of the studies done fulfilled inclusion criteria.24 Despite this, recent reviews have mentioned that the several small scale randomized controlled trials (RCTs) involving methylphenidate are among the strongest studies in pharmacologic therapy in brain injury. Overall, these reviews suggest that methylphenidate treatment leads to significant improvements for speed of processing, and certain areas of attention and concentration, such as attentiveness. It has also been suggested that those with slower baseline information processing demonstrate a greater drug response. Thus there is relatively strong evidence that methylphenidate improves speed of processing and attention in traumatic brain injury patients.33,34 Dosages that have been used in the literature include 20 mg or 0.3 mg/kg.10,35
Donepezil and other cholinergic medications: Donepezil, galantamine, and rivastigmine, all cholinergic agonists, have not been as rigorously studied as methylphenidate. There has been one small RCT and a few small case series that concluded an increase in sustained attention and short-term memory in donepezil up to 10 mg/day. One open label study of galantamine, donepezil, and rivastigmine demonstrated some positive response in attention and vigilance, but no difference in side effects. A single large rivastigmine trial failed to show benefit in all groups; however, those with more significant memory impairments had a trend toward improvement.36 Overall, there has been moderate evidence of the use of cholinergic medications to enhance attention and concentration.37
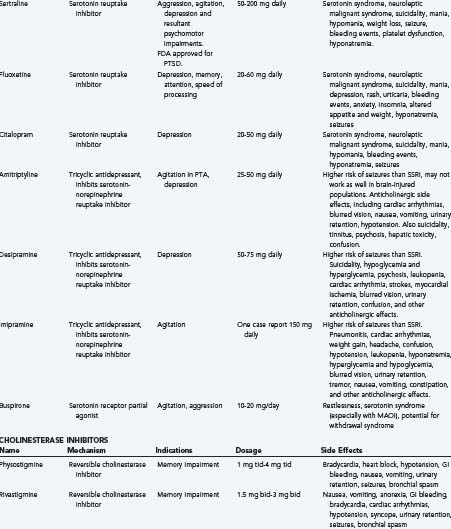
Amantadine: Amantadine is a dopamine receptor agonist and N-methyl-D-aspartate (NMDA) NMDA receptor antagonist. Case studies have suggested positive response with short-term memory, attention, planning, impulsivity, and disinhibition.38 There has been one small double-blind, crossover RCT that did not demonstrate significant difference in rate of cognitive improvement.39 Despite conflicting information, recent reviews in 2008 suggest “at doses of 200 to 400 mg/day, amantadine appears to safely improve arousal and cognition in patients with TBI,” though they warn that more studies are necessary.37,40 Small case reports have suggested some benefit for other dopaminergic agents, such as bromocriptine, selegiline, and levodopa, but the data are still very limited with regard to these agents.
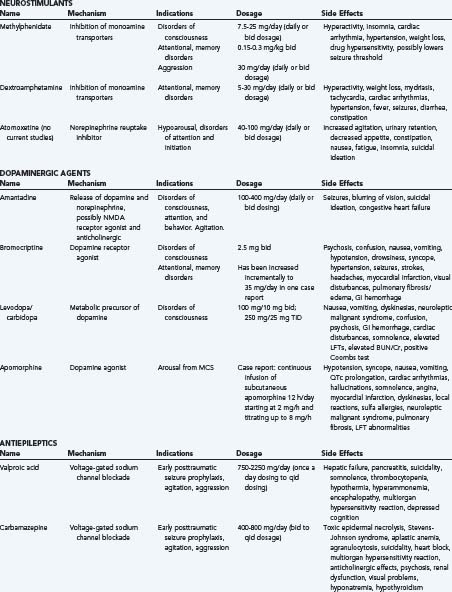
Sertraline and other SSRIs: It is well known that depression may confound cognitive impairment in TBI. Recent consensus opinion encourages the use of antidepressants in TBI, though notes that there may not be a strong effect on cognition. They suggest the use of sertraline as a first-line agent because it does not have as many side effects. One small, nonrandomized study without control suggested that fluoxetine may have improved attentional task and working memory in chronic TBI patients.35,41
Modafinil and atomoxetine: Although there have not been any human studies on the effect of atomoxetine on cognition in TBI patients at this time, animal studies have been promising.42 Modafinil has been studied for underarousal and fatigue in TBI, but has not been studied in cognition.43,44 Still, one recent review suggests that, given their effect on the dorsolateral prefrontal cortex, and mechanisms of action, atomoxetine and modafinil may be useful for attention and arousal in TBI patients.35
Warnings: Although one class I study has suggested that valproate does not impair or improve cognitive function, many other medications used in brain injury may inhibit or impair cognitive function. For example, although no studies have been done on topiramate and TBI, some authors suggest that topiramate use may cause neuropsychological problems in patients who have underlying cognitive and behavioral problems.45 The use of neuroleptics, such as haloperidol, is controversial in brain-injured patients because they may increase the length of PTA and have a negative impact on cognitive recovery.46 In general, practitioners should be wary of neuroactive medications in the brain-injured patient, and seek to eliminate medications with cognitive side effects.
Nonpharmacologic Therapy in TBI
A complete review of therapies is beyond the scope of this chapter, but some mention of various therapies should be made here. As with pharmacologic therapy, there are very few class I studies involving therapy in brain-injured patients. Currently, there are several Cochrane protocols out that are reviewing music therapy, fatigue management, vocational rehabilitation, acupuncture and cognitive rehabilitation. Currently, a Cochrane review of speech-language therapy for dysarthria in stroke and TBI patients has noted that there were no studies that met inclusion.47 A Cochrane review of multidisciplinary rehabilitation suggested that (1) different patients with different problems will need different interventions, (2) patients with moderate to severe brain injury should be routinely screened to assess needs for rehabilitation, (3) intensive intervention appears to lead to earlier gains, (4) patients discharged from in-patient rehabilitation should have access to out-patient community services, (5) mild brain injury patients benefit from follow-up and appropriate education and advice, and (6) although difficult to do, further longitudinal studies should be done to address further questions with practice-based evidence.48
Dysautonomia
Definition
Following TBI, a subset of patients experience dysautonomia, a syndrome characterized by severe paroxysmal changes in heart rate, respirations, blood pressure, temperature regulation (including diaphoresis), and muscle tone.49 Dysautonomia has been called various other names, including autonomic dysfunction syndrome, sympathetic storming, hypothalamic-midbrain dysregulation syndrome, acute midbrain syndrome, and diencephalic epilepsy.
Incidence and Effects
Dysautonomia in particular is associated with severe TBI. Other associations include severe diffuse axonal injury, preadmission hypoxia, younger age at time of injury, and possibly brainstem injury.50 In the intensive care setting, the incidence of dysautonomia ranges from 8% to 33%, falling to 5% in the rehabilitation setting. This decreased incidence is consistent with the observation that over time paroxysmal autonomic overactivity gradually settles, coinciding with neurological recovery. TBI patients with dysautonomia, however, are more likely to experience significant long-term disability, both cognitive and physical, and have longer hospital and rehabilitation stays; length of ICU stay is similar to the nondysautonomic group.50,51 A discrepancy in reported incidence is evidence of variability in criteria used in research and highlights the need for standardization in the literature.
Mechanisms
Evidence suggests that the disconnection syndrome can result from structural and/or functional disconnection. Structural damage results from diffuse and focal injury sustained to the central nervous system in TBI patients. In contrast, functional disconnections may occur because of neurotransmitter abnormalities or through alterations of the functional environment (e.g., raised intracranial pressure [ICP]).52
Anecdotally, various afferent stimuli have been identified as dysautonomic triggers including endotracheal tube suctioning, Foley catheter manipulation, passive movements (e.g., repositioning patient), and muscle stretch.49
Symptoms
Identifying dysautonomia in the intensive care unit (ICU) or rehabilitation setting should be based on the operational definition of paroxysmal increases in at least five of the following seven clinical features: heart rate greater than 120 beats/min, respiratory rate greater than 30, temperature, systolic blood pressure greater than 160, sweating/flushing/piloerection, muscle tone, and posturing (e.g., decerebrate or decorticate). By definition, several of these components will occur simultaneously and typically to a marked degree.53,54 The higher metabolic demand in the posturing dysautonomic patient, combined with postinjury gastrointestinal abnormalities results in an estimated 25% loss of body weight because of a highly catabolic state. This malnutrition increases the risk of infections and the development of critical illness neuropathy. In addition, dystonic posturing in the setting of weight loss increases the risk of developing pressure injury and contracture.55
Treatment
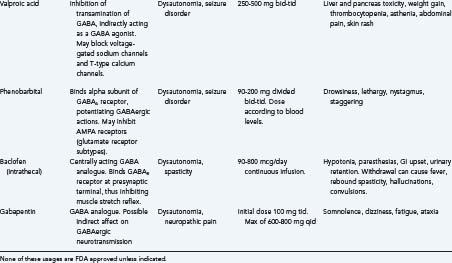
A wide range of medications has been used to treat patients with this disorder and have anecdotal support in the literature. These include morphine, α-blockers (predominantly clonidine), β-blockers (predominantly propranolol and metoprolol), anticonvulsants (valproic acid and phenobarbital), dopamine agonists (in particular bromocriptine), and benzodiazepines.56 Much of the therapeutic approach has been directed at reducing efferent activity and more recent literature has suggested benefits from both intrathecal baclofen and gabapentin administration. Both are GABA analogues and it is possible that both act to normalize modulation of afferent stimuli in dysautonomic patients by enabling inhibitory pathways within the spinal cord, thus preventing the development of the efferent arm of the syndrome.55
Agitation in Traumatic Brain Injury
Agitation and aggression can be common problems of posttraumatic brain injury. In one retrospective study in Australia, up to 70% of patients admitted for rehabilitation post-TBI demonstrated agitation. These patients had a longer duration of PTA, increased length of stay, and reduced functional independence at discharge.57 However, the incidence of agitation may vary from study to study as its definition remains unclear; for example, is it limited to the period before clearing PTA or not? Does it need to be accompanied by physical aggression or anger? A survey of experts in 1997 demonstrated a large amount of variation in definitions of agitation.58 In 2005 Lombard and associates suggested that posttraumatic agitation be defined as a state of aggression during posttraumatic amnesia in the absence of other physical, medical, or psychiatric causes, thereby distinguishing it from psychomotor agitation that occurs later in recovery.59 They also suggested that it be quantified on the Agitated Behavior Scale, where a score of 21 or higher is defined as being agitated, so that the effect of interventions can be quantified. In a recent Cochrane review, Fleminger and associates distinguished the term “agitation” from “aggression,” where aggression occurs after the period of PTA, in later stages of recovery,60 and noted that treatment options may differ between the two states.
Treatment of Agitation
β-blockers
β-blockers are postulated to work by modulating the hyperadrenergic state in TBI patients. Of medications included in the recent Cochrane review by Fleminger and associates, β-blockers had the most data to support usage for aggression and agitation in TBI. There have been two randomized controlled clinical trials on patients with propranolol; one study to evaluate effectiveness in agitation and the other to look at chronic aggression in post-TBI patients. In these studies, doses of propranolol ranged from 60 mg/day to up to 520 mg/day, the maximum dose of which exceeds standard daily dosages. Smaller RCT studies also support usage of other β-blockers, such as pindolol for agitation as well. β-blockers seem to be relatively safe for use in TBI patients; in fact, some recent reviews and retrospective studies have suggested that β-blocker administration may improve mortality after TBI,61 though a Cochrane review by Ker and coworkers suggests that more data would be needed to ascertain the risk of use in acute brain injury.62 Medication side effects include hypotension and bradycardia.
Medications to Avoid: Antipsychotics
Recently, there has been increasing concern with the use of antipsychotic medications in TBI patients. TBI experts often avoid these medications more than nonspecialized physicians.58 While there have been case studies suggesting decreases in agitation with medication administration, animal studies with typical antipsychotics have had slower motor recoveries. Typical antipsychotics such as haloperidol have been shown to correlate with longer periods of posttraumatic amnesia in human studies, although some speculate that this may be caused by increased usage of antipsychotic in more severe injuries. Patients who are tapered off chronic typical antipsychotic medications have improved neuropsychiatric studies. While atypical antipsychotics such as olanzapine, clozapine, and risperidone have not had similar effects in animal models, recent work has suggested that high doses of risperidone may have similar cognitive effects in humans as haloperidol.
Sleep Disturbance
One of the most common post-TBI sequelae is sleep disturbance, which occurs in as many as 30% to 70% of patients after TBI.63 Previous studies have suggested an increased risk of sleep disturbance in patients with milder injuries, depression, fatigue, pain,64 anxiety,65 and female gender.66 It has been postulated that sleep disturbance may be correlated with cognitive impairment; however, the evidence has been inconclusive.67–70
It has been demonstrated that subjective and objective measures of sleep disturbance do not correlate well.71,72 However, the TBI patient may have one of a myriad of sleep disturbances, from insomnia, which is the most prevalent, to circadian rhythm disturbances, to obstructive sleep apnea, to periodic leg movement syndrome. Obstructive sleep apnea (OSA) has been reported to occur as frequently as 23% of cases in one study, and periodic leg movement syndrome in 7%.73 Therefore, asking about excessive snoring, leg movements, and confirmatory polysomnography may be useful to diagnose and tailor treatment for these diseases.
Treatment of insomnia begins with elimination of other factors (such as OSA or medications). Good sleep hygiene—regular sleeping hours, avoidance of naps, etc.—is important. Cognitive behavioral therapy was found to be useful for patients with insomnia, though larger studies are needed for confirmation of efficacy.74 There has been a relative lack of research on the pharmacology of sleep in TBI patients. As mentioned above, benzodiazepines should not be taken by TBI patients because of possible long-term effects on cognition. Similarly, experts have avoided the use of GABAA receptor 1 subtype agonists (zolpidem, zopiclone, zaleplon) because they are known to occasionally cause unexpected responses in patients with cognitive deficits. Trazodone is frequently used by experienced practitioners; however, there are no published studies on trazodone for insomnia in TBI patients. Similarly, there are no published studies on melatonin or ramelteon in the TBI population.75 Lastly, modafinil for excessive daytime sleepiness was studied in a single-center, double-blind, placebo-controlled crossover trial of 53 patients in an inpatient rehabilitation setting more than 1 year after their injuries. Doses up to 400 mg did not have any effect on daytime sleepiness or fatigue.43
Posttraumatic Epilepsy
Brain injury is a very common cause of acquired epilepsy, accounting for 30% of new-onset seizure patients between the ages of 15 to 34 years, and in up to 6% of all age groups. Seizures can occur in up to 50% of certain populations of brain injury survivors.76 Posttraumatic seizures are defined by the time period after injury:
Most studies are focused on attempting to find prophylactic treatments for posttraumatic epilepsy, but none have been proved effective. Overall, though there has been some question as to whether phenytoin prevents early seizures,10 most studies suggest that phenytoin and carbamazepine have been shown to reduce early seizures. They do not, however, reduce late seizures.77 Magnesium has been tested with early phenytoin and has had no benefit. Valproate was compared with phenytoin, and though not statistically significant, was not as effective for treating early seizures, and had no positive effect on late seizures.78 Levetiracetam has been compared with phenytoin for early seizures, and seems to be as effective in preventing clinical early seizures though they did have increased abnormal findings on an electroencephalogram (EEG).79 Steroids have not been shown to decrease risk of seizures.10
Currently, the most recent practice parameters set by the American Academy of Neurology recommend that for adult patients with severe TBI (prolonged loss of consciousness or amnesia, intracranial hematoma or brain contusion on CT scan, and/or depressed skull fracture), prophylactic treatment with phenytoin, beginning with an IV loading dose, should be initiated as soon as possible after injury to decrease the risk of posttraumatic seizures occurring in the first 7 days. Prophylactic treatment with phenytoin, carbamazepine, or valproate should not routinely be used to decrease the risk of posttraumatic seizures beyond the first 7 days after injury.80 These are consistent with older practice parameters set out by the Brain Trauma Foundation.81 Unfortunately, recent reviews have stated that there are no studies to evaluate a treatment specifically for posttraumatic epilepsy, and treatment recommendations are to treat it as epilepsy caused by any other etiology.
TBI and VTE Prophylaxis
Venous thromboembolism (VTE) is by definition the occurrence of deep venous thrombosis (DVT) with or without pulmonary embolism (PE). Pulmonary embolism is associated with a high mortality and prophylactic treatment significantly reduces the incidence of VTE. In TBI, concerns about extending hemorrhage may lead to reluctance to use anticoagulant prophylaxis.82
Retrospective analyses have shown that risk factors for developing VTE in brain injury patients include extended ICU stay, need for mechanical ventilation, age over 45, male sex, coexisting spinal cord injury, and pelvic or femur fractures.83,84 Patients with severe TBI often have multiple associated fractures—predisposing to immobility—and elevated levels of circulating tissue factor, which may increase the likelihood of a hypercoagulable state.
A retrospective review of 88 patients with isolated TBI found that 25% developed DVT despite the use of prophylactic therapy with sequential compression devices in 100% of the patients and low-molecular-weight heparin (LMWH) in 42%.84 In a recent study of more than 1900 subjects by Carlile and colleagues, the incidence of DVT was examined. These investigators noted that of the 1000 patients screened via Doppler examination upon admission to inpatient rehabilitation the incidence of DVT was 1.6% in patients on prophylaxis and 1.8% in patients without prophylaxis, suggesting its use does not conclusively reduce venous thromboembolism. These also demonstrated a relative safety in the use of prophylactic anticoagulation at least in the postacute setting.85 The impact of developing DVT on outcome in TBI patients is still under investigation, though there is literature to suggest that functional outcomes of TBI patients are not compromised.86
The Brain Trauma Foundation guidelines recommend the use of compression stockings until patients are ambulatory. It is also recommended that mechanical prophylaxis be combined with pharmacologic prophylaxis but there is still insufficient evidence to support a specific drug or regimen.87
Though studies are lacking in the TBI population, a double-blind RCT found that following elective neurosurgery, LMWH administered within 24 hours in combination with compression stockings is more effective than use of the stockings alone in preventing VTE. The incidence of intracranial bleeding was similar in both groups.88 Another double-blind RCT showed that LMWH appears more effective than UFH in patients after major trauma, again without a major difference in hemorrhagic complications.89 The benefit of IVC filter placement is yet to be convincingly shown; a prospective study in a large trauma group found that the incidence of PE was not altered by the placement of IVC filters. Currently filter placement is reserved for patients that fail pharmacologic prophylaxis or have a definite contraindication to anticoagulation. Its routine use in other patients is not advised.82
Treatment
Significant practice diversity exists in the event of VTE occurring despite prophylactic therapy. Hospital surveys have shown that 75% of centers initiate full anticoagulation with LMWH or UFH followed by long-term oral warfarin. Nearly 90% of centers used IVC filters in combination with anticoagulation or as a single treatment.90 This diversity of therapeutic approaches points to the need for prospective clinical trials.
Spasticity
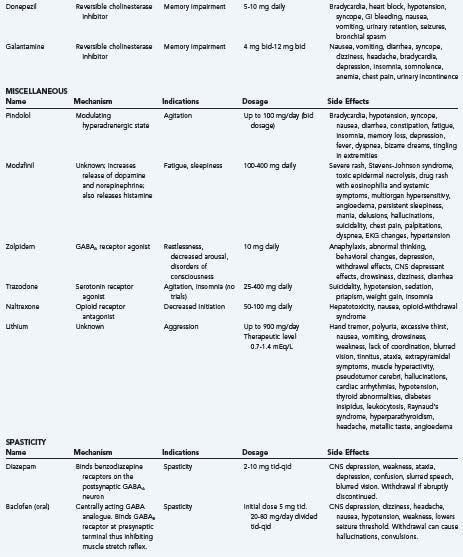
Spasticity is a velocity-dependent increase in muscle resistance to passive range of motion. It can be a common and disabling consequence of TBI. There is no class I evidence for the treatment of this disorder, therefore much has been inferred from other conditions for which there is more literature such as the spinal cord injury (SCI) population.10
Mechanism
The stretch reflex arc is the basic neural circuit that underlies the problem of spasticity. The arc consists of an afferent and efferent limb. The afferent limb originates in the muscle spindle and is carried in sensory neurons to the dorsal horn of the spinal cord. Here, synapse with a motor neuron occurs and the efferent limb exits via the anterior spinal root innervating the contractile muscle fibers. The contraction of agonist muscles must simultaneously be complemented by the relaxation of antagonist muscles, which occurs via an inhibitory neuron within the spinal cord gray matter. This basic loop is modulated by multiple synaptic influences that include descending cerebral pathways and various interspinal neurons. The treatment of spasticity aims to interrupt this arc at one or more points.91
Assessment
Several measures of hypertonicity exist and it is important to note that the definition of spasticity can have a significant impact on the validity of the assessment tool. A useful summary can be found in the review by Platz and coworkers.92 For routine assessment and clinical trials, the Ashworth Scale demonstrates good interrater reliability (Table 342-2).
Treatment
Spasticity management is a multidisciplinary activity. Goals and available treatment modalities must be discussed with both the patient and caregivers and management for each patient is individualized. Before initiating treatment for spasticity any underlying provocative factors (e.g., poor posture, constipation, incontinence, limb pain, and skin or tissue damage) should be addressed because these may have a profound influence on the disorder).93
Medications
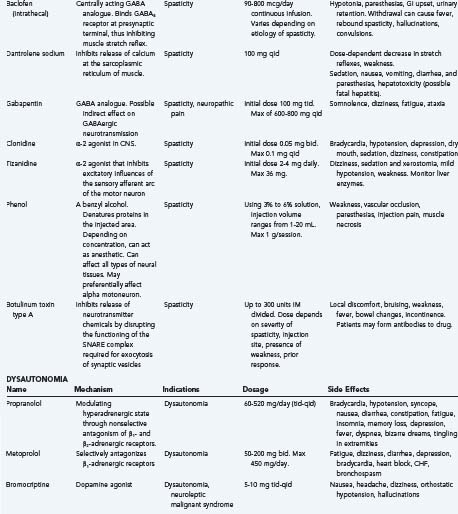
Commonly used medications include baclofen, tizanidine, dantrolene, clonidine, and gabapentin.
Clonidine, another α-2 agonist lacks studies that prove efficacy. It should be used as an alternative or adjunct if other therapies prove ineffective.91
Forty percent of patients are unable to tolerate oral agents because of side effects or do not develop an adequate antispastic effect before side effects occur.93 In this population, cognitive side effects should be considered with all of the above agents.
Chemical Blocks
Chemical blocks can be aimed at either the nerve or the muscle. Phenol has been injected into the nerve or muscle to lessen inappropriate muscle contraction. Phenol acts by inactivating the efferent limb of the spinal reflex arc. Unfortunately, chronic dysesthesia limits the use of this treatment.10
In recent years there has been a progressive shift to more widespread use of botulinum toxin type A. The toxin is taken up by the presynaptic terminal and is a potent neurotoxin that inhibits the release of neurotransmitter chemicals by disrupting the functioning of the SNARE (Soluble NSF attachment protein) complex required for exocytosis of synaptic vesicles. The resultant muscle weakness reduces muscle tone, but over the course of 3 to 4 months the presynaptic terminal sprouts and reestablishes muscle fiber communication, returning muscle tone and necessitating repeat treatments. Highly selective blockade can be achieved by using electromyography to select individual spastic muscles. Few side effects are reported and relate mostly to the injection site. Newer formulations are less antigenic and resistance is becoming less common.93
Surgical Intervention
Surgery can correct deformities induced by spasticity and improve function. This option tends to be reserved for the most refractory cases and procedures include tendon lengthening, tenotomy, and tendon transfer. More destructive procedures such as myelotomy, cordotomy, or cordectomy have been attempted to interrupt the reflex arc, but results are variable. Osteotomies may correct secondary orthopedic deformities.91
Dysphagia
Normal Swallowing
Normal swallowing is a complex process with three distinct phases. In the oral phase the cohesive bolus of food is passed to the back of the tongue voluntarily, triggering a swallowing reflex. The pharyngeal phase follows as food is transferred to the esophagus. Lastly, in the esophageal phase the bolus is shuttled via peristalsis into the stomach. This process requires the coordination of approximately 50 paired muscles and cranial nerves V through XII (excluding VIII). Volitional swallowing is thought to be controlled in the frontal lobe; additionally, adequate alertness is a necessary precondition for safe swallowing.94
Etiology and Associations
Following TBI, between 25% to 61% of patients experience swallowing impairment that is associated with severity of injury, cognitive state, and ventilatory status. The majority of these patients—up to 90%—recover the ability to feed orally with the greatest improvement occurring within 6 months of the initial trauma, following the pattern of motor recovery.95
Mechanisms of Dysphagia
Traumatic brain injury patients have neurological impairment affecting all three phases of swallowing, normal behavior, and the cognitive processes critical to a normal swallow. Abnormal muscle tone, reflexes, and sensory deficits all contribute to swallowing impairments in these patients. Marginal sensation is responsible for the poor motor response to an oral bolus. Loss of the sensory response in the pharynx dramatically impairs reflex swallowing ability and several of these abnormalities often occur in concert. Aspiration risk is increased because of delay in reflex swallowing mechanisms and reduction in tongue control. In the setting of aspiration, the physiologic response is reflex coughing, but neurological patients show a high percentage of silent aspiration, up to 60%.95 As is common in this population, the need for tracheostomy and prolonged ventilation can also augment the risk of abnormal swallowing.94
Assessment and Treatment
Clinically, dysphagia should be a concern in any patient with alteration in speech or voice quality, a cough related to swallowing, or an impairment in the gag reflex. Of note, approximately one third of normal patients do not have a gag reflex and this sign has been poorly correlated with aspiration risk. Because many TBI patients have severe speech impairments, assessment of speech and voice quality is difficult in this population.96 The difficulty of clinical assessment and high frequency of dysphagia direct that all patients should be assumed to have swallowing dysfunction. Failure to diagnose and treat patients with dysphagia place them at high risk for aspiration pneumonia, malnutrition, and dehydration and can lead to prolongation of hospital and rehabilitation stays.
Video fluoroscopy is the “gold standard” evaluation method and it is important that speech and language pathology is involved in the care of patients at a very early stage.97 Data suggest that almost half of patients that receive speech and language pathology intervention in a health care setting have dysphagia secondary to a neurological process and that more than two thirds will improve.98 The goal of therapy is to achieve safe and effective swallowing and to prevent respiratory and nutritional complications. Diet at discharge can range from normal to exclusive gastrostomy feeding. Return to unrestricted dieting has been found to occur typically within 120 days of rehabilitation. By approximately 50 days, half of patients have reached this milestone.99
Nutrition
Metabolism
Contrary to the previously held notion that comatose patients have very low metabolism, more recent work has shown that metabolism is actually increased by an average of 140% in these critically ill individuals.100 Patients that are paralyzed still have requirements above normal but less than nonparalyzed patients, suggesting that muscle tone has a significant role to play. Nitrogen excretion from protein metabolism also significantly increases for as long as a month after TBI with related depletion in muscle mass and depressed immune function. The higher metabolic rate has to be taken into consideration in the planning of caloric replacement.101 Evidence-based guidelines, however, are lacking to support enhancing caloric intake to meet the accelerated metabolic needs. Should the need to measure metabolic rates in individual patients arise, the preferred method is indirect calorimetry.
Route
Two routes exist to provide nutrition to head injured patients, enteral and parenteral. Unfortunately, upper gastrointestinal dysfunction is common following TBI with a frequency from 44% to 100% acutely. Little data exists to inform about the longer-term course of gastrointestinal intolerance. Enteral feeding can be either gastric or postpyloric. The incidence of pneumonia and aspiration with both routes is similar, but postpyloric feeds are better tolerated in patients in whom gastric dysfunction is pronounced.102
Parenteral feeding carries an increased risk of bacteremia and septicemia, possibly from catheter-related infections and possibly from inactivity of the gut. As such, enteral nutrition is clearly the preferential route for nutritional support in the TBI population.103
Timing
Timing of nutritional support is influenced by the route chosen for feeding. Data suggest that in patients who are fed via the parenteral route, feeding is typically started early—between 1 and 3 days from the time of injury. For enteral gastric feeding, nutritional support is typically held until bowel sounds are appreciated, usually from 3 to 5 days after initial injury. Enteral jejunal feeding, however, can typically be started earlier, even in the face of poor gastric emptying. Data suggest that in patients in whom parenteral feeding is used typically are started earlier, between 1 and 3 days from the time of injury.104 Early enteral nutrition has many advantages, including minimizing intestinal mucosal atrophy, hence preserving the integrity of the intestinal defense against bacterial translocation.105 This leads to a decrease in infectious complications, preserves normal gut flora, and prevents malnutrition.102 Even in patients that are unable to tolerate nutritional support to meet their caloric needs, trophic feeds at lower, better-tolerated rates serve to maintain the integrity of the intestine.
Outcome studies have shown increased mortality rates in patients who are underfed or patients in whom feeding is delayed. Therefore the goal should be to have full caloric intake in place before postinjury day 7. While consensus on early initiation of feeds exists, recommendations of other aspects of nutrition in the TBI population are vague. Specific attention should be paid to ensure adequate fluid requirements are met and to select an appropriate formula with the assistance and involvement of dietary specialists. Electrolyte monitoring to maintain homeostasis is crucial since electrolyte and glucose shifts may be detrimental to the patient. All patients being fed enterally should have aspiration precautions as standard practice and interactions between feedings and medications as well as medication reactions that might impact the gut (e.g., diarrhea, should constantly be borne in mind).103
Based on the above observations, the Brain Trauma Foundation Guidelines suggest protein and calorie replacements within 72 hours postinjury.106
Posttraumatic Hydrocephalus
Definition and Epidemiology
Posttraumatic hydrocephalus (PTH) is the commonest neurosurgical complication in the rehabilitation setting in patients with severe TBI.10 The reported incidence in the literature varies widely from 0.7% to 29%, reflective of variable definitions of the condition. If imaging criteria of ventriculomegaly are used the reported incidence is higher, ranging between 30% to 86%.107
Symptoms
Posttraumatic hydrocephalus is typically recognized in the rehabilitation setting or during neurosurgical follow-up. Several different clinical syndromes exist, all of which fall into two general categories: patients either plateau in a trajectory of previous clinical improvement or clinically deteriorate. The clinical syndromes include (1) obtundation, (2) psychomotor retardation, (3) memory impairment, (4) gait impairment, (5) incontinence, (6) behavioral disturbances, and (7) emotional disturbances.108 Clinical deterioration may involve several symptoms; alterations in consciousness and behavior are typically the most indicative of this change.109 Symptoms of hydrocephalus are not infrequently masked by the degree of brain injury sustained, making diagnosis difficult when the patient is too severely injured to display hydrocephalic symptoms or has atypical symptoms.
Investigations
Ventricular enlargement is a common finding in the postacute phase of TBI. The controversy is to distinguish brain atrophy-related ventriculomegaly from active, symptomatic ventricular dilation. Typically PTH develops within 3 months from TBI, whereas ventriculomegaly because of cortical atrophy evolves over 6 or more months. Further, cortical atrophy appears to correlate with cerebral hypoxia and anoxia and diffuse axonal injury, whereas PTH is more clearly related to CSF blockage over the convexities.109 A high level of clinical suspicion for PTH must be maintained and any suspicion explored with CT imaging, and if indicated, lumbar puncture (LP). If CSF pressure on LP is consistently above 18 cm H2O, the diagnosis seems likely. On imaging, ventriculomegaly is a typical finding but not always a good predictor of therapeutic response to interventions. Ventricular dilation in the absence of prominent sulci and gyri (i.e., the absence of brain atrophy) is most suggestive of PTH.
Several studies have attempted to describe predictive tests for selection of the most appropriate candidates for treatment. Measurements of CSF dynamics may help formulate the diagnosis as might assessment of cerebral aqueduct flow void on MRI. SPECT scanning showing temporal lobe hypoperfusion may also be a useful indicator of patients that will respond to surgery. However, these methods remain ill-defined and as yet lack strong data to support their utility. Simple CT calculations of ventricular to biparietal ratios may be an effective and easy method to evaluate for hydrocephalus.108
Treatment
In patients where high clinical suspicion exists or where testing is confirmatory for PTH, ventricular shunting is the definitive treatment. In patients who have undergone decompressive craniotomy, shunt implantation is most likely to improve outcome when combined with concomitant cranioplasty. Clinical improvement might be relatively rapid or might take several days to weeks. Shunt placement is associated with its own complications with an incidence of 20% to 40% for shunt failure and 8% to 23% for infection. Shunt failure may be seen with all types of shunts and is usually the result of delayed outflow or peritoneal catheter obstruction. The timing of treatment is another controversial issue; however, existing data suggests that patients who have been symptomatic for less than 6 months before implantation have a more favorable prognosis. Posttraumatic hydrocephalus is a prognostic factor for late functional recovery and for the development of posttraumatic epilepsy.109
Peripheral Neuropathy
To our knowledge, the rate of peripheral neuropathy in adult TBI patients has not been reported, but in children the rates reach about 8%.110 Most common causes include direct trauma from the inciting incident, or compression neuropathy from positioning during neurosurgery or during recovery during hospitalization. Common compressive neuropathies are ulnar or common peroneal neuropathies. However, there have been case reports of sciatic neuropathy because of heterotopic ossification111 and retroperitoneal hematoma.112 Evaluation of peripheral neuropathies often require EMG studies; while EMG/NCS may be abnormal within 1 week of nerve injury, it often takes 3 weeks to develop denervation changes that will help localize the lesion.113
Depression
TBI seems to increase the risk for development of psychiatric disorders, including major depression, bipolar affective disorder, general anxiety disorder, PTSD, panic disorder, and obsessive-compulsive disorder.10 About 25% of TBI patients (in some studies up to 60%)10 meet the DSM-IV criteria of major depression, with the most common symptoms being fatigue, distractibility, anger/irritation, or rumination.114 Depression has been associated with preinjury unemployment and poverty,114 and premorbid psychiatric history, including substance abuse.115 It can be difficult to tease out whether the neurobehavioral symptoms such as fatigue, sadness, irritability, loss of interest, sleep disturbance, psychomotor retardation, poor concentration, and memory dysfunction associated with depression are due to TBI-related injury or postinjury depression.
Neuroanatomically, older literature has stated that in acute TBI, depression may be correlated with left dorsolateral frontal lesions and or left basal ganglia lesions, and less strongly with right parietal-occipital lesions. Pure cortical lesions were associated with a decrease in incidence of depression. Older longitudinal studies in chronic TBI did not show any correlation between lesion location and symptoms, and depressive symptoms were only associated with lesion location over the first 3 months. However, further studies have not supported these data and currently some authors remain skeptical of these findings.116
Neurochemically, most researchers currently uphold the bioamine hypothesis. It is postulated that the frontal lobe, a site that is frequently involved in TBI, contains noradrenergic and serotoninergic projections from the brainstem to the cortex. Interruption of these projections is thought to lead to disruption of cortical aminergic function, possibly leading to downregulation of noradrenergic and serotoninergic receptors.116 Thus most treatments for depression target these pathways.
Treatment of depression should initially rule out medication-related symptoms, and neuroendocrine dysfunction. Patients should be screened for substance abuse because up to 12% of patients may have alcohol addiction or dependence. Patients should also be screened for suicidal tendencies because TBI patients have 3 to 4 times the risk of the normal population, with risk increasing for patients with cerebral contusions or hemorrhages.10 Referral to a psychiatrist or psychologist is important for multimodal treatment. In a telephone survey questioning preferred type of treatment modalities, the majority of patients stated that they preferred exercise or counseling over other treatments. Patients who had a history of antidepressant use or previous outpatient mental health treatment were more likely to prefer antidepressants.117
Pharmacologic treatment of depression generally focuses on selective serotonin reuptake inhibitors (SSRIs). Although anticholinergic medications (imipramine, nortriptyline, amitriptyline, and doxepin) have had some debatable success in treating post-TBI depression, clinicians are generally wary of their anticholinergic side effects and effects of lowering the seizure threshold.116 Because of this, SSRIs are the mainstay of treatment despite a relative lack of data regarding their efficacy. There has been one randomized controlled trial of sertraline (25 to 200 mg) for depression in TBI, with nonsignificant, but positive treatment effects in both experimental (59%) and placebo (32%) groups.118 Another smaller, randomized controlled trial with both methylphenidate and sertraline (25 to 100 mg) suggested that both methylphenidate and sertraline did have significant effects on depressive symptoms.119 Other prospective, nonrandomized trials on sertraline, fluoxetine, and citalopram have similarly had confusing results.10 However, overall, authors suggest that sertraline or citalopram (>20 mg) may be appropriate medications to treat post-TBI related depression.10,120
Apathy
Apathy is commonly defined as a state of indifference or lack of concern. In the TBI patient, the definition can be further modified as reduced goal-directed behavior because of lack of motivation. It has been associated with other frontal lobe and prefrontal cortex–directed functions, such as acquisition/memory, executive function, and psychomotor speed.121 In one prospective study, approximately 11% of TBI patients had apathy without depression and 60% of patients exhibited apathy and depression.122 As expected, apathy is associated with poor recovery and outcomes123 and can be another area of intervention in the TBI patient.
A recent Cochrane review on interventions for apathy in the TBI patient only found one randomized controlled trial in the literature. This study evaluated cranial electrotherapy stimulation; however, no statistical comparisons between treatment and placebo groups were reported, and no follow-up studies were done. There have been case reports and small case series evaluating dopaminergic agonists, such as selegiline, bromocriptine, and amantadine, or cholinergic medications such as donepezil that reported promising results.124 A recent review by Deb and coworkers also suggests that psychostimulants, such as methylphenidate, may be useful treatments, although studies overall did not target apathy exclusively.125 In conclusion, these treatments may be beneficial for patients with apathy, though more research needs to be done before formal recommendations can be made.
Fecal Incontinence
Incontinence, referring to the inadvertent loss of stool as the result of failure of normal continence mechanisms, is often overlooked or misinterpreted in patients with TBI. The incidence varies and has been reported to be as high as 68% acutely and 5% after 1 year.97 In addition to the personal and social implications, fecal incontinence has several medical complications including skin irritation, pressure ulceration, increased infection risk, and greater nursing demand.
In the TBI population, incontinence is more likely in older patients. This likely reflects the greater cognitive and physical limitations in older adults, though there may be some contributions from age-related physiologic changes in gastrointestinal motility and continence. More severe TBI is related to an increased incidence of fecal incontinence, likely a functional deficit reflecting impaired cognitive and motor ability. Because of the relation between the skull and anterior fossa, frontal lobe injury is a common occurrence in TBI. Located in the frontal lobes is the frontal defecation center, which is believed to modulate social control of defecation, overriding the reflex need to defecate. This region is poorly functional in infantile brains (yet to be myelinated) and in senile brains (because of degenerative change) and is likely to be injured in many individuals with moderate to severe TBI. Improvement in individuals that subsequently regain continence attests in part to the recovery of this portion of frontal lobe function.126
Urinary Incontinence
Urinary disturbance, like fecal disturbance is associated with poor functional outcome following TBI. Incidence has been reported as 62% acutely and 18% at 6 month follow-up.97
Bladder dysfunction may occur from injury at various levels of neural control. Local pelvic and bladder damage may affect muscle or lower motor neuron inputs. Spinal cord damage is seen in polytraumas and the contribution of supraspinal centers must be considered. The exact anatomic location responsible for micturition control remains in debate, although, as with fecal control, it appears to be localized in the frontal lobe. In addition, patients may have communication deficits that interfere with their ability to express the need to urinate or be unable to physically use toilet facilities. Because incontinence as a clinical syndrome is nonspecific, any patient being evaluated requires urodynamic studies to differentiate the various neurological disturbances that may produce symptoms.127 Leary and coworkers found that in the rehabilitation setting, 90% of patients were given multidisciplinary goals addressing self-care and mobility but only 3.5% were given multidisciplinary goals for bowel and bladder function.128 Because significant improvement is seen following rehabilitation directed at these functions, it is important that focus is directed at correcting bowel and bladder abnormalities.
Headache
Definition
Headache is a common physical manifestation of TBI and either produces new primary symptoms or worsens preexisting headache syndromes in patients with a prior headache history.129 Headaches are classified according to the International Classification of Headache Disorders (ICHD), which provides clinical criteria to categorize primary and secondary headaches. According to these criteria, posttraumatic headache is either acute or chronic. The headache must begin within 2 weeks and resolve within 2 months to meet acute criteria. Chronic posttraumatic headache requires the symptoms to continue for more than 8 weeks. The criteria also indicate that the causative trauma should be significant, causing abnormal clinical, neurological, or laboratory examination but also notes that mild head trauma may produce symptoms even in the absence of other signs of brain dysfunction.130
Incidence
Headache is the most common somatic complaint among TBI patients.131 The estimated incidence is 30% to 90% of patients with mild or moderate head injury with apparently a lower incidence in patients with severe TBI, although this remains a topic of investigation.132 The presence of posttraumatic headache has not been correlated consistently with injury severity. Premorbid headache history, however, has been shown to increase the risk of posttraumatic headache.133 Because acute posttraumatic headache resolves within a few weeks, the majority of cases are represented by chronic posttraumatic headache.130 No specific headache type defines posttraumatic headaches; all the features typical of primary headaches may occur. The majority, up to 85% in some series, experience tension types of headaches, with up to 21% meeting criteria for a migraine. In addition, cervicogenic headaches are found in many patients, though figures for incidence are disparate because of variable diagnostic criteria and range between 8% and 88%. It is also noteworthy that chronic posttraumatic headache is not an isolated phenomenon, occurring instead as a part of the posttraumatic/postconcussive syndrome with its associated somatic, psychological, and cognitive complaints.132
Clinical Features
Posttraumatic headaches take on the features of the primary headache disorder they resemble. Tension types of headaches, for example, often show bilateral discomfort, occasionally in a band-like distribution and are steady as opposed to throbbing. A posttraumatic migraine often has the typical features of photophobia and phonophobia but usually in a milder form. Unlike tension types of headaches, these tend to be unilateral and throbbing. A cervicogenic headache is almost always posttraumatic with cervical spine pain radiating to the occiput or even the front of the head. It also is unilateral and should be suspected where an examination demonstrates limited range of cervical motion.132
Therapy
Following TBI, headaches are treated much the same way as primary headache disorders. Acute therapy consists of NSAIDS, acetaminophen, and triptan medication for migraine-like symptoms. Symptomatic therapy with antiemetics should be aggressive. Should headaches become chronic and frequent, prophylactic therapy may become needed using either β-blockers, calcium-channel blockers, antiepileptic medications, or antidepressants. The choice of a prophylactic agent should be guided by the psychological and cognitive symptoms present at the time of treatment. Patients that have experienced only mild TBI will be able to participate in associated therapies such as biofeedback, stress management, therapeutic exercise, and neuromuscular reeducation.134
Neuropathic Pain
Central neuropathic pain may be defined as pain initiated or caused by a primary lesion or primary dysfunction of the CNS. The basic underlying mechanism is neuronal hyperexcitability.135
Mechanisms
Nociception is the result of detection of intense stimuli by nociceptors, specialized sensory receptors. These receptors typically respond to the usual sensory inputs, but are distinguished by being high threshold. Once stimulated they transfer action potentials to the spinal cord and from there to the brain. Central pain occurs in the absence of such a stimulus and in the presence of reduced nociceptive thresholds so that usually innocuous stimuli produce pain. This hyperexcitability may be the result of enhanced facilitation or impaired inhibition of central processes coupled with cellular pathophysiology that includes electrolyte imbalances and abnormalities of neurotransmitter release.136 The development of neuropathic pain involves not just neuronal pathways but also peripheral components such as Schwann cells, satellite cells in the dorsal root ganglia, and the immune system, as well as microglia and astrocytes in the spine.
Manifestations of central sensitization include allodynia (reduced sensory threshold for pain), hyperalgesia (increased responsiveness of sensory neurons to stimuli), and secondary hyperalgesia (expansion of the territory by which a sensory neuron is activated).136,137
Complex Regional Pain Syndrome
Pain due to polytraumatic injuries presents several rehabilitation challenges. Due to cognitive impairments, evaluating and monitoring pain may be difficult. Commonly used pain therapies may potentially interfere with the cognition needed to participate adequately in rehabilitation.131 Complex regional pain syndrome (CRPS) is a painful disorder that often develops as a disproportionate reaction to trauma.
The disorder typically afflicts the limbs and is characterized by severe pain, allodynia, hyperalgesia, disorders of blood flow and sweat regulation, edema, and trophic changes to the skin and its organs.139 CRPS can be stratified into two types. Type 1, the commonest, is defined by the presence of major nerve damage, which is not seen in type 2.
The presence of finger agnosia, elements of neglect, and delay in performing imagined movements all point to a disturbance of parietal lobe function in these patients. The role of the parietal cortex in maintenance of internal representation suggests that altered sensory perception may also occur as a consequence in this disorder.140
Epidemiology
Existing data on population-based incidence rates varies dramatically between 5.5 and 26.2 per 100,000 person years. The difference likely results from variations in case definition and methodology and highlights the need for uniform nomenclature and diagnostic criteria. Certain demographic characteristics appear common. Women are affected at an approximate ratio of 3.5 to 1 compared with men, and most cases are in groups over age 40. Data also show a predominance in Caucasian and Japanese populations.141
Etiology
Reports of CRPS occur after a wide subset of events including common injuries, cardiovascular events, malignancies, infections, and medication administration. Spontaneous onset is rare but has been reported. Fractures—upper extremity more so than lower—are the commonest precipitating events and classically involve the wrist. Severity of injury is unrelated to the risk of developing CRPS. There have been studies that make a correlation to intra-articular fractures and complaints of tightness and pressure during casting as positive predictive factors. However, fracture repositioning and the use of external fixation have not been associated.141
Mechanisms
Several different forms of pain are distinguished in CRPS including neuropathic, inflammatory, nociceptive, and functional. Classically CRPS has been regarded as a CNS disorder with the major pathogenesis being the dysregulation of sympathetic function. Recently, however, the contribution of peripheral mechanisms including inflammation and hypoxia has been better appreciated. Alterations have been found in small nerve fibers innervating blood vessels and the integument, as well as aberrations in vascular and muscular tissue and other deep somatic tissues thought to arise as a result of damage by initial inflammation.139
The exact mechanism remains unclear but several possibilities have been entertained. Hypoxia triggers inflammatory responses and nociceptive input may lead to sensitization and alteration in cortical organization of sensory and motor units. Dorsal horn neuropeptides may facilitate sensitization by binding to NMDA receptors, whereas adrenergic receptors may have their sensitivity and expression altered through the effect of inflammation on nociceptive fibers. Sympathetic dysfunction may result from central sympathetic hyperactivity or hypersensitivity of peripheral adrenergic receptors. Sympathetic dysfunction may also cause hypoxia by impairing nutritive blood flow. Cytokine alterations affect the nitric oxide and endothelin balance and severe pain impairs movement, possibly resulting in inflammatory mediators and free radicals accumulating, thus preventing desensitization. Severe chronic pain causes psychological distress, which in turn influences sympathetic outflow and circulating catecholamine levels. While many of these concepts are hypotheses, their existence points to the likely multifactorial nature of this disease and the circular interaction between many of the factors involved.141
Diagnosis
Evaluation and diagnosis of CRPS is typically clinical, without a gold standard for confirmation. Three-phase bone scintigraphy may provide important information for the common osseous changes. Pathologic uptake in metacarpal and metacarpophalangeal bones has been proposed as a sensitive and specific marker. The best timing is in the subacute phase (i.e., under 1 year). Plain x-rays, bone densitometry, and MRI may be abnormal but are not sensitive or specific for this condition. Similarly, autonomic testing may reveal abnormal sweating patterns in the affected limbs, but this too is nonspecific.142
Treatment
Pharmacologic and interventional options exist. It is important to note that traditional pain therapies appear to offer little benefit. A clearer understanding of mechanisms will permit more effective approaches to the care of these patients and therapies targeted to sensory-motor integration appear promising.140
Conservative Measures
The importance of a conservative treatment regimen to augment the aforementioned treatment options cannot be overemphasized. Physical and occupational therapy and somatic integration therapies should have realistic goals and the treatment algorithm must involve an interdisciplinary approach. Finally, the clinical suspicion of CRPS should prompt the initiation of treatment as early as possible.142
Heterotopic Ossification
Heterotopic ossification (HO) refers to the formation of extraosseous lamellar bone in the soft tissues. It typically involves proximal large joints, and in the TBI population has a range of incidence between 10% to 77%, with only 20% of these being clinically significant.97 The condition is typically diagnosed during the first 6 months postinjury, with a peak in the second month, but may be present many weeks before it is clinically recognized.
Clinical Features
The hip is the commonest location, followed by the knee and elbow with 30% of patients having two joints affected. In the most severe forms, complete ankylosis of the joint occurs with massive bone formation.143
Pathogenesis
Debate exists as to the exact mechanism of heterotopic bone formation, but four factors appear necessary. First, an inciting event; second, an inflammatory stimulus that arises from the site of injury (likely in the form of cytokine activity); third, the arrival of mesenchymal progenitor cells; and fourth, the differentiation of these cells into osteoblasts and chondroblasts. TBI results in accelerated fracture healing and reduced collagen degradation, which may also predispose to abnormal bone formation. Acute TBI patients are often hyperventilated to reduce ICP with resulting respiratory alkalosis.144 The alkaline pH may promote calcium precipitation, which is an essential component of the bony matrix.
Investigations
The current gold standard is a triple-phase bone scan with the first and second phases becoming positive within 3 weeks after injury and the third phase 1 to 4 weeks later. CT scans and plain radiographs will show maturing bone and elevated serum alkaline phosphatase levels may be measured, but their utility is debated because of a poor correlation between levels and clinical syndrome. Calcium and phosphate are usually normal in the serum.143
Treatment
Etidronate is considered a common first-line therapy for the management of HO and has been repeatedly shown to halt or slow the progression of disease in the SCI population. In the TBI population, there has been one prospective controlled trial that found a lower incidence of HO in the treatment versus control group.145
The prophylactic use of NSAIDs, particularly indomethacin; COX2 inhibitors; and ibuprofen have been studied in patients with SCI and hip arthroplasty and have provided evidence that the early use of anti-inflammatory agents reduces the likelihood of developing HO by up to a magnitude of three.145–147 These findings have not yet been translated in the TBI population.
Surgery is typically reserved for patients with significant skin breakdown, pain, or marked loss of function and is usually not considered until 18 months after development of HO to ensure full maturation of the newly formed bone. Based on several studies, however, there is only limited evidence that excision improves clinical outcomes in the TBI population.145
Baguley IJ, Heriseanu RE, Cameron ID, et al. A critical review of the pathophysiology of dysautonomia following traumatic brain injury. Neurocrit Care. 2008;8:293-300.
Dobscha SK, Clark ME, Morasco BJ, et al. Systematic review of the literature on pain in patients with polytrauma including traumatic brain injury. Pain Med. 2009;10:1200-1217.
Draper K, Ponsford J. Cognitive functioning ten years following traumatic brain injury and rehabilitation. Neuropsychology. 2008;22:618-625.
Formisano R, Bivona U, Catani S, et al. Post-traumatic headache: facts and doubts. J Headache Pain. 2009;10:145-152.
Fugate LP, Spacek LA, Kresty LA, et al. Definition of agitation following traumatic brain injury: I. A survey of the brain injury special interest group of the American Academy of Physical Medicine and Rehabilitation. Arch Phys Med Rehabil. 1997;78:917-923.
Gordon WA, Zafonte R, Cicerone K, et al. Traumatic brain injury rehabilitation: state of the science. Am J Phys Med Rehabil. 2006;85:343-382.
Krakau K, Omne-Ponten M, Karlsson T, et al. Metabolism and nutrition in patients with moderate and severe traumatic brain injury: a systematic review. Brain Inj. 2006;20:345-367.
Lombard LA, Zafonte RD. Agitation after traumatic brain injury: considerations and treatment options. Am J Phys Med Rehabil. 2005;84:797-812.
Mazzini L, Campini R, Angelino E, et al. Posttraumatic hydrocephalus: a clinical, neuroradiologic, and neuropsychologic assessment of long-term outcome. Arch Phys Med Rehabil. 2003;84:1637-1641.
Menon DK, Zahed C. Prediction of outcome in severe traumatic brain injury. Curr Opin Crit Care. 2009;15:437-441.
Mysiw WJ, Bogner JA, Corrigan JD, et al. The impact of acute care medications on rehabilitation outcome after traumatic brain injury. Brain Inj. 2006;20:905-911.
Nakase-Richardson R, Sepehri A, Sherer M, et al. Classification schema of posttraumatic amnesia duration-based injury severity relative to 1-year outcome: analysis of individuals with moderate and severe traumatic brain injury. Arch Phys Med Rehabil. 2009;90:17-19.
Orff HJ, Ayalon L, Drummond SP. Traumatic brain injury and sleep disturbance: a review of current research. J Head Trauma Rehabil. 2009;24:155-165.
Riggio S, Wong M. Neurobehavioral sequelae of traumatic brain injury. Mt Sinai J Med. 2009;76:163-172.
Safaz I, Alaca R, Yasar E, et al. Medical complications, physical function and communication skills in patients with traumatic brain injury: a single centre 5-year experience. Brain Inj. 2008;22:733-739.
Satkunam LE. Rehabilitation medicine: 3. Management of adult spasticity. CMAJ. 2003;169:1173-1179.
Stiver SI, Wintermark M, Manley GT. Motor trephine syndrome: a mechanistic hypothesis. Acta Neurochir Suppl. 2008;102:273-277.
Tang ME, Lobel DA. Severe traumatic brain injury: maximizing outcomes. Mt Sinai J Med. 2009;76:119-128.
Tenovuo O. Pharmacological enhancement of cognitive and behavioral deficits after traumatic brain injury. Curr Opin Neurol. 2006;19:528-533.
The Brain Trauma Foundation, The American Association of Neurological Surgeons, The Joint Section on Neurotrauma and Critical Care. Role of antiseizure prophylaxis following head injury. J Neurotrauma. 2000;17:549-553.
Wagner RH, Cifu DX, Keyser-Marcus L. Functional outcome of individuals with traumatic brain injury and lower extremity deep venous thrombosis. J Head Trauma Rehabil. 1999;14:558-566.
Whyte J, Katz D, Long D, et al. Predictors of outcome in prolonged posttraumatic disorders of consciousness and assessment of medication effects: a multicenter study. Arch Phys Med Rehabil. 2005;86:453-462.
Wieloch T, Nikolich K. Mechanisms of neural plasticity following brain injury. Curr Opin Neurobiol. 2006;16:258-264.
1 Thurman DJ, Sniezek JE, Johnson D, et al. Guidelines for Surveillance of Central Nervous System Injury. Atlanta: Centers for Disease Control and Prevention; 1995.
2 Langlois JA, Rutland-Brown W, Thomas KE. Traumatic brain injury in the United States: emergency department visits, hospitalizations, and deaths. Atlanta: Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; 2004.
3 Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil. 2006;21:375-378.
4 Okie S. Traumatic brain injury in the war zone. N Engl J Med. 2005;352:2043-2047.
5 Department of Health and Human Services (US) and Centers for Disease Control and Prevention. Non-fatal sports- and recreation-related traumatic brain injuries treated in emergency departments—United States, 2002-2005. MMWR. 2007;56(29):733-737.
6 Finkelstein E, Corso PS, Miller TR. The Incidence and Economic Burden of Injuries in the United States. Oxford, UK: Oxford University Press; 2006.
7 Zasler ND, Katz DI, Zafonte RD. Brain Injury Medicine Principles and Practice. New York: Demos; 2007.
8 Nakase-Richardson R, Sepehri A, Sherer M, et al. Classification schema of posttraumatic amnesia duration-based injury severity relative to 1-year outcome: analysis of individuals with moderate and severe traumatic brain injury. Arch Phys Med Rehabil. 2009;90:17-19.
9 McNett M. A review of the predictive ability of Glasgow Coma Scale scores in head-injured patients. J Neurosci Nurs. 2007;39:68-75.
10 Gordon WA, Zafonte R, Cicerone K, et al. Traumatic brain injury rehabilitation: state of the science. Am J Phys Med Rehabil. 2006;85:343-382.
11 Sherer M, Struchen MA, Yablon SA, et al. Comparison of indices of traumatic brain injury severity: Glasgow Coma Scale, length of coma and post-traumatic amnesia. J Neurol Neurosurg Psychiatry. 2008;79:678-685.
12 Katz DI, Alexander MP. Traumatic brain injury. Predicting course of recovery and outcome for patients admitted to rehabilitation. Arch Neurol. 1994;51:661-670.
13 Menon DK, Zahed C. Prediction of outcome in severe traumatic brain injury. Curr Opin Crit Care. 2009;15:437-441.
14 Levin HS. Neuroplasticity following non-penetrating traumatic brain injury. Brain Inj. 2003;17:665-674.
15 Wieloch T, Nikolich K. Mechanisms of neural plasticity following brain injury. Curr Opin Neurobiol. 2006;16:258-264.
16 Rosenzweig MR. Effects of differential experience on brain and cognition throughout. In: Broman SH, Fletcher JM, editors. The Changing Nervous System: Neurobehavioral Consequences of Early Brain Disorders. New York: Oxford University Press; 1999:25-56.
17 Buchli AD, Schwab ME. Inhibition of Nogo: a key strategy to increase regeneration, plasticity and functional recovery of the lesioned central nervous system. Ann Med. 2005;37:556-567.
18 Hummel FC, Cohen LG. Drivers of brain plasticity. Curr Opin Neurol. 2005;18:667-674.
19 Whyte J, Katz D, Long D, et al. Predictors of outcome in prolonged posttraumatic disorders of consciousness and assessment of medication effects: a multicenter study. Arch Phys Med Rehabil. 2005;86:453-462.
20 Giacino JT, Zasler ND. Outcome after severe traumatic brain injury: coma, the vegetative state, and the minimally responsive state. J Head Trauma Rehabil. 1995;10:40-56.
21 Giacino JT, Zasler ND, Katz DI, et al. Development of practice guidelines for assessment and management of the vegetative and minimally conscious states. J Head Trauma Rehabil. 1997;12:79-89.
22 Richer E, Tell L. Indications, efficacy and tolerance of drug therapy in view of improving recovery of consciousness following a traumatic brain injury. Ann Readapt Med Phys. 2003;46:177-183.
23 Willis C, Lybrand S, Bellamy N. Excitatory amino acid inhibitors for traumatic brain injury. Cochrane Database Syst Rev. 2004;CD003986.
24 Forsyth RJ, Jayamoni B, Paine TC. Monoaminergic agonists for acute traumatic brain injury. Cochrane Database Syst Rev. 2006;CD003984.
25 Lombardi F, Taricco M, De TA, et al. Sensory stimulation for brain injured individuals in coma or vegetative state. Cochrane Database Syst Rev. 2002;CD001427.
26 Swaine BR, Sullivan SJ. Longitudinal profile of early motor recovery following severe traumatic brain injury. Brain Inj. 1996;10:347-366.
27 Watson MJ. Path tree analysis as a tool for describing motor recovery following acquired brain injury: an exploratory study. Disabil Rehabil. 2007;29:231-243.
28 Stiver SI, Wintermark M, Manley GT. Motor trephine syndrome: a mechanistic hypothesis. Acta Neurochir Suppl. 2008;102:273-277.
29 Draper K, Ponsford J. Cognitive functioning ten years following traumatic brain injury and rehabilitation. Neuropsychology. 2008;22:618-625.
30 Green RE, Colella B, Christensen B, et al. Examining moderators of cognitive recovery trajectories after moderate to severe traumatic brain injury. Arch Phys Med Rehabil. 2008;89:S16-S24.
31 Christensen BK, Colella B, Inness E, et al. Recovery of cognitive function after traumatic brain injury: a multilevel modeling analysis of Canadian outcomes. Arch Phys Med Rehabil. 2008;89:S3-S15.
32 Gordon WA, Zafonte R, Cicerone K, et al. Traumatic brain injury rehabilitation: state of the science. Am J Phys Med Rehabil. 2006;85:343-382.
33 Rees L, Marshall S, Hartridge C, et al. Cognitive interventions post acquired brain injury. Brain Inj. 2007;21:161-200.
34 Willmott C, Ponsford J. Efficacy of methylphenidate in the rehabilitation of attention following traumatic brain injury: a randomised, crossover, double blind, placebo controlled inpatient trial. J Neurol Neurosurg Psychiatry. 2009;80:552-557.
35 Tenovuo O. Pharmacological enhancement of cognitive and behavioral deficits after traumatic brain injury. Curr Opin Neurol. 2006;19:528-533.
36 Silver JM, Koumaras B, Chen M, et al. Effects of rivastigmine on cognitive function in patients with traumatic brain injury. Neurology. 2006;67:748-755.
37 Liepert J. Pharmacotherapy in restorative neurology. Curr Opin Neurol. 2008;21:639-643.
38 Kraus MF, Maki PM. Effect of amantadine hydrochloride on symptoms of frontal lobe dysfunction in brain injury: case studies and review. J Neuropsychiatry Clin Neurosci. 1997;9:222-230.
39 Schneider WN, Drew-Cates J, Wong TM, et al. Cognitive and behavioural efficacy of amantadine in acute traumatic brain injury: an initial double-blind placebo-controlled study. Brain Inj. 1999;13:863-872.
40 Sawyer E, Mauro LS, Ohlinger MJ. Amantadine enhancement of arousal and cognition after traumatic brain injury. Ann Pharmacother. 2008;42:247-252.
41 Gordon WA, Zafonte R, Cicerone K, et al. Traumatic brain injury rehabilitation: state of the science. Am J Phys Med Rehabil. 2006;85:343-382.
42 Reid WM, Hamm RJ. Post-injury atomoxetine treatment improves cognition following experimental traumatic brain injury. J Neurotrauma. 2008;25:248-256.
43 Jha A, Weintraub A, Allshouse A, et al. A randomized trial of modafinil for the treatment of fatigue and excessive daytime sleepiness in individuals with chronic traumatic brain injury. J Head Trauma Rehabil. 2008;23:52-63.
44 Elovic E. Use of Provigil for underarousal following TBI. J Head Trauma Rehabil. 2000;15:1068-1071.
45 Tang V, Warden J, Cullen N, et al. Topiramate in traumatic brain injury: adverse effects on cognitive function. J Head Trauma Rehabil. 2007;22:409-410.
46 Mysiw WJ, Bogner JA, Corrigan JD, et al. The impact of acute care medications on rehabilitation outcome after traumatic brain injury. Brain Inj. 2006;20:905-911.
47 Sellars C, Hughes T, Langhorne P. Speech and language therapy for dysarthria due to non-progressive brain damage. Cochrane Database Syst Rev. 2005;CD002088.
48 Turner-Stokes L, Disler PB, Nair A, et al. Multi-disciplinary rehabilitation for acquired brain injury in adults of working age. Cochrane Database Syst Rev. 2005;CD004170.
49 Baguley IJ, Heriseanu RE, Nott MT, et al. Dysautonomia after severe traumatic brain injury: evidence of persisting overresponsiveness to afferent stimuli. Am J Phys Med Rehabil. 2009;88:615-622.
50 Baguley IJ, Nicholls JL, Felmingham KL, et al. Dysautonomia after traumatic brain injury: a forgotten syndrome? J Neurol Neurosurg Psychiatry. 1999;67:39-43.
51 Baguley IJ, Heriseanu RE, Felmingham KL, et al. Dysautonomia and heart rate variability following severe traumatic brain injury. Brain Inj. 2006;20:437-444.
52 Baguley IJ, Heriseanu RE, Cameron ID, et al. A critical review of the pathophysiology of dysautonomia following traumatic brain injury. Neurocrit Care. 2008;8:293-300.
53 Srinivasan S, Lim CC, Thirugnanam U. Paroxysmal autonomic instability with dystonia. Clin Auton Res. 2007;17:378-381.
54 Baguley IJ, Slewa-Younan S, Heriseanu RE, et al. The incidence of dysautonomia and its relationship with autonomic arousal following traumatic brain injury. Brain Inj. 2007;21:1175-1181.
55 Baguley IJ, Heriseanu RE, Gurka JA, et al. Gabapentin in the management of dysautonomia following severe traumatic brain injury: a case series. J Neurol Neurosurg Psychiatry. 2007;78:539-541.
56 Baguley IJ, Cameron ID, Green AM, et al. Pharmacological management of dysautonomia following traumatic brain injury. Brain Inj. 2004;18:409-417.
57 Nott MT, Chapparo C, Baguley IJ. Agitation following traumatic brain injury: an Australian sample. Brain Inj. 2006;20:1175-1182.
58 Fugate LP, Spacek LA, Kresty LA, et al. Definition of agitation following traumatic brain injury: I. A survey of the brain injury special interest group of the American Academy of Physical Medicine and Rehabilitation. Arch Phys Med Rehabil. 1997;78:917-923.
59 Lombard LA, Zafonte RD. Agitation after traumatic brain injury: considerations and treatment options. Am J Phys Med Rehabil. 2005;84:797-812.
60 Fleminger S, Greenwood RJ, Oliver DL. Pharmacological management for agitation and aggression in people with acquired brain injury. Cochrane Database Syst Rev. 2006;CD003299.
61 Tran TY, Dunne IE, German JW. Beta blockers exposure and traumatic brain injury: a literature review. Neurosurg Focus. 2008;25:E8.
62 Ker K, Blackhall K. Beta-2 receptor antagonists for acute traumatic brain injury. Cochrane Database Syst Rev. 2008;CD006686.
63 Ouellet MC, Beaulieu-Bonneau S, Morin CM. Insomnia in patients with traumatic brain injury: frequency, characteristics, and risk factors. J Head Trauma Rehabil. 2006;21:199-212.
64 Orff HJ, Ayalon L, Drummond SP. Traumatic brain injury and sleep disturbance: a review of current research. J Head Trauma Rehabil. 2009;24:155-165.
65 Rao V, Spiro J, Vaishnavi S, et al. Prevalence and types of sleep disturbances acutely after traumatic brain injury. Brain Inj. 2008;22:381-386.
66 Clinchot DM, Bogner J, Mysiw WJ, et al. Defining sleep disturbance after brain injury. Am J Phys Med Rehabil. 1998;77:291-295.
67 Mahmood O, Rapport LJ, Hanks RA, et al. Neuropsychological performance and sleep disturbance following traumatic brain injury. J Head Trauma Rehabil. 2004;19:378-390.
68 Makley MJ, Johnson-Greene L, Tarwater PM, et al. Return of memory and sleep efficiency following moderate to severe closed head injury. Neurorehabil Neural Repair. 2009;23:320-326.
69 Parsons LC, Ver BD. Sleep-awake patterns following cerebral concussion. Nurs Res. 1982;31:260-264.
70 Prigatano GP, Stahl ML, Orr WC, et al. Sleep and dreaming disturbances in closed head injury patients. J Neurol Neurosurg Psychiatry. 1982;45:78-80.
71 Hossain JL, Reinish LW, Heslegrave RJ, et al. Subjective and objective evaluation of sleep and performance in daytime versus nighttime sleep in extended-hours shift-workers at an underground mine. J Occup Environ Med. 2004;46:212-226.
72 Ouellet MC, Morin CM. Subjective and objective measures of insomnia in the context of traumatic brain injury: a preliminary study. Sleep Med. 2006;7:486-497.
73 Castriotta RJ, Wilde MC, Lai JM, et al. Prevalence and consequences of sleep disorders in traumatic brain injury. J Clin Sleep Med. 2007;3:349-356.
74 Ouellet MC, Morin CM. Efficacy of cognitive-behavioral therapy for insomnia associated with traumatic brain injury: a single-case experimental design. Arch Phys Med Rehabil. 2007;88:1581-1592.
75 Larson EB, Zollman FS. The effect of sleep medications on cognitive recovery from traumatic brain injury. J Head Trauma Rehabil. 2010;25:61-67.
76 Lowenstein DH. Epilepsy after head injury: an overview. Epilepsia. 2009;50(Suppl 2):4-9.
77 Schierhout G, Roberts I. Anti-epileptic drugs for preventing seizures following acute traumatic brain injury. Cochrane Database Syst Rev. 2001;CD000173.
78 Temkin NR. Preventing and treating posttraumatic seizures: the human experience. Epilepsia. 2009;50(Suppl 2):10-13.
79 Jones KE, Puccio AM, Harshman KJ, et al. Levetiracetam versus phenytoin for seizure prophylaxis in severe traumatic brain injury. Neurosurg Focus. 2008;25:E3.
80 Chang BS, Lowenstein DH. Practice parameter: antiepileptic drug prophylaxis in severe traumatic brain injury: report of the quality standards subcommittee of the American Academy of Neurology. Neurology. 2003;60:10-16.
81 The Brain Trauma Foundation, The American Association of Neurological Surgeons, The Joint Section on Neurotrauma and Critical Care. Role of antiseizure prophylaxis following head injury. J Neurotrauma. 2000;17:549-553.
82 Vergouwen MD, Roos YB, Kamphuisen PW. Venous thromboembolism prophylaxis and treatment in patients with acute stroke and traumatic brain injury. Curr Opin Crit Care. 2008;14:149-155.
83 Page RB, Spott MA, Krishnamurthy S, et al. Head injury and pulmonary embolism: a retrospective report based on the Pennsylvania trauma outcomes study. Neurosurgery. 2004;54:143-148.
84 Denson K, Morgan D, Cunningham R, et al. Incidence of venous thromboembolism in patients with traumatic brain injury. Am J Surg. 2007;193:380-383.
85 Carlile M, Nicewander D, Yablon SA, et al. Prophylaxis for venous thromboembolism during rehabilitation for traumatic brain injury: a multicenter observational study. J Trauma. 2010;68:916-923.
86 Wagner RH, Cifu DX, Keyser-Marcus L. Functional outcome of individuals with traumatic brain injury and lower extremity deep venous thrombosis. J Head Trauma Rehabil. 1999;14:558-566.
87 Bratton SL, Chestnut RM, Ghajar J, et al. Guidelines for the management of severe traumatic brain injury. V. Deep vein thrombosis prophylaxis. J Neurotrauma. 2007;24(Suppl 1):S32-S36.
88 Agnelli G, Piovella F, Buoncristiani P, et al. Enoxaparin plus compression stockings compared with compression stockings alone in the prevention of venous thromboembolism after elective neurosurgery. N Engl J Med. 1998;339:80-85.
89 Geerts WH, Jay RM, Code KI, et al. A comparison of low-dose heparin with low-molecular-weight heparin as prophylaxis against venous thromboembolism after major trauma. N Engl J Med. 1996;335:701-707.
90 Carlile MC, Yablon SA, Mysiw WJ, et al. Deep venous thrombosis management following traumatic brain injury: a practice survey of the traumatic brain injury model systems. J Head Trauma Rehabil. 2006;21:483-490.
91 Satkunam LE. Rehabilitation medicine: 3. Management of adult spasticity. CMAJ. 2003;169:1173-1179.
92 Platz T, Eickhof C, Nuyens G, et al. Clinical scales for the assessment of spasticity, associated phenomena, and function: a systematic review of the literature. Disabil Rehabil. 2005;27:7-18.
93 Ward AB. Spasticity treatment with botulinum toxins. J Neural Transm. 2008;115:607-616.
94 Morgan AS, Mackay LE. Causes and complications associated with swallowing disorders in traumatic brain injury. J Head Trauma Rehabil. 1999;14:454-461.
95 Terre R, Mearin F. Evolution of tracheal aspiration in severe traumatic brain injury-related oropharyngeal dysphagia: 1-year longitudinal follow-up study. Neurogastroenterol Motil. 2009;21:361-369.
96 Terre R, Mearin F. Prospective evaluation of oro-pharyngeal dysphagia after severe traumatic brain injury. Brain Inj. 2007;21:1411-1417.
97 Safaz I, Alaca R, Yasar E, et al. Medical complications, physical function and communication skills in patients with traumatic brain injury: a single centre 5-year experience. Brain Inj. 2008;22:733-739.
98 Wheeler-Hegland K, Frymark T, Schooling T, et al. Evidence-based systematic review: oropharyngeal dysphagia behavioral treatments. Part V—applications for clinicians and researchers. J Rehabil Res Dev. 2009;46:215-222.
99 Hansen TS, Engberg AW, Larsen K. Functional oral intake and time to reach unrestricted dieting for patients with traumatic brain injury. Arch Phys Med Rehabil. 2008;89:1556-1562.
100 Mackay LE, Morgan AS, Bernstein BA. Factors affecting oral feeding with severe traumatic brain injury. J Head Trauma Rehabil. 1999;14:435-447.
101 Tang ME, Lobel DA. Severe traumatic brain injury: maximizing outcomes. Mt Sinai J Med. 2009;76:119-128.
102 Krakau K, Omne-Ponten M, Karlsson T, et al. Metabolism and nutrition in patients with moderate and severe traumatic brain injury: a systematic review. Brain Inj. 2006;20:345-367.
103 Cook AM, Peppard A, Magnuson B. Nutrition considerations in traumatic brain injury. Nutr Clin Pract. 2008;23:608-620.
104 Perel P, Yanagawa T, Bunn F, et al. Nutritional support for head-injured patients. Cochrane Database Syst Rev. 2006;CD001530.
105 Yanagawa T, Bunn F, Roberts I, et al. Nutritional support for head-injured patients. Cochrane Database Syst Rev. 2002;CD001530.
106 The Brain Trauma Foundation, The American Association of Neurological Surgeons, The Joint Section on Neurotrauma and Critical Care. Nutrition. J Neurotrauma. 2000;17:539-547.
107 Guyot LL, Michael DB. Post-traumatic hydrocephalus. Neurol Res. 2000;22:25-28.
108 Wen L, Wan S, Zhan RY, et al. Shunt implantation in a special sub-group of post-traumatic hydrocephalus—patients have normal intracranial pressure without clinical representations of hydrocephalus. Brain Inj. 2009;23:61-64.
109 Mazzini L, Campini R, Angelino E, et al. Posttraumatic hydrocephalus: a clinical, neuroradiologic, and neuropsychologic assessment of long-term outcome. Arch Phys Med Rehabil. 2003;84:1637-1641.
110 Philip PA, Philip M. Peripheral nerve injuries in children with traumatic brain injury. Brain Inj. 1992;6:53-58.
111 Safaz I, Alaca R, Bozlar U, et al. Bilateral sciatic nerve entrapment due to heterotopic ossification in a traumatic brain-injured patient. Am J Phys Med Rehabil. 2008;87:65-67.
112 Groah SL, Cifu DX. The rehabilitative management of the traumatic brain injury patient with associated femoral neuropathy. Arch Phys Med Rehabil. 1995;76:480-483.
113 Winfree CJ, Kline DG. Intraoperative positioning nerve injuries. Surg Neurol. 2005;63:5-18.
114 Seel RT, Kreutzer JS, Rosenthal M, et al. Depression after traumatic brain injury: a National Institute on Disability and Rehabilitation Research Model Systems multicenter investigation. Arch Phys Med Rehabil. 2003;84:177-184.
115 Fedoroff JP, Starkstein SE, Forrester AW, et al. Depression in patients with acute traumatic brain injury. Am J Psychiatry. 1992;149:918-923.
116 Rosenthal M, Christensen BK, Ross TP. Depression following traumatic brain injury. Arch Phys Med Rehabil. 1998;79:90-103.
117 Fann JR, Jones AL, Dikmen SS, et al. Depression treatment preferences after traumatic brain injury. J Head Trauma Rehabil. 2009;24:272-278.
118 Ashman TA, Cantor JB, Gordon WA, et al. A randomized controlled trial of sertraline for the treatment of depression in persons with traumatic brain injury. Arch Phys Med Rehabil. 2009;90:733-740.
119 Lee H, Kim SW, Kim JM, et al. Comparing effects of methylphenidate, sertraline and placebo on neuropsychiatric sequelae in patients with traumatic brain injury. Hum Psychopharmacol. 2005;20:97-104.
120 Silver JM, McAllister TW, Arciniegas DB. Depression and cognitive complaints following mild traumatic brain injury. Am J Psychiatry. 2009;166:653-661.
121 Andersson S, Bergedalen AM. Cognitive correlates of apathy in traumatic brain injury. Neuropsychiatry Neuropsychol Behav Neurol. 2002;15:184-191.
122 Kant R, Duffy JD, Pivovarnik A. Prevalence of apathy following head injury. Brain Inj. 1998;12:87-92.
123 Gray JM, Shepherd M, McKinlay WW, et al. Negative symptoms in the traumatically brain-injured during the first year postdischarge, and their effect on rehabilitation status, work status and family burden. Clin Rehabil. 1994;8:188-197.
124 Lane-Brown A, Tate R. Interventions for apathy after traumatic brain injury. Cochrane Database Syst Rev. 2009;CD006341.
125 Deb S, Crownshaw T. The role of pharmacotherapy in the management of behaviour disorders in traumatic brain injury patients. Brain Inj. 2004;18:1-31.
126 Foxx-Orenstein A, Kolakowsky-Hayner S, Marwitz JH, et al. Incidence, risk factors, and outcomes of fecal incontinence after acute brain injury: findings from the Traumatic Brain Injury Model Systems national database. Arch Phys Med Rehabil. 2003;84:231-237.
127 Moiyadi AV, Devi BI, Nair KP. Urinary disturbances following traumatic brain injury: clinical and urodynamic evaluation. NeuroRehabilitation. 2007;22:93-98.
128 Leary SM, Liu C, Cheesman AL, et al. Incontinence after brain injury: prevalence, outcome and multidisciplinary management on a neurological rehabilitation unit. Clin Rehabil. 2006;20:1094-1099.
129 Nampiaparampil DE. Prevalence of chronic pain after traumatic brain injury: a systematic review. JAMA. 2008;300:711-719.
130 Formisano R, Bivona U, Catani S, et al. Post-traumatic headache: facts and doubts. J Headache Pain. 2009;10:145-152.
131 Dobscha SK, Clark ME, Morasco BJ, et al. Systematic review of the literature on pain in patients with polytrauma including traumatic brain injury. Pain Med. 2009;10:1200-1217.
132 Solomon S. Posttraumatic headache. Med Clin North Am. 2001;85:987-996.
133 Riggio S, Wong M. Neurobehavioral sequelae of traumatic brain injury. Mt Sinai J Med. 2009;76:163-172.
134 Gottesman RF, Komotar R, Hillis AE. Neurologic aspects of traumatic brain injury. Int Rev Psychiatry. 2003;15:302-309.
135 Gray P. Pregabalin in the management of central neuropathic pain. Expert Opin Pharmacother. 2007;8:3035-3041.
136 Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci. 2007;10:1361-1368.
137 Freeman R. The treatment of neuropathic pain. CNS Spectr. 2005;10:698-706.
138 Nicholson BD. Evaluation and treatment of central pain syndromes. Neurology. 2004;62:S30-S36.
139 Groeneweg G, Huygen FJ, Coderre TJ, et al. Regulation of peripheral blood flow in complex regional pain syndrome: clinical implication for symptomatic relief and pain management. BMC Musculoskelet Disord. 2009;10:116.
140 McCabe CS, Cohen H, Hall J, et al. Somatosensory conflicts in complex regional pain syndrome type 1 and fibromyalgia syndrome. Curr Rheumatol Rep. 2009;11:461-465.
141 de MM, Sturkenboom MC, Huygen FJ. Current understandings on complex regional pain syndrome. Pain Pract. 2009;9:86-99.
142 Hsu ES. Practical management of complex regional pain syndrome. Am J Ther. 2009;16:147-154.
143 van Kuijk AA, Geurts AC, van Kuppevelt HJ. Neurogenic heterotopic ossification in spinal cord injury. Spinal Cord. 2002;40:313-326.
144 Andermahr J, Elsner A, Brings AE, et al. Reduced collagen degradation in polytraumas with traumatic brain injury causes enhanced osteogenesis. J Neurotrauma. 2006;23:708-720.
145 Cullen N, Bayley M, Bayona N, et al. Management of heterotopic ossification and venous thromboembolism following acquired brain injury. Brain Inj. 2007;21:215-230.
146 Banovac K, Williams JM, Patrick LD, et al. Prevention of heterotopic ossification after spinal cord injury with indomethacin. Spinal Cord. 2001;39:370-374.
147 Banovac K, Williams JM, Patrick LD, et al. Prevention of heterotopic ossification after spinal cord injury with COX-2 selective inhibitor (rofecoxib). Spinal Cord. 2004;42:707-710.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 342 Rehabilitation of Patients with Traumatic Brain Injury
Accidents happen. Brain injury.
Unsolicited. Unanticipated. Unwelcome.
In the blink of an eye, everything changes.
—www.biama.org (Brain Injury Association of Massachusetts)
Definitions and Epidemiology
This excludes lacerations or contusions of the face, eye, ear or scalp, or fractures of the facial bones without other criteria listed above; as well as birth trauma, primary anoxic, inflammatory, infectious, toxic or metabolic encephalopathies which are not complications of head trauma, neoplasms or ischemic/hemorrhagic stroke without associated trauma.1
Using data collected from 1999-2001, the U.S. Centers for Disease Control and Prevention estimated that the annual incidence of TBI was approximately 1.4 million people per year.2 This number has remained relatively steady, as the most recent data from 2003 estimated an incidence of 1,565,000 with an overall mortality rate of 17.5/100,000 people. TBI accounts for an estimated 1.1 million emergency department visits, 235,000 hospitalizations, and 50,000 deaths in the United States per year.3 Morbidity rates remain high and are estimated at about 5.3 million Americans; in other words, about 2% of the population needs long-term help to perform activities of daily living because of TBI.3 Despite being quite impressive, these numbers likely underestimate the current impact of TBI on the American population for two reasons: (1) a new influx of returning veterans from Iraq and Afghanistan, in which incidence of TBI likely approaches 22%, if not higher4; and (2) likely underreporting of mild brain injury (concussion). For example, though sports-related concussion has been estimated at an annual incidence of 1.6 to 3.8 million,5 only 200,000 such patients are treated in emergency departments per year.3
Patients between the ages of 15 and 44 years make up about 45% of cases, with males making up about 60% of the population. Most recent data from 2003 still suggest that falls remain the most common cause of injury (32%), with motor vehicle accidents as the second most common cause (19.2%). Because patients are often injured in the prime of their lives, the socioeconomic impact can be devastating. Direct and indirect costs of TBI in the United States were estimated to be $60 billion in 2000.6 Thus despite advances in treatment of TBI, it remains a significant cause of morbidity and mortality in the United States.
Prognosis in TBI
| Category | Description |
|---|---|
| 1. Death | Death |
| 2. Persistent vegetative state | Prolonged unconsciousness with no verbalization or following of commands. Absent awareness of self and environment. Patient may open eyes and have reflexive actions. Sleep-wake cycles are present. |
| 3. Severe disability | Patient unable to be independent for any 24-hour period due to physical or mental disability |
| 4. Moderate disability | Patient can travel by public transportation and work in a sheltered environment. There may be residual deficits that do not interfere with independent daily life. |
| 5. Good recovery | Return to normal life with minor or no residual deficits. |
Some important predictive factors are listed below:
The best predictor of outcomes after TBI is the length of posttraumatic amnesia (PTA). The longer the duration of PTA, the worse outcomes generally are. Clinically, resolution of PTA occurs when patients are able to incorporate daily events into working memory. Patients with PTA of less than 24 hours generally have a quick and full recovery with few exceptions, and patients with PTA of more than 4 weeks generally have permanent deficits. However, more stringent “threshold” criteria have been used in previous textbooks to preserve hope for caregivers. These criteria state that severe disability (on GOS) is unlikely with PTA of less than 2 months, and good recovery (on GOS) is unlikely when PTA is more than 3 months.7 It has been shown that 21% patients with PTA between 1 and 2 months did return to work within a year, whereas only 9% of patients with PTA of more olthan 70 days were able to return to work within a year.8
Length of coma, defined as time from injury to follow commands, can be an important milestone as well. Generally, severe disability (GOS) is likely when length of coma is more than 2 weeks. Good recovery (GOS) is unlikely when length of coma is more than 4 weeks. For more information, please see the section on Disorders of Consciousness.
The best Glasgow Coma Scale (GCS) score within 24 hours correlates with outcomes, with lower scores associated with worse outcomes, and with the best motor response subset as the most accurate predictor of outcomes. Unfortunately, there are no “threshold” criteria because patients with severe injury can have good recovery. Indeed, recent reviews of GCS in outcomes of TBI suggest that other measures, such as age and pupillary response, improve its predictive capacity.9
Neuroimaging has become increasingly important in predicting outcome. It has been suggested that an adequate threshold marker is the presence of bilateral brainstem lesions; good recovery (GOS) is unlikely when bilateral brainstem lesions are present on an early MRI. CT scan findings of subarachnoid hemorrhage, midline shift, extradural hematoma, bilateral injury, or transventricular injury correlate with worse outcomes, though a full range of outcomes is still possible with these injuries.7
Research continues in this field to delineate other possible markers of prognosis and outcome. Biomarkers such as S100, genetic markers such as APOE 4 status, the ratio of total brain volume to third ventricle size, T2 gradient echo MRI lesions, PET scanning, diffusion tensor imaging, magnetic resonance spectroscopy, and somatosensory evoked potential responses are currently under study and may provide important prognostic information in the future.10
Given the uncertainty surrounding various predictors, several authors are currently trying to mathematically model outcomes of TBI using predictive variables.11–13 One such model, based on a large population study, is available at http://www.tbi-impact.org/. It must be noted that these models are still undergoing refinement, though they may also provide important prognostic information in the future.
Mechanisms of Recovery
Introduction
Much basic science research has been done on the mechanisms of neural recovery, particularly in stroke literature. Exact comparisons may be difficult for TBI because the nature of the injury is often diffuse but it seems likely that the basic mechanisms of recovery are similar. Trauma disrupts neuronal connections including the connectivity and organization of networks mediating executive functions and sensory and motor responses.14 In addition to the focal damage of TBI, dysfunction can occur in areas anatomically removed, likely because of diaschisis involving tissue hypometabolism, neurovascular uncoupling, and aberrant neurotransmission.15 In addition to local repair, neuroplasticity is thought to be a crucial feature of recovery from TBI with research from as far back as the 1960s demonstrating how brain structure and chemistry can be affected by environmental manipulation.16
Overview
Recovery of neurological function is dynamic and multifaceted but often limited partly because of the inability for dramatic anatomic reorganization in the brain following lesions. Three distinct phases of recovery have been described. The first involved activation of cell repair and reversal of diaschisis; the second, plasticity of existing neuronal pathways to produce a functional change; the third, neuroanatomic plasticity to produce nascent connections. Where damage is focal, recovery primarily engages ipsilateral brain tissue, but with more severe TBI it is likely that contralateral brain regions are also recruited.16 The recovery process begins immediately and extends over the course of many months to years.
Therapeutic Targets
One area of interest is growth promoters and inhibitors. When administered within days of the insult, functional recovery is promoted by growth factors through stimulation of neuronal plasticity. Several agents are currently in clinical trials and are diverse in nature, including erythropoietin, granulocyte colony stimulating factor, statin drugs, and phosphodiesterase-5 inhibitors.15 By contrast, growth inhibitors impair recovery with particular insights being gained into the effects of growth cone extension. Myelin-associated proteins, including Nogo-A, oligodendrocyte myelin glycoprotein, and myelin-associated glycoprotein (MAG), activate the inhibitory Nogo-66 receptor to prevent axonal sprouting.17 Intravenous or intrathecal delivery of antibodies or peptides aimed at either the Nogo protein or receptor dramatically enhance functional recovery after neural injury in animal models.
Finally, therapy may in the future focus on brain activation with early studies showing that amphetamine and possibly L-dopa enhanced recovery of function in animal models. The suggestion was that adrenergic and cholinergic neurotransmitter systems are critical to the recovery process, but human trials have had limited results.18 Brain tissue can also be activated by physical and environmental means, for example, repetitive physical activity or sensory experiences. As part of environmental stimulation, social interaction is felt to be important in that it induces multiple biologic effects on the brain.
While many questions remain about the mechanisms of recovery in TBI, the current sophisticated molecular tools available for researchers continue to yield insights and hopefully will lead to promising new therapies.15
Disorders of Consciousness
Up to 15% of patients with severe TBI are unable to respond consistently to commands at 4 weeks after injury, and the majority of these will have impairments of consciousness.19 There has been much confusion surrounding the different states of consciousness. By definition:
Vegetative state (VS)—arousable neurobehavioral unresponsiveness. Patients have periods of eye opening, either spontaneously or in response to external stimuli; subcortical responses to external stimuli (including posturing, tachycardia, diaphoresis with pain); return of autonomic functions (sleep-wake cycle, normalization of respiratory and digestive functions); roving eye movements without concomitant visual tracking ability.20
Minimally conscious state (MCS)—a condition of severely altered consciousness in which the patient demonstrates minimal but definite behavioral evidence of self- or environmental awareness. The Aspen workgroup proposed that one or more of the following should be present: (1) following simple commands; (2) manipulation of objects; (3) gestural or verbal yes/no responses; (4) intelligible verbalization; or (5) stereotypical movements that occur in a meaningful relationship to the eliciting stimulus and are not attributable to reflexive activity. These responses must be reproducible or occur on a sustained basis.21
Amantadine, bromocriptine, amphetamine, and methylphenidate and other neurostimulants have been proposed to help with arousal and vigilance.22 There has been a case report of a patient in MCS who regained full consciousness after treatment with amantadine who declined when the medication was withdrawn.10 However, Cochrane Reviews on monoaminergic agonists and excitatory amino acid inhibitors have not found good randomized trials to review.23,24 Similarly, another recent Cochrane Review determined that there have not been any good studies to support the use of sensory stimulation in MCS or VS patients.25 Unfortunately, most research surrounding treatment of disorders of consciousness in brain injury is poor, involving case reports or small numbers of patients without controls. More research is needed to further clarify treatment algorithms in the future.
Motor Recovery
As with everything in brain injury, motor recovery is highly individualized and varies from person to person. Though it seems that the majority of motor recovery happens in the first 6 months after injury, it is possible for patients to continue gaining function after that. Swaine and coworkers studied a small population of patients with severe TBI (GCS ≤ 8) for 6 weeks after injury, and demonstrated that, while the percentage of patients with basic primitive reflexes (asymmetric tonic neck reflex, flexor withdrawal) stayed relatively stable, equilibrium reactions, protective reactions, sitting reactions, and simple motor skills slowly increased over time. The sequence of recovery—reactions in sitting appeared before those in kneeling and before those in standing. Recovery of equilibrium reactions tended to occur before development of protective reactions. Similarly, with motor skills, rolling ability recovered before ability to sit unsupported, followed by the ability to stand, then walk independently, then climb stairs, then hop on one foot, then the ability to run. Surprisingly, some simple motor skills (rolling, sitting, even walking unsupported) seemed to recover faster than equilibrium. Also surprisingly, patients did not necessarily lose primitive reflexes before recovering simple motor tasks. Of note, in this case series, 60% of patients by 6 weeks could sit independently and 56% of patients could walk without assistance.26 Watson and associates have suggested a path-tree analysis instead of a simple linear progression of motor skills and demonstrated that while there is an underlying pattern of recovery, there are many routes that a patient can follow.27
Motor Trephine Syndrome
Following craniectomy, patients may suffer delayed or insidious headache, fatigue, and mood disturbance. A few may have delayed contralateral monoparesis that is reversible with cranioplasty—the motor trephine syndrome. In one retrospective review, up to 26% of patients with decompressive craniectomy developed delayed contralateral upper extremity weakness that improved with cranioplasty, with full recovery 1 month after cranioplasty. Weakness developed an average of 4.5 months later, was predominantly distal upper extremity proximal, causing difficulty with grip and extension of metacarpals, and was slowly progressive. There was no sensory or language impairment. These patients more often had blossoming of cerebral contusions after craniectomy and cerebrospinal fluid (CSF) flow disturbances (hygromas and hydrocephalus), and had a longer time to cranioplasty than patients without motor trephine syndrome. Mechanistically, it has been proposed that motor trephine syndrome is caused by disturbances of CSF flow, leading to edema in brain parenchyma. Cranioplasty allows egress of CSF, which resolves brain edema and restores function.28
Cognitive Dysfunction in Traumatic Brain Injury
Cognitive complaints are common after TBI, and often are due to a combination of multiple factors. The initial injury surely can result in cognitive impairments; however, medical issues, medications, sleep disturbance, emotional and psychiatric issues often complicate management. Studies have shown that there are significant deficits in attention, processing speed, executive functioning, and memory compared with controls, even up to 10 years postinjury.29 Cognitive recovery after injury improves with younger age and better outcomes correlate with higher premorbid IQs.30 Recovery follows an asymptotic trajectory, with the majority of recovery occurring in the first 6 months. Different domains of cognition recover at different rates; for example, the domains of memory and manual dexterity improved faster compared to executive function and word knowledge.31
Treatment of Cognitive Dysfunction
Overall, there have been very few class I studies to guide treatment of cognitive dysfunction in brain injury.32 However, some possible targets and treatments are outlined below (Table 342-1).
Methylphenidate: Recently, a Cochrane review concluded that there was not enough evidence to warrant using a monoaminergic agonist to improve recovery after TBI because none of the studies done fulfilled inclusion criteria.24 Despite this, recent reviews have mentioned that the several small scale randomized controlled trials (RCTs) involving methylphenidate are among the strongest studies in pharmacologic therapy in brain injury. Overall, these reviews suggest that methylphenidate treatment leads to significant improvements for speed of processing, and certain areas of attention and concentration, such as attentiveness. It has also been suggested that those with slower baseline information processing demonstrate a greater drug response. Thus there is relatively strong evidence that methylphenidate improves speed of processing and attention in traumatic brain injury patients.33,34 Dosages that have been used in the literature include 20 mg or 0.3 mg/kg.10,35
Donepezil and other cholinergic medications: Donepezil, galantamine, and rivastigmine, all cholinergic agonists, have not been as rigorously studied as methylphenidate. There has been one small RCT and a few small case series that concluded an increase in sustained attention and short-term memory in donepezil up to 10 mg/day. One open label study of galantamine, donepezil, and rivastigmine demonstrated some positive response in attention and vigilance, but no difference in side effects. A single large rivastigmine trial failed to show benefit in all groups; however, those with more significant memory impairments had a trend toward improvement.36 Overall, there has been moderate evidence of the use of cholinergic medications to enhance attention and concentration.37
Amantadine: Amantadine is a dopamine receptor agonist and N-methyl-D-aspartate (NMDA) NMDA receptor antagonist. Case studies have suggested positive response with short-term memory, attention, planning, impulsivity, and disinhibition.38 There has been one small double-blind, crossover RCT that did not demonstrate significant difference in rate of cognitive improvement.39 Despite conflicting information, recent reviews in 2008 suggest “at doses of 200 to 400 mg/day, amantadine appears to safely improve arousal and cognition in patients with TBI,” though they warn that more studies are necessary.37,40 Small case reports have suggested some benefit for other dopaminergic agents, such as bromocriptine, selegiline, and levodopa, but the data are still very limited with regard to these agents.
Sertraline and other SSRIs: It is well known that depression may confound cognitive impairment in TBI. Recent consensus opinion encourages the use of antidepressants in TBI, though notes that there may not be a strong effect on cognition. They suggest the use of sertraline as a first-line agent because it does not have as many side effects. One small, nonrandomized study without control suggested that fluoxetine may have improved attentional task and working memory in chronic TBI patients.35,41
Modafinil and atomoxetine: Although there have not been any human studies on the effect of atomoxetine on cognition in TBI patients at this time, animal studies have been promising.42 Modafinil has been studied for underarousal and fatigue in TBI, but has not been studied in cognition.43,44 Still, one recent review suggests that, given their effect on the dorsolateral prefrontal cortex, and mechanisms of action, atomoxetine and modafinil may be useful for attention and arousal in TBI patients.35
Warnings: Although one class I study has suggested that valproate does not impair or improve cognitive function, many other medications used in brain injury may inhibit or impair cognitive function. For example, although no studies have been done on topiramate and TBI, some authors suggest that topiramate use may cause neuropsychological problems in patients who have underlying cognitive and behavioral problems.45 The use of neuroleptics, such as haloperidol, is controversial in brain-injured patients because they may increase the length of PTA and have a negative impact on cognitive recovery.46 In general, practitioners should be wary of neuroactive medications in the brain-injured patient, and seek to eliminate medications with cognitive side effects.
Nonpharmacologic Therapy in TBI
A complete review of therapies is beyond the scope of this chapter, but some mention of various therapies should be made here. As with pharmacologic therapy, there are very few class I studies involving therapy in brain-injured patients. Currently, there are several Cochrane protocols out that are reviewing music therapy, fatigue management, vocational rehabilitation, acupuncture and cognitive rehabilitation. Currently, a Cochrane review of speech-language therapy for dysarthria in stroke and TBI patients has noted that there were no studies that met inclusion.47 A Cochrane review of multidisciplinary rehabilitation suggested that (1) different patients with different problems will need different interventions, (2) patients with moderate to severe brain injury should be routinely screened to assess needs for rehabilitation, (3) intensive intervention appears to lead to earlier gains, (4) patients discharged from in-patient rehabilitation should have access to out-patient community services, (5) mild brain injury patients benefit from follow-up and appropriate education and advice, and (6) although difficult to do, further longitudinal studies should be done to address further questions with practice-based evidence.48
Dysautonomia
Definition
Following TBI, a subset of patients experience dysautonomia, a syndrome characterized by severe paroxysmal changes in heart rate, respirations, blood pressure, temperature regulation (including diaphoresis), and muscle tone.49 Dysautonomia has been called various other names, including autonomic dysfunction syndrome, sympathetic storming, hypothalamic-midbrain dysregulation syndrome, acute midbrain syndrome, and diencephalic epilepsy.
Incidence and Effects
Dysautonomia in particular is associated with severe TBI. Other associations include severe diffuse axonal injury, preadmission hypoxia, younger age at time of injury, and possibly brainstem injury.50 In the intensive care setting, the incidence of dysautonomia ranges from 8% to 33%, falling to 5% in the rehabilitation setting. This decreased incidence is consistent with the observation that over time paroxysmal autonomic overactivity gradually settles, coinciding with neurological recovery. TBI patients with dysautonomia, however, are more likely to experience significant long-term disability, both cognitive and physical, and have longer hospital and rehabilitation stays; length of ICU stay is similar to the nondysautonomic group.50,51 A discrepancy in reported incidence is evidence of variability in criteria used in research and highlights the need for standardization in the literature.
Mechanisms
Evidence suggests that the disconnection syndrome can result from structural and/or functional disconnection. Structural damage results from diffuse and focal injury sustained to the central nervous system in TBI patients. In contrast, functional disconnections may occur because of neurotransmitter abnormalities or through alterations of the functional environment (e.g., raised intracranial pressure [ICP]).52
Anecdotally, various afferent stimuli have been identified as dysautonomic triggers including endotracheal tube suctioning, Foley catheter manipulation, passive movements (e.g., repositioning patient), and muscle stretch.49
Symptoms
Identifying dysautonomia in the intensive care unit (ICU) or rehabilitation setting should be based on the operational definition of paroxysmal increases in at least five of the following seven clinical features: heart rate greater than 120 beats/min, respiratory rate greater than 30, temperature, systolic blood pressure greater than 160, sweating/flushing/piloerection, muscle tone, and posturing (e.g., decerebrate or decorticate). By definition, several of these components will occur simultaneously and typically to a marked degree.53,54 The higher metabolic demand in the posturing dysautonomic patient, combined with postinjury gastrointestinal abnormalities results in an estimated 25% loss of body weight because of a highly catabolic state. This malnutrition increases the risk of infections and the development of critical illness neuropathy. In addition, dystonic posturing in the setting of weight loss increases the risk of developing pressure injury and contracture.55
Treatment
A wide range of medications has been used to treat patients with this disorder and have anecdotal support in the literature. These include morphine, α-blockers (predominantly clonidine), β-blockers (predominantly propranolol and metoprolol), anticonvulsants (valproic acid and phenobarbital), dopamine agonists (in particular bromocriptine), and benzodiazepines.56 Much of the therapeutic approach has been directed at reducing efferent activity and more recent literature has suggested benefits from both intrathecal baclofen and gabapentin administration. Both are GABA analogues and it is possible that both act to normalize modulation of afferent stimuli in dysautonomic patients by enabling inhibitory pathways within the spinal cord, thus preventing the development of the efferent arm of the syndrome.55
Agitation in Traumatic Brain Injury
Agitation and aggression can be common problems of posttraumatic brain injury. In one retrospective study in Australia, up to 70% of patients admitted for rehabilitation post-TBI demonstrated agitation. These patients had a longer duration of PTA, increased length of stay, and reduced functional independence at discharge.57 However, the incidence of agitation may vary from study to study as its definition remains unclear; for example, is it limited to the period before clearing PTA or not? Does it need to be accompanied by physical aggression or anger? A survey of experts in 1997 demonstrated a large amount of variation in definitions of agitation.58 In 2005 Lombard and associates suggested that posttraumatic agitation be defined as a state of aggression during posttraumatic amnesia in the absence of other physical, medical, or psychiatric causes, thereby distinguishing it from psychomotor agitation that occurs later in recovery.59 They also suggested that it be quantified on the Agitated Behavior Scale, where a score of 21 or higher is defined as being agitated, so that the effect of interventions can be quantified. In a recent Cochrane review, Fleminger and associates distinguished the term “agitation” from “aggression,” where aggression occurs after the period of PTA, in later stages of recovery,60 and noted that treatment options may differ between the two states.
Treatment of Agitation
β-blockers
β-blockers are postulated to work by modulating the hyperadrenergic state in TBI patients. Of medications included in the recent Cochrane review by Fleminger and associates, β-blockers had the most data to support usage for aggression and agitation in TBI. There have been two randomized controlled clinical trials on patients with propranolol; one study to evaluate effectiveness in agitation and the other to look at chronic aggression in post-TBI patients. In these studies, doses of propranolol ranged from 60 mg/day to up to 520 mg/day, the maximum dose of which exceeds standard daily dosages. Smaller RCT studies also support usage of other β-blockers, such as pindolol for agitation as well. β-blockers seem to be relatively safe for use in TBI patients; in fact, some recent reviews and retrospective studies have suggested that β-blocker administration may improve mortality after TBI,61 though a Cochrane review by Ker and coworkers suggests that more data would be needed to ascertain the risk of use in acute brain injury.62 Medication side effects include hypotension and bradycardia.
Medications to Avoid: Antipsychotics
Recently, there has been increasing concern with the use of antipsychotic medications in TBI patients. TBI experts often avoid these medications more than nonspecialized physicians.58 While there have been case studies suggesting decreases in agitation with medication administration, animal studies with typical antipsychotics have had slower motor recoveries. Typical antipsychotics such as haloperidol have been shown to correlate with longer periods of posttraumatic amnesia in human studies, although some speculate that this may be caused by increased usage of antipsychotic in more severe injuries. Patients who are tapered off chronic typical antipsychotic medications have improved neuropsychiatric studies. While atypical antipsychotics such as olanzapine, clozapine, and risperidone have not had similar effects in animal models, recent work has suggested that high doses of risperidone may have similar cognitive effects in humans as haloperidol.
Sleep Disturbance
One of the most common post-TBI sequelae is sleep disturbance, which occurs in as many as 30% to 70% of patients after TBI.63 Previous studies have suggested an increased risk of sleep disturbance in patients with milder injuries, depression, fatigue, pain,64 anxiety,65 and female gender.66 It has been postulated that sleep disturbance may be correlated with cognitive impairment; however, the evidence has been inconclusive.67–70
It has been demonstrated that subjective and objective measures of sleep disturbance do not correlate well.71,72 However, the TBI patient may have one of a myriad of sleep disturbances, from insomnia, which is the most prevalent, to circadian rhythm disturbances, to obstructive sleep apnea, to periodic leg movement syndrome. Obstructive sleep apnea (OSA) has been reported to occur as frequently as 23% of cases in one study, and periodic leg movement syndrome in 7%.73 Therefore, asking about excessive snoring, leg movements, and confirmatory polysomnography may be useful to diagnose and tailor treatment for these diseases.
Treatment of insomnia begins with elimination of other factors (such as OSA or medications). Good sleep hygiene—regular sleeping hours, avoidance of naps, etc.—is important. Cognitive behavioral therapy was found to be useful for patients with insomnia, though larger studies are needed for confirmation of efficacy.74 There has been a relative lack of research on the pharmacology of sleep in TBI patients. As mentioned above, benzodiazepines should not be taken by TBI patients because of possible long-term effects on cognition. Similarly, experts have avoided the use of GABAA receptor 1 subtype agonists (zolpidem, zopiclone, zaleplon) because they are known to occasionally cause unexpected responses in patients with cognitive deficits. Trazodone is frequently used by experienced practitioners; however, there are no published studies on trazodone for insomnia in TBI patients. Similarly, there are no published studies on melatonin or ramelteon in the TBI population.75 Lastly, modafinil for excessive daytime sleepiness was studied in a single-center, double-blind, placebo-controlled crossover trial of 53 patients in an inpatient rehabilitation setting more than 1 year after their injuries. Doses up to 400 mg did not have any effect on daytime sleepiness or fatigue.43
