[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Figure 11-1 The cell cycle.
Table 11-1
CDKs, Activating Cyclins, and Select Substrates
| CDK |
Cyclin Partner |
Substrate |
| CDKi (CDC2) |
A and B |
Lamins, histone Hi |
| CDK2 |
E and A |
Rb, P107, P130, Cdt1, CP110 |
| CDK3 |
C |
Rb |
| CDK4 |
D |
Rb, P107, P130, SMAD2, and SMAD3 |
| CDK6 |
D |
Rb, P107, P130, SMAD2, and SMAD3 |
| CDK7 (CAK) |
H |
CDK1-CDK6, RNA pol 11 |
CDK, Cyclin-dependent kinase.
Shortly after the identification of CDK7/cyclin H as CAK, CDK7/cyclin H was shown to be the previously identified activity referred to as
TFIIH, 5 demonstrating that CAK (CDK7/cyclin H) not only contributes to CDK activation but is also implicated in transcriptional regulation. TFIIH phosphorylates multiple serine/threonine residues located in the carboxyl-terminal domain (CTD) of the largest subunit of RNA polymerase II (RNAPII), thereby contributing to increased transcriptional initiation.
5,6 CDK7 is also conserved in budding yeast. However, in yeast, CDK7 does not contribute to CDK activation. Rather, it is solely a regulator of RNA polymerase activity. Bona fide CAK activity in yeast is contributed by a distinct protein, CAK1.
7,8
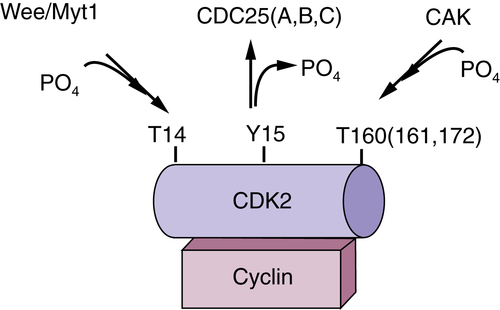
Figure 11-2 Regulation of cyclin-dependent kinase (CDK) CDKs are binary enzymes composed of a catalytic subunit, CDK, and a regulatory subunit, cyclin. Activation requires phosphorylation of a C-terminal threonine by the CDK-activating enzyme, CAK. In contrast, phosphorylation of N-terminal threonine and tyrosine residues inhibits adenosine triphosphate (ATP) binding and thus CDK activity.
CDK phosphorylation is not solely an activating event. Phosphorylation of N-terminal threonine and tyrosine residues near the ATP binding pocket is inhibitory. Phosphorylation of threonine 14/tyrosine 15 is catalyzed by two enzymes, Wee1 and Myt1 (see
Figure 11-2). Although Wee1 is a cytosolic enzyme, Myt1 is localized to endoplasmic reticulum structures.
9 The significance of the differential localization of Wee1 versus Myt1 remains to be established. Threonine 14/tyrosine 15 is located adjacent to the ATP binding pocket of CDKs, providing a structural basis for how phosphorylation of these residues prevents ATP binding.
10 Both threonine and tyrosine residues are conserved in CDK1-3, but only the tyrosine residue is present in CDK4-6. Although phosphorylation of CDK1-2 contributes to the timing of their activation during a normal cell cycle, the CDK4/6 enzymes appear to be subject to this inhibitory phosphorylation only when cells incur DNA damage.
11 In mammalian cells, the removal of N-terminal inhibitory phosphates is catalyzed by any one of three highly related dual-specificity protein phosphatases: CDC25A, CDC25B, or CDC25C.
12 In contrast, yeast cells harbor a single CDC25 isoform that carries out all relevant functions. CDC25 isoforms are expressed in a cell cycle–dependent manner, and the A-B-C designation corresponds to their order of expression during the cell cycle. CDC25 A is expressed first, with its expression peaking at the G
1/S boundary. CDC25B expression follows that of CDC25A, with the highest levels detected during S phase. Finally, CDC25C is expressed during late G
2 and mitosis. From this expression pattern, substrate specificity was inferred, with CDC25A targeting the G
1 CDK
S (CDK4/6 or CDK2-cyclin E), CDC25B regulating the S-phase CDKs (CDK2-cyclin A), and CDC25C regulating mitotic CDKs (CDK1-cyclin B). Consistent with this hypothesis, inhibition of CDC25A resulted in increased CDK2-cyclin E tyrosine phosphorylation.
13 Also consistent
with substrate specificity being determined by the timing of expression, CDC25 enzymes do not exhibit any specificity toward distinct CDK substrates in vitro. However, timing of expression is not the sole determinant. Deletion of CDC25B or CDC25C, or even the combined deletion does not impair mouse development or cell proliferation in vitro.
14 It appears from this analysis that CDC25A expression is sufficient to drive cell cycle expression.
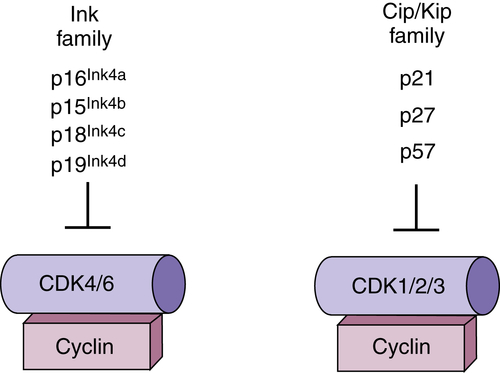
Figure 11-3 Regulation of cyclin-dependent kinases (CDKs) by polypeptide inhibitors Two distinct families of CDK inhibitors (CKIs) regulate CDK activity. The Cip/ Kip family binds with varying affinities to all CDK/cyclin complexes, but have the greatest inhibitory activity toward CDK2. The Ink4 family (inhibitor of CDK4/6) binds specifically to CDK4/6 and has no capacity to directly regulate other CDKs.
CDK Regulation by Small-Polypeptide Inhibitors
In addition to CDK regulation via phosphorylation, CDKs are subject to direct regulation by small-polypeptide inhibitory proteins referred to as
CDK inhibitors, or CKIs (
Figure 11-3 ).
15 These regulators bind directly to and inactivate CDK-cyclin complexes. There are two families of CKIs that have distinct biochemical activities. The
Ink4 (inhibitors of CDK4) family proteins bind exclusively to G
1 CDKs CDK4 and CDK6. Binding can directly inhibit an active CDK4/6-cyclin complex, or Ink4 protein can bind to monomeric CDK4/6 and prevent cyclin association. The second family,
Cip/Kip family proteins, bind to a broad range of CDK-cyclin complexes but functionally appear to be negative regulators of CDK2 complexes.
Ink4 Family
The Ink4 family of proteins consists of four members: p16
Ink4a, p15
Ink4b, p18
Ink4c, and p19
Ink4d. All four proteins bind exclusively to and inhibit D-type cyclin-dependent kinases CDK4 and CDK6. The founding member of the Ink4a family was discovered as a protein that interacted with CDK4 in co-immunoprecipitation experiments,
16 subsequently identified as MTS1.
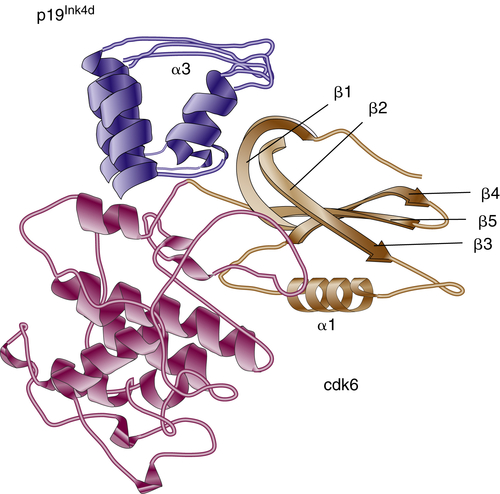
Figure 11-4 Three-dimensional structure of the p19Ink4d/Cdk6 complex p19lnk4d is dark blue, apart from helix α3, which is light blue. The C-terminal domain of Cdk6 is dark brown, whereas the N-terminal domain, which undergoes extensive movement, is light brown.
Ink4 proteins are homologous in primary structure, sharing the presence of four or five ankyrin repeats, which are responsible for protein-protein interactions with CDK4/6. Each repeat consists of an extended strand connected by a
helix-loop-helix (HLH) motif to the next extended strand. The crystal structure of the p19
Ink4d-CDK6 complex has been solved and provided valuable details about the mechanism of CDK inhibition by Ink proteins (
Figure 11-4 ).
17 α-Helices and β-turns of p19
Ink4d ankyrin repeats form a “cap” over the N-terminal domain of CDK6 and induce its spatial movement away from the C terminus. This event inhibits productive ATP binding but does not interfere with the formation of CDK-cyclin complex. As expected from their structure, all four Ink proteins exhibit similar biochemical activities toward CDK4 and CDK6. Interestingly, a short peptide that was derived from one of the ankyrin motifs had the ability to bind and inhibit CDK4, implying the importance of these domains in Ink4 functionality.
18 Despite similar biochemical activities and comparable tertiary structures of Ink proteins, their regulation is distinct. p16
Ink4a is not expressed in most tissues. Rather, it is induced in response to expression of oncogenic or transforming proteins and during cellular senescence. Several oncogenes as well as tumor suppressors regulate p16
Ink4a expression. For example, overexpression of Ras increases p16
Ink4a levels in primary rodent cells.
19 Inactivation of the retinoblastoma susceptibility protein, Rb, or tumor suppressor p53 can also promote p16
Ink4a expression.
20 In contrast, p15
Ink4b expression is regulated by growth-inhibitory factors
(anti-mitogens) such as TGF-β. Only p18
Ink4c and p19
Ink4d expression seems to be regulated during various phases of the cell cycle, with expression peaking during S phase.
21 The expression patterns of Ink4 proteins are also differentially regulated during development.
The structure of the genomic Ink4a locus is unique. Transcription through this locus gives rise to two biochemically distinct proteins, p16
Ink4a and p19
ARF, as a result of alternative exon utilization.
22 Although p16
Ink4a regulates CDK4/CDK6 activity, thereby indirectly regulating the Rb tumor suppressor, p19
ARF regulates the p53 tumor suppressor.
23 p19
ARF acts by attenuating Mdm2-mediated degradation of p53 and is known as an activator of the p53 pathway. Therefore, loss of p19
ARF leads to impairment of p53 signaling. Elimination of the Ink4a/ARF genetic locus in mice makes the animals highly prone to tumor development.
24
Cip/Kip Family
The Cip/Kip family of CKIs includes three members: p21
Cip1, p27
Kip1, and p57
Kip2. Unlike the Ink4 family of CKIs, Cip/Kip inhibitors bind to and efficiently inhibit various CDKs. Members of the Cip/Kip family are highly homologous and share approximately 50% of their sequences. The amino terminus of both p21
Cip1 and p27
Kip1 contains an RXL (where X is typically basic) sequence that is responsible for binding to cyclins and is called the
cyclin-binding motif. Cip/Kip inhibitors also contain a domain that is responsible for the binding to CDKs (N-terminal in p21
Cip1 and p27
Kip1).
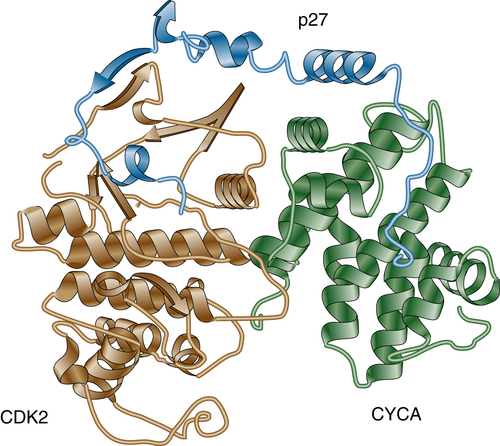
25 The crystal structure of the p27
Kip1/cyclin A/cdk2 complex (
Figure 11-5 ) revealed that p27
Kip1 binds CDK2 at its N terminus and inserts into the catalytic cleft, thus mimicking ATP.
26 On cyclin A/CDK2, p27
Kip1 binds to the groove of the cyclin box. Because both Ink and Cip/Kip proteins occupy almost the same binding sites on CDKs, binding is mutually exclusive. For example, in vitro, p15
Ink4b inhibits binding of p27
Kip1. However, in cells, which protein gets to the CDK first is often determined by the coordinated cellular localization of the inhibitors and cyclin-CDK complexes.
Figure 11-5 Cyclin A/CDK2/p27KIP1 complex Crystal structure of the inhibited ternary cyclin A/CDK2/p27Kip1 complex.
p27
Kip1 is responsible for induction and maintenance of the quiescent state. p27
Kip1 expression is induced in response to growth factor withdrawal and on contact inhibition in cell cultures.
27 p27
Kip1 levels are decreased on addition of the mitogens by various mechanisms described in subsequent paragraphs. Overexpression of p27
Kip1 in cells leads to cell cycle arrest in G
1 phase. Unlike p27
Kip1, p21
Cip1 is present at high levels in cycling cells, keeping CDK activities under tight control. p21
Cip1 levels are induced in response to DNA damage and genotoxic stress as a result of activation of p53. Similar to p21
Cip1 and p27
Kip1, induction of p57
Kip2 can mediate cell cycle arrest in G
1 phase. In addition, p57
Kip2 also participates in the M-to-G
1 transition through activation by p73.
28 Abrogation of p73 or its downstream effector p57
KIP2 perturbs mitotic progression and transition to G
1 phase.
29
Transcriptional Regulation by the E2F Transcription Factors
E2F was originally identified as a cellular DNA binding activity that regulated expression of the viral E2 promoter.
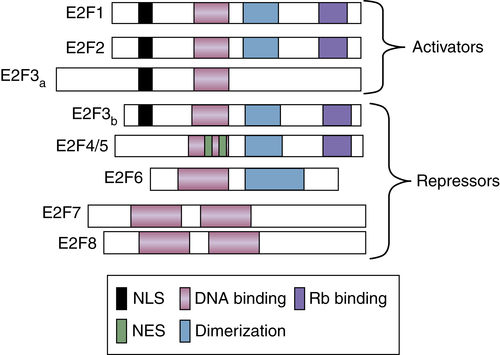
30,31 Since this seminal work, molecular analysis has revealed that the E2F activity is encoded by a family of DNA binding proteins, which includes transcriptional activators and repressors. Mammalian cells encode eight known E2F proteins (E2F1-8;
Figure 11-6 ). Further complication
ensues from the fact that E2F associates with DNA as a heterodimer; the two known heterodimeric partners for E2F are DP1 and DP2. Indeed, the founding member, E2F1, can drive S-phase entry in the absence of growth factor stimulation.
32 The ability of E2F1 to drive S phase derives from its role in the regulation of genes whose protein products play essential roles in S-phase progression. Established E2F targets include components of DNA replication complexes (MCMs) and S-phase cyclins (E and A).
33 E2F family members were initially considered requisite regulators of S-phase entry. E2F1, E2F2, and E2F3 accumulate during G
1 phase and play critical roles in promoting expression of S-phase targets. Strikingly, E2F4 through E2F7 function as transcriptional repressors;
34 E2F3b, an alternatively spliced isoform of E2F3, is also a transcriptional repressor. The E2F repressors function to maintain cells in a quiescent or resting state. In addition to DP1, E2F complexes are further modulated by members of the retinoblastoma protein (pRb) family (pRb, p107, p130;
Figure 11-7 ). The Rb family member functions to recruit chromatin-remodeling enzymes, such as histone deacetylases, to E2F complexes. As a consequence, increased activity of E2F1 through E2F3 requires dissociation of “pRb” from the E2F/DP1 heterodimer. As illustrated in the following sections, the G
1 CDK/cyclin kinase triggers this through direct phosphorylation of the associated pRb family member.
35
Figure 11-6 E2F family of transcription factors There are eight members of the E2F family of transcription factors. E2FS are classified as transcriptional activators or repressors. Functional domains are indicated by differential shading.
In addition to the regulation of S-phase entry and progression, E2F transcriptional activators can trigger apoptosis or cell suicide. The mechanisms whereby E2F induces cell death remain unclear. However, pro-apoptotic genes have been identified as E2F target genes. Examples include the p19ARF protein, which is a known regulator of the p53 tumor suppressor. In addition, E2F can increase expression of pro-apoptotic proteins Puma, Noxa, and Bim and repress the anti-apoptotic Bcl2 family protein, Mcl1.
Two new E2F family members recently identified, E2F7 and E2F8, provide an important constraint against excessive E2F1 activation. Unlike the other mammalian E2Fs, E2F7 and E2F8 have two DNA-binding domains and do not require a DP partner to bind to DNA and as such are classified as atypical E2F family members.
36 These atypical E2Fs bind to the consensus E2F recognition sequence and can repress expression from a subgroup of cell cycle–regulated E2F targets. An E2F7 and E2F8 double knockout is an embryonic lethal resulting from massive apoptosis; this phenotype can be bypassed by removing E2F1 or p53.
37 Our current level of understanding underscores E2F7 and E2F8 as a distinct arm of the E2F network involved in repression of transcription during S-G
2 and control of the E2F1-p53 apoptotic axis.
G1 Regulation/Restriction Point Control
During the first Gap phase or G
1, cells prepare for DNA replication. They must synthesize proteins necessary to replicate their genome, and once these are made, assemble the various components of the DNA replication machinery on chromatin at so-called origins of replication. This is
coordinated with nutrient and growth factor availability to ensure that the cell is in an environment that supports cell division. The G
1 phase of the cell cycle is unique in that it represents the only time wherein cells are sensitive to signals from their extracellular environment. These signals are in the form of adhesion to substratum and growth factors. Cells require growth factor–dependent signals up to a point in late G
1 referred to as the
restriction point (“start” in yeast).
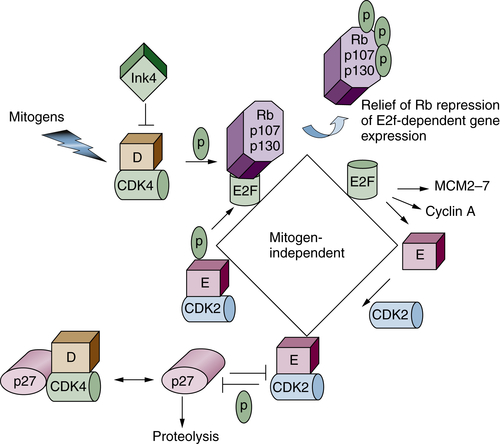
Figure 11-7 Restriction point control Progression through G1 phase requires growth factor–mediated (mitogenic) signals. Mitogens promote the activation of the initial cyclin-dependent kinase (CDK; cyclin D/CDK4) complex, which phosphorylates Rb family proteins (inactivating signal). The CDK4 enzyme also binds to Cip/Kip CDK inhibitors (CKIs), thereby sequestering these proteins to facilitate CDK2 activation. Rb phosphorylation releases the transcription activating E2FS (E2F1-E2F3), which promote transcription of downstream targets such as cyclin E, A, and MCM proteins. Cyclin E binds to CDK2, and this active complex maintains Rb in an inactive state. Active cyclin E/CDK2 also targets its own inhibitor (p27Kip1) for proteolysis via site-specific phosphorylation. The complete activation of CDK2, first by cyclin E and then by cyclin A, marks passage through the restriction point. Once past this point, cells no longer require growth factor stimulation for progression through the remainder of that cell division.
Progression through G1 phase is driven by the collective activities of two distinct CDKs. The first is CDK4 or CDK6 in combination with a D-type cyclin. Mammalian cells encode three distinct D cyclins (D1, D2, D3), which are expressed in a tissue-specific manner. Whereas CDK4 and CDK6 are constitutively expressed, D cyclins are expressed in response to growth factor signaling. Following accumulation of active cyclin D/CDK4 or CDK6, the CDK2 kinase in combination with cyclin E accumulates to facilitate the transition from G1 to S phase.
A key protein that regulates G
1-phase progression in the mammalian cell cycle is retinoblastoma protein, Rb. The Rb family consists of three members, Rb, p107, and p130. In quiescent cells, Rb proteins associate with E2F transcription factors to repress E2F-dependent transcription. E2F targets include genes responsible for regulation of cell cycle and DNA replication, such as cyclins E and A (see
Figure 11-7). Rb activity is regulated at the level of posttranslational modification, specifically phosphorylation. Hypophosphorylated Rb is active and binds to E2F, thereby silencing E2F-dependent activity. Hypophosphorylated Rb family proteins therefore play a central role in maintaining cells in a resting or quiescent state. Quiescent cells reenter the cell cycle in response to mitogenic growth factors. Growth factor signaling induces the expression of D-type cyclins at transcriptional and posttranslational levels,
38 leading to activation of cyclin D–dependent kinases CDK4 and -6 and subsequent Rb phosphorylation. Cyclin D/CDK4 or -6 complexes also have a kinase-independent function. They sequester p21
Cip1 and p27
Kip1 CDK1s from CDK2 kinases and allow activation of basal CDK2/cyclin E kinases, which further phosphorylate Rb family proteins. Phosphorylation of Rb promotes its dissociation from E2F, allowing transcriptional activation of E2F targets such as cyclin E. The E2F-dependent spike in cyclin E, and thus CDK2/cyclin E activity, represents the transition from mitogen-dependent to mitogen-independent cell cycle progression (or passage through the restriction point). In addition to maintaining Rb proteins in a hyperphosphorylated (inactive) state, the activation of cyclin E/CDK2 promotes proteasome-dependent degradation of its own inhibitor p27
Kip1 (described in a subsequent section). These changes, which include cyclin D/CDK4/6 and cyclin E/CDK2 activation, Rb phosphorylation, and destruction of p27
Kip1, render cells with decreased mitogen dependency and are irreversibly committed to enter S phase of the cell cycle.
Regulation of DNA Replication (S Phase)
Early experimentation, which relied on techniques wherein two cells (generally one human and one rodent cell) in distinct phases of the cell cycle are fused together (one cytoplasm containing the two distinct nuclei), revealed that chromosomes were competent for duplication in G1 and S phases. For example, fusing an S-phase cell with a G1-phase cell could enforce replication of a G1 cell; in contrast, fusion of a cell in G2 phase with an S-phase cell could not enforce replication of G2 chromosomes. It was inferred from these experiments that S-phase cells contained a factor that triggered replication initiation and that G1 chromosomes were prepared or “licensed” for this initiating activity. Research efforts have shed light on the molecular basis of regulated replication initiation.
Although DNA is actively replicated during S phase, cells must prepare DNA for replication during the preceding G
1 phase. During G
1 phase,
origins (chromatin positions where DNA polymerase complexes initiate replication) must first be established or “licensed.” Licensing refers to the formation of the pre-RC (pre-replication complex) at origins of replication (
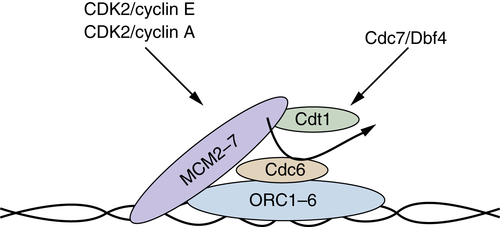
Figure 11-8 ). Initially, the origin of replication complex (ORC) must be associated with chromatin to act as a landing pad on which the pre-RC is formed. Unlike most components of the pre-RC, ORC remains constitutively bound to DNA. In budding yeast, ORC acts as a sequence-specific DNA binding complex; however, in fission yeast and mammalian cells, no sequence specificity has been elucidated for ORC. The next step is the recruitment
of
Cdc6 to the ORC. Cdc6 subsequently recruits the MCM complex and Cdt1. However, MCMs are not stably bound at this point. Stable loading of the MCM2-7 helicase complex requires ATP hydrolysis by CDC6, which also results in release of Cdt1.
39 At the G
1/S boundary additional factors are recruited, including
MCM10, which functions to recruit
Cdc45 and subsequently, DNA polymerase
α and primase.
Figure 11-8 Prereplication complex Prereplication complexes (pre-RCs) form during mid- to late G1 phase, and once they are formed, origins of replication are considered licensed for replication. Origins are recognized first by the hexameric origin of replication complex (ORC1-ORC6), which serves as a landing pad for recruitment of the remaining components. Following ORC recognition, Cdt1 and CDC6 function in a concerted fashion to recruit the MCM2-MCM7 complex, which is considered the replicative helicase. At the beginning of S phase, additional factors (MCM10, CDC45, and polymerases) are recruited and replication can initiate in a fashion dependent on the CDK2 and CDC7 kinase activities.
Like G
1 phase, both the G
1/S transition and S-phase progression are driven by cyclin-dependent kinases (CDK2/cyclin E and CDK2/cyclin A, respectively), along with the activity of a distinct CDK-like protein kinase,
Cdc7/Dbf4. The precise substrates that must be phosphorylated for the firing of origins remain to be conclusively identified. Substrates identified so far include ORC1, MCM2, MCM3, MCM4, and Rad18.
40,41 Not all origins fire simultaneously, but they are temporally regulated. Origins can be grouped generally into those that initiate at the beginning of S phase, “early,” and those that fire toward the middle to end of S-phase, “late.” The temporal control of firing most likely reflects local controls (chromatin structure modifications) and activation of the complex via phosphorylation.
Paradoxically, although origin firing requires CDK activity, CDK2 activity is also essential for inhibition of a second-round DNA replication (re-replication) within the same cell cycle. Although the precise mechanisms whereby CDK2 prevents replication are still under intense investigation, one way it achieves this goal is through direct regulation of Cdt1 levels. On release from the pre-RC, Cdt1 is subject to ubiquitin-mediated proteolysis. Ubiquitination of Cdt1 is in turn facilitated by CDK2-dependent phosphorylation, which targets it to ubiquitinating machinery.
42 In addition to Cdt1, MCM complexes dissociate from DNA during replication. Whether this dissociation reflects dislodgment from chromatin by polymerases or also reflects a CDK-dependent function remains to be established (
Table 11-2 ).
When DNA is replicated, it is essential to retain memory of epigenetic marks in daughter cells. Histone methylation and acetylation during initiation of DNA replication contribute to this memory as well as proper replication. The histone methyltransferase PR-Set7 has been implicated in catalyzing histone monomethylation at lysine 20 (H4K20 me1) at replication origins, and this methylation is required for initiation of replication.
43 According to a model of epigenetic recruitment at the start of replication, histone acetylation plays a major role in DNA replication origin complex recruitment and H4K12Ac, H3K56Ac accompany the generation of the origin replication complex.
44 DNA processivity factor proliferating cell nuclear antigen (PCNA) is loaded on both DNA strands during replication and also provides a link between replication and epigenetic memory. NP95 (also referred as UHRF1 and ICBP90) in mammals and VIM1 in
Arabidopsis thaliana binds to hemimethylated DNA and DNMT1. NP95 recruits DNMT1 to replicating DNA. Another factor, LSH, has been shown to connect DNA methylation to replication in collaboration with NP95. Histone chaperones are required for proper recruitment of histones at DNA. H3.1-H4 chaperone chromatin assembly factor 1 (CAF1) interacts with PCNA, histone deacetylase (HDAC), and Lys methyltransferase. CAF1 regulates nucleosome assembly during DNA replication through proper deposition of H3.1-H4.
45 Another chaperone ASF1 assists CAF1 by supplying newly synthesized histones.
46 Histone methylation levels are transiently reduced during S-phase.
47 It has been recently proposed that TrxG and PcG proteins are associated with DNA during replication.
48 G2/M Transition Regulation
The Kinases of Mitosis
The transition from the second Gap phase (G
2) to mitosis (prophase, metaphase, anaphase, telophase) is regulated by CDK1 (formerly Cdc2) in association primarily with cyclin B.
49 Like other CDKs, CDK1 is relatively stable, and activation depends first on accumulation of cyclin B.
Mitotic cyclins accumulate during S phase and associate with CDK1; however, this complex is maintained in an inactive form via two mechanisms. In the first, Wee/Myt1-dependent phosphorylation of Thr-14/Tyr15 prevents ATP binding. The second mechanism relies on active transport of CDK1/cyclin complexes out of the nucleus. Onset of mitosis is triggered by dephosphorylation of CDK1 by a CDC25 isoform and increased nuclear transport/decreased nuclear exit of CDK1/cyclin complexes. Substrates for CDK1/cyclin B include APC20 (a component of the E3 ligase that ultimately degrades cyclin B), microtubule effectors, microtubule motor proteins, and tubulin itself.
50 From this and related work, it is clear that CDK1-dependent phosphorylation plays a significant role in the formation and regulation of cellular mitotic structures.
Table 11-2
Regulators of DNA Replication and Function
| Cdt1 |
Associates with MCM2-MCM7 and, in concert with Cdc6, facilitates MCM loading on origins. |
| Cdc6 |
Functions to recruit and load the MCM complex in an ATPase-dependent manner. |
| CdC45 |
Associates with the MCM and is responsible for recruitment of DNA polymerase α, primase, and replication protein A. |
| MCM2-7 |
Minichromosome maintenance proteins. Hetero-hexameric complex composed of six distinct but related proteins (MCM2-MCM7). The MCM complex functions as the putative replicative helicase. |
| MCM10 |
Structurally distinct from MCM2-MCM7; functions to recruit CDC45. |
| Orc |
Origin recognition complex. Hetero-hexameric complex that binds directly to DNA and functions as a protein landing pad on which the replication complexes form. |
| Origin |
Functionally defined in mammalian cells as regions of chromatin where DNA replication initiates. |
| Cdc7/Dbf4 |
The Cdc7 protein kinase, like cyclin-dependent kinases (CDKs), requires an allosteric activator, Dbf4. The Cdc7/Dbf4 kinase phosphorylates components of the replication complexes to initiate DNA replication. |
| Pre-RC |
The prereplication forms during G1 and contains ORC1-ORC6, Cdc6, MCM2-7. Replication ensues at S phase on recruitment of DNA polymerase and phosphorylation by both the Cdc7/Dbf4 and CDK2-cyclin A protein kinases. |
MCM, Minichromosome maintenance; ORC, origin of replication complex.
In addition to CDK1, a second family of kinases, called polo-like kinases (PLKs), also contributes to mitotic progression. In mammalian cells, there are five PLKs (PLK1-PLK5) with PLK1 being the human homolog of the founding member, Polo, from
Drosophila. 51 PLKs are serine/threonine kinases. Structurally, they consist of an N-terminal kinase domain and a C terminus with one (PLK3) or two (PLK1-PLK3) “polo box” domains. Current models suggest that PLKs are not constitutively active kinases. Rather, PLK substrates are first phosphorylated by CDKs (e.g., CDK1/cyclin B). Phosphorylation by CDKs is thought to provide a docking site for the polo box domain. Binding of the polo box results in a conformation change in PLK resulting in its activation, whereupon it phosphorylates additional critical residues within the substrate. Alternative models suggest that PLKs are activated through phosphorylation by an upstream kinase, such as CDK1/cyclin B. Although CDK1 can indeed phosphorylate PLK1 in vitro, the functional significance of phosphorylation has not been established. Importantly, neither model is mutually exclusive, and both regulatory mechanisms could contribute to the regulation of PLK activity in cells.
Like CDKs, substrates for PLKs are still being elucidated. As alluded to in previous sections, many PLK substrates may also be CDK substrates. Substrates of PLK1 include CDC25C and Wee1. The consequence of PLK phosphorylation depends on the substrate. Whereas PLK-dependent phosphorylation of CDC25C promotes its activation during mitosis, phosphorylation of Wee1 promotes Wee1 destruction.
Entry into Mitosis
Entry into mitosis requires the nuclear accumulation of active CDK1/cyclin B kinase. During interphase, activity is low. During G2, cyclin B accumulates as a consequence of increased gene expression and decreased protein degradation. Newly accumulated cyclin B is free to associate with CDK1. However, these complexes are maintained in the cytoplasm and are inactive as a consequence of the combined activities of Wee1 and Myt1. Activation of CDK1/cyclin B at the G2/M boundary is triggered through CAK-dependent phosphorylation of Thr161 in the T loop and dephosphorylation of Thr14/Tyr15 by CDC25. The initial dephosphorylation is likely catalyzed by CDC25B. The activated CDK1/cyclin B then targets CDC25C and Wee1 to promote CDC25C activity and Wee1 destruction, respectively, thereby forming an amplification loop that drives mitotic progression. The accumulation of CDK1/cyclin B in the nucleus is facilitated by phosphorylation of cyclin B near its nuclear export signal, which thereby impedes nuclear exit. PLK1 contributes to mitotic entry and progression by facilitating these processes. PLK1 can phosphorylate cyclin B just outside the NES (serine 133), thereby preventing nuclear exit. Like the CDK1/cyclin B kinase, PLK1 can also phosphorylate both CDC25C and Wee1, again contributing to CDC25C activation and Wee1 destruction and thereby ensuring full CDK1/cyclin B activation.
Chromosome Cohesion
G
2 phase and the beginning of mitosis are denoted by a 4-N DNA content. Following DNA replication and prior to cell division (cytokinesis), cells must maintain the integrity and proximity of the recently duplicated chromosomes (sister chromatids). Before segregation, sister chromatids are held together or “glued” by a multiprotein complex called Cohesin.
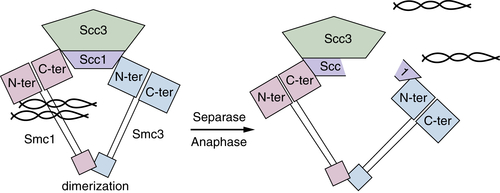
52,53 The cohesin complex ensures that sister chromatids are recognized and properly aligned during metaphase. Once aligned, segregation ensues following proteolytic cleavage of cohesin components. Cohesin is composed of four subunits, Smc1/3 and Scc1/3. Smc1 and Smc3 heterodimerize in a head-to-head, tail-to-tail fashion to form a ring structure in an ATP-dependent manner. The Scc1/3 subunits associate with the Smc heads to complete the structure (
Figure 11-9 ). The Scc1 subunit contacts both Smc1 and 3 and likely stabilizes the ring structure. Models suggest that the cohesin ring has a diameter of approximately 50 nm, sufficiently large to encircle two sister chromatids.
54 Cohesin is envisioned to function by binding and encircling DNA, thereby “gluing” sister chromatids together until released.
Exit from Mitosis
During mitotic prophase, chromosome structures are again altered by a complex called
condensin, which serves to package chromosomes before mitotic division.
55 The mitotic spindle also forms during prophase. The mitotic spindle is a bilaterally symmetric microtubule organizing center shaped like a football. Each half of the spindle contains a
centrosome and three distinct sets of microtubules (astral, kinetochore, and polar); the kinetochore microtubules are those
that attach to chromosomes at the kinetochores to facilitate movement to opposite poles before cytokinesis. PLKs are also implicated in the formation of mitotic spindles.
56 Loss-of-function experiments in multiple organisms (yeast to mammalian cells) result in the formation of monopolar spindles. During metaphase, the chromosomes align along the “metaphase plate” in preparation for cell division. Anaphase is marked by segregation of chromosomes to opposite poles. The proteolytic
cleavage of the Scc1 protein by a protease called
separase triggers the opening of the cohesin ring, thereby allowing chromosome segregation. Anaphase is also marked by the loss of CDK1 activity, which results from proteolytic destruction of cyclin B and cyclin A. The loss of cohesin and mitotic cyclins is coordinated by a multisubunit E3 ubiquitin ligase called the anaphase-promoting complex/cyclosome (APC/C; see subsequent sections).
Figure 11-9 Chromosomes are held together by a complex called cohesin Smc1 and Smc3 form a protein ring that is held together by a dimerization “hinge” region that encircles chromatids. The Scc1 and Scc3 subunits interact with the Smc “heads,” which retain intrinsic ATPase activity essential for separation of heads to allow DNA to enter. Once all chromatids are aligned during mitosis, Scc1 is cleaved by a protease called Separase to open the ring and allow movement to opposite spindles.
Mitotic Checkpoint
The primary goal of mitosis is to ensure that each daughter cell receives one chromosome complement after cellular division. During mitosis it means that a cell divides only after chromosomes are attached to the microtubules of the mitotic spindle. The
mitotic checkpoint, or
spindle assembly checkpoint, is activated as cells enter mitosis, in prometaphase, where it is triggered by
unattached kinetochores, leading to the delay of anaphase onset. Thus, the role of the proteins that are involved in mitotic checkpoint signaling is to sense the attachment and/or tension at kinetochores.
57 These proteins are often found to be kinetochore-associated and comprise the mitotic checkpoint complex.
58 The mitotic checkpoint complex includes BubR1 and Mps1 kinases, CENP-E (
centromere
protein
E), Mad (mitotic arrest deficiency proteins)-1 and -2, and others. The mission of mitotic checkpoint kinases is to signal regulatory proteins to inhibit the entry to anaphase. Models suggest that unattached kinetochores lead to phosphorylation of Mad1/2 proteins, which are then directed to the APC/C, resulting in the inhibition of its ubiquitin-ligating activity. This action ensures that chromosomes are accurately distributed to daughter cells. In human neoplasia, the mitotic checkpoint can be inactivated through mutations in components of MCC,
59 contributing to aberrant mitotic divisions and the appearance of aneuploid cells (genetic instability).
Regulated Proteolysis in Cell Cycle Control
Levels of cyclins and CKIs are tightly regulated throughout the cell cycle. This degree of regulation is achieved by coupling the rate of gene expression with regulated proteolysis, which occurs through the ubiquitin proteasome system. The ubiquitin polypeptide consists of 76 residues and is covalently attached to proteins destined for degradation. Attachment occurs through a reversible isopeptide linkage between the carboxy terminus of ubiquitin and lysine residue in the sequence of protein. The name ubiquitin derives from early observations of its ubiquitous expression. Indeed, ubiquitin is a highly conserved protein throughout evolution from yeast to humans.
Modification of proteins (
ubiquitination) with ubiquitin polypeptides requires a conserved series of enzymes. This system includes the ubiquitin-activating enzyme (E1) that performs ATP-dependent activation of ubiquitin. There is only one known E1 enzyme encoded in the human genome. The E1 passes activated ubiquitin to the ubiquitin-conjugating enzyme (E2), of which there are more than 30.
60 In the final stage of ubiquitination, the E2 acts together with an E3, ubiquitin ligase, to attach mono- or polyubiquitin chains onto the target protein. The E3 ligase acts as the specificity factor that determines substrate recognition and thus comprises the largest group. Once a substrate is polyubiquitinated (four or more tandem ubiquitin molecules on a single lysine within the substrate) it is targeted to the 26S proteasome for degradation.
There are two primary E3 ubiquitin ligases involved in the cell cycle that regulate key cell cycle proteins such as cyclins and CKIs. Both sets of ligases belong to a broader E3 subfamily, either the Skp1-Cul1-F-box (SCF) protein ubiquitin ligases or the APC/C. These two systems are structurally similar. However, as one would expect, they target
distinct substrates in a cell cycle–specific manner and are differentially regulated.
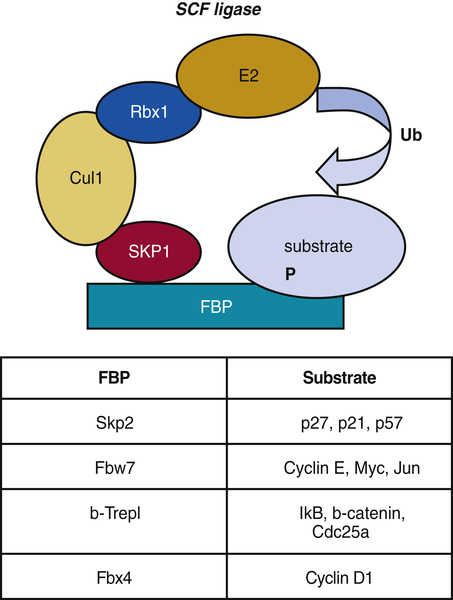
Figure 11-10 The Skp1-Cul1-F-box (SCF) E3 ligase F-box protein, or FBP, acts as a specificity component of SCF E3 ligase that recognizes mostly phosphorylated substrates. Further assembly of SKP1-Cul1-Rbx1 components of SCF complex brings E2 ligases and substrates in close proximity for further ubiquitination. Examples of FBPs and their substrates are indicated in the table.
SCF Ligases
The SCF complex consists of variable and invariable components. The core components employed by all SCF ligases include a scaffold protein Cul1; a ring-finger protein, Rbx1/Roc1; and adaptor protein Skp1 (
Figure 11-10 ). The variable component of the SCF ligase, which determines substrate specificity, is the F-box protein (FBP). FBPs bind Skp1 through an F-box motif initially identified in cyclin F and the substrate bringing the two within close proximity. There are approximately 70 F-box proteins reported in mammals.
61 F-box proteins are classified accordingly to various protein-protein interaction domains that they use to bind to substrates. WD40 repeats give the name to the FBXW class of F-box proteins—leucine-rich repeats (LRRs)—to the FBXL class and F-box proteins that recognize the substrates through other/unknown protein interaction domains belong to the FBXO (F-box only) class. Structurally, FBPs are organized in a fashion that allows them to recognize diverse substrates. Although substrate recognition by FBPs is generally regulated by phosphorylation of the substrate, recognition by one FBP, FBXL2, is determined at least in part by substrate modification with sugar moieties (N-glycans).
62 Thus, the activity of SCF seems to be constant, but the ability to bind to the target protein is regulated.
One of the most rigorously studied FBPs that is involved in cell cycle regulation is Skp2. Although discovered as cyclin A–associated protein, it has since been implicated in the degradation of CKIs: p27
Kip1, p21
Cip1, and p57
Kip2. Skp2 deletion in mice suggests that p27
Kip1 is a bona fide target for Skp2-mediated degradation, because these mice exhibited striking p27
Kip1 accumulation.
63 The binding of Skp2 to p27
Kip1 requires the phosphorylation of Thr187 by cyclin E/A/CDK2 in p27
Kip1. This binding occurs with high affinity only in the presence of another protein, called Cks1.
64 On binding of SCF
skp2/Cks1, phosphorylated p27
Kip1 is ubiquitinated and undergoes proteasome-dependent degradation in late G
1 and early S phases of the cell cycle. Fbw7, another FBP that has been implicated in the degradation of cell cycle key molecules, targets cyclin E, Myc, and c-Jun for degradation.
65 SCF complexes generally regulate proteins involved in G
1 to late S phase, at which point the APC/C is activated and regulates M-phase activities.
FBXO4 is an FBP that specifically directs ubiquitination of cyclin D1. FBXO4 dimerization requires GSK3β-mediated phosphorylation of FBXO4 Ser12, which triggers ligase activation at the G
1/S transition. 14-3-3ε facilitates FBXO4 dimerization, and 14-3-3ε interaction is dependent on Ser8, which is frequently mutated in human cancer, and phosphorylation of Ser12.
66 Recent work revealed the tumor suppressor function of FBXO4. FBXO4 mutations have been identified in human esophageal carcinoma and gastrointestinal stromal tumors.
67,68 Loss of FBXO4 results in cyclin D1 stabilization and nuclear accumulation throughout cell division. FBXO4
+/− and FBXO4
−/− mice succumb to multiple tumor phenotypes including lymphomas, histiocytic sarcomas, and, less frequently, mammary and hepatocellular carcinomas.
69
APC/C Ligase
Structurally the APC/C ligase is similar to the SCF complex. The core components are Rbx1/Roc1-related ring-finger protein, APC11, a Cul1-related scaffold protein, APC2, and 11 additional proteins with required but essentially unknown functions.
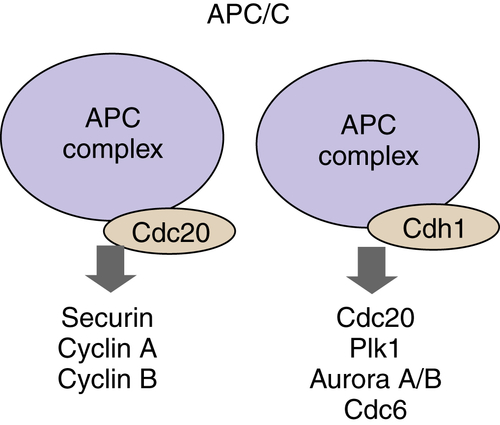
70 Two components determine substrate specificity similar to SCF FBPs function: cell division cycle 20 (Cdc20) and Cdh1 (
Figure 11-11 ). APC/C ligases recognize specific sequences in target proteins called the destruction box (D-box) and the Ken box. These short-peptide
sequences are recognized by the Cdh1 and Cdc20 specificity adaptors and therefore facilitate recruitment of the active APC/C.
Figure 11-11 The anaphase-promoting complex/cyclosome (APC/C) APC/C ubiquitin ligase is a multiprotein complex that is active in the M through G1 phases of the cell cycle. The subunits that are responsible for the recognition of substrates by APC are Cdc20 and Cdh1.
APC/C is active from anaphase through early G
1 phase. However, the regulation of APC/C activity is distinct from SCF ligases. The Cdc20 subunit of APC/C, APC/C
Cdc20, itself undergoes activating phosphorylation events by CDK1/cyclin B. APC/C
Cdc20 can also be phosphorylated and activated by PLK1 and inactivated by PKA. The activity of APC/C
Cdc20 is regulated by protein-protein interactions. Mitotic spindle checkpoint proteins Mad1/Mad2 bind to and inhibit APC/C
Cdc20 function, thereby delaying the onset of anaphase. The substrates of APC/C
Cdc20 ligase include securin, a protein associated with the mitotic protease separase that allows sister chromatid separation, cyclins A and B. When cyclin B is degraded, CDK1 activity declines, contributing to the activation of APC/C
Cdh1; active APC/C
Cdh1 proceeds to fully ubiquitinate cyclin B molecules, eliminating CDK1 activity. The switch of Cdc20 specificity component of APC/C complex to Cdh1 in late M phase also leads to degradation of Cdc20 itself, Plk1, Aurora A/B kinases, and others (reviewed in).
70 APC/C
dh1 remains active during early G
1 phase where it also ubiquitinates Skp2, permitting p27
Kip1 and p21
Cip1 accumulation, as described earlier.
Sumoylation
Sumoylation is another form of posttranslational modification that regulates the cellular localization of modified proteins. Small ubiquitin-like modifiers (SUMOs) are ubiquitin-like polypeptides that become covalently conjugated to cellular proteins in a manner similar to ubiquitination. Ras induces sumoylation and nuclear accumulation of cyclin D1, thereby inhibiting Rb phosphorylation.
71 The lysine residue, K33, of cyclin D1 is a key site for this newly identified regulation.
71
Integration of Growth Factor Signals During G1 Phase by the Ras Small GTP-Binding Protein
Growth factor–dependent signaling promotes the expression and accumulation of factors essential for cell growth (mass accumulation), cell survival, and cell cycle progression. With regard to the cell cycle, growth factor signaling converges on G1-phase components. Entry to and progression through G1 phase of the cell cycle requires activation of signal transduction pathways via extracellular growth factors. G1 progression requires G1 CDK/cyclin complexes to accumulate and become activated and conversely that CKIs be destroyed. Although this is accomplished through numerous pathways, the molecular basis for Ras-dependent signals in G1-phase progression is understood with the greatest detail.
Extracellular growth factors promote the guanosine triphosphate (GTP) loading of Ras, its active form. Active Ras-GTP intersects with the cell cycle via the regulation of cyclin D1 expression and activation of the CDK4/6 kinase. Ras-GTP subsequently triggers the activation of multiple independent signaling pathways including canonical MAP kinase signaling Raf, mitogen-activated protein kinase-kinases (MEK1 and -2), and the sustained activation of extracellular signal-regulated protein kinases (ERKs or MAPK). This pathway contributes to cyclin D1 gene expression.
72 Ras-GTP triggers the activation of a second related, small-GTP binding protein, Rho; activation of Rho also plays a critical role in growth factor–dependent cyclin D1 expression during G
1 phase. A third pathway activated by Ras involves PI-3K and Akt (PKB). The activation of this pathway contributes to increased translation of a multitude of proteins, including cyclin D1 by virtue of the ability of Akt to regulate translation initiation.
73 Active Akt also inactivates glycogen synthase kinase-3β (GSK-3β) by site-specific phosphorylation. Active GSK-3β kinase phosphorylates cyclin D1, thereby promoting cyclin D1 ubiquitination and proteolysis.
74 Thus inactivation of GSK-3β is a critical step necessary for cyclin D1 accumulation during G
1 phase.
For cells to progress through G1 phase, growth factor signaling must promote increased G1 cyclin accumulation and suppress accumulation of the cell cycle inhibitor p27Kip1. Active Ras also plays a central role in the regulation of p27Kip1 in G1 phase by decreasing the efficiency of p27Kip1 translation and increasing the kinetics of p27Kip1 proteolysis. Ras-dependent regulation of p27Kip1 translation and degradation requires Rho signaling. The concerted increase in cyclin D1 accumulation and decrease in p27Kip1 accumulation provides a threshold of CDK4/cyclin D1 activity that is necessary and sufficient for restriction point passage and commitment to S-phase entry.
Deregulation of G1 Restriction Point Control in Cancer
In G1 phase, cells make the decision to either progress through the restriction point and enter S phase or enter G0. These decisions are based on extracellular signals that the cell receives and on the integrity of signaling machinery that detects these signals. Deregulation of G1 progression is a frequent occurrence in cancer, through mutations or deregulated expression of CDKs, cyclins, or CKIs. Loss- or gain-of-function mutations in upstream regulators of the CDK kinases also occur in cancer. In this section, we discuss some alterations found in cell cycle regulators in cancer.
Cyclin D–dependent kinases are a primary point of control for the progression through G
1 phase and are linked to cancer progression. Cyclin D1 overexpression is a hallmark of breast and esophageal cancers.
75 In many cases this upregulation is due to cyclin D1 gene amplifications, but it can also result from increased transcription.
75 In addition to gene expression alterations, decreased cyclin D1 proteolysis is implicated in deregulated cyclin D/CDK4 activity in breast and esophageal cancers. Cyclin D1 overexpression also occurs as a consequence of chromosomal translocations. Amplifications encompassing the
CDK4 and
CDK2 genes have been reported in large B-cell lymphomas, lung tumors, and cervical carcinomas. Downstream targets of cyclin D/CDK4/6 kinases, Rb proteins, are also targeted in cancer. Mutations and deletions in the
Rb gene are common events in tumors; inactivation of Rb alleviates a cell need for CDK4/6 kinase and thus relieves some cellular dependence on growth factor signals.
76 As one might anticipate, Cip/Kip inhibitors can also function as tumor suppressor proteins in mouse model systems, and, consistent with this work, their expression is deregulated in human cancers. p53, the main transcriptional regulator of p21
Cip1, is often lost or mutated during tumorigenesis. Reduced p27
Kip1 levels alone or together with increased cyclin E expression are associated with poor prognosis in breast and ovarian carcinomas. Inactivation of p16
Ink4a occurs frequently in lung, bladder, and breast carcinomas, as well as leukemia (reviewed in Ref.
24).
In addition to alterations in the expression and integrity of cell cycle genes in cancers, attenuation of their regulatory pathways also occurs. These include signaling pathways (Ras), transcription factors (myc), and components of ubiquitin ligases. Skp2, the specificity component of the SCF ligase for p27
Kip1, is upregulated in a variety of tumors, including colon, lung, breast, prostate, and lymphoma,
70 where it decreases p27
Kip1. Another F-box protein, Fbw7, which regulates degradation of cyclin E, is mutated in ovarian and breast cancers.
Altered functionality of cyclin D1 ubiquitin ligase can lead to increased cyclin D1 expression and ultimately to tumorigenesis. Cyclin ubiquitination requires both FBXO4 and a specificity co-factor, αB-crystallin.
77 αB-crystallin expression is lost or downregulated in breast cancer and melanoma cell lines, which correlates with decreased cyclin D1 proteolysis.
78,79 Primary esophageal carcinomas, which are known to frequently overexpress cyclin D1, exhibit hemizygous, missense mutations of FBXO4.
80 Mutations and deregulation of the expression of regulators of mitosis are also observed in human malignancy. Increased accumulation of Cdc20 (APC/C) is observed in lung and gastric tumor cell lines. Mutations in PLK1 are found in human cancer cell lines, and its attenuated expression is observed in colorectal, endometrial, and breast carcinomas.
Targeting the Cell Cycle as a Therapeutic Modality
Dysregulated cell division is a hallmark of cancer progression.
81 Therefore, the use of agents targeting the cell cycle machinery has long been considered as an ideal strategy for cancer therapy.
82 Cell cycle–based agents can be grouped into categories that reflect their molecular targets: CDK inhibitors, checkpoint inhibitors, and mitotic inhibitors. These drugs target the abnormal expression of CDKs, mitotic kinases/kinesins, or affect the cellular checkpoints, resulting in cell-cycle arrest followed by induction of apoptosis in cancer cells.
Targeting CDKs
The rationale for targeting CDKs in anticancer therapy is based on both their role in catalyzing cell division and, in certain cases, the frequency of their perturbation in human malignancy.
83 The rationale suggests that inhibition of CDKs would selectively block tumor growth without compromising normal cells, given that most normal tissues are postmitotic. During the past two decades, numerous CDK inhibitors have been identified as antitumor agents. These drugs have been classified as pan-CDK inhibitors or selective CDK inhibitors. First-generation CDK inhibitors such as flavopiridol, olomoucine, and roscovitine generally did not meet expectations following preclinical studies, showing low activity or no response in the clinical trials. Second-generation CDK inhibitors, such as aminothiazole SNS-032, pyrazole-3-carboxamide AT7519, and synthetic flavone P276-00, have recently been shown to be promising drug candidates in preclinical and clinical trials.
SNS-032, a potent and selective CDK 2, 7, and 9 inhibitor, is currently in phase I clinical study in B-cell malignancies including chronic lymphocytic leukemia, mantle-cell lymphoma (MCL), and multiple myeloma, providing data supporting the ongoing clinical trials. A phase I study of
pan-CDK inhibitor AT7519 was carried out in 28 patients with refractory solid tumors. Four patients showed stable disease and one had a prolonged partial response.
84 A phase II study to treat patients with relapsed or refractory multiple myeloma has been initiated. P276-00 is a highly specific inhibitor of CDK2. Confirmed stable disease has been observed in a phase I study in patients with advanced refractory neoplasms. Phase I/II studies are being performed to evaluate P276-00 efficacy in combination with gemcitabine in patients with pancreatic cancers or in combination with radiation in patients with squamous-cell carcinoma of the head and neck.
Other selective inhibitors such as CDK4/6 inhibitor fascaplysin, CDK4 inhibitor ryuvidine, CDK2 inhibitors purvalanol A and NU2058, and CDK5 inhibitor BML-259 are commercially available and broadly used in the research; however, they had not been entered into clinical trials. The highly selective CDK4/6 inhibitor PD 0332991 is a novel, orally administered inhibitor, which shows potential single-agent activity in clinical trials. PD 0332991 induces G
1 cell cycle arrest by blocking phosphorylation of Rb at CDK4/6-specific sites. Phase I study has been conducted in patients with Rb-expressing advanced solid tumors, relapsed MCL, or refractory non-Hodgkin’s lymphoma. Dose-limiting toxicities relate mainly to myelosuppression.
85–87 A therapeutic response to PD 0332991 has also been seen in inoperable growing teratoma.
88 A phase II study of PD 0332991 in patients with recurrent Rb-positive glioblastoma is currently ongoing.
Targeting Cell Cycle Checkpoints
Targeting the S and G
2 checkpoints is also attractive for cancer therapy because loss of G
1 checkpoint control is a common feature of cancer cells,
89 making them more reliant on the S and G
2 checkpoints to prevent DNA damage–triggered cell death. Various molecules such as CHK1, CHK2, PP2A, Wee1, and cell division cycle 25 (CDC25) have been suggested as the key targets for checkpoint abrogation.
90 Numerous checkpoint inhibitors have entered into clinical trials, most of which focused on CHK1. Among all the checkpoint inhibitors, UCN-01 is most clinically advanced,
91–93 but after phase II trials it was discontinued because of dose-limiting toxicities and a lack of convincing efficacy. The newer, more specific inhibitors of CHK are still under investigation.
90,94
Targeting Regulators of Mitosis
Critical mediators of mitosis that are also implicated in tumorigenesis include three distinct protein kinase families (CDKs, Aurora kinases, and PLKs) and specific mitotic kinesins. Overexpression/amplification of Aurora kinases has been observed in cancer cells and contributes to dysregulated spindle formation, compromised spindle checkpoint, and cytokinesis failure; collectively, these anomalies contribute to cellular aneuploidy.
95 Aurora kinase inhibitors are being pursued, and many of them are already in clinical development.
Like Aurora kinases, PLK1 is often overexpressed in human tumors but not in healthy, nondividing cells. This makes PLK inhibitors an attractive, selective target for cancer drug development. PLK inhibitors interfere with different stages of mitosis, such as centrosome maturation, spindle formation, chromosome separation, and cytokinesis. They induce mitotic chaos and severely perturb cell cycle progression, eventually leading to cancer cell death.
96 Numerous PLK inhibitors are being tested to evaluate their therapeutic potential in oncology.
Kinesins either have a single function or are functionally involved at different stages of the mitotic phase, making them potential anticancer targets.
97 Compounds that inhibit mitotic kinesins EG5 and centromere-associated protein E (CENP-E) have entered clinical trials. Additional mitotic kinesins are currently at varying stages of drug development, raising the possibility of kinesin as a successful target for cancer therapy.
Conclusions
Significant advances have been made in the understanding of the molecular basis of cell cycle regulation. Conceptually, it was anticipated that understanding the basic mechanisms and regulators would permit scientists to ask how they contribute to organismal development and/or cancer progression. Indeed, these questions are now being addressed through targeted deletion of individual genes in the mouse genome. G
1 cyclins and CDKs have been removed from the mouse genome by targeted deletion to evaluate the role of these molecules in organismal development and basic cell growth. Although genetic deletion strains in mice have revealed unique properties of each molecule, what has been most striking is the revelation that no one cyclin or CDK is absolutely essential for development.
98 Thus, although each mammalian CDK is considered to have distinct substrates, in an intact cell there is sufficient redundancy to permit loss of any one complex.
The identification of the critical regulators of cell division has also facilitated the development of antiproliferative therapies through the design of small-molecule inhibitors of the CDKs. Given that deregulated growth control is a fundamental property of cancer, the development of small molecules that inhibit the molecular machine that drives cell cycle transitions is a conceptually attractive therapeutic option. The continued investigation of components of the cell cycle machine will undoubtedly continue to contribute fundamental insights into cell growth control and potentially provide additional insights into diseases that alter growth properties.
References
1. Pines J. , Hunter T. Human cyclins A and B1 are differentially located in the cell and undergo cell cycle-dependent nuclear transport . J Cell Biol . 1991 ; 115 : 1 – 17 .
2. Brown N.R. , Noble M.E. , Endicott J.A. et al. The crystal structure of cyclin A . Structure . 1995 ; 3 : 1235 – 1247 .
3. Jeffrey P.D. , Russo A.A. , Polyak K. et al. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex . Nature . 1995 ; 376 : 313 – 320 .
4. Makela T.P. , Tassan J.P. , Nigg E.A. et al. A cyclin associated with the CDK-activating kinase MO15 . Nature . 1994 ; 371 : 254 – 257 .
5. Shiekhattar R. , Mermelstein F. , Fisher R.P. et al. Cdk-activating kinase complex is a component of human transcription factor TFIIH . Nature . 1995 ; 374 : 283 – 287 .
6. Feaver W.J. , Svejstrup J.Q. , Henry N.L. et al. Relationship of CDK-activating kinase and RNA polymerase II CTD kinase TFIIH/TFIIK . Cell . 1994 ; 79 : 1103 – 1109 .
7. Kaldis P. , Sutton A. , Solomon M.J. The Cdk-activating kinase (CAK) from budding yeast . Cell . 1996 ; 86 : 553 – 564 .
8. Espinoza F.H. , Farrell A. , Erdjument-Bromage H. et al. A cyclin-dependent kinase-activating kinase (CAK) in budding yeast unrelated to vertebrate CAK . Science . 1996 ; 273 : 1714 – 1717 .
9. Baldin V. , Ducommun B. Subcellular localisation of human wee1 kinase is regulated during the cell cycle. J Cell Sci . 108 Part . 1995 ; 6 : 2425 – 2432 .
10. Atherton-Fessler S. , Parker L.L. , Geahlen R.L. et al. Mechanisms of p34cdc2 regulation . Mol Cell Biol . 1993 ; 13 : 1675 – 1685 .
11. Terada Y. , Tatsuka M. , Jinno S. et al. Requirement for tyrosine phosphorylation of Cdk4 in G1 arrest induced by ultraviolet irradiation . Nature . 1995 ; 376 : 358 – 362 .
12. Nilsson I. , Hoffmann I. Cell cycle regulation by the Cdc25 phosphatase family . Prog Cell. Cycle Res . 2000 ; 4 : 107 – 114 .
13. Sandhu C. , Donovan J. , Bhattacharya N. et al. Reduction of Cdc25A contributes to cyclin E1-Cdk2 inhibition at senescence in human mammary epithelial cells . Oncogene . 2000 ; 19 : 5314 – 5323 .
14. Ferguson A.M. , White L.S. , Donovan P.J. et al. Normal cell cycle and checkpoint responses in mice and cells lacking Cdc25B and Cdc25C protein phosphatases . Mol Cell Biol . 2005 ; 25 : 2853 – 2860 .
15. Sherr C.J. , Roberts J.M. CDK inhibitors: positive and negative regulators of G1-phase progression . Genes Dev . 1999 ; 13 : 1501 – 1512 .
16. Serrano M. , Hannon G.J. , Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4 . Nature . 1993 ; 366 : 704 – 707 .
17. Brotherton D.H. , Dhanaraj V. , Wick S. et al. Crystal structure of the complex of the cyclin D-dependent kinase Cdk6 bound to the cell-cycle inhibitor p19INK4d . Nature . 1998 ; 395 : 244 – 250 .
18. Boice J.A. , Fairman R. Structural characterization of the tumor suppressor p16, an ankyrin-like repeat protein . Protein Sci . 1996 ; 5 : 1776 – 1784 .
19. Serrano M. , Lin A.W. , McCurrach M.E. et al. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a . Cell . 1997 ; 88 : 593 – 602 .
20. Shapiro G.I. , Edwards C.D. , Kobzik L. et al. Reciprocal Rb inactivation and p16INK4 expression in primary lung cancers and cell lines . Cancer Res . 1995 ; 55 : 505 – 509 .
21. Thullberg M. , Bartkova J. , Khan S. et al. Distinct versus redundant properties among members of the INK4 family of cyclin-dependent kinase inhibitors . FEBS Lett . 2000 ; 470 : 161 – 166 .
22. Quelle D.E. , Zindy F. , Ashmun R.A. et al. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest . Cell . 1995 ; 83 : 993 – 1000 .
23. Sherr C.J. Tumor surveillance via the ARF-p53 pathway . Genes Dev . 1998 ; 12 : 2984 – 2991 .
24. Serrano M. , Lee H. , Chin L. et al. Role of the INK4a locus in tumor suppression and cell mortality . Cell . 1996 ; 85 : 27 – 37 .
25. Chen J. , Jackson P.K. , Kirschner M.W. et al. Separate domains of p21 involved in the inhibition of Cdk kinase and PCNA . Nature . 1995 ; 374 : 386 – 388 .
26. Russo A.A. , Jeffrey P.D. , Patten A.K. et al. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex . Nature . 1996 ; 382 : 325 – 331 .
27. Polyak K. , Kato J.Y. , Solomon M.J. et al. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest . Genes Dev . 1994 ; 8 : 9 – 22 .
28. Roeb W. , Boyer A. , Cavenee W.K. et al. PAX3-FOXO1 controls expression of the p57Kip2 cell-cycle regulator through degradation of EGR1 . Proc Natl Acad Sci U S A . 2007 ; 104 : 18085 – 18090 .
29. Merlo P. , Fulco M. , Costanzo A. et al. A role of p73 in mitotic exit . J Biol Chem . 2005 ; 280 : 30354 – 30360 .
30. Kovesdi I. , Reichel R. , Nevins J.R. Role of an adenovirus E2 promoter binding factor in E1A-mediated coordinate gene control . Proc Natl Acad Sci U S A . 1987 ; 84 : 2180 – 2184 .
31. Yee A.S. , Reichel R. , Kovesdi I. et al. Promoter interaction of the E1A-inducible factor E2F and its potential role in the formation of a multi-component complex . EMBO J . 1987 ; 6 : 2061 – 2068 .
32. Johnson D.G. , Schwarz J.K. , Cress W.D. et al. Expression of transcription factor E2F1 induces quiescent cells to enter S phase . Nature . 1993 ; 365 : 349 – 352 .
33. Ohtani K. , DeGregori J. , Nevins J.R. Regulation of the cyclin E gene by transcription factor E2F1 . Proc Natl Acad Sci U S A . 1995 ; 92 : 12146 – 12150 .
34. Pierce A.M. , Schneider-Broussard R. , Philhower J.L. et al. Differential activities of E2F family members: unique functions in regulating transcription . Mol Carcinog . 1998 ; 22 : 190 – 198 .
35. Kato J. , Matsushime H. , Hiebert S.W. et al. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4 . Genes Dev . 1993 ; 7 : 331 – 342 .
36. Vlieghe K. , Boudolf V. , Beemster G.T. et al. The DP-E2F-like gene DEL1 controls the endocycle in Arabidopsis thaliana . Curr Biol . 2005 ; 15 : 59 – 63 .
37. Li J. , Ran C. , Li E. et al. Synergistic function of E2F7 and E2F8 is essential for cell survival and embryonic development . Dev Cell . 2008 ; 14 : 62 – 75 .
38. Sherr C.J. , Matsushime H. , Roussel M.F. Regulation of CYL/cyclin D genes by colony-stimulating factor 1 . Ciba Found Symp . 1992 ; 170 : 209 – 219; discussion 219-226 .
39. Tye B.K. MCM proteins in DNA replication . Annu Rev Biochem . 1999 ; 68 : 649 – 686 .
40. Lin D.I. , Aggarwal P. , Diehl J.A. Phosphorylation of MCM3 on Ser-112 regulates its incorporation into the MCM2-7 complex . Proc Natl Acad Sci U S A . 2008 ; 105 : 8079 – 8084 .
41. Vaziri C. , Masai H. Integrating DNA replication with trans-lesion synthesis via Cdc7 . Cell Cycle . 2010 ; 9 : 4818 – 4823 .
42. Li X. , Zhao Q. , Liao R. et al. The SCF(Skp2) ubiquitin ligase complex interacts with the human replication licensing factor Cdt1 and regulates Cdt1 degradation . J Biol Chem . 2003 ; 278 : 30854 – 30858 .
43. Tardat M. , Brustel J. , Kirsh O. et al. The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells . Nat Cell Biol . 2010 ; 12 : 1086 – 1093 .
44. Liu J. , McConnell K. , Dixon M. et al. Analysis of model replication origins in Drosophila reveals new aspects of the chromatin landscape and its relationship to origin activity and the prereplicative complex . Mol Biol Cell . 2011 ; 23 : 200 – 212 .
45. Polo S.E. , Roche D. , Almouzni G. New histone incorporation marks sites of UV repair in human cells . Cell . 2006 ; 127 : 481 – 493 .
46. Mello J.A. , Sillje H.H. , Roche D.M. et al. Human Asf1 and CAF-1 interact and synergize in a repair-coupled nucleosome assembly pathway . EMBO Rep . 2002 ; 3 : 329 – 334 .
47. Xu M. , Wang W. , Chen S. et al. A model for mitotic inheritance of histone lysine methylation . EMBO Rep . 2012 ; 13 : 60 – 67 .
48. Petruk S. , Sedkov Y. , Johnston D.M. et al. TrxG and PcG proteins but not methylated histones remain associated with DNA through replication . Cell . 2012 ; 150 : 922 – 933 .
49. Morgan D.O. Principles of CDK regulation . Nature . 1995 ; 374 : 131 – 134 .
50. Kotani S. , Tanaka H. , Yasuda H. et al. Regulation of APC activity by phosphorylation and regulatory factors . J Cell Biol . 1999 ; 146 : 791 – 800 .
51. Clay F.J. , McEwen S.J. , Bertoncello I. et al. Identification and cloning of a protein kinase-encoding mouse gene, Plk, related to the polo gene of . Drosophila. Proc Natl Acad Sci U S A . 1993 ; 90 : 4882 – 4886 .
52. Uhlmann F. , Lottspeich F. , Nasmyth K. Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1 . Nature . 1999 ; 400 : 37 – 42 .
53. Nasmyth K. Separating sister chromatids . Trends Biochem Sci . 1999 ; 24 : 98 – 104 .
54. Ivanov D. , Nasmyth K. A topological interaction between cohesin rings and a circular minichromosome . Cell . 2005 ; 122 : 849 – 860 .
55. Hirano T. , Kobayashi R. , Hirano M. Condensins, chromosome condensation protein complexes containing XCAP-C, XCAP-E and a Xenopus homolog of the Drosophila Barren protein . Cell . 1997 ; 89 : 511 – 521 .
56. Lee K.S. , Yuan Y.L. , Kuriyama R. et al. Plk is an M-phase-specific protein kinase and interacts with a kinesin-like protein, CHO1/MKLP-1 . Mol Cell Biol . 1995 ; 15 : 7143 – 7151 .
57. Ault J.G. , Nicklas R.B. Tension, microtubule rearrangements, and the proper distribution of chromosomes in mitosis . Chromosoma . 1989 ; 98 : 33 – 39 .
58. Foley E.A. , Kapoor T.M. Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore . Nat Rev Mol Cell Biol . 2013 ; 14 : 25 – 37 .
59. Cahill D.P. , Lengauer C. , Yu J. et al. Mutations of mitotic checkpoint genes in human cancers . Nature . 1998 ; 392 : 300 – 303 .
60. Zhang X.D. , Matunis M.J. Ub in charge: regulating E2 enzyme nuclear import . Nat Cell Biol . 2005 ; 7 : 12 – 14 .
61. Nakayama K.I. , Nakayama K. Regulation of the cell cycle by SCF-type ubiquitin ligases . Semin Cell Dev Biol . 2005 ; 16 : 323 – 333 .
62. Yoshida Y. , Chiba T. , Tokunaga F. et al. E3 ubiquitin ligase that recognizes sugar chains . Nature . 2002 ; 418 : 438 – 442 .
63. Nakayama K. , Nagahama H. , Minamishima Y.A. et al. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication . EMBO J . 2000 ; 19 : 2069 – 2081 .
64. Spruck C. , Strohmaier H. , Watson M. et al. A CDK-independent function of mammalian Cks1: targeting of SCF(Skp2) to the CDK inhibitor p27Kip1 . Mol Cell . 2001 ; 7 : 639 – 650 .
65. Tetzlaff M.T. , Yu W. , Li M. et al. Defective cardiovascular development and elevated cyclin E and Notch proteins in mice lacking the Fbw7 F-box protein . Proc Natl Acad Sci U S A . 2004 ; 101 : 3338 – 3345 .
66. Barbash O. , Lee E.K. , Diehl J.A. Phosphorylation-dependent regulation of SCF(Fbx4) dimerization and activity involves a novel component, 14-3-3varepsilon . Oncogene . 2011 ; 30 : 1995 – 2002 .
67. Barbash O. , Zamfirova P. , Lin D.I. et al. Mutations in Fbx4 inhibit dimerization of the SCF(Fbx4) ligase and contribute to cyclin D1 overexpression in human cancer . Cancer Cell . 2008 ; 14 : 68 – 78 .
68. Kang G. , Lee J. , Jang K.T. et al. Multiplex mutation screening by mass spectrometry in gastrointestinal stromal tumours . Pathology . 2012 ; 44 : 460 – 464 .
69. Vaites L.P. , Lee E.K. , Lian Z. et al. The Fbx4 tumor suppressor regulates cyclin D1 accumulation and prevents neoplastic transformation . Mol Cell Biol . 2011 ; 31 : 4513 – 4523 .
70. Nakayama K.I. , Nakayama K. Ubiquitin ligases: cell-cycle control and cancer . Nat Rev Cancer . 2006 ; 6 : 369 – 381 .
71. Wang X.D. , Lapi E. , Sullivan A. et al. SUMO-modified nuclear cyclin D1 bypasses Ras-induced senescence . Cell Death Differ . 2006 ; 18 : 304 – 314 .
72. Filmus J. , Robles A.I. , Shi W. et al. Induction of cyclin D1 overexpression by activated ras . Oncogene . 1994 ; 9 : 3627 – 3633 .
73. Shi Y. , Sharma A. , Wu H. et al. Cyclin D1 and c-myc internal ribosome entry site (IRES)-dependent translation is regulated by AKT activity and enhanced by rapamycin through a p38 MAPK- and ERK-dependent pathway . J Biol Chem . 2005 ; 280 : 10964 – 10973 .
74. Diehl J.A. , Cheng M. , Roussel M.F. et al. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization . Genes Dev . 1998 ; 12 : 3499 – 3511 .
75. Steeg P.S. , Zhou Q. Cyclins and breast cancer . Breast Cancer Res Treat . 1998 ; 52 : 17 – 28 .
76. Hall M. , Peters G. Genetic alterations of cyclins, cyclin-dependent kinases, and Cdk inhibitors in human cancer . Adv Cancer Res . 1996 ; 68 : 67 – 108 .
77. Lin D.I. , Barbash O. , Kumar K.G. et al. Phosphorylation-dependent ubiquitination of cyclin D1 by the SCF(FBX4-alphaB crystallin) complex . Mol Cell . 2006 ; 24 : 355 – 366 .
78. Hu R. , Aplin A.E. AlphaB-crystallin is mutant B-RAF regulated and contributes to cyclin D1 turnover in melanocytic cells . Pigment Cell Melanoma Res . 2011 ; 23 : 201 – 209 .
79. Chelouche-Lev D. , Kluger H.M. , Berger A.J. et al. AlphaB-crystallin as a marker of lymph node involvement in breast carcinoma . Cancer . 2004 ; 100 : 2543 – 2548 .
80. Barbash O. , Diehl J.A. SCF(Fbx4/alphaB-crystallin) E3 ligase: when one is not enough . Cell Cycle . 2008 ; 7 : 2983 – 2986 .
81. Hanahan D. , Weinberg R.A. Hallmarks of cancer: the next generation . Cell . 2011 ; 144 : 646 – 674 .
82. Schwartz G.K. , Shah M.A. Targeting the cell cycle: a new approach to cancer therapy . J Clin Oncol . 2005 ; 23 : 9408 – 9421 .
83. Chen Y.N. , Sharma S.K. , Ramsey T.M. et al. Selective killing of transformed cells by cyclin/cyclin-dependent kinase 2 antagonists . Proc Natl Acad Sci U S A . 1999 ; 96 : 4325 – 4329 .
84. Mahadevan D. , Plummer R. , Squires M.S. et al. A phase I pharmacokinetic and pharmacodynamic study of AT7519, a cyclin-dependent kinase inhibitor in patients with refractory solid tumors . Ann Oncol . 2011 ; 22 : 2137 – 2143 .
85. Leonard J.P. , LaCasce A.S. , Smith M.R. et al. Selective CDK4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma . Blood . 2012 ; 119 : 4597 – 4607 .
86. Flaherty K.T. , Lorusso P.M. , Demichele A. et al. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer . Clin Cancer Res . 2012 ; 18 : 568 – 576 .
87. Schwartz G.K. , LoRusso P.M. , Dickson M.A. et al. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1) . Br J Cancer . 2011 ; 104 : 1862 – 1868 .
88. Vaughn D.J. , Flaherty K. , Lal P. et al. Treatment of growing teratoma syndrome . N Engl J Med . 2009 ; 360 : 423 – 424 .
89. Massague J. G1 cell-cycle control and cancer . Nature . 2004 ; 432 : 298 – 306 .
90. Curtin N.J. DNA repair dysregulation from cancer driver to therapeutic target . Nat Rev Cancer . 2012 ; 12 : 801 – 817 .
91. Li T. , Christensen S.D. , Frankel P.H. et al. A phase II study of cell cycle inhibitor UCN-01 in patients with metastatic melanoma: a California Cancer Consortium trial . Invest New Drugs . 2012 ; 30 : 741 – 748 .
92. Fracasso P.M. , Williams K.J. , Chen R.C. et al. A Phase 1 study of UCN-01 in combination with irinotecan in patients with resistant solid tumor malignancies . Cancer Chemother Pharmacol . 2011 ; 67 : 1225 – 1237 .
93. Kummar S. , Gutierrez M.E. , Gardner E.R. et al. A phase I trial of UCN-01 and prednisone in patients with refractory solid tumors and lymphomas . Cancer Chemother Pharmacol . 2010 ; 65 : 383 – 389 .
94. Garrett M.D. , Collins I. Anticancer therapy with checkpoint inhibitors: what, where and when? Trends Pharmacol Sci . 2011 ; 32 : 308 – 316 .
95. Cheung C.H. , Coumar M.S. , Chang J.Y. et al. Aurora kinase inhibitor patents and agents in clinical testing: an update (2009-10) . Expert Opin Ther Pat . 2011 ; 21 : 857 – 884 .
96. Schoffski P. Polo-like kinase (PLK) inhibitors in preclinical and early clinical development in oncology . Oncologist . 2009 ; 14 : 559 – 570 .
97. Rath O. , Kozielski F. Kinesins and cancer . Nat Rev Cancer . 2012 ; 12 : 527 – 539 .
98. Sherr C.J. , Roberts J.M. Living with or without cyclins and cyclin-dependent kinases . Genes Dev . 2004 ; 18 : 2699 – 2711 .
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Figure 11-1 The cell cycle.
Table 11-1
CDKs, Activating Cyclins, and Select Substrates
| CDK |
Cyclin Partner |
Substrate |
| CDKi (CDC2) |
A and B |
Lamins, histone Hi |
| CDK2 |
E and A |
Rb, P107, P130, Cdt1, CP110 |
| CDK3 |
C |
Rb |
| CDK4 |
D |
Rb, P107, P130, SMAD2, and SMAD3 |
| CDK6 |
D |
Rb, P107, P130, SMAD2, and SMAD3 |
| CDK7 (CAK) |
H |
CDK1-CDK6, RNA pol 11 |
CDK, Cyclin-dependent kinase.
Shortly after the identification of CDK7/cyclin H as CAK, CDK7/cyclin H was shown to be the previously identified activity referred to as
TFIIH, 5 demonstrating that CAK (CDK7/cyclin H) not only contributes to CDK activation but is also implicated in transcriptional regulation. TFIIH phosphorylates multiple serine/threonine residues located in the carboxyl-terminal domain (CTD) of the largest subunit of RNA polymerase II (RNAPII), thereby contributing to increased transcriptional initiation.
5,6 CDK7 is also conserved in budding yeast. However, in yeast, CDK7 does not contribute to CDK activation. Rather, it is solely a regulator of RNA polymerase activity. Bona fide CAK activity in yeast is contributed by a distinct protein, CAK1.
7,8
Figure 11-2 Regulation of cyclin-dependent kinase (CDK) CDKs are binary enzymes composed of a catalytic subunit, CDK, and a regulatory subunit, cyclin. Activation requires phosphorylation of a C-terminal threonine by the CDK-activating enzyme, CAK. In contrast, phosphorylation of N-terminal threonine and tyrosine residues inhibits adenosine triphosphate (ATP) binding and thus CDK activity.
CDK phosphorylation is not solely an activating event. Phosphorylation of N-terminal threonine and tyrosine residues near the ATP binding pocket is inhibitory. Phosphorylation of threonine 14/tyrosine 15 is catalyzed by two enzymes, Wee1 and Myt1 (see
Figure 11-2). Although Wee1 is a cytosolic enzyme, Myt1 is localized to endoplasmic reticulum structures.
9 The significance of the differential localization of Wee1 versus Myt1 remains to be established. Threonine 14/tyrosine 15 is located adjacent to the ATP binding pocket of CDKs, providing a structural basis for how phosphorylation of these residues prevents ATP binding.
10 Both threonine and tyrosine residues are conserved in CDK1-3, but only the tyrosine residue is present in CDK4-6. Although phosphorylation of CDK1-2 contributes to the timing of their activation during a normal cell cycle, the CDK4/6 enzymes appear to be subject to this inhibitory phosphorylation only when cells incur DNA damage.
11 In mammalian cells, the removal of N-terminal inhibitory phosphates is catalyzed by any one of three highly related dual-specificity protein phosphatases: CDC25A, CDC25B, or CDC25C.
12 In contrast, yeast cells harbor a single CDC25 isoform that carries out all relevant functions. CDC25 isoforms are expressed in a cell cycle–dependent manner, and the A-B-C designation corresponds to their order of expression during the cell cycle. CDC25 A is expressed first, with its expression peaking at the G
1/S boundary. CDC25B expression follows that of CDC25A, with the highest levels detected during S phase. Finally, CDC25C is expressed during late G
2 and mitosis. From this expression pattern, substrate specificity was inferred, with CDC25A targeting the G
1 CDK
S (CDK4/6 or CDK2-cyclin E), CDC25B regulating the S-phase CDKs (CDK2-cyclin A), and CDC25C regulating mitotic CDKs (CDK1-cyclin B). Consistent with this hypothesis, inhibition of CDC25A resulted in increased CDK2-cyclin E tyrosine phosphorylation.
13 Also consistent
with substrate specificity being determined by the timing of expression, CDC25 enzymes do not exhibit any specificity toward distinct CDK substrates in vitro. However, timing of expression is not the sole determinant. Deletion of CDC25B or CDC25C, or even the combined deletion does not impair mouse development or cell proliferation in vitro.
14 It appears from this analysis that CDC25A expression is sufficient to drive cell cycle expression.
Figure 11-3 Regulation of cyclin-dependent kinases (CDKs) by polypeptide inhibitors Two distinct families of CDK inhibitors (CKIs) regulate CDK activity. The Cip/ Kip family binds with varying affinities to all CDK/cyclin complexes, but have the greatest inhibitory activity toward CDK2. The Ink4 family (inhibitor of CDK4/6) binds specifically to CDK4/6 and has no capacity to directly regulate other CDKs.
CDK Regulation by Small-Polypeptide Inhibitors
In addition to CDK regulation via phosphorylation, CDKs are subject to direct regulation by small-polypeptide inhibitory proteins referred to as
CDK inhibitors, or CKIs (
Figure 11-3 ).
15 These regulators bind directly to and inactivate CDK-cyclin complexes. There are two families of CKIs that have distinct biochemical activities. The
Ink4 (inhibitors of CDK4) family proteins bind exclusively to G
1 CDKs CDK4 and CDK6. Binding can directly inhibit an active CDK4/6-cyclin complex, or Ink4 protein can bind to monomeric CDK4/6 and prevent cyclin association. The second family,
Cip/Kip family proteins, bind to a broad range of CDK-cyclin complexes but functionally appear to be negative regulators of CDK2 complexes.
Ink4 Family
The Ink4 family of proteins consists of four members: p16
Ink4a, p15
Ink4b, p18
Ink4c, and p19
Ink4d. All four proteins bind exclusively to and inhibit D-type cyclin-dependent kinases CDK4 and CDK6. The founding member of the Ink4a family was discovered as a protein that interacted with CDK4 in co-immunoprecipitation experiments,
16 subsequently identified as MTS1.
Figure 11-4 Three-dimensional structure of the p19Ink4d/Cdk6 complex p19lnk4d is dark blue, apart from helix α3, which is light blue. The C-terminal domain of Cdk6 is dark brown, whereas the N-terminal domain, which undergoes extensive movement, is light brown.
Ink4 proteins are homologous in primary structure, sharing the presence of four or five ankyrin repeats, which are responsible for protein-protein interactions with CDK4/6. Each repeat consists of an extended strand connected by a
helix-loop-helix (HLH) motif to the next extended strand. The crystal structure of the p19
Ink4d-CDK6 complex has been solved and provided valuable details about the mechanism of CDK inhibition by Ink proteins (
Figure 11-4 ).
17 α-Helices and β-turns of p19
Ink4d ankyrin repeats form a “cap” over the N-terminal domain of CDK6 and induce its spatial movement away from the C terminus. This event inhibits productive ATP binding but does not interfere with the formation of CDK-cyclin complex. As expected from their structure, all four Ink proteins exhibit similar biochemical activities toward CDK4 and CDK6. Interestingly, a short peptide that was derived from one of the ankyrin motifs had the ability to bind and inhibit CDK4, implying the importance of these domains in Ink4 functionality.
18 Despite similar biochemical activities and comparable tertiary structures of Ink proteins, their regulation is distinct. p16
Ink4a is not expressed in most tissues. Rather, it is induced in response to expression of oncogenic or transforming proteins and during cellular senescence. Several oncogenes as well as tumor suppressors regulate p16
Ink4a expression. For example, overexpression of Ras increases p16
Ink4a levels in primary rodent cells.
19 Inactivation of the retinoblastoma susceptibility protein, Rb, or tumor suppressor p53 can also promote p16
Ink4a expression.
20 In contrast, p15
Ink4b expression is regulated by growth-inhibitory factors
(anti-mitogens) such as TGF-β. Only p18
Ink4c and p19
Ink4d expression seems to be regulated during various phases of the cell cycle, with expression peaking during S phase.
21 The expression patterns of Ink4 proteins are also differentially regulated during development.
The structure of the genomic Ink4a locus is unique. Transcription through this locus gives rise to two biochemically distinct proteins, p16
Ink4a and p19
ARF, as a result of alternative exon utilization.
22 Although p16
Ink4a regulates CDK4/CDK6 activity, thereby indirectly regulating the Rb tumor suppressor, p19
ARF regulates the p53 tumor suppressor.
23 p19
ARF acts by attenuating Mdm2-mediated degradation of p53 and is known as an activator of the p53 pathway. Therefore, loss of p19
ARF leads to impairment of p53 signaling. Elimination of the Ink4a/ARF genetic locus in mice makes the animals highly prone to tumor development.
24
Cip/Kip Family
The Cip/Kip family of CKIs includes three members: p21
Cip1, p27
Kip1, and p57
Kip2. Unlike the Ink4 family of CKIs, Cip/Kip inhibitors bind to and efficiently inhibit various CDKs. Members of the Cip/Kip family are highly homologous and share approximately 50% of their sequences. The amino terminus of both p21
Cip1 and p27
Kip1 contains an RXL (where X is typically basic) sequence that is responsible for binding to cyclins and is called the
cyclin-binding motif. Cip/Kip inhibitors also contain a domain that is responsible for the binding to CDKs (N-terminal in p21
Cip1 and p27
Kip1).
25 The crystal structure of the p27
Kip1/cyclin A/cdk2 complex (
Figure 11-5 ) revealed that p27
Kip1 binds CDK2 at its N terminus and inserts into the catalytic cleft, thus mimicking ATP.
26 On cyclin A/CDK2, p27
Kip1 binds to the groove of the cyclin box. Because both Ink and Cip/Kip proteins occupy almost the same binding sites on CDKs, binding is mutually exclusive. For example, in vitro, p15
Ink4b inhibits binding of p27
Kip1. However, in cells, which protein gets to the CDK first is often determined by the coordinated cellular localization of the inhibitors and cyclin-CDK complexes.
Figure 11-5 Cyclin A/CDK2/p27KIP1 complex Crystal structure of the inhibited ternary cyclin A/CDK2/p27Kip1 complex.
p27
Kip1 is responsible for induction and maintenance of the quiescent state. p27
Kip1 expression is induced in response to growth factor withdrawal and on contact inhibition in cell cultures.
27 p27
Kip1 levels are decreased on addition of the mitogens by various mechanisms described in subsequent paragraphs. Overexpression of p27
Kip1 in cells leads to cell cycle arrest in G
1 phase. Unlike p27
Kip1, p21
Cip1 is present at high levels in cycling cells, keeping CDK activities under tight control. p21
Cip1 levels are induced in response to DNA damage and genotoxic stress as a result of activation of p53. Similar to p21
Cip1 and p27
Kip1, induction of p57
Kip2 can mediate cell cycle arrest in G
1 phase. In addition, p57
Kip2 also participates in the M-to-G
1 transition through activation by p73.
28 Abrogation of p73 or its downstream effector p57
KIP2 perturbs mitotic progression and transition to G
1 phase.
29
Transcriptional Regulation by the E2F Transcription Factors
E2F was originally identified as a cellular DNA binding activity that regulated expression of the viral E2 promoter.
30,31 Since this seminal work, molecular analysis has revealed that the E2F activity is encoded by a family of DNA binding proteins, which includes transcriptional activators and repressors. Mammalian cells encode eight known E2F proteins (E2F1-8;
Figure 11-6 ). Further complication
ensues from the fact that E2F associates with DNA as a heterodimer; the two known heterodimeric partners for E2F are DP1 and DP2. Indeed, the founding member, E2F1, can drive S-phase entry in the absence of growth factor stimulation.
32 The ability of E2F1 to drive S phase derives from its role in the regulation of genes whose protein products play essential roles in S-phase progression. Established E2F targets include components of DNA replication complexes (MCMs) and S-phase cyclins (E and A).
33 E2F family members were initially considered requisite regulators of S-phase entry. E2F1, E2F2, and E2F3 accumulate during G
1 phase and play critical roles in promoting expression of S-phase targets. Strikingly, E2F4 through E2F7 function as transcriptional repressors;
34 E2F3b, an alternatively spliced isoform of E2F3, is also a transcriptional repressor. The E2F repressors function to maintain cells in a quiescent or resting state. In addition to DP1, E2F complexes are further modulated by members of the retinoblastoma protein (pRb) family (pRb, p107, p130;
Figure 11-7 ). The Rb family member functions to recruit chromatin-remodeling enzymes, such as histone deacetylases, to E2F complexes. As a consequence, increased activity of E2F1 through E2F3 requires dissociation of “pRb” from the E2F/DP1 heterodimer. As illustrated in the following sections, the G
1 CDK/cyclin kinase triggers this through direct phosphorylation of the associated pRb family member.
35
Figure 11-6 E2F family of transcription factors There are eight members of the E2F family of transcription factors. E2FS are classified as transcriptional activators or repressors. Functional domains are indicated by differential shading.
In addition to the regulation of S-phase entry and progression, E2F transcriptional activators can trigger apoptosis or cell suicide. The mechanisms whereby E2F induces cell death remain unclear. However, pro-apoptotic genes have been identified as E2F target genes. Examples include the p19ARF protein, which is a known regulator of the p53 tumor suppressor. In addition, E2F can increase expression of pro-apoptotic proteins Puma, Noxa, and Bim and repress the anti-apoptotic Bcl2 family protein, Mcl1.
Two new E2F family members recently identified, E2F7 and E2F8, provide an important constraint against excessive E2F1 activation. Unlike the other mammalian E2Fs, E2F7 and E2F8 have two DNA-binding domains and do not require a DP partner to bind to DNA and as such are classified as atypical E2F family members.
36 These atypical E2Fs bind to the consensus E2F recognition sequence and can repress expression from a subgroup of cell cycle–regulated E2F targets. An E2F7 and E2F8 double knockout is an embryonic lethal resulting from massive apoptosis; this phenotype can be bypassed by removing E2F1 or p53.
37 Our current level of understanding underscores E2F7 and E2F8 as a distinct arm of the E2F network involved in repression of transcription during S-G
2 and control of the E2F1-p53 apoptotic axis.
G1 Regulation/Restriction Point Control
During the first Gap phase or G
1, cells prepare for DNA replication. They must synthesize proteins necessary to replicate their genome, and once these are made, assemble the various components of the DNA replication machinery on chromatin at so-called origins of replication. This is
coordinated with nutrient and growth factor availability to ensure that the cell is in an environment that supports cell division. The G
1 phase of the cell cycle is unique in that it represents the only time wherein cells are sensitive to signals from their extracellular environment. These signals are in the form of adhesion to substratum and growth factors. Cells require growth factor–dependent signals up to a point in late G
1 referred to as the
restriction point (“start” in yeast).
Figure 11-7 Restriction point control Progression through G1 phase requires growth factor–mediated (mitogenic) signals. Mitogens promote the activation of the initial cyclin-dependent kinase (CDK; cyclin D/CDK4) complex, which phosphorylates Rb family proteins (inactivating signal). The CDK4 enzyme also binds to Cip/Kip CDK inhibitors (CKIs), thereby sequestering these proteins to facilitate CDK2 activation. Rb phosphorylation releases the transcription activating E2FS (E2F1-E2F3), which promote transcription of downstream targets such as cyclin E, A, and MCM proteins. Cyclin E binds to CDK2, and this active complex maintains Rb in an inactive state. Active cyclin E/CDK2 also targets its own inhibitor (p27Kip1) for proteolysis via site-specific phosphorylation. The complete activation of CDK2, first by cyclin E and then by cyclin A, marks passage through the restriction point. Once past this point, cells no longer require growth factor stimulation for progression through the remainder of that cell division.
Progression through G1 phase is driven by the collective activities of two distinct CDKs. The first is CDK4 or CDK6 in combination with a D-type cyclin. Mammalian cells encode three distinct D cyclins (D1, D2, D3), which are expressed in a tissue-specific manner. Whereas CDK4 and CDK6 are constitutively expressed, D cyclins are expressed in response to growth factor signaling. Following accumulation of active cyclin D/CDK4 or CDK6, the CDK2 kinase in combination with cyclin E accumulates to facilitate the transition from G1 to S phase.
A key protein that regulates G
1-phase progression in the mammalian cell cycle is retinoblastoma protein, Rb. The Rb family consists of three members, Rb, p107, and p130. In quiescent cells, Rb proteins associate with E2F transcription factors to repress E2F-dependent transcription. E2F targets include genes responsible for regulation of cell cycle and DNA replication, such as cyclins E and A (see
Figure 11-7). Rb activity is regulated at the level of posttranslational modification, specifically phosphorylation. Hypophosphorylated Rb is active and binds to E2F, thereby silencing E2F-dependent activity. Hypophosphorylated Rb family proteins therefore play a central role in maintaining cells in a resting or quiescent state. Quiescent cells reenter the cell cycle in response to mitogenic growth factors. Growth factor signaling induces the expression of D-type cyclins at transcriptional and posttranslational levels,
38 leading to activation of cyclin D–dependent kinases CDK4 and -6 and subsequent Rb phosphorylation. Cyclin D/CDK4 or -6 complexes also have a kinase-independent function. They sequester p21
Cip1 and p27
Kip1 CDK1s from CDK2 kinases and allow activation of basal CDK2/cyclin E kinases, which further phosphorylate Rb family proteins. Phosphorylation of Rb promotes its dissociation from E2F, allowing transcriptional activation of E2F targets such as cyclin E. The E2F-dependent spike in cyclin E, and thus CDK2/cyclin E activity, represents the transition from mitogen-dependent to mitogen-independent cell cycle progression (or passage through the restriction point). In addition to maintaining Rb proteins in a hyperphosphorylated (inactive) state, the activation of cyclin E/CDK2 promotes proteasome-dependent degradation of its own inhibitor p27
Kip1 (described in a subsequent section). These changes, which include cyclin D/CDK4/6 and cyclin E/CDK2 activation, Rb phosphorylation, and destruction of p27
Kip1, render cells with decreased mitogen dependency and are irreversibly committed to enter S phase of the cell cycle.
Regulation of DNA Replication (S Phase)
Early experimentation, which relied on techniques wherein two cells (generally one human and one rodent cell) in distinct phases of the cell cycle are fused together (one cytoplasm containing the two distinct nuclei), revealed that chromosomes were competent for duplication in G1 and S phases. For example, fusing an S-phase cell with a G1-phase cell could enforce replication of a G1 cell; in contrast, fusion of a cell in G2 phase with an S-phase cell could not enforce replication of G2 chromosomes. It was inferred from these experiments that S-phase cells contained a factor that triggered replication initiation and that G1 chromosomes were prepared or “licensed” for this initiating activity. Research efforts have shed light on the molecular basis of regulated replication initiation.
Although DNA is actively replicated during S phase, cells must prepare DNA for replication during the preceding G
1 phase. During G
1 phase,
origins (chromatin positions where DNA polymerase complexes initiate replication) must first be established or “licensed.” Licensing refers to the formation of the pre-RC (pre-replication complex) at origins of replication (
Figure 11-8 ). Initially, the origin of replication complex (ORC) must be associated with chromatin to act as a landing pad on which the pre-RC is formed. Unlike most components of the pre-RC, ORC remains constitutively bound to DNA. In budding yeast, ORC acts as a sequence-specific DNA binding complex; however, in fission yeast and mammalian cells, no sequence specificity has been elucidated for ORC. The next step is the recruitment
of
Cdc6 to the ORC. Cdc6 subsequently recruits the MCM complex and Cdt1. However, MCMs are not stably bound at this point. Stable loading of the MCM2-7 helicase complex requires ATP hydrolysis by CDC6, which also results in release of Cdt1.
39 At the G
1/S boundary additional factors are recruited, including
MCM10, which functions to recruit
Cdc45 and subsequently, DNA polymerase
α and primase.
Figure 11-8 Prereplication complex Prereplication complexes (pre-RCs) form during mid- to late G1 phase, and once they are formed, origins of replication are considered licensed for replication. Origins are recognized first by the hexameric origin of replication complex (ORC1-ORC6), which serves as a landing pad for recruitment of the remaining components. Following ORC recognition, Cdt1 and CDC6 function in a concerted fashion to recruit the MCM2-MCM7 complex, which is considered the replicative helicase. At the beginning of S phase, additional factors (MCM10, CDC45, and polymerases) are recruited and replication can initiate in a fashion dependent on the CDK2 and CDC7 kinase activities.
Like G
1 phase, both the G
1/S transition and S-phase progression are driven by cyclin-dependent kinases (CDK2/cyclin E and CDK2/cyclin A, respectively), along with the activity of a distinct CDK-like protein kinase,
Cdc7/Dbf4. The precise substrates that must be phosphorylated for the firing of origins remain to be conclusively identified. Substrates identified so far include ORC1, MCM2, MCM3, MCM4, and Rad18.
40,41 Not all origins fire simultaneously, but they are temporally regulated. Origins can be grouped generally into those that initiate at the beginning of S phase, “early,” and those that fire toward the middle to end of S-phase, “late.” The temporal control of firing most likely reflects local controls (chromatin structure modifications) and activation of the complex via phosphorylation.
Paradoxically, although origin firing requires CDK activity, CDK2 activity is also essential for inhibition of a second-round DNA replication (re-replication) within the same cell cycle. Although the precise mechanisms whereby CDK2 prevents replication are still under intense investigation, one way it achieves this goal is through direct regulation of Cdt1 levels. On release from the pre-RC, Cdt1 is subject to ubiquitin-mediated proteolysis. Ubiquitination of Cdt1 is in turn facilitated by CDK2-dependent phosphorylation, which targets it to ubiquitinating machinery.
42 In addition to Cdt1, MCM complexes dissociate from DNA during replication. Whether this dissociation reflects dislodgment from chromatin by polymerases or also reflects a CDK-dependent function remains to be established (
Table 11-2 ).
When DNA is replicated, it is essential to retain memory of epigenetic marks in daughter cells. Histone methylation and acetylation during initiation of DNA replication contribute to this memory as well as proper replication. The histone methyltransferase PR-Set7 has been implicated in catalyzing histone monomethylation at lysine 20 (H4K20 me1) at replication origins, and this methylation is required for initiation of replication.
43 According to a model of epigenetic recruitment at the start of replication, histone acetylation plays a major role in DNA replication origin complex recruitment and H4K12Ac, H3K56Ac accompany the generation of the origin replication complex.
44 DNA processivity factor proliferating cell nuclear antigen (PCNA) is loaded on both DNA strands during replication and also provides a link between replication and epigenetic memory. NP95 (also referred as UHRF1 and ICBP90) in mammals and VIM1 in
Arabidopsis thaliana binds to hemimethylated DNA and DNMT1. NP95 recruits DNMT1 to replicating DNA. Another factor, LSH, has been shown to connect DNA methylation to replication in collaboration with NP95. Histone chaperones are required for proper recruitment of histones at DNA. H3.1-H4 chaperone chromatin assembly factor 1 (CAF1) interacts with PCNA, histone deacetylase (HDAC), and Lys methyltransferase. CAF1 regulates nucleosome assembly during DNA replication through proper deposition of H3.1-H4.
45 Another chaperone ASF1 assists CAF1 by supplying newly synthesized histones.
46 Histone methylation levels are transiently reduced during S-phase.
47 It has been recently proposed that TrxG and PcG proteins are associated with DNA during replication.
48 G2/M Transition Regulation
The Kinases of Mitosis
The transition from the second Gap phase (G
2) to mitosis (prophase, metaphase, anaphase, telophase) is regulated by CDK1 (formerly Cdc2) in association primarily with cyclin B.
49 Like other CDKs, CDK1 is relatively stable, and activation depends first on accumulation of cyclin B.
Mitotic cyclins accumulate during S phase and associate with CDK1; however, this complex is maintained in an inactive form via two mechanisms. In the first, Wee/Myt1-dependent phosphorylation of Thr-14/Tyr15 prevents ATP binding. The second mechanism relies on active transport of CDK1/cyclin complexes out of the nucleus. Onset of mitosis is triggered by dephosphorylation of CDK1 by a CDC25 isoform and increased nuclear transport/decreased nuclear exit of CDK1/cyclin complexes. Substrates for CDK1/cyclin B include APC20 (a component of the E3 ligase that ultimately degrades cyclin B), microtubule effectors, microtubule motor proteins, and tubulin itself.
50 From this and related work, it is clear that CDK1-dependent phosphorylation plays a significant role in the formation and regulation of cellular mitotic structures.
Table 11-2
Regulators of DNA Replication and Function
| Cdt1 |
Associates with MCM2-MCM7 and, in concert with Cdc6, facilitates MCM loading on origins. |
| Cdc6 |
Functions to recruit and load the MCM complex in an ATPase-dependent manner. |
| CdC45 |
Associates with the MCM and is responsible for recruitment of DNA polymerase α, primase, and replication protein A. |
| MCM2-7 |
Minichromosome maintenance proteins. Hetero-hexameric complex composed of six distinct but related proteins (MCM2-MCM7). The MCM complex functions as the putative replicative helicase. |
| MCM10 |
Structurally distinct from MCM2-MCM7; functions to recruit CDC45. |
| Orc |
Origin recognition complex. Hetero-hexameric complex that binds directly to DNA and functions as a protein landing pad on which the replication complexes form. |
| Origin |
Functionally defined in mammalian cells as regions of chromatin where DNA replication initiates. |
| Cdc7/Dbf4 |
The Cdc7 protein kinase, like cyclin-dependent kinases (CDKs), requires an allosteric activator, Dbf4. The Cdc7/Dbf4 kinase phosphorylates components of the replication complexes to initiate DNA replication. |
| Pre-RC |
The prereplication forms during G1 and contains ORC1-ORC6, Cdc6, MCM2-7. Replication ensues at S phase on recruitment of DNA polymerase and phosphorylation by both the Cdc7/Dbf4 and CDK2-cyclin A protein kinases. |
MCM, Minichromosome maintenance; ORC, origin of replication complex.
In addition to CDK1, a second family of kinases, called polo-like kinases (PLKs), also contributes to mitotic progression. In mammalian cells, there are five PLKs (PLK1-PLK5) with PLK1 being the human homolog of the founding member, Polo, from
Drosophila. 51 PLKs are serine/threonine kinases. Structurally, they consist of an N-terminal kinase domain and a C terminus with one (PLK3) or two (PLK1-PLK3) “polo box” domains. Current models suggest that PLKs are not constitutively active kinases. Rather, PLK substrates are first phosphorylated by CDKs (e.g., CDK1/cyclin B). Phosphorylation by CDKs is thought to provide a docking site for the polo box domain. Binding of the polo box results in a conformation change in PLK resulting in its activation, whereupon it phosphorylates additional critical residues within the substrate. Alternative models suggest that PLKs are activated through phosphorylation by an upstream kinase, such as CDK1/cyclin B. Although CDK1 can indeed phosphorylate PLK1 in vitro, the functional significance of phosphorylation has not been established. Importantly, neither model is mutually exclusive, and both regulatory mechanisms could contribute to the regulation of PLK activity in cells.
Like CDKs, substrates for PLKs are still being elucidated. As alluded to in previous sections, many PLK substrates may also be CDK substrates. Substrates of PLK1 include CDC25C and Wee1. The consequence of PLK phosphorylation depends on the substrate. Whereas PLK-dependent phosphorylation of CDC25C promotes its activation during mitosis, phosphorylation of Wee1 promotes Wee1 destruction.
Entry into Mitosis
Entry into mitosis requires the nuclear accumulation of active CDK1/cyclin B kinase. During interphase, activity is low. During G2, cyclin B accumulates as a consequence of increased gene expression and decreased protein degradation. Newly accumulated cyclin B is free to associate with CDK1. However, these complexes are maintained in the cytoplasm and are inactive as a consequence of the combined activities of Wee1 and Myt1. Activation of CDK1/cyclin B at the G2/M boundary is triggered through CAK-dependent phosphorylation of Thr161 in the T loop and dephosphorylation of Thr14/Tyr15 by CDC25. The initial dephosphorylation is likely catalyzed by CDC25B. The activated CDK1/cyclin B then targets CDC25C and Wee1 to promote CDC25C activity and Wee1 destruction, respectively, thereby forming an amplification loop that drives mitotic progression. The accumulation of CDK1/cyclin B in the nucleus is facilitated by phosphorylation of cyclin B near its nuclear export signal, which thereby impedes nuclear exit. PLK1 contributes to mitotic entry and progression by facilitating these processes. PLK1 can phosphorylate cyclin B just outside the NES (serine 133), thereby preventing nuclear exit. Like the CDK1/cyclin B kinase, PLK1 can also phosphorylate both CDC25C and Wee1, again contributing to CDC25C activation and Wee1 destruction and thereby ensuring full CDK1/cyclin B activation.
Chromosome Cohesion
G
2 phase and the beginning of mitosis are denoted by a 4-N DNA content. Following DNA replication and prior to cell division (cytokinesis), cells must maintain the integrity and proximity of the recently duplicated chromosomes (sister chromatids). Before segregation, sister chromatids are held together or “glued” by a multiprotein complex called Cohesin.
52,53 The cohesin complex ensures that sister chromatids are recognized and properly aligned during metaphase. Once aligned, segregation ensues following proteolytic cleavage of cohesin components. Cohesin is composed of four subunits, Smc1/3 and Scc1/3. Smc1 and Smc3 heterodimerize in a head-to-head, tail-to-tail fashion to form a ring structure in an ATP-dependent manner. The Scc1/3 subunits associate with the Smc heads to complete the structure (
Figure 11-9 ). The Scc1 subunit contacts both Smc1 and 3 and likely stabilizes the ring structure. Models suggest that the cohesin ring has a diameter of approximately 50 nm, sufficiently large to encircle two sister chromatids.
54 Cohesin is envisioned to function by binding and encircling DNA, thereby “gluing” sister chromatids together until released.
Exit from Mitosis
During mitotic prophase, chromosome structures are again altered by a complex called
condensin, which serves to package chromosomes before mitotic division.
55 The mitotic spindle also forms during prophase. The mitotic spindle is a bilaterally symmetric microtubule organizing center shaped like a football. Each half of the spindle contains a
centrosome and three distinct sets of microtubules (astral, kinetochore, and polar); the kinetochore microtubules are those
that attach to chromosomes at the kinetochores to facilitate movement to opposite poles before cytokinesis. PLKs are also implicated in the formation of mitotic spindles.
56 Loss-of-function experiments in multiple organisms (yeast to mammalian cells) result in the formation of monopolar spindles. During metaphase, the chromosomes align along the “metaphase plate” in preparation for cell division. Anaphase is marked by segregation of chromosomes to opposite poles. The proteolytic
cleavage of the Scc1 protein by a protease called
separase triggers the opening of the cohesin ring, thereby allowing chromosome segregation. Anaphase is also marked by the loss of CDK1 activity, which results from proteolytic destruction of cyclin B and cyclin A. The loss of cohesin and mitotic cyclins is coordinated by a multisubunit E3 ubiquitin ligase called the anaphase-promoting complex/cyclosome (APC/C; see subsequent sections).
Figure 11-9 Chromosomes are held together by a complex called cohesin Smc1 and Smc3 form a protein ring that is held together by a dimerization “hinge” region that encircles chromatids. The Scc1 and Scc3 subunits interact with the Smc “heads,” which retain intrinsic ATPase activity essential for separation of heads to allow DNA to enter. Once all chromatids are aligned during mitosis, Scc1 is cleaved by a protease called Separase to open the ring and allow movement to opposite spindles.
Mitotic Checkpoint
The primary goal of mitosis is to ensure that each daughter cell receives one chromosome complement after cellular division. During mitosis it means that a cell divides only after chromosomes are attached to the microtubules of the mitotic spindle. The
mitotic checkpoint, or
spindle assembly checkpoint, is activated as cells enter mitosis, in prometaphase, where it is triggered by
unattached kinetochores, leading to the delay of anaphase onset. Thus, the role of the proteins that are involved in mitotic checkpoint signaling is to sense the attachment and/or tension at kinetochores.
57 These proteins are often found to be kinetochore-associated and comprise the mitotic checkpoint complex.
58 The mitotic checkpoint complex includes BubR1 and Mps1 kinases, CENP-E (
centromere
protein
E), Mad (mitotic arrest deficiency proteins)-1 and -2, and others. The mission of mitotic checkpoint kinases is to signal regulatory proteins to inhibit the entry to anaphase. Models suggest that unattached kinetochores lead to phosphorylation of Mad1/2 proteins, which are then directed to the APC/C, resulting in the inhibition of its ubiquitin-ligating activity. This action ensures that chromosomes are accurately distributed to daughter cells. In human neoplasia, the mitotic checkpoint can be inactivated through mutations in components of MCC,
59 contributing to aberrant mitotic divisions and the appearance of aneuploid cells (genetic instability).
Regulated Proteolysis in Cell Cycle Control
Levels of cyclins and CKIs are tightly regulated throughout the cell cycle. This degree of regulation is achieved by coupling the rate of gene expression with regulated proteolysis, which occurs through the ubiquitin proteasome system. The ubiquitin polypeptide consists of 76 residues and is covalently attached to proteins destined for degradation. Attachment occurs through a reversible isopeptide linkage between the carboxy terminus of ubiquitin and lysine residue in the sequence of protein. The name ubiquitin derives from early observations of its ubiquitous expression. Indeed, ubiquitin is a highly conserved protein throughout evolution from yeast to humans.
Modification of proteins (
ubiquitination) with ubiquitin polypeptides requires a conserved series of enzymes. This system includes the ubiquitin-activating enzyme (E1) that performs ATP-dependent activation of ubiquitin. There is only one known E1 enzyme encoded in the human genome. The E1 passes activated ubiquitin to the ubiquitin-conjugating enzyme (E2), of which there are more than 30.
60 In the final stage of ubiquitination, the E2 acts together with an E3, ubiquitin ligase, to attach mono- or polyubiquitin chains onto the target protein. The E3 ligase acts as the specificity factor that determines substrate recognition and thus comprises the largest group. Once a substrate is polyubiquitinated (four or more tandem ubiquitin molecules on a single lysine within the substrate) it is targeted to the 26S proteasome for degradation.
There are two primary E3 ubiquitin ligases involved in the cell cycle that regulate key cell cycle proteins such as cyclins and CKIs. Both sets of ligases belong to a broader E3 subfamily, either the Skp1-Cul1-F-box (SCF) protein ubiquitin ligases or the APC/C. These two systems are structurally similar. However, as one would expect, they target
distinct substrates in a cell cycle–specific manner and are differentially regulated.
Figure 11-10 The Skp1-Cul1-F-box (SCF) E3 ligase F-box protein, or FBP, acts as a specificity component of SCF E3 ligase that recognizes mostly phosphorylated substrates. Further assembly of SKP1-Cul1-Rbx1 components of SCF complex brings E2 ligases and substrates in close proximity for further ubiquitination. Examples of FBPs and their substrates are indicated in the table.
SCF Ligases
The SCF complex consists of variable and invariable components. The core components employed by all SCF ligases include a scaffold protein Cul1; a ring-finger protein, Rbx1/Roc1; and adaptor protein Skp1 (
Figure 11-10 ). The variable component of the SCF ligase, which determines substrate specificity, is the F-box protein (FBP). FBPs bind Skp1 through an F-box motif initially identified in cyclin F and the substrate bringing the two within close proximity. There are approximately 70 F-box proteins reported in mammals.
61 F-box proteins are classified accordingly to various protein-protein interaction domains that they use to bind to substrates. WD40 repeats give the name to the FBXW class of F-box proteins—leucine-rich repeats (LRRs)—to the FBXL class and F-box proteins that recognize the substrates through other/unknown protein interaction domains belong to the FBXO (F-box only) class. Structurally, FBPs are organized in a fashion that allows them to recognize diverse substrates. Although substrate recognition by FBPs is generally regulated by phosphorylation of the substrate, recognition by one FBP, FBXL2, is determined at least in part by substrate modification with sugar moieties (N-glycans).
62 Thus, the activity of SCF seems to be constant, but the ability to bind to the target protein is regulated.
One of the most rigorously studied FBPs that is involved in cell cycle regulation is Skp2. Although discovered as cyclin A–associated protein, it has since been implicated in the degradation of CKIs: p27
Kip1, p21
Cip1, and p57
Kip2. Skp2 deletion in mice suggests that p27
Kip1 is a bona fide target for Skp2-mediated degradation, because these mice exhibited striking p27
Kip1 accumulation.
63 The binding of Skp2 to p27
Kip1 requires the phosphorylation of Thr187 by cyclin E/A/CDK2 in p27
Kip1. This binding occurs with high affinity only in the presence of another protein, called Cks1.
64 On binding of SCF
skp2/Cks1, phosphorylated p27
Kip1 is ubiquitinated and undergoes proteasome-dependent degradation in late G
1 and early S phases of the cell cycle. Fbw7, another FBP that has been implicated in the degradation of cell cycle key molecules, targets cyclin E, Myc, and c-Jun for degradation.
65 SCF complexes generally regulate proteins involved in G
1 to late S phase, at which point the APC/C is activated and regulates M-phase activities.
FBXO4 is an FBP that specifically directs ubiquitination of cyclin D1. FBXO4 dimerization requires GSK3β-mediated phosphorylation of FBXO4 Ser12, which triggers ligase activation at the G
1/S transition. 14-3-3ε facilitates FBXO4 dimerization, and 14-3-3ε interaction is dependent on Ser8, which is frequently mutated in human cancer, and phosphorylation of Ser12.
66 Recent work revealed the tumor suppressor function of FBXO4. FBXO4 mutations have been identified in human esophageal carcinoma and gastrointestinal stromal tumors.
67,68 Loss of FBXO4 results in cyclin D1 stabilization and nuclear accumulation throughout cell division. FBXO4
+/− and FBXO4
−/− mice succumb to multiple tumor phenotypes including lymphomas, histiocytic sarcomas, and, less frequently, mammary and hepatocellular carcinomas.
69
APC/C Ligase
Structurally the APC/C ligase is similar to the SCF complex. The core components are Rbx1/Roc1-related ring-finger protein, APC11, a Cul1-related scaffold protein, APC2, and 11 additional proteins with required but essentially unknown functions.
70 Two components determine substrate specificity similar to SCF FBPs function: cell division cycle 20 (Cdc20) and Cdh1 (
Figure 11-11 ). APC/C ligases recognize specific sequences in target proteins called the destruction box (D-box) and the Ken box. These short-peptide
sequences are recognized by the Cdh1 and Cdc20 specificity adaptors and therefore facilitate recruitment of the active APC/C.
Figure 11-11 The anaphase-promoting complex/cyclosome (APC/C) APC/C ubiquitin ligase is a multiprotein complex that is active in the M through G1 phases of the cell cycle. The subunits that are responsible for the recognition of substrates by APC are Cdc20 and Cdh1.
APC/C is active from anaphase through early G
1 phase. However, the regulation of APC/C activity is distinct from SCF ligases. The Cdc20 subunit of APC/C, APC/C
Cdc20, itself undergoes activating phosphorylation events by CDK1/cyclin B. APC/C
Cdc20 can also be phosphorylated and activated by PLK1 and inactivated by PKA. The activity of APC/C
Cdc20 is regulated by protein-protein interactions. Mitotic spindle checkpoint proteins Mad1/Mad2 bind to and inhibit APC/C
Cdc20 function, thereby delaying the onset of anaphase. The substrates of APC/C
Cdc20 ligase include securin, a protein associated with the mitotic protease separase that allows sister chromatid separation, cyclins A and B. When cyclin B is degraded, CDK1 activity declines, contributing to the activation of APC/C
Cdh1; active APC/C
Cdh1
Buy Membership for Hematology, Oncology and Palliative Medicine Category to continue reading.
Learn more here
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
The Molecular Basis of Cancer Expert Consult - Online