Regulation Of Growth Hormone And Action (Secretagogues)
John J. Kopchick, Gabriel Á. Martos-Moreno, MáRta Korbonits, Bruce D. Gaylinn, Ralf Nass and Michael O. Thorner
GH-Induced Signal Transduction
Signal Transducers and Activators of the Transcription Signaling Pathway
Mitogen-Activated Protein Kinase Signaling Pathway
Insulin Receptor Substrate/PI3K-AKT Signaling Pathway
Protein Kinase C Signaling Pathway
Suppressors of Cytokine Signaling, Protein Tyrosine Phosphatases, and Src Kinases
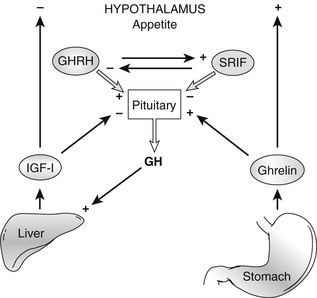
GHRH, Ghrelin, and GH Secretagogues
Growth Hormone Releasing Hormone
Since the initial discovery of a growth-promoting principle from the pituitary gland in 1921,1 human growth hormone (hGH) has been identified and isolated and has entered clinical use in growth hormone (GH)-deficient patients. In 1979, hGH complementary DNA (cDNA) was cloned and expressed2; subsequently (in 1985), recombinant (r)hGH was generated and approved for clinical use, which has increased its availability and utilization, even in non–GH-deficient states.
Major scientific goals during the past few decades have been to establish the mechanism by which GH interacts with its receptor (GHR) and to identify downstream intracellular signaling pathways. Another important goal has been to understand the regulation of pulsatile GH secretion from the pituitary. In the 1960s, Reichlin3,4 proposed the existence of a hypothalamic GH-releasing hormone (GHRH) that regulates the release of GH. GHRH was characterized, isolated, and sequenced using a single human pancreatic tumor. The receptor for GHRH was cloned from a cDNA library derived from the tumor of an acromegalic patient, through proposed homology to the secretin/vasoactive intestinal polypeptide (VIP) receptor family.
Growth Hormone
Growth hormone (GH), chorionic somatomammotropin (CS), placental lactogen (PL), and prolactin (PRL) belong to a family of hormones thought to have evolved from a common precursor.5 The hGH family members are encoded by genes located in the long arm of chromosome 17 that span ≈2.0 kilobases (kb) and contain five exons and four intervening sequences. The translation start and stop codons are located in exons 1 and 5, respectively.5
Each member of the GH family of proteins contains ≈200 amino acids, with two (GH) or three (PRL) disulfide bonds and a molecular mass of ≈22,000, with similar sedimentation and diffusion coefficients. The amino acid composition and sequence of the molecules are comparable, ranging from ≈60% to 90% in amino acid sequence identity.6 GHs are synthesized as precursor proteins, that is, they contain aminoterminal secretory signal peptides.5
The hGH gene family consists of hGH, a GH variant termed hGH-V, hCS, and hPRL. Unlike hGH, which is expressed primarily (although not exclusively) in the pituitary, hGH-V encodes a glycosylated protein that is expressed in the placenta and is found in the serum during pregnancy. It differs from pituitary hGH in 13 of 191 amino acid residues5 and, like hGH, it promotes growth. Another variant of hGH, termed 20 kDa (20K), has been found in the pituitary and blood. It is produced by alternative splicing of the hGH precursor messenger RNA (mRNA) and lacks amino acids 32 to 46 (Fig. 16-1).7
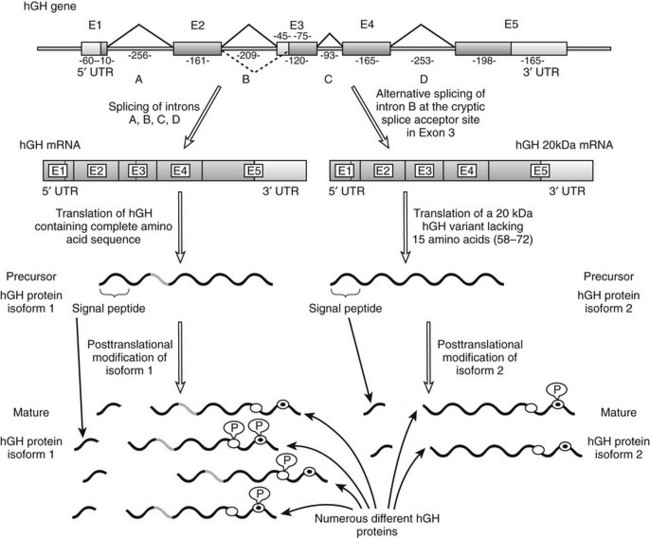
FIGURE 16-1 Schematic representation of the human GH1 gene showing the alternative splicing of exon 3. This gene contains five exons and four introns; exons are numbered E1 through E5 and introns are shown as A through D. Lengths of these introns and exons are shown in kilobases. As is shown on the left-hand side, normal gene transcription and precursor RNA splicing produces a messenger RNA that is translated to produce a growth hormone (GH) precursor of 217 amino acids (isoform 1). The mature protein, as a result of post-translational cleavage of the signal peptide (which allows it to exit the cell), contains 191 amino acids and has a molecular weight of 22 kDa. As is shown on the right-hand side, alternative precursor RNA splicing gives rise to a variant GH of 20 kDa (isoform 2) that lacks 15 amino acids from the beginning of exon 3 (amino acid residues 58 through 72). This alternative splicing reaction (indicated by the dashed line) takes place because of the presence of a 3′ cryptic alternative splice acceptor site in exon 3. Each GH isoform can undergo further post-translational modification. Several such variants of human (h)GH have been described, including variable phosphorylation of residues Ser132 (white circles) and Ser176 (white circles with black dots). mRNA, Messenger RNA; UTR, untranslated region; P, phosphate group. (From Kopchick JJ, Sackmann-Sala L, Ding J: Primer: molecular tools used for the understanding of endocrinology, Nat Clin Pract Endocrinol Metab 3[4]:355–368, 2007.)
A family of genes that encodes several transcription factors, including POU1F1 (POU domain, class 1, transcription factor 1, formerly called PIT1) and PROP1 (prophet of PIT1), have been identified and cloned, and have been found to have a major influence on the development of GH-producing cells. Expression of these genes is important in differentiation of pituitary cell lines to somatotrophs that ultimately synthesize and release GH.8 Expression and secretion of GH by somatotrophs are controlled by nutrition, sleep, exercise, and several hormones, as well as by hypothalamic peptides such as GHRH and SS, and GH secretagogues, including ghrelin. This topic is thoroughly covered in this chapter.
GH Activities
Hyposecretion of GH during childhood and adolescence leads to a GH-deficient state associated with dwarfism, whereas hypersecretion of GH before the end of puberty leads to gigantism. These disorders are due to the lack or excess of the growth-promoting action of GH on the bone growth plate. In contrast, during adulthood, when linear growth has already been completed, GH deficiency does not affect growth; however, it does affect body composition, carbohydrate and lipid metabolism, cardiovascular risk profile, and quality of life.9,10 Hypersecretion of GH in adults, mainly derived from pituitary adenomas, results in a clinical condition known as acromegaly that is characterized by soft tissue enlargement, most of which occurs in the acral regions, and involves abnormal growth of several organs, including the heart, liver, and kidneys. Together, these pathologic changes lead to life-threatening conditions, including diabetes mellitus, cardiovascular disease, and sleep apnea.11
In healthy adults, GH displays several metabolic effects, including those noted on protein and fat, although its major effects are exerted on carbohydrate metabolism. Insulin is the main hormone that controls substrate metabolism during the fed state; however, during fasting, when insulin secretion is suppressed, this function shifts to GH.12 Nevertheless, the specific impact of GH on carbohydrate metabolism is not fully understood, with two contradictory actions described: acute or early insulin-like activities, and chronic or late anti-insulin effects. The chronic effect is also described as the diabetogenic activity of GH; acute insulin-like activities include hypoglycemia and increased glucose and amino acid transport and metabolism, with increased protein synthesis,13 increased glycogenesis, and increased lipogenesis.10 These insulin-like activities are seen primarily in vitro or under special in vivo circumstances, and have been suggested to be secondary to an immediate increase in pancreatic insulin release caused by GH.14
The anti-insulin activities of GH in animals were discovered many decades ago,15 when GH was found to inhibit the action of insulin, with associated rises in serum glucose levels. This activity was also shown in humans in the 1960s,16 and 19% to 56% of individuals with acromegaly develop type 2 diabetes that results from chronically elevated circulating insulin levels and subsequent insulin resistance. This increase in insulin results in an increased rate of triglyceride production, along with an altered lipoprotein profile.11
The anti-insulin effect of GH has been found to occur after relatively long periods of GH treatment, that is, after chronic exposure, both in cultured cells and in vivo, or in acromegalic patients who overproduce GH. This diabetogenic effect, which is thought to represent a major physiologic effect of GH, includes hyperglycemia secondary to an increase in hepatic glucose output following enhanced gluconeogenesis and glycolysis, hyperinsulinemia, and decreased glucose transport, as well as increased lipolysis. This last effect results in an increase in serum levels of nonesterified fatty acids, which further enhances the insulin-resistant state.10,17,18
The diabetogenic effect of GH is exerted directly by GH-induced intracellular signaling through the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway in humans, as well as in rodents and cultured cell lines. Indeed, GH was recently shown to upregulate the p85α regulatory subunit of phosphoinositide (PI)-3 kinase expression and, thus, the activity of PI-3 kinase in white adipose tissue (WAT),19 skeletal muscle, and liver.20 This upregulation of p85α results in relative inhibition of the insulin signaling pathway, and ultimately insulin resistance accompanied by low levels of adiponectin, an insulin-sensitizing adipokine.19,20 However, other studies have questioned the role of PI-3 kinase in GH-induced insulin resistance in human muscle tissue.21
Mouse models of GH action have added significantly to the understanding of the physiologic effects of GH. These models include giant GH transgenic mice, dwarf GH antagonist (GHA) transgenic mice, and GHR gene deleted or knockout mice (GHR−/−) (Fig. 16-2). GH transgenic mice are giant, lean, and insulin resistant, and die prematurely as the result of kidney, liver, and heart problems. GHA transgenic mice are dwarf, have low levels of IGF-1, are somewhat insulin sensitive, and possess normal life spans. GHR−/− mice are dwarf and obese, express extremely low levels of IGF-1, are extremely insulin sensitive, and have extended longevity.22–24 The fact that GHR−/− mice are obese and yet insulin sensitive challenges the dogmatic notion that obesity is directly related to insulin resistance. However, contrary to what has been found in these mouse models, lipolysis induction by GH and its effect on body composition can indirectly exert a beneficial effect on insulin sensitivity, as is seen in GH-deficient patients treated with rGH.25 The effect of obesity on insulin resistance therefore may reside in the fat depot affected by GH.
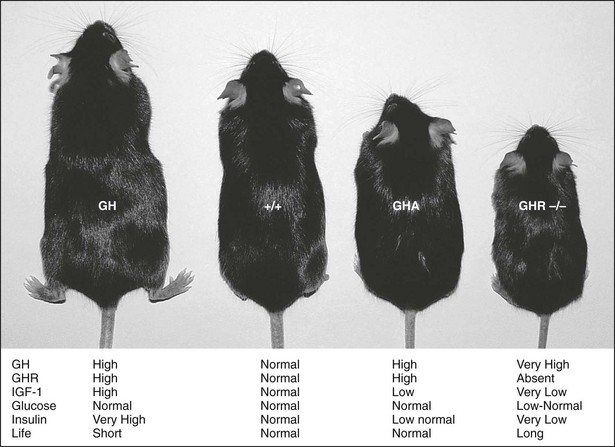
FIGURE 16-2 GH transgenic, wild-type (+/+), GH antagonist (GHA) transgenic, and GHR/BP gene-disrupted (GHR−/−) mice. A wild-type mouse is shown second from the left (+/+). The GHR/BP (−/−) mouse is approximately half the size of the normal, wild-type (+/+) mouse and is slightly smaller than the GHA transgenic mice. General endocrine values, including life span (life), are noted.
One of the major physiologic effects of GH is its influence on body composition and adipose tissue distribution. As stated above, GH transgenic mice are giant and possess a lean phenotype. In contrast, GHR−/− mice are dwarf and obese.26,27 It is surprising that a nonuniform distribution of adipose tissue in these mouse models was discovered. In GHR−/− mice, the subcutaneous and retroperitoneal depots are increased relative to control mice.26,28 Also, GHR−/− mice exhibit major decreases in the numbers of intraperitoneal adipocytes, whereas subcutaneous adipocyte number is increased relative to controls.26,28 This differential effect of GH on adipose depots is an exciting new finding in the GH field that may help to resolve the mechanisms of GH-induced insulin resistance.
The non–growth-related roles of GH have widened the clinical utilization of recombinant (r)hGH. Initially, rhGH was indicated for GH-deficient children. In addition, rhGH is now indicated for growth promotion during childhood and adolescence in several conditions associated with growth impairment in the absence of GH deficiency. These indications include Turner’s and Prader-Willi syndromes, chronic renal insufficiency, SHOX gene defects, and children born small for gestational age (SGA) without catch-up growth. Two further indications—idiopathic short stature and Noonan’s syndrome—have been approved in the United States, but not in Europe, for specific brands of rhGH. In addition, rhGH treatment has been approved for GH-deficient adults, after its beneficial effects on body composition, metabolic parameters, and quality of life in these individuals were documented.29
Involvement of GH in cancer was, and still is, a controversial issue. Data from the Pfizer International Metabolic Database (KIMS) database provide no evidence that administration of rhGH to humans causes or promotes cancer.30 In addition, in a long-term study in mice and rats, administration of rGH had no effect on the incidence of cancer.31 However, Swanson and colleagues have shown that GH signaling is important in mouse prostate32 and mammary33 carcinogenesis. These investigators crossed the GHR−/− mouse with the C3(1)/TAg mouse, in which males develop prostatic intraepithelial neoplasia (PIN) and females develop mammary carcinomas driven by the large T antigen (TAg). (In both sexes, carcinogenesis is known to progress to invasive prostate carcinoma in a manner similar to the process observed in humans.) Progeny of this cross were genotyped, and TAg/GHR+/+ and TAg/GHR−/− mice were compared. In both prostate and mammary cancer models, carcinogenesis was significantly slowed in animals harboring TAg but lacking GHR (TAg/GHR−/−) compared with TAg mice expressing wild-type GHR (TAg/GHR+/+).
Swanson’s group has also shown that the spontaneous dwarf rat (SDR), which lacks GH as the result of a point mutation in the GH gene, is resistant to chemically induced mammary carcinogenesis or TAg-driven prostate cancer.34 This model is significant in that the SDR differs from the Sprague-Dawley rat only by this single point mutation. Exposure of the Sprague-Dawley rat to N-methyl-N-nitrosourea (MNU) is one of the most commonly used and thoroughly characterized models for human breast cancer, particularly for the ability of hormones to regulate tumor growth, and is considered to be an excellent model of human breast cancer. Finally, this group has reported that the SDR can be made vulnerable to MNU-induced mammary carcinogenesis by treatment with GH, and that once mammary tumors were established, cessation of GH treatment induced rapid and dramatic regression of mammary tumors.35
Through analysis of converging results from epidemiologic research and in vivo carcinogenesis models, Pollak and coworkers have established an association between the GH/IGF-1 axis and cancer, showing that high levels of circulating IGF-1 are associated with a modest increase in the risk for several common cancers, such as colorectal, prostate, and breast cancer.36 Based on these findings, experimental pharmacologic strategies that reduce IGF-1 receptor (IGF-1R) signaling are currently under development. In addition, when mice that express a GH antagonist were exposed to chemically induced breast cancer, significant suppression of the development of breast cancer was noted.37 Finally, a GH antagonist inhibited the growth of human meningiomas and colorectal and breast carcinomas in xenograft experiments in mice, suggesting this class of drugs as potential therapeutic agents through blockade of GHR-mediated signal transduction pathways.38
Recently, Lobie and colleagues have shown that hGH is synthesized at a number of extrapituitary sites, with autocrine hGH expression found in certain human carcinoma cell lines. This autocrine GH was found to stimulate survival, proliferation, migration, and invasion of human microvascular endothelial cells. Furthermore, recent studies have demonstrated that autocrine hGH is a wild-type orthotopically expressed oncogene for immortalized human mammary epithelial cells. Thus, autocrine and paracrine hGH may play a key role in angiogenic and lymphangiogenic processes in tumor neovascularization.39 It is important to note that the GH antagonist (described above) inhibits some of these processes.40 Thus, the association of GH with the initiation and progression of a variety of cancers is controversial, requiring further study before any conclusions can be drawn. Nonetheless, the GH antagonist (Pegvisomant) and any interventions that downregulate the GH-IGF axis may be possible treatment options for several types of cancer.
To explain these various GH actions, several hypotheses have been presented, including (1) the existence of multiple GHRs; (2) the presence of multiple “active centers” in the GH molecule; and (3) the presence of small, active GH fragments (≈90 fragments have been studied) with a variety of activities.41 Data related to these hypotheses are presented in the following section.
Structure of GH
The crystal structure of the GH molecule, in particular, porcine (p)GH, was solved in 1987 (Fig. 16-3).42 GH was found to be an elongated molecule with approximate dimensions of 55 × 35 × 35 Å. The molecule contains four α helices, which are tightly packed as antiparallel bundles aligned in an up-up-down-down orientation, and contain 54% of the 191 amino acids of GH. The molecule also contains a “large loop” between residues 33 and 75, a “smaller loop” between residues 129 and 154, and a “small loop” located at the carboxyterminus.42 In 1992, the crystal structure of hGH, along with the GH binding protein (GHBP), was solved.43 Again, four GH α helices were detected: α helix I (residues 9 through 34), α helix II (residues 72 through 92), α helix III (residues 106 through 128), and α helix IV (residues 155 through 184). Two small mini-helices, residues 38 through 47 and 64 through 79, were also found in the large loop between α helices I and II.43
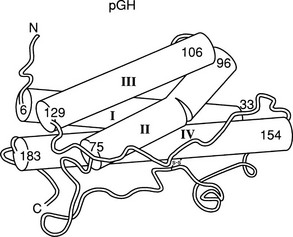
FIGURE 16-3 Crystal representation of porcine (pGH) at the 2.8 Å resolution level. Four α helices are depicted (cylindrical rods). The nonhelical region is shown as a thin tube. Also, one of the two disulfide bonds is shown; the other is hidden behind helix IV. The amino (A) and carboxyl (C) termini are located in the upper left and lower left corners, respectively. (Modified from Abdel-Meguid SS, et al: Three-dimensional structure of a genetically engineered variant of porcine growth hormone, Proc Natl Acad Sci U S A 84[18]:6434–6437, 1987.)
Bovine GH has four Cys residues located at positions 53, 164, 181, and 189, which are conserved among all GH, PRL, and placental lactogen molecules.6 The four Cys residues form two disulfide bridges in bGH (three in PRL) that are located between Cys 53 and Cys 164, which results in a large loop, and between Cys 181 and Cys 189, which forms a small C-terminal loop. Conservation of the Cys residues among members of the GH family may indicate that these residues are important for the structural integrity and biological activity of the molecules. Thus, disulfide bonds, as well as the third α helix, constitute potentially important elements for the activity of this molecule.
Structure/Function Studies of GH
Multiple studies have been developed to assess the importance of the disulfide bonds in the activity of GH, including bond splitting and site-directed mutagenesis techniques targeting the Cys residues involved in disulfide bond formation. Results derived from these experiments were contrasting but suggested that the biological significance of the disulfide bond integrity may be species specific, and that the integrity of the large loop, but not that of the small loop, is essential for the growth-enhancing activity of GH.44 However, the effect of GH on lipid metabolism was unchanged.45
Information about functional domains of GH obtained through fragment experiments was limited because the overall conformation of the protein is not maintained. In the early 1990s, a novel approach toward understanding the structure of GH was employed using recombinant DNA techniques termed “homologue scanning.” Cloned DNA sequences encoding hPRL, which possess minimal GHR binding affinity, were substituted for corresponding regions of hGH, and the PRL/GH chimeric molecules were assayed for their ability to bind PRLR or GHR. This approach was very effective in defining the receptor binding domains of hGH.46 It was found that the GHR binding domains in hGH are located mainly in the NH2-terminal portion of α helix I, a loop region between amino acid residues 54 and 74, and the COOH-terminal portion of α helix IV.46 However, these experiments could not identify the specific residues involved in the ligand/receptor interaction.
Following the GH homologue scanning studies, a more refined approach was applied to the structure/binding relationships of GH and GHR. In this approach, Ala codons were systematically substituted for many codons in the GH gene, including those encoding residues found in α helix I, the large loop, and α helix IV. This “alanine scanning” approach was used to define specific amino acids residues important for GHR binding.47 It was reported that amino acid residues 10, 54, 56, 58, 64, and 68, which were in the loop region, and 171, 172, 175, 178, 182, and 185, in the C terminus, are involved in GHR binding.46,47 The scanning mutagenesis studies largely ignored the third α helix of GH because of the fact that amino acid substitutions in this region resulted in little change in receptor binding affinity.
THE THIRD α HELIX OF GH
The search for a growth-related domain in GH was pioneered by Sonenberg’s group in the late 60s and early 70s. Their main finding was that a short sequence, generated by tryptic digestion of bGH and containing residues 96 through 133, retained low but significant bone growth–stimulating activity, whereas segments 1 through 95 and 134 through 191 had much less activity. It is interesting to note that the tryptic peptide, 96-133, contains the third α helix of GH. Subsequently, it was reported that an hGH fragment (1-134) was fully active in an assay using the IM-9 strain of human lymphocyte assay.48
Recombinant hormones possessing different portions of GH, PRL, or PL also have been generated and analyzed. hGH 1-134 was linked to hPL 141-191 and hPL 1-134 was linked to hGH 141-191 through a Cys53-Cys165 disulfide bond.49 These recombinant hormones then were tested for both their immunoreactivities and their receptor binding properties; recombinant hGH (1-134)-hPL (141-191) retained hGH immunoreactivity and full GHR binding ability but had little hPL activity; on the other hand, recombinant hPL (1-134)-hGH (141-191) possessed a large quantity of hPL immunoreactivity and PRLR binding characteristics, with negligible hGH activity. These observations showed that the immunoreactivity and biological activity of hormones were determined primarily by the NH2-terminal fragment (residues 1 through 134), while the carboxyl-terminus appears to have little effect in determining the specificity of biological activity. Together these results suggested that GH activity could be ascribed to different regions of the GH molecule, and that the 96-133 segment might be an “active core” required for growth promotion.49
These two lines of evidence laid the foundation for the structure/function studies of the third α helix of GH. By combining site-specific mutagenesis of the GH gene with the ability of resulting bGH analogues to enhance the growth of transgenic mice, we have reported a growth-promoting region of GH localized in the third α helix (50-55), which is not a perfectly amphiphilic helix because of the presence of Glu 117, Gly 119, and Ala 122, as stated previously. To convert the imperfect third amphiphilic α helix of GH to a “perfectly amphiphilic” α helix, we substituted Glu 117, Gly 119, and Ala 122 with Leu, Arg, and Asp, respectively.50 The resulting GH analogue was bound to GHRs with the same affinity as native GH. However, when the Glu117Leu, Gly119Arg, Ala122Asp GH analogue (termed M8) was assayed for its ability to enhance growth in transgenic mice, this GH analogue did not enhance growth but suppressed it, resulting in mice with a dwarf phenotype. This was the first report of a GH analogue that antagonized the action of endogenous GH and, thus, the first report of a GHR antagonist.50
In a subsequent study, we extended this observation by performing individual amino acid substitutions. Substitution of Leu 117 for Glu resulted in a GH analogue that behaved identically to native GH,51 so we concluded that residue 117 of bGH is not likely to be involved in growth-promoting activity. In contrast, the bGH analogue Gly119Arg was found to bind to GHRs with the same affinity as native GH, but transgenic mice that expressed this analogue were about one-half the size of their non-transgenic littermates.51 This was the second report of a GHR antagonist. We further confirmed this observation by generating hGH-Gly120Arg dwarf transgenic mice.54 Also, several other amino acids were substituted for bGH Gly119 and were found to act as GHR antagonists.55 Finally, substitution of Asp for Ala at residue 122 results in a bGH analogue that binds to GHRs but does not enhance (or suppress) growth in transgenic mice and may be acting as a partial agonist. Together, these studies were the first to document the discovery of GHR antagonists.51–53
It is important to note that GH analogues with amino acid substitutions that resulted in changes in growth-promoting activity are located within a region of nine amino acids, that is, between Asp 115 and Leu 123,55 which form two turns of third helix. In viewing the side chains of these potentially important amino acids, it appears that Gly 119 and Ala 122, two amino acids with relatively small side chains, form a “hinge-like” or “cleft” structure that, as stated previously, has been shown to exist in the crystal structure of hGH,43 near the center of this α helix, primarily as the result of Gly 119 (Fig. 16-4). We postulated that this cleft is important for the growth-promoting activity of the GH molecule, and that Gly may be the only residue that is tolerable at this position.51 Extension of this model would yield the prediction that any other amino acid substitution at this position would decrease the flexibility of the molecule and/or “fill” the cleft, which ultimately would result in decreased biological activity. Finally, Asp 115 and Leu 123, amino acids with negatively charged (Asp) and long (Leu) side chains, respectively, flank the cleft and may be involved in the interaction with GHR.
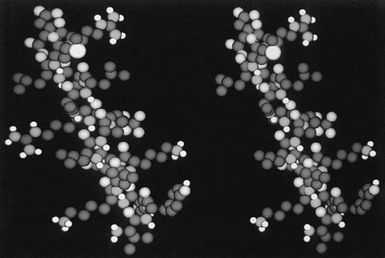
FIGURE 16-4 A space-filling model of the third α helix of GH (right) and a GH antagonist (left). The amino terminal of the helices is located on the top of the figure, and the COOH end at the bottom. Note the cleft that is located in the middle of the wild-type helix (right) and the occupancy of this cleft with the side chain of Arg (left). (From Chen WY, et al: Glycine 119 of bovine growth hormone is critical for growth-promoting activity, Mol Endocrinol 5[12]:1845–1852, 1991.)
To further substantiate the importance of the cleft in the third α helix, we designed a bGH analogue with a deletion at Gly 119 (Δ119). Transgenic mice that expressed this analogue demonstrated a phenotype similar to that of their littermates.55 These data provide supportive evidence for the importance of the cleft structure in the third α helix. It is interesting to point out that all bGH analogues tested in this study were able to bind to the GHR with an affinity similar to bGH, including Δ119 (deletion mutation, inactive analog), SAP (scrambled helix, weak antagonist), and Gly119Arg (potent antagonist).
To accommodate all of the data related to amino acid substitutions in GH, including those derived from the alanine scanning studies47 and those directed at the third α helix of GH,50 we proposed the second target hypothesis for GH action.50 In this model, residues in α helices I and IV and the large loop region interact with the GHR, as reported by Cunningham.47 Additionally, we postulated that the cleft region in the third α helix interacts with an unidentified target, and the tripartite complex is the functional unit responsible for the induction of GH action.49
GH Antagonists
Gly 119 is conserved among all members of the GH family, including PRL and PL.6 Gly is unique among amino acids in that it possesses a single hydrogen atom as a side chain. The absolute conservation of this amino acid within a strong α-helical forming region of helix III of GH implies a crucial role for this residue.
As stated above, when bGH Gly 119 or hGH Gly 120 was replaced with a variety of amino acids and the mutated genes were expressed in transgenic mice, dwarf animals resulted.50,51,54 We also tested for the ability of the GH-substituted molecules to inhibit GH-dependent conversion of mouse preadipocytes to adipocytes. The bGH Gly119Arg or hGH Gly120Arg analogues were found to inhibit this reaction by 50% at equal molar concentrations of GH and analogues, thereby defining them as GHR antagonists.56,57 A confirmatory study on the generation of a GH antagonist by substitution of arginine for glycine at position 120 in hGH was subsequently reported.58
Later, Chihara and coworkers reported that another hGH gene mutation resulted in a “natural” GH antagonist.59 They also reported another hGH gene mutation that encoded an inactive GH60 in patients with reduced growth and short stature. In the first case, the codon for Arg 77 was found to be mutated so as to encode Cys, and the resulting molecule inhibited GH-stimulated JAK2 phosphorylation in vitro.59 Unexpectedly, the expression of this GH analogue, hGH Arg77Cys, in transgenic mice resulted in giant animals (Stevens and Kopchick, unpublished results). In the second case, the mutation resulted in the substitution of Gly for Asp acid at codon 112, found to be within Site 2 of the GH molecule60 (see later). No additional data have been reported on these GH gene mutations, which may encode a GHR antagonist and/or inactive GH.
In vitro and in vivo studies of hGH antagonists (GHAs) have demonstrated that they possess great potential to counteract the pathologic conditions of excess hGH in clinical settings, which include acromegaly, diabetic nephropathy, diabetic retinopathy, and, as stated before, certain cancers. Additionally, when GH (giant) and GHA (dwarf) mice are crossed, the resulting offspring are intermediate in size, suggesting that GHAs may overcome the growth-enhancing properties of GH.61
The GHA (hGH-Gly120Lys), like wild-type GH, has a short half-life,62 which limits its utility in the clinical setting. To increase the serum half-life of the molecule, the hGH antagonist has been modified by the addition of polyethylene-glycol (PEG); hGH-Gly120Lys with four to six PEGs has a half-life of approximately 18 hours after single intravenous (iv), intraperitoneal (ip), or subcutaneous (sc) injection.62 When mice received a daily sc single injection of various doses (0.25 to 4 mg/kg) of Gly120Lys-PEG or vehicle for 5 days, a significant, dose-dependent suppression of IGF-1 became obvious, starting at day 3. The maximum suppression (up to 70%) of IGF-1 production was achieved by 1 mg/kg dosing at day 6 after the first injection. Hepatic GHRs were significantly increased on day 8, also in a dose-dependent manner (Chen et al., unpublished data). These results suggest that exogenous administration of Gly120Lys-PEG can dramatically decrease serum IGF-1 levels.
These mouse data led to the development of the first hGH antagonist for the treatment of individuals with acromegaly. This hGH antagonist included eight amino acid substitutions at Site 1 of GH and the original G120K substitution at Site 2, along with four to five PEG additions. This molecule was termed B2036-PEG and was also called Pegvisomant. Pegylation of the molecule reduces clearance and therefore increases the serum half life and reduces immunogenicity, as well as its interaction with the GHBP.63 Thus, relatively high doses of Pegvisomant are required to lower serum IGF-1. Also, Pegvisomant binds to a pre-formed receptor dimer64 with an affinity similar to wild-type G120K and induces internalization but not subsequent GH-dependent intracellular signaling63,65 (Fig. 16-5). Thus, the importance of the eight amino acid substitutions in Pegvisomant is not one of increased GHR binding characteristics. However, because two of the amino acid substitutions at GH Site 1 involved Lys residues, and because Lys are potential pegylation sites, the importance of the changes may be that GH Site 1 cannot be pegylated. Thus, the molecule would be able to interact with the GHR.63
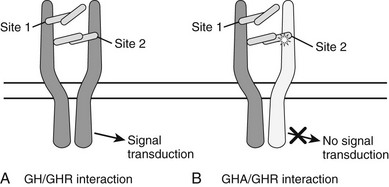
FIGURE 16-5 A, The one-GH/two-GHR model of GH action. The GH molecule and its four α helices are represented by the horizontal cylinders. The pre-formed GHR is shown as crossing the cellular membrane (dark horizontal lines). The interaction of GH with the pre-formed GHR at Site 1 and Site 2 is indicated. Not shown are the several N-linked glycosylation sites on the GHR. B, The interaction of a GH antagonist (A) with the pre-formed GHR dimer. The GHA and its four α helices are represented by horizontal cylinders. The Gly-to-Lys change in the third α helix is depicted by a star. The interaction of GHA with the pre-formed GHR at Site 1 and Site 2 is indicated. An improper or nonfunctional GHA/GHR dimer is indicated by the light shade of gray in the second GHR molecule found in the pre-formed dimer. Not shown are the several N-linked glycosylation sites on the GHR.
Debate has occurred over the mechanism of action of the GH antagonist. Fuh et al.58 suggested that the antagonist prevented the formation of the receptor dimer. However, because the GHR is pre-dimerized,63–65 the GHAs do not prevent GHR dimerization but prevent proper or functional GHR dimerization and subsequent signal transduction that ultimately results in decreased IGF-1 levels.66
Somavert (Pegvisomant for injection) has been approved for use in acromegalic individuals in the United States, Europe, and Japan. Data describing the results of Pegvisomant in acromegalic individuals, as well as their quality of life after treatment, have been put forth.67,68
Co-Crystallization of GH With The GHR
As was stated previously, when the crystal structure of hGH complexed with GHBP was solved,43 two identical GHBP molecules were found to interact with one GH molecule. This observation of two GHRs interacting with one GH molecule (Fig. 16-6) is one of the most fundamental findings in the GH molecular endocrinology field. Thus, the co-crystallization of one GH molecule with two GHBPs supported the GH second target hypothesis of GH action,50 that is, the “second target” was another GHR. It should be pointed out that reagents used in these co-crystallization studies include non-glycosylated, bacterially synthesized GHBP, and not a membrane-associated and glycosylated full-length GHR. Also, GH has not been found in a GHBP dimer in vivo.
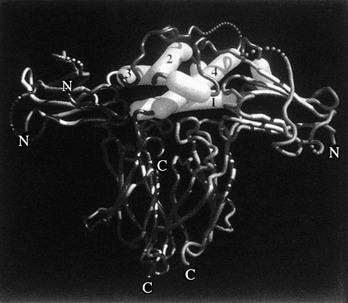
FIGURE 16-6 Representation of the GH/GHR co-crystal structure. The GH α helices are indicated as cylinders and are labeled 1, 2, 3, and 4. α helices 1 and 4 (Site 1) are shown interacting with one GHBP; α helix 3 (Site 2) is shown interacting with a second GHBP. (From de Vos AM, Ultsch M, Kossiakoff AA: Human growth hormone and extracellular domain of its receptor: crystal structure of the complex, Science 255[5042]:306–312, 1992.)
Growth Hormone Receptor
Growth hormone receptors (GHRs) have been found on the cell surfaces of many tissues throughout the body, including liver, muscle, adipose, and kidney, and in early embryonic and fetal tissue. Although most GHRs reside on the cell surface and in the endoplasmic reticulum, pronounced nuclear localization is noted in many cells.69 Evidence for the importance of the GHR in growth has come from studies on individuals expressing different mutations located throughout the GHR gene that result in the dwarf phenotype and a GH-insensitive state, also termed Laron syndrome. Later, another proof of the importance of the GHR in growth was shown with disruption of the GHR and GHBP genes in mice.70 These mice are approximately half the size of wild-type mice and are obese; they express very low levels of IGF-1 and insulin and high levels of GH, and they have an extended life span.24,71 Whether data on extended life span from GHR-/- mice extends to humans is not known. More research on this controversial subject will ensue.
The extracellular portion of the GHR consists of two fibronectin type III domains, each containing seven β-strands, arranged to form a sandwich of two antiparallel β-sheets.43 Stabilizing the GHR structure is a salt bridge between Arg 39 and Asp 132, and hydrogen bonds between Arg 43 and Glu 169.43 Also, the GHR contains seven cysteine residues in its extracellular domain43; the six in the GH binding domain form three disulfide bonds in the active signaling conformation, and help the receptor to maintain its correct three-dimensional structure.72 Van den Eijnden and coworkers have suggested, after studying the effects of replacing the Cys with Ser and Ala residues, that the middle disulfide bond, Cys83-Cys94, is important for ligand binding, whereas removal of disulfide bond Cys108-Cys122 has little effect on GH-induced intracellular signaling.72 The GHR has a half-life of approximately 1 hour and is degraded continuously even in the absence of GH through two known mechanisms: endocytosis and ectodomain cleavage. The reader is referred to excellent reviews on the GHR.69,73,74
In addition to the membrane-bound GHR, a soluble form exists that is composed of a portion of the extracellular domain, GHBP. In mice and rats, it is encoded by an additional exon, Exon 8A, and is produced by alternative splicing of the GHR precursor mRNA. In other vertebrates, it is believed that it is generated by proteolytic cleavage of the extracellular domain of the GHR. A metalloprotease tumor necrosis factor (TNF)-α converting enzyme (TACE/ADAM-17) acts on surface GHR to generate the GHBP.75 The function of the GHBP is not fully understood, but it may modulate GH activity by enhancing its half-life or reducing its availability to bind the GHR. The reader is referred to a review paper on GHBPs for further information.76
GH/GHR Interaction
Examination of the 2.8 Å crystal structure of the complex between GH and the extracellular domain of the GHR produced by Escherichia coli (hGHBP) demonstrated that the complex consisted of one molecule of GH and two molecules of the GHR (Fig. 16-6).43 Furthermore, the crystal structure reveals how a nonsymmetrical molecule, that is, GH, binds to two copies of the GHR, with one of the GHRs binding with high affinity to the GH molecule at Site 1, and with a weaker binding between Site 2 of GH and a second GHR.
Amino acid residues in the hGHR (actually hGHBP) involved in contact with hGH have been determined from co-crystallization analyses of the GH/GHR complex.43 The major binding determinants in the GH molecule (Site 1), located in the two mini-helices between α helices 1 and 2 (amino acids 60 through 63) and between the center and carboxyterminus of helix IV (amino acids 168 through 174), match to GHR amino acids 40 through 45 and 101 through 106, respectively, and with GHR Trp 169 interacting with hGH residues 171 through 179 in α helix IV. Site 2 GH residues are important for contact and dimerization and only Phe 1, Ile 4, and Asp 116, whereas the significant binding determinants in the GHR are similar as for GHR Site 1, especially Trp 104 and Trp 109.77 Of particular interest is the close encounter with hGH Gly 120 and GHR Trp 104 in this Site 2 interaction.
The model for GHR activation postulates that GH induces GHR dimerization and, consequently, activation and signaling in which most amino acid residues in the two binding interfaces act in an additive fashion.78 In addition, the GHR contains a GH-induced dimerization domain in which Cys 241 undergoes GH-induced intermolecular disulfide bonding, thus bridging together two GHRs. Eight hGHR amino acid residues are involved in the salt bridge and the hydrogen bond interactions across the extracellular dimerization domain.43 Of these eight residues, five are important for GH/GHR-mediated signal transduction, namely, Ser 145, His 150, Asp 152, Try 200, and Ser 201, but not Leu 146 or Thr 147. This study, as well as others using monoclonal antibodies to induce a GH response,79 suggests that a GH-induced conformational change in the GHR is required for a full biological response. Additionally, subtle but significant differences between the 1hGH/2GHR43 and 1hGH/1GHR80 co-crystal structures suggest that a conformational change does occur in the one ligand/two receptor complex.
Although most if not all of these GH/GHBP interactive studies use a non-glycosylated, bacterially expressed GHBP, and not the membrane bound GHR, the interaction of one GH with two GHBPs has been extrapolated to the in vivo interaction of GH with the GHR. This finding has led to the theory of a sequential binding mechanism in which hGH binds to two GHRs.77 In this model, hGH must first bind to one GHR using a high-affinity receptor binding site, which subsequently allows binding of the second receptor. The binding Site 1 of hGH is located at residues identified by Ala scanning mutagenesis studies,47 that is, α helix I, the loop between amino acids 54 through 74 and α helix four. Binding Site 2 is located at the N-terminus (Ile 4), and the third α helix, namely, Gly 120. The model predicts that a Typ 104 residue of the GHR “fits” into the “cleft” of the third α helix of GH. Chen et al.56,57 have proposed that the reason that hGH-Gly120Arg acts as a GH antagonist is because substitution of Arg for Gly at position 120 blocked or inhibited the “second” GHR from properly interacting with binding Site 2 on the GH molecule. This would inhibit proper or functional GH-induced GHR dimerization. These data nicely supported the “cleft” theory pertaining to the interaction of hGH Gly 120 with a second target.56,57 The importance of GH-induced GHR dimerization in humans has been supported by the finding of an adenine-to-guanine mutation in the hGH gene that results in the conversion of Asp112 to Gly. In the heterozygous state, this mutation is believed to be the cause of dwarfism in a female child, with the encoded hGH analogue binding to hGHR in vitro, but not inducing GHR dimerization and JAK2 and STAT5 activation.60
Later studies have dramatically changed the GH-induced dimerization theory. The work of Gent et al.,64 who used coimmunoprecipitation and epitope-tagged truncated GHRs, conclusively showed that ligand-independent GHR dimerization occurs in the endoplasmic reticulum and on cell membranes independent of GH binding.64 In addition, studies with GHAs (bGH Gly119Arg, hGH Gly120Lys; B2036 and Pegvisomant) have demonstrated that these antagonists exist in a complex with two GHRs and are internalized properly.63,65 Thus, results showed that GHAs prevent neither GHR dimerization nor internalization, but they interfere with proper GHR dimerization. This has led to the proposed model of GH binding to a constitutively homodimerized GHR, causing a structural change that results in activation of JAK2 and signal transduction.69
The mechanism by which GH binding converts the inactive pre-dimerized GHR to its active conformation remains uncertain. However, it has been shown that the composition and/or length of the transmembrane domain does not affect GH-induced GHR dimerization.81 Also, through the mutation of Site 2 in GHR, this site has been shown to be not essential for GHR to achieve its active conformation (identified by GHR disulfide linkages) and to trigger signal transduction. Additionally, the extracellular domain of GHR shows substantial flexibility to achieve active conformation in response to GH and will even accommodate GH-GH dimers.82 The interaction of GH with the GHR has been proposed to cause repositioning of the intracellular domain of GHR, in which rotation of individual GHR molecules ultimately triggers GH-induced intracellular signal transduction.83 This model, developed by Waters and colleagues over the past few years, has greatly influenced and stimulated research on the interaction of GH with GHRs and represents a seminal finding in the GH field.83
Finally, the mechanisms responsible for GHR turnover (proteolysis) have been shown to influence GH sensitivity. As was stated before, cleavage of the extracellular domain of the GHR is catalyzed by a transmembrane ectoenzyme termed TACE (tumor necrosis-α cleaving enzyme, also known as ADAM-17).75 GHR proteolysis renders cells less sensitive to subsequent GH-induced signaling by downregulating GHR abundance. Recent in vivo studies indicate that this mechanism of receptor downregulation may also mediate desensitization to GH in vivo84 and in pathophysiologic states in which it may be advantageous to limit the anabolic effects of GH (e.g., sepsis). It is interesting to note that the GHR remnant that results from metalloproteolysis is further cleaved within the transmembrane domain by an enzyme termed γ-secretase, leading to release of the intracellular domain into the cytosol and its accumulation in the nucleus. The consequences of this inducible nuclear localization of the GHR intracellular domain are as yet unknown, but may suggest novel GHR-dependent signaling pathways.
We have only touched on a few of the structural characteristics of the GHR. For a more exhaustive review of this subject, the reader is referred to an excellent review by Brooks, Waters, and coworkers.69
Clinical Manisfestations of Mutations in The GHR
GH-insensitive individuals with a dwarf phenotype were first described by Laron. The molecular defect in these Laron syndrome (LS) individuals has been found in the GHR. Since the initial series of GHR mutations was documented in LS individuals, many other sites of GHR mutation have been documented.85 Because these individuals are GH insensitive, treatment with IGF-1 is the only option.85,86
In 2004, Dos Santos et al.87 reported that a rather frequent polymorphism of the GHR gene was associated with increased responsiveness to GH. This GHR gene mutation resulted in removal of exon 3 and has been termed the d3 GHR allele.87 Children born SGA, children with idiopathic short stature, girls with Turner’s syndrome, and children with severe GH deficiency all in the presence of a d3 GHR polymorphism have a greater response to exogenous rhGH administration than do similar individuals who express the full-length GHR. These findings suggest that a subpopulation of individuals with the d3 GHR allele may have increased sensitivity to GH. Also, in a study of acromegalic individuals,88 the wild-type, full-length GHR was found to be homozygous in 50% of patients, while 50% had at least one d3 GHR allele. The GHR genotype (specifically, the deletion of exon 3) was hypothesized to modulate the relationship between GH and IGF-1 serum concentrations in these acromegalic individuals.88
GH-Induced Signal Transduction
The molecular mechanisms by which GH transmits its signals via its receptor have been largely elucidated by experiments in cultured cells or hypophysectomized rats. However, GH-induced in vivo tissue-specific signal transduction systems are still largely unknown, although data are beginning to emerge in this area, as is knowledge about modulators of GHR.21,89,90 Several GH-mediated intracellular signal transduction pathways are summarized below, some of which may overlap with signal transduction intermediates induced by insulin and other hormones, thus providing opportunities for “biological cross-talk” between these molecules. Several reviews on the subject have been presented.89,90 It is interesting to note that one of the hormones involved in this cross-talk is GH-induced IGF-1. Recently, examination of GH signaling in murine 3T3-F442A pre-adipocytes revealed that GH induces formation of a complex that includes the GHR, JAK2, and the IGF-1 receptor (IGF-1R).91 Even though both the GHR and JAK2 in the complex are tyrosine phosphorylated, formation of this complex does not depend on tyrosine phosphorylation of any of the partners. It is interesting to note that co-treatment of cells with the combination of GH and IGF-1 resulted in enhancement of downstream signals compared with GH alone. This suggests functional consequences of the complex formation. Further evidence for the idea that the GHR and the IGF-1R may function in parallel has come from studies in primary mouse osteoblasts in which the IGF-1R gene is flanked by loxP sites that enable excision of the gene when Cre recombinase is expressed. When IGF-1R was deleted from these cells, GH-induced STAT5 activation was substantially diminished, suggesting that the IGF-1R, even when not engaged by IGF-1, contributes to optimal GH-induced GHR signaling.92
Janus Kinase Activation
In the early 1990s, GH treatment of responsive cells was found to induce association of a tyrosine kinase with the GHR. This kinase was later identified as a 121 kDA protein and was found to be a member of the Janus kinase (JAK) family of proteins, in particular JAK2. Activation of JAKs appears to be an initial step in one of the GH-induced signal transduction systems. Although three JAK molecules are involved in GH/GHR signal transduction, JAK2 exhibits the greatest degree of activation. GH-dependent JAK2 activation requires interaction between JAK2 and the membrane-proximal, proline-rich motif (termed Box 1) located in the intracellular region of GHR. Because the GHR itself has no intrinsic kinase activity, it is thought that co-localization of two JAK2 molecules by the dimerized GHR results in transphosphorylation of one JAK2 by the other, leading to JAK2 activation. Activated JAK2, in turn, is thought to phosphorylate GHR on multiple tyrosine residues, providing docking sites for cytosolic components of at least three distinct signaling pathways: the STAT, MAPK, and PI3K pathways.90,93,94 An overview of these pathways is presented in Fig. 16-7.
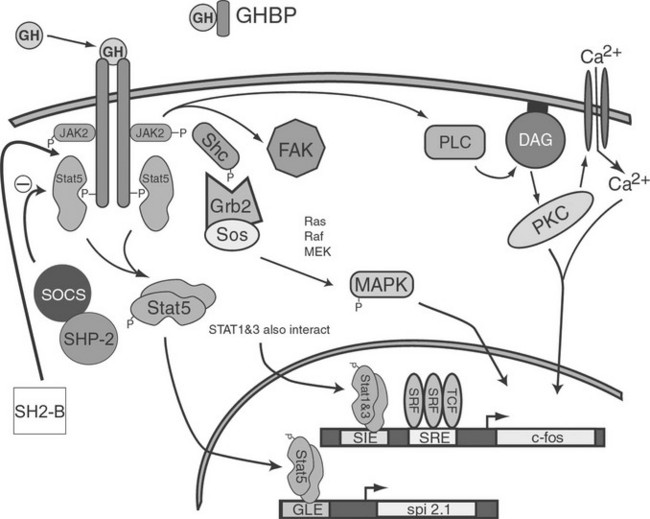
Signal Transducers and Activators of The Transcription Signaling Pathway
Many of the physiologic effects of GH result from transcriptional regulation of a variety of genes. Several different signaling pathways contribute to this regulation, but the pathway that was discovered in the mid-1990s, and perhaps the most universal pathway implicated in GH action, involves signal transducers and activators of transcription (STAT) proteins. Upon phosphorylation, cytoplasmic STAT proteins form homodimers or heterodimers, translocate into the nucleus, bind DNA, and activate transcription.95
GH-dependent tyrosyl phosphorylation requiring JAK2 activation has been demonstrated for STAT1, STAT3, and STAT5 (a and b). In addition, STAT5 activation requires regions of GHR not involved in JAK2 activation, suggesting that STAT5 also interacts directly with GHR. As was previously stated, the docking of STAT5 with the GHR requires phosphorylated tyrosine residues presumably mediated by JAK2. The tyrosine residues found to be phosphorylated and important in STAT5 docking and subsequent activation have been reported. Although STAT5 has been found to directly associate with the GHR, STAT1 and STAT3 probably do not interact with the GHR but with JAK2 instead. STAT1, STAT3, STAT5, and possibly STAT4 have also been identified in GH-induced DNA binding complexes of several genes, and their presence is required for maximum transcriptional gene activation.90,94
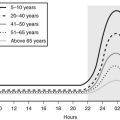
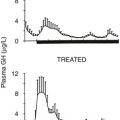
In liver and other GH-responsive tissues, STAT5 activation following the stimulation of cell surface GHRs by plasma GH occurs intermittently, reflecting the pulsatile nature of pituitary GH secretion. GH secretion and consequently the temporal patterns of plasma GH levels differ between males and females, with more frequent episodes of pituitary GH release and shorter plasma GH interpulse intervals in females as compared with males, both in rodents and in humans.96 This sex difference or dimorphism in plasma GH profiles leads to sex differences in hepatic STAT5 signaling and liver gene expression. Thus, in male rat liver, intracellular STAT5 signaling is intermittent and is followed by downregulation of hepatic STAT5 signaling and resetting of the intracellular signaling apparatus in time for STAT5 to respond to the next plasma GH pulse. In females, more frequent plasma GH pulsation leads to partial desensitization of hepatic STAT5 activation, with the peak level of active STAT5 in the nucleus being substantially lower than in males.97
Additional studies of these gender differences have shown that male mice with a targeted disruption of the STAT5b gene display two striking phenotypes that are not seen (or are much less dramatic) in female mice: (1) Body growth rates are reduced in STAT5b-deficient males beginning just prior to puberty; and (2) sex-specific liver gene expression is abolished, with more than 1000 STAT5b-dependent, sex-specific genes identified in the liver.98 These genes are important for physiologic processes such as lipid metabolism and steroid hormone metabolism, and include genes that code for phenomone-binding proteins, cytochromes P-450, and other enzymes that metabolize steroids, drugs, and environmental chemicals. The loss of liver sexual dimorphism that is seen in global STAT5b-knockout male mice and in liver-specific STAT5a/STAT5b–double knockout male mice involves downregulation of ≈90% of male-specific genes and upregulation (de-repression) of ≈60% of female-specific genes.98,99 The mechanisms that underlie these effects of STAT5b on liver sexual dimorphism are complex and most likely involve both direct and indirect effects of STAT5b on sex-specific genes.100 Sex-dependent effects of GH on liver gene expression may be important in humans as well, as CYP3A4, the major catalyst of human hepatic drug metabolism, shows sex-specific expression (female > male)101 and a pattern of GH regulation similar to that of several mouse Cyp3a genes. These findings have important implications for the sex-dependent metabolism and pharmacokinetics observed with a wide range of drugs in humans.
Mitogen-Activated Protein Kinase Signaling Pathway
Another GH-inducible pathway that ultimately culminates in transcriptional regulation of a number of genes involves activation of two mitogen-activated protein kinases (MAPKs), termed extracellular signal regulated kinase (ERK) 1 and ERK2.102 This pathway was first described for insulin-mediated signal transduction. The pathway most likely begins with GH-stimulated binding of adapter protein (SHC) family members to phosphorylated residues in both GHR and JAK2, followed by phosphorylation of the SHCs by JAK2. Subsequently, the tyrosyl phosphorylated SHC proteins interact with growth factor receptor bound 2 (GRB2) that, in turn, interacts with son of sevenless (SOS).103 Finally, GH activates RAS GTPase, RAF kinase, and MAP-ERK kinase (MEK).103 These studies, as well as those by Winston and Hunter,104 implicate GH as the inducer of the SHC-GRB2-SOS-RAS-RAF-MEK pathway for activation of MAPK. GH also activates insulin receptor substrates (IRS)-1 and IRS-2,105 which can lead to activation of the RAS-MEK signaling pathway.
GH activates the S6 kinase, p90RSK, most likely via MAPK. p90RSK, in turn, can phosphorylate a transcription factor, termed serum response factor (SRF), that binds to the GH-responsive serum response element (SRE) of the c-fos promoter/enhancer.106 GH may activate another protein, the ternary complex factor p62TCF/ELK1, which interacts with SRF to bind SRE but is directly activated by ERKs 1 and 2.107 Further evidence that MAPKs are involved in the GH-dependent transcriptional regulation of c-fos comes from the observation that the same regions of GHR required for activation of MAPK are also required for c-fos gene induction.108 As mentioned earlier, STAT proteins are also involved in c-fos gene regulation, demonstrating a convergence of at least two divergent GH signaling pathways in the regulation of a single gene. MAPK activation, also inducible by a number of growth factors, may represent a common signal transduction system, whereas activation of STAT proteins (in particular STAT5) may be more specific to GH.102
Based on recent mutational studies, disruption of the confomational change induced by GH on the F′G′ loop of the GHR (residues 216 through 221) causes specific impairment of ERK signaling without affecting STAT activation in FDC-P1 cells.109 This finding suggests that movements of this F′G′ loop could determine the signaling choice after GH binding to GHR. It is interesting to note that this conformational change in the GHR is not induced after binding of the human GH antagonist (hGHA) to the GHR.
Insulin Receptor Substrate/PI3K-AKT Signaling Pathway
In addition to sharing some MAPK pathway intermediates with insulin, GH activates members of an additional insulin signaling pathway: insulin receptor substrate (IRS)-1 and IRS-2. Although the nature of the interaction between the IRS molecules and the GHR/JAK2 complex is not clear, it does appear that JAK2 activation results in tyrosyl phosphorylation of IRS-1 and IRS-2, which is involved in the insulin-like effects of GH. Phosphoinositol-3-kinase (PI3K) is also involved in the insulin-like effects of GH, in that a GH-induced interaction between the regulatory subunit of PI3K and tyrosyl phosphorylated IRS-1 and IRS-2 has been demonstrated in adipocytes.19 The ability of the PI3K-AKT pathway to promote cell proliferation and differentiation and to prevent apoptosis has been well documented.
A role for GH induction of the PI3K-AKT pathway in GH regulation of IGF-1 expression cannot be ruled out, because inhibition of this pathway results in reduction of GH-induced IGF-1 expression in mouse cells. Nevertheless, targeted gene disruption studies of the p85α regulatory subunit of PI3K, as well as the downstream effector of PI3K-AKT, indicate that, although the PI3K-AKT pathway is essential for survival and normal growth, its effects are not necessarily direct functions of GH action.110 In contrast, and as stated previously, upregulation of the p85α regulatory subunit of PI3K by GH has been suggested as the possible mechanism of its diabetogenic effect in adipose tissue, skeletal muscle, and liver,19,20 although this mechanism is not unanimously accepted in human tissue.21
Protein Kinase C Signaling Pathway
One pathway for PKC activation involves GH-induced 1,2-diacylglycerol (DAG) production by phospholipase C (PLC) that is possibly coupled to GHR via a G protein.111 Another proposed pathway for PKC activation involves the IRS/PI3K.111 This proposal is supported by our finding that GH promotes activation and translocation of a PK isoform, PKC-ε, from the cytosol to plasma membrane, suggesting that GH-dependent activation of PKC may involve the IRS/PI3′-kinase pathway.112
Suppressors of Cytokine Signaling, Protein Tyrosine Phosphatases, and Src Kinases
Several molecules have been implicated as inhibiting GH-induced intracellular signaling. Suppressors of cytokine signaling (SOCS) proteins are important in the regulation of GHR/JAK2 signaling. The SOCS family has eight members, and GH induces SOCS 1, 2, and 3; conversely, SOCS 1, 2, 3, and 7 proteins downregulate GH signaling by inhibiting JAK2 and STAT molecules, and have been postulated as the intracellular mediators of cytokine inhibition of GH action.113 In addition to SOCS proteins, several protein tyrosine phosphatases are involved in termination of GH-activated STAT signaling. PTP-H1 modulates GHR signaling and systemic growth through IGF-1 secretion.114 Finally, the possible involvement of another family of kinases (Src kinases) in GH signaling and STAT/ERK activation has been ruled out recently through the absence of signs of impairment in GH-induced STAT/ERK activation after blockage of Src kinase.115 Work continues in the area of GH-induced signaling, and, as stated earlier, data generated from human tissue are desperately needed.
GHRH, Ghrelin, and GH Secretagogues
Ghrelin, a 28 amino acid peptide, was discovered as an endogenous ligand for the GH secetagogue receptor. It is secreted predominantly into the circulation from the stomach and has strong GH-releasing effects. It amplifies the spontaneous secretion of GH. Ghrelin exerts its central GH-releasing effects via stimulation of GHRH secreting neurons and by functional somatostatin antagonism at both the hypothalamic and pituitary levels. Ghrelin has also been shown to play a significant role in energy homeostasis. It is present in hypothalamic neurons in the arcuate nucleus, and it activates NPY- and AGRP-containing neurons, as well as the midbrain dopamine reward circuitry, to stimulate orexigenesis. Clinical studies suggest that under fed conditions, the acylated form of circulating ghrelin increases the amplitude of GH secretion pulses. Based on its orexigenic and GH-releasing effects, ghrelin has emerged as a crucial factor, connecting physiologic systems that regulate energy balance, nutritional partitioning, and growth (Fig. 16-8).
Growth Hormone–Releasing Hormone
In 1982, growth hormone–releasing hormone (GHRH) became the last of the originally proposed hypophysiotropic factors to be identified structurally. Its existence had been proposed by Reichlin because selective hypothalamic lesions yielded a GH deficiency state and growth failure,3,4 although it was finally isolated from GHRH-producing abdominal tumors rather than from the hypothalamus. In a patient with acromegaly and Turner’s syndrome, the acromegaly was due to somatotroph hyperplasia, as it persisted despite transsphenoidal surgery.116 Two different teams isolated a 40 amino acid peptide from a 5 cm tumor in the tail of the patient’s pancreas, GHRH(1-40)-OH, then designated GH-releasing factor (GH-RF or GRF).117–119 Simultaneously, the Guillemin laboratory sequenced three GHRH peptides from a different tumor: GHRH(1-44)-NH2, GHRH(1-40)-OH, and GHRH(1-37)-OH.120,121 Complete amino acid sequence identity occurred apart from varying C-terminal extensions, indicating possible peptide processing prior to release. The full biological activity resided in residues 1 to 29,122 and by sequence homology, the peptides were members of the glucagon-secretin family. These peptides eventually were demonstrated to fulfill all the requirements of a hypophysiotropic growth hormone–releasing hormone.
Molecular and Cellular Biology
GHRH is produced predominantly by neurons in the arcuate nucleus of the hypothalamus, which send processes to the median eminence, where GHRH is released into the pituitary portal circulation. GHRH then stimulates the pulsatile release of GH from somatotrophs of the anterior pituitary.123 Both GHRH(1-44)-NH2 and GHRH(1-40)-OH can be found in the human hypothalamus124 and in pituitary tumors of acromegalic patients.125 GHRH is also produced in other tissues, where it may serve autocrine or paracrine roles.
GHRH Peptide
GHRH is a member of a family of homologous peptides that in humans includes secretin, glucagon, glucagon-like peptides (GLP-1, GLP-2), vasoactive intestinal peptide (VIP), pituitary adenylate cyclase–activating peptide (PACAP), PACAP-related peptide (PRP), peptide histidine-methionine (PHM, known as PHI in other species where the C-terminal residue is isoleucine), and glucose-dependent insulinotrophic polypeptide (GIP, also called gastric inhibitory peptide).126,127 These peptides are thought to have arisen from a common ancestor through a series of gene duplications127; as the result of sequence and structural similarities, they can interact at each other’s receptors to a varying extent.126 The N-terminal (1-29) residues of GHRH are required for receptor binding in the human122 and are 62% identical in the mouse (the most divergent known mammal) and are less conserved in more distant species, including birds, fish, and even protochordate invertebrates.128–130 This is in contrast to related peptides such as PACAP, VIP, and glucagon, which are 100% identical in many mammals and better than 90% identical in more distant vertebrates.127,130 Indeed, because of sequence similarities between PRP and GHRH in non-mammalian vertebrates, some of the peptides from non-mammalian species originally named as GHRH or GHRH-like are now seen to be orthologues of PRP, and not GHRH.131,132 The relative activities of these PRPs at GHRH receptors appear to differ between species131,133; specific receptors that respond preferentially to PRPs have been identified in fish,134 but a corresponding PRP receptor gene appears to be absent in mammals,134 and human PRP has no activity at the human GHRH receptor.
It has been proposed that the active tertiary structure of the GHRH peptide is an amphiphilic α helix that runs from residue 4 onward.135,136 This helical structure, with polar and hydrophobic faces, is presumably stabilized when the peptide is bound to its receptor but is not stable in aqueous solution.137 Circulating GHRH is inactivated rapidly in vivo by dipeptidylaminopeptidase IV (DPP-IV) acting at Ala2,138 and more slowly by oxidation at Met27.122 Medicinal chemists have used the GHRH scaffold to develop peptidic GHRH analogues with increased stability and potency. These efforts have utilized combinations of strategies that include increasing the stability of the α helix with helix-forming residues or ring structures; prolonging the half-life of the peptide in the circulation by introducing unnatural amino acids or polyethylene glycol (PEG) residues to decrease DPP, tryptic, and chymotryptic protease susceptibility; and replacing the oxidizable methionine.122,139–143 An analogue of GHRH has also been developed that couples to endogenous circulating albumin when injected, thus conferring an extended half-life,144,145 and modified GHRH sequences have been expressed in muscle tissue by electrophoration of injected plasmids.146 Substituting the alanine at position 2 with d-arginine was found to produce a GHRH antagonist,147 and subsequently, more stable, higher-affinity versions of this type of antagonist have been developed148 that may prove useful in blocking the mitogenic effects of GHRH.149 Although GHRH acts as a low-affinity agonist at the VIP receptor, GHRH analogues such as N-Ac-Tyr1 and D-Phe2GHRH(1-29)-NH2 have been developed as VIP antagonists.150
Gene and mRNA
Complementary DNA (cDNA) probes have allowed identification of the single-copy GHRH gene on human chromosome 20. Human, rat, and mouse genes span approximately 10 kb of DNA and include five exons. The third exon encodes residues 1 through 31, which are sufficient for the known biological activities of GHRH. However, the human mRNA encodes a 108 amino acid precursor protein, the middle region of which is processed to form the mature GHRH peptide. Brain-, placenta-, and gonad-specific forms of GHRH mRNA have been isolated,151–153 the messages for which are initiated at different gene promotors and result in mRNAs of different sizes, although the encoded precursor protein remains identical. Immunologic evidence shows that the C-terminal fragment of the precursor protein is processed into an additional peptide known as GHRH-related peptide (GHRH-RP), which is expressed in the human hypothalamus,154 where its role is not known, and in rat testis, where it is reported to regulate Sertoli cell function.155 An additional, alternatively spliced mRNA found in rat placenta but not hypothalamus encodes the normal GHRH but includes an altered GHRH-RP.156
Tissue Distribution
In humans and a number of other species, GHRH immunoreactivity is present in the basal hypothalamus, appropriate anatomically for release into the pituitary portal vessels.157 GHRH cell bodies directing processes to the median eminence originate from both the perifornical nucleus158 and the arcuate (rat) or infundibular (human) nucleus.157 GHRH perikarya are also found in the ventromedial nucleus,159 electrical stimlation of which can induce increased release of GH.160 A reciprocal innervation occurs between GHRH and somatostatin neurons in the rat hypothalamus,161 providing the potential for direct communication between the major stimulatory and inhibitory neurons governing GH release. This relationship may participate in the ultradian oscillation of hypothalamic GHRH and somatostatin mRNA.162 GHRH neurons also directly express somatostatin receptors.163 A number of other brain regions outside of the hypothalamus contain immunoreactive GHRH.158,164,165 The ontogeny of GHRH neurons suggests that they appear in the human fetus at between 18 and 29 weeks’ gestation,166 and in the rat on embryonic day 18, reaching adult levels by postnatal day 30.167
Much evidence for GHRH is evident outside the central nervous system in a number of cell types and tissues in humans and in rodents, but its function outside the GH axis remains to be established. mRNA for GHRH, immunoactive GHRH, or bioactive GHRH content is reported in the anterior pituitary, ovaries, testes, placenta, leukocytes, adrenal medulla, pancreas, and gastrointestinal tract,168 and in tumors associated with the GH axis,125,169–171 as well as many other tumor types, including human breast, endometrium, and ovarian.172 Indeed, trace amounts of GHRH mRNA have been found in most rat tissues examined by reverse transcriptase polymerase chain reaction (RT-PCR).173 Studies in the somatotrope found immunoreactive GHRH in secretory granules and in the cell nuclei.174 Additional data demonstrate somatotrope uptake of labeled GHRH into secretory granules, lysosomes, and the nuclear membrane.175
GHRH Receptor
GHRH acts through a high-affinity G protein–coupled receptor (GHRH-R) found on the anterior pituitary somatotroph and coupled to cyclic adenosine monophosphate (cAMP).176 The receptor was cloned from human pituitary tumor and from rat and mouse pituitary,177–179 and it was found to be a member of the G protein–coupled receptor family B, also called the secretin family; the rat and human protein sequences are 82% identical.177 The GHRH-R protein has 47%, 42%, 35%, and 28% sequence identity with receptors for VIP, secretin, calcitonin, and parathyroid hormone, respectively.177,178 The isolated cDNAs encoded a 423 amino acid protein that has seven putative transmembrane domains and a 108 residue extracellular N-terminal domain (after signal peptide cleavage) containing one glycosylation site. The GHRH-R sequence predicts ten extracellular cysteine residues that also are found in the secretin, VIP, and PACAP receptor subgroup; eight of these ten are also conserved in most members of the wider receptor family B. These cysteine residues are proposed to form sulfhydral cross-links that stabilize an extracellular domain involved in hormone binding.180
Cloned pituitary GHRH-R expressed in cell lines demonstrated saturable, high-affinity, GHRH-specific binding and stimulated the accumulation of intracellular cAMP in response to physiologic concentrations of GHRH.177–179 Unlike some related receptors that could signal through both cAMP : PKA and phospholipase C : IP3:PKC pathways, only cAMP activation could be detected, although stimulation of phospholipid turnover was noted in somatotrope cells. A specific GHRH antagonist blocked both binding and second messenger responses.
Data from the cloned receptor were consistent with photoaffinity cross-linking studies of GHRH-R in sheep pituitary membranes that revealed high-affinity binding sites with an apparent molecular weight of 55 kDa and one glycosylation site. After deglycosylation and taking into account the mass of the coupled GHRH analogue, the molecular weight (MW) of the native ovine receptor protein was estimated at 42 kDa,181 in agreement with the prediction from the human cDNA sequence of 45 kDa, assuming cleavage of a signal peptide.178 Further, the binding characteristics of the natural sheep receptor and the cloned human receptor are largely in agreement with a single high-affinity site with a Kd of ≈0.2 nM.
Various radiolabeled forms of GHRH bind to membranes of the pituitary, thymus, and spleen. The dissociation constants estimated in these studies vary wildly from 41 pM182 to 590 nM.183 No binding was measurable in three nonfunctional pituitary adenomas, although consistent GHRH binding to five acromegalic adenomas was seen, with dissociation constants averaging 0.3 nM.184
Studies that attempted to delineate the receptor’s GHRH binding domains using chimeric receptor constructs180 or GHRH cross-linking185 suggest that although the large N-terminal extracellular domain plays a major role in GHRH binding, other domains are also essential for ligand selectivity and binding. In vitro studies of a naturally occurring receptor truncation mutant186 and a receptor truncation resulting from alternative splicing187 show a dominant negative effect on GHRH signaling, suggesting that the receptor may function as a dimer .
GHRH Receptor mRNA and Gene
Two GHRH-R mRNA transcripts of approximately 2.5 and 4 kb were identified in rat pituitary, as were 2.0 and 2.1 kb in mouse and 3.5 kb in ovine pituitary.177–179 Further, in mouse, the receptor is expressed in a spatial and temporal pattern that corresponds to expression of GH.179 In the mouse, the first evidence of POU1F1 (a pituitary specific transcription factor, also called Pit-1 or GHF1) expression occurred at embryonic day 14.5, while transcripts encoding the cloned receptor first appeared on embryonic day 16.5.179 Mutations that cause a loss of POU1F1 expression, such as in dw/dw mice, lead to a lack of GHRH-R gene expression and somatotrope hypoplasia.179
The human GHRH-R gene is divided into 13 exons separated by variably sized introns that spread its length to over 15 kb, the complete sequence of which is known.188 Fluorescent in situ hybridization localized the gene to human chromosome 7p14-15.189,190 Studies of the promoter region of the receptor gene found no traditional initiator motifs such as a TATA box.191,192 Putative binding sites for several transcription factors, including POU1F1, Oct-1, Brn-2, NF-1, cAMP response element (CRE), and estrogen receptor response elements (EREs), were identified. An in vitro reporter system demonstrated that expression was enhanced by POU1F1 and glucocorticoids, and was inhibited by estrogen.191 POU1F1 stimulation is consistent with previous studies in Snell and Jackson dwarf mice showing POU1F1 dependence of receptor expression179; five different POU1F1 sites have been shown to contribute to this POU1F1 dependence.193 The glucocorticoid effect on the promotor may be the mechanism for glucocorticoid upregulation of GHRH binding sites194 and receptor mRNA195,196 in rats. The estrogen inhibition of promotor transcription is consistent with observed sexual dimorphism in receptor mRNA expression.197 Studies have suggested that GHRH-R expression is upregulated by GHRH itself198–200; although a putative CRE that could explain this effect was found in the receptor gene promoter, in vitro regulation of the promoter by forskolin could not be demonstrated.191
The structure of the rat GHRH-R gene201 closely matches that of the human but includes an additional exon that would predict an alternatively spliced receptor message that encodes 41 additional amino acids in the third intracellular loop. Indeed, both rat and mouse GHRH-R cDNA clones for this long form have been isolated,177,179 although analysis of rat pituitary mRNA by PCR found evidence of the shorter form only.177 Alternative splicing at the homologous site in the PACAP receptor results in functional receptors that differ in their relative signaling through cAMP and phospholipase C second messenger pathways.202 When the long form of the rat GHRH-R is stably expressed in cell lines, it binds GHRH, but no intracellular signaling could be detected.201
In mice, evidence is seen of an alternative splice variant that encodes a receptor devoid of the first transmembrane domain.179 Alternatively spliced GHRH mRNA encoding a receptor lacking the last two transmembrane domains has been reported in human pituitary tumors and in normal pituitary.203 In the rat, an alternative splice replacing the last five amino acid residues at the C terminus with a new 17-residue sequence has been found by PCR204; this receptor variant appears to signal cAMP normally. No functional role for any of these alternatively spliced GHRH-R messages has been established, although it has been proposed that a truncated receptor variant expressed in tumors can act as a dominant negative in inhibiting GHRH signaling.187,205
Synthetic GHRH antagonists can inhibit the growth and proliferation of a variety of human tumors and tumor cell lines,206–208 which is consistent both with the hypothesis that GHRH can act as a local autocrine/paracrine factor in the stimulation of cell growth209 and with the mitogenic actions of GHRH at the somatotroph.210 The mechanism underlying this action has been unclear because a full-length GHRH receptor could not be detected in the cell lines that responded to GHRH antagonist.208 However, many tumor cells and also normal human prostate express low levels of alternatively spliced forms of GHRH receptor messages not found in the pituitary.208,211 Indeed, a receptor splice variant with an alternative N-terminal domain (SV1) may be the site of action for the antiproliferative effects of GHRH antagonists,212–214 as well as for ligand-independent stimulation of tumor growth.215
GHRH-R mutations resulting in dwarfism have been identified in mouse and humans. The first such mutation was found in the little mouse, a dwarf strain that has an inherited autosomal defect resulting in low levels of GH and pituitary hypoplasia, but is still responsive to exogenous GH. Pituitary cells from these mice would not respond to GHRH but could release GH in response to other activators of cAMP, suggesting a receptor defect.216 After cloning of the GHRH-R cDNA, the mouse gene was localized to the midregion of chromosome 6.217,218 Subsequent sequencing of the receptor demonstrated an Asp-to-Gly point mutation at residue 60 of the receptor’s extracellular domain, which is highly conserved in related receptors, mutation of which resulted in complete loss of cAMP signaling. Additional studies demonstrated that this mutant receptor was properly expressed and localized in the cell membrane, but was unable to bind GHRH.219
In humans, a variety of loss-of-function GHRH-R mutations have been identified in patients with GH deficiency and proportionate short stature.220 Two such mutations have been found in large kindreds—one in three distantly related families from India, Pakistan, and Sri Lanka, and another in a Brazilian kindred with more than 100 affected individuals.221 Additional receptor mutations continue to be identified, and it is now suggested that 10% of all human familial isolated GH deficiency type 1 is caused by GHRH-R defects.222 Affected individuals are homozygous for point mutations or deletions in the receptor protein coding region, at an intron splice junction, or in the gene’s promoter. In addition, some affected individuals are compound heterozygotes with two different loss-of-function mutations. Individuals heterozygous for an inactivating missense point mutation of the GHRH-R gene did not show a significant reduction in adult stature or in serum IGF-1, but changes in body composition and possibly an increase in insulin sensitivity were reported.223 In contrast to this, two different receptor truncations have been shown to have dominant negative properties in vitro186,187; thus truncation mutations could potentially affect heterozygotes. It is proposed that activating receptor mutations could be associated with acromegaly or adenoma, but screening of pituitary tumors for activating mutations so far has yielded only ambiguous224 or negative results.225,226
GHRH Intracellular Signaling
At the somatotrope, GHRH activates many of the classical signaling systems, including cAMP, calmodulin, calcium mobilization, and phospholipid pathways, indicating a significant commitment of the somatotrope to respond to GHRH. As with many secretory cells, GHRH-accelerated GH release requires both calcium227 and calmodulin.228,229 Intracellular calcium is elevated within seconds of a GHRH stimulus, both in pituitary cells230 and in thymocytes.231 This calcium response is dependent on influx of extracellular calcium rather than on the release of intracellular stores.232 cAMP also signals to the cell nucleus, regulating gene expression via multiple transcription factors.233
cAMP Metabolism
At the somatotrope, GHRH stimulates cAMP accumulation and GH release, and these responses are blocked by somatostatin.234,235 Glucococorticoid pretreatment enhances both the potency and the efficacy of GHRH in driving cAMP accumulation and GH release236; this steroid is necessary for GHRH-induced cAMP accumulation after several days in culture. Adenylate cyclase activity in membranes of normal rat pituitary or human acromegalic tumor237 is enhanced by GHRH in a guanine nucleotide– and calmodulin-sensitive manner.229 Pertussis toxin enhances GHRH-initiated cAMP accumulation and GH release.234 The spontaneous reduction in GHRH-stimulated cAMP levels that occurs over time can be blocked by cycloheximide,234 and the stimulatory ability of GHRH is potentiated by protein kinase C activation.238,239 This indicates that another receptor system that stimulates C kinase may directly enhance the productivity of the GHRH-R–coupling protein-adenylate cyclase complex,240 a candidate for which is the ghrelin (growth hormone secretagogue) receptor.239
Phospholipids
GHRH increases phosphatidylinositol labeling241 and free arachidonate levels242 in the pituitary. Although in most systems, no effect of GHRH on polyphosphoinositide hydrolysis is detectable,243 a report shows that in a specific subclass of porcine somatotropes (low-density somatotropes), GHRH stimulates both cAMP- and inositol phosphate–dependent second messenger pathways.244 Other metabolic pathways involving phospholipid metabolism may be activated by or may modulate GHRH activity.238,245 cAMP metabolism can be dissociated from GH release after GHRH with some phospholipid metabolic enzyme inhibitors, indicating that they may act distal to the cAMP system to evoke exocytosis.238
Mitogenic Signaling
In vivo, insufficient GHRH signaling during development through a GHRH-R defect218 or as the result of GHRH antisera administration246 results in somatotrope hypoplasia. Excess GHRH signaling through tumor expression,116 Gs mutation,247 or transgenic overexpression248 stimulates somatotrope hyperplasia. In vitro, GHRH is a mitogenic signal for somatotrope proliferation.210 The MAPK pathway is a potential mechanism for this action. GHRH dose dependently stimulates tyrosine phosphorylation of MAPK.249–251
GH mRNA and Release Dynamics in Culture
GHRH stimulates the level of GH mRNA,252 the release of newly synthesized GH253 and total GH (stored plus released),254 and the proliferation of somatotropes in vitro.210 The GHRH effect on the somatotrope varies according to the anatomic location of the somatotrope within the pituitary,255 and the GH-releasing effect is further enhanced by acute administration of glucocorticoids,256 possibly through increased GHRH binding.194 Like glucocorticoids, triiodothyronine,257 ghrelin,258 and ghrelin mimetics239 can amplify GHRH-stimulated GH secretion. In contrast, IGF-1256 and somatostatin259 are noncompetitive inhibitors of GHRH-accelerated GH release in vitro.
Accelerated GH release occurs immediately after exposure to GHRH234 and remains elevated for the duration of the GHRH pulse,234 albeit at declining rates of release after about 10 minutes.260 This spontaneous decline could occur without GH content depletion260 and could be blocked by cycloheximide, suggesting the participation of a rapidly turning over inhibitory protein.229 The reciprocal interaction of GHRH and somatostatin, as suggested neuroanatomically and by pituitary portal blood measurements,168 results in a greater mass of GH release per GHRH pulse. This has been demonstrated in perifusion culture.261
Picomolar to nanomolar concentrations of GHRH that likely are present in pituitary portal blood262,263 regulate a graded GH response from the somatotrope.118 GHRH also stimulates modest prolactin release at low GHRH concentrations in vitro,264,265 as well as the secretion of a protein known as peptide 23 (identical to pancreatitis-associated protein and a member of the C-type lectin supergene family).266,267 Because GHRH can interact with VIP receptors in intestinal epithelium268 and GH3 cells,269 it is possible that pharmacologic or pathologic levels of GHRH can activate this and other receptor types.
Animal Studies
The pulsatile release of GH is influenced by numerous factors, including nutrition, body composition, metabolism, age, sex steroids, adrenal corticoids, thyroid hormones, and renal and hepatic functions.168 A major common pathway for these factors is seen in their effects on GHRH release from the hypothalamus through direct actions on GHRH neurons, and also through interplay with somatostatin neurons. These effects may be mediated through other factors such as ghrelin, catecholamines, interleukin-1, somatostatin, opioids, leptin, inhibin, and neuropeptide Y (NPY).270,271
GHRH Effects on the GH Axis
GHRH was first demonstrated to stimulate GH release in vivo in anesthetized rats.272,273 These GH responses to GHRH could be enhanced by passive immunization against somatostatin or blocked by passive immunization against rat GHRH.168 It was soon found that GHRH could enhance GH secretion in every vertebrate species tested, including mammals, birds, and fish.
GHRH is necessary for endogenous pulsatile GH secretion, as anti-GHRH antisera treatment eliminated these pulses in rats and sheep.168 However, rhythmic GH secretion persists in an amplitude-miniaturized version in the absence of a GHRH-R signal, at least in men.274 This observation is supported by a gender-specific difference after administration of a GHRH antagonist, with no change in basal secretion in men but a significant decrease in women.275 Antisera to GHRH also decreased statural growth276 and GHRH-R mRNA expression199 in the rat. Conversely, GHRH administered over several days to weeks enhanced body or organ growth and function in experimental animals.277 The effect is particularly striking in mice transgenic for GHRH.278,279 Pulses of GHRH are measured in pituitary portal plasma of unanesthetized sheep with peak values of 25 to 40 pg/mL for a period of 71 minutes.263 Temporal analysis of GH pulses in sheep suggested a complex regulation that can be explained only partially by GHRH and somatostatin pulses and ghrelin.239,280
During development, basal GH responses to exogenous GHRH decrease from postnatal day 1 to day 28 in the rhesus monkey.281 Passive immunization against GHRH shows that endogenous GHRH is an active secretagogue up to day 9.282 In the rat, GHRH injections do not increase GH levels at postnatal day 2,283 whereas stimulatory responses of similar magnitude are measured at postnatal days 10, 30, and 75, as well as at 14 months.284 Likewise, GHRH injections given over 5 days elevate GH biosynthesis in rat pituitaries at postnatal day 10.285 Rat pituitary GHRH-R mRNA expression was highest in early gestation, declined to a nadir at 12 days of age, increased at the onset of sexual maturation, and then declined with aging.286
Significant changes in GHRH status are noted during aging. In the hypothalamus, a reduction in GHRH gene expression and content is seen,287 as is a decrease in GHRH binding to pituitary in 18-month-old rats.288 GHRH-R mRNA is correspondingly decreased in 18-month-old rats but can be brought back toward levels observed in younger animals through treatment with GHRH.289 Decreased GHRH-R expression may contribute to the diminished pituitary response to GHRH in aged male rats290 and humans.291
The GHRH system is also strongly influenced by gender (or gonadal steroids). Hypothalamic GHRH mRNA levels are greater in male than female rats292,293 and are reduced by orchidectomy and increased by testosterone treatment in intact294 or castrated295 male rats. Estradiol has no effect on hypothalamic GHRH mRNA.296 The ability of GHRH to elicit a GH response in vivo varies during the rat estrous cycle297; it is of greater magnitude in the male than in the female284,298 and is strongly sex steroid dependent.298 Somatotropes from male rats likewise have greater cAMP and GH responses to GHRH than do those from female rats when studied in static299 or perifusion298 cultures. Furthermore, the intact and castrate male rat treated with testosterone yields the greatest quantity of GHRH-responsive somatotropes,298 as does direct testosterone treatment of cultured somatotropes.300 Gender differences can be measured at the level of the single somatotrope using the hemolytic plaque assay301; testosterone (administered in vivo) increases secretory capacity and recruits a subpopulation of somatotropes, while estradiol has the opposite effects.302 GHRH receptor message levels are dramatically lower in female than in male rats,168 and estrogen acts at the receptor gene to inhibit GHRH-R mRNA expression.191
In addition to sex steroid effects, free fatty acids303 and GH itself304 reduce the in vivo release of GH in response to GHRH. GH treatment decreases hypothalamic GHRH content in intact rats293 and after hypophysectomy GHRH levels in the hypothalamus rise,305 suggesting feedback regulation of GHRH by GH. Thyroxine replacement can restore hypothalamic immunoreactive GHRH levels reduced by thyroidectomy in rats,168 and triiodothyronine and cortisol can protect against reduced GHRH-stimulated GH release in hypothyroid306 or adrenalectomized rats,307 respectively. Indeed, long-term glucocorticoids in vivo decrease GHRH expression in GHRH neurons of the arcuate nucleus.308
Months of excess GHRH exposure in transgenic mice is associated with increased pituitary mass and mammosomatotrope hyperplasia278 that eventually results in adenoma formation after 12 months.309 This is reminiscent of the clinical findings in patients with ectopic GHRH secretion. What was surprising was the rapidity of this effect; GHRH infusions were capable of inducing enlargement of the anterior pituitary within days in intact, normal rats.310 The dose range of this acute effect has yet to be defined, and this observation does not address the potential risks of replacement of GHRH in deficiency states. In pituitary allograph studies, in orchidectomized hamsters, exogenous GHRH maintains somatotrope size without affecting the percent of GH cells.311
GHRH Effects on Functions Outside of the GH Axis
GHRH stimulates gastrin release and epithelial cell proliferation in the digestive tract.312 It also stimulates insulin, glucagon, and somatostatin secretion from the pancreas.313–315 GHRH enhances non–rapid eye movement sleep in rats.316 GHRH antisera317 or GHRH antagonists318 inhibit sleep, and sleep deprivation enhances hypothalamic GHRH mRNA levels.319 GHRH-Ab treatment of female rats results in osteopenic effects,320 and plasmid-mediated GHRH expression in an animal model of cancer cachexia was able to prevent weight loss.321 Most of these activities, including control of GH status, suggest that GHRH predominantly acts as a nutrient-partitioning hormone, to regulate body composition.
Circulating GHRH in Humans
Following the initial synthesis of GHRH, radioimmunoassays (RIAs) were used to demonstrate that, contrary to hopes that GHRH in the peripheral circulation would be principally of hypothalamic origin and its measurement would thus serve as an index of hypothalamic secretion, most circulating GHRH is not of hypothalamic origin, but instead comes from the gut.322 Further, an RIA ideally would measure intact biologically active hormone. However, as the result of cleavage by DPP-IV, GHRH(1-44)-NH2 has a very short half-life in the circulation of 6.8 minutes, and the metabolite GHRH(3-44)NH2 appears within 1 minute of an IV injection of GHRH(1-44)NH2.323 The biological activity of GHRH(3-44)NH2 is less than 10−3 that of GHRH(1-44)NH2.138 Unfortunately, most RIAs measure GHRH(1-44)-NH2 and GHRH(3-44)-NH2 with equal efficiency, and measurements therefore do not reflect biological activity in the circulation. Most assays are directed to the midportion of the GHRH molecule and therefore do not distinguish between different circulating forms. Besides the RIA, more sensitive enzyme immunoassays for GHRH measurement have been developed.324 One of the few indications for measuring serum GHRH is a GHRH-producing tumor.
GHRH Levels in Acromegaly
Interest in the frequency of ectopic GHRH as a cause of acromegaly has been intense. Two extensive studies have addressed this issue. In a study of 80 patients with acromegaly, 76 had GHRH levels in the normal range325; of the four with elevated levels, one was known to harbor a GHRH-secreting tumor. Extensive evaluation of the other three failed to determine a source for the GHRH. In a second study, 3 of 177 patients with acromegaly exhibited elevated serum levels of GHRH.326 In all cases, GHRH levels were markedly elevated (i.e., in the nanogram per milliliter range), and patients were known to have had previous GHRH-secreting tumors. Thus, although apparently rare, ectopic secretion of GHRH must be considered as a possible cause of acromegaly, and measurement of peripheral GHRH seems prudent as a part of the evaluation. Because it is known that the release of ectopic hormones may be intermittent, and because only 300 pg/mL of GHRH is necessary to stimulate GH release in normal subjects, a single normal or modestly elevated GHRH determination may not exclude ectopic GHRH-associated acromegaly.
The subject of GHRH-producing tumors has been reviewed previously.327,328 GHRH-producing tumors associated with acromegaly are rare. Unique features of patients with acromegaly harboring tumors secreting GHRH included young age, female preponderance, foregut derivation of the tumors, benign biological behavior, small secretory granules in the tumor, and frequent association with multiple endocrine neoplasia type 1 (MEN-1) syndrome. The pancreas and the lung are common primary sites. GHRH-containing tumors that are not associated with acromegaly include those of the gut and thymus, small cell carcinoma of the lung, and medullary carcinoma of the thyroid. Several tumors are plurihormonal. In contrast to somatotroph adenoma as seen in patients with classic acromegaly, the hypophyseal lesion represents somatotroph hyperplasia in acromegalic patients with GHRH-producing tumor. This finding indicates that GHRH not only increases somatotroph secretory activity, but causes somatotroph proliferation. Studies of GHRH-producing tumors are of fundamental importance for obtaining insight into endocrine activity of pituitary somatotrophs and the pathogenesis of GH-secreting pituitary adenomas associated with acromegaly; the importance of GHRH in the origin of acromegaly is still unresolved. Preliminary evidence suggests that the amount of GHRH mRNA expression seen in somatotroph adenomas is associated with the progression and aggressiveness of these tumors.171 The GHRH receptor mRNA is specifically expressed in GH-producing adenomas and somatotrophs.170 To address whether GHRH can produce tumors, transgenic mice expressing the human GHRH gene have been developed. These animals, which had been exposed to excessive quantities of GHRH throughout development and life, developed mammosomatotrope or somatotrope adenomas.329 The significance of these observations in terms of human disease is unclear, and additional studies are needed. Early studies have investigated the beneficial effects of GHRH antagonists in animals and humans. It is interesting to note that in one single reported clinical case, ectopic GHRH secretion was associated with empty sella syndrome.330
Treatment of transgenic mice overexpressing the human GHRH gene with GHRH antagonists resulted in suppression of GH and secretion of IGF-1.331 The relationship between GHRH-secreting tumors and MEN-1 syndrome is controversial; additional studies are required to elucidate whether they represent two distinct entities, or whether GHRH-producing tumors accompanied by acromegaly are only forme fruste manifestations of MEN-1 syndrome. Several cases of acromegaly due to ectopic GHRH secretion associated with MEN-1 syndrome have been described.332–336
Eutopic GHRH Secretion
Occasionally, hypothalamic gangliocytomas may be associated with acromegaly; immunocytochemical staining of such tumors for GHRH has been described. It has been suggested that these tumors should be considered as an unusual cause of acromegaly.337 On occasion, these tumors are intrasellar; in such cases, the observation that somatotrophs are in close anatomic association with neurons suggests that GHRH not only stimulates GH secretion but may also cause adenoma formation. An intrasellar gangliocytoma with somatotroph adenoma has been described, which was strongly positive for gastrin and weakly positive for GHRH. Because gastrin administered intracerebroventricularly increases GH secretion, it has been suggested that gastrin release may act in a paracrine fashion on gangliocytoma to enhance GHRH secretion and thus cause somatotrope adenoma.338
GHRH Levels in GH-Deficient Children
Many reports describing serum and cerebrospinal fluid (CSF) concentrations of GHRH in children with various forms of growth deficiency have appeared. In 22 children with the diagnosis of constitutional short stature (defined as 2 to 3 SD below the predicted mean height for age, peak levels of GH in excess of 10 µg/L during at least one provocation test, and bone age approximating chronological age), basal GHRH levels (8 to 148 pg/mL) were no different from those noted in normal children.339 In addition, in five of nine children, GHRH levels rose twofold 15 minutes after administration of levodopa (500 mg po). In another study of 16 children with idiopathic delayed puberty, the peak serum GHRH concentration following levodopa was 41 ± 10 pg/mL, and this compared with 96 ± 25 pg/mL in children with constitutional short stature.340 Similarly, in patients with hypothalamic hypopituitarism, no increase in circulating GHRH levels was seen after levodopa, a finding that contrasts with responses in normal subjects.341,342 These patients with hypothalamic hypopituitarism do respond to exogenous GHRH administration. Insulin-induced hypoglycemia increased circulating GHRH levels in normal subjects, but not in six patients with isolated GH deficiency.343 In ten children with short stature, GHRH levels increased at 15 minutes after hypoglycemia from 10 ± 0.5 to 17.1 ± 3.1 pg/mL.344 No increase in GHRH was evident after arginine, even though hypoglycemia alone or arginine alone increased GH concentrations.
However, in contrast to children with constitutional short stature, five children with GH deficiency associated with hypothalamic germinomas were reported to have undetectable concentrations of GHRH in the CSF.345 In addition, children with idiopathic GH deficiency have GHRH present in the CSF but at concentrations lower than those in normal children (15.1 ± 1.0 vs. 29.3 ± 2.0 pg/mL; mean ± SEM).
GHRH as A Diagnostic Agent
Until 1985, the use of cadaveric GH was strictly controlled and was limited to use in children with short stature due to severe GH deficiency. Dynamic tests of GH reserve were therefore of clinical significance in pediatric endocrinology and were performed primarily for academic interest in adults with hypothalamic-pituitary disease. With the advent of recombinant human GH, now available in unrestricted quantities, and its approval for use in adults with GH deficiency resulting from hypothalamic-pituitary disease in the United States, Europe, and Australia, the need to diagnose GH deficiency safely and effectively has become an important issue in adult endocrinology.346
Many of the tests used to determine GH status are hazardous under certain circumstances (the insulin tolerance test [ITT] is contraindicated in the elderly and in patients with ischemic heart disease or a history of seizures) or are associated with unpleasant side effects (glucagon causes nausea and delayed hypoglycemia, clonidine is associated with drowsiness and hypotension, and arginine causes dizziness and phlebitis). Tests that are effective in children, such as those using arginine or clonidine as the stimulus, are less effective at releasing GH in adults.347 The ITT is frequently quoted as the gold standard investigation for diagnosing GH deficiency in children and adults, but many endocrinologists shy away from this procedure because of concerns about the effects of hypoglycemia in their patients, and because the test has to be supervised by a physician for its duration.
Administration of GHRH to Normal Individuals
The effects of GHRH and its analogues given as a bolus have been studied in healthy men, women, and children.348–358 Following an IV injection of GHRH, GH levels begin to rise within 5 minutes and reach a peak at between 30 and 60 minutes. The ability of GHRH to release GH is dose dependent,357 the maximal response being observed following a dose of 1 µg/kg or higher.352 In adults, the GH response to GHRH is similar in men and women, although women are more sensitive to GHRH than men; the dose of GHRH required for half-maximal GH secretion is 0.4 µg/kg in men and 0.2 µg/kg in women. The GH response to GHRH in women is not altered by the changes in sex steroid hormones that occur during the menstrual cycle.356
The effect of pubertal development on the GH response to GHRH is slight.359,360 When 68 prepubertal children were compared with 66 children at various stages of puberty, no overall difference in GH response to GHRH was observed.359 In a more detailed study that examined children at each stage of puberty, a slight decrease in GH response to GHRH was seen in boys during midpuberty. Although a similar decrease was not observed in girls, the GH response to GHRH did not differ significantly between the sexes during puberty.360 In prepubertal children who are being evaluated for poor growth, priming the hypothalamic-pituitary-GH axis with sex steroids can normalize a suboptimal GH response to some stimuli. The GH response to GHRH is not affected by priming with estrogen,361 suggesting that sex steroids assert their effects at the hypothalamus, either reducing somatostatin tone or increasing GHRH release—not at the pituitary.
The ability of GHRH to release GH is similar in prepubertal and pubertal children, and in young adults. However, over the course of the adult life span, the magnitude of the GH peak following GHRH declines with increasing age.348,362 The likely mechanism for the age-related decline in response to GHRH is an increase in somatostatin tone.363 Support for this is found in studies that utilize GHRH in combination with pyridostigmine or arginine.364,365 These agents have been used alone as diagnostic tests for GH deficiency, producing a GH pulse by reducing somatostatin tone.366,367 Arginine and pyridostigmine act synergistically with GHRH, producing large GH pulses similar to those seen in younger adults.
The GHRH Test
With any test used in clinical practice, it is important to determine what constitutes a normal response. Accordingly, several studies have addressed this question for the GHRH test. Ranke et al.368 defined the normal range of GH responses to GHRH in 86 children with a normal GH axis as 11.8 to 172.4 µg/L, by determining the mean response ±2 SD. This study also concluded that a GH response of less than 10 µg/L should be used as the diagnostic threshold for GH deficiency in children.
The diagnostic threshold of 10 µg/L has been used in other studies utilizing the GHRH test369–371 and now is generally accepted in pediatric practice. This peak is similar to that used to define GH deficiency when most stimuli are used in children, although it is recognized that different stimuli are not equal in their ability to release GH.359 Such thresholds are defined arbitrarily, despite attempts to rationalize them.372 This may reflect the fact that the diagnosis of GH deficiency in a child depends primarily on the clinical finding of poor growth, and that the stimulation test is used to confirm the presence of GH deficiency.
In adults, the diagnostic threshold used to identify patients with GH deficiency is lower than that used in pediatric practice. For example, a GH peak of less than 3 µg/L during an ITT is considered to be indicative of severe GH deficiency that warrants therapy with exogenous GH. If arginine were to be used as the stimulus, the diagnostic cutoff would be even lower than this.373 In adults, the GHRH test is generally a more potent stimulus of GH secretion, resulting in higher pulses than those seen following ITT, arginine, or glucagon.374 This would suggest that the diagnostic cutoff for severe GH deficiency might be higher than 3 µg/L when the GHRH test is used in adults, but this has not been determined.
The GHRH Test in Children With Short Stature:
Normal Hypothalamic-Pituitary-GH Axis.: The GH response elicited by GHRH in children with short stature due to a variety of causes has been well characterized.368,369,375–383 Children who have constitutional short stature with no underlying pathology have a normal GH peak following GHRH administration,377,383 but the timing of that peak may be delayed.378 Children who are short as a result of intrauterine growth retardation also have a normal GH response to GHRH.
GH Deficiency.: Children with GH deficiency defined by the use of conventional tests frequently have a greater response to GHRH than to other tests of GH status.375 In a study of prepubertal children undergoing investigation of abnormal growth, subjects were divided into groups according to their responses to conventional tests. GH status was considered normal if the peak GH response to a conventional test was greater than 10 µg/L; partial and severe GH deficiency were defined as a peak GH response of between 5 and 10 µg/L and less than 5 µg/L, respectively. When a diagnostic cutoff of 10 µg/L is assumed for the GHRH test, 76% of children with partial and 39% with severe GH deficiency had a GH peak greater than 10 µg/L during the GHRH test. Conversely, 10% of children considered to have a normal GH axis had a peak GH response less than 10 µg/L—a figure consistent with the findings of other studies. This study demonstrated a considerable discordance between conventional tests of GH status and the GHRH test. It also indicated that, in a significant proportion of children with GH deficiency diagnosed clinically and confirmed by conventional tests, somatotroph function may be preserved, with the cause of impaired GH status being hypothalamic rather than pituitary dysfunction.
Patients who develop GH deficiency following cranial irradiation also exhibit discordance between the GHRH test and conventional tests of GH status. The GH response to GHRH in such patients was greater than the response to the ITT and the arginine test in 80% of patients in one study.384 This suggests that radiation primarily affects the hypothalamic mechanisms that regulate GH secretion from the anterior pituitary. The magnitude of the GH peak during the GHRH test decreases as time from radiation increases. This may be a direct effect of radiation on the somatotrophs or the indirect effect of long-standing GHRH deficiency, which depletes the available GH pool within the somatotroph. The latter is supported by the observation that priming with GHRH for several days prior to the GHRH test can significantly increase the GH response to GHRH.385
GHRH and the Diagnosis of GH Deficiency in Adults
Limitations of the GHRH Test: The use of GHRH alone as a diagnostic test for GH deficiency is limited by several factors. The discordance observed between the results of the GHRH test and the results of other stimulation tests such as the ITT can lead to difficulties in interpreting the results of the GHRH test. Considerable interindividual and intraindividual variation has been noted in results of the GHRH test. The coefficient of variation for the GHRH test has been reported as 60% for children386 and 45% for adults,374 although the variability diminishes with increasing age in adults.387 The sensitivity of the GHRH test is relatively poor in children with short stature and varies with the severity of the GH deficiency as determined by other tests of GH status. In one study, the sensitivity of the GHRH test was 24% in patients with GH insufficiency (GH peak 5.0 to 10.0 µg/L to conventional tests) and 61% in patients with severe GH deficiency (GH peak <5.0 µg/L).375 The specificity of the GHRH test has been reported to be 85% to 90%.359,375 The ability to interpret the GHRH test is confounded further by the inhibition of the GH response in obese subjects.388,389 This may be particularly important when patients with GH deficiency are assessed, because they have abnormal body composition with a propensity to abdominal obesity. The GH response to GHRH in obese patients with pituitary tumors is reduced, making it difficult to define their GH status accurately, particularly when the GH deficiency is isolated.390
Tests Utilizing GHRH in Combination With Other Agents
The observation that GHRH acts synergistically with agents that reduce somatostatin tone has led to the development of tests combining GHRH with arginine, pyridostigmine, or clonidine. GHRH acts synergistically with these agents, producing large GH pulses, the magnitude of which frequently exceeds 50 µg/L in healthy subjects.359,374,391 The addition of these agents to GHRH increases the reproducibility of the test and improves diagnostic accuracy.
GHRH in combination with pyridostigmine or arginine causes profound release of GH. In one study of normal children and adolescents, the normal range for GHRH plus pyridostigmine was 22.6 to 90.0 µg/L (n = 94), and that of GHRH plus arginine, 22.4 to 108 µg/L (n = 81).359 The results of these tests were not influenced by pubertal stage in either girls or boys.359,392 However, a study by Maghnie et al.393 concludes that in patients with acquired childhood-onset GH deficiency, the number of pituitary hormone deficits and patient age affect the GH response to the combined GHRH and arginine test. Additional study suggests that body mass index (BMI) and age do have an impact on the GHRH arginine test in children with idiopathic GH deficiency.394 Both studies were conducted with a small number of patients.
In children with GH deficiency, the GHRH-pyridostigmine test, used with the diagnostic threshold of 20 µg/L, confirmed the diagnosis in 100% of patients with organic GH deficiency (caused by craniopharyngioma) and in 80% of patients with idiopathic GH deficiency.395 GHRH in combination with arginine has been extensively evaluated in adults with pituitary disease in the hope that it will provide a safer alternative to the ITT, which currently is considered to be the gold standard investigation of GH status in adults. In assessing GH secretion, no difference in sensitivity and specificity has been noted in young adults among the Arg + GHRH test, the PD + GHRH test, and the ITT.396 The combination of GHRH with arginine was chosen over pyridostigmine because the side effects with pyridostigmine were unpleasant and the GH response to GHRH plus arginine is not affected by age.397 In adults, the GH response to GHRH and arginine is not affected by gender or age, as similar results are achieved in male and female subjects and in young and old adults.391,397,398 Across the adult life span, the third percentile limit of GH response to GHRH plus arginine is 16.5 µg/L, and the first percentile limit is 9 µg/L.
Adults with GH deficiency all had a peak GH response to GHRH plus arginine that fell below 16.5 µg/L, and 92% of patients had a peak below 9 µg/L. GH peaks achieved during the GHRH-arginine test correlate well with those obtained during the ITT, although the absolute GH response to GHRH plus arginine is considerably greater than that to the ITT. Of seven patients who had achieved a peak GH greater than 9 µg/L during the GHRH-arginine test, six had achieved a GH peak greater than the diagnostic threshold for the ITT (3 µg/L). Thus, the authors proposed that the diagnostic cutoff for the GHRH-arginine test should be 9 µg/L for severe GH deficiency in adults and 16.5 µg/L for GH insufficiency. Comparison of the sensitivity and specificity of six different tests—ITT, arginine (ARG), levodopa (L-DOPA), ARG + L-DOPA, ARG + GHRH, and IGF-I measurement—in the diagnosis of GH deficiency found the greatest diagnostic accuracy with the ARG + GHRH test and the ITT. The former was preferred over the latter because it produced fewer side effects.399 The GRS consensus of 2007400 suggested that GHRH + Arg, GHRH + Ghrelin, and glucagon tests are as sensitive as the ITT in GHD, provided that appropriate cutoff limits are considered. Clinical practice guidelines of the Endocrine Society401 suggest that ITT and the GHRH-arginine tests have sufficient sensitivity and specificity to establish the diagnosis of GHD. In the clinical situation of radiation-induced GH deficiency, ITT should be used instead of the GHRH-arginine test.402 GHRH directly stimulates the pituitary; therefore it can give a falsely normal GH response in patients with GHD of hypothalamic origin (e.g., those having received irradiation of the hypothalamic-pituitary region). In patients with new-onset GHD (within 10 years) (e.g., that due to irradiation), testing with GHRH-arginine may be misleading. The guidelines also suggest that in the presence of deficiencies of three or more pituitary axes, the occurrence of GHD is very likely, and provocative testing is optional. The validated cutoff level for GHD in adults for the ITT and the glucagon test is a peak GH response <3 µg/L; this has not been validated in obesity. For the GHRH + Arg test, the following cutoff levels have been validated, depending on the BMI: <25 kg/m2, peak GH <11 µg/L; 25 to 30 kg/m2, peak GH <8 µg/L; >30 kg/m2, peak GH <4 µg/L.403 The cutoff level validated for the GHRH + GHRP-6 is 10 µg/L in lean patients and 5.0 µg/L in obese patients.404,405
GHRH Therapy in GH-Deficient Children
The potential of GHRH as a therapeutic agent for GH-deficient children has been examined in several studies. Most GH-deficient children with short stature and growth failure have a disorder of hypothalamic regulation of the pituitary rather than a defect of the somatotroph. In these children, injections of GHRH, as well as other GHSs, might be a useful treatment option. One of the first studies published in 1985 reported the use of GHRH (1-40)-OH administered sc with a peristaltic pump every 3 hours to two children with organic hypopituitarism (post-traumatic, hydrocephalus).406 Children received 3-hourly doses of 1 or 3 µg/kg GHRH for 6 months. Both children increased their growth rate by 1.5- to 6.0-fold compared with the growth rate before administration of GHRH(1-40)-OH. The rationale for using this dose regimen was based on the observation that, in growing children, five to nine pulses of GH are detected every 24 hours.
Since that time, several studies have been performed to evaluate the benefits of GHRH therapy in GH-deficient children with different GHRH injection regimens and different doses.385,407–423 However, the groups of children who were treated were not homogeneous, and their diagnoses varied from GH deficiency and short stature to normal variant short stature without GH deficiency. The GHRH preparations used included GHRH(1-40)-OH, GHRH(1-44)-NH2, and GHRH(1-29)-NH2.
GHRH Given by Pump
Response to the administration of GH with a pump varied from 71% to 100%. The growth rate on GHRH therapy varied from 6.2 to 10 cm/yr over the first 6 months and was maintained for up to 5 years. Growth velocity appears to be related to the total daily dosage, which ranged from 10 to 2150 µg/day. So far, no studies have evaluated whether it is the frequency of administration or the total daily dose that has the greatest impact on therapeutic outcome. The first study that looked at administration by pump or single injections of GHRH was published in 1988.407 It described the effects of different routes of GHRH administration in 24 GH-deficient children. GHRH (1-40) was given either by pump every 3 hours sc or every 3 hours sc overnight only. Alternatively, GHRH was given twice daily at a dose of 1 to 4 µg/kg per dose for 6 months. In all three circumstances, the growth velocity increased between 1.8- and 2.9-fold, with greatest effects seen during pump therapy with injections given every 3 hours.
Unfortunately, no long-term comparative studies have examined the growth-promoting effects of GHRH with GH. Two short-term studies (6 months’ treatment) provided conflicting results: One suggested a comparable growth response with GHRH similar to that with GH treatment,408 whereas the other suggested that GHRH was less effective.409
GHRH Given sc Twice a Day407,410–417
The most impressive results with GHRH injections given twice daily were achieved in a multicenter study using GHRH(1-44)-NH2 in 20 GH-deficient children.414 All children responded with accelerated growth velocity, which increased from 3.6 cm/yr before treatment to 8.6 cm after 6 months and decreased to 8.1 cm after 12 months. Ogilvy-Stuart et al.417 reported a significant beneficial effect of GHRH(1-29) given at a dose of 30 µg/kg/day over 1 year, which resulted in a 1.8-fold increase in growth velocity per year in nine children with radiation-induced GH deficiency.
The effects of GHRH(1-29) therapy in growth retardation caused by chronic renal failure were examined in nine children by Pasqualini and coworkers.416 Children were treated conservatively or with dialysis, or underwent renal transplantation, and GHRH(1-29) 52 µg/kg/day was administered for 3 to 6 months. Growth velocity increased from 3.8 to 8 cm/yr.
GHRH Given sc Once a Day
Several groups have investigated the effects of GHRH given sc by once-daily injection.385,418–423 A study investigating 110 GH-deficient children treated with a dose of 30 µg/kg/day of GHRH(1-29)423 showed a significant increase in linear growth velocity, with 4.1 cm/yr measured before treatment and 7.2 cm/yr after 1 year of treatment (Fig. 16-9). The largest number of children was investigated in the GHRH European Multicenter Study,420 which reported on the treatment of 111 GH-deficient children. Height velocity increased from 3.8 cm/yr before to 6 cm/yr during 6 months of treatment. The group of Bozzola419 reported an increase in growth velocity from 3.5 cm/yr to 7.3 cm/yr after 6 months of treatment in 10 GH-deficient children with GHRH(1-44). Optimal growth velocity was observed during the first 9 months of therapy in most studies.418 Lanes and Carrillo422 reported that therapy with GHRH(1-29) in 16 prepubertal GH-deficient children given once daily sc over 12 to 24 months resulted in a significant increase in growth velocity.
FIGURE 16-9 The effect of 6 and 12 months’ growth hormone–releasing hormone (1-29) treatment in growth hormone–deficient children. The height velocities at baseline (triangle) and after 6 months (circle) and 12 months (diamond) of treatment are plotted by increasing baseline values. (From Thorner M, et al: Once daily subcutaneous growth hormone–releasing hormone therapy accelerates growth in growth hormone-deficient children during the first year of therapy. Geref International Study Group, J Clin Endocrinol Metab 81[3]:1189–1196, 1996.)
Which Children Should be Considered for GHRH Therapy?
Adverse Effects: Development of antibodies during GHRH treatment has been described.407,420,423 Treatment-related adverse events, including local injection reactions characterized by pain, swelling, or redness, and headache, flushing, dysphagia, dizziness, hyperactivity, and urticaria, have been reported. Few data on thyroid function have been reported in children with GH deficiency. One study reported an increase in thyroid replacement requirements in one of seven patients424; another study reported a 5% incidence of hypothyroidism.407 The mechanism underlying the change in thyroid function is unclear, but it has been hypothesized that an increase in GH levels results in an increase in somatostatin tone with subsequent inhibition of TSH secretion.425
GHRH Treatment in Adults
Optimal GHRH therapy requires the functional integrity of somatotroph cells.426 Although in GH-deficient adults this is no longer the case, older adults in whom somatotroph hyposecretion is thought to be caused by decreased activity of GHRH-secreting neurons427 still have an intact hypothalamic-pituitary axis. In addition, the pituitary GH-releasable pool in the elderly is comparable with that in young adults.
In fact, the few available studies dealing with the administration of GHRH in adults have been performed in the elderly to investigate whether GHRH treatment could counteract the age-dependent decline in GH. Twice-daily injections of GHRH(1-29) for 2 weeks,428 as well as continuous GHRH(1-44) infusions for 2 weeks, in healthy older men429 partially reversed the age-related decrease in GH, as well as IGF-1 levels. Another study, performed over 6 weeks,430 suggested that administration of GHRH to the elderly might attenuate some effects of aging on muscle strength.
The half-life of GHRH after iv injection is about 10 to 12 minutes, which is one of the limitations of the use of GHRH as a therapeutic. The long-acting analogue of GHRH, CJC-1295, contains full biological activity of GHRH(1-44)NH2. It binds to endogenous serum albumin after sc administration, which prolongs its duration of action, and it is modified by substitution of four amino acids, which makes the compound resistant to proteolytic cleavage. Subcutaneous injection of CJC-1293, another GHRH analogue, in healthy adults resulted in increased GH and IGF-1 levels 1 week after injection, which was explained mainly by the increase in trough levels.431 Another study432 showed that in healthy volunteers aged 21 to 61 years, elevated GH and IGF-1 serum concentrations were present for 6 and 14 days, and that tachyphylaxis did not occur with this compound. The sc administration of another compound, PEG-GHRH, increased GH levels in healthy adults, an effect that was present for 12 hours.433 Overall, additional studies are necessary to assess the safety profile of these compounds and its short-term and long-term effects on body composition.
Ghrelin and Growth Hormone Secretagogues (GHS)
Opiates have long been recognized to stimulate GH secretion. The GHS story evolved from the seminal observation by Bowers and colleagues that met-enkephalin analogues, such as GH-releasing peptide-6, which lacked analgesic activity, preserved their GH-releasing activity.434 Numerous peptide and non-peptide analogues active in GH release were developed (for reviews on analogues see references 435 and 436), and a group at Merck, using one of its synthetic compounds (MK-0677), succeeded in cloning the GHS receptor, at that point an orphan receptor.437 A natural ligand for this receptor, ghrelin, was identified in 1999 as the result of a “reverse pharmacology” process.438 Although the highest concentration of the receptor is found in the hypothalamus and the pituitary, ghrelin was identified from the stomach. At the same time, the ghrelin mRNA sequence was identified from the stomach by another group and was named motilin-related peptide m46.439 Ghrelin, well conserved over 400 million years, has been detected in a number of tissues, including small amounts in the pituitary and hypothalamus, and is a new member of the brain-gut peptides.440 Following earlier reports on positive effects on the appetite of some GHSs, the profound GH-independent orexigenic effects of ghrelin were recognized.441
Another peptide has been identified arising from the proghrelin molecule (Fig. 16-10) named obestatin.442 Considerable controversy surrounds the physiologic effects and the target receptor of this circulating peptide,443 with suggestions of having anorectic effects and inhibitory effects on gastric motility, and GPR39 being the obestatin receptor. Later studies could not reproduce the originally published obestatin activities, and the controversy remains unresolved.
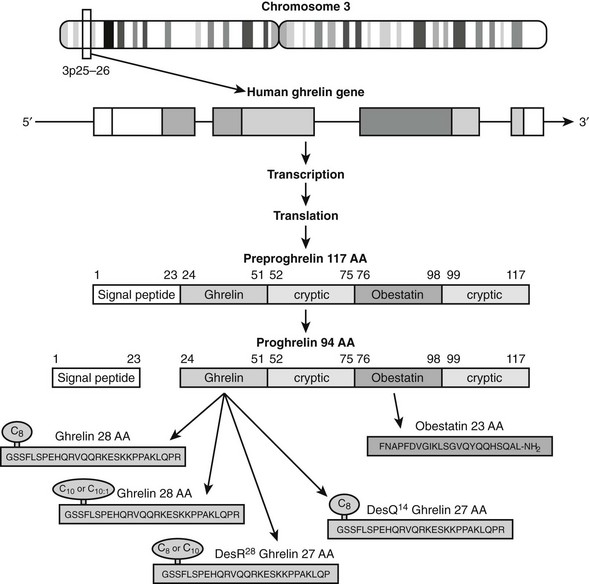
FIGURE 16-10 Schematic diagram of the preproghrelin polypeptide with the post-translational steps that lead up to the cleavage of two products, ghrelin and obestatin. Variants of the ghrelin peptide and its lipid modification are also shown (see text). Numbers represent amino acids (AA); subscript number next to C represents the length of the lipid moiety.
Ghrelin Structure
Ghrelin is a 28 amino acid peptide with a unique fatty acid chain modification on the N-terminal third amino acid (see Fig. 16-10). The acylation of the hydroxyl group of Ser3 is necessary for the activities mediated by the GHS-R1a receptor such as calcium mobilization, GH release, and appetite effects. In contrast, the proliferative, anti-apoptotic, lipogenic, and cardiovascular effects of ghrelin do not appear to require acylation.440 As is shown in Fig. 16-10, several forms of ghrelin are found in the circulation. Amino acid Gln14 may be omitted owing to variations in intron splicing, and Arg28 may be removed as the result of variations in cleavage from the prohormone. Also, the acylation at Ser3 can be 8 or 10 carbons long and occasionally includes one double bond. Yet, all of these acylated variants appear to have comparable biological activity.
Studies of ghrelin processing have shown that prohormone convertase 1/3 (PC1/3) is the endoprotease responsible for the production of ghrelin from proghrelin.444 It is interesting to note that in the absence of PC1/3, ghrelin was still acylated normally, consistent with a model in which acylation takes place on the nascent peptide before cleavage. The enzyme that acylates ghrelin has been identified recently445,446 and is a member of the family of membrane-bound O-acyl transferases (MBOATs) that was named GOAT (ghrelin O-acyl transferase). The mRNA for GOAT was found in stomach, intestine, and pancreas, consistent with the place where ghrelin is found. GOAT was the only MBOAT capable of acylating ghrelin in vitro, and a GOAT knockout animal had no detectable circulating acyl-ghrelin,446 suggesting that GOAT is the physiologic mechanism of ghrelin acylation. Because no substrate for GOAT other than ghrelin is known, GOAT is being investigated as a specific pharmacologic target for the modulation of ghrelin activity.447
Assays for Ghrelin
Most published studies have used single-antibody ghrelin assays that recognize an epitope unique to acyl-ghrelin (active ghrelin), or common to both acyl- and des-acyl ghrelin (total ghrelin). Akamizu et al.448 report that 40% to 60% of the signal detected at a single-site, total ghrelin assay is due to inactive ghrelin fragments. Two-site sandwich assays can have greater specificity and can avoid cross-reactivity with peptide fragments.449 These two-site assays report lower levels of ghrelin but see greater percent suppression upon eating,450 suggesting greater physiologic relevance.
Acyl-ghrelin is deacylated quickly by esterase activity in the blood, and it is likely that it is also degraded by other mechanisms. For assay of acyl-ghrelin, blood samples should be collected to chilled tubes with esterase inhibitor, and cold plasma should be separated and acidified promptly.450 Acidification to pH ≈3 inhibits esterase, but if insufficient acid is used, the esterase activitys returns upon neutralization for assay, and if too much acid is used (pH ≤2), the acylation can be chemically cleaved.451
Ghrelin measurement is further complicated by the strong basic charge and hydrophobic acylation of ghrelin, which can cause it to stick to surfaces. In the circulation, ghrelin, but not des-acyl ghrelin, may be predominantly bound to carrier proteins, with ghrelin having specific lipoprotein interactions not seen with des-acyl ghrelin; ghrelin antisera differ in their ability to detect these bound forms. Together, these factors can affect the assay of ghrelin, as when two popular commercial RIA kits were compared on the same sample set, a 10-fold difference in measured total ghrelin levels was observed.452
Serious concerns have also been expressed about the specificity of available acyl-ghrelin assays. In one study examining patients with anorexia nervosa,453 the acyl-ghrelin levels measured in the same sample set were increased, decreased, or unchanged in subjects with anorexia relative to normal controls, depending on which of three different active ghrelin assays was used.
Sites of Ghrelin Synthesis
In the stomach, ghrelin is synthethized in the X/A-like cells, which represent about 20% of the chromogranin A–immunoreactive endocrine cells. Circulating ghrelin levels are reduced after gastrectomy in rats and humans, but levels increase over time postoperatively.440 Outside the intestinal tract, ghrelin expression has been identified in a number of tissues, at the mRNA or protein level, or both. In the hypothalamus, ghrelin peptide has been detected by immunostaining and was localized in the internuclear space between the hypothalamic nuclei and the ependymal layer of the third ventricle.440 Ghrelin was also seen in the axon terminals, and these axons innervated the arcuate, ventromedial, paraventricular, and dorsomedial nucleus and the lateral hypothalamus, as well as outside the hypothalamus in the bed nucleus of the stria terminalis, amygdala, thalamus, and habenula.454 Circulating ghrelin might reach the hypothalamic nuclei directly from the bloodstream (arcuate nucleus) or by crossing the blood-brain barrier,455 but peripheral (vagal) connections to the brain stem may also play an important role in the effects of ghrelin (Fig. 16-11). Within the pituitary, ghrelin immunostaining was co-localized to prolactin, growth hormone, and thyroid-stimulating hormone (TSH)-secreting cells, but not to adrenocorticotropic hormone (ACTH) or gonadotroph cells. Ghrelin mRNA and/or protein expression has been described in all normal human tissues studied, as well as in different tumors, including pituitary adenomas, neuroendocrine tumors, thyroid and medullary thyroid carcinomas, and endocrine tumors of the pancreas and lung. Ghrelin peptide has been shown to be expressed in pituitary, immune cells, lung, placenta, testis, and kidney, and by cyclic expression in the ovary. In the pancreas, controversial reports show expression in β and α cells, or in a new islet cell type, the ε cell.
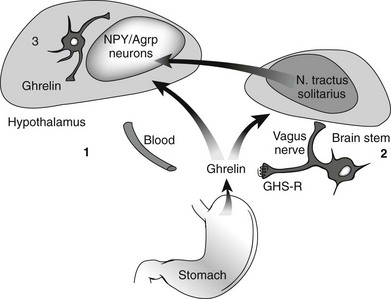
FIGURE 16-11 Ghrelin exerts its effects in the hypothalamus via three different pathways462: (1) Ghrelin synthesized in the stomach reaches the arcuate nucleus via the bloodstream and possibly other brain areas via active transport through the blood-brain barrier438,455; (2) ghrelin synthesized in the periphery is stimulating vagal afferents, which has been shown to express GHS-R, and vagal connections reach the nucleus tractus solitarius in the brain stem, which then communicates with the hypothalamus645; (3) controversy has arisen over whether ghrelin cells are present in the hypothalamus,521,522,566 but some data suggest that ghrelin is synthesized locally in the hypothalamus and has direct connections with NPY/AGRP and other hypothalamic cells.454
Regulation of Ghrelin
Factors shown to be important in ghrelin regulation are listed in Table 16-1. In the human, ghrelin levels are high during short-term caloric restriction and fall immediately after food intake. Higher levels are observed during the night.456,457 However, after prolonged fasting, acyl-ghrelin levels are lower when compared with fed conditions.450 Similar findings are reported when only total ghrelin is measured.458 Chronic regulation is influenced by body weight, with high levels in subjects with low BMI and low levels in obese subjects.459 Insulin appears to be an important regulator of ghrelin levels, as both insulin resistance and hyperinsulinemia predict low ghrelin levels in a population consisting of insulin-resistant and insulin-sensitive subjects who have equal BMI.460 Several studies suggest that ghrelin levels are similar in males and females, but some data have showed higher ghrelin levels in females. Estrogen elevates the peak overnight production rate of acylated ghrelin. Other studies suggest that combined estrogen-progestin supplementation enhances GH responsiveness to ghrelin in postmenopausal women. In addition, estradiol-dependent augmentation of the orexigenic potency of ghrelin has been reported.
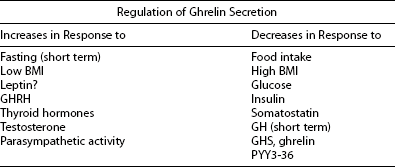
Physiology and Molecular Biology
Following the identification of GHRP-6, GHRP-2, and the first non-peptide analogues of Merck, a large number of pharmaceutical companies and research groups started to actively develop GHSs, resulting in the identification of many new peptide and non-peptide compounds.435,436 Ghrelin effects are similar to a number of recognized effects of GHSs; however, different GHSs seem to differ in their relative potencies for these effects, suggesting differing cross-reactivities at possible known or unknown receptor subtypes, and raising hopes for new compounds tailored specifically for these sites. An example of specific effect is the activity of hexarelin on the lipid scavanger receptor CD36; this is not shared by ghrelin or some other GHSs.461 Although ghrelin and some GHS compounds are reported to elevate prolactin, ACTH, and cortisol, others may have greater specificity for appetite, sleep, and cardiac function.462 The effects of GH release and appetite can be separated, as a pharmacologic antagonist of GHS-R1a–induced GH release can still induce appetite and weight increase,463,464 while des-acyl ghrelin can also stimulate appetite possibly via the orexin pathway.465
GHS Receptor
With the use of S35-MK-0677, two types of GHS-R cDNA (types 1a and 1b) were identified.437 The human type 1a cDNA encodes a 366 amino acid seven-transmembrane receptor of the rhodopsin family and has a relatively limited tissue distribution, including the hypothalamus, the hippocampus (dentate gyrus, CA2 and CA3 regions), the pars compacta of the substantia nigra, the ventral tegmental area, the dorsal and medial raphe nuclei, Edinger-Westphal nucleus, and pons and medulla oblongata, as well as peripheral tissues, such as the thyroid gland, intestine, pancreas, kidney, heart, and aorta, and various endocrine tumors of the pituitary, pancreas, lung, and stomach.462 Growth hormone secretagogue receptor (GHS-R) established a new branch in the rhodopsin family of G protein–coupled receptors, with other related receptors later identified, including the motilin receptor. The type 1b GHS-R cDNA represented an alternatively spliced message from the same gene but encoded only 289 amino acids, representing the first five transmembrane domains, and appeared to be nonfunctional.437 Although the mRNA of the type 1b form is widely detectable in tissues, it is unclear whether the type 1b receptor is expressed as a protein, although it is conceivable that this truncated form could interact with the full-length receptor and modulate its activity.
Ghrelin is bound primarily on presynaptic axon terminals, suggesting that the receptor is regulating neurotransmission via presynaptic localization. Ghrelin and the GHSs act on somatotrophs through a phospholipase C–inositol triphosphate–PKC signaling pathway distinct from the cAMP-protein kinase A (PKA) pathway activated by GHRH.438 Data suggest, however, that cAMP levels are elevated when ghrelin and GH secretagogues are co-administered with GHRH, but the exact mechanism of this synergism is not known. The GHS-R, similar to other G protein–coupled receptors, shows desensitization in its calcium and GH effects, although long-term human studies with ghrelin mimetics showed sustained elevation of IGF-1 levels for 1 to 2 years.466,467
The GHS-R was found to be highly constitutively active in a ligand-independent manner in transfected COS-7 and HEK293 cells. Although ghrelin and GHSs further increased inositol phosphate turnover, a low-potency antagonist (substance P antagonist) was found to be a high-affinity inverse agonist.468 These data open the possibility for development of inverse agonist compounds to oppose the effects of activation of the receptor.
Regulation of the GHS Receptor
Studies using the dw/dw rat model or normal rats to investigate the effects of GH on hypothalamic and pituitary GHS-R expression suggested that GHS-Rs are involved in feedback regulation of GH. Whether these effects occur directly at the pituitary level or are mediated indirectly through the hypothalamus has yet to be determined. In addition, expression of the pituitary GHS-R mRNA seems to be sex dependent, whereas the hypothalamic expression of this receptor showed no significant sex difference. GHRH appears to positively regulate the pituitary GHS-R in rats. GHS-R has been found to be upregulated by GHRH, GH deficiency, estrogen, glucocorticoids, and thyroid hormones.462
Evidence for Alternative Ligands and Receptor Subtypes
Evidence for alternative ligands for the GHS receptor is controversial. Adenosine has been proposed to act as a partial agonist at the GHS-R,469,470 although others refute this.471,472 Cortistatin, a 14 amino acid peptide neuropeptide with similarity to somatostatin, has been reported to bind to GHR-Rs in the pituitary.473 Recently, it was proposed that GHRH binds directly to the GHS-R expressed in vitro, activating calcium mobilization and enhancing ghrelin binding. The authors suggest positive cooperativity between two distinct binding sites on the GHS-R—one for ghrelin and one for GHRH.474
Animals with the known GHS-R knocked out showed neither an appetite change nor GH release in response to acyl-ghrelin.475 However, these GHS-R knockout animals continue to respond to des-acyl ghrelin, and it is possible that other actions of acyl-ghrelin may be transduced at additional sites. Alternative binding sites for ghrelin, des-acyl ghrelin, and some GHSs have been identified in the pituitary, thyroid, heart, and other tissues, and ghrelin has been found to be associated with a high-density lipoprotein in plasma.462 Hexarelin-like GHSs but not ghrelin can act at a macrophage scavenger receptor CD36.476
EFFECTS OF GHRELIN AND GHSs
In recent years, the metabolic effects of ghrelin came to the forefront of research as opposed to the originally described classical pituitary hormone effects on GH, ACTH, and prolactin release. Ghrelin has been described to have effects on feeding, glucose and lipid metabolism, gastric acid secretion, gastric motility, and cell proliferation, as well as on sleep, anxiety, and memory (Table 16-2). Data from ghrelin and GHS-R knockout mice suggest that alternative pathways can compensate for a number of the known effects of ghrelin (see later), but subtle abnormalities are detected under various experimental conditions (Table 16-3).

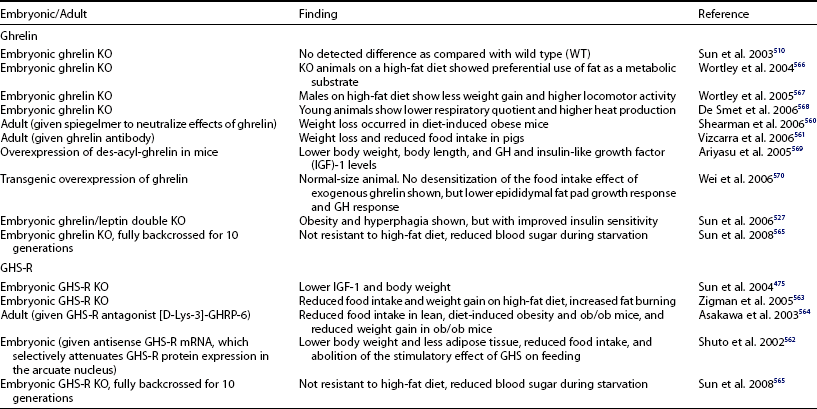
GH-Related Effects
The GH-releasing effect of ghrelin acts via a dual mechanism involving the hypothalamus and the pituitary. In in vitro studies with rat pituitary cultures, ghrelin has been shown to specifically activate the GHS-R and to stimulate GH release438 via the phopspholipase C–diacylglycerol–IP3–Ca2+–protein kinase 3 pathway. At the level of individual pituitary cells examined with the reverse hemolytic plaque assay, GHSs increase the number of GH-secreting cells without altering the amount of GH released per cell. In contrast, GHRH increases both the amount of GH secreted per cell and the number of GH-secreting cells, while somatostatin predominantly acts to decrease the number of secreting cells; this is the opposite of the effect of GHSs and supports the view that GHSs act as functional antagonists of somatostatin.462 The effect of GHSs in vivo is much stronger than the in vitro effect; IV administration of ghrelin to freely moving rats caused a dose-dependent increase in GH release, but the effect depends on the presence of GHRH. Although hypothalamic activation can be seen in the lit/lit mouse, which has an inactivating mutation of GHRH-R, no GH release is observed. It is interesting to note that in humans with GHRH-R mutations, a very small but significant GH response can be observed with sensitive GH assays, while the ACTH and prolactin-releasing effects of GHSs are intact.505 The efficacy of GHSs or ghrelin is greatly attenuated following administration of anti-GHRH serum in rats or a GHRH antagonist in humans. Pituitary stalk lesions cause attenuation of GHS or ghrelin effects, but less so if the lesion occurred recently and the pituitary somatotrophs are not atrophic because of long-term GHRH deficiency. Coadministration of GHS with GHRH causes a synergistic GH release, and this effect can be used for diagnostic purposes to diagnose adult GH deficiency. Continuous ghrelin administration leads to attenuation of the effect; intermittent administration causes long-lasting effects. GHSs do not stimulate GH synthesis in adult somatotroph cells, although a positive effect on GH synthesis was shown in infant rat pituitary. Both peripherally and centrally administered GHSs and ghrelin stimulate the hypothalamic arcuate nucleus GHS-R and directly act at the pituitary GHS-R to release GH. However, blockade of the afferent fibers of the vagus nerve abolished peripheral ghrelin-induced GH release. Therefore, the exact mechanism of GH release elucidated by peripheral ghrelin remains to be clarified.
In humans, the GH rise after an equivalent dose (1 µg/kg) of IV bolus GHRH, hexarelin, and ghrelin shows significantly different responses, with ghrelin being the most effective (peak mean GH ± SEM: 26.7 ± 8.7 µg/L, 68.4 ± 14.7 µg/L, and 92.1 ± 16.7 µg/L, respectively). Studies in rodents found no change in the magnitude of the ghrelin-induced GH release with age.506 No age or gender dependence in the ACTH, cortisol, and prolactin effects and in the glucose (increase) and insulin (decrease) effects of ghrelin have been described.507 Reduced GH release is observed more often in obese subjects than in normal subjects in response to ghrelin or to GHSs. Food intake decreases the effects of ghrelin on GH release in both animal and human studies.
Ghrelin, a natural ligand for GHS-R, potently stimulates GH release when administered exogenously. An increasing set of data, albeit not all data, suggests that ghrelin not only potently stimulates GH release when administered exogenously, it also plays a role in physiologic GH regulation. Both SC and intracerebroventricular infusions of the rat GHS-R antagonist BIM-28163508 did not alter the pulsatile pattern of GH secretion but lowered the GH pulse amplitude. In humans, a GHS-R missense mutation, which impairs the constitutive activity of the GHS-R, is associated with short stature.509 Consistent with these results, GHS-R–null mice have lower IGF-1 levels when compared with wild-type animals,475 and ghrelin-null mice also show a tendency to lower IGF-1 concentrations when compared with control animals510; however, this did not reach statistical significance. This body of data implies that endogenous ghrelin plays a role in GH regulation. In a patient with ghrelin-secreting pancreatic tumor and 50 times higher ghrelin levels, GH and IGF levels were in the normal range, although desensitization and downregulation of the GHS-R are possible in this situation, and it was not tested whether the ghrelin secreted by the tumor was in the active acylated form. Several clinical studies have found a relationship between ghrelin and GH levels511–513; others have not.514–516 All of these studies measured total ghrelin and did not distinguish between acyl-ghrelin, des-acyl ghrelin, and inactive fragments. Studies measuring acyl-ghrelin levels every 20 minutes with a single-site assay found no relationship with GH under fed or fasting conditions.517 In a study using a two-site ghrelin assay and more frequent sampling (every 10 minutes for 24 hours), a close association between full-length acyl-ghrelin levels and amplitudes of individual GH secretory events was demonstrated, suggesting that acyl-ghrelin modulates GH release.518
Other Hormone-Related Effects
Feeding Effect: Ghrelin stimulates feeding in both animal and human studies.440 Similar effects were shown earlier for GHSs.519 Ghrelin acts via stimulating NPY/AGRP cells and orexin cells and indirectly inhibiting pro-opiomelanocortin (POMC) cells in the hypothalamus.462 The vagus also seems to play an important role in the feeding effects, as vagotomy inhibits the feeding effects of peripheral ghrelin. Therefore, the feeding effect of ghrelin could occur via three possible mechanisms (Fig. 16-11): (1) via peripheral ghrelin reaching the arcuate nucleus and stimulating orexigenic cells; (2) via GHS-R in the peripheral vagal afferents and the nucleus tractus solitarius; or (3) via locally produced ghrelin-stimulating arcuate and lateral hypothalamus orexigenic cells, and or inhibiting anorectic cells, although it has to be emphasized that some studies were not able to detect ghrelin and/or ghrelin cells in the hypothalamus.520–522 The mechanism of ghrelin effects on appetite involves the metabolic enzyme adenosine monophosphate-induced protein kinase (AMPK).523 A recently suggested pathway for the orexigenic effect of ghrelin includes the activation of Ca2+ signaling in NPY neurones in the arcuate nucleus (ARC). The Ca2+ rise leads to CAMKK2 activation, which can stimulate hypothalamic AMPK. AMPK activation leads to increased mitochondrial oxidation and activation of the uncoupling protein 2 (UCP2), which can regulate NPY/AgRP neuronal activity, and ultimately to stimulation of appetite (see Fig. 16-12 and review524).
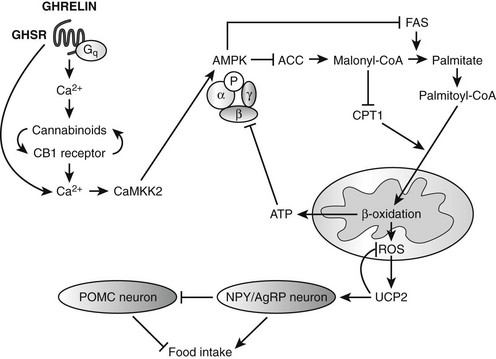
FIGURE 16-12 Schematic diagram showing the proposed molecules involved in the appetite-inducing effect of ghrelin. (GHS-R, Growth hormone secretagogue receptor; CaMKK, calmodulin kinase kinase; CB1, cannabinoid receptor type 1; AMPK, AMP-activated protein kinase; ACC, acetyl-coenzyme A carboxylase; malonyl-CoA, malonyl coenzyme A; FAS, fatty acid synthase; CPT1, carnitil palmitoyl transferase 1; ROS, reactive oxygen species; UCP2, uncoupled protein 2; NPY, neuropeptite Y; AgRP, agouti-related peptide; POMC, pro-opiomelanocortin.) (Redrawn based on results from several studies reviewed in Kola and Korbonits 2009.524)
Other Effects of Ghrelin
Most animal and human studies have observed that ghrelin inhibits insulin levels, leading to increased glucose levels.525 Ghrelin has been shown to inhibit AMPK activity in liver, which could lead to disinhibition of phosphoenolpyruvate carboxykinase (PEPCK) and activation of gluconeogenesis.526 The most powerful proof of the important effect of ghrelin on carbohydrate mechanism comes from the double mutant ghrelin/leptin knockout mice, as these mice are obese similar to leptin knockouts, but ablation of ghrelin improves the diabetic phenotype.527
That ghrelin analogues increase body fat in rodents, despite their GH-releasing effect, has been elegantly demonstrated by Lall et al.528 Ghrelin specifically stimulates fat accumulation and has been shown to have a direct stimulatory effect on adipocyte proliferation and on peroxisome-proliferator–activated receptor-γ (PPAR-γ) synthesis. Ghrelin inhibits AMPK activity in fat tissue, which could explain its adipogenic effect.526
GHSs and ghrelin have GH-independent effects on the cardiovascular system. Ghrelin is effective in improving cardiac performance in chronic heart failure.529,530 Ghrelin prevents increased sympathetic tone and reduces mortality after myocardial infarction.487 Ghrelin stimulates AMPK in cardiac tissue, which could be important in terms of its positive inotropic and anti-ischemic effects.526 Ghrelin has vasodilator effects, and the density of ghrelin binding is increased in atherosclerosis.531 Ghrelin inhibits apoptosis via an MAPK- and Akt-dependent pathway in cardiomyocytes. Desoctanoyl ghrelin has also been shown to produce similar effects, and it has been shown to bind to H9c2 cells that do not express GHS-R1a mRNA.532 It seems that at least some of the cardiovascular effects of ghrelin and hexarelin on cardiac function are different; this is apparently due to the specific actions of hexarelin at the CD36 receptor.476
Given the fact that the reproductive axis is highly dependent on nutritional status, ghrelin, acting at central and peripheral levels, could be one of the signaling mechanisms linking nutritional status to the hypothalamo-pituitary-gonadal axis.533 Both animal and human studies have shown that ghrelin can inhibit luteinizing hormone (LH) pulsatility, possibly via the central nervous system (CNS).478
Both GHSs and ghrelin have been shown to promote sleep in humans, although GHRP-2 showed an increase in stage 2 sleep, and MK-0677 and ghrelin increase slow wave sleep. It is interesting to note that in lit/lit mice with a nonfunctional GHRH receptor, ghrelin increased food intake but had no effect on sleep, suggesting that GHRH is involved in the sleep effect of ghrelin.534
GHS-R has been identified in the hippocampus, and ghrelin administration increases anxiety.535,536 On the other hand, ghrelin defends against depressive symptoms of chronic stress.503 These findings suggest that ghrelin may have a role in mediating neuroendocrine and behavioral responses to stressors, and that the stomach might play an important role in the regulation of anxiety.
Ghrelin Levels in Pathologic Conditions
Ghrelin is negatively correlated with weight, and higher ghrelin levels were found in patients on a low-calorie diet457,537 and in patients suffering from anorexia due to cancer,538,539 cardiac disease,540 or anorexia nervosa.541 Patients with anorexia nervosa have high ghrelin levels, and weight gain decreases ghrelin concentrations.542,543 Neonates with intrauterine growth retardation have higher ghrelin levels than normal neonates, and the increased orexigenic drive could contribute to postnatal catch-up growth.544
Obese subjects have lower ghrelin levels than lean subjects.545 Weight loss through dieting increases circulating ghrelin levels.457,537 Data suggest that ghrelin depends on body weight, probably via regulation by insulin, and does not depend on fat mass or fat distribution.
The most effective treatment for morbid obesity is gastric surgery with Roux-en-Y gastric bypass or adjustable gastric banding. It has long been observed that Roux-en-Y gastric bypass has effects beyond restriction of the capacity of the stomach, and long-term reduction in appetite is achieved. It has been suggested that decreased ghrelin levels, probably due to the loss of stomach tissue involved in food processing, could be the cause of loss of appetite after gastric bypass surgery. However, other studies were not able to reproduce uniformly these data. A review of 18 prospective and cross-sectional studies summarized the results so far as being ambiguous and inconsistent, with insufficient evidence to support a firm conclusion of any sort about the relationship between ghrelin levels and bariatric surgery546; more recent studies have still resulted in mixed outcomes regarding ghrelin levels.
Diabetes Mellitus and Insulin Resistance
Patients with insulin resistance (type 2 diabetes, polycystic ovary syndrome) have lower ghrelin levels than BMI-matched controls.440 The relationship between higher insulin concentration (and/or the pathologic process of insulin resistance) and lower ghrelin levels cannot be determined by these data. Low ghrelin levels appeared to be associated with high blood pressure independent of BMI in a population-based study as well as in pregnant women.
Prader-Willi Syndrome
Although all forms of human obesity, including simple obesity, congenital leptin deficiency, leptin receptor or melanocortin-4 receptor mutations, or hypothalamic obesity from craniopharyngioma, show low ghrelin levels, patients with Prader-Willi syndrome (PWS) have inappropriately high ghrelin levels, in the range of patients with anorexia nervosa.547–550 PWS is the most common syndromal cause of genetic obesity, caused by loss of expression of imprinted genes on the paternally inherited chromosome 15q11-q13, and characterized by life-threatening childhood-onset hyperphagia and obesity, as well as GH deficiency and hypogonadism, thought to be due to hypothalamic abnormalities.551 Hyperghrelinemia, which is three to four times higher than in BMI-matched controls, is reduced after food intake in parallel to normal subjects.552 Somatostatin treatment of PWS patients inhibited ghrelin levels, but no change in the hyperphagia was noted.553 It remains to be determined whether ghrelin is directly involved in the pathogenesis of the different symptoms in PWS.
Acromegaly and Growth Hormone Deficiency
Ghrelin levels increase after surgery in acromegalic subjects in inverse correlation with GH, IGF-1, and insulin levels, suggesting that one or a combination of these hormones results in suppressed ghrelin levels in active acromegaly.554,555 GH-deficient patients have low or normal ghrelin levels compared with BMI-matched subjects, and again the increased percent body fat can play a role in determining ghrelin levels. GH replacement causes a reduction in ghrelin levels.556
Hyperthyroidism
Ghrelin levels are reduced in hyperthyroidism, suggesting that ghrelin is not involved in the hyperphagia of hyperthyroidism.557 Because known factors suppressing ghrelin levels—high BMI, high insulin, or somatostatin—cannot play a role here, and because the decreased body weight of hyperthyroidism should rather stimulate ghrelin levels, it is assumed that thyroid hormones have a direct inhibitory effect on ghrelin.
Renal Disease
Ghrelin levels are elevated in chronic renal failure and show a reduction after a single course of hemodialysis.558 Unpublished data (T. Kuppusamy, R. Gupta, J. Patrie, M.O. Thorner, J. Liu, B. Gaylinn, and W.K. Bolton) suggest that the reduction on dialysis is predominantly des-acyl and not acyl-ghrelin, consistent with evidence that acyl- but not des-acyl ghrelin circulates in a bound form.559
Models of Ghrelin or Ghrelin Receptor Deficiency
Research into the functional importance of ghrelin has included animal models that had a genetic or an acquired deficiency of ghrelin or ghrelin receptor (see Table 16-3). The data about ghrelin deficiency are somewhat controversial, depending on the timing of onset of ghrelin deficiency and on appropriate backcrossing of the genetically modified animals. Embryonic knockout animals show very little if any difference in energy metabolism, possibly only in insulin sensitivity, and no difference in pituitary function (see Table 16-3). It is interesting to note that the leptin-ghrelin double knockout model shows obesity and hyperphagia but improved insulin release (via reduction of levels of UCP2) and insulin sensitivity, suggesting an important role for ghrelin in β cell physiology.527 An acquired ghrelin deficiency resulted in diet-induced obese mice eating less and storing less fat, and ultimately having a lower body weight and body fat mass as compared with control or vehicle groups,560 while pigs showed reduced food intake and weight loss after ghrelin immunoneutralization.561 The prominent phenotype in acquired ghrelin deficiency as opposed to the mild phenotype of the embryonic knockouts suggests that prenatal ablation of the ghrelin gene produced more subtle phenotypic effects as a consequence of the plasticity within the system, as other metabolic pathways had been able to develop in a way that enabled them to compensate for loss of ghrelin. However, this compensation was not possible when the effects of ghrelin were neutralized in adult life.560 This suggests that ghrelin is an important player in the field of energy homeostasis and appetite regulation, as part of a bigger network of regulatory molecules, which can only compensate for its loss if this occurs early in development.
A selective hypothalamic knockout model using antisense GHS-R mRNA expression under the tyrosine hydroxylase control showed lower GH and IGF-1 levels in female animals.562
GHS-R knockout mice have lower body weight475,563 and lower IGF-1 values.475 In keeping with this, a study into the effects of antagonizing the GHS-R receptor (in adult mice) found that this decreased feeding in both lean and obese mice lowered body weight gain and reduced the rate of gastric emptying.564 It is interesting to note that a recent study backcrossing knockout animals to 10 generations found that knockout animals are not resistant to high-fat diet–induced obesity, and the only abnormality described is reduced blood sugar during starvation.565
Clinical Implications of Ghrelin and GHS
Since early studies reported the GH-releasing capabilities of GHSs in humans, interest in their potential as diagnostic agents has increased. Peptide and non-peptide GHSs are powerful stimulators of GH release, effective when administered IV, sc, intranasally,571 or PO.572 These agents typically cause GH release in excess of that observed following GHRH or during ITT.
GHS Actions in Healthy Subjects
GHSs cause the release of GH in a dose-dependent fashion.573 They are more potent than GHRH: 1 µg/kg of GHRP-6 peptide results in significantly greater GH release than the same dose of GHRH. The effect of GHSs on GH release is more reproducible than the effect of GHRH. The peptide GHSs (e.g., GHRP-6, GHRP-1, GHRP-2, hexarelin) and the non-peptide GHSs (e.g., MK-0677) differ in terms of their pharmacokinetics. MK-0677 has been developed specifically as an orally active agent. The peptidyl GHSs are active po, but only at doses several hundred times higher than that required when IV is administered.
An intact hypothalamic-pituitary axis is vital in facilitating the maximal effect of GHSs on GH release. GHRH and somatostatin both influence the action of GHSs, augmenting and diminishing the magnitude of the GH pulse, respectively. When GHRH is administered in combination with GHSs, the effect is synergistic, the magnitude of the GH pulse being greater than that obtained from the sum of the two agents administered separately.574–577 The presence of GHRH is required for GHSs to exert their effects on GH secretion. In a family from the remote Valley of Sind in Pakistan, a missense mutation in the GHRH receptor that changes the glycine at residue 72 in the extracellular domain to a stop codon results in a phenotype analogous to the little mouse.505 When members of the family that were homozygous for the mutation were challenged with hexarelin, no GH response was detectable. In addition, studies performed in children who are GH deficient as the result of pituitary stalk transsection show that they are unresponsive to GHSs.578 Thus, somatotroph exposure to GHRH is necessary for GHSs to exert their full action.
Manipulation of somatostatin tone also affects the GH response to GHSs. When hexarelin was given to subjects in combination with somatostatin, the amount of GH released was significantly reduced.579 When arginine was administered to the elderly, a group proposed to have increased somatostatin tone, GH levels following the administration of GHRP-6 increased significantly, to levels seen in younger subjects.580
GHSs demonstrate GH-releasing activity during the neonatal period at a level that persists during prepubertal life.581 During puberty, increased GH response to GHSs persists into adult life.582–584 Subsequently, over the course of the adult life span, the GH response to GHSs declines, in line with the reduction in spontaneous GH secretion.575,580,585
The response to GHSs does not vary with sex, apart from during puberty, when girls exhibit a greater response to GHSs than do boys.583 The response in adult women is similar to that observed in men. Women who received GHSs at various times during the menstrual cycle achieved similar peaks, whether studied during the early follicular, late follicular, or luteal phase.586 Several studies suggest that estrogen and estrogen-progestin supplementation has an impact on the GH response to ghrelin.587,588
GHSs AND THE DIAGNOSIS OF GH DEFICIENCY
The combination of GHRH and GHS is the most potent stimulus of GH release known and provides a promising alternative to ITT in the diagnosis of GH deficiency. In normal subjects, the lower limit of the normal range (third percentile) of responses to GHRH and hexarelin was 55.5 µg/L, which was considerably higher than the lower limit of normal following GHRH and arginine (third percentile = 17.5 µg/L). The response to GHS plus GHRH is reproducible within an individual; GH levels attained are similar among individuals and do not appear to decline with age.577 Similar to GHRH+Arg, the combined GHRH+GHRP-6 is partially refractory to the inhibitory effects of glucose and free fatty acid load, as well as of rGH.589 Perhaps the most important feature of the GHS-GHRH test is its ability to discriminate GH-deficient patients from normal subjects. In two studies, the combination of GHS and GHRH had a specificity and a sensitivity of 100%.590,591 The combination of GHS and GHRH is safe and well tolerated; side effects are similar to those seen when the two agents are administered separately. Chihara et al.592 compared the effects of a single iv GHRP-2 injection versus a classical ITT in patients with severe GHD. Severe GHD could be diagnosed with high reliability in all subjects using a threshold of 15 µg/L. Since the discovery of the natural ligand of the GHS receptor ghrelin in 1999, several studies have been performed to evaluate the acute effects of ghrelin on GH release, while the use of ghrelin or a ghrelin mimetic alone has not been suggested for diagnostic use in GH deficiency, these used in combination with GHRH form a very effective test. Aimaretti et al593 showed that ghrelin given IV in a dose of 1 µg/kg to adults with isolated GH deficiency increased GH levels significantly higher when compared with the ITT and the combined test of GHRH and arginine. These data suggest that ghrelin itself should be a good agent to be used in a provocative test in the future; however, normative data are still missing.
The GHRH and GHS Test in Difficult Diagnostic Situations
Obesity
Spontaneous and stimulated GH secretion is reduced in obese subjects.389 The exact cause of the hyposomatotropism is uncertain, but a variety of mechanisms have been suggested. Among the hypotheses put forward are increased somatostatin tone, a reduction in the secretion of GHRH or of the natural ligand for the GHS receptor, and any combination of these.594 What is known for certain is that the GH response to a number of stimuli, including GHRH,388 GHRH and arginine,595 GHRP-6,596 and hexarelin,597 is inversely correlated with body fat mass, specifically, abdominal fat mass. The diminished GH response can make it difficult to accurately define the GH status of obese patients with hypothalamic-pituitary disease, particularly those in whom GH deficiency may be the only hormone abnormality.
The combination of GHRH with either arginine598 or GHS596 administered to an obese subject causes a GH response far greater than any other stimulus, although the GH level does not quite reach that seen in normal controls. These tests are useful tools in the differentiation of true GH deficiency from hyposomatotropism caused by obesity. The validated cutoff level for growth hormone deficiency (GHD) in adults for the ITT and the glucagon test is a peak GH response <3 µg/L, which has not been validated in obesity. For the GHRH+Arg test, the following cutoff levels have been validated, depending on the BMI: <25 kg/m2, a peak GH <11 µg/L; 25 to 30 kg/m2, a peak GH <8 µg/L; >30 kg/m2, a peak GH <4 µg/L.403 The cutoff level validated for the GHRH+GHRP-6 is 10 µg/L in lean patients and 5.0 µg/L in obese patients.404,405
Syndromes of Glucocorticoid Excess
In rats, glucocorticoids potentiate GHRH action and enhance spontaneous GH secretion. In normal humans, a biphasic effect results from pharmacologic doses of glucocorticoids. When normal men were treated with a single IV bolus of dexamethasone 4 mg, and 3 hours later were challenged with a bolus injection of GHRH, the peak GH response to GHRH increased from 9.9 ± 2.0 µg/L to 29.2 ± 5.7 µg/L. When the dexamethasone dose was increased to 8 mg IV 12 hours before a GHRH bolus, the peak GH response to GHRH was attenuated to 3 ± 1.1 µg/L. These results suggest an acute stimulatory response followed by a later inhibitory effect.599 Pretreatment with pyridostigmine before administration of GHRH partially reversed the effects of 48 hours of dexamethasone therapy, suggesting that somatostatin tone may be increased by glucocorticoids.600
Patients with glucocorticoid excess caused by Cushing’s syndrome or by exogenous steroids have markedly impaired GH secretion.601 This may result from the combined effects of chronic exposure to glucocorticoids and the changes in body composition associated with Cushing’s syndrome, particularly the central adiposity. The suppression of GH in these patients may persist for up to 1 year after resolution of the glucocorticoid excess,602 which may give rise to misinterpretation of a patient’s GH status. GHRH and GHRP-6 have been administered to patients with Cushing’s syndrome separately and in combination. The effect of GHRH in these patients was almost abolished and the response to GHRP-6 was considerably reduced compared with controls. The combination of GHRH and GHRP-6 was considerably more potent than either GHRH or GHRP-6 used alone, but the GH peaks were only 20% of those seen in normal subjects.603 Similarly, the GH-releasing effects of ghrelin are significantly reduced in patients with Cushing’s disease.604,605 An earlier study had shown that the response to GHRH and pyridostigmine increased threefold following 7 days’ priming with GHRH.606 These data suggest that the effects of chronic glucocorticoid excess are caused primarily by reduced GHRH secretion. Whether the combination of GHRH and GHS will be able to predict which patients with Cushing’s disease will recover normal GH secretion is a matter for further study.
Nascif et al.607 demonstrate that the effects of GHSs, GHRP-6, GHRH, and ghrelin in hyperthyroid patients were significantly greater with ghrelin than with GHRP-6. A significant decrease in GH responsiveness to ghrelin, GHRP-6, and GHRH was also noted in the hyperthyroid group compared with controls.
THERAPEUTIC POTENTIAL OF GHSs
Since the discovery of GHRPs in 1976, 5 years before the discovery of GHRH,608 several different types of GHSs have been developed, including a series of nonpeptidyl GHSs.609–613 The concept that these agents amplify the pulsatile GH secretory pathway, instead of overriding normal physiology, has made this group of drugs the target of intensive research. The initial enthusiasm that accompanied the concept of orally available peptidergic and non-peptidergic GHSs, however, has been mitigated by the controversial results of several studies suggesting that no benefits are evident in terms of changes in body composition, as well as a report of tolerance.614 Others could not find development of tolerance and described beneficial effects on body composition.615
Therapeutic Potential of GHSs in Children
The use of GHSs in children with growth retardation has been thought to have therapeutic potential. Several studies have proved the GH-releasing effects of these compounds, peptidergic as well as non-peptidergic, in short-term infusion studies in children. As a GH stimulus, they are as potent as, or even more potent than, GHRH. Loche and coworkers616 demonstrated that IV bolus infusions of hexarelin, 2 µg/kg body weight, can enhance GH release in short-statured children (familial short stature and constitutional delay of growth).
In a trial performed by Mericq et al.,617 GHRP-2 was administered subcutaneously to six prepubertal children of short stature with GH deficiency, defined as a GH response of less than 7 µg/L to at least two standard provocative tests and a growth velocity of 4 cm/year or less. Agents were administered for 6 months at increasing doses (0.3, to 1.0, to 3.0 µg/kg/day). At months 7 and 8, the children received GHRP-2, 3 µg/kg/day, together with GHRH, 3 µg/kg/day. The maximal overnight GH and GH peak amplitude showed a progressive increase at the higher doses. Growth velocities were increased when compared with baseline (5.3 ± 0.8 vs. 3.0 ± 0.5 cm/year; P < .05). During the long treatment period, GHRP-2 injections were well tolerated. However, the study was not placebo controlled.
To date, only two studies have reported the effects of non-peptidergic GHSs in children.618,619 In a short-term pilot study of 8 days, MK-0677 increased GH, IGF-1, and IGFBP-3 in some children with GH deficiency. In the following long-term double-blind placebo-controlled study by Yu et al.,618 with the GHS MK-0677 and 94 previously untreated, prepubertal GH-deficient children (height <5th percentile, growth velocity <25th percentile, peak GH <10 ng/mL on two tests), the GHS was well tolerated. The children were treated for 6 months with 0.4 mg/kg/day or 0.8 mg/kg/day. Mean growth velocity increased by more than 3 cm/year at 6 months.618
Similar results have been reported620 in a group of prepubertal non–GH-deficient children. In this study, eight prepubertal children with constitutional short stature were treated with hexarelin administered three times daily intranasally. After treatment for up to 8 months, the growth velocity increased significantly (mean ± SD) from 5.3 ± 0.8 to 8.3 ± 1.7 cm/year in this group. Whether these changes would translate to increased adult height in these children after a longer therapy period is unclear to date.
Therapeutic Potential of GHSs in Adults
GH therapy has been approved in adults in the United States with organic GH deficiency346 caused by hypothalamic-pituitary disease and by the AIDS-related wasting syndrome.621 Because adults with GH deficiency very often do not have an intact hypothalamic-pituitary axis, only some of these patients probably would benefit from the use of GHSs.622,623 A synthetic GHRH(1-44) analogue that has an increased half-life compared with the native GHRH has been shown to have sustained positive effects on visceral adipose tissue and lipid profiles in patients with HIV.624,625 Currently, no data have been derived by investigating the potential positive effects of ghrelin or ghrelin mimetics treatment in the AIDS-related wasting syndrome, even though this agent might be a suitable candidate for this type of disease based on the GH-releasing and anorexic effects of ghrelin and ghrelin mimetics.
Another potential field of research is the catabolic state of severe illness. It seems that in the prolonged catabolic state of critical severe illness, a relative hyposomatotropism occurs, which seems to be of hypothalamic origin in part.626 Infusion studies with GHRP-2 showed that given alone or together with GHRH, the somatotrophs can respond in this condition. In addition, a significant responsiveness to GH seems to occur under GHRP infusion to these patients.626–628 A 5 day infusion of GHRP-2 and thyrotropin-releasing hormone (TRH) in protracted critical illness not only reactivated the blunted GH and TSH secretion but also showed metabolic effectiveness in this condition.629 A double-blind placebo-controlled study630 in a small number of healthy volunteers showed that diet-induced nitrogen wasting can be reduced after 7 days of treatment with an oral GH secretagogue (MK-0677). Studies by Neary et al.631 suggest that ghrelin is able to increase energy intake in cancer patients. Similar data in animal cancer models support the anticachectic effect of ghrelin.632 A 3 week preliminary study conducted in patients with pulmonary cachexia showed promising effects on lean body mass and respiratory muscle strength with daily ghrelin IV infusions.633 However, the study was not blinded and was not placebo controlled. This study also showed that ghrelin administration decreased circulating plasma norepinephrine levels (i.e., sympathetic nerve activity633). The same group of researchers found beneficial cardiac and muscle effects in patients with congestive heart failure (CHF) after daily IV ghrelin administration for 3 weeks.634 These findings support the concept of GHS use in the catabolic state. Further evaluation of the merits of the use of GHSs in adult patients on maintenance hemodialysis is warranted.635
The use of GHSs for treatment of critically ill patients has to be evaluated carefully. A previous study636 showed that treatment with high doses of GH (0.07 to 0.13 mg/kg/day) leads to an increase in morbidity and mortality in these patients.
Another potential field of application for GHSs is seen in the normal older population, as there is an age-dependent decrease in GH secretion.363 Some of the changes in body composition seen in the elderly resemble those seen in GH-deficient adults.363 Studies investigating the effects of GH treatment in the elderly have shown that GH treatment in the elderly might have beneficial effects on lean body mass, adipose tissue, and bone mineral density.637 In addition, the releasable GH pool of the pituitary is preserved in the elderly.
The few existing studies investigating the effects of GHSs in the elderly have shown conflicting results. One study, using the peptidergic GHS hexarelin, could not show a beneficial effect on body composition after 16 weeks of SC treatment. The same investigators reported a partial and reversible attenuation of the GH response to hexarelin, measured 4 weeks after cessation of hexarelin therapy. IGF-1 did not change significantly.614
Studies performed with an oral non-peptidergic GHS (MK-0677) resulted in a significant increase in IGF-1 concentration compared with that of young adults after 4 weeks of treatment with 25 mg given once a day. This increase was accompanied by an increase in the mean 24 hour GH concentration, GH pulse height, and interpulse nadir concentrations of these volunteers. No significant changes in pulse number were observed. No desensitization of the hypothalamic-pituitary GH axis occurred. Despite the fact that GHSs have been shown to have ACTH- and PRL-releasing activity, no change in cortisol secretion was found in this study. PRL levels rose slightly but remained within the normal range. Fasting insulin levels, as well as glucose levels, have been reported to increase under MK-0677 treatment. Results of a double-blind placebo-controlled trial show that MK-0677 is able to significantly increase IGF-1 levels in healthy older men and women for 2 years.467 In the same study, fat-free mass increased by 1.1 kg in the treatment group after 1 year of treatment; however, strength and function did not change significantly.467 Total appendicular skeletal mass increased significantly after 1 year of treatment, and a small but significant decrease in LDL levels was noted after 1 year of treatment with MK-677.467 Similar changes in body composition were found with the orally active ghrelin mimetic capromorelin466 after 6 months of treatment. The group described an improvement in functional parameters after 6 and 12 months. In a randomized, placebo-controlled study with 292 postmenopausal women, the effects of a GHS (MK-0677) on bone mineral density (BMD) alone and in combination with an antiresorptive agent (alendronate) resulted in increased BMD at the femoral neck when given together.638 In a 6 month study in healthy older adults recovering from hip fracture treatment with MK-0677, greater improvement relative to placebo was noted in some lower extremity measures. Overall performance measures did not show a significant change.639 The interpretation of such functional data is difficult, and a consensus is needed to define meaningful end points. These divergent results underline once again that each GHS is unique in terms of its bioavailability and specificity profiles.
Whether the use of GHSs in obesity will be of any benefit is questionable and requires further careful evaluation. GH-releasing effects in obesity are decreased when compared with a normal population.388,640 Eight weeks of treatment with the GHS MK-0677 at a dose of 25 mg/day in 24 obese men showed no change in total or visceral fat.615 Of interest is the fact that the fat-free mass increased significantly and IGF-1 levels increased by approximately 40% in this study.
Other ghrelin agonists such as TZP-101 and RC-1291, which can be given orally, are currently under evaluation for possible use in gastrointestinal motility.641 The latter has been evaluated for possible use in cancer cachexia.642 The compound showed promising effects in animal studies on cisplatin-induced hyperalgesia and cisplatin-induced cachexia. Previous animal studies in an animal melanoma model showed promising anti-cachectic effects of ghrelin.632 In animal studies, MK-0677 resulted in increased production of cytotoxic T lymphocytes and enhanced resistance to initiation of tumor growth and metastases.643 EPO1572, another ghrelin agonist, has been shown to effectively release GH.644 The results of long-term studies with MK-677467 suggest that this intervention is safe and well tolerated. However, before a final conclusion about the beneficial effects of these compounds can be reached, studies with carefully selected target populations and outcome parameters are recommended.