Regulation of Breathing
After reading this chapter you will be able to:
 Identify where the structures that regulate breathing are located.
Identify where the structures that regulate breathing are located.


 Identify the effect of various reflexes on breathing.
Identify the effect of various reflexes on breathing.
 Describe how the central and peripheral chemoreceptors differ in the way they regulate breathing.
Describe how the central and peripheral chemoreceptors differ in the way they regulate breathing.





 Describe the characteristics of abnormal breathing patterns.
Describe the characteristics of abnormal breathing patterns.
Medullary Respiratory Center
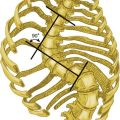
Animal experiments show that transecting the brainstem just below the medulla (Figure 14-1, level IV) stops all ventilatory activity. However, breathing continues rhythmically after the brainstem is transected just above the pons (see Figure 14-1, level I). Until more recently, physiologists thought that separate inspiratory and expiratory neuron “centers” in the medulla were responsible for the cyclic pattern of breathing. Researchers believed that inspiratory and expiratory neurons fired by self-excitation and that they mutually inhibited one another. More recent evidence shows that inspiratory and expiratory neurons are anatomically intermingled and do not inhibit one another.1 No clearly separate inspiratory and expiratory centers exist. Instead, the medulla contains several widely dispersed respiratory-related neurons, as shown in Figure 14-1. The dorsal respiratory groups (DRGs) contain mainly inspiratory neurons, whereas the ventral respiratory groups (VRGs) contain both inspiratory and expiratory neurons.
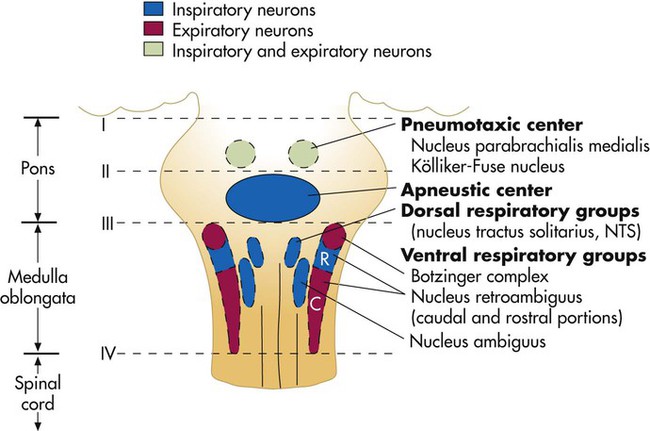
Dorsal Respiratory Groups
As shown in Figure 14-1, DRG neurons are mainly inspiratory neurons located bilaterally in the medulla. These neurons send impulses to the motor nerves of the diaphragm and external intercostal muscles, providing the main inspiratory stimulus.1 Many DRG nerves extend into the VRGs, but few VRG fibers extend into the DRGs. Reciprocal inhibition is an unlikely explanation for rhythmic, spontaneous breathing.1
Ventral Respiratory Groups
VRG neurons are located bilaterally in the medulla in two different nuclei and contain inspiratory and expiratory neurons (see Figure 14-1). Some inspiratory VRG neurons send motor impulses through the vagus nerve to the laryngeal and pharyngeal muscles, abducting the vocal cords and increasing the diameter of the glottis. Other VRG inspiratory neurons transmit impulses to the diaphragm and external intercostal muscles. Still other VRG neurons have mostly expiratory discharge patterns and send impulses to the internal intercostal and abdominal expiratory muscles.
The exact origin of the basic rhythmic pattern of ventilation is unknown. No single group of pacemaker cells has been identified. Two predominant theories of rhythm generation are the pacemaker hypothesis and the network hypothesis.2 The pacemaker hypothesis holds that certain medullary cells have intrinsic pacemaker properties (i.e., rhythmic self-exciting characteristics) and that these cells drive other medullary neurons. The network hypothesis suggests that rhythmic breathing is the result of a particular pattern of interconnections between neurons dispersed throughout the rostral VRG, the pre-Bötzinger complex, and the Bötzinger complex. This hypothesis assumes that certain populations of inspiratory and expiratory neurons inhibit one another and that one of the neuron types fires in a self-limiting way, such that it becomes less responsive the longer it fires. There is no definitive proof of either hypothesis; the precise origin of respiratory rhythm generation remains elusive.2
Inspiratory Ramp Signal
The inspiratory muscles do not receive an instantaneous burst of signals from the dorsal and ventral inspiratory neurons. Rather, the firing rate of DRG and VRG inspiratory neurons increases gradually at the end of the expiratory phase, creating a ramp signal (Figure 14-2). The inspiratory muscles contract steadily and smoothly, gradually expanding the lungs rather than filling them in an abrupt inspiratory gasp. During exercise, various reflexes and receptors influence the medullary neurons, steepening the ramp signal and filling the lungs more rapidly.
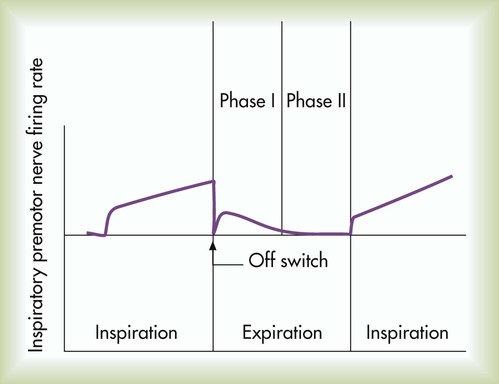
During quiet breathing, inspiratory neurons fire with increasing frequency for approximately 2 seconds and then abruptly switch off, allowing expiration to proceed for approximately 3 seconds.3 At the start of expiration, inspiratory neurons again fire briefly, retarding the early phase of expiration (see Figure 14-2). The inhibitory neurons that switch off the inspiratory ramp signal are controlled by the pneumotaxic center and pulmonary stretch receptors, which are discussed later in this chapter.
Pontine Respiratory Centers
If the brainstem is transected above the medulla (see Figure 14-1, level III), spontaneous respiration continues, although in a more irregular pattern. The pons does not promote rhythmic breathing; rather, it modifies the output of the medullary centers. Figure 14-1 shows two groups of neurons in the pons: (1) the apneustic center and (2) the pneumotaxic center.
Pneumotaxic Center
The pneumotaxic center is a bilateral group of neurons located in the upper pons (see Figure 14-1). The pneumotaxic center controls the “switch-off” point of the inspiratory ramp, controlling inspiratory time. Strong pneumotaxic signals increase the respiratory rate, and weak signals prolong inspiration and increase tidal volumes. The exact nature of the interaction between the pneumotaxic and apneustic centers is poorly understood. They apparently work together to control the depth of inspiration.3
Reflex Control Of Breathing
Hering-Breuer Inflation Reflex
The Hering-Breuer inflation reflex, described by Hering and Breuer in 1868, is generated by stretch receptors located in the smooth muscle of both large and small airways. When lung inflation stretches these receptors, they send inhibitory impulses through the vagus nerve to the DRG neurons, stopping further inspiration. In this way, the Hering-Breuer reflex has an effect similar to that of the pneumotaxic center. In adults, the Hering-Breuer reflex is activated only at large tidal volumes (≥800 to 1000 ml) and apparently is not an important control mechanism in quiet breathing.2 This reflex is important, however, in regulating respiratory rate and depth during moderate to strenuous exercise.
Deflation Reflex
Sudden collapse of the lung stimulates strong inspiratory effort. This inspiratory effort may be the result of decreased stretch receptor activity, or it may be caused by the stimulation of other receptors, such as the irritant receptors and J-receptors (discussed later). Although it is unclear which receptors are involved, it is clear that the vagus nerve is the pathway (as it is for the Hering-Breuer reflex) and that the effect is hyperpnea.1 The deflation reflex is probably responsible for the hyperpnea observed with pneumothorax (air in the pleural space).
Head Paradoxical Reflex
In 1889, Head observed that if the Hering-Breuer reflex is blocked by cooling the vagus nerve, lung hyperinflation causes a further increase in inspiratory effort—the opposite of the Hering-Breuer reflex. The receptors for this reflex are called rapidly adapting receptors because they stop firing promptly after a volume change occurs. The Head reflex may help maintain large tidal volumes during exercise and may be involved in periodic deep sighs during quiet breathing. Periodic sighs help prevent alveolar collapse, or atelectasis. The Head reflex also may be responsible for the first breaths of a newborn.1
Peripheral Proprioceptors
Proprioceptors in muscles, tendons, and joints and pain receptors in muscles and skin send stimulatory signals to the medullary respiratory center. Such stimuli increase medullary inspiratory activity and cause hyperpnea.4 For this reason, moving the limbs, slapping or splashing cold water on the skin, and other painful stimuli stimulate ventilation in patients with respiratory depression.
Proprioceptors in joints and tendons may be important in initiating and maintaining increased ventilation at the beginning of exercise. Passive limb movement around a joint increases breathing rate in both anesthetized animals and unanesthetized humans.4
Muscle Spindles
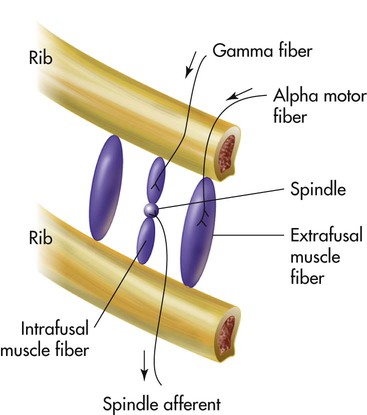
Muscle spindles in the diaphragm and intercostal muscles are part of a reflex arc that helps the muscles adjust to an increased load. Muscle spindles are sensing elements located on intrafusal muscle fibers, arranged parallel to the main extrafusal muscle fibers (Figure 14-3). The extrafusal fibers that elevate the ribs are innervated by different motor fibers (alpha fibers) than the fibers that innervate the intrafusal spindle fibers (gamma fibers). When the main extrafusal muscle fiber and the intrafusal fibers contract simultaneously, the sensing element (spindle) of the intrafusal muscle fiber stretches and sends impulses over spindle afferent nerves directly to the spinal cord (see Figure 14-3). The spindle’s afferent (sensory) nerve synapses directly with the alpha motor neuron in the spinal cord, sending impulses back to the main extrafusal muscle. A single synapse reflex arc is created. Alpha motor neuron impulses cause the main extrafusal muscle fibers to contract with greater force, shortening the nearby intrafusal fibers. The stretch-sensitive spindle is unloaded, and its impulses cease. In this way, inspiratory muscle force adjusts to the load imposed by decreased lung compliance or increased airway resistance.
Chemical Control Of Breathing
The body maintains the proper amounts of oxygen (O2), carbon dioxide (CO2), and hydrogen ions (H+) in the blood mainly by regulating ventilation. Physiologic mechanisms that monitor these substances in the blood allow ventilation to respond appropriately to maintain homeostasis. An increase in blood H+ concentration stimulates specialized nerve structures called chemoreceptors. Consequently, the chemoreceptors transmit impulses to the medulla, increasing ventilation. Centrally located chemoreceptors in the medulla respond to H+, which normally arises from dissolved CO2 in the cerebrospinal fluid (CSF). Peripherally located chemoreceptors in the fork of the common carotid arteries and the aortic arch are also sensitive to H+ and indirectly to CO2. These receptors are also indirectly sensitive to hypoxemia because hypoxemia increases the sensitivity of the peripheral chemoreceptors to H+.2
Central Chemoreceptors
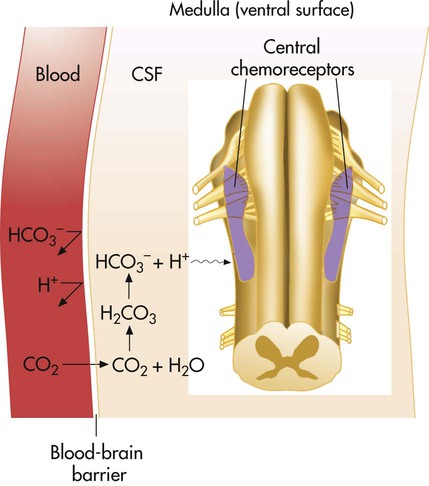
Hydrogen ions, not CO2 molecules, stimulate highly responsive chemosensitive nerve cells, located bilaterally in the medulla. Nevertheless, these central chemoreceptors are extremely sensitive to CO2 in an indirect fashion. The chemoreceptors are not in direct contact with arterial blood (Figure 14-4). Instead, they are bathed in the CSF, separated from the blood by a semipermeable membrane called the blood-brain barrier. This membrane is almost impermeable to H+ and HCO3−, but it is freely permeable to CO2. When PaCO2 increases, CO2 diffuses rapidly through the blood-brain barrier into the CSF. In the CSF, CO2 reacts with water (H2O) to form H+ and HCO3− (see Figure 14-4). The H+ generated in this fashion stimulates the central chemoreceptors, which stimulate the medullary inspiratory neurons. PaCO2 is indirectly the primary minute-to-minute controller of ventilation. CO2 diffusing from the blood into the CSF increases [H+] almost instantly, exciting the chemoreceptors within seconds. Alveolar ventilation increases by approximately 2 to 3 L/min for each 1-mm Hg increase in PaCO2.5
Peripheral Chemoreceptors
The peripheral chemoreceptors are small, highly vascular structures known as the carotid and aortic bodies. The carotid bodies are located bilaterally in the bifurcations of the common carotid arteries. The aortic bodies are found in the arch of the aorta. These neural structures increase their firing rates in response to increased arterial [H+] regardless of its origin (i.e., whether from fixed acid accumulation or increased CO2). The carotid bodies send their impulses to the respiratory centers in the medulla via the glossopharyngeal nerve, whereas the aortic bodies send their impulses over the vagus nerve. The carotid bodies exert much more influence over the respiratory centers than the aortic bodies do, especially with respect to arterial hypoxemia and acidemia.1
Response to Decreased Arterial Oxygen
Traditionally, it was believed that the carotid bodies directly sense low PaO2, implying that arterial hypoxemia represents an independent drive to breathe—the “hypoxic drive.” Although the peripheral chemoreceptors fire more frequently in the presence of arterial hypoxemia, they do so only because hypoxemia makes them more sensitive to H+.2 That is, when PaO2 is low, carotid body sensitivity to a given [H+] increases; in this way, hypoxia increases ventilation for any given pH. Conversely, elevated PaO2 (hyperoxia) decreases carotid body sensitivity to [H+]. The carotid bodies respond to arterial hypoxemia only because hypoxia makes them more sensitive to [H+]. This means that if the arterial [H+] is extremely low (high pH), as in severe alkalemia, hypoxemia has little effect on the carotid bodies.2 Simply stated, the ultimate effect of hypoxemia is to increase the sensitivity of the peripheral chemoreceptors to the given blood [H+], which increases their firing rate and brings about increased ventilation.
Because of their extremely high blood flow rates, the carotid bodies respond to decreased arterial partial pressure of O2 (in the indirect way just described) rather than to an actual decrease in arterial O2 content. That is, the extraction of O2 by the carotid bodies from each unit of rapidly flowing blood is so small that their O2 needs are met entirely by dissolved O2 in the plasma. This is why conditions associated with low arterial O2 content but normal PaO2 (e.g., anemia and carbon monoxide poisoning) do not stimulate ventilation.5
When pH and PaCO2 are normal (pH = 7.40 and PaCO2 = 40 mm Hg), the nerve-impulse transmission rate of the carotid bodies does not increase significantly until the PaO2 decreases to about 60 mm Hg.5 If PaO2 decreases further from 60 mm Hg to 30 mm Hg, the rate of impulse transmission increases sharply and linearly because hypoxemia makes the carotid bodies much more sensitive to a pH of 7.40. A decrease in PaO2 from 60 mm Hg to 30 mm Hg corresponds to the sharpest decrease in O2 content on the O2-hemoglobin equilibrium curve (i.e., the steepest part of the curve). Arterial hypoxemia does not stimulate ventilation greatly until the PaO2 decreases to less than 60 mm Hg. O2 plays no role in the drive to breathe in healthy individuals at sea level. High altitude causes a healthy person’s ventilation to increase because low barometric pressure decreases the inspired PO2 and the arterial PO2, which increases the sensitivity of peripheral chemoreceptors to their H+ environment. The resulting increase in ventilation is less than expected, however, because hyperventilation decreases PaCO2 and increases arterial pH. The increased pH depresses the medullary respiratory center, counteracting the excitatory effect of a low PaO2 on peripheral chemoreceptors. Hypoxemia-mediated hyperventilation may be impossible in certain conditions, such as severe chronic obstructive pulmonary disease (COPD), in which lung mechanics are so deranged that the stimulatory effect of hypoxemia on ventilation fails to decrease PaCO2 regardless of the patient’s effort. In such instances, there is no alkalosis to counteract the stimulatory effects of hypoxemia on ventilation.
 Rule of Thumb
Rule of ThumbResponse to Increased PaCO2 and Hydrogen Ions
For a given increase in PaCO2 or [H+], the carotid bodies are less responsive than the central chemoreceptors. The peripheral chemoreceptors account for only 20% to 30% of the ventilatory response to hypercapnia.5 However, they respond to increased arterial [H+] more rapidly than do the central chemoreceptors. The explanation is that, in contrast to the central chemoreceptors, the carotid bodies are exposed directly to arterial blood. The body’s initial ventilatory response to metabolic acidosis is fairly quick, even though H+ crosses the blood-brain barrier with difficulty.
As stated earlier, hypoxemia increases the sensitivity of the peripheral chemoreceptors to H+ and to PaCO2. Conversely, high PaO2 (hyperoxia) decreases the peripheral chemoreceptors’ PCO2 sensitivity to almost zero.2 This means that when the PaO2 is high, the ventilatory response to PaCO2 is mainly due to the central chemoreceptors, which are unaffected by hypoxemia.
Because the only effect of hypoxia on the peripheral chemoreceptors is to increase their sensitivity to arterial [H++]—and indirectly to PaCO2—the following statements are true: (1) High PO2 renders the peripheral chemoreceptors almost unresponsive to PCO2, and (2) low PaCO2 renders the peripheral chemoreceptors almost unresponsive to hypoxemia.2 Coexisting arterial hypoxemia, acidemia, and high PaCO2 (i.e., asphyxia) maximally stimulate the peripheral chemoreceptors.
 Problem
ProblemIndividuals with chronic hypercapnia secondary to advanced COPD have depressed ventilatory responses to acute increases in arterial CO2, partly because of their altered acid-base status and partly because their deranged lung mechanics prevents them from increasing their ventilation adequately.1 The altered acid-base status arises from the preexisting high levels of blood buffer base, a compensatory response to chronic respiratory acidosis (see Chapter 13).
 Rule of Thumb
Rule of ThumbOxygen-Associated Hypercapnia
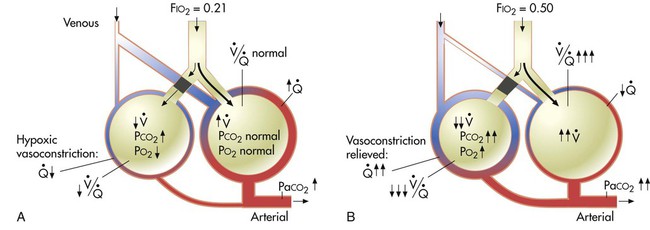
PaCO2 of chronically hypercapnic patients with COPD often increases acutely after these patients are given O2. The reason for this phenomenon continues to be a subject of much debate and misunderstanding. The traditional explanation for this phenomenon is that O2 breathing removes the hypoxic ventilatory stimulus and induces hypoventilation, but this explanation is probably overly simplified. (This explanation has been challenged only in the context of chronically hypercapnic and hypoxemic patients with COPD; no investigator has suggested a different kind of explanation for why severely hypoxemic, hyperventilating patients with no history of chronic hypercapnia decrease their ventilation and increase their PaCO2 values after breathing supplemental O2. That is, if one accepts that hypoxemia stimulates ventilation as earlier described, it logically follows that the removal of that stimulus via O2 administration would cause ventilation to decrease and PaCO2 to increase; however, when a chronically hypercapnic patient is involved, many investigators completely discount this mechanism as playing a causative role.) Nevertheless, the reduction in minute ventilation after O2 breathing in patients with advanced COPD is not always severe enough to account for the increased PaCO2.6,7 Some investigators suggest that O2 breathing worsens the ventilation/perfusion ratio (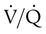 ) relationships in the lungs and is responsible for the increase in PaCO2.6,7 Other investigators have suggested that O2-induced hypercapnia is caused by the combined effects of hypoxic stimulus removal and redistribution of
) relationships in the lungs and is responsible for the increase in PaCO2.6,7 Other investigators have suggested that O2-induced hypercapnia is caused by the combined effects of hypoxic stimulus removal and redistribution of 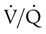 relationships in the lungs.8,9
relationships in the lungs.8,9
Breathing O2 worsens the already poor 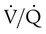 relationships in patients with COPD because it abolishes hypoxic pulmonary vasoconstriction in poorly ventilated lung regions. As a result, vascular resistance of underventilated regions decreases, and they receive more blood flow, drawing blood away from well-ventilated regions (Figure 14-5). At the same time that poorly ventilated regions receive more blood flow, they become even less ventilated as O2-rich inspired gas washes out resident nitrogen gas, making these alveoli more subject to absorption atelectasis (i.e., O2 may be absorbed by the pulmonary circulation more rapidly than the slowed ventilation can replenish it—notice the further decreased
relationships in patients with COPD because it abolishes hypoxic pulmonary vasoconstriction in poorly ventilated lung regions. As a result, vascular resistance of underventilated regions decreases, and they receive more blood flow, drawing blood away from well-ventilated regions (Figure 14-5). At the same time that poorly ventilated regions receive more blood flow, they become even less ventilated as O2-rich inspired gas washes out resident nitrogen gas, making these alveoli more subject to absorption atelectasis (i.e., O2 may be absorbed by the pulmonary circulation more rapidly than the slowed ventilation can replenish it—notice the further decreased 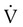 in Figure 14-5, B). As a result, inspired gas tends to flow preferentially to the already well-ventilated alveoli (see Figure 14-5, B), increasing their
in Figure 14-5, B). As a result, inspired gas tends to flow preferentially to the already well-ventilated alveoli (see Figure 14-5, B), increasing their 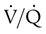 . The increased
. The increased 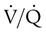 in these alveoli is exaggerated further as a greater proportion of the cardiac output than before is redirected to poorly ventilated alveoli, whose vascular resistance is reduced by O2 breathing.
in these alveoli is exaggerated further as a greater proportion of the cardiac output than before is redirected to poorly ventilated alveoli, whose vascular resistance is reduced by O2 breathing.
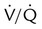 mismatches. A, Low
mismatches. A, Low 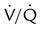 unit (left) is hypoxic and hypercapnic while breathing ambient air; this induces pulmonary vasoconstriction. B, Breathing 50% O2 predisposes the poorly ventilated unit to absorption atelectasis, further decreasing its ventilation, and simultaneously relieves hypoxic vasoconstriction, increasing its blood flow. These events (1) lower the poorly ventilated unit’s
unit (left) is hypoxic and hypercapnic while breathing ambient air; this induces pulmonary vasoconstriction. B, Breathing 50% O2 predisposes the poorly ventilated unit to absorption atelectasis, further decreasing its ventilation, and simultaneously relieves hypoxic vasoconstriction, increasing its blood flow. These events (1) lower the poorly ventilated unit’s 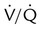 ratio further and (2) divert blood flow away from and ventilation toward already well-ventilated units. The latter increases alveolar dead space (high
ratio further and (2) divert blood flow away from and ventilation toward already well-ventilated units. The latter increases alveolar dead space (high 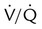 ).
). Although some investigators believe the mechanisms just described explain why O2 administration is associated with hypercapnia in COPD, other studies support an equally important role for O2 suppression of the hypoxic ventilatory stimulus.8,9 These studies show that O2 administration in acutely ill, chronically hypercapnic patients with COPD significantly reduces ventilation and increases the PaCO2 level needed to stimulate ventilation.
Central Chemoreceptor Response to Acute Carbon Dioxide Increase in Chronic Hypercapnia
As discussed earlier, the kidneys compensate for the acidic effects of chronic hypercapnia by increasing the plasma bicarbonate level, keeping the medullary chemoreceptor pH environment in the normal range. This does not mean that the medullary chemoreceptors cannot respond to further acute increases in PaCO2. A sudden elevation in PaCO2 immediately crosses the blood-brain barrier into the CSF, generating H+ that subsequently stimulates the medullary chemoreceptors. The resulting ventilatory response is depressed, however, for chemical and mechanical reasons: (1) The blood’s increased buffering capacity (high HCO3− level) in chronic hypercapnia prevents arterial pH from decreasing as sharply as it would in normal conditions, and (2) abnormal breathing mechanics impair the lung’s ability to increase ventilation appropriately. To illustrate the blood’s changed buffering capacity, compare a healthy person (pH = 7.40, PaCO2 = 40 mm Hg, HCO3− = 24 mEq/L) with a chronically hypercapnic person (pH = 7.38, PaCO2 = 60 mm Hg, HCO3− = 34 mEq/L). A sudden 30-mm Hg increase in PaCO2 of both individuals causes the healthy person’s arterial pH to decrease to 7.21 and the hypercapnic person’s pH to decrease to only 7.24. (These values are calculated using the Henderson-Hasselbalch equation, assuming a 1-mEq/L increase in plasma HCO3− concentration for each acute 10-mm Hg increase in PaCO2.) The central chemoreceptors of a chronically hypercapnic patient experience less stimulation than the central chemoreceptors of normal individuals for the same increase in PaCO2. Several investigators have confirmed the reduced ventilatory response to CO2 in chronic hypercapnia.10,11
 Rule of Thumb
Rule of Thumb
Ventilatory Response To Exercise
Strenuous exercise can increase CO2 production and O2 consumption by 20-fold.3 Ventilation normally keeps pace with CO2 production, keeping PaCO2, PaO2, and arterial pH constant. Because arterial blood gases do not change during normal exercise, some other mechanism must be responsible for the increased ventilation in healthy individuals during exertion.
The exact mechanism responsible for this increase in ventilation is not well understood. Especially mysterious is the abrupt increase in ventilation at the onset of exercise, long before any chemical or humoral changes can occur in the body. Two predominating theories for this phenomenon are the following: (1) When the cerebral motor cortex sends impulses to exercising muscles, it apparently sends collateral excitatory impulses to the medullary respiratory centers; (2) exercising limbs moving around their joints stimulate proprioceptors, which transmit excitatory impulses to the medullary centers.1,3 Evidence also suggests that the sudden increase in ventilation at the onset of exercise is a learned response.1,3 With repeated experience, the brain may learn to anticipate the proper amount of ventilation required to maintain normal blood gases during exercise.
Abnormal Breathing Patterns
Commonly described abnormal breathing patterns include Cheyne-Stokes respiration, Biot respiration, apneustic breathing, and central neurogenic hypoventilation and hyperventilation. In Cheyne-Stokes respiration, respiratory rate and tidal volume gradually increase and then gradually decrease to complete apnea (absence of ventilation), which may last several seconds. Tidal volume and breathing frequency gradually increase again, repeating the cycle. This pattern occurs when cardiac output is low, as in congestive heart failure, delaying the blood transit time between the lungs and the brain.4 In this instance, changes in respiratory center PCO2 lag behind changes in PaCO2.
Biot respiration is similar to Cheyne-Stokes respiration except that tidal volumes are of identical depth. It occurs in patients with increased intracranial pressure (ICP), but the mechanism for this pattern is unclear.4
Apneustic breathing indicates damage to the pons. Central neurogenic hyperventilation is characterized by persistent hyperventilation driven by abnormal neural stimuli. It is related to midbrain and upper pons damage associated with head trauma, severe brain hypoxia, or lack of blood flow to the brain.12 Conversely, central neurogenic hypoventilation means the respiratory centers do not respond appropriately to ventilatory stimuli, such as CO2. It also is associated with head trauma and brain hypoxia as well as narcotic suppression of the respiratory center.12
Carbon Dioxide And Cerebral Blood Flow
CO2 plays an important role in regulating cerebral blood flow. Its effect is mediated through the formation of H+ by CO2.13 Increased PCO2 dilates cerebral vessels, increasing cerebral blood flow, whereas decreased PCO2 constricts cerebral vessels and reduces cerebral blood flow. In patients with traumatic brain injury (TBI), the brain swells acutely; this increases the ICP in the rigid skull to such high levels that blood supply to the brain might be cut off, causing cerebral hypoxia (ischemia). That is, high ICP may exceed cerebral arterial pressure and stop blood flow to the brain.
Mini Clini
Mechanical Hyperventilation of a Patient With Traumatic Brain Injury
 Problem
ProblemMore than 40 years ago, clinical investigators showed that the volume of the swollen brain could be reduced by decreasing the PaCO2. Since then, mechanical hyperventilation has been a cornerstone in managing increased ICP associated with TBI.13 Hyperventilation decreases ICP by causing cerebral vasoconstriction, ultimately reducing cerebral blood volume. This subject is not without controversy; hyperventilation-induced cerebral vasoconstriction has the potential to reduce cerebral blood flow to levels that cause cerebral hypoxia (ischemia). Over the last decade, this concern has dampened enthusiasm for hyperventilation in TBI. Both the proponents and the opponents of hyperventilation recognize that TBI poses an ischemic threat to the brain; proponents believe that the reduction of cerebral blood flow ultimately improves cerebral oxygenation by reducing the ICP, which helps sustain the cerebral perfusion pressure. Opponents point out that no other hypoxic organ in the body is treated by reducing its blood flow and O2 supply. (Hyperventilation in this context is generally defined as PaCO2 < 35 mm Hg.13)
The debate centers around the question of whether a hyperventilation-induced decrease in cerebral blood flow creates an additional hypoxic insult to the already ischemic brain and whether patients managed in this way have better clinical outcomes than patients in whom hyperventilation is not instituted. A comprehensive review of the subject published in 2005 concluded that hyperventilation produced no advantage in long-term clinical outcome of TBI compared with ventilation that maintained PaCO2 in the normal range.13 The authors concluded that in TBI, hyperventilation therapy should be considered only for patients with high ICPs; no benefit can be expected if ICP is normal. They further concluded that hyperventilation is most appropriate in the second or third day after injury because cerebral blood flow is lowest in the first 24 hours after injury, and the risk of inducing ischemia through hyperventilation is greatest during this time. The authors advise against the hyperventilation of TBI patients to PaCO2 less than 30 mm Hg because of the increased danger of cerebral ischemia. Finally, hyperventilation is effective for only about 24 to 48 hours because compensatory renal elimination of bicarbonate in the face of alkalemia restores the acid-base balance, negating the vasoconstrictive effect of hypocapnia. In any case, hypoventilation in patients with head trauma and increased ICPs is especially dangerous because hypercapnia dilates cerebral vessels and increases ICP further. Even opponents of hyperventilation generally maintain PaCO2 of TBI patients in the low-normal range around 35 mm Hg.