[level-membership-for-neurosurgery-category]
CHAPTER 328 Regeneration and Repair
Historically, neurosurgical interventions for traumatic brain injury (TBI) have centered on strategies to reduce sequelae of the primary brain insult. Despite progress in understanding the pathophysiology of TBI, clinical neuroprotection trials targeting these secondary injury mechanisms have failed to show consistent improvement in the outcomes of head-injured patients (at least 21 multicenter clinical trials have been conducted since 1985).1,2 In addition to local neuronal destruction resulting from focal injury, mechanical forces secondarily induce a progressive cascade of related events that contribute to neuronal death, including ischemia, brain edema, diffuse axonal injury (DAI), excitotoxicity, radical-mediated damage, mitochondrial dysfunction, and dysregulation of calcium homeostasis.3,4 In response to these processes, however, the injured brain activates a limited program for plasticity and cellular regeneration. Demonstration of these latent mechanisms in animal and human studies has spawned great interest in developing techniques to facilitate repair of the neuronal circuitry damaged by TBI.
Restorative strategies represent a shift in therapeutic goals from focusing solely on salvaging acutely threatened tissue to introducing interventions that support spontaneous and directed functional recovery (Fig. 328-1). Much of the evidence for spontaneous recovery of the damaged cerebrum comes from the stroke literature, where studies in animal models have provided cellular and molecular information, whereas systems-level data are increasingly being obtained from neuroimaging or neurophysiology studies in patients. Clinically, several basic principles of recovery have been indentified: most spontaneous recovery occurs within 3 to 6 months, cognitive deficits are more likely than motor deficits to show further gain beyond this point, the rate of recovery is inversely proportional to the severity of the deficit, and recovery patterns vary between types of deficits in the same patient.5 These observations suggest multiple mechanisms of recovery whose effectiveness varies with the type and location of the physical injury, as well as with the type and severity of the clinical findings. These mechanisms can be generalized to three basic categories: plasticity of intact networks, repair of damaged circuitry, and replacement of lost neurons. This chapter describes the opportunities and obstacles inherent in potential therapeutic strategies for repair and regeneration after TBI, including manipulation of endogenous neurogenesis, cell transplantation, gene therapy, neurorehabilitation, and electrophysiologic manipulation.
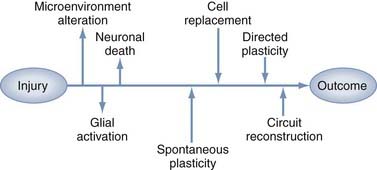
Trauma-Induced Plasticity
The brain activates a dramatic response to traumatic injury that is initially manifested as changes in the cellular microenvironment. These changes involve inflammation, disruption of physical structure and protein expression in the extracellular matrix (ECM), and altered expression of trophic factors. Although some of these mechanisms attenuate acute damage at the expense of future regenerative capacity, others retain the potential to participate in therapeutic interventions. Plasticity occurs when injury induces reorganization of cortical connections, but plasticity is not always synonymous with functional recovery. Even though processes such as synaptic sprouting, unmasking of dormant circuits, and the development of new polysynaptic connections can enable function, plasticity can also restrict function, as occurs when posttraumatic epileptic seizure foci or neuropathic pain is produced.6 This dichotomy is most evident in the developing brain, where increased plasticity has traditionally been thought to confer an advantage for recovery from TBI. Recovery also strongly depends on the type of injury,7 and the relationship between age and functional outcome is different within pediatric and adult age groups.8 These points reiterate the fact that plasticity is a complex series of molecular, cellular, and physiologic events that must be carefully orchestrated for both optimal maturation of the developing brain and optimal functional recovery from traumatic injury.9
The differences between injury responses in the developing and mature brain are also quite important when one considers that functional recovery after injury may require recapitulation of developmental events to produce neuronal circuit reconnections. Work in animal models has shown that focal damage in the adult brain can lead to a number of molecular and cellular changes, in both perilesional and remote brain regions, that are normally seen only in the developing brain.10 These distinctions should be kept in mind when considering how plasticity is altered by injury-induced microenvironmental changes in neurotransmission, gene and protein expression, cell death, neurogenesis, and neural connectivity.
The Injury Microenvironment: A Double-Edged Sword
In addition to local neuronal destruction resulting from the primary mechanical insult, TBI induces a progressive cascade of delayed secondary events that contribute to neuronal death, including ischemia, “wallerian degeneration” secondary to DAI, excitotoxicity, free radical–mediated damage, mitochondrial dysfunction, and dysregulation of calcium homeostasis. Injury may occur in the form of focal damage, as typically occurs after acute subdural hematoma or contusion, or it may be diffuse with widespread delayed neuronal loss, as typically occurs after DAI. Focal damage is characteristically seen around hemorrhagic lesions such as contusions within the gray matter or at gray-white matter junctions. These lesions are usually located at the frontal and temporal poles and in the orbital frontal cortex.11 Such focal neuronal death may occur as a result of both rapid necrotic and slower apoptotic mechanisms.12,13 Among diffuse injury sites, the hippocampus is known to be especially vulnerable in humans, with neuronal loss occurring in more than 80% of fatal TBIs, even in the absence of elevated intracranial pressure.14,15 After the acute phase of focal TBI and brain ischemic events, hippocampal neurons may be the most vulnerable neurons in the brain in that they show the earliest evidence of TBI-induced degeneration in experimental models.16 These hippocampal changes correlate with the profound memory impairment that is seen in both human and animal models of TBI.17,18 In the two most frequently used experimental TBI models, the lateral fluid percussion (LFP) and controlled cortical impact (CCI) injury models, selective neuronal death has also been well described in the hippocampus.19
The slow diffuse neuronal loss that is often seen in DAI, involving apoptotic morphology and wallerian degeneration, was previously thought to be an inevitable result of axonal interruption, as seen with primary axotomy of central nervous system (CNS) neurons. More recently, animal studies of DAI have shown that despite the proximity of traumatic axonal injury to the neuronal somata, neuronal cell bodies do not always show a pathologic progression to cell death and in fact some neurons and axons exhibit changes suggestive of reorganization and potential repair.20 These data suggest that injured neurons may be amenable to rescue through augmentation of endogenous plasticity. Therapeutic interventions, however, must account for changes in the injury microenvironment that encompass barriers to both functional reconnection and opportunities for repair (Table 328-1), as discussed in the following sections.
TABLE 328-1 Microenvironmental Factors and Potential Role in Neuroplasticity
| RESPONSE | DISABLING ROLE | ENABLING ROLE |
|---|---|---|
| Inflammation | Cytokine release Complement activation |
Glial activation Microglial activation |
| Reactive gliosis | Neurite inhibition Dysmyelination |
Neurotrophin secretion Neurotransmitter regulation |
| ECM deposition | Neuropil distortion | Growth permission |
| MMP activation | Synapse dysregulation | Substrate guidance |
| Neurogenesis | Seizure potential | Cell replacement |
ECM, extracellular matrix; MMP, matrix metalloproteinase.
Inflammatory Mechanisms and Gliosis
TBI produces a neuroinflammatory condition of the CNS in which rupture of the blood-brain barrier leads to accumulation of leukocytes from the systemic circulation and subsequent initiation of the immune functions of native glia.21 Acute inflammatory cytokine-mediated events such as monocyte/macrophage-mediated phagocytosis22 and complement-mediated cytolysis23 are now well recognized early after TBI and persist at least for several days and weeks. This response is partially represented in the form of “microglial stars” and “perivascular cuffing” in human pathologic specimens.24 Additionally, in a primate cortical lesion model, microglia and macrophages remain reactive in the cortex near the lesion and along sites of wallerian degeneration (corpus callosum and corticospinal tracts) for at least 12 months.25 The expression of brain-derived neurotrophic factor (BDNF) by a subpopulation of microglia/macrophages at this late time point suggests a possible neurotrophic role for microglia in long-term recovery.
Both inflammatory mechanisms and necrotic cell death trigger reactive gliosis, the astrocytic response to injury that consists of astrocyte hypertrophy, activation of fibrillary processes, and penumbral proliferation.26 Staining of glial fibrillary acidic protein (GFAP) increases dramatically in many areas of the brain days, weeks, and months after severe TBI.27 Upregulation of intermediate filaments, a hallmark of reactive astrocytes, was traditionally thought to be an obstacle to brain plasticity. In mice lacking GFAP and vimentin, however, the cellular processes of reactive astrocytes fail to exhibit the characteristic hypertrophy after entorhinal cortex lesions, as well as the initial increased loss of hippocampal neuronal synapses.28 Although these findings suggested that reactive astrocytes may play a beneficial role in the acute stage after a CNS insult, within a few days of injury, normal animals showed decreased synaptogenesis in comparison to animals lacking the two intermediate filament proteins, thus indicating a prohibitive effect of astrocytic hypertrophy at subacute stages.
The findings just presented support the idea that the beneficial effects of astrocytes at the site of an injury probably occur early in the injury response whereas subacute formation of the glial scar hinders regeneration. The beneficial effects of an astrocytic response include secretion of neurotrophic factors, regulation of metabolic factors (particularly important in times of stress), and maintenance of homeostatic levels of neurotransmitters.29 Astrocytes play a central role in neurometabolic coupling involving glutamate-stimulated aerobic glycolysis, a process that undergoes adaptations in parallel to synaptic plasticity.30 In both immature and adult TBI models, abnormalities have been reported in glutamatergic, GABAergic (transmitting γ-aminobutyric acid [GABA]), cholinergic, and aminergic transmission, whereas dysfunctional neurotransmission has been clinically implicated in pediatric memory impairment and attention problems.9 The detrimental effects of glial scars include inhibition of neurite outgrowth, inhibition of the remyelination of axons in white matter lesions, and hindrance of remyelination by inhibiting the differentiation of glial precursors into oligodendrocytes.29
Neural Connectivity
CNS neurons are intrinsically capable of axonal regeneration; however, this capacity rapidly diminishes postnatally at the onset of axonal myelination by mature oligodendrocytes. This loss of regenerative capacity can at least in part be attributed to the restriction of axonal regeneration and neuroplasticity by several CNS myelin proteins that inhibit neurite outgrowth, including Nogo-A, myelin-associated glycoprotein (MAG), and oligodendrocyte myelin.31 In neurons, Nogo-A is expressed during development and adulthood, whereas in oligodendrocytes, expression is initiated postnatally just before the appearance of myelin basic protein and myelination.32 With regard to TBI models, Nogo-A is upregulated after fluid percussion injury (FPI),33 and intraventricular infusion of an anti-Nogo antibody has been associated with improved postinjury cognitive recovery.34,35 Recently, it has become clear that the production of other inhibitory molecules, such as proteoglycans, is equally as important in creating a nonpermissive environment for axons attempting to regenerate.36
Mechanical obstruction of axonal regeneration by concomitant deposition of ECM molecules, which distorts the architecture of the neuropil, has also been well described. Conversely, levels of two ECM molecules critical to neural development, fibronectin and laminin, increase after human and experimental brain injury and may play a protective or reparative role in the injury response.37 The family of matrix metalloproteinases (MMPs) that regulate ECM molecules also performs essential functions during neuroplasticity in both developing and adult nervous systems, including substrate guidance during neuritogenesis and the establishment of boundaries for axonal terminal fields. In animals injured by entorhinal cortex lesions, MMPs regulate reactive synaptogenesis in a time- and injury-dependent manner to the extent that their inhibition interferes with emergence of the capacity for long-term potentiation in the sprouting crossed temporodentate pathway38 and produces deficits in spatial learning.39
The pervasive memory, cognitive, and motor deficits apparent in patients with mild to moderate TBI, sometimes occurring in the absence of frank neuronal cell loss, suggests that TBI alters the balance between excitatory and inhibitory neurotransmission in surviving neurons and disrupts the synaptic circuitry in the hippocampus.40 This effect occurs in a temporal and regional fashion such that in some pathways, injury may induce either increased excitability41 or inhibition42 of dentate gyrus granule cell neurons, thereby potentially affecting the ability of these cells to resist the synchronized bursting characteristic of seizure activity. In addition, the hippocampus ipsilateral to the cortical contusion is capable of a significant plasticity response, but synapse replacement in this area does not necessarily result in significant improvement in spatial learning.43
Gene and Protein Expression
Gene and protein profiling strategies have been used to study plasticity-related gene expression after experimental TBI. Several studies have shown that expression of neurotrophic factors is significantly altered after experimental TBI. RNA studies have demonstrated that some neurotrophic compounds such as nerve growth factor (NGF) and BDNF are upregulated44,45 whereas others such as NT-3 are downregulated.46,47 Data from the CCI model suggest that fibroblast growth factor 2 (FGF-2) is also upregulated, which stimulates posttraumatic neurogenesis and preserves granule cell layer volume, effects that are increased with FGF-2 supplementation via gene transfer.48 S100B is a neurotrophic and neuroprotective calcium-binding protein produced primarily by astrocytes that despite a correlation between increased cerebrospinal fluid levels and poor prognosis in TBI patients, has a beneficial effect on neuronal maintenance, neurogenesis, and cognitive performance in rodent TBI models.49
These alterations are not simple issues of upregulation versus downregulation because they occur with temporal, subregional, and age-related specificity. For instance, BDNF levels after experimental TBI are increased to a greater extent in aged animals than in young animals.50 BDNF is an activity-related molecule closely associated with experience-dependent plasticity and developmental regulation, and it may play a compensatory role after mild TBI in the developing rodent brain, where despite reductions in BDNF mRNA and protein expression in the ipsilateral cortex and hippocampus, contralateral upregulation occurs up to 2 weeks after injury.51 NGF-induced protein B, a transcription factor, is upregulated in a severity-dependent fashion in the immature brain, but it is increased independent of severity in adults. In contrast, the synaptic activity–inhibiting, calcium-binding protein synaptotagmin IV increases only in developing animals after severe injury and not at all in adults.52 Although these examples illustrate the complexity of the molecular injury response, they also suggest many opportunities to improve our understanding of important differences between developmental and injury-related plasticity.
Regional intrinsic differences in gene expression before and after injury are also likely to define the pathology and recovery of function after TBI. The differential vulnerability of the hippocampus to TBI as compared with the high degree of plasticity of the cortex, which may underlie the reason why memory dysfunction lingers in comparison to motor and sensory recovery, has been suggested by the fact that the majority of mRNA molecules that change after injury were found to be exclusively altered in either the hippocampus or the frontal cortex.53 Within the hippocampus itself, gene expression appears to be influenced by both injury severity and time since the insult. Many genes encoding molecules for cellular signaling, synaptic plasticity, metabolism, ion channels, and transporters that are initially upregulated after severe injury are downregulated after moderate injury, although moderate injury is associated with an increasing number of responsive genes as a function of time after injury as opposed to a decreasing number after severe injury.54 Overall, there is still much to discover about region- and time-specific microenvironmental dynamics, all of which combine to form various posttraumatic brain niches.
Endogenous Neurogenesis after Traumatic Brain Injury
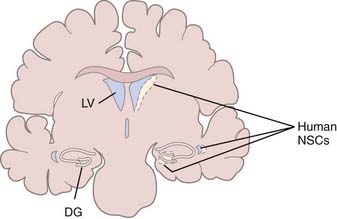
TBI induces changes in the specialized microenvironment that regulates the proliferation of neural stem cells (NSCs) and differentiation of neuronal progenitor cells (NPCs). These neurogenic niches are found in the human dentate gyrus of the hippocampus and the subventricular zone (SVZ) of the lateral ventricular walls (Fig. 328-2).55 The best-characterized neurogenic region in the adult human brain is the SVZ lining the lateral ventricle, where a ribbon of SVZ astrocytes have been identified that proliferate in vivo and behave as multipotent progenitor cells in vitro.56 Unlike the situation in rodents and primates, where neurons born in the SVZ migrate in chains through the rostral migratory stream to replace interneurons of the olfactory bulb,57–59 there is no evidence of chains of migrating neuroblasts in the human SVZ.60,61 It has been estimated that in normal humans, less than 1% of astrocytes within the SVZ ribbon are in division,56 and although these endogenous NSCs can be expanded in culture,62 their response to injury in patients has not been studied. Less well characterized in the human brain are proliferating NPCs in the hippocampus,63 which have also been demonstrated to be multipotent progenitor cells in vitro.64
Rat models have been used to investigate TBI-induced neurogenesis and have shown that the number of NPCs in both the SVZ and hippocampus are substantially increased after TBI (Fig. 328-3), a process that is more robust in juvenile animals.65 Newborn NPCs from the SVZ appear to have the potential to migrate to areas of focal cortical damage, whereas hippocampal neurogenesis occurs in the setting of diffuse local damage. Nevertheless, in adult and aged animals, the number of new neurons developing after TBI remains small in comparison to astrocyte and oligodendrocyte differentiation, as assessed by bromodeoxyuridine (BrdU) staining.
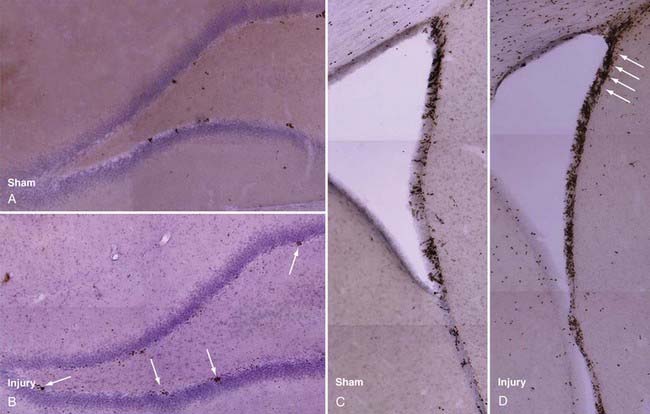
Subventricular Zone Neurogenesis after Trauma
In common with models of seizure, ischemia, and deafferentation injury, experimental TBI induces an increase in SVZ NPC proliferation and a secondary redirected streaming of these precursors into the injured zone.66 Studies demonstrate up to twofold increases in ipsilateral SVZ NPC proliferation within the first 2 to 5 days after FPI67–70 and as much as a fourfold increase after the more focal CCI up to 14 days after injury.71,72 Contralateral NPC proliferation in the SVZ also increases, apparently to a lesser extent, after injury in both models.69,70,72 Newborn cells colabeled with BrdU and neuronal phenotypic markers have been observed in the cortex, striatum, and corpus callosum as long as 45 days after injury. Patch clamp studies have demonstrated that such BrdU cells exhibit neuronal electrophysiologic properties,73 and anatomic integration of these new neurons into host tissue has been shown by retrograde tracer labeling and synaptophysin triple-label immunohistochemical techniques.74
Migration of Subventricular Zone Neural Progenitor Cells to the Site of Injury
Glial cell proliferation has long been appreciated at sites of focal TBI damage, but more recently, it has been shown that some glia migrate into the lesion site from the SVZ and that these cells arise from NPCs. After the intraventricular injection of fluorescent microspheres to label BrdU-positive subventricular cells, migration from the SVZ to the cortex surrounding a cortical contusion impact site has been observed.71 In the SVZ, corpus callosum, striatum, and cortex, many of these cells expressed doublecortin, a marker of immature neuroblasts, thus suggesting that local neurogenesis could be driven by injury-specific extracellular signaling pathways. In the pericontusional cortex, expression of the mature neuronal marker NeuN was seen in cells tagged before injury by intraventricular infusion of a lipophilic dye, a finding suggesting that these cells were derived from SVZ NPCs that migrated to the site of injury.75
Hippocampal Neurogenesis after Brain Trauma
Although selectively more vulnerable to TBI, the injured hippocampus may have a unique ability to locally replace damaged neurons inasmuch as the dentate gyrus is normally an area of active endogenous neurogenesis. In humans, in vivo dentate gyrus neurogenesis was demonstrated on histologic sections obtained from patients who had been administered BrdU for diagnostic purposes.63 Studies from rodents also demonstrated that cells with an astrocyte phenotype residing in the adult subgranular zone of the dentate gyrus continually generate cells with a neuroblast phenotype that migrate into the granule cell layer.76 Although some of these new neurons die by apoptosis, many of these new neurons demonstrate functional integration within the hippocampal circuitry by sending dendrites into the molecular layer and axons into the CA3 region,77,78 and they additionally exhibit mature neuronal electrophysiologic characteristics.79
Moderate and severe TBI is always associated with learning and memory deficits—the “functional hallmarks” of TBI. This is usually accompanied by histologic damage in the form of cell loss, especially in the dentate gyrus and CA1 layers. Because neurogenesis in this region has been linked to enhanced learning and memory functions,80 it is reasonable to hypothesize that injury-induced neurogenesis may function in the replacement of damaged neurons and thus contribute to repair of neuronal circuits and restoration of neurological function in patients with TBI. After LFP, cell proliferation increases in the rodent dentate gyrus threefold to fourfold beginning as early as 2 days after injury, peaks during the first week after injury, and returns to baseline levels by day 35 after injury.67,81 Similarly, CCI may induce up to a sixfold increase in dividing cells in the ipsilateral dentate gyrus and a significant but smaller increase in the contralateral dentate gyrus.48,72,82–84 A significant increase in the number of new cells that express neuronal markers has been reported after longer maturation periods. TBI can initially induce a fourfold increase in the number of BrdU-labeled cells that colabel for NeuN, a mature neuronal phenotypic marker, nearly half of which migrate to the inner half of the granular cell layer and persist 10 weeks after injury.74 Others have reported a fivefold increase in the number of injury-generated double-labeled neurons, seen as late as 60 days after injury.84 The central issue remains, however: do these new neurons functionally integrate with the host hippocampal tissue and exhibit appropriate electrophysiology?
To begin to answer these questions, hippocampal neurons born after injury, which express NeuN and calbindin, have been labeled by stereotactic retrograde tracer injections of the neuronal tracer fluorogold into the CA3 region, which is one target zone for the granular cell neuronal projections.74 After 2 weeks, about 30% of the BrdU-positive cells in the dentate gyrus were labeled with fluorogold, a finding suggestive of connectivity with the CA3 target region, and these double-labeled cells were increased fourfold by FPI. Labeled cell bodies were also frequently enveloped by a synaptophysin “lattice,” as described previously,78 thus suggesting the formation of synapses onto the somata of newborn neurons.
In terms of measuring whether posttraumatic neurogenesis is functional on a broader scale that affects behavior, memory function, as assessed by Morris water maze tasks, is decreased in association with procedures that abolish the proliferation of new cells, such as the administration of antimitotic drugs.84a Low-dose irradiation, which has been shown to markedly reduce neurogenesis, also significantly reduces the ability to recover spatial memory function after experimental TBI.85 Additionally, inactivation of a subset of adult hippocampal NSCs in mice was shown to produce both a significant reduction in overall stem cell proliferation and a selective decrement in spatial learning.85a
Cortical Neurogenesis after Brain Trauma
Although the hippocampus is selectively damaged in TBI, the greatest density of neuronal loss occurs with focal parenchymal contusions arising in locations that vary with the heterogeneity of both the primary injury and subsequent mechanism of secondary injury. As discussed earlier, the injured brain may induce migration of neuroblasts from the SVZ into cortical lesions, but latent NPCs may potentially be activated at sites of cortical injury in the mammalian brain. In songbirds such as zebra finches, it has long been known that some cortical projection neurons are lost and regenerated each year from SVZ precursors in response to evolutionary pressure.86 Macklis and colleagues suggested the possibility that endogenous neural precursors from the cortex itself in rodents may differentiate into corticothalamic neurons in layer VI of the anterior cortex after targeted neuronal death and survive at least long enough to form appropriate long-distance corticothalamic connections.87 In humans, the existence of such NPCs in nongerminal cortex has been suggested from some isolation and culture studies, including the isolation and continuous culture of putative neuroblasts from neocortical tissue surgically resected from TBI patients at the time of decompressive craniotomy for high intracranial pressure,88 as well as the isolation of neurosphere-generating cells from similar cortical tissue.89 After CCI in rodents, the formation of neurospheres from neocortical cells has also been observed, but only when the injured tissue was isolated at 3 days after injury, which corresponds to a peak in nestin expression in pericontusional cortex.90 Induction of nestin expression in the cortex adjacent to the CCI injury has also been reported at 7 days after injury,84 thus suggesting the possible activation of resident cortical NPCs, although in both cases these cells could alternatively have migrated from the SVZ.
Effect of Age on Posttraumatic Neurogenesis
Juvenile mammals usually recover cerebral function to a greater extent than adults do after TBI. Because cell proliferation in the dentate gyrus decreases with age in normal rats,91 it has been hypothesized that age-related differences in hippocampal neurogenesis after TBI may be responsible in part for the greater cognitive recovery typically seen in young mammals.81 Juvenile rats generate nearly twice the number of new cells in the subgranular zone than adults do in the first 2 days after LFP injury, although this response appears to be related to a greater level of basal juvenile neurogenesis because the percent increases in cell number above baseline are similar in young and old animals. Additionally, more newly generated cells appear to differentiate into mature neurons over time in juveniles. Typical granule cell neurons have been shown to functionally integrate by 14 days after generation in the normal adult rodent brain77 and become critically involved in the learning response within this same period.92 Fewer cognitive deficits and better recovery are observed during the first 2 weeks after injury in juvenile rats than in adults.93 This difference may therefore result in part from a greater ability of younger animals to generate new hippocampal granule cell neurons in response to injury. The mechanism by which neuronal differentiation is enhanced in younger animals is not clear, but answers to this question may aid discovery of therapeutic interventions to optimize neurogenesis after TBI.
Despite a constant decline in NPC proliferation in the dentate gyrus from adolescence through senescence,91,94,95 the adult brain upregulates proliferation to almost the same extent as the juvenile brain after TBI, thus indicating that NPCs conserve the ability to respond to proliferative signals. Although injury signals increase the proliferative rate of NPCs, the increased presence of actual newly generated neurons in younger brains may reflect greater integrity of the neurogenic niche mechanisms required for differentiation or survival of new neurons. One possible mechanism, similar to that hypothesized for the effects of an enriched environment on hippocampal neurogenesis (a survival-promoting effect of newborn cells that is selective for neurons),96 is a greater level of specific neurotrophin support97 that is either not present or not induced in adults. All of the putative mechanisms just described, however, would be nearly irrelevant without clinical evidence of functional recovery in the human brain after TBI.
Evidence of Functional Recovery
Spontaneous recovery from TBI varies with the heterogeneity of the injury, and consequently, a wide range of outcomes are produced: in some vegetative patients, functional magnetic resonance imaging (fMRI) demonstrates appropriate cortical responses to spoken commands that are indistinguishable from normal responses,98 whereas in patients with mild TBI, diffusion tensor imaging demonstrates variations in microstructural white matter integrity that are associated with persistent impairment in memory and attentional control.99 Approximately 85% of recovery from TBI occurs within 6 months after injury,100 and functional neuroimaging techniques have increasingly been used as a starting point to understanding the appropriate anatomic correlates.
Functional Imaging
The molecular events described in the previous section are difficult to measure in TBI patients, and therefore functional neuroimaging techniques have been used to investigate tissue function and neurophysiologic responsiveness after injury. Most TBI-related studies to date have involved fMRI of stroke patients. These studies suggest that the behavioral improvements seen in the weeks after injury are supported by compensatory reorganization of surviving neurostructural elements as follows: increased activity in brain regions distant from but connected to the injured zone, increased activity in the contralesional hemisphere (reduced laterality), and shifts in representational maps within intact cortical regions surrounding the injury zone.5 Each of these events may be a target for therapeutic intervention, although stroke studies assessing sensorimotor, language, and right hemisphere attentional deficits suggest that the final behavioral outcome is related to the degree of activity in the corresponding primary cortex.5
The motor system has been studied more extensively than other systems as a model for basic plasticity phenomena because control of motor behavior is so easily evident, but understanding the neurophysiology of motor function and the integral role of sensory input in motor control is fundamental for understanding general plasticity mechanisms.101 Transcranial magnetic brain stimulation (TMS) is used to study brain plasticity by applying a brief and intense magnetic field directly to the scalp and recording motor evoked potential responses to somatotopically map the cortical motor output. Magnetoencephalography is an additional imaging technique that spatially identifies the synchronous firing of neurons from restricted cortical areas in relation to either spontaneous cerebral activity or external stimuli, thereby allowing precise three-dimensional localization of the firing neuronal pool. By using these methods, it is possible to demonstrate lesion- or use-dependent changes in cortical maps. Combined-modality functional imaging studies suggest that reorganization of motor output is ongoing for several months after brain injury, and the mode and degree of motor recovery may largely depend on functional compensation by spared parallel descending pathways, activation of the contralateral premotor cortex, and cerebellar activation.101
Effect of Neuronal Stimulation
Cortical reorganization after neurological injury may be classified as function-enabling or function-disabling plasticity. The former leads to behavioral improvement, such as changes in cortical representation and functional gain with the use of an affected extremity, whereas the latter results in deterioration of function, such as the appearance of seizures after brain injury.102 Injury-induced microenvironmental pathways are dynamic and responsive to neuronal stimulation triggered by external factors such as physical activity and environmental enrichment. The clinical effects of stimulation-triggered neuroplasticity have been examined mainly at the behavioral level. Neurorehabilitation interventions based on this concept have shown some success, although more work is needed to elucidate the mechanisms underlying functional recovery. The positive effect of motor practice and intensive training on system reorganization in the injured brain has been demonstrated after constraint-induced movement therapy103 and intensive language training,104 whereas noninvasive cortical stimulation and modulation of somatosensory input have shown that motor and the somatosensory cortices display an interconnected capacity for plasticity.105 N-methyl-D-aspartate (NMDA) receptor activation and GABAergic inhibition have been indentified as mechanisms operating in use-dependent plasticity in the intact human motor cortex, thus suggesting similarities in the mechanisms underlying this form of plasticity and long-term potentiation, with GABA agonists and NMDA and muscarinic antagonists exerting a deleterious effect on human plasticity and drugs with adrenergic or dopaminergic function enhancing use-dependent plasticity.106
Animal studies measuring the effects of environmental complexity on recovery from injury have begun to demonstrate molecular-level, stimulation-triggered, pro-neuroplastic events. Exposure to environmental complexity enhances recovery of cognitive function after FPI107 and reverses motor deficits in the setting of reduced scar formation.108 Neurotrophin upregulation, such as increased NGF gene expression and protein levels in the hippocampus of animals exposed to an enriched environment,109 probably plays a role in the enhanced posttraumatic plasticity. Exercise has also been shown to block injury-related increases in the neurite-inhibiting proteins MAG and Nogo-A via a BDNF-dependent pathway.110 The timing of exposure to environmental stimulation is important. Injury severity appears to determine the time window for exercise-induced increases in BDNF, synapsin I, and cyclic adenosine monophosphate response element–binding protein, with delayed onset of stimulation being required after severe injury. Additionally, environmental enrichment after TBI enhances dendritic branching, promotes the survival of progenitor cells, and decreases dopamine transporter levels.111 Investigation of gene-environment interactions to identify molecular targets for the development of therapeutic agents that mimic or enhance the beneficial effects of experimental environmental stimulation will be necessary to bridge these experimental findings to the human condition.
Developing Therapies for Functional Recovery after Traumatic Brain Injury
Several neuroplastic processes identified at the molecular level in animal models and suggested by neuroimaging studies at the clinical level are being developed as surgical strategies for brain repair (Table 328-2). Based on preclinical gene and cell therapy experiments in primates, significant progress has been achieved in developing restorative neurosurgical strategies for movement disorders.112 Achieving similar progress in treating TBI, however, will be much more difficult because unlike the clearly identified network defect in Parkinson’s disease, the neuronal loss in TBI occurs across multiple anatomic and physiologic regions and involves the interruption of multiple brain circuits with varying severity. Opportunities for initiating potential restorative therapies are therefore present at multiple neuroanatomic sites (Fig. 328-4).
TABLE 328-2 Potential Restorative Therapies for Traumatic Brain Injury
| MODALITY | GOAL |
|---|---|
| ECM manipulation and bioscaffolds | Circuit reconstruction Circuit reorganization |
| Neurogenesis | Cell replacement Circuit reconstruction |
| Cell transplantation | |
| Expanded NPCs | Cell replacement |
| Autologous NPCs | Cell replacement |
| Autologous MSCs | Neurotrophic support |
| OPCs | Circuit reconstruction |
| Neurorehabilitation (TMS, DCS, CIT) | Circuit reorganization |
| Gene therapy | Gene replacement Gene overexpression Gene silencing |
| Deep brain stimulation | Circuit reorganization |
CIT, constraint-induced movement therapy; DCS, direct current stimulation; ECM, extracellular matrix; MSCs, mesenchymal stem cells; NPCs, neuronal progenitor cells; OPCs, oligodendrocyte precursors; TMS, transcranial magnetic brain stimulation.
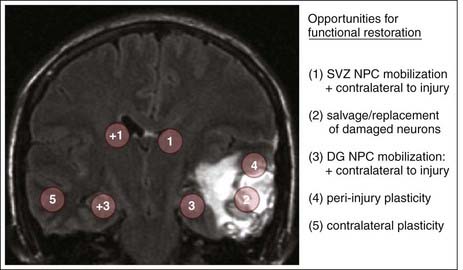
Enhancing Neoneurogenesis after Brain Trauma
Assuming that the adult human brain is capable of generating new neurons in response to injury, as is clearly the case in rodents, what steps could be taken to clinically enhance the production and functional integration of these cells? First, the functional magnitude of the neurogenic response to injury is relatively small; whether this is due to failure of new neurons to develop at a sufficiently rapid rate or failure to survive to time points that allow sufficient integration into the host environment is not clear. Many of the various overlapping local interactions among growth factors, extracellular proteins, metalloproteinases, neurotransmitters, hormones, and angiogenesis, which define the neurogenic niches of the adult brain,113 are altered after TBI. Understanding the effects of the disease process on these factors is necessary for developing strategies to influence adult neurogenesis for therapeutic purposes.114
We currently do not know whether TBI-induced neurogenesis contributes at all to functional recovery or whether recovery is due solely to other processes such as sprouting, dendritic arborization, or functional synaptic plasticity. In studies addressing this question, intraventricular administration of FGF and epidermal growth factor (EGF) into the lateral ventricle after FPI resulted in an increased rate of memory recovery and concomitant increases in the numbers of new hippocampal neurons colabeled with NeuN. Similarly, intraventricular administration of S100B resulted in a twofold increase in the percentage of newly generated cells coexpressing NeuN in the dentate gyrus at 35 days after injury.68,115 When water maze performance was compared with the percentage of BrdU-NeuN–colabeled cells, improved cognitive recovery correlated strongly with the number of newly generated neurons. Likewise, the cognitive recovery of animals receiving other pro-neurogenic factors after CCI has been evaluated.116 Animals administered erythropoietin for 14 days after injury had a significantly increased percentage of newly generated cells that differentiated into mature neurons in the granular cell layers of both the contralateral and ipsilateral dentate gyrus, and these animals also performed significantly better than did nontreated injured animals in water maze testing. Direct evidence, however, of improved cognitive function resulting from increased production of new neurons after TBI has not been reported.
Additional factors in the neurogenic niche, such as Noggin, bone morphogenetic proteins, sonic hedgehog, Notch, transforming growth factor-α, Eph/ephrins, and vascular endothelial growth factor (VEGF),113 have been less well studied in brain injury models. Upregulation of VEGF is observed after CCI117 and has been implicated in neurogenesis and neuromigration in a rodent model of focal cerebral ischemia.118 With better understanding of the developmental signaling molecules and morphogens mentioned earlier, mobilization of endogenous NSCs or progenitors for glial or neural repair may become possible via targeted infusion, expression, or silencing of certain niche factors.55
If the rate of “neoneurogenesis” is a limiting factor for cognitive recovery, could neurotrophin administration be used clinically for TBI? Although the optimal combinations of neurotrophins remain unknown in rodents, studies with single mitogens support this strategy.42 If prevention of apoptotic cell death over the weeks and months that follow TBI is required, additional classic neuroprotective drug strategies may be needed. For example, experimental neuroinflammation has been shown to inhibit hippocampal neurogenesis, an effect that can be reversed with the administration of minocycline,119 which inhibits microglial activation and reduces apoptotic cell loss. Additionally, indomethacin, a common nonsteroidal anti-inflammatory drug, blocks the effects of endotoxin- and irradiation-induced inflammation on hippocampal neurogenesis120 and enhances neurogenesis after experimental stroke.121 In contrast, hippocampal neurogenesis induced by an enriched environment is associated with the recruitment of T cells and the activation of microglia and can be restored in immune-deficient mice by administering T cells recognizing myelin basic protein.122 The dual nature of this microenvironmental process illustrates the complexity of selecting molecular targets for therapeutic intervention.
Cell Transplantation
The clinical trials undertaken thus far involving cell transplantation for Parkinson’s disease and Huntington’s disease have all used primary fetal tissue containing NSCs and NPCs, for which there was abundant preclinical work in primates that has been extensively reviewed in the literature.123,124 These clinical studies have shown that human fetal mesencephalic dopaminergic neurons can reinnervate the striatum, release dopamine, and integrate in the brain in a manner that induces symptomatic relief.125,126 Clinical outcomes have been variable, however, and future cell transplantation trials will probably use culture-expanded NSCs rather than primary fetal cells as graft material. The ability to extensively characterize, modify, and standardize NSCs should allow greater therapeutic predictability after transplantation of these cells. The only such cells to be transplanted in patients are hNT neurons, which are terminally differentiated, postmitotic, nontumorigenic neurons derived from a human teratocarcinoma cell line (NTera-2). Transplantation of these cells has produced partial recovery of neurological function in some patients after basal ganglia stroke.127,128
Here, however, it is important to note the major differences between cell grafting to the striatum for Parkinson’s disease and cell grafting for TBI. One of the putative targets for TBI, the hippocampus, is a neurogenic niche55,113 and is therefore more likely to permit the survival and appropriate differentiation of transplanted NPCs. In addition, trauma-related factors that alter the integration of adult newborn neurons into the hippocampal circuitry will also be relevant for the integration of transplanted NPCs. In contrast, focally damaged areas of the neocortex offer a less welcoming milieu for the survival of transplanted neuronal cells, and transplanted cells may be less likely to survive in this hostile environment acutely after TBI.129 Strategies for clinical transplantation in TBI will probably vary with patients’ individual anatomic pathology. Graft location will also be tied to the desired functional role of the grafted cells. Replacement neurons, remyelinating oligodendrocytes, and supporting chaperone cells are all potentially useful cell types whose relative value may change depending on the clinical situation. The source and characteristics of various cell types available for transplantation are well reviewed in the literature,73,129 and the following discussion focuses on recent, clinically applicable cell transplantation data from TBI models.
Neuronal Progenitor Cell Grafts in Traumatic Brain Injury Models
The earliest cell-suspension transplantation studies in TBI involved grafting hNT cells to the peri-injured cortex of FPI animals.131,132 Despite the survival of transplanted cells in the traumatically injured brain, hNT cells had no significant effect on posttraumatic neurological motor or cognitive function. More recently, hNT neurons transduced to overexpress NGF and transplanted to the medial septal nuclei 24 hours after CCI resulted in improved water maze performance at 4 weeks after injury,133 but only in animals with NGF-secreting grafts. These results illustrate that predifferentiated hNT neurons have not shown the ability for neuronal integration after TBI but do function as support cells, presumably via the trophic action of NGF on medial septal neurons and preservation of the septohippocampal pathway.133
NPC transplantation studies have also demonstrated behavioral improvements in injured animals despite a lack of neuronal differentiation. Shear and colleagues assessed the long-term survival, migration, differentiation, and functional significance of fetal-derived NPCs transplanted into the ipsilateral striatum of CCI mice up to 1 year after transplantation.134 NPCs were observed to migrate throughout the injured hippocampus and adjacent cortical regions, but the majority of the transplanted cells expressed NG2, an oligodendrocyte progenitor cell marker, whereas none expressed neuronal markers. Transplanted animals did show significant improvements in spatial learning, which suggested release of neurotrophic factors by the transplanted cells. The grafted cells also appeared to respond to intrinsic cues by migrating to the ipsilateral hippocampus, an effect that had previously been facilitated by implanting cells within a fibronectin matrix.135 In addition, enhanced migration has been reported via EGF receptor transduction of immortalized fetal-derived NSCs (C17.2), although these cells also resulted in behavioral improvement despite a lack of neuronal differentiation when transplanted into the corpus callosum of CCI animals.136 C17.2 grafts express NeuN up to 13 weeks after hippocampal-cortical transplantation in animals that showed significantly improved motor function but without an improvement in cognitive function.137 Similar improvement in sensorimotor but not cognitive function has been observed after the transplantation of fetal-derived NPCs to the periphery of the injured cortex in CCI mice, where cells also showed phenotypic evidence of neuronal differentiation.138 Alternatively, improved cognition was observed with perilesional transplantation of C17.2 cells transduced with glial-derived neurotrophic factor (GDNF), which also resulted in increased cell migration from the graft site.139 Taken together, these data suggest that separate processes exist for restoring cognitive and motor function, with cognitive recovery being associated with the proximity of cells or growth factors to the hippocampus and motor recovery possibly being associated with cells capable of neuronal differentiation near the site of cortical injury.
The transplantation of undifferentiated cells is a passive approach that attempts to allow the injury microenvironment to guide appropriate phenotypic differentiation. The aforementioned results illustrate the complexity of hypothesizing which cell type will be optimal, and few studies have involved the transplantation of specific neuronal subtypes in TBI models. Inhibitory cells may sustain primary damage at the time of impact, or they may undergo functional changes that are secondary to early pathogenic levels of neuroexcitation42; thus, inhibitory interneurons are possible graft candidates. Murine ES cells predifferentiated into GABAergic neurons produced motor recovery up to 5 weeks after grafting below the cortical injury cavity, whereas cells predifferentiated to astrocytes did not provide any benefit.140 However, whether these cells released significant amounts of GABA or perhaps a neurotrophin was not investigated. Other authors have transplanted human fetal-derived NSCs, primed to differentiate into cholinergic neurons, into the hippocampus of immunosuppressed FPI rats141 and found improvements in water maze performance. The transplanted cells expressed the neuronal marker microtubule-associated protein 2 (MAP2), and although the markers of specific neuronal phenotypes were not assayed, release of GDNF by the grafted cells was suggested by immunohistochemistry and increased detection of GDNF in microdialysis samples.
In contrast to the limited results just mentioned, point-to-point reconstruction of damaged motor circuits in the adult brain has been suggested in experiments in which fetal cortical tissue was grafted into aspiration-damaged adult brain.142 The transplanted fetal motor cortical neurons extended long-distance projections to appropriate cortical and subcortical targets and appeared to develop reciprocal synaptic contacts. These results from the use of homotopic tissue are several magnitudes better than those obtained thus far with the use of expanded NPCs. Fetal grafts from visual cortical tissue did not reconstitute the damaged pathways, thus further suggesting that only appropriately committed neurons are able to specifically recognize the molecular cues expressed after injury. Ultimately, cells engineered for replacement therapy will probably require genetic and epigenetic modification to recapitulate maturation down a desired lineage pathway.
Possibility of Autologous Neural Stem Cell Transplantation
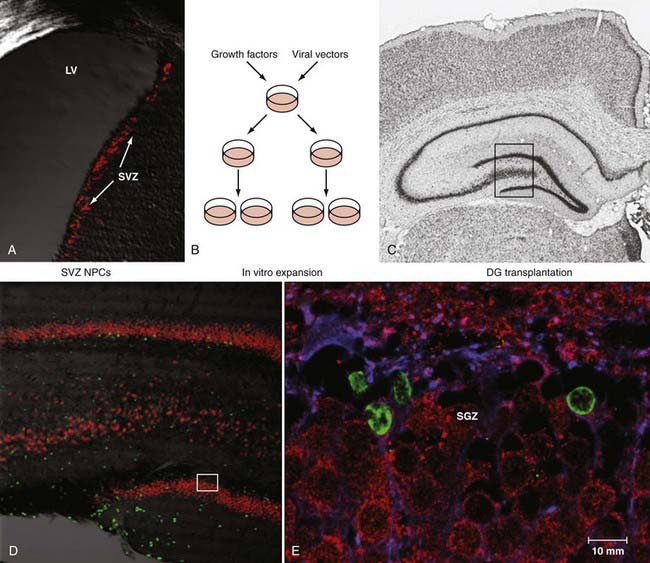
Because of immunosuppression, allogenicity, availability, and potential neoplasia issues related to the use of NSCs from other sources, autologous NPCs would be an attractive source of cells for first attempts to repopulate the injured brain at sites of focal damage or within the hippocampus. To achieve this feat in practice, NPCs need to be concentrated from the source tissue, enriched or purified, expanded ex vivo, and directed toward a desired lineage. For human use, this must be achieved at a good laboratory practice standard, and the product must be shown to be safe for transplantation. As an initial model, SVZ NPCs from the adult rat have been expanded in culture and allogenically transplanted to the adult rat hippocampus to test their ability to transdifferentiate to neurons appropriate to the hippocampal niche (Fig. 328-5). By 2 weeks after transplantation, nearly 80% of the new cells that were located in the granule cell layer had the immunohistochemical profile of granule cell neurons, thus suggesting the ability of adult SVZ cells to transdifferentiate to locally appropriate neurons in the hippocampus.143 Human neurosphere-derived NPCs have been transplanted into immunosuppressed FPI rats. Both graft cell proliferation and more robust incorporation into the host hippocampus were seen after injury than in noninjured, transplanted animals. Surprisingly, human autologous transplantation of tissue containing putative NPCs harvested from contusion zones and reintroduced into TBI patients has been reported.144,145
An alternative strategy has been suggested by the laboratory of Chopp and colleagues, who performed several studies to evaluate the therapeutic use of bone marrow–derived cells to treat experimental TBI and stroke, beginning with the pericontusional transplantation of whole bone marrow146 and followed by the intravenous147 and intra-arterial148 administration of bone marrow–derived mesenchymal stem cells (MSCs). In all cases, cells were reported to survive, migrate to areas of injury, and produce some measurable neurological benefit. The therapeutic benefits did not result from replacement of injured tissue by exogenous cells but instead were presumed to work indirectly via paracrine stimulation with an array of growth and neurotrophic factors that result in the production of various restorative factors from host astrocytes (see Chopp and colleagues149 for review). In response to MSC treatment, neurogenesis, angiogenesis, and synaptogenesis appear to be amplified, the glial scar surrounding an ischemic lesion reduced, and white matter bundles increased in size in the rodent stroke model. Progenitor and premature oligodendrocytes are also increased in the striatum and corpus callosum, and the boundary of the ischemic lesion appears to become encapsulated by axonal extension.150
If these results are upheld in clinical trials, autologous MSC administration could potentially become a cornerstone therapy for generating microenvironments within the injured brain that amplify brain plasticity in preparation for the application of more specifically targeted restorative agents. A phase I clinical trial to evaluate the use of autologous bone marrow–derived mononuclear cells to treat children with isolated severe TBI (postresuscitation Glasgow Coma Scale score between 5 and 8) is scheduled to reach completion in 2009.151 Alternatively, understanding the mechanisms involved in the neuroplastic response may allow the development of agents that mimic the effects of MSCs without requiring stem cell harvest and reimplantation.
Oligodendrocyte Replacement
Very little is known about the role of abnormal myelination after TBI, in contrast to spinal cord injury, where it has been shown to affect 20% to 30% of axons that appear to be in continuity. Ultrastructural studies have shown frequent poorly myelinated axons and disorganization of myelin layers after TBI, but quantization studies are lacking. If dysmyelination is important in TBI, oligodendrocyte progenitor cell transplantation or endogenous activation strategies may eventually be worthy of therapeutic trials. The presence of oligodendrocyte precursors in adult human white matter152 suggests that these cells may be readily available for mobilization near sites of injury. Cografting NPCs with olfactory ensheathing cells (OECs), which guide olfactory receptor axons to the olfactory bulb, has also been proposed.153 This follows a great surge of interest in transplanting OECs in patients with spinal cord injury,154 which has occurred in a phase I clinical trial.155 Additionally, strategies to retard the molecular and structural inhibition of axonal regeneration, such as silencing the intracellular signaling pathways of neurite inhibitors and blocking the neuronal receptors of myelin-associated inhibitors,31 may be necessary to allow the integration of axons from salvaged, newborn, or transplanted neurons introduced therapeutically into the injured brain. In addition, seeding transplanted cells onto synthetic bioscaffolds may also facilitate the formation of new connections across damaged tissue.156
Gene Therapy
Gene therapy for brain disorders is one of the most promising frontiers in the practice of restorative neurosurgery. There are significant experimental gene therapy initiatives under way that have led to currently active clinical trials involving the direct intracerebral delivery of viral vectors for treating neurodegenerative movement disorders, and these treatments have been reported to be safe and well tolerated.112 It is conceivable that many of the neuroplastic molecular interactions discussed earlier could be targeted for direct genetic manipulation. Patterns of gene expression that vary in injury severity and recovery time suggest that the pathophysiologic cascade induced by TBI is accompanied by molecular responses that may offer windows for therapeutic interventions involving gene therapy.54
Over the past decade, in work to develop direct intraparenchymal CED of viral vectors for treating Parkinson’s disease, multiple studies have demonstrated the efficacy of CED for gene delivery to specified brain regions. Specifically, robust gene expression in neurons, preferentially transduced by adeno-associated virus type 2 vectors, has resulted in translation of this technology to human application.157 In addition, the ability to induce cortical gene expression after thalamic CED158 via thalamocortical projections suggests that damaged cortical regions may be indirectly and safely targeted with this approach. Additionally, an intraoperative MRI (iMRI) platform to guide infusions in real time is in preclinical development and will provide rapid feedback on the physical and anatomic diffusion parameters important for optimizing gene transfer and reducing the potential for adverse effects.159
Cortical Stimulation and Neurorehabilitation
Cortical neuronal stimulation by direct and indirect methods is used experimentally in the clinical rehabilitation setting. Neuronal stimulation by TMS is achieved via electromagnetic induction of a current that passes through the scalp and generates action potentials in cortical neurons lying beneath the stimulating coil. Depending on the stimulus location and parameters, repetitive TMS can either enhance or decrease activity in the stimulated region. Transcranial direct current stimulation (DCS) can also induce an intracerebral current flow sufficiently large to achieve changes in cortical excitability and has been applied to humans noninvasively to induce focal and reversible shifts in cortical excitability. TMS and DCS can enhance the beneficial effects of motor training, visuomotor coordination, implicit motor learning, skilled finger movements, probabilistic classification learning, working memory, and sleep-dependent consolidation of declarative memories in healthy subjects, and there is evidence that TMS and DCS are successful adjuvants to traditional interventions in stroke neurorehabilitation.105 A phase I clinical trial is currently under way to assess TMS for the treatment of impairment of consciousness after severe TBI.160
Neurorehabilitation may aid in optimizing the functional integration of newly generated cells and inhibiting the formation of irrelevant neural connections.161 Primate studies have suggested that repeated, task-specific practice with injury-affected limbs induces cortical reorganization and correlative functional improvement. Constraint-induced movement therapy (CIT), which induces repeated limb use, elicits adaptive cortical reorganization measured by TMS after stroke in humans103 and is effective in improving upper limb use and function after TBI. Timing of the initiation of neurorehabilitation is quite important in that early, intense rehabilitative measures that target an impaired forelimb can be detrimental to anatomic and behavioral recovery.162
With regard to cell transplantation, grafts are not transplanted in their final form but must develop appropriately to establish the correct pattern of reconnections, as is also the case for endogenously generated neurons in the injured brain. This growth process is probably influenced by neuronal stimulation, and significant functional improvement may depend on the external activation of relevant circuitry. Indeed, environmental enrichment and involuntary exercise have been shown to induce morphologic and cellular changes in transplanted cells that are associated with plasticity.163
Deep Brain Stimulation
Studies demonstrating the preservation of large-scale cerebral networks in patients in the minimally conscious state (MCS),164,165 a condition characterized by intermittent evidence of awareness of self or the environment, suggest residual functional capacity in these patients that could potentially be augmented by therapeutic interventions. Hypothesizing that further recovery in some MCS patients is limited by chronic underactivation of potentially recruitable large-scale networks, Schiff and colleagues showed that bilateral deep brain electrical stimulation (DBS) of the central thalamus resulted in behavioral responsiveness in one patient who had previously remained in MCS for 6 years after TBI.166 The effects of DBS were interpreted as compensating for a loss of arousal regulation normally controlled by the frontal lobe in the intact brain. This result, in an appropriately selected MCS patient, stands in contrast to an earlier large multicenter series of patients in a vegetative state implanted with DBS systems in the centromedian thalamus, in whom arousal responses occurred without significant clinical improvement.167 Confirmation of this finding in other patients with evidence of similar baseline function (intact language processing on fMRI) would suggest that DBS may potentially open avenues for functional restoration in carefully selected patients.
Ethical Issues
Potential neurorestorative studies in MCS patients highlight the ethical issues inherent in developing experimental therapies for TBI patients with significant cognitive deficits. With regard to severe brain injury, recovery of consciousness and cognitive or physical function often comes in degrees, although the use of neurorestorative therapies for these patients may be justified when the result is restoration of conscious awareness of self and improvement in cognition or motor ability. Whether a certain therapy is in the best interests of a brain-injured individual will depend not only on the degree to which these functions are restored but also on the individual’s psychological response to the outcome.168 Consideration of how individuals with severe brain injury might benefit from or be harmed by innovative but invasive interventions in the brain will remain critical for determining when and in which patients an experimental technique should be initiated.
Alvarez-Buylla A, Lim DA. For the long run: maintaining germinal niches in the adult brain. Neuron. 2004;41:683.
Cohen AS, Pfister BJ, Schwarzbach E, et al. Injury-induced alterations in CNS electrophysiology. Prog Brain Res. 2007;161:143.
Cramer SC. Repairing the human brain after stroke: I. Mechanisms of spontaneous recovery. Ann Neurol. 2008;63:272.
Floyd CL, Lyeth BG. Astroglia: important mediators of traumatic brain injury. Prog Brain Res. 2007;161:61.
Gaillard A, Prestoz L, Dumartin B, et al. Reestablishment of damaged adult motor pathways by grafted embryonic cortical neurons. Nat Neurosci. 2007;10:1294.
Glannon W. Neurostimulation and the minimally conscious state. Bioethics. 2008;22:337.
Kells AP, Hadaczek P, Yin D, et al. Efficient gene therapy–based method for the delivery of therapeutics to primate cortex. Proc Natl Acad Sci U S A. 2009;106:2407.
Lim DA, Huang YC, Alvarez-Buylla A. The adult neural stem cell niche: lessons for future neural cell replacement strategies. Neurosurg Clin N Am. 2007;18:81.
Niogi SN, Mukherjee P, Ghajar J, et al. Structural dissociation of attentional control and memory in adults with and without mild traumatic brain injury. Brain. 2008;131:3209.
Nithianantharajah J, Hannan AJ. Enriched environments, experience-dependent plasticity and disorders of the nervous system. Nat Rev Neurosci. 2006;7:697.
Owen AM, Coleman MR. Functional neuroimaging of the vegetative state. Nat Rev Neurosci. 2008;9:235.
Povlishock JT, Katz DI. Update of neuropathology and neurological recovery after traumatic brain injury. J Head Trauma Rehabil. 2005;20:76.
Richardson RM, Sun D, Bullock MR. Neurogenesis after traumatic brain injury. Neurosurg Clin N Am. 2007;18:169.
Richardson RM, Varenika V, Forsayeth JR, et al. Future applications: gene therapy. Neurosurg Clin N Am. 2009;20:205.
Sanai N, Berger MS, Garcia-Verdugo JM, et al. Comment on “Human neuroblasts migrate to the olfactory bulb via a lateral ventricular extension. ”. Science. 2007;318;:393. author reply 393
Schiff ND, Giacino JT, Kalmar K, et al. Behavioural improvements with thalamic stimulation after severe traumatic brain injury. Nature. 2007;448:600.
Schiff ND, Rodriguez-Moreno D, Kamal A, et al. fMRI reveals large-scale network activation in minimally conscious patients. Neurology. 2005;64:514.
Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146.
Sun D, McGinn MJ, Zhou Z, et al. Anatomical integration of newly generated dentate granule neurons following traumatic brain injury in adult rats and its association to cognitive recovery. Exp Neurol. 2007;204:264.
van Praag H, Schinder AF, Christie BR, et al. Functional neurogenesis in the adult hippocampus. Nature. 2002;415:1030.
1 Tolias CM, Bullock MR. Critical appraisal of neuroprotection trials in head injury: what have we learned? NeuroRx. 2004;1:71.
2 Maas AI, Marmarou A, Murray GD, et al. Prognosis and clinical trial design in traumatic brain injury: the IMPACT study. J Neurotrauma. 2007;24:232.
3 Povlishock JT, Katz DI. Update of neuropathology and neurological recovery after traumatic brain injury. J Head Trauma Rehabil. 2005;20:76.
4 Sahuquillo J, Poca MA, Amoros S. Current aspects of pathophysiology and cell dysfunction after severe head injury. Curr Pharm Des. 2001;7:1475.
5 Cramer SC. Repairing the human brain after stroke: I. Mechanisms of spontaneous recovery. Ann Neurol. 2008;63:272.
6 Kwakkel G, Kollen B, Lindeman E. Understanding the pattern of functional recovery after stroke: facts and theories. Restor Neurol Neurosci. 2004;22:281.
7 Levin HS. Neuroplasticity following non-penetrating traumatic brain injury. Brain Inj. 2003;17:665.
8 Niedzwecki CM, Marwitz JH, Ketchum JM, et al. Traumatic brain injury: a comparison of inpatient functional outcomes between children and adults. J Head Trauma Rehabil. 2008;23:209.
9 Giza CC, Prins ML. Is being plastic fantastic? Mechanisms of altered plasticity after developmental traumatic brain injury. Dev Neurosci. 2006;28:364.
10 Cramer SC, Chopp M. Recovery recapitulates ontogeny. Trends Neurosci. 2000;23:265.
11 Gennarelli TA, Graham DI. Neuropathology of the head injuries. Semin Clin Neuropsychiatry. 1998;3:160.
12 Smith FM, Raghupathi R, MacKinnon MA, et al. TUNEL-positive staining of surface contusions after fatal head injury in man. Acta Neuropathol. 2000;100:537.
13 Clark RS, Kochanek PM, Watkins SC, et al. Caspase-3 mediated neuronal death after traumatic brain injury in rats. J Neurochem. 2000;74:740.
14 Kotapka MJ, Graham DI, Adams JH, et al. Hippocampal pathology in fatal human head injury without high intracranial pressure. J Neurotrauma. 1994;11:317.
15 Kotapka MJ, Graham DI, Adams JH, et al. Hippocampal pathology in fatal non-missile human head injury. Acta Neuropathol. 1992;83:530.
16 McIntosh TK, Saatman KE, Raghupathi R, et al. The Dorothy Russell Memorial Lecture. The molecular and cellular sequelae of experimental traumatic brain injury: pathogenetic mechanisms. Neuropathol Appl Neurobiol. 1998;24:251.
17 Umile EM, Sandel ME, Alavi A, et al. Dynamic imaging in mild traumatic brain injury: support for the theory of medial temporal vulnerability. Arch Phys Med Rehabil. 2002;83:1506.
18 Smith DH, Okiyama K, Thomas MJ, et al. Evaluation of memory dysfunction following experimental brain injury using the Morris water maze. J Neurotrauma. 1991;8:259.
19 Morales DM, Marklund N, Lebold D, et al. Experimental models of traumatic brain injury: do we really need to build a better mousetrap? Neuroscience. 2005;136:971.
20 Singleton RH, Zhu J, Stone JR, et al. Traumatically induced axotomy adjacent to the soma does not result in acute neuronal death. J Neurosci. 2002;22:791.
21 Morganti-Kossmann MC, Satgunaseelan L, Bye N, et al. Modulation of immune response by head injury. Injury. 2007;38:1392.
22 Kelley BJ, Lifshitz J, Povlishock JT. Neuroinflammatory responses after experimental diffuse traumatic brain injury. J Neuropathol Exp Neurol. 2007;66:989.
23 Stahel PF, Morganti-Kossmann MC, Kossmann T. The role of the complement system in traumatic brain injury. Brain Res Brain Res Rev. 1998;27:243.
24 Adams JH, Doyle D, Ford I, et al. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology. 1989;15:49.
25 Nagamoto-Combs K, McNeal DW, Morecraft RJ, et al. Prolonged microgliosis in the rhesus monkey central nervous system after traumatic brain injury. J Neurotrauma. 2007;24:1719.
26 Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146.
27 Floyd CL, Lyeth BG. Astroglia: important mediators of traumatic brain injury. Prog Brain Res. 2007;161:61.
28 Wilhelmsson U, Li L, Pekna M, et al. Absence of glial fibrillary acidic protein and vimentin prevents hypertrophy of astrocytic processes and improves post-traumatic regeneration. J Neurosci. 2004;24:5016.
29 Carmen J, Magnus T, Cassiani-Ingoni R, et al. Revisiting the astrocyte-oligodendrocyte relationship in the adult CNS. Prog Neurobiol. 2007;82:151.
30 Magistretti PJ. Neuron-glia metabolic coupling and plasticity. J Exp Biol. 2006;209:2304.
31 Walmsley AR, Mir AK. Targeting the Nogo-A signalling pathway to promote recovery following acute CNS injury. Curr Pharm Des. 2007;13:2470.
32 Huber AB, Weinmann O, Brosamle C, et al. Patterns of Nogo mRNA and protein expression in the developing and adult rat and after CNS lesions. J Neurosci. 2002;22:3553.
33 Marklund N, Fulp CT, Shimizu S, et al. Selective temporal and regional alterations of Nogo-A and small proline-rich repeat protein 1A (SPRR1A) but not Nogo-66 receptor (NgR) occur following traumatic brain injury in the rat. Exp Neurol. 2006;197:70.
34 Marklund N, Bareyre FM, Royo NC, et al. Cognitive outcome following brain injury and treatment with an inhibitor of Nogo-A in association with an attenuated downregulation of hippocampal growth-associated protein-43 expression. J Neurosurg. 2007;107:844.
35 Lenzlinger PM, Shimizu S, Marklund N, et al. Delayed inhibition of Nogo-A does not alter injury-induced axonal sprouting but enhances recovery of cognitive function following experimental traumatic brain injury in rats. Neuroscience. 2005;134:1047.
36 Fitch MT, Silver J. CNS injury, glial scars, and inflammation: inhibitory extracellular matrices and regeneration failure. Exp Neurol. 2008;209:294.
37 Tate CC, Tate MC, LaPlaca MC. Fibronectin and laminin increase in the mouse brain after controlled cortical impact injury. J Neurotrauma. 2007;24:226.
38 Reeves TM, Prins ML, Zhu J, et al. Matrix metalloproteinase inhibition alters functional and structural correlates of deafferentation-induced sprouting in the dentate gyrus. J Neurosci. 2003;23:10182.
39 Falo MC, Fillmore HL, Reeves TM, et al. Matrix metalloproteinase-3 expression profile differentiates adaptive and maladaptive synaptic plasticity induced by traumatic brain injury. J Neurosci Res. 2006;84:768.
40 Cohen AS, Pfister BJ, Schwarzbach E, et al. Injury-induced alterations in CNS electrophysiology. Prog Brain Res. 2007;161:143.
41 Lowenstein DH, Thomas MJ, Smith DH, et al. Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J Neurosci. 1992;12:4846.
42 Reeves TM, Lyeth BG, Phillips LL, et al. The effects of traumatic brain injury on inhibition in the hippocampus and dentate gyrus. Brain Res. 1997;757:119.
43 Scheff SW, Price DA, Hicks RR, et al. Synaptogenesis in the hippocampal CA1 field following traumatic brain injury. J Neurotrauma. 2005;22:719.
44 Oyesiku NM, Evans CO, Houston S, et al. Regional changes in the expression of neurotrophic factors and their receptors following acute traumatic brain injury in the adult rat brain. Brain Res. 1999;833:161.
45 Truettner J, Schmidt-Kastner R, Busto R, et al. Expression of brain-derived neurotrophic factor, nerve growth factor, and heat shock protein HSP70 following fluid percussion brain injury in rats. J Neurotrauma. 1999;16:471.
46 Yang K, Perez-Polo JR, Mu XS, et al. Increased expression of brain-derived neurotrophic factor but not neurotrophin-3 mRNA in rat brain after cortical impact injury. J Neurosci Res. 1996;44:157.
47 Hicks RR, Numan S, Dhillon HS, et al. Alterations in BDNF and NT-3 mRNAs in rat hippocampus after experimental brain trauma. Brain Res Mol Brain Res. 1997;48:401.
48 Yoshimura S, Teramoto T, Whalen MJ, et al. FGF-2 regulates neurogenesis and degeneration in the dentate gyrus after traumatic brain injury in mice. J Clin Invest. 2003;112:1202.
49 Kleindienst A, Ross Bullock M. A critical analysis of the role of the neurotrophic protein S100B in acute brain injury. J Neurotrauma. 2006;23:1185.
50 Shah SA, Prough DS, Garcia JM, et al. Molecular correlates of age-specific responses to traumatic brain injury in mice. Exp Gerontol. 2006;41:1201.
51 Griesbach GS, Hovda DA, Molteni R, et al. Alterations in BDNF and synapsin I within the occipital cortex and hippocampus after mild traumatic brain injury in the developing rat: reflections of injury-induced neuroplasticity. J Neurotrauma. 2002;19:803.
52 Giza CC, Prins ML, Hovda DA, et al. Genes preferentially induced by depolarization after concussive brain injury: effects of age and injury severity. J Neurotrauma. 2002;19:387.
53 Rall JM, Matzilevich DA, Dash PK. Comparative analysis of mRNA levels in the frontal cortex and the hippocampus in the basal state and in response to experimental brain injury. Neuropathol Appl Neurobiol. 2003;29:118.
54 Li HH, Lee SM, Cai Y, et al. Differential gene expression in hippocampus following experimental brain trauma reveals distinct features of moderate and severe injuries. J Neurotrauma. 2004;21:1141.
55 Lim DA, Huang YC, Alvarez-Buylla A. The adult neural stem cell niche: lessons for future neural cell replacement strategies. Neurosurg Clin N Am. 2007;18:81.
56 Sanai N, Tramontin AD, Quinones-Hinojosa A, et al. Unique astrocyte ribbon in adult human brain contains neural stem cells but lacks chain migration. Nature. 2004;427:740.
57 Kornack DR, Rakic P. The generation, migration, and differentiation of olfactory neurons in the adult primate brain. Proc Natl Acad Sci U S A. 2001;98:4752.
58 Pencea V, Bingaman KD, Freedman LJ, et al. Neurogenesis in the subventricular zone and rostral migratory stream of the neonatal and adult primate forebrain. Exp Neurol. 2001;172:1.
59 Alvarez-Buylla A, Garcia-Verdugo JM. Neurogenesis in adult subventricular zone. J Neurosci. 2002;22:629.
60 Quinones-Hinojosa A, Sanai N, Soriano-Navarro M, et al. Cellular composition and cytoarchitecture of the adult human subventricular zone: a niche of neural stem cells. J Comp Neurol. 2006;494:415.
61 Sanai N, Berger MS, Garcia-Verdugo JM, et al. Comment on “Human neuroblasts migrate to the olfactory bulb via a lateral ventricular extension.”. Science. 2007;318:393.
62 Walton NM, Sutter BM, Chen HX, et al. Derivation and large-scale expansion of multipotent astroglial neural progenitors from adult human brain. Development. 2006;133:3671.
63 Eriksson PS, Perfilieva E, Bjork-Eriksson T, et al. Neurogenesis in the adult human hippocampus. Nat Med. 1998;4:1313.
64 Roy NS, Wang S, Jiang L, et al. In vitro neurogenesis by progenitor cells isolated from the adult human hippocampus. Nat Med. 2000;6:271.
65 Richardson RM, Sun D, Bullock MR. Neurogenesis after traumatic brain injury. Neurosurg Clin N Am. 2007;18:169.
66 Parent JM. Injury-induced neurogenesis in the adult mammalian brain. Neuroscientist. 2003;9:261.
67 Rice AC, Khaldi A, Harvey HB, et al. Proliferation and neuronal differentiation of mitotically active cells following traumatic brain injury. Exp Neurol. 2003;183:406.
68 Kleindienst A, McGinn MJ, Harvey HB, et al. Enhanced hippocampal neurogenesis by intraventricular S100B infusion is associated with improved cognitive recovery after traumatic brain injury. J Neurotrauma. 2005;22:645.
69 Chirumamilla S, Sun D, Bullock MR, et al. Traumatic brain injury induced cell proliferation in the adult mammalian central nervous system. J Neurotrauma. 2002;19:693.
70 Urrea C, Castellanos DA, Sagen J, et al. Widespread cellular proliferation and focal neurogenesis after traumatic brain injury in the rat. Restor Neurol Neurosci. 2007;25:65.
71 Ramaswamy S, Goings GE, Soderstrom KE, et al. Cellular proliferation and migration following a controlled cortical impact in the mouse. Brain Res.. 2005;1053:38.
72 Lu D, Mahmood A, Zhang R, et al. Upregulation of neurogenesis and reduction in functional deficits following administration of DEtA/NONOate, a nitric oxide donor, after traumatic brain injury in rats. J Neurosurg. 2003;99:351.
73 Schouten JW, Fulp CT, Royo NC, et al. A review and rationale for the use of cellular transplantation as a therapeutic strategy for traumatic brain injury. J Neurotrauma. 2004;21:1501.
74 Sun D, McGinn MJ, Zhou Z, et al. Anatomical integration of newly generated dentate granule neurons following traumatic brain injury in adult rats and its association to cognitive recovery. Exp Neurol. 2007;204:264.
75 Salman H, Ghosh P, Kernie SG. Subventricular zone neural stem cells remodel the brain following traumatic injury in adult mice. J Neurotrauma. 2004;21:283.
76 Seri B, Garcia-Verdugo JM, McEwen BS, et al. Astrocytes give rise to new neurons in the adult mammalian hippocampus. J Neurosci. 2001;21:7153.
77 Hastings NB, Gould E. Rapid extension of axons into the CA3 region by adult-generated granule cells. J Comp Neurol. 1999;413:146.
78 Markakis EA, Gage FH. Adult-generated neurons in the dentate gyrus send axonal projections to field CA3 and are surrounded by synaptic vesicles. J Comp Neurol. 1999;406:449.
79 van Praag H, Schinder AF, Christie BR, et al. Functional neurogenesis in the adult hippocampus. Nature. 2002;415:1030.
80 Leuner B, Gould E, Shors TJ. Is there a link between adult neurogenesis and learning? Hippocampus. 2006;16:216.
81 Sun D, Colello RJ, Daugherty WP, et al. Cell proliferation and neuronal differentiation in the dentate gyrus in juvenile and adult rats following traumatic brain injury. J Neurotrauma. 2005;22:95.
82 Emery DL, Fulp CT, Saatman KE, et al. Newly born granule cells in the dentate gyrus rapidly extend axons into the hippocampal CA3 region following experimental brain injury. J Neurotrauma. 2005;22:978.
83 Dash PK, Mach SA, Moore AN. Enhanced neurogenesis in the rodent hippocampus following traumatic brain injury. J Neurosci Res. 2001;63:313.
84 Kernie SG, Erwin TM, Parada LF. Brain remodeling due to neuronal and astrocytic proliferation after controlled cortical injury in mice. J Neurosci Res. 2001;66:317.
84a Seigers R, Schagen SB, Coppens CM, et al. Methotrexate decreases hippocampal cell proliferation and induces memory deficits in rats. Behav Brain Res. 2009;201:279-284.
85 Rola R, Raber J, Rizk A, et al. Radiation-induced impairment of hippocampal neurogenesis is associated with cognitive deficits in young mice. Exp Neurol. 2004;188:316-330.
85a Zhang CL, Zou Y, He W, et al. A role for adult TLX-positive neural stem cells in learning and behaviour. Nature. 2008;451:1004.
86 Alvarez-Buylla A. Neurogenesis and plasticity in the CNS of adult birds. Exp Neurol. 1992;115:110.
87 Magavi SS, Leavitt BR, Macklis JD. Induction of neurogenesis in the neocortex of adult mice. Nature. 2000;405:951.
88 Richardson RM, Holloway KL, Bullock MR, et al. Isolation of neuronal progenitor cells from the adult human neocortex. Acta Neurochir (Wien). 2006;148:773.
89 Arsenijevic Y, Villemure JG, Brunet JF, et al. Isolation of multipotent neural precursors residing in the cortex of the adult human brain. Exp Neurol. 2001;170:48.
90 Itoh T, Satou T, Hashimoto S, et al. Isolation of neural stem cells from damaged rat cerebral cortex after traumatic brain injury. Neuroreport. 2005;16:1687.
91 Kuhn HG, Dickinson-Anson H, Gage FH. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci. 1996;16:2027.
92 Shors TJ, Miesegaes G, Beylin A, et al. Neurogenesis in the adult is involved in the formation of trace memories. Nature. 2001;410:372.
93 Prins ML, Lee SM, Cheng CL, et al. Fluid percussion brain injury in the developing and adult rat: a comparative study of mortality, morphology, intracranial pressure and mean arterial blood pressure. Brain Res Dev Brain Res. 1996;95:272.
94 Driscoll I, Howard SR, Stone JC, et al. The aging hippocampus: a multi-level analysis in the rat. Neuroscience. 2006;139:1173.
95 Heine VM, Maslam S, Joels M, et al. Prominent decline of newborn cell proliferation, differentiation, and apoptosis in the aging dentate gyrus, in absence of an age-related hypothalamus-pituitary-adrenal axis activation. Neurobiol Aging. 2004;25:361.
96 Kempermann G, Kuhn HG, Gage FH. Experience-induced neurogenesis in the senescent dentate gyrus. J Neurosci. 1998;18:3206.
97 Olson AK, Eadie BD, Ernst C, et al. Environmental enrichment and voluntary exercise massively increase neurogenesis in the adult hippocampus via dissociable pathways. Hippocampus. 2006;16:250.
98 Niogi SN, Mukherjee P, Ghajar J, et al. Structural dissociation of attentional control and memory in adults with and without mild traumatic brain injury. Brain. 2008;131:3209.
99 Owen AM, Coleman MR. Functional neuroimaging of the vegetative state. Nat Rev Neurosci. 2008;9:235.
100 Maas AI, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008;7:728.
101 Rossini PM, Dal Forno G. Neuronal post-stroke plasticity in the adult. Restor Neurol Neurosci. 2004;22:193.
102 Cecatto RB, Chadi G. The importance of neuronal stimulation in central nervous system plasticity and neurorehabilitation strategies. Funct Neurol. 2007;22:137.
103 Liepert J, Bauder H, Wolfgang HR, et al. Treatment-induced cortical reorganization after stroke in humans. Stroke. 2000;31:1210.
104 Meinzer M, Flaisch T, Breitenstein C, et al. Functional re-recruitment of dysfunctional brain areas predicts language recovery in chronic aphasia. Neuroimage. 2008;39:2038.
105 Hummel FC, Cohen LG. Drivers of brain plasticity. Curr Opin Neurol. 2005;18:667.
106 Celnik PA, Cohen LG. Modulation of motor function and cortical plasticity in health and disease. Restor Neurol Neurosci. 2004;22:261.
107 Hamm RJ, Temple MD, O’Dell DM, et al. Exposure to environmental complexity promotes recovery of cognitive function after traumatic brain injury. J Neurotrauma. 1996;13:41.
108 Maegele M, Lippert-Gruener M, Ester-Bode T, et al. Multimodal early onset stimulation combined with enriched environment is associated with reduced CNS lesion volume and enhanced reversal of neuromotor dysfunction after traumatic brain injury in rats. Eur J Neurosci. 2005;21:2406.
109 Pham TM, Winblad B, Granholm AC, et al. Environmental influences on brain neurotrophins in rats. Pharmacol Biochem Behav. 2002;73:167.
110 Chytrova G, Ying Z, Gomez-Pinilla F. Exercise normalizes levels of MAG and Nogo-A growth inhibitors after brain trauma. Eur J Neurosci. 2008;27:1.
111 Nithianantharajah J, Hannan AJ. Enriched environments, experience-dependent plasticity and disorders of the nervous system. Nat Rev Neurosci. 2006;7:697.
112 Richardson RM, Larson PS, Bankiewicz KS. Gene and cell delivery to the degenerated striatum: status of preclinical efforts in primate models. Neurosurgery. 2008;63:629.
113 Alvarez-Buylla A, Lim DA. For the long run: maintaining germinal niches in the adult brain. Neuron. 2004;41:683.
114 Lie DC, Song H, Colamarino SA, et al. Neurogenesis in the adult brain: new strategies for central nervous system diseases. Annu Rev Pharmacol Toxicol. 2004;44:399.
115 Kleindienst A, Harvey HB, Rice AC, et al. Intraventricular infusion of the neurotrophic protein S100B improves cognitive recovery after fluid percussion injury in the rat. J Neurotrauma. 2004;21:541.
116 Lu D, Mahmood A, Qu C, et al. Erythropoietin enhances neurogenesis and restores spatial memory in rats after traumatic brain injury. J Neurotrauma. 2005;22:1011.
117 Skold MK, von Gertten C, Sandberg-Nordqvist AC, et al. VEGF and VEGF receptor expression after experimental brain contusion in rat. J Neurotrauma. 2005;22:353.
118 Wang Y, Jin K, Mao XO, et al. VEGF-overexpressing transgenic mice show enhanced post-ischemic neurogenesis and neuromigration. J Neurosci Res. 2007;85:740.
119 Ekdahl CT, Claasen JH, Bonde S, et al. Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci U S A. 2003;100:13632.
120 Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760.
121 Hoehn BD, Palmer TD, Steinberg GK. Neurogenesis in rats after focal cerebral ischemia is enhanced by indomethacin. Stroke. 2005;36:2718.
122 Ziv Y, Ron N, Butovsky O, et al. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci. 2006;9:268.
123 Dunnett SB, Nathwani F, Bjorklund A. The integration and function of striatal grafts. Prog Brain Res. 2000;127:345.
124 Kendall AL, Hantraye P, Palfi S. Striatal tissue transplantation in non-human primates. Prog Brain Res. 2000;127:381.
125 Bjorklund A, Dunnett SB, Brundin P, et al. Neural transplantation for the treatment of Parkinson’s disease. Lancet Neurol. 2003;2:437.
126 Lindvall O, Bjorklund A. Cell therapy in Parkinson’s disease. NeuroRx. 2004;1:382.
127 Kondziolka D, Steinberg GK, Wechsler L, et al. Neurotransplantation for patients with subcortical motor stroke: a phase 2 randomized trial. J Neurosurg. 2005;103:38.
128 Kondziolka D, Wechsler L, Goldstein S, et al. Transplantation of cultured human neuronal cells for patients with stroke. Neurology. 2000;55:565.
129 Molcanyi M, Riess P, Bentz K, et al. Trauma-associated inflammatory response impairs embryonic stem cell survival and integration after implantation into injured rat brain. J Neurotrauma. 2007;24:625.
130 Richardson RM, Fillmore HL, Holloway KL, et al. Progress in cerebral transplantation of expanded neuronal stem cells. J Neurosurg. 2004;100:659.
131 Muir JK, Raghupathi R, Saatman KE, et al. Terminally differentiated human neurons survive and integrate following transplantation into the traumatically injured rat brain. J Neurotrauma. 1999;16:403.
132 Philips MF, Muir JK, Saatman KE, et al. Survival and integration of transplanted postmitotic human neurons following experimental brain injury in immunocompetent rats. J Neurosurg. 1999;90:116.
133 Watson DJ, Longhi L, Lee EB, et al. Genetically modified NT2N human neuronal cells mediate long-term gene expression as CNS grafts in vivo and improve functional cognitive outcome following experimental traumatic brain injury. J Neuropathol Exp Neurol. 2003;62:368.
134 Shear DA, Tate MC, Archer DR, et al. Neural progenitor cell transplants promote long-term functional recovery after traumatic brain injury. Brain Res. 2004;1026:11.
135 Tate MC, Shear DA, Hoffman SW, et al. Fibronectin promotes survival and migration of primary neural stem cells transplanted into the traumatically injured mouse brain. Cell Transplant. 2002;11:283.
136 Boockvar JA, Schouten J, Royo N, et al. Experimental traumatic brain injury modulates the survival, migration, and terminal phenotype of transplanted epidermal growth factor receptor–activated neural stem cells. Neurosurgery. 2005;56:163.
137 Riess P, Zhang C, Saatman KE, et al. Transplanted neural stem cells survive, differentiate, and improve neurological motor function after experimental traumatic brain injury. Neurosurgery. 2002;51:1043.
138 Hoane MR, Becerra GD, Shank JE, et al. Transplantation of neuronal and glial precursors dramatically improves sensorimotor function but not cognitive function in the traumatically injured brain. J Neurotrauma. 2004;21:163.
139 Bakshi A, Shimizu S, Keck CA, et al. Neural progenitor cells engineered to secrete GDNF show enhanced survival, neuronal differentiation and improve cognitive function following traumatic brain injury. Eur J Neurosci. 2006;23:2119.
140 Becerra GD, Tatko LM, Pak ES, et al. Transplantation of GABAergic neurons but not astrocytes induces recovery of sensorimotor function in the traumatically injured brain. Behav Brain Res. 2007;179:118.
141 Gao J, Prough DS, McAdoo DJ, et al. Transplantation of primed human fetal neural stem cells improves cognitive function in rats after traumatic brain injury. Exp Neurol. 2006;201:281.
142 Gaillard A, Prestoz L, Dumartin B, et al. Reestablishment of damaged adult motor pathways by grafted embryonic cortical neurons. Nat Neurosci. 2007;10:1294.
143 Richardson RM, Broaddus WC, Holloway KL, et al. Heterotypic neuronal differentiation of adult subependymal zone neuronal progenitor cells transplanted to the adult hippocampus. Mol Cell Neurosci. 2005;28:674.
144 Zhu J, Zhou L, Xing Wu F. Tracking neural stem cells in patients with brain trauma. N Engl J Med. 2006;355:2376.
145 Zhu J, Wu X, Zhang HL. Adult neural stem cell therapy: expansion in vitro, tracking in vivo and clinical transplantation. Curr Drug Targets. 2005;6:97.
146 Mahmood A, Lu D, Yi L, et al. Intracranial bone marrow transplantation after traumatic brain injury improving functional outcome in adult rats. J Neurosurg. 2001;94:589.
147 Mahmood A, Lu D, Chopp M. Intravenous administration of marrow stromal cells (MSCs) increases the expression of growth factors in rat brain after traumatic brain injury. J Neurotrauma. 2004;21:33.
148 Lu D, Li Y, Wang L, et al. Intraarterial administration of marrow stromal cells in a rat model of traumatic brain injury. J Neurotrauma. 2001;18:813.
149 Chopp M, Li Y, Zhang ZG. Mechanisms underlying improved recovery of neurological function after stroke in the rodent after treatment with neurorestorative cell-based therapies. Stroke. 2009;40(3 suppl):S143.
150 Li Y, Chen J, Zhang CL, et al. Gliosis and brain remodeling after treatment of stroke in rats with marrow stromal cells. Glia. 2005;49:407.
151 Harting MT, Baumgartner JE, Worth LL, et al. Cell therapies for traumatic brain injury. Neurosurg Focus. 2008;24(3-4):E18.
152 Nunes MC, Roy NS, Keyoung HM, et al. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat Med. 2003;9:439.
153 Ao Q, Wang AJ, Chen GQ, et al. Combined transplantation of neural stem cells and olfactory ensheathing cells for the repair of spinal cord injuries. Med Hypotheses. 2007;69:1234.
154 Ibrahim A, Li Y, Li D, et al. Olfactory ensheathing cells: ripples of an incoming tide? Lancet Neurol. 2006;5:453.
155 Feron F, Perry C, Cochrane J, et al. Autologous olfactory ensheathing cell transplantation in human spinal cord injury. Brain. 2005;128:2951.
156 Park KI, Teng YD, Snyder EY. The injured brain interacts reciprocally with neural stem cells supported by scaffolds to reconstitute lost tissue. Nat Biotechnol. 2002;20:1111.
157 Eberling JL, Jagust WJ, Christine CW, et al. Results from a phase I safety trial of hAADC gene therapy for Parkinson disease. Neurology. 2008;70:1980.
158 Kells AP, Hadaczek P, Yin D, et al. Efficient gene therapy–based method for the delivery of therapeutics to primate cortex. Proc Natl Acad Sci U S A. 2009;106:2407.
159 Richardson RM, Varenika V, Forsayeth JR, et al. Future applications: gene therapy. Neurosurg Clin N Am. 2009;20:205.
160 Pape TL, Rosenow J, Lewis G. Transcranial magnetic stimulation: a possible treatment for TBI. J Head Trauma Rehabil. 2006;21:437.
161 Polgar S, Borlongan CV, Koutouzis TK, et al. Implications of neurological rehabilitation for advancing intracerebral transplantation. Brain Res Bull. 1997;44:229.
162 Schallert T, Leasure JL, Kolb B. Experience-associated structural events, subependymal cellular proliferative activity, and functional recovery after injury to the central nervous system. J Cereb Blood Flow Metab. 2000;20:1513.
163 Dobrossy MD, Dunnett SB. Morphological and cellular changes within embryonic striatal grafts associated with enriched environment and involuntary exercise. Eur J Neurosci. 2006;24:3223.
164 Boly M, Faymonville ME, Peigneux P, et al. Auditory processing in severely brain injured patients: differences between the minimally conscious state and the persistent vegetative state. Arch Neurol. 2004;61:233.
165 Schiff ND, Rodriguez-Moreno D, Kamal A, et al. fMRI reveals large-scale network activation in minimally conscious patients. Neurology. 2005;64:514.
166 Schiff ND, Giacino JT, Kalmar K, et al. Behavioural improvements with thalamic stimulation after severe traumatic brain injury. Nature. 2007;448:600.
167 Tsubokawa T, Yamamoto T, Katayama Y, et al. Deep-brain stimulation in a persistent vegetative state: follow-up results and criteria for selection of candidates. Brain Inj. 1990;4:315.
168 Glannon W. Neurostimulation and the minimally conscious state. Bioethics. 2008;22:337.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 328 Regeneration and Repair
Historically, neurosurgical interventions for traumatic brain injury (TBI) have centered on strategies to reduce sequelae of the primary brain insult. Despite progress in understanding the pathophysiology of TBI, clinical neuroprotection trials targeting these secondary injury mechanisms have failed to show consistent improvement in the outcomes of head-injured patients (at least 21 multicenter clinical trials have been conducted since 1985).1,2 In addition to local neuronal destruction resulting from focal injury, mechanical forces secondarily induce a progressive cascade of related events that contribute to neuronal death, including ischemia, brain edema, diffuse axonal injury (DAI), excitotoxicity, radical-mediated damage, mitochondrial dysfunction, and dysregulation of calcium homeostasis.3,4 In response to these processes, however, the injured brain activates a limited program for plasticity and cellular regeneration. Demonstration of these latent mechanisms in animal and human studies has spawned great interest in developing techniques to facilitate repair of the neuronal circuitry damaged by TBI.
Restorative strategies represent a shift in therapeutic goals from focusing solely on salvaging acutely threatened tissue to introducing interventions that support spontaneous and directed functional recovery (Fig. 328-1). Much of the evidence for spontaneous recovery of the damaged cerebrum comes from the stroke literature, where studies in animal models have provided cellular and molecular information, whereas systems-level data are increasingly being obtained from neuroimaging or neurophysiology studies in patients. Clinically, several basic principles of recovery have been indentified: most spontaneous recovery occurs within 3 to 6 months, cognitive deficits are more likely than motor deficits to show further gain beyond this point, the rate of recovery is inversely proportional to the severity of the deficit, and recovery patterns vary between types of deficits in the same patient.5 These observations suggest multiple mechanisms of recovery whose effectiveness varies with the type and location of the physical injury, as well as with the type and severity of the clinical findings. These mechanisms can be generalized to three basic categories: plasticity of intact networks, repair of damaged circuitry, and replacement of lost neurons. This chapter describes the opportunities and obstacles inherent in potential therapeutic strategies for repair and regeneration after TBI, including manipulation of endogenous neurogenesis, cell transplantation, gene therapy, neurorehabilitation, and electrophysiologic manipulation.
Trauma-Induced Plasticity
The brain activates a dramatic response to traumatic injury that is initially manifested as changes in the cellular microenvironment. These changes involve inflammation, disruption of physical structure and protein expression in the extracellular matrix (ECM), and altered expression of trophic factors. Although some of these mechanisms attenuate acute damage at the expense of future regenerative capacity, others retain the potential to participate in therapeutic interventions. Plasticity occurs when injury induces reorganization of cortical connections, but plasticity is not always synonymous with functional recovery. Even though processes such as synaptic sprouting, unmasking of dormant circuits, and the development of new polysynaptic connections can enable function, plasticity can also restrict function, as occurs when posttraumatic epileptic seizure foci or neuropathic pain is produced.6 This dichotomy is most evident in the developing brain, where increased plasticity has traditionally been thought to confer an advantage for recovery from TBI. Recovery also strongly depends on the type of injury,7 and the relationship between age and functional outcome is different within pediatric and adult age groups.8 These points reiterate the fact that plasticity is a complex series of molecular, cellular, and physiologic events that must be carefully orchestrated for both optimal maturation of the developing brain and optimal functional recovery from traumatic injury.9
The differences between injury responses in the developing and mature brain are also quite important when one considers that functional recovery after injury may require recapitulation of developmental events to produce neuronal circuit reconnections. Work in animal models has shown that focal damage in the adult brain can lead to a number of molecular and cellular changes, in both perilesional and remote brain regions, that are normally seen only in the developing brain.10 These distinctions should be kept in mind when considering how plasticity is altered by injury-induced microenvironmental changes in neurotransmission, gene and protein expression, cell death, neurogenesis, and neural connectivity.
The Injury Microenvironment: A Double-Edged Sword
In addition to local neuronal destruction resulting from the primary mechanical insult, TBI induces a progressive cascade of delayed secondary events that contribute to neuronal death, including ischemia, “wallerian degeneration” secondary to DAI, excitotoxicity, free radical–mediated damage, mitochondrial dysfunction, and dysregulation of calcium homeostasis. Injury may occur in the form of focal damage, as typically occurs after acute subdural hematoma or contusion, or it may be diffuse with widespread delayed neuronal loss, as typically occurs after DAI. Focal damage is characteristically seen around hemorrhagic lesions such as contusions within the gray matter or at gray-white matter junctions. These lesions are usually located at the frontal and temporal poles and in the orbital frontal cortex.11 Such focal neuronal death may occur as a result of both rapid necrotic and slower apoptotic mechanisms.12,13 Among diffuse injury sites, the hippocampus is known to be especially vulnerable in humans, with neuronal loss occurring in more than 80% of fatal TBIs, even in the absence of elevated intracranial pressure.14,15 After the acute phase of focal TBI and brain ischemic events, hippocampal neurons may be the most vulnerable neurons in the brain in that they show the earliest evidence of TBI-induced degeneration in experimental models.16 These hippocampal changes correlate with the profound memory impairment that is seen in both human and animal models of TBI.17,18 In the two most frequently used experimental TBI models, the lateral fluid percussion (LFP) and controlled cortical impact (CCI) injury models, selective neuronal death has also been well described in the hippocampus.19
The slow diffuse neuronal loss that is often seen in DAI, involving apoptotic morphology and wallerian degeneration, was previously thought to be an inevitable result of axonal interruption, as seen with primary axotomy of central nervous system (CNS) neurons. More recently, animal studies of DAI have shown that despite the proximity of traumatic axonal injury to the neuronal somata, neuronal cell bodies do not always show a pathologic progression to cell death and in fact some neurons and axons exhibit changes suggestive of reorganization and potential repair.20 These data suggest that injured neurons may be amenable to rescue through augmentation of endogenous plasticity. Therapeutic interventions, however, must account for changes in the injury microenvironment that encompass barriers to both functional reconnection and opportunities for repair (Table 328-1), as discussed in the following sections.
TABLE 328-1 Microenvironmental Factors and Potential Role in Neuroplasticity
| RESPONSE | DISABLING ROLE | ENABLING ROLE |
|---|---|---|
| Inflammation | Cytokine release Complement activation |
Glial activation Microglial activation |
| Reactive gliosis | Neurite inhibition Dysmyelination |
Neurotrophin secretion Neurotransmitter regulation |
| ECM deposition | Neuropil distortion | Growth permission |
| MMP activation | Synapse dysregulation | Substrate guidance |
| Neurogenesis | Seizure potential | Cell replacement |
ECM, extracellular matrix; MMP, matrix metalloproteinase.
Inflammatory Mechanisms and Gliosis
TBI produces a neuroinflammatory condition of the CNS in which rupture of the blood-brain barrier leads to accumulation of leukocytes from the systemic circulation and subsequent initiation of the immune functions of native glia.21 Acute inflammatory cytokine-mediated events such as monocyte/macrophage-mediated phagocytosis22 and complement-mediated cytolysis23 are now well recognized early after TBI and persist at least for several days and weeks. This response is partially represented in the form of “microglial stars” and “perivascular cuffing” in human pathologic specimens.24 Additionally, in a primate cortical lesion model, microglia and macrophages remain reactive in the cortex near the lesion and along sites of wallerian degeneration (corpus callosum and corticospinal tracts) for at least 12 months.25 The expression of brain-derived neurotrophic factor (BDNF) by a subpopulation of microglia/macrophages at this late time point suggests a possible neurotrophic role for microglia in long-term recovery.
Both inflammatory mechanisms and necrotic cell death trigger reactive gliosis, the astrocytic response to injury that consists of astrocyte hypertrophy, activation of fibrillary processes, and penumbral proliferation.26 Staining of glial fibrillary acidic protein (GFAP) increases dramatically in many areas of the brain days, weeks, and months after severe TBI.27 Upregulation of intermediate filaments, a hallmark of reactive astrocytes, was traditionally thought to be an obstacle to brain plasticity. In mice lacking GFAP and vimentin, however, the cellular processes of reactive astrocytes fail to exhibit the characteristic hypertrophy after entorhinal cortex lesions, as well as the initial increased loss of hippocampal neuronal synapses.28 Although these findings suggested that reactive astrocytes may play a beneficial role in the acute stage after a CNS insult, within a few days of injury, normal animals showed decreased synaptogenesis in comparison to animals lacking the two intermediate filament proteins, thus indicating a prohibitive effect of astrocytic hypertrophy at subacute stages.
The findings just presented support the idea that the beneficial effects of astrocytes at the site of an injury probably occur early in the injury response whereas subacute formation of the glial scar hinders regeneration. The beneficial effects of an astrocytic response include secretion of neurotrophic factors, regulation of metabolic factors (particularly important in times of stress), and maintenance of homeostatic levels of neurotransmitters.29 Astrocytes play a central role in neurometabolic coupling involving glutamate-stimulated aerobic glycolysis, a process that undergoes adaptations in parallel to synaptic plasticity.30 In both immature and adult TBI models, abnormalities have been reported in glutamatergic, GABAergic (transmitting γ-aminobutyric acid [GABA]), cholinergic, and aminergic transmission, whereas dysfunctional neurotransmission has been clinically implicated in pediatric memory impairment and attention problems.9 The detrimental effects of glial scars include inhibition of neurite outgrowth, inhibition of the remyelination of axons in white matter lesions, and hindrance of remyelination by inhibiting the differentiation of glial precursors into oligodendrocytes.29
Neural Connectivity
CNS neurons are intrinsically capable of axonal regeneration; however, this capacity rapidly diminishes postnatally at the onset of axonal myelination by mature oligodendrocytes. This loss of regenerative capacity can at least in part be attributed to the restriction of axonal regeneration and neuroplasticity by several CNS myelin proteins that inhibit neurite outgrowth, including Nogo-A, myelin-associated glycoprotein (MAG), and oligodendrocyte myelin.31 In neurons, Nogo-A is expressed during development and adulthood, whereas in oligodendrocytes, expression is initiated postnatally just before the appearance of myelin basic protein and myelination.32 With regard to TBI models, Nogo-A is upregulated after fluid percussion injury (FPI),33 and intraventricular infusion of an anti-Nogo antibody has been associated with improved postinjury cognitive recovery.34,35 Recently, it has become clear that the production of other inhibitory molecules, such as proteoglycans, is equally as important in creating a nonpermissive environment for axons attempting to regenerate.36
Mechanical obstruction of axonal regeneration by concomitant deposition of ECM molecules, which distorts the architecture of the neuropil, has also been well described. Conversely, levels of two ECM molecules critical to neural development, fibronectin and laminin, increase after human and experimental brain injury and may play a protective or reparative role in the injury response.37 The family of matrix metalloproteinases (MMPs) that regulate ECM molecules also performs essential functions during neuroplasticity in both developing and adult nervous systems, including substrate guidance during neuritogenesis and the establishment of boundaries for axonal terminal fields. In animals injured by entorhinal cortex lesions, MMPs regulate reactive synaptogenesis in a time- and injury-dependent manner to the extent that their inhibition interferes with emergence of the capacity for long-term potentiation in the sprouting crossed temporodentate pathway38 and produces deficits in spatial learning.39
The pervasive memory, cognitive, and motor deficits apparent in patients with mild to moderate TBI, sometimes occurring in the absence of frank neuronal cell loss, suggests that TBI alters the balance between excitatory and inhibitory neurotransmission in surviving neurons and disrupts the synaptic circuitry in the hippocampus.40 This effect occurs in a temporal and regional fashion such that in some pathways, injury may induce either increased excitability41 or inhibition42 of dentate gyrus granule cell neurons, thereby potentially affecting the ability of these cells to resist the synchronized bursting characteristic of seizure activity. In addition, the hippocampus ipsilateral to the cortical contusion is capable of a significant plasticity response, but synapse replacement in this area does not necessarily result in significant improvement in spatial learning.43
Gene and Protein Expression
Gene and protein profiling strategies have been used to study plasticity-related gene expression after experimental TBI. Several studies have shown that expression of neurotrophic factors is significantly altered after experimental TBI. RNA studies have demonstrated that some neurotrophic compounds such as nerve growth factor (NGF) and BDNF are upregulated44,45 whereas others such as NT-3 are downregulated.46,47 Data from the CCI model suggest that fibroblast growth factor 2 (FGF-2) is also upregulated, which stimulates posttraumatic neurogenesis and preserves granule cell layer volume, effects that are increased with FGF-2 supplementation via gene transfer.48 S100B is a neurotrophic and neuroprotective calcium-binding protein produced primarily by astrocytes that despite a correlation between increased cerebrospinal fluid levels and poor prognosis in TBI patients, has a beneficial effect on neuronal maintenance, neurogenesis, and cognitive performance in rodent TBI models.49
These alterations are not simple issues of upregulation versus downregulation because they occur with temporal, subregional, and age-related specificity. For instance, BDNF levels after experimental TBI are increased to a greater extent in aged animals than in young animals.50 BDNF is an activity-related molecule closely associated with experience-dependent plasticity and developmental regulation, and it may play a compensatory role after mild TBI in the developing rodent brain, where despite reductions in BDNF mRNA and protein expression in the ipsilateral cortex and hippocampus, contralateral upregulation occurs up to 2 weeks after injury.51 NGF-induced protein B, a transcription factor, is upregulated in a severity-dependent fashion in the immature brain, but it is increased independent of severity in adults. In contrast, the synaptic activity–inhibiting, calcium-binding protein synaptotagmin IV increases only in developing animals after severe injury and not at all in adults.52 Although these examples illustrate the complexity of the molecular injury response, they also suggest many opportunities to improve our understanding of important differences between developmental and injury-related plasticity.
Regional intrinsic differences in gene expression before and after injury are also likely to define the pathology and recovery of function after TBI. The differential vulnerability of the hippocampus to TBI as compared with the high degree of plasticity of the cortex, which may underlie the reason why memory dysfunction lingers in comparison to motor and sensory recovery, has been suggested by the fact that the majority of mRNA molecules that change after injury were found to be exclusively altered in either the hippocampus or the frontal cortex.53 Within the hippocampus itself, gene expression appears to be influenced by both injury severity and time since the insult. Many genes encoding molecules for cellular signaling, synaptic plasticity, metabolism, ion channels, and transporters that are initially upregulated after severe injury are downregulated after moderate injury, although moderate injury is associated with an increasing number of responsive genes as a function of time after injury as opposed to a decreasing number after severe injury.54 Overall, there is still much to discover about region- and time-specific microenvironmental dynamics, all of which combine to form various posttraumatic brain niches.
Endogenous Neurogenesis after Traumatic Brain Injury
TBI induces changes in the specialized microenvironment that regulates the proliferation of neural stem cells (NSCs) and differentiation of neuronal progenitor cells (NPCs). These neurogenic niches are found in the human dentate gyrus of the hippocampus and the subventricular zone (SVZ) of the lateral ventricular walls (Fig. 328-2).55 The best-characterized neurogenic region in the adult human brain is the SVZ lining the lateral ventricle, where a ribbon of SVZ astrocytes have been identified that proliferate in vivo and behave as multipotent progenitor cells in vitro.56 Unlike the situation in rodents and primates, where neurons born in the SVZ migrate in chains through the rostral migratory stream to replace interneurons of the olfactory bulb,57–59 there is no evidence of chains of migrating neuroblasts in the human SVZ.60,61 It has been estimated that in normal humans, less than 1% of astrocytes within the SVZ ribbon are in division,56 and although these endogenous NSCs can be expanded in culture,62 their response to injury in patients has not been studied. Less well characterized in the human brain are proliferating NPCs in the hippocampus,63 which have also been demonstrated to be multipotent progenitor cells in vitro.64
Rat models have been used to investigate TBI-induced neurogenesis and have shown that the number of NPCs in both the SVZ and hippocampus are substantially increased after TBI (Fig. 328-3), a process that is more robust in juvenile animals.65 Newborn NPCs from the SVZ appear to have the potential to migrate to areas of focal cortical damage, whereas hippocampal neurogenesis occurs in the setting of diffuse local damage. Nevertheless, in adult and aged animals, the number of new neurons developing after TBI remains small in comparison to astrocyte and oligodendrocyte differentiation, as assessed by bromodeoxyuridine (BrdU) staining.
Subventricular Zone Neurogenesis after Trauma
In common with models of seizure, ischemia, and deafferentation injury, experimental TBI induces an increase in SVZ NPC proliferation and a secondary redirected streaming of these precursors into the injured zone.66 Studies demonstrate up to twofold increases in ipsilateral SVZ NPC proliferation within the first 2 to 5 days after FPI67–70 and as much as a fourfold increase after the more focal CCI up to 14 days after injury.71,72 Contralateral NPC proliferation in the SVZ also increases, apparently to a lesser extent, after injury in both models.69,70,72 Newborn cells colabeled with BrdU and neuronal phenotypic markers have been observed in the cortex, striatum, and corpus callosum as long as 45 days after injury. Patch clamp studies have demonstrated that such BrdU cells exhibit neuronal electrophysiologic properties,73 and anatomic integration of these new neurons into host tissue has been shown by retrograde tracer labeling and synaptophysin triple-label immunohistochemical techniques.74
Migration of Subventricular Zone Neural Progenitor Cells to the Site of Injury
Glial cell proliferation has long been appreciated at sites of focal TBI damage, but more recently, it has been shown that some glia migrate into the lesion site from the SVZ and that these cells arise from NPCs. After the intraventricular injection of fluorescent microspheres to label BrdU-positive subventricular cells, migration from the SVZ to the cortex surrounding a cortical contusion impact site has been observed.71 In the SVZ, corpus callosum, striatum, and cortex, many of these cells expressed doublecortin, a marker of immature neuroblasts, thus suggesting that local neurogenesis could be driven by injury-specific extracellular signaling pathways. In the pericontusional cortex, expression of the mature neuronal marker NeuN was seen in cells tagged before injury by intraventricular infusion of a lipophilic dye, a finding suggesting that these cells were derived from SVZ NPCs that migrated to the site of injury.75
Hippocampal Neurogenesis after Brain Trauma
Although selectively more vulnerable to TBI, the injured hippocampus may have a unique ability to locally replace damaged neurons inasmuch as the dentate gyrus is normally an area of active endogenous neurogenesis. In humans, in vivo dentate gyrus neurogenesis was demonstrated on histologic sections obtained from patients who had been administered BrdU for diagnostic purposes.63 Studies from rodents also demonstrated that cells with an astrocyte phenotype residing in the adult subgranular zone of the dentate gyrus continually generate cells with a neuroblast phenotype that migrate into the granule cell layer.76 Although some of these new neurons die by apoptosis, many of these new neurons demonstrate functional integration within the hippocampal circuitry by sending dendrites into the molecular layer and axons into the CA3 region,77,78 and they additionally exhibit mature neuronal electrophysiologic characteristics.79
Moderate and severe TBI is always associated with learning and memory deficits—the “functional hallmarks” of TBI. This is usually accompanied by histologic damage in the form of cell loss, especially in the dentate gyrus and CA1 layers. Because neurogenesis in this region has been linked to enhanced learning and memory functions,80 it is reasonable to hypothesize that injury-induced neurogenesis may function in the replacement of damaged neurons and thus contribute to repair of neuronal circuits and restoration of neurological function in patients with TBI. After LFP, cell proliferation increases in the rodent dentate gyrus threefold to fourfold beginning as early as 2 days after injury, peaks during the first week after injury, and returns to baseline levels by day 35 after injury.67,81 Similarly, CCI may induce up to a sixfold increase in dividing cells in the ipsilateral dentate gyrus and a significant but smaller increase in the contralateral dentate gyrus.48,72,82–84 A significant increase in the number of new cells that express neuronal markers has been reported after longer maturation periods. TBI can initially induce a fourfold increase in the number of BrdU-labeled cells that colabel for NeuN, a mature neuronal phenotypic marker, nearly half of which migrate to the inner half of the granular cell layer and persist 10 weeks after injury.74 Others have reported a fivefold increase in the number of injury-generated double-labeled neurons, seen as late as 60 days after injury.84 The central issue remains, however: do these new neurons functionally integrate with the host hippocampal tissue and exhibit appropriate electrophysiology?
To begin to answer these questions, hippocampal neurons born after injury, which express NeuN and calbindin, have been labeled by stereotactic retrograde tracer injections of the neuronal tracer fluorogold into the CA3 region, which is one target zone for the granular cell neuronal projections.74 After 2 weeks, about 30% of the BrdU-positive cells in the dentate gyrus were labeled with fluorogold, a finding suggestive of connectivity with the CA3 target region, and these double-labeled cells were increased fourfold by FPI. Labeled cell bodies were also frequently enveloped by a synaptophysin “lattice,” as described previously,78 thus suggesting the formation of synapses onto the somata of newborn neurons.
In terms of measuring whether posttraumatic neurogenesis is functional on a broader scale that affects behavior, memory function, as assessed by Morris water maze tasks, is decreased in association with procedures that abolish the proliferation of new cells, such as the administration of antimitotic drugs.84a Low-dose irradiation, which has been shown to markedly reduce neurogenesis, also significantly reduces the ability to recover spatial memory function after experimental TBI.85 Additionally, inactivation of a subset of adult hippocampal NSCs in mice was shown to produce both a significant reduction in overall stem cell proliferation and a selective decrement in spatial learning.85a
Cortical Neurogenesis after Brain Trauma
Although the hippocampus is selectively damaged in TBI, the greatest density of neuronal loss occurs with focal parenchymal contusions arising in locations that vary with the heterogeneity of both the primary injury and subsequent mechanism of secondary injury. As discussed earlier, the injured brain may induce migration of neuroblasts from the SVZ into cortical lesions, but latent NPCs may potentially be activated at sites of cortical injury in the mammalian brain. In songbirds such as zebra finches, it has long been known that some cortical projection neurons are lost and regenerated each year from SVZ precursors in response to evolutionary pressure.86 Macklis and colleagues suggested the possibility that endogenous neural precursors from the cortex itself in rodents may differentiate into corticothalamic neurons in layer VI of the anterior cortex after targeted neuronal death and survive at least long enough to form appropriate long-distance corticothalamic connections.87 In humans, the existence of such NPCs in nongerminal cortex has been suggested from some isolation and culture studies, including the isolation and continuous culture of putative neuroblasts from neocortical tissue surgically resected from TBI patients at the time of decompressive craniotomy for high intracranial pressure,88 as well as the isolation of neurosphere-generating cells from similar cortical tissue.89 After CCI in rodents, the formation of neurospheres from neocortical cells has also been observed, but only when the injured tissue was isolated at 3 days after injury, which corresponds to a peak in nestin expression in pericontusional cortex.90 Induction of nestin expression in the cortex adjacent to the CCI injury has also been reported at 7 days after injury,84 thus suggesting the possible activation of resident cortical NPCs, although in both cases these cells could alternatively have migrated from the SVZ.
Effect of Age on Posttraumatic Neurogenesis
Juvenile mammals usually recover cerebral function to a greater extent than adults do after TBI. Because cell proliferation in the dentate gyrus decreases with age in normal rats,91 it has been hypothesized that age-related differences in hippocampal neurogenesis after TBI may be responsible in part for the greater cognitive recovery typically seen in young mammals.81 Juvenile rats generate nearly twice the number of new cells in the subgranular zone than adults do in the first 2 days after LFP injury, although this response appears to be related to a greater level of basal juvenile neurogenesis because the percent increases in cell number above baseline are similar in young and old animals. Additionally, more newly generated cells appear to differentiate into mature neurons over time in juveniles. Typical granule cell neurons have been shown to functionally integrate by 14 days after generation in the normal adult rodent brain77 and become critically involved in the learning response within this same period.92 Fewer cognitive deficits and better recovery are observed during the first 2 weeks after injury in juvenile rats than in adults.93 This difference may therefore result in part from a greater ability of younger animals to generate new hippocampal granule cell neurons in response to injury. The mechanism by which neuronal differentiation is enhanced in younger animals is not clear, but answers to this question may aid discovery of therapeutic interventions to optimize neurogenesis after TBI.
Despite a constant decline in NPC proliferation in the dentate gyrus from adolescence through senescence,91,94,95 the adult brain upregulates proliferation to almost the same extent as the juvenile brain after TBI, thus indicating that NPCs conserve the ability to respond to proliferative signals. Although injury signals increase the proliferative rate of NPCs, the increased presence of actual newly generated neurons in younger brains may reflect greater integrity of the neurogenic niche mechanisms required for differentiation or survival of new neurons. One possible mechanism, similar to that hypothesized for the effects of an enriched environment on hippocampal neurogenesis (a survival-promoting effect of newborn cells that is selective for neurons),96 is a greater level of specific neurotrophin support97 that is either not present or not induced in adults. All of the putative mechanisms just described, however, would be nearly irrelevant without clinical evidence of functional recovery in the human brain after TBI.
Evidence of Functional Recovery
Spontaneous recovery from TBI varies with the heterogeneity of the injury, and consequently, a wide range of outcomes are produced: in some vegetative patients, functional magnetic resonance imaging (fMRI) demonstrates appropriate cortical responses to spoken commands that are indistinguishable from normal responses,98 whereas in patients with mild TBI, diffusion tensor imaging demonstrates variations in microstructural white matter integrity that are associated with persistent impairment in memory and attentional control.99 Approximately 85% of recovery from TBI occurs within 6 months after injury,100 and functional neuroimaging techniques have increasingly been used as a starting point to understanding the appropriate anatomic correlates.
