Chapter 12 Rasmussen’s Encephalitis
Introduction
Rasmussen’s encephalitis (RE) is an acquired progressive unihemispheric disease characterized by intractable focal seizures, often in the form of epilepsia partialis continua (EPC) with motor and cognitive deterioration. Neuroimaging shows the progressive damage of the affected hemisphere, and histopathology is consistent with a T-cell dominated encephalitis with activated microglial cells and reactive astrogliosis.1
The etiopathogenesis of RE is still not fully understood; nevertheless, it has been considered a chronic inflammatory disease since its original description in 1958.2 This definition stems from the features of its clinical course, histopathology, and the reported efficacy of immunomodulatory treatments in delaying disease progression.
Clinical Features in Typical RE
The disease is sporadic, affecting both males and females. RE typically starts in childhood or early adolescence, with a mean age of presentation at 6 years. The previous personal history is uneventful, but in about half of the patients, a trivial febrile illness in the months preceding the onset of seizures or a remote head trauma have been reported.
In its typical form, the onset of RE is marked by epileptic seizures, either focal or secondary-generalized. In most patients, seizures are isolated, but in a proportion of cases, the presenting symptom is status epilepticus (SE) or, more rarely, EPC.3–4 In a few patients, slowly progressive hemiparesis may precede seizure onset.4–5 A rarely reported presenting symptom is hemidystonia or hemiathetosis;6 this modality of presentation is not surprising, given the early damage of the basal ganglia in RE (see later discussion), and mild movement disorders are likely to be frequently overlooked or underreported, as suggested by Andermann.7
EPILEPSY AND EEG FEATURES
The main characteristic of seizures in RE is their polymorphism in a given patient. Besides simple motor seizures that are almost always present, the patient may experience postural, versive, somatosensory, autonomic, visual, auditory, and limbic seizures (Figure 12-1). This polymorphism, which may be evident from the onset and is invariably present during the disease course, can be due to the multifocal—albeit hemispheric—origin of the seizures, or to the progressive enlargement of the original epileptic zone. Seizures are refractory to antiepileptic (AE) treatment, their frequency usually increases rapidly, and partial SE may recur.3–4
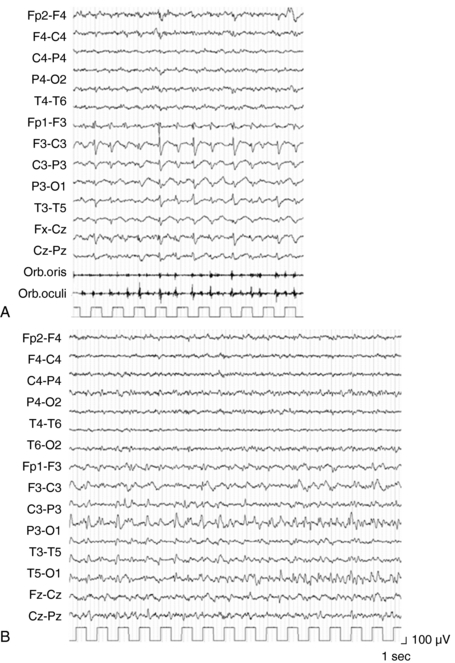
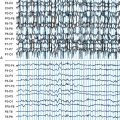
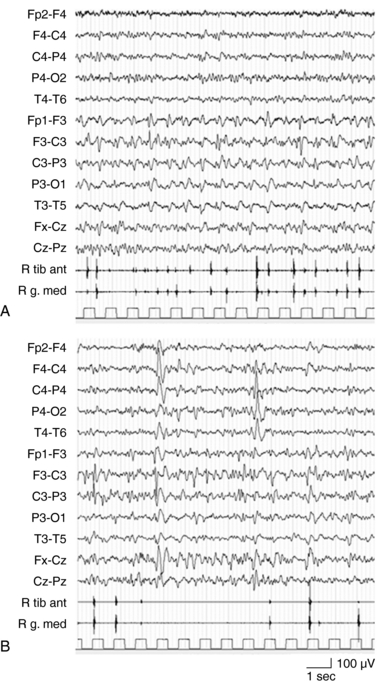
EPC, defined as spontaneous regular or irregular clonic twitching confined to one part of the body (Figure 12-2), aggravated by action or sensory stimuli, and persisting during sleep,8 occurs in most of the patients (56 to 92% according to different series) at some time during the disease course, unresponsive to AE drugs.3–4,9
The striking EEG feature at onset is the presence of slow focal activity mainly involving the temporal and central leads on the affected hemisphere, associated with few epileptic abnormalities on the same regions. Additional distinctive features, in the first few months, are the early evidence of ictal and interictal hemispheric multifocality, the presence of subclinical ictal discharges, and the unilateral impoverishment of the background activity and of sleep spindles (Figure 12-2).4,10
During the disease course, the background activity further flattens, and sleep organization deteriorates further; epileptic and slow activity tend to increase and spread within the affected hemisphere and the unaffected one. With time, the contralateral abnormal activity, either resulting from diffusion or seemingly asynchronous, may become more frequent than that recorded in the affected hemisphere (i.e., “false lateralization”).11 In any case, the seizure onset, albeit multifocal, is closely unilateral; the recording of seizure from the supposed unaffected hemisphere strongly questions the diagnosis of RE.
EPC, like in other conditions, is often not clearly related to electroencephalogram (EEG) changes recorded by scalp electrodes; however, auxiliary neurophysiologic techniques may contribute to define the cortical origin of the jerks. Back-averaging may identify the spikes preceding the motor phenomena, and somatosensory-evoked potentials of the rolandic cortex are often abnormally enlarged.12
NEUROLOGIC SYMPTOMS OTHER THAN EPILEPSY
Hemiparesis invariably develops during the disease course. As already mentioned, the motor deficit may be the symptom of onset, and exceptionally remains the one and only manifestation of RE.5 In the first stages of the disease, hemiparesis may be limited to the postictal phase, but rapidly becomes constant, albeit fluctuating in severity as it worsens with increasing seizure activity. With time, hemiparesis, sometimes associated with a dystonic component, stabilizes. In an Italian series of 12 patients, focal motor deficits appeared from 15 days to 24 months after the first seizure and invariably worsened to severe hemiparesis, leading three patients to be wheelchair bound within 3 years of onset.4
Movement disorders, which seldom mark the onset of RE, are rarely reported during the disease course, probably because they are misinterpreted or overlooked in patients whose clinical picture is dominated by epilepsy and hemiparesis. Additional neurologic symptoms include hemianopia, cortical sensory loss, and aphasia when the dominant hemisphere is affected.13
Cognitive impairment is another constant feature of RE, and as for motor deficits, its appearance may be subtle. Behavioral changes, with irritability, emotional lability, or hyperactivity, often herald the first signs of mental decline, which consist mainly of memory and attention disorders and learning difficulties. In the Italian series, these symptoms were detected within 4 to 36 months after the first seizure and progressively worsened until surgery. At the time of surgery, performed between 7 months and 14 years after disease onset, the mean IQ was 61.3 ± 15.1 (range 44–87).4 In most patients, the progression of mental impairment seems to correlate with the severity of epilepsy and particularly with the bilateral spreading of EEG epileptic abnormalities (personal experience), or with the appearance of asynchronous contralateral foci.14
The natural history of RE is summarized in the seminal paper by Oguni from the Montreal Neurological Institute3 and in the more recent report from the Bonn group, correlating the clinical course with the development of brain damage as documented by serial MRIs.9 Both groups recognize three stages that are virtually comparable. In the “prodromal stage,” lasting from 0 months to 8 years, seizures manifest at low frequency, and rarely, mild hemiparesis may be present. The “acute stage,” which in a significant number of cases appears to be the initial clinical manifestation, lasts 4 to 8 months and is characterized by frequent seizures, EPC (not always present), and rapid neurological deterioration. Finally, the patient enters the “residual stage,” with stable neurological deficits and persisting, albeit less frequent, seizures.
Atypical RE
ADOLESCENT- AND ADULT-ONSET RE
A limited, but increasing number of patients with adult-onset RE have been reported, accounting for about 10% of all RE reported cases.12,15 The overall features in childhood and adult onset are similar with regard to clinical, electrophysiological, and neuroimaging findings. The main differences consist of a more frequent posterior onset and a milder, more protracted, clinical course. Motor and mental deterioration are less severe than in typical childhood RE, hemispheric damage less pronounced, and quality of life somewhat preserved. However, in a number of cases, late-onset RE runs a malignant course, comparable to that of childhood-onset disease. In a recent paper Villani and coworkers identified two distinct patterns of disease presentation, one characterized by focal motor epilepsy (the “epileptic” phenotype), and the other by focal cortical myoclonus (the “myoclonic” phenotype). Unilateral neurologic deficits and brain atrophy were progressive in both phenotypes, but they were more prominent and detected earlier in the “epileptic” phenotype.12
RE PROTRACTED VARIANTS
A few RE patients are reported to have an acceptable seizure frequency and not invalidating motor deficits.15 This was already described in the pioneering study by Aguilar and Rasmussen,16 who reported that some patients may have mild neurologic deficits, despite the neuropathological evidence of active encephalitis.
RE ASSOCIATED WITH OTHER DISEASES (DOUBLE PATHOLOGY)
Patients affected by preexisting brain lesions account for about 10% of RE cases.1 This dual pathology has been documented in low-grade tumors, cortical dysplasia, tuberous sclerosis, vascular malformations, or old ischemic lesions. The association of structural abnormalities and inflammation is still a matter of investigation. The hypothesis that structural or acquired brain lesions, and the resulting epilepsy, may alter the blood–brain barrier permeability, allowing the entry of compounds with immunogenic or inflammatory potential, is conceivable but, at least to date, not demonstrated.17
The association of RE with definite autoimmune diseases, such as SLE,18 linear scleroderma,19 Parry–Romberg syndrome,20 although rarely reported, is noteworthy, as it provides a further hint for the role of immune-mediated mechanisms in the pathogenesis of RE.
BILATERAL RE
The term “bilateral RE” has been used in the literature to include two different conditions: (1) the secondary involvement of the unaffected hemisphere, as indicated by spreading of focal seizures from the original focus, by the appearance of interictal epileptic abnormalities, and by the mild brain atrophy, that most probably results from Wallerian degeneration of commissure fibers;21 (2) a familiar22 and few sporadic cases of early-onset malignant epilepsy with neuropathology consistent with chronic encephalitis resembling RE;23–24 and (3) rare adult-onset cases.25
The frequently observed secondary involvement of the unaffected hemisphere, of course, does not rule out the diagnosis of RE because in no cases is there evidence of truly contralateral disease. It should also be underlined that in RE patients surgically treated, even after a very protracted disease, by hemispherectomy (or hemispheric disconnection), no relapse of seizures from the contralateral hemisphere was ever recorded. Conversely, in “bilateral RE,” symptoms and, when available, MRI findings pointed to bilateral brain damage within the first months of the disease. In our opinion, from a pragmatic point of view, these cases should be kept separate from true RE (and probably named differently), given the different clinical picture, MRI findings, and, overall, the ensuing therapeutic approaches.
Pathogenesis of RE
The etiology of RE remains unknown, but our understanding of its pathogenesis improved considerably in recent years. The initiating event triggering RE remains unknown. A viral etiology was first proposed, based on the features of lymphocyte infiltration and microglial reaction within the brain and on similarities with known forms of viral encephalitis.2 However, no attempt of isolation of a pathogenic virus has been successful.26 The interest in RE has been renewed by histopathological and experimental studies showing the involvement of the immune system in the pathogenesis of the disease, providing evidence for both humoral and cellular factors.
In 1994, Rogers and coworkers observed that rabbits immunized with a recombinant fragment of the glutamate receptor (GluR3) developed seizures and that the histopathological examination of their brain showed inflammatory changes reminiscent of those found in patients with RE. In parallel, the same authors detected antiGluR3 antibodies in three patients with RE and observed clinical improvement after plasma exchange in one of them.27 Further reports of transient clinical improvement after removal of antibodies have been subsequently published.13,28–29 These preliminary findings fostered the search for such autoantibodies in larger series of patients with RE. AntiGluR3 antibodies, at first assayed by ELISA techniques, were found in serum and cerebrospinal fluid (CSF) not only in a proportion of patients with RE, but also in other forms of epilepsy, particularly in the catastrophic epilepsy of children. Therefore, their presence is not specific for RE but seems to correlate with epilepsy and its severity.30–31 However, the importance of antiGluR3 antibodies has been recently questioned by means of different laboratory techniques (ELISA, Western blot, immunoprecipitation, immunohistochemistry, and electrophysiology).32 Two different mechanisms by which antiGluR3 antibodies might exert their damage to the brain have been proposed. Plasma or serum of GluR3-immunized rabbits promoted the death of cortical neurons in vitro by a complement-mediated mechanism. Anti-IgG and anti-MAC antibodies were localized on neuronal cell bodies of cortex sections resected from RE patients,33–35 leading to the complement activation hypothesis. Other authors reported that sera and IgG fractions of GluR3-immunized rabbits elicited rapid, reversible, and voltage-independent opening of cationic channels in cultured neurons, promoting excytotoxic damage.36–37 Other antibodies have been described, such as to Munc-18 (a cytosolic protein), to NMDA-GluRε2 and anti-α7nAChR in a limited number of patients,38–40 but their actual role is still to be elucidated. Nevertheless, the analysis of immunoglobulin gene rearrangement in brain samples from four RE patients provided evidence for a restricted profile.41
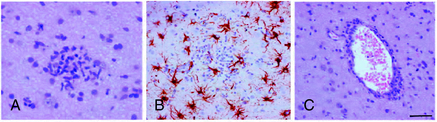
The role of T cells in the pathogenesis of RE has been emphasized by several pathological studies. The main pathological features of RE include brain inflammation dominated by T cells, microglial activation, microglial nodules, neuronal loss, and astrogliosis in the affected hemisphere (Figure 12-3).42,43 The extent of T-cell infiltration and microglial activation is inversely correlated with duration of the disease, being more prominent in the earliest stages while in the latest gliosis predominates.44 The majority of infiltrating T cells have been characterized as CD8+ cells containing granules positive for granzyme B, and part of these cells were observed in close contact with MHC class I positive neurons,45 a set of features considered evidence of a cytotoxic T-cell reaction against neuronal cells. The same authors could not detect deposits of immunoglobulins or signs of complement activation in the same brain specimens. Recently, the importance of astrocytes has been investigated in detail in RE.46 In RE, astrocytic apoptosis and loss were detected both in the cortex and the white matter; interestingly, as already observed by previous studies on T cells, granzyme B+ lymphocytes were found in close contact with astrocytes with granules polarized toward the astrocytic membranes. The authors therefore suggested that astrocytes might be a target for cytotoxic T cells, ultimately leading to astrocytic degeneration. Considering their multiple functional roles, degeneration of astrocytes might enhance neuronal loss as well as contribute to seizure induction and maintenance.
Laboratory Findings Other Than EEG
NEUROIMAGING
The refinement of imaging techniques has highly contributed to the recognition of RE, even in the early stages of the disease, and better characterization of the type and sequence of brain pathologic changes (Figure 12-4).
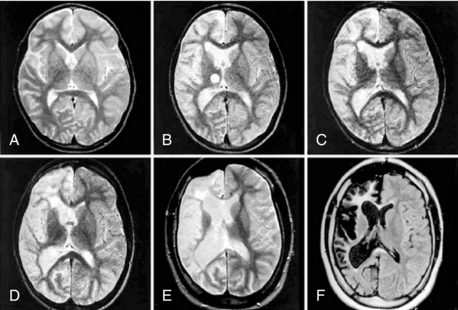
Recent studies focused on the early signs of the disease and demonstrated that within the first few months, most patients disclose a combination of changes that, albeit not diagnostic, are typical enough to raise the suspicion of RE.4,47 These early signs include, (1) mild focal cortical atrophy, always involving the insular and periinsular regions; (2) ipsilateral ventricle enlargement; (3) increased cortical and/or subcortical T2 and FLAIR signal; and (4) T2 hyperintensity or atrophy of the head of the caudate nucleus. The described changes are preceded, as evaluated in the few cases with a very early examination, by focal, transient, cortical swelling. Gadolinium enhancement is not observed.
During the progression of the disease, unilateral cortical and caudate atrophy progressively worsens, whereas signal abnormalities are less evident. In a study comparing MRI and histopathology findings, Bien and coworkers demonstrated that increased MRI signal correlates with an active inflammatory process, as witnessed by the high density of inflammatory cells and reactive astrocytes (i.e., with the presence of T cells, microglial nodules, and GFAP-positive astrocytes). Conversely, MRI atrophic areas without signal abnormalities correlate with tissue loss, which mostly occurs during the first year after the “acute disease stage.”44 The sovratentorial tissue loss is accompanied by atrophy of the contralateral, but seldom global, cerebellar hemisphere of the brainstem and thinning of the corpus callosum.47
The role of functional imaging, PET, SPECT, and MR spectroscopy (MRS) is still under study. These techniques may demonstrate, even in the very early stages, areas of abnormal metabolism (PET), or perfusion (SPECT), and reduced N-acetylaspartate (NAA), but are not useful in defining the suspected inflammatory nature of the disease.48–49 Moreover, PET and SPECT findings are highly influenced by seizure activity; MRS may show increased lactate, also resulting from seizure activity, potentially misleading the diagnosis. For all these reasons, data obtained by these techniques must be cautiously interpreted and integrated with clinical and MRI findings. In clinical practice, the usefulness of PET, SPECT, and MRS should be limited to confirming the unilateral brain damage.
BLOOD AND CSF TESTS
CSF may be normal or show nonspecific abnormalities, such as a mild increase in the white cell count and proteins. Oligoclonal bands are found in about half the patients.4,50 In summary, blood and CSF analysis are not suitable in confirming RE, but must be carried out to exclude CNS infections or other disorders.
BRAIN BIOPSY
Histopathology study is not required in all RE cases because other criteria allow the correct diagnosis, even in early stages, in most cases (see following discussion). It has to be considered, moreover, that histology shows nonspecific patchy chronic inflammatory changes, and that normal and abnormal tissue elements may be located in very close apposition.42–43 Therefore, false-negative results may be obtained, particularly if small tissue samples are taken. In the few patients in which biopsy is required (Table 12-1), an open biopsy, in a noneloquent area where there is increased T2/FLAIR signal on MRI, should include meninges, gray, and white matter.44
TABLE 12–1 Diagnostic Criteria for RE by a Two-step Approach
| 1° step: Check for the features of Part A. | |
| 2° step: If features of Part 1 are not fulfilled, check criteria of Part B. | |
| Part A: | |
| 1. Clinical | Focal seizures (with or without epilepsia partialis continua) and unilateral cortical deficit(s) |
| 2. EEG | Unihemispheric slowing with or without epileptiform activity and unilateral seizure onset |
| 3. MRI | |
* Clinical progression: at least two sequential clinical evaluation must document a neurological deficit that must increase over the time. MRI progression: at least two sequential MRI studies must show hemiatrophy, that must increase over time.
Modified from Bien CG, Granata T, Antozzi C, et al. Pathogenesis, diagnosis and treatment of Rasmussen encephalitis. A European consensus statement. Brain. 2005;128: 454-471.
Diagnostic Clues and Differential Diagnosis
In patients with a long-lasting disease, the diagnosis is rather simple, and differential diagnosis is limited to a few conditions; conversely, the recognition of RE in its early stages, before progressive neurologic deficits and brain atrophy have developed, can be a challenge. In the last years, efforts have been made to identify the early manifestations of RE that could prompt a reasonably secure diagnosis and, consequently, an early targeted treatment.4,9 Diagnostic criteria including typical and atypical RE have been recently reviewed by a European consensus (Table 12-1); however, it should be underlined that the correct diagnosis of RE needs a wide clinical experience supported by multidisciplinary expertise.
The diagnostic workup must exclude, on a clinical and/or laboratory basis, conditions characterized by unilateral neurologic syndromes or EPC, as well as inflammatory diseases mimicking RE. Among these conditions, the following must be considered: (1) focal or hemispheric dysplasia, neoplasia, and neurocutaneous syndromes (in particular Sturge-Weber syndrome), stroke, and hemiconvulsion-hemiplegia-epilepsy syndrome; (2) progressive neurometabolic or degenerative syndromes, namely MELAS and other mitochondriopathies, lipofuscinoses, and Alpers disease; (3) metabolic disorders, including type 1 diabetes mellitus, associated with anti-GAD 65-antibodies, and renal or hepatic encephalopathy; (4) cerebral vasculitis in systemic diseases, such as lupus erythematosus or Derry’s vasculitis; (5) rare infective disorders such as cat-scratch disease, but also HIV infection; and (6) finally, there are a few reports of severe epilepsy and EPC in bone marrow–transplanted patients; this possibility must be kept in mind giving the increasing indications to this treatment.
A summary of differential diagnosis with related clinical and laboratory investigations are summarized in the mentioned European consensus statement.1
Treatment
RE is an inflammatory and most probably immunomediated disease, running a progressive course, characterized by severe epilepsy and worsening motor and mental deficits. These features must be kept in mind in the therapeutic decision process. The management of RE, at least theoretically, should therefore include the treatment of both seizures and the underlying inflammation. In addition, therapeutic efficacy must be assessed on both seizure reduction and neurologic improvement and weighted against side effects and potential residual deficits. Beside surgery, which is unanimously considered the only treatment able to stop seizures and disease progression (see following discussion), different drug treatments have been employed, based on the proposed etiopathogenesis of RE. Antiviral and interferon treatment was reported in a few papers in the nineties, but the paucity of data does not allow any conclusions on their effect. On the contrary, an increasing number of reports on the use of immunomodulatory treatments have accumulated in the last decade, indicating their potential role in selected patients and circumstances. The mechanisms by which these treatments may act in RE are probably complex and include, besides the antiinflammatory and immunomodulating effects, the regulation of BBB permeability (i.e., steroids) and a direct antiepileptic effect (steroids and IVIG).17
IMMUNOMODULATORY TREATMENTS
The rarity of disease, the variability of disease severity, the fluctuating course, and differences in treatment schedules employed by different centers, together with the lack of controlled clinical trials due to ethical reasons, make the evaluation of treatment efficacy very arduous. Nevertheless, case reports accumulated in the last years allow the evaluation of different treatments on the basis of which prospective studies have started. As a general statement, in the vast majority of cases, as per literature reports and personal experience, immunomodulation has only a transient and partial effect on seizure activity and progression of symptoms and should therefore be limited to patients that are not, or not yet, suitable for surgery. These include late-onset (adolescent or adult) RE with slower and milder course than the typical childhood-onset form; patients with dominant hemisphere involvement and slow progression when worsening of motor and language functions resulting from surgery are not accepted from the patient and family; suspected RE in which neurologic deterioration and hemispheric atrophy have not yet occurred; and proven or suspected bilateral RE. Nevertheless, the reports of transient efficacy of immunomodulation in some of these patients are in keeping with pathogenetic data suggesting an inflammatory brain process. In some of these patients, immunomodulation with corticosteroids, immunosuppressants, and IVIG or plasma treatment may be of help in the management of acute deterioration of the clinical picture or in slowing down its progression.
Corticosteroids are the most widely used, and in our personal experience the most effective therapy, not only as chronic treatment, but also in halting epileptic status or reducing the intensity of EPC. In the latter condition, methylprednisolone should be used in high-dose pulses (i.e., 400 mg/m2/day or, in children, 20 mg/kg/day tapered over 10 to 12 days).50–51 Some evidence exists that steroid treatment is more effective when started close to the onset of the disease.51–52 As far as long-term steroid therapy is concerned, it has been recommended to start with boluses of intravenous methylprednisolone and then shift to 1 to 2 mg/kg/day of oral prednisone,50–51 to be slowly tapered to the minimal effective dosage.
IVIg efficacy has been reported in a limited proportion of children and adults with RE.53–55 The recommended dosing scheme consists of five consecutive infusions of 0.4 g/kg/day followed by a monthly dose of 0.4 to 2.0 g/kg distributed over 1 to 5 consecutive days; the assessment of IVIG efficacy may require as long as six courses. The combination of steroids and IVIg may also be considered50–51 when the two treatments alone are ineffective.
Immunomodulation by Plasmapheresis (PE) or Protein A Immunoadsorption (PAI)
Plasmatic treatment has been used with the rationale of removing potentially pathogenetic circulating antibodies (namely anti GluR3). A dramatic effect in blocking status epilepticus and neurologic deterioration has been reported in a few patients,28,51 whereas evidence of long-term efficacy has been only exceptionally reported.13 For this reason, as well as for technical issues (particularly in children) and costs, PEX or PAI should be reserved to phases of acute deterioration, or to assess the residual motor and mental abilities before surgery.
Immunosuppression Tacrolimus
The only trial so far reported on the use of immunosuppressive agents is that of Bien and coworkers.56 The authors tested the effect of tacrolimus, a T-cell inhibiting drug, in seven RE patients and compared their outcome with that of twelve “historical” untreated patients. Despite no significant effect on seizure frequency, tacrolimus-treated patients had a better outcome in terms of motor and mental functions, as well as on the rate of brain atrophy, to the point that none of these patients was submitted to brain surgery.
Conflicting or limited results, hindering any conclusion, have been reported for cyclophosphamide and azathioprine.28,51,57
ANTIEPILEPTIC DRUGS
It is common experience that conventional antiepileptic drugs, including the more recent compounds, have only partial effect on seizures, and no mono- or poly-therapy has been reported to be superior to others.58 AEDs should be therefore chosen on the basis of the empirical demonstration of efficacy and tolerability, avoiding heavy politherapy to limit drug interactions and potential side effects. High-dose phenobarbital remains, in our personal experience, one of the more effective treatments in reducing seizure frequency and the intensity of EPC.
SURGERY
RE was first described in a patient surgically treated for intractable focal seizures and hemiparesis. Fifty years later, despite the advances in understanding the pathogenesis of RE and attempts to find rational medical therapies, surgery, and namely the surgical exclusion of the affected hemisphere, still remains inevitable in almost all patients, even in those who experienced transient benefit from immunomodulation.51–52
Hemispherectomy, and in the more recent years hemispherotomy (consisting in deafferentation of the hemisphere with very limited removal of tissue), is effective in halting seizures, as well as motor and mental deterioration, in over 80% of patients. Following surgery, most patients recover, at least in part, from neurologic decline, with significant improvement in both patients and family quality of life.59–62
1. Bien CG, Granata T, Antozzi C, et al. Pathogenesis, diagnosis and treatment of Rasmussen encephalitis. A European consensus statement. Brain. 2005;128:454-471.
2. Rasmussen T, Olszewski L, Lloyd-Smith D. Focal seizures due to chronic localized encephalitis. Neurology. 1958;8:435-445.
3. Oguni H, Andermann F, Rasmussen TB. The natural history of the syndrome of chronic encephalitis and epilepsy: a study of the MNI series of forty-eight cases. In: Andermann F, editor. Chronic Encephalitis and Epilepsy: Rasmussen’s Syndrome. Boston: Butterworth-Heinemann; 1991:7-35.
4. Granata T, Gobbi G, Spreafico R, et al. Rasmussen’s encephalitis: early characteristics allow diagnosis. Neurology. 2003;60:422-425.
5. Bien CG, Elger CE, Leitner Y, et al. Slowly progressive hemiparesis in childhood as a consequence of Rasmussen encephalitis without or with delayed-onset seizure. Eur J Neurol. 2007;14:387-390.
6. Frucht S. Dystonia, athetosis, and epilepsia partialis continua in a patient with late-onset Rasmussen’s encephalitis. Mov Disord. 2002;17:609-612.
7. Andermann F. Rasmussen syndrome and movement disorder. Mov Disord. 2002;17:437-438.
8. Bancaud J, Bonis A, Trottier S, Talairach J, Dulac O. Continuous partial epilepsy: syndrome and disease. Rev Neurol. 1982;138:803-814.
9. Bien CG, Widman G, Urbach H, et al. The natural history of Rasmussen’s encephalitis. Brain. 2002;125:1751-1759.
10. So N, Gloor P. Electroencephalographic and electrocorticographic findings in chronic encephalitis of the Rasmussen type. In: Andermann F, editor. Chronic Encephalitis and Epilepsy: Rasmussen’s Syndrome. Boston: Butterworth-Heinemann; 1991:37-45.
11. Andrews PI, McNamara JO, Lewis DV. Clinical and electroencephalographic correlates in Rasmussen’s encephalitis. Epilepsia. 1997;38:189-194.
12. Villani F, Pincherle A, Antozzi C, et al. Adult-onset Rasmussen’s encephalitis: anatomical-electrographic-clinical features of 7 Italian cases. Epilepsia. 2006;47(S5):41-46.
13. Antozzi C, Granata T, Aurisano N, et al. Long-term selective IgG immunoadsorption improves Rasmussen’s encephalitis. Neurology. 1998;51:302-305.
14. Longaretti F, Dunkley C, Varadkar S, Boyd S, Cross JH. Early EEG findings in children with Rasmussen’s encephalitis versus focal cortical dysplasia. Dev Med Child Neurol. 2006;48:24.
15. Hart YM, Andermann F, Fish DR, et al. Chronic encephalitis and epilepsy in adults and adolescents: a variant of Rasmussen’s syndrome? Neurology. 1997;48:418-424.
16. Aguilar MJ, Rasmussen T. Role of encephalitis in pathogenesis of epilepsy. Arch Neurol. 1960;2:663-676.
17. Vezzani A, Granata T. Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia. 2005;46:1724-1743.
18. Lascelles K, Dean AF, Robinson RO. Rasmussen’s encephalitis followed by lupus erythematosus. Dev Med Child Neurol. 2002;44:572-574.
19. Pupillo G, Andermann F, Dubeau F. Linear scleroderma and intractable epilepsy: neuropathologic evidence for a chronic inflammatory process. Ann Neurol. 1996;39:277-278.
20. Shah JR, Juhasz C, Kupsky WJ, et al. Rasmussen encephalitis associated with Parry-Romberg syndrome. Neurology. 2003;61:395-397.
21. Larionov S, König R, Urbach H, Sassen R, Elger CE, Bien CG. MRI brain volumetry in Rasmussen encephalitis: the fate of affected and “unaffected” hemispheres. Neurology. 2005;64:885-887.
22. Silver K, Andermann F, Meagher-Villemure K. Familial epilepsia partialis continua with chronic encephalitis: another variant of Rasmussen syndrome? Arch Neurol. 1988;55:733-735.
23. Tobias SM, Robitaille Y, Hickey WF, Rhodes CH, Nordgren R, Andermann F. Bilateral Rasmussen encephalitis: postmortem documentation in a five-year-old. Epilepsia. 2003;44:127-130.
24. Andermann F, Farrel K. Early onset Rasmussen syndrome: a malignant, often bilateral form of the disorder. Epilepsy Res. 2006;70(suppl 1):S259-S262.
25. McLachlan RS, Girvin JP, Blume WT, Reichman H. Rasmussen’s chronic encephalitis in adults. Arch Neurol. 1993;50:269-274.
26. Atkins MR, Terrell W, Hulette CM. Rasmussen’s syndrome: a study of potential viral etiology. Clin Neuropathol. 1995;14:7-12.
27. Rogers SW, Andrews PI, Gahring LC, et al. Autoantibodies to glutamate receptor GluR3 in Rasmussen’s encephalitis. Science. 1994;265:648-651.
28. Andrews PI, Dichter MA, Berkovic SF, Newton MR, McNamara JO. Plasmapheresis in Rasmussen’s encephalitis. Neurology. 1996;46:242-246.
29. Palcoux JB, Carla H, Tardieu M, et al. Plasma exchange in Rasmussen’s encephalitis. Ther Apher. 1997;1:79-82.
30. Wiendl H, Bien CG, Bernasconi P, et al. GluR3 antibodies: prevalence in focal epilepsy but no specificity for Rasmussen’s encephalitis. Neurology. 2001;57:1511-1514.
31. Mantegazza R, Bernasconi P, Baggi F, et al. Antibodies against GluR3 peptides are not specific for Rasmussen’s encephalitis but are also present in epilepsy patients with severe, early onset disease and intractable seizures. J Neuroimmunol. 2002;131:179-185.
32. Watson R, Jiang Y, Bermudez I, et al. Absence of antibodies to glutamate receptor type 3 (GluR3) in Rasmussen encephalitis. Neurology. 2004;63:43-50.
33. He XP, Patel M, Whitney KD, Janumpalli S, Tenner A, McNamara JO. Glutamate receptor GluR3 antibodies and death of cortical cells. Neuron. 1998;20:153-163.
34. Whitney KD, Andrews PI, McNamara JO. Immunoglobulin G and complement immunoreactivity in the cerebral cortex of patients with Rasmussen’s encephalitis. Neurology. 1999;53:699-708.
35. Whitney KD, McNamara JO. GluR3 autoantibodies destroy neural cells in a complement-dependent manner modulated by complement regulatory proteins. J Neurosci. 2000;20:7307-7316.
36. Twyman RE, Gahring LC, Spiess J, Rogers SW. Glutamate receptor antibodies activate a subset of receptors and reveal an agonist binding site. Neuron. 1995;14:755-762.
37. Levite M, Fleidervish IA, Schwarz A, Pelled D, Futerman AH. Autoantibodies to the glutamate receptor kill neurons via activation of the receptor ion channel. J Autoimmun. 1999;13:61-72.
38. Yang R, Puranam RS, Butler LS, et al. Autoimmunity to munc-18 in Rasmussen’s encephalitis. Neuron. 2000;28:375-383.
39. Takahashi Y, Mori H, Mishina M, et al. Autoantibodies and cell-mediated autoimmunity to NMDA-type GluRepsilon2 in patients with Rasmussen’s encephalitis and chronic progressive epilepsia partialis continua. Epilepsia. 2005;46(S5):152-158.
40. Watson R, Jepson JE, Bermudez I, et al. Alpha7-acetylcholine receptor antibodies in two patients with Rasmussen encephalitis. Neurology. 2005;65:1802-1804.
41. Baranzini SE, Laxer K, Saketkhoo R, et al. Analysis of antibody gene rearrangement, usage, and specificity in chronic focal encephalitis. Neurology. 2002;58:709-716.
42. Robitaille Y. Neuropathologic aspects of chronic encephalitis. In: Andermann F, editor. Chronic Encephalitis and Epilepsy: Rasmussen’s Syndrome. Boston: Butterworth-Heinemann; 1991:79-110.
43. Pardo CA, Vining EP, Guo L, Skolasky RL, Carson BS, Freeman JM. The pathology of Rasmussen syndrome: stages of cortical involvement and neuropathological studies in 45 hemispherectomies. Epilepsia. 2004;45:516-526.
44. Bien CG, Urbach H, Deckert M, et al. Diagnosis and staging of Rasmussen’s encephalitis by serial MRI and histopathology. Neurology. 2002;58:250-257.
45. Bien CG, Bauer J, Deckwerth TL, et al. Destruction of neurons by cytotoxic T cells: a new pathogenic mechanism in Rasmussen’s encephalitis. Ann Neurol. 2002;51:311-318.
46. Bauer J, Elger CE, Volkmar HH, et al. Astrocytes are a specific immunological target in Rasmussen’s encephalitis. Ann Neurol. 2007;62:67-80.
47. Chiapparini L, Granata T, Farina L, et al. Diagnostic imaging in 13 cases of Rasmussen’s encephalitis: can early MRI suggest the diagnosis? Neuroradiology. 2003;45:171-183.
48. Fiorella DJ, Provenzale JM, Edward CR, Crain BJ, Al Sugair A. (18)F-fluorodeoxyglucose positron emission tomography and MR imaging findings in Rasmussen encephalitis. Am J Neuroradiol. 2001;22:1291-1299.
49. Fogarasi A, Hegyi M, Neuwirth M, et al. Comparative evaluation of concomitant structural and functional neuroimages in Rasmussen’s encephalitis. J Neuroimaging. 2003;13:339-345.
50. Hart YM, Cortez M, Andermann F, et al. Medical treatment of Rasmussen’s syndrome (chronic encephalitis and epilepsy): effect of high-dose steroids or immunoglobulins in 19 patients. Neurology. 1994;44:1030-1036.
51. Granata T, Fusco L, Gobbi G, et al. Experience with immunomodulatory treatments in Rasmussen’s encephalitis. Neurology. 2003;61:1807-1810.
52. Bahi-Buisson N, Villanueva V, Bulteau C, et al. Long term response to steroid therapy in Rasmussen encephalitis. Seizure. 2007;16:485-492.
53. Leach JP, Chadwick DW, Miles JB, Hart IK. Improvement in adult-onset Rasmussen’s encephalitis with long-term immunomodulatory therapy. Neurology. 1999;52:738-742.
54. Villani F, Spreafico R, Farina L, et al. Positive response to immunomodulatory therapy in an adult patient with Rasmussen’s encephalitis. Neurology. 2001;56:248-250.
55. Granata T. Rasmussen’s syndrome. Neurol Sci. 2003;S4:239-243.
56. Bien CG, Gleissner U, Sassen R, Widman G, Urbach H, Elger CE. An open study of tacrolimus therapy in Rasmussen’s encephalitis. Neurology. 2004;62:2106-2109.
57. Krauss GL, Campbell ML, Roche KW, Huganir RL, Niedermeyer E. Chronic steroid-responsive encephalitis without autoantibodies to glutamate receptor GluR3. Neurology. 1996;46:247-249.
58. Dubeau F, Sherwin AL. Pharmacologic principles in the management of chronic focal encephalitis. In: Andermann F, editor. Chronic Encephalitis and Epilepsy: Rasmussen’s Syndrome. Boston: Butterworth-Heinemann; 1991:179-192.
59. Villemure J-G, Andermann F, Rasmussen TB. Hemispherectomy for the treatment of epilepsy due to chronic encephalitis. In: Andermann F, editor. Chronic Encephalitis and Epilepsy: Rasmussen’s Syndrome. Boston: Butterworth-Heinemann; 1991:235-241.
60. Vining EP, Freeman JM, Brandt J, Carson BS, Uematsu S. Progressive unilateral encephalopathy of childhood (Rasmussen’s syndrome): a reappraisal. Epilepsia. 1993;34:639-650.
61. Thomas P, Zifkin B, Ghetâu G, Delalande O. Persistence of ictal activity after functional hemispherectomy in Rasmussen syndrome. Neurology. 2003;60:140-142.
62. Tubbs RS, Nimje SM, Oajes WJ. Long-term follow-up in children with functional hemispherectomy for Rasmussen encephalitis. Childs Nerv Syst. 2005;21:461-465.