Chapter 10 The Spectrum of Epilepsies Associated with Generalized Spike-and-Wave Patterns
Introduction
The generic term epilepsy embraces a particular group of paroxysmal brain disorders, but by itself is of limited clinical value. Intense clinical and genetic research over the last few decades has identified a large number of well-defined epilepsy syndromes with different clinical, EEG, neuropsychological, and neuroimaging profiles, natural history and prognosis, and conditions that ultimately require different management. There is evidence that early treatment can reduce the risk of seizure recurrence,1 and its efficacy depends largely on the appropriate drug choice in relation to the particular clinical syndrome.2 Therefore, identification of the particular form or syndrome is the cornerstone of meaningful, optimal management of the individual patient.2,3
THE INTERNATIONAL CLASSIFICATION OF EPILEPTIC SEIZURES AND SYNDROMES: BASIC CONCEPTS AND DEFINITIONS
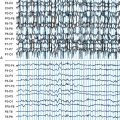
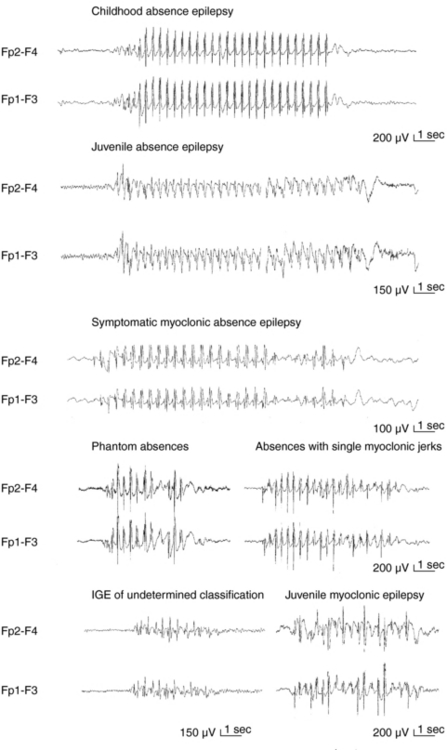
Apart from assisting patients’ clinical management and prognostication, recognition of different syndromes enables effective communication between clinicians and clinical and genetic research. The current classification of epileptic seizures4 and epilepsies5 of the International League Against Epilepsy (ILAE) and also the new ILAE proposal6,7 are essentially organized around two dichotomies in terms of their topography between generalized and localized (partial or focal) and in terms of their etiology between idiopathic epilepsies (genetically determined) and symptomatic epilepsies (due to cerebral pathology). The ILAE classification is currently under revision, but is still valid. Presumed symptomatic epilepsies, in which no certain etiology can be demonstrated, are sometimes referred to as “cryptogenic.” Generalized epilepsies manifest with generalized seizures, whose first ictal clinical changes reflect involvement of both hemispheres and in which initial EEG changes are bilateral, as, for example, the typical absences (TA) that are associated with 3-Hz GSW (Figure 10-1). Localization-related epilepsies (also called focal or partial) manifest with focal (or partial) seizures, in which the initial activation of a group of neurons is limited to a part of one hemisphere5; the EEG shows focal abnormalities, but GSW discharges may occur as a result of secondary bilateral synchrony (SBS).8
CONVENTIONS AND LIMITATIONS OF THE ILAE CLASSIFICATION SYSTEM AND SYNDROMIC DIAGNOSIS IN CLINICAL PRACTICE
Identification of the seizure type(s) is the quintessential element of an epileptic syndrome and the first step for its definition.2 However, it is not always straightforward, particularly on historical evidence (accounts of patients and witnesses) only. TA in idiopathic generalized epilepsy (IGE), for example, may share some principal clinical features with limbic complex partial seizures (CPS) (such as unresponsiveness and automatisms), and one must turn to either their onset (presence or absence of aura) or immediately post-termination (presence or absence of confusion) to make an educated clinical hypothesis. Interictal EEG studies will assist diagnosis by showing focal epileptiform activity or GSW, but even so the rate of misdiagnosis is high with the generalized epilepsies being erroneously diagnosed as focal rather than the converse.9,10 Important reasons for such misdiagnosis include usually short-lived “focal” clinical symptoms and signs, such as unilateral jerks and rotatory seizures in juvenile myoclonic epilepsy (JME),10–13 versive absences,14 and in addition, well-recognized asymmetrical or focal interictal EEG changes.11,15,16 Such electroclinical focalities within the generalized seizures are interpretable within the model of generalized corticoreticular epilepsy, but stand uncomfortably within the inflexible and simplified generalized versus focal dichotomy of the ILAE seizure and syndrome classification system.17 Focal epilepsies may (also) present with rapidly generalized convulsions and infrequently with interictal GSW discharges as in SBS.
In this chapter, we will discuss primarily the various syndromes of IGE, whose GSW is the defining electrographic trait. From the brief foreword, it follows that GSW discharges do not always indicate IGE or indeed generalized epilepsies, and Table 10-1 includes all the main epilepsies and syndromes that are associated with 3–4 Hz GSW. The interictal EEG alone cannot be used for establishing or excluding the diagnosis of epilepsy (including IGEs) or provide oversimplified clues for reliable syndromic diagnosis; this can only be based on a comprehensive electroclinical synthesis for the individual patient, without which EEG interpretation may be completely meaningless and even misleading.2,17
TABLE 10–1 Epilepsy Syndromes and Conditions Associated with GSW Discharges
| Seizures Types | Interictal EEG | |
|---|---|---|
| IGE | TA, MS, GTCS, rarely tonic | Normal background 2.5–4 Hz regular GSW, nonlocalizing focal |
| Absence syndromes | TA, late GTCS, rarely MS (in JAE) | |
| CAE, JAE | ||
| Phantom TA | Brief TA, GTCS; frequent ASE | |
| Myoclonic syndromes | 3-6 Hz irregular GPSW with fragmentations | |
| BMEI | MS | |
| JME | MS, GTCS, TA (35%) | |
| EMA/PMA | TA, GTCS, rarely MS and absence status epilepticus | |
| GTCS only | GTCS | GSW at 2.5–6Hz, regular or irregular ± fragmentations |
| Mixed/unclassified | TA, MS, GTCS, tonicabsences | |
| Generalized Cryptogenic/Symptomatic Epilepsies | ||
IGE
Notwithstanding the aforementioned diagnostic limitations and pitfalls, GSW discharges are the characteristic EEG building blocks of all seizures in IGE that include typical absences (TA), myoclonic seizures (MS), and GTCS. EEG background activity is normal. IGE comprise a group of genetically determined epilepsies, unrelated to any structural brain pathology, and associated with normal neurological and neuropsychological status. Suitable AED include valproic acid VPA (arguably the first choice), clonazepam (CLZ), ethosuximide (ESX), lamotrigine (LTG), levetiracetam (LEV), topiramate (TPM), and zonisamide (ZNS) (with specific indications against main seizure types),18–20 whereas carbamazepine (CBZ), oxcarbazepine (OCBZ), vigabatrin (VGB), tiagabine (TGB), gabapentin (GBP), pregabalin (PGB), and phenytoin (PHT) are generally contraindicated, as they can cause seizure worsening.2,20–23
BENIGN MYOCLONIC EPILEPSY IN INFANCY (BMEI)2,24–26
This is the earliest form of IGE with a prevalence of about 1% to 2% of epilepsies that start before the age of 4 years. MS start between 4 months to 3 to 4 years and are typically favored by sleep. Two-thirds of the affected children are boys. MS are frequently described by parents as “spasms” or “head-nodding” attacks, and indeed differentiation from spasms may be difficult from description, so ictal video EEG recordings with dedicated EMG electrodes have to be employed. MS are associated with brief bursts of GSW, but interictal EEG is normal. MS may be provoked by photic stimulation in 20% and by unexpected acoustic or tactile stimuli in 10% of children; the latter may be easier to control. Simple febrile seizures occur in 10%, but there are no other seizure types. BMEI responds to VPA, and remission usually occurs within 1 year from onset, but 10 to 20% may develop infrequent GTCS in their early teens. Differential diagnosis includes nonepileptic conditions, such as hypnagogic jerks and benign nonepileptic myoclonus and epileptic syndromes, including severe myoclonic epilepsy of infancy Dravet syndrome), familial infantile myoclonic epilepsy (mapped to 16p13), infantile spasms (West syndrome), epilepsy with myoclonic-astatic seizures (MAE), and the myoclonic form of Lennox-Gastaut syndrome (LGS).
EPILEPSY WITH MYOCLONIC-ASTATIC SEIZURES (MAE)2,27,28
MAE is a genetically determined (some patients fall into the spectrum of generalized epilepsy with febrile seizures plus [GEFS+] nonlesional generalized epilepsy that affects previously normal children between 2 and 5 years of age, but has variable course ranging from good responsiveness to treatment, with normal cognitive functions and even remission, to nonprogressive epileptic encephalopathy with seizure intractability and cognitive impairment. Therefore it clinically overlaps with both IGE (see also the 2001 ILAE proposal, where it features within the IGE6) and cryptogenic/symptomatic generalized epilepsies (see relevant following section). Prevalence is around 1to 2% of all childhood epilepsies, onset ranges between 7 months and 6 years, peaking between 2 and 4 years, and two-thirds of patients are boys. Myoclonic-astatic seizures are the defining seizure symptom and consist of a myoclonic jerk immediately followed by loss of muscle tone. Either component can cause falls, head nodding, or bending of the knees. More than half of patients have brief absences, often with myoclonic jerks, facial myoclonias, and atonia. In two-thirds of patients, febrile and nonfebrile GTCS precede myoclonic astatic seizures by several months. Episodes of absence status may occur, usually introduced by inappropriate treatment such as CBZ; they are frequent in the cryptogenic form. Myoclonic-atonic seizures are associated with discharges of irregular GSW or polyspike waves at >2.5 Hz, with the atonic part of the seizure corresponding to the slow wave of the GSW complex and being associated with diffuse electromyography (EMG) paucity. VPA at high doses is the first drug of choice, combined if needed with LEV, ESX, CLZ, or sulthiame. MAE shares many clinical features with cryptogenic Lennox-Gastaut syndrome, particularly its myoclonic form.29
CHILDHOOD ABSENCE EPILEPSY (CAE)2,30–32
TA are associated with severe impairment of consciousness (unresponsiveness and interruption of ongoing activities) and are usually multiple per day, hence the term pyknolepsy. They have abrupt onset and termination and last from 4 to 20 sec (mainly around 10 sec). Automatisms occur in two-thirds of the seizures but are not stereotyped. Mild myoclonic elements of eyes, eyebrows, and eyelids may feature at the onset of an absence. More severe and sustained myoclonic jerks of facial ro limb muscles indicate other IGEs. TA are nearly always provoked by hyperventilation, even in the clinic. Other types of seizures are incompatible with CAE except for febrile seizures and solitary or infrequent GTCS (usually in adolescence after absences have remitted).
The interictal EEG may show characteristic rhythmic posterior delta activity and brief, usually regular GSW. Monotherapy with VPA ESX, or LTG is successful in most cases, and remission occurs before the age of 12 years with less than 10% of patients developing infrequent GTCS in adolescence or adult life.33 As in all IGEs, VGB, TGB, CBZ, and GBP can produce worsening of absences. CAE is not the only IGE absence syndrome in the first decade of life, and meticulous differentiation from other absence syndromes of worse prognosis is clinically important.
EPILEPSY WITH MYOCLONIC ABSENCES (E-MA)34,35
This rare type of generalized epilepsy was described by Tassinari36,37 and is characterized by myoclonic absences that are associated with 3-Hz regular GSW activity like the typical absences in childhood or juvenile absence epilepsies, but also with bilateral myoclonic jerks of the arms and legs that are time locked to the spike component of the ictal discharge and have a superimposed tonic contraction resulting in a characteristic stepwise upward abduction of the arms. Other seizure types may include GTCS, clonic seizures, and rarely simple absences without myoclonus. It affects less than 1% of children with epileptic disorders, mostly boys (70%), between 5 months and 13 years (median 7 years). Etiology is heterogeneous, and only one-third of patients are of normal neurological and mental state (idiopathic form). These retain normal EEG background. The others are symptomatic, with learning difficulties manifested before or after seizure onset. Early control of absences may secure normal development. The main task of differential diagnosis here is to distinguish between idiopathic and symptomatic cases (see later discussion).
JUVENILE ABSENCE EPILEPSY (JAE)2,38–40
The prevalence of JAE is around 2 to 3% of all epilepsies and around 8 to 10% of IGE in adults after the age of 20 years. About 70% of the patients start having absences between ages of 9 and 13 years, but age at onset may range from 5 to 20 years. Males and females are equally affected. TA are similar to those of CAE, but milder, and duration varies from 4 to 30 sec or longer. Typically infrequent GTCS and random, rather than early morning, myoclonic jerks appear in 80% and 20% of patients, respectively, 1 to 10 years after the onset of TA. Focal epileptiform abnormalities and abortive asymmetrical bursts of spike/multiple spikes are common in the interictal EEG, whereas ictal GSW discharges are typically regular at 3 Hz. Differential diagnosis involves CAE (because of the age overlap), JME (because of possible MS), and limbic CPS because of ictal automatisms during prolonged absences (30 sec or more), which make distinction very difficult on clinical grounds only. VPA is the drug of choice, but LTG and LEV are possible alternatives. Monotherapy with ESX is not recommended because the natural history of the syndrome includes GTCS, against which ESX provides no protection. JAE are a lifelong disorder, although seizures are controlled in 70 to 80% of patients, particularly if appropriate treatment is initiated early.
JUVENILE MYOCLONIC EPILEPSY (JME)2,41–44
JME is the quintessence of myoclonic IGE and affects 8 to 10% of patients with epilepsies without any sex difference. Inheritance is probably complex and polygenic with susceptibility loci in chromosomes 6p11-12 (EJM1) and 15q14 (EJM2). MS are the defining seizure, as they occur in all patients, typically on awakening. They start between 8 to 26 years (with peak in early teens), may occur in volleys preceding a GTCS, and are typically activated by sleep deprivation, awakening, fatigue and mental stress and excitement, and excessive alcohol intake. Triggers include photosensitivity (present in one-third of patients) and less frequently movements or intension of movement (praxis-induced), whereas reading-induced jerks are rare but well documented. TA occur in around one-third of patients between 5 and 16 years of age (peak at 10 years) and are usually mild and inconspicuous, therefore detectable mainly by video EEG. The combination of more prolonged and frequent TA and random MS favor. JAE rather than JME. GTCS occur in nearly all patients, usually months after onset of myoclonic jerks. In untreated patients, interictal EEG is usually abnormal, with generalized discharges of irregular 3- to 6-Hz spike/polyspike waves. Focal abnormalities occur in 30 to 40% of patients. A normal EEG in a patient suspected of having JME should prompt an EEG during sleep and on awakening. MS are associated with generalized, frequently asymmetrical, multiple spikes, and TA are brief with 3- to 6-Hz GSW and multiple spikes and irregular intradischarge frequency with fragmentations. Susceptibility to all seizures is probably permanent, although patients show improvement after the age of 40. Despite appropriate treatment, seizures become intractable in about 20% of patients, with those who have all three types of seizures being more likely to show pharmacoresistance. Misdiagnosis is common resulting in avoidable morbidity. Factors responsible for misdiagnosis include lack of familiarity with JME, failure to elicit a history of jerks, misinterpretation of absences or jerks as focal seizures, and high prevalence of focal EEG abnormalities. An uncertain number of patients with MS only may escape diagnosis, as they do not seek medical advice. Optimal management includes appropriate education and advice, focusing mainly on seizure precipitants. VPA is the most effective AED but with significant concerns regarding women. Control and open studies indicate that LEV is probably one of the most promising new AEDs. Clonazepam is effective for persistent myoclonic jerks, but may deprive patients from the early morning volley that alerts for an impending GTCS.45
IGE WITH GTCS ONLY (IGE/GTCS)2,46,47
By definition, this syndrome manifests with GTCS only without TA and MS throughout the history on clinical and EEG basis, but many accept rare MS or brief TA. Therefore, the true prevalence is uncertain. EEG-wise, the interictal occurrence of GSW discharges should also be a diagnostic prerequisite. The age at onset ranges from 6 to 47 years with a peak around 16 to 17 years. GTCS can occur on awakening (usual pattern), during relaxation or leisure, during sleep, and randomly. Seizure precipitants include sleep deprivation, fatigue, and excessive alcohol consumption, whereas about 15% show EEG photosensitivity. IGE with GTCS on awakening is probably a lifelong disorder with high rate of relapse on withdrawal of treatment. GTCS become rarer and more random with time, either as a result of the evolution of the disease or drug-induced modifications, and precipitants less potent. Differential diagnosis includes JME (if one accepts rare MS), with which shares the typical circadian distribution, and the syndrome of phantom absences (if one accepts TA). Differentiation from cryptogenic generalized and focal epilepsies with secondarily GTCS or secondary bilateral synchrony employs clinical and EEG criteria, and patients with poorly understood nocturnal seizures and without clear EEG indicators should not be included here. CBZ, OXC, PB, and PHT can be helpful in some patients provided that TA and MS are not part of the clinical picture. If these seizures also occur, only VPA, LEV, TPM, and LTG are recommended.
EYELID MYOCLONIA WITH ABSENCES (ELMA OR JEAVONS SYNDROME)2,49–52
This is the most distinctive reflex IGE syndrome, characterized by (a) eyelid myoclonia with and without absences; (b) eye closure-induced seizures, EEG paroxysms, or both; and (c) photosensitivity. The age at onset ranges between 2 and 14 years (with peak at 6 to 8 years), and its prevalence is around 3% of adults with epileptic disorders and 13% of those with absence IGE syndromes. There is a twofold female preponderance. Eyelid myoclonia, the clinical hallmark of this syndrome, includes brief but marked, episodic jerking of the eyelids, the eyebrows, and often of the eyeballs and head, which may be time locked to GSW and polyspike discharges, and may or may not be associated with clinically apparent impairment of awareness (eyelid myoclonia with or without absences). These seizures are brief (3 to 6 sec) and occur mainly after eye closure, many times per day, and especially on awakening. Spontaneous or photically induced GTCS are probably inevitable in the long term and are easily provoked by sleep deprivation, alcohol, and inappropriate AED modifications. Myoclonic jerks of the limbs may occur. Absence Status Epilepticus (ASE) occurs in one-fifth of patients with prominent but discontinuous eyelid myoclonia and usually mild impairment of consciousness. Video-EEG is important for the diagnosis. Photoparoxysmal responses occur in all untreated young patients, but photosensitivity declines in adulthood. ELMA is particularly resistant to AED treatment (VPA, LEV, and CLZ are the main choices), and prognosis is guarded. Despite the sometimes extreme photosensitivity, self-induction is rare.
THE SYNDROME OF “DE NOVO ABSENCE-LIKE STATUS OF LATE ONSET”
De novo absence-like status is a form of nonconvulsive status epilepticus that occurs for the first time in nonepileptic patients of middle or old age as a result of an exogenous trigger and bears electroclinical resemblance to idiopathic absence status epilepticus.59–61 Distinction from idiopathic ASE is clinically important, as biological substrates and by implication management and prognosis are different. Patients with idiopathic ASE need appropriate prophylactic treatment with AED; for those with de novo absence-like status, elimination of the offending factor may suffice, and the overall prognosis is excellent with little risk for recurrence.60 Triggers include acute withdrawal of chronic psychotropic medication (mainly benzodiazepines60,62), but also tricyclic antidepressants,63 major neuroleptics and lithium, acute or chronic alcoholism and metabolic imbalance, and toxic or other pharmacological agents.60 However, relying only on apparent triggers may be misleading, and one has to bear in mind that in some patients with IGE, episodes of true absence status may be precipitated after discontinuation of a benzodiazepine (taken as antiepileptic medication such as clobazam) or moderate alcohol consumption. The underlying pathophysiology of de novo absence-like status is uncertain, and therefore its nosological taxonomy has been controversial. It appears that the triggering or facilitating potential of these factors becomes particularly effective when acting on brains compromised already by vascular or degenerative processes, as is more frequently the case in elderly people. A contribution of a genetically determined liability cannot be excluded.
Other Epileptic Syndromes and Conditions Associated with GSW Activity
CRYPTOGENIC/SYMPTOMATIC GENERALIZED EPILEPTIC SYNDROMES AND EPILEPTIC ENCEPHALOPATHIES
Angelman Syndrome (Partial Monosomy 15q)
This is a severe neurodevelopmental disorder caused by the failure of the expression of the maternal copy of the imprinted UBE3A gene on chromosome 15q11-q13. Characteristics include developmental delay with happy disposition and inappropriate laughter, mainly motor speech impairment, ataxia, hyperactivity, dysmorphic features, and seizures in up to 90% of patients with onset from 3 months to 20 years, usually in early childhood. Seizure activity may improve with age. Seizure types include myoclonic, atypical absences mostly associated with rhythmic triphasic delta waves of high amplitude,64 convulsive generalized and unilateral clonic, and complex partial with eye deviation and vomiting suggesting occipital origin.65 The interictal EEG shows rather characteristic large monomorphic slow spike-wave activity at 2 Hz over the posterior areas that becomes diffuse symmetrical bilateral and synchronous.
Ring Chromosome 20 Syndrome (r20S)
This rare chromosomal syndrome is characterized by variable psychomotor delay, behavioral disturbance, and frequent episodes of nonconvulsive status epilepticus (NCSE), but there are no dysmorphisms, and this may delay the diagnosis. Some patients may have normal intelligence. Focal seizures with affective symptomatology (usually terror), frontal discharges, and atypical absences with 2- to 3-Hz bilateral rhythmic slow activity and some autonomic features66 may be part of the clinical picture, which becomes typical after the age of 8 years.67 As a rule, seizures are resistant to treatment. Interictal EEG may be normal or show bilateral frontocentral theta activity. The NCSE in r20S is mainly an absence state with an associated myoclonic component. Differentiation from IGE may be difficult in patients with regular spike-wave activity, mild retardation or normal intellect, and normal or almost normal interictal EEG. Middle-aged patients may be misdiagnosed as de novo absence-like status if previous episodes are not identified, and frontal lobe status should also be considered in view of the affective and autonomic symptomatology.
Severe Myoclonic Epilepsy in Infancy—Dravet Syndrome
First described by Dravet in 1978,68 this syndrome is a genetically determined channelopathy69 and a syndromic phenotype that can occur within the frame of GEFS+.70 The first seizure is typically triggered by fever during infancy and is prolonged unilateral or generalized clonic or tonic-clonic; other nonfebrile convulsive seizures follow soon. Generalized myoclonic seizures associated with brief 3Hz GSW with polyspikes and segmental myoclonus appear in the 2nd or 3rd year, and atypical absences occur in up to 80 to 90% of the children, either concurrently with the myoclonic seizures or later. Focal and tonic seizures also occur. Episodes of absence status occur in at least 30 to 40% of cases and may persist for hours and even days (obtundation status). Photosensitivity occurs in up to 42% of children and may persist.71
Cryptogenic/Symptomatic Form of Epilepsy with Myoclonic-Astatic Seizures
As opposed to the idiopathic form of MAE (see in IGE section earlier), the cryptogenic variant is characterized by frequent (up to 90% of children) episodes of generalized myoclonic/absence status associated with chaotic EEG with asynchronous SW discharges that resemble hypsarrhythmia,29 tonic seizures during sleep, progressive slowing of the EEG background, and unfavorable outcome. These children continue with seizures, severe cognitive impairment, and behavioral abnormalities, whereas ataxia and motor and linguistic disturbances may emerge. Treatment is similar to the idiopathic form with intravenous benzodiazepines being used for nonconvulsive status epilepticus. In general, myoclonic astatic seizures occur in many childhood epilepsies, particularly epileptic encephalopathies, and differential diagnosis includes benign or progressive myoclonic epilepsies, Dravet syndrome, Lennox-Gastaut syndrome, atypical benign focal epilepsy of childhood, atypical evolutions of rolandic epilepsy, Panayiotopoulos syndrome, and nonepileptic myoclonus of various neurological disorders.
Lennox-Gastaut Syndrome (LGS)
LGS is a severe cryptogenic or symptomatic epileptic encephalopathy with tonic, atonic, atypical absences and other seizures, slow GSW (up to 2 to 2.5 Hz) and fast rhythms during sleep, and neuropsychological decline. Onset occurs between 3 and 10 years. The slow GSW and the lack of a prominent myoclonus distinguish cryptogenic LGS from MAE; however, some forms with predominant myoclonic-atonic seizures (known as myoclonic variant of LGS) resemble MAE and may have better outcome.29 Status epilepticus occurs in up to 90% of patients and is usually nonconvulsive.
EPILEPTIC ENCEPHALOPATHY WITH CONTINUOUS SPIKE-AND-WAVE DURING SLEEP (CSWS) INCLUDING LANDAU-KLEFFNER SYNDROME (LKS)
This is an age-related and partly reversible childhood disorder characterized by generalized and partial seizures mainly occurring during sleep and typical or atypical absences during awake state, global or selective regression of high cognitive functions, motor impairment. The EEG is characteristic with diffuse or regional spike-wave activity which is nearly continuous and occurs during non-REM sleep.2 There are no tonic seizures, and the kind of neuropsychological impairment relates to the topography of the continuous discharges. CSWS becomes apparent a couple of years after the onset of epilepsy and is associated with seizure worsening including also diurnal episodes of absence status. Seizures and CSWS gradually resolve, but only 25% of the children attain acceptable social and professional levels. In LKS, focal spike-wave activity heavily involves the “speech cortex” and cannot be considered generalized.
IDIOPATHIC FOCAL EPILEPSIES OF CHILDHOOD
Due perhaps to the very nature of “idiopathic focal or system epileptogenesis”, both benign rolandic epilepsy with centrotemporal spikes (BECTS) and Panayiotopoulos syndrome (PS) manifest with bilateral regional or diffuse epileptogenicity, distinctly different to the focal ictogenesis of the symptomatic focal epilepsies.72 GSW discharge may occur, sometimes associated with mild impairment of consciousness (absences). In addition, both BECTS and PS may occasionally evolve into atypical electroclinical forms that include continuous spike-wave during sleep, absences with inhibitory phenomena, and episodes of atypical absence status, with favorable outcome in most children.73–78
SYMPTOMATIC OR CRYPTOGENIC FOCAL EPILEPSIES
GSW activity associated with some impairment of consciousness may occur in frontal lobe epilepsies, but also in some patients with malformations of cortical development including temporal lobe dysembryoplastic neuroepithelial tumors.79 Diagnosis here is based on the existence of other seizure types, and the possibility of coexistence of IGE and focal lobar epilepsy should be also considered.80
1. First Seizure Trial Group (FIR.S.T. Group). Randomized clinical trial on the efficacy of antiepileptic drugs in reducing the risk of relapse after a first unprovoked tonic-clonic seizure. Neurology. 1993;43(3 Pt 1):478-483.
2. Panayiotopoulos CP. A Clinical Guide to Epileptic Syndromes and Their Treatment, 2nd. London: Springer, 2007.
3. Panayiotopoulos CP. Evidence-based epileptology, randomized controlled trials, and SANAD: a critical clinical view. Epilepsia. 2007;48(7):1268-1274.
4. Commission of Classification and Terminology of the International League Against Epilepsy. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia. 1981;22:489-501.
5. Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia. 1989;30:389-399.
6. Engel JJr. A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: Report of the ILAE Task Force on Classification and Terminology. Epilepsia. 2001;42:796-803.
7. Engel JJr. Report of the ILAE classification core group. Epilepsia. 2006;47(9):1558-1568.
8. Blume WT, Pillay N. Electrographic and clinical correlates of secondary bilateral synchrony. Epilepsia. 1985;26(6):636-641.
9. Panayiotopoulos CP, Chroni E, Daskalopoulos C, Baker A, Rowlinson S, Walsh P. Typical absence seizures in adults: clinical, EEG, video-EEG findings and diagnostic/syndromic considerations. J Neurol Neurosurg Psychiatr. 1992;55(11):1002-1008.
10. Usui N, Kotagal P, Matsumoto R, Kellinghaus C, Luders HO. Focal semiologic and electroencephalographic features in patients with juvenile myoclonic epilepsy. Epilepsia. 2005;46(10):1668-1676.
11. Panayiotopoulos CP, Tahan R, Obeid T. Juvenile myoclonic epilepsy: factors of error involved in the diagnosis and treatment. Epilepsia. 1991;32(5):672-676.
12. Topcuoglu MA, Saygi S, Ciger A. Rotatory seizures in juvenile myoclonic epilepsy. Clin Neurol Neurosurg. 1997;99(4):248-251.
13. So GM, Thiele EA, Sanger T, Schmid R, Riviello JJJr. Electroencephalogram and clinical focalities in juvenile myoclonic epilepsy. J Child Neurol. 1998;13(11):541-545.
14. Ferrie CD. Idiopathic generalized epilepsies imitating focal epilepsies. Epilepsia. 2005;46(Suppl 9):91-95.
15. Aliberti V, Grunewald RA, Panayiotopoulos CP, Chroni E. Focal electroencephalographic abnormalities in juvenile myoclonic epilepsy. Epilepsia. 1994;35(2):297-301.
16. Lombroso CT. Consistent EEG focalities detected in subjects with primary generalized epilepsies monitored for two decades. Epilepsia. 1997;38(7):797-812.
17. Koutroumanidis M, Smith S. Use and abuse of EEG in the diagnosis of idiopathic generalized epilepsies. Epilepsia. 2005;46(Suppl 9):96-107.
18. Hitiris N, Brodie MJ. Evidence-based treatment of idiopathic generalized epilepsies with older antiepileptic drugs. Epilepsia. 2005;46(Suppl 9):149-153.
19. Bergey GK. Evidence-based treatment of idiopathic generalized epilepsies with new antiepileptic drugs. Epilepsia. 2005;46(Suppl 9):161-168.
20. Shorvon S, editor. Handbook of Epilepsy Treatment, 2nd, Oxford: Blackwell Science, 2005.
21. Chaves J, Sander JW. Seizure aggravation in idiopathic generalized epilepsies. Epilepsia. 2005;46(Suppl 9):133-139.
22. Thomas P, Valton L, Genton P. Absence and myoclonic status epilepticus precipitated by antiepileptic drugs in idiopathic generalized epilepsy. Brain. 2006;129(Pt 5):1281-1292.
23. Kwan P. Pro-myoclonic anti-epileptic drugs to avoid in IGE with myoclonic jerks. In: Panayiotopoulos CP, editor. Idiopathic Generalised Epilepsies with Myoclonic Jerks, 2. Oxford: Medicinae; 2007:189-196.
24. Darra F, Fiorini E, Zoccante L, et al. Benign myoclonic epilepsy in infancy (BMEI): a longitudinal electroclinical study of 22 cases. Epilepsia. 2006;47(Suppl 5):31-35.
25. Fejerman N. Benign myoclonic epilepsy in infancy. In: Panayiotopoulos CP, editor. Idiopathic Generalised Epilepsies with Myoclonic Jerks, 2. Oxford: Medicinae; 2007:55-59.
26. Capovilla G, Beccaria F, Gambardella A, Montagnini A, Avantaggiato P, Seri S. Photosensitive benign myoclonic epilepsy in infancy. Epilepsia. 2007;48(1):96-100.
27. Neubauer BA, Hahn A, Doose H, Tuxhorn I. Myoclonic-astatic epilepsy of early childhood—definition, course, nosography, and genetics. Adv Neurol. 2005;95:147-155.
28. Oguni H, Hayashi K, Imai K, et al. Idiopathic myoclonic-astatic epilepsy of early childhood—nosology based on electrophysiologic and long-term follow-up study of patients. Adv Neurol. 2005;95:157-174.
29. Kaminska A, Ickowicz A, Plouin P, Bru MF, Dellatolas G, Dulac O. Delineation of cryptogenic Lennox-Gastaut syndrome and myoclonic astatic epilepsy using multiple correspondence analysis. Epilepsy Res. 1999;36(1):15-29.
30. Loiseau P, Panayiotopoulos CP. Childhood absence epilepsy [ILAE Web site]. 2004 Available at: http://www.ilae-epilepsy org/Visitors/Centre/ctf/childhood_absence.html. Accessed September 18, 2007. Epileptic syndromes [Epilepsy Web site]. Available at: www.epilepsy.org/ctf.
31. Grosso S, Galimberti D, Vezzosi PS, et al. Childhood absence epilepsy: evolution and prognostic factors. Epilepsia. 2005;46(11):1796-1801.
32. Hirsch E, Panayiotopoulos CP. Childhood absence epilepsy and related syndromes. In: Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, editors. Epileptic Syndromes in Infancy, Childhood and Adolescence. 4th. Montrouge, France: John Libbey Eurotext; 2005:315-335.
33. Posner E. Pharmacological treatment of childhood absence epilepsy. Expert Rev Neurother. 2006;6(6):855-862.
34. Genton P, Bureau M. Epilepsy with myoclonic absences. CNS Drugs. 2006;20(11):911-916.
35. Tassinari A, Rubolli R, Michellluchi R. Epilepsy with myoclonic absences [ILAE Web site]. 2006. Available at: http://www.ilae-epilepsy org/ctf/over_frame.html. Accessed September 18, 2008.
36. Tassinari CA, Lyagoubi S, Santos V, et al. [Study on spike and wave discharges in man. II. Clinical and electroencephalographic aspects of myoclonic absences]. Rev Neurol (Paris). 1969;121(3):379-383.
37. Bureau M, Tassinari CA. Myoclonic absences: the seizure and the syndrome. Adv Neurol. 2005;95:175-183.
38. Obeid T. Clinical and genetic aspects of juvenile absence epilepsy. J Neurol. 1994;241(8):487-491.
39. Trinka E, Baumgartner S, Unterberger I, et al. Long-term prognosis for childhood and juvenile absence epilepsy. J Neurol. 2004;251(10):1235-1241.
40. Tovia E, Goldberg-Stern H, Shahar E, Kramer U. Outcome of children with juvenile absence epilepsy. J Child Neurol. 2006;21(9):766-768.
41. Panayiotopoulos CP, Obeid T, Tahan AR. Juvenile myoclonic epilepsy: a 5-year prospective study. Epilepsia. 1994;35(2):285-296.
42. Calleja S, Salas-Puig J, Ribacoba R, Lahoz CH. Evolution of juvenile myoclonic epilepsy treated from the outset with sodium valproate. Seizure. 2001;10(6):424-427.
43. Genton P, Gelisse P. Juvenile myoclonic epilepsy. Arch Neurol. 2001;58(9):1487-1490.
44. Zifkin B, Andermann E, Andermann F. Mechanisms, genetics, and pathogenesis of juvenile myoclonic epilepsy. Curr Opin Neurol. 2005;18(2):147-153.
45. Obeid T, Panayiotopoulos CP. Clonazepam in juvenile myoclonic epilepsy. Epilepsia. 1989;30(5):603-606.
46. Wolf P. Epilepsy with grand mal on awakening. In: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, editors. Epileptic Syndromes in Infancy, Childhood and Adolescence. London: John Libbey & Company; 1992:329-341.
47. Janz D. Epilepsy with grand mal on awakening and sleep-waking cycle. Clin Neurophysiol. 2000;111(Suppl 2):S103-S110.
48. Panayiotopoulos CP. Syndromes of idiopathic generalized epilepsies not recognized by the International League Against Epilepsy. Epilepsia. 2005;46(Suppl 9):57-66.
49. Duncan JS, Panayiotopoulos CP, editors. Eyelid Myoclonia with Absences. London: John Libbey & Company Ltd, 1996.
50. Panayiotopoulos CP. Eyelid myoclonia with and without absences [ILAE Web site]. 2006. Available at: http://www.ilae-epilepsy org/ctf/over_frame.html. Accessed September 18, 2007.
51. Covanis A. Eyelid myoclonia and absences: Jeavons syndrome (with video-EEG sequences). In: Panayiotopoulos CP, editor. Idiopathic Generalised Epilepsies with Myoclonic Jerks, 2. Oxford: Medicinae; 2007:88-92.
52. Joshi CN, Patrick J. Eyelid myoclonia with absences: routine EEG is sufficient to make a diagnosis. Seizure. 2007;16(3):254-260.
53. Panayiotopoulos CP, Koutroumanidis M, Giannakodimos S, Agathonikou A. Idiopathic generalised epilepsy in adults manifested by phantom absences, generalised tonic-clonic seizures, and frequent absence status. J Neurol Neurosurg Psychiatr. 1997;63(5):622-627.
54. Agathonikou A, Panayiotopoulos CP, Giannakodimos S, Koutroumanidis M. Typical absence status in adults: diagnostic and syndromic considerations. Epilepsia. 1998;39(12):1265-1276.
55. Panayiotopoulos CP, Ferrie CD, Koutroumanidis M, Rowlinson S, Sanders S. Idiopathic generalised epilepsy with phantom absences and absence status in a child. Epileptic Disord. 2001;3(2):63-66.
56. Panayiotopoulos CP, Ferrie CD, Giannakodimos S, Robinson RO. Perioral myoclonia with absences: a new syndrome. In: Wolf P, editor. Epileptic Seizures and Syndromes. London: John Libbey & Company Ltd; 1994:143-153.
57. Bilgic B, Baykan B, Gurses C, Gokyigit A. Perioral myoclonia with absence seizures: a rare epileptic syndrome. Epileptic Disord. 2001;3(1):23-27.
58. Baykan B, Noachtar S. Perioral myoclonia with absences: an overlooked and misdiagnosed generalized seizure type. Epilepsy Behav. 2005;6(3):460-462.
59. Thomas P, Beaumanoir A, Genton P, Dolisi C, Chatel M. “De novo” absence status of late onset: report of 11 cases [see comments]. Neurology. 1992;42(1):104-110.
60. Thomas P, Andermann F. Late-onset absence status epilepticus is most often situation-related. In: Malafosse A, Genton P, Hirsch E, Marescaux C, Broglin D, Bernasconi R, editors. Idiopathic Generalized Epilepsies. London: John Libbey & Company Ltd; 1994:95-109.
61. Koutroumanidis M. NCSE in the idiopathic and other generalized epilepsies (including absence status). In: Kaplan P, Drislane F, ed. Nonconvulsive Status Epilepticus. New York: Demos Medical Publishing. In press.
62. Dunne JW, Summers QA, Stewart-Wynne EG. Non-convulsive status epilepticus: a prospective study in an adult general hospital. Q J Med. 1987;62(238):117-126.
63. Bourrat C, Garde P, Boucher M, Fournet A. [Prolonged absence states in aged patients without epileptic antecedents]. [French]. Revue Neurologique. 1986;142(8-9):696-702.
64. Laan LA, Renier WO, Arts WF, et al. Evolution of epilepsy and EEG findings in Angelman syndrome. Epilepsia. 1997;38(2):195-199.
65. Viani F, Romeo A, Viri M, et al. Seizure and EEG patterns in Angelman’s syndrome. J Child Neurol. 1995;10(6):467-471.
66. Canevini MP, Sgro V, Zuffardi O, et al. Chromosome 20 ring: a chromosomal disorder associated with a particular electroclinical pattern. Epilepsia. 1998;39(9):942-951.
67. Ville D, Kaminska A, Bahi-Buisson N, et al. Early pattern of epilepsy in the ring chromosome 20 syndrome. Epilepsia. 2006;47(3):543-549.
68. Dravet C. Les epilepsies graves de l’enfant. Vie Med. 1978;8:543-548.
69. Claes L, Del Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. 2001;68(6):1327-1332.
70. Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain. 1997;120(Pt 3):479-490.
71. Dravet C, Bureau M, Guerrini R, Giraud N, Roger J. Severe myoclonic epilepsy in infants. In: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, editors. Epileptic Syndromes in Infancy, Childhood and Adolescence. London: John Libbey & Company; 1992:75-78.
72. Koutroumanidis M. Panayiotopoulos syndrome: an important electroclinical example of benign childhood system epilepsy. Epilepsia. 2007;48(6):1044-1053.
73. Fejerman N. Atypical evolutions of benign partial epilepsies in children. Int Pediatr. 1996;11(6):351-356.
74. Fejerman N, Caraballo R, Tenembaum SN. Atypical evolutions of benign localization-related epilepsies in children: are they predictable? Epilepsia. 2000;41(4):380-390.
75. Caraballo RH, Astorino F, Cersosimo R, Soprano AM, Fejerman N. Atypical evolution in childhood epilepsy with occipital paroxysms (Panayiotopoulos type). Epileptic Disord. 2001;3(3):157-162.
76. Prats JM, Garaizar C, Garcia-Nieto ML, Madoz P. Antiepileptic drugs and atypical evolution of idiopathic partial epilepsy. Pediatr Neurol. 1998;18(5):402-406.
77. Ferrie CD, Koutroumanidis M, Rowlinson S, Sanders S, Panayiotopoulos CP. Atypical evolution of Panayiotopoulos syndrome: a case report [published with video-sequences]. Epileptic Disord. 2002;4(1):35-42.
78. Grosso S, Balestri M, Di Bartolo RM, et al. Oxcarbazepine and atypical evolution of benign idiopathic focal epilepsy of childhood. Eur J Neurol. 2006;13(10):1142-1145.
79. Raymond AA, Fish DR, Sisodiya SM, Alsanjari N, Stevens JM, Shorvon SD. Abnormalities of gyration, heterotopias, tuberous sclerosis, focal cortical dysplasia, microdysgenesis, dysembryoplastic neuroepithelial tumour and dysgenesis of the archicortex in epilepsy. Clinical, EEG and neuroimaging features in 100 adult patients. Brain. 1995;118:629-660.
80. Koutroumanidis M, Hennessy MJ, Elwes RD, Binnie CD, Polkey CE. Coexistence of temporal lobe and idiopathic generalized epilepsies. Neurology. 1999;53(3):490-495.