Chapter 8 Cortical Myoclonus and Epilepsy: Overlap and Differences
Introduction
The term myoclonus is used to describe a brief and jerky involuntary movement involving antagonist muscles and originating from brief active contractions of muscles (positive myoclonus) or, more rarely, from brief interruptions of ongoing electromyographic activity (negative myoclonus).1 Clinically, myoclonus may be classified as “focal” if it involves a restricted, usually distal, group of muscles; “multifocal” when asynchronous focal jerks involve different body areas; or “generalized” when jerks involve most body segments in an apparently synchronous manner.
Myoclonus is spontaneous if occurring irrespective from external triggering situations or can be induced by movement (action myoclonus) or by sensory or visual stimuli (reflex myoclonus). Finally, regarding periodicity, myoclonus may be either rhythmic or arrhythmic.
Many conditions in which myoclonus is a prominent manifestation are known, allowing an etiological classification1,2 in which four major clinical syndrome categories are identified: (1) physiologic myoclonus (sleep-related, hiccup, myoclonus induced by anxiety or exercise); (2) essential myoclonus (individuals without other neurological signs); (3) epileptic myoclonus (conditions in which the predominant element is epilepsy); and (4) symptomatic myoclonus (conditions in which the predominant element is encephalopathy).
Neurophysiological characteristics can be used to divide myoclonus into six main physiological categories, including cortical, cortical-subcortical, subcortical-supraspinal, spinal, and peripheral.3
The term epileptic myoclonus is still confusing. Some authors define as epileptic myoclonus what occurs within the setting of epilepsy.4 Others define as epileptic myoclonus those forms in which a paroxysmal depolarization shift is thought to be the underlying neurophysiological substrate, irrespective of which population of neurons (cortical or subcortical) is primarily involved.5 In our opinion, epileptic myoclonus can be comprehensively defined as an elementary electroclinical manifestation of epilepsy involving descending neurons, whose spatial (spread) or temporal (self-sustained repetition) amplification can trigger overt epileptic activity.6
Frequently, the electroencephalogram (EEG) correlate of epileptic myoclonus can be detected only by using jerk-locked (EEG or magnetoencephalogram) averaging. Yet, many patients with cortical myoclonus have rhythmic electromyogram (EMG) bursts at relatively high frequency (especially those with minipolymyoclonus, cortical tremor, Angelman syndrome or autosomal dominant cortical myoclonus, and epilepsy), which make it difficult to identify a cortical correlate. Recent works have demonstrated in these cases the role of EEG-EMG coherence by means of frequency analysis in demonstrating common cortical drives.7,8,9
Myoclonus can be only one component of a seizure (myoclonic jerks heralding a generalized tonic-clonic seizure in juvenile myoclonic epilepsy or in progressive myoclonus epilepsies), the only seizure manifestations (myoclonic jerks of benign myoclonic epilepsy), one of multiple seizure types (as observed in myoclonic-astatic epilepsy), or the basis of a movement-related disorder (action myoclonus in progressive myoclonus epilepsies). However, the relationships between myoclonus and epilepsy have been elucidated only in part, and the neurophysiological bases, nosology, and electroclinical characteristics of myoclonus in the setting of specific epilepsy syndromes are only partially understood. According to available evidence, epileptic myoclonus can be classified neurophysiologically as cortical (positive and negative), secondarily generalized, thalamocortical and reticular reflex myoclonus.10 Cortical epileptic myoclonus constitutes a fragment of partial or symptomatic generalized epilepsy; thalamocortical epileptic myoclonus is a fragment of idiopathic generalized epilepsy.5 Reflex reticular myoclonus, which does not have a time-locked EEG correlate, represents the clinical counterpart of hypersynchronous activity of neurons in the brainstem reticular formation. In the following sections, major epilepsy or neurological syndromes featuring different forms of epileptic myoclonus are described.
Cortical Myoclonus (CM)
CM originates from abnormal neuronal discharges in the sensorimotor cortex. Abnormally firing motoneurons may be primarily hyperexcitable or may be driven by abnormal inputs originating from hyperexcitable parietal11 or occipital12 neurons. Each jerk originates from the discharge of a small group of cortical motoneurons, somatotopically connected to a group of contiguous muscles. A cortical potential, time locked to the myoclonic potential and localized on the contralateral sensorimotor region, can be demonstrated by EEG, magnetoencephalogram, or jerk-locked averaging.13–16 Facilitation of interhemispheric and intrahemispheric spread of CM activity through transcallosal or corticocortical pathways seems to play a major role in producing generalized or bilateral myoclonus.17
In patients with cortical reflex myoclonus (CRM), appropriate stimuli administered to a resting somatic segment produce a reflex muscle response (jerk) with a latency of around 50 msec (C-reflex).18 A similar response is only observed in normal subjects during voluntary contraction. Somatosensory evoked potentials (SEPs) of giant amplitude are typically seen in association with CRM.19–22 The striking resemblance in latency and morphology of the giant SEPs to the myoclonus-related cortical spikes suggests that both originate from common cortical mechanisms.15
In the typical forms of CRM, the reflex jerk in the hand has a latency of ∼50 msec, and the CRT has a mean duration of about 7 msec.23 Typical CRM can be observed in patients with focal cortical lesions,18 spinocerebellar degeneration,14,24,25 multiple system atrophy,25,26,27 cerebral anoxia,14,28 childhood metabolic degenerations such as neuronal ceroid lipofuscinosis and sialidosis,11,19 Alzheimer’s disease,29,30 Down syndrome, and mitochondrial disorders.31,32,33
Epileptic negative myoclonus (ENM) is characterized by brief (50 to 400 msec) muscle inhibitions with focal, multifocal, or bilateral distribution and time locked to sharp-wave or spike-wave discharges on the contralateral central areas.34,35 ENM has a wide etiological spectrum ranging from idiopathic to symptomatic forms due to cortical dysplastic lesions.36 It may occasionally be precipitated by an adverse reaction to antiepileptic drugs.37,38,39 Previous studies35,40,41 hypothesized a cortical origin of ENM. Epileptic activity associated with ENM was described in the premotor42,43,44 and postcentral somatosensory cortex.40,45 Through cortical electrical stimulation studies it was suggested that negative motor areas might be present in the lateral and mesial portion of frontal lobe, encompassed in the supplementary sensorimotor area (SMA).46 However, no subsequent confirmatory studies are available.
A possible role of the SMA in ENM was also proposed in a recent study47 in which electrical stimulation of the SMA constantly evoked ENM, with no preceding positive myoclonus, as it was instead observed following stimulation of the premotor, primary motor, and sensorimotor cortex.
EPILEPTIC SYNDROMES AND NEUROLOGICAL DISORDERS WITH CM
Cortical Action/Reflex Myoclonus
Progressive Myoclonus Epilepsies (PMEs)
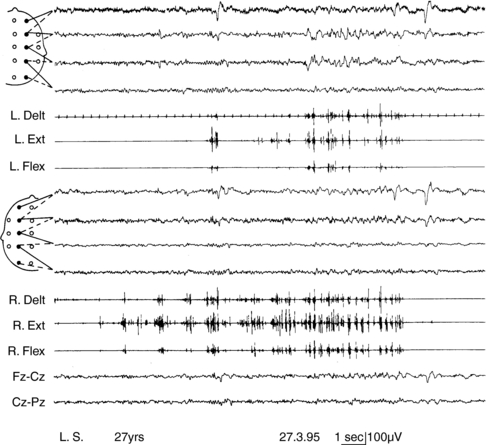
Progressive myoclonus epilepsies represent a clinically and etiologically heterogeneous group of diseases with a progressive course, characterized by myoclonus, generalized, tonic-clonic seizures, and neurological deterioration.48 Onset is most frequently in late childhood or adolescence.49 Different forms are known, including Unverricht-Lundborg disease, Lafora disease, neuronal ceroid-lipofuscinosis, type III Gaucher disease, infantile and juvenile GM2-gangliosidosis, some mitochondrial encephalopathies, sialidosis, dentatorubropallidoluysian atrophy, and action myoclonus-renal failure (AMRF).49,50 Causative genes have been identified for most PMEs.51,52 Onset features comprise myoclonus and rare generalized tonic-clonic seizures, as in idiopathic myoclonic epilepsies.53,54 The tonic-clonic seizures can occur without any warning or after a long buildup of myoclonic jerks. The EEG shows generalized polyspike and spike-and-wave discharges, frequently precipitated by photic stimulation. Background EEG activity becomes progressively slower.53 Cortical reflex myoclonus is common to all PME syndromes, in which it is manifested with the classic combination of action myoclonus (Figure 8-1), spontaneous jerks, giant SEPs, C-reflex at rest, and the premyoclonus spike. According to Cantello et al.,55 focal subcortical reflex myoclonus can also be demonstrated in these patients. Initially mild, myoclonus becomes increasingly disabling during the course. Severe action myoclonus has a devastating impact on the patients’ level of autonomy.
Rett Syndrome
Rett syndrome is an X-linked dominant disorder, with an estimated prevalence of 1 in 10,000 to 15,000 females, making it one of the most common causes of severe mental retardation in females. Mutations in the exons 1 to 4 of the methyl CpG binding protein 2 gene (MECP2)56 have been identified in roughly 75 to 80% of girls with classical Rett syndrome;57 in MECP2-negative patients additional screening using (multiplex ligation probe amplification) MLPA enables detection of large deletions in nearly half of the remaining patients with the syndrome.58 The clinical phenotype includes progressive cognitive deterioration leading to dementia, autistic features, truncal ataxia/apraxia, loss of purposeful hand movements, breathing abnormalities, stereotypies, extrapyramidal signs, and epilepsy. A form of CRM characterized by prolonged C-reflex (65 ± 5 msec) latency has been described in affected girls.59 Myoclonus is multifocal and arrhythmic, and major myoclonic seizures are not seen in these patients. A positive potential, localized on the contralateral centroparietal area, precedes myoclonus with a latency of 34 ± 7 msec for the forearm muscle compatible with corticomotoneuronal conduction. The N20-P30 and P30-N35 components of the SEPs have significantly increased amplitude. In addition, the latency of the N20 component is delayed, and the N20-P30-N35 interval is significantly increased and has expanded morphology. It is probable that in Rett syndrome the following sequence of events occurs: slight delay in central conduction of the impulse afferent to the sensorimotor cortex (N20), slowing of the processing of the afferent impulse (interval N20-P30; mean = 11 msec), delay in corticocortical transmission to the precentral neurons subserving movement of the stimulated body segment (latency increase P30 – C reflex; mean = 32 msec), and rapid descending volley to the spinal motoneurons. Intracortical conduction time could be particularly prolonged because of the synaptic abnormalities that have been observed.60
Huntington’s Disease
In Huntington’s disease, action myoclonus is a rare manifestation, but a few patients have been described in whom CRM was the presenting symptom.33,61 Seizures are an infrequent complication and are mainly seen with juvenile onset, rarely presenting with a typical PME syndrome.62
Postanoxic Encephalopathy
Postanoxic encephalopathy is characterized by dysarthria, ataxia, pyramidal signs, rigidity, epilepsy, and myoclonus, which is usually spontaneous and action-induced, multifocal and generalized, and extremely disabling. Electromyographic silent periods following the jerks contribute to producing postural lapses.63 Postanoxic myoclonus may be cortical in origin, involving the sensorimotor cortex and rapidly conducting pyramidal pathways.14,28 More rarely it may have brainstem origin, either as an exaggerated startle reflex or as reticular reflex myoclonus.64,65 Forty percent of patients with postanoxic myoclonus suffer from generalized epileptic seizures.
Focal Cortical Repetitive Myoclonus
Epilepsia Partialis Continua
Epilepsia partialis continua, or Kojewnikow’s syndrome,66 is characterized by almost continuous, focal, rhythmic (around 1 to 2 Hz) muscle jerks, which are observed both while awake and asleep, for periods ranging from hours to days, or rarely years.48 Unilateral somatomotor seizures are constantly associated. Two types of epilepsia partialis continua have been identified.67 The first type is due to fixed epileptogenic lesions involving the motor cortex. Causative factors include ischemia, posttraumatic head injury, cortical dysplasia, tumors, and vascular malformations.67–70 A stable motor deficit, predating seizure onset, and nonprogressive evolution are usual features. A second type of epilepsia partialis continua is observed in Rasmussen’s syndrome. Onset occurs during childhood, with continuous focal jerking and intractable homolateral motor or generalized seizures. Progressive hemiparesis, hemianopia, and eventually, cognitive deterioration follow. Magnetic resonance imaging (MRI) shows progressive atrophy of the affected hemisphere. Pathological studies reveal inflammation with perivascular infiltrates and microglia nodules.71 A viral etiology was originally hypothesized. More recently, the role of antibody-mediated mechanisms and more recently cell-mediated immunity have been hypothesized72,73,74 with inconclusive results. An analogous form of progressive epilepsia partialis continua has been observed in some children with mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke (MELAS).75
Rhythmic High-Frequency Cortical Myoclonus (Cortical Tremor)
Cortical tremor is a form of rhythmic myoclonus, presenting as postural or action tremor in some patients with progressive myoclonus epilepsy (PME),76,77 in Angelman syndrome, and in different forms of autosomal dominant epilepsy.78–83 The nosologic boundaries between epilepsia partialis continua and this peculiar form of repetitive myoclonus are unclear.
Angelman Syndrome
Angelman syndrome is a neurogenetic disorder deriving from a defect in maternal chromosome 15q11 to q13. Seventy percent of patients present a cytogenetic or molecular deletion encompassing three subunits of receptor α for gamma-aminobutyric acid (GABRB3, GABRA5, and GABRG3) and the gene UBE3A. Uniparental paternal disomy for chromosome 15, or mutations in the imprinting center or in the UBE3A gene, are more rarely found. Patients have microbrachycephaly, severe to moderate mental retardation, absence of speech, inappropriate paroxysmal laughter, epilepsy, ataxic gait, tremor, and jerky movements. Neurophysiologic investigations reveal a spectrum of manifestations of myoclonus.84 All patients present with rapid distal jerking of fluctuating amplitude, which causes a sort of coarse distal tremor combined with dystonic limb posturing. Jerks occur at rest in prolonged runs. In addition, the majority of patients have myoclonic and absence seizures, as well as episodes of myoclonic status. Bilateral jerks of myoclonic absences show rhythmic repetition at ∼ 2.5 Hz and are time locked with a cortical spike. Interside latency of both spikes and jerks is consistent with transcallosal spread, and spike-to-jerk latency indicates propagation through rapid conduction corticospinal pathway. A contralateral, central premyoclonic potential is uncovered by jerk-locked averaging. SEPs are normal, and C-reflex is absent.
Familial Adult Myoclonic Epilepsy and Autosomal Dominant Cortical Reflex Myoclonus and Epilepsy
A form of autosomal dominant epilepsy with cortical myoclonus manifested as cortical tremor has been described in several families, mostly of Japanese origin,79,85 and given the acronym BFAME (benign familial adult myoclonic epilepsy) or FAME (familial adult myoclonic epilepsy). Affected patients present homogeneous characteristics, including (a) autosomal dominant inheritance; (b) adult onset (mean age 38 years, range: 19 to 73); (c) nonprogressive course; (d) distal, rhythmic myoclonus enhanced during posture maintenance (cortical tremor); (e) rare, apparently generalized, seizures often preceded by worsening of myoclonus; (f) absence of other neurological signs; (g) generalized interictal spike-and-wave discharges; (h) photoparoxysmal response; (i) giant SEPs and hyperexcitability of the C-reflex (Figure 8-2); and (j) cortical EEG potential time locked to the jerks. The original Japanese families linked to chromosome 8q23.3-q24.86 However, European families with a similar phenotype did not link to the same locus.85,87,88
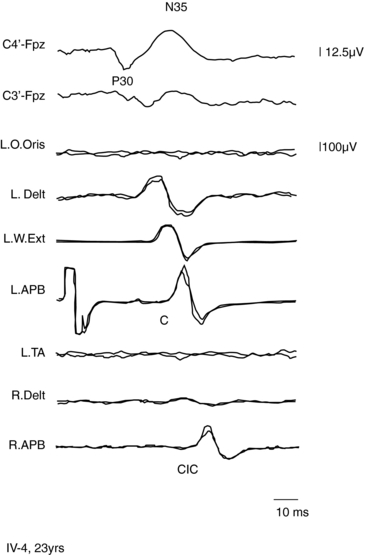
Autosomal dominant cortical reflex myoclonus and epilepsy (ADCME)81 has been described in patients with a homogeneous syndromic core, including an association of nonprogressive cortical reflex myoclonus, manifested as semicontinuous rhythmic distal jerking (cortical tremor), generalized tonic-clonic convulsions (GTCs) preceded in some patients by generalized myoclonic jerks, and generalized EEG abnormalities. Age at onset of cortical tremor and of GTCs overlapped in a given individual but varied between individuals, ranging from 12 to 50 years. This clinical picture shares some features with FAME86; however, all ADCME patients had in addition focal frontotemporal EEG abnormalities, and some individuals also had focal seizures, of variable severity, starting around the same age as the other manifestations. The pattern of inheritance is autosomal dominant with high penetrance. Linkage analysis identified a critical region in chromosome 2p11.1-q12.2.81 Three Italian families with familial adult myoclonic epilepsy have been described, possibly linked to 2p11.1-q12.2, suggesting a possible allelism with ADCME.88,89
Recently two novel families with adult myoclonic epilepsy have been reported,82,83 neither linking to known loci. In the first report, clinical photosensitivity was a prominent feature; in the second report photosensitivity was not a prominent feature, but cerebellar ataxia, dementia, and progression of symptoms were observed, raising doubts about the nosology of this disorder.
Epileptic Syndromes with Secondarily Generalized Epileptic Myoclonus
Severe Myoclonic Epilepsy in Infancy (SMEI), or Dravet Syndrome
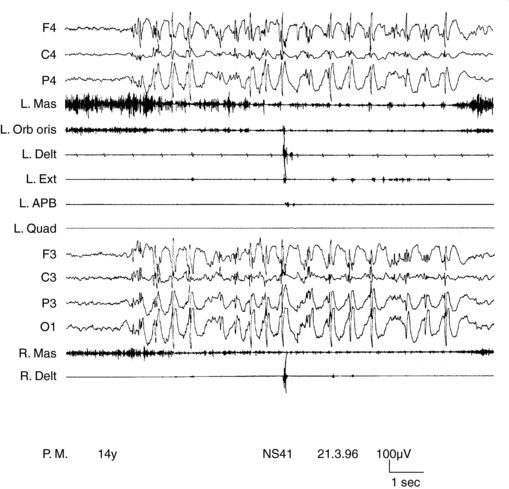
SMEI, or Dravet syndrome, is observed in 6 to 7% of children with seizure onset in the first year of life90 and is a severe form of epilepsy, characterized by multiple seizure types and unfavorable prognostic outlook. Its classification as a form of myoclonic epilepsy is controversial because myoclonus, although present in most children, can be a transient phenomenon and often does not represent a hallmark of the syndrome. A subgroup of children does not exhibit myoclonic seizures at all.90,91 SMEI represents the prototype of an epileptic encephalopathy in which onset of severe, prolonged seizures precedes deterioration of cerebral functions.92 Mutations of the SCN1A gene are observed in about 80% of cases.93 Onset of epilepsy occurs during the first year of life with prolonged generalized or unilateral, clonic or tonic-clonic seizures during fever, often evolving to status. They rapidly become associated with similar nonfebrile attacks. By the third to fourth year of life, resistant myoclonic, partial seizures and atypical absences also appear. EEG, normal at the beginning, subsequently shows multifocal and generalized abnormalities. Early photosensitivity is seen in some children. Neurological development appears delayed from the second year of life onward. Two main types of myoclonus have been described. Almost all children show arrhythmic, distal jerks, manifested as twitching of fingers, whereas some also have generalized jerks. Demonstrating a premyoclonic potential for multifocal jerks may be difficult, even using jerk-locked averaging. Generalized jerks have an obvious EEG correlate, which appear to originate from spread of CM activity, when small time differences are measured (Figure 8-3).10,54
Lennox-Gastaut Syndrome
Lennox-Gastaut syndrome (LGS) has a prevalence of 2 to 3% in children with epilepsy and is often observed in the brain damaged.94 Typical seizures start at 3 to 5 years of age as tonic, atonic, or atypical absences.94 A previous form of epilepsy, especially West syndrome, is frequently observed. Associated seizure types are myoclonic, generalized tonic-clonic, and rarely, focal. Epilepsy is drug resistant, and episodes of status are frequent. Interictal EEG shows abnormal background activity, slow (1.5 to 2.5 Hz) generalized spike waves, and often multifocal abnormalities. During sleep, all patients show typical rhythmic discharges around 10 Hz, accompanying tonic seizures or without apparent clinical correlate. Myoclonus is not a prominent feature of Lennox-Gastaut syndrome,95 but some patients exhibit generalized myoclonic jerks that seem to be produced by a secondary generalization of focal CM.54,96 Minipolymyoclonus, a term used to describe distal, small focal jerks, frequently leading to individual tiny finger movements, is observed in some patients with LGS8,97 in which back-averaged EEG shows a bilateral frontal negative slow wave, with 20 to 500 msec latency. In other patients, a sharper bilateral frontal negativity is demonstrable, leading the jerks by 40 to 70 msec.97 Minipolymyoclonus is strongly similar to the pattern of distal myoclonus observed in Angelman syndrome.8,84
Epileptic Syndromes and Neurological Disorders with Subcortical-Cortical (Thalamocortical) Myoclonus
IDIOPATHIC (PRIMARY) GENERALIZED EPILEPTIC MYOCLONUS
Generalized epileptic myoclonus is spontaneous, is predominantly arrhythmic, and has an inconstant axial predominance. Patients may present with simple head nodding or raised shoulders or may stagger or fall. The generalized jerks appear to originate from afferent volleys from subcortical structures that act synchronously on a hyperexcitable cortex.5,98 As a consequence, muscles from both sides are activated synchronously, as in reticular myoclonus, and muscles innervated by the cranial nerves are involved through a rostrocaudal pattern of activation, as in cortical myoclonus. The EEG correlate is a generalized spike wave. The negative peak of the spike precedes the generalized jerks by 20 to 75 msec.
Idiopathic Generalized Epilepsies
Benign Myoclonic Epilepsy of Infancy (BME)
BME affects 0.4 to 2% of all children with seizure onset by age 3 years.99,100,101 Age at onset ranges between 4 months and 5 years in children with normal development. Some patients, however, can have mild cognitive impairment.102,103 Seizures consist of generalized myoclonic jerks, which are brief, isolated, or repeated in small series. If the child is standing or sitting, the jerks often cause nodding with upward gaze deviation and eyelid myoclonus, accompanied by slight arm abduction or elbow bending. Staggering may occur, especially up to the second year of life, when walking is still unstable. If falls occur, the child collapses on the buttocks and then gets up immediately. In most cases, the jerks occur many times per day. A few patients may have generalized tonic-clonic seizures in adolescence.103 Treatment had been withdrawn in most patients aged more than 6 years at follow-up.102 However, the use of the term benign is questionable according to the most recent ILAE (International League Against Epilepsy) definitions in that outcome is often judged only in retrospect, and children with the same clinical presentation at onset might have cognitive or behavioral sequelae.104,105 About 10% of children with BME have photic-induced jerks.102 Some have both spontaneous and reflex myoclonus, the latter triggered by tactile or sudden acoustic stimuli.106 Neurophysiology of myoclonus reveals symmetric, rostrocaudal muscle activation and a premyoclonus negative spike preceding jerks by 30 ± 2 msec.6 Duration of the myoclonic jerk is roughly 100 msec.
Juvenile Myoclonic Epilepsy (JME)
JME has a prevalence of between 3.4 and 11.9% and represents the most common form of idiopathic generalized epilepsy (23.3%).107 The syndrome is genetically heterogeneous and in most cases is presumed to be polygenic. However, mutations of three different genes have been identified in rare families having dominant (CLCN2 and GABRA1)108,109,110 or recessive (EFHC1) forms of the syndrome.111 Onset occurs at around age 14, with generalized myoclonus and generalized tonic-clonic seizures. Myoclonic jerks constitute the initial symptom in 54% of patients. They are characteristically concentrated in the minutes following awakening and are bilateral, single or repetitive, arrhythmic, and more pronounced in the upper limbs. If intense, they may result in falls, but are too brief to be accompanied by loss of consciousness. Facial or lingual and perioral jerks, usually isolated, may be precipitated by talking in some patients,112 a phenomenon analogous to the jerking observed in patients with primary reading epilepsy. In 5% of patients, generalized jerks are also triggered by intermittent photic stimulation. Severe increase in frequency of jerks may herald episodes of myoclonic status epilepticus, which have become rarer113,114 with improved treatment. However, drug withdrawal or inappropriate drug choice are among the main factors that may precipitate symptoms.115 Generalized tonic-clonic seizures are present in 84% of patients and represent the initial symptom in 35% of cases. They are often preceded by a buildup of generalized myoclonic jerks. In 27% of patients, absences are also present, occurring infrequently (less than several times per week). Treatment with valproic acid in monotherapy or in association with clonazepam leads to total control of seizures in 80% of patients.107 Discontinuation of drug therapy is followed by a high rate of relapse (90%).107
Neurophysiologic analysis of myoclonus in JME indicates that muscles from both sides are activated synchronously, and those innervated by the cranial nerves are involved through a rostrocaudal pattern of activation. The EEG correlate is a generalized spike- or polyspikes-wave at 3 to 5 Hz, in which the negative peak of the spike precedes the generalized jerk by 10 to 30 msec.6 Duration of the EEG transient is ∼100 msec, and that of the myoclonic potential is less than 100 msec. In a recent work,116 a lateralized onset of the EEG transient has been suggested on the basis of an interside latency (9.5 ± 1.7 msec) that was thought to be compatible with transcallosal spread. However, it remains to be explained why this supposed focal trigger constantly spreads to constantly generate a generalized phenomenon, without producing any focal jerking (as usually seen, for example in patients with PME, who constantly exhibit both).
Myoclonic-Astatic Epilepsy (MAE)
MAE has its onset between 2 and 6 years of age. Seizure types include massive myoclonus and atonic falls, atypical absences, generalized clonic or tonic-clonic seizures, and episodes of status epilepticus with erratic myoclonus and clouding of consciousness.117 Interictal EEG, often normal at onset, can become very disorganized.104 Outcome is unpredictable. Remission within a few months or years with normal cognition is possible, even after a severe early course.118,119 About 30% of children experience long-lasting intractability and cognitive impairment.120 A few children with MAE have been shown to have inherited SCN1A and GABRG2 gene mutations from parents with generalized epilepsy with febrile seizures plus.121 However, the genetics of MAE is complex. Myoclonus in MAE manifests as bilateral, synchronous whole body jerks, consistent with the hypothesis of a thalamocortical origin.54 The jerks, lasting around 100 msec, are preceded by a negative EEG potential by around 30 msec.96 Myoclonic status in MAE has neurophysiological characters of erratic CM with multifocal jerking, increase in muscle tone, and clouding of consciousness. Nonconvulsive status may be precipitated by carbamazepine (Figures 8-4 and 8-5).6,37 Treatment is primarily with valproate and ethosuximide, often in combination. Lamotrigine, topiramate, and benzodiazepines might be useful in some patients.
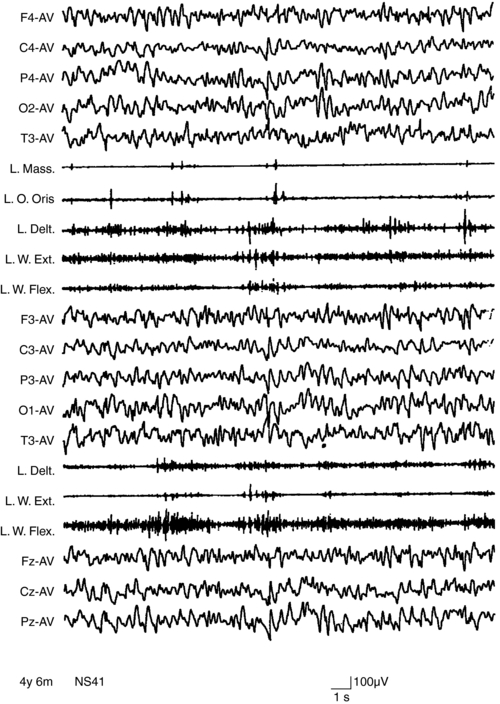
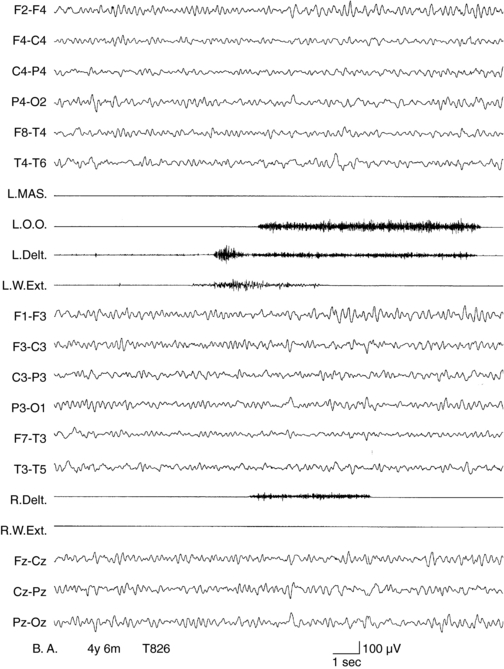
Figure 8–5 Same patient as in Figure 8-4. Carbamazepine was briskly suspended after the recording of status myoclonicus shown in Figure 8-4. Status quickly resolved in the following days to disappear 4 days later, as shown in this recording, EEG shows rhythmic background, no paroxysmal discharges, and no myoclonic potentials are recorded on EMG channels.
Epileptic Syndromes with Myoclonus of Unclear Neurophysiologic Characterization
EARLY MYOCLONIC ENCEPHALOPATHY
Early myoclonic encephalopathy is a rare syndrome classified among the generalized symptomatic epilepsies with nonspecific etiology.48 Its causes are multiple and include some inborn errors of metabolism, such as methylmalonic acidemia and nonketotic hyperglycinemia. Onset is in the neonatal period or during the first month of life with severe myoclonus, followed by partial seizures and tonic spasms. Myoclonus has a multifocal distribution, leading to the description of “erratic.” Neurological development is severely delayed, with hypotonia, impaired alertness, and, often, vegetative state.122 The EEG is characterized by suppression bursts. Erratic myoclonus generally does not have an EEG correlate.122
MYOCLONIC STATUS IN FIXED ENCEPHALOPATHIES
This condition is seen exclusively in severe encephalopathies with profound cognitive impairment and hypotonia and is characterized by recurrent, prolonged, and drug-resistant episodes of myoclonic status.123 Partial motor seizures, myoclonic absences, generalized myoclonus, and, rarely, unilateral or generalized clonic seizures can be associated. Myoclonic status is characterized by almost continuous absences accompanied by erratic, distal, multifocal, frequent myoclonic jerks, at times more rhythmic and diffuse. It is extremely important to recognize this condition and to differentiate it from a progressive encephalopathy.
EPILEPSY WITH MYOCLONIC ABSENCES
Onset is at about age 7 years with absences recurring many times a day, accompanied by bilateral rhythmic jerks, involving the shoulders, arms, or legs and, eventually, by a mild axial tonic contraction. Consciousness is cloudy but not completely interrupted.124 Ictal EEG shows bilateral, synchronous, and symmetric spike-wave discharges at 3 Hz and myoclonic jerks at the same frequency. Absences are often resistant to treatment. Evolution is variable featuring cognitive impairment in some patients, transition to a different type of epilepsy, or at times full recovery without sequelae.
The physiology of myoclonus in myoclonic absences is difficult to study as jerks appear against a background of increased muscle tone.6 Tassinari and coworkers125 found a constant relationship between the spike-and-wave complex and the jerk, with the positive spike of the spike-and-wave complex being followed by a myoclonic jerk by a latency of 15 to 40 msec (proximal muscles).
EYELID MYOCLONIA WITH AND WITHOUT ABSENCES
Eyelid myoclonia with absences are characterized by prominent jerking of the eyelids with upward deviation of the eyes. Some authors126,127 emphasized the severity of eyelid jerking in these patients as compared with the slight flicker of the eyelids seen in typical absences. The phenomenon may be so short (1 to 2 seconds) that it may be impossible to find out whether there is concomitant lapse of consciousness. The intensity of the jerking justifies the inclusion of this condition within the group of myoclonic epilepsies, more so as the myoclonic phenomena are difficult to control and persist into adulthood, whereas the absences are relatively easily controlled. A marked photosensitivity and self-stimulation are features that eyelid myoclonia, with and without absences, share with other myoclonic epilepsies of infancy and childhood.
MYOCLONIC SEIZURES INDUCED BY PHOTIC STIMULI
Myoclonic attacks can be induced by photic stimuli. Jeavons and Harding126 found that only 1.5% of pure photosensitive epilepsies (i.e., epilepsies induced exclusively by exposure to visual stimuli without any spontaneous attacks) were myoclonic. Visually induced generalized myoclonic jerks are usually symmetrical and predominate in the upper limbs. In most cases, they are mild, only producing head nodding and slight arm abduction. More generalized jerks, involving the face, trunk, and legs, may occasionally cause the patient to fall. The relationship of myoclonic jerks to the stimulus is complex. Sometimes there is no definite time relationship. On other occasions, the jerks may be repeated rhythmically with the same frequency as the stimulus or at one of its subharmonics.128 The jerks are associated in the EEG recording with the photoparoxysmal response, consisting of a bilateral polyspike or polyspike-and-wave discharge.128,129
Spontaneous seizures are said to occur mainly, but not exclusively, when the polyspike-wave discharge persists after discontinuation of the stimulation (prolonged photoconvulsive response).130 Myoclonic attacks can be provoked by television watching, especially when the patients are close to the screen and while playing video games. Some patients, especially, but not exclusively, mentally retarded, induce the myoclonic attacks by waving a hand between their eyes and a source of light, flickering their eyelids in front of a light source, or staring at patterned surfaces or by similar maneuvers.126,131,132 There is no clear-cut nosologic distinction between eyelid myoclonia and photic-induced myoclonus.
Reticular Reflex Myoclonus
Reticular myoclonus presents most of the clinical and neurophysiological characteristics of epileptic myoclonus, although it lacks a time-locked EEG correlate.5
Clinically, myoclonic jerks are generalized, mostly involving proximal and flexor muscles, spontaneous, or induced by somatosensory, auditory, and visual stimuli, or by movement.5,64 Reticular myoclonus seems to originate from the brainstem reticular formation, as involvement of muscles innervated by XIth cranial nerve (trapezius and sternocleidomastoid muscles) precedes that of orbicularis oris (VIIth cranial nerve) and masseter (Vth cranial nerve).64 EEG discharges have a wide distribution and greater amplitude at the vertex and can follow the onset of the jerks, suggesting that they are projected and not directly responsible for the myoclonic jerks.5 SEPs have normal amplitude. In reflex reticular myoclonus, both slow and fast conducting pathways have been observed.20 Reticular myoclonus has been described in postanoxic encephalopathy and can appear alongside cortical myoclonus in some patients.14,24,65 In patients with progressive myoclonus epilepsy, Cantello described a form of focal subcortical reflex myoclonus whose latencies might be consistent with origin in the reticular formation.55 Clinically, there are both multifocal and generalized jerks. Neurophysiological study of reticular reflex myoclonus is, however, difficult, especially because of the coexistence of cortical myoclonus in most patients. As a consequence, its neurophysiological correlates and relationships with epilepsy are poorly understood. Electrophysiological recordings in the urea-induced myoclonus in the rat, which is considered to be a model of reticular reflex myoclonus, have demonstrated neuronal activity resembling paroxysmal depolarization shift in the nucleus reticularis gigantocellularis.133
Antiepileptic Drug-Induced Myoclonus
Antiepileptic drugs can aggravate or induce myoclonus or myoclonic seizures, either because of paradoxical reaction or inappropriate choice. Carbamazepine and vigabatrin have been reported to worsen or precipitate myoclonic seizures.134,135 De novo appearance of myoclonic jerks was described in children or young adults with cryptogenic or symptomatic partial epilepsy treated with add-on vigabatrin.136,137 Carbamazepine should be avoided in MAE, because it can trigger episodes of myoclonic status.6 Adolescents with juvenile absence epilepsy may experience myoclonic status if treated with carbamazepine when their absence seizures are misdiagnosed as complex partial seizures.138 Exacerbation of epileptic negative myoclonus has been reported in children with benign rolandic epilepsy after carbamazepine treatment.37,38
Lamotrigine may be useful in some children with myoclonic astatic epilepsy,139 but it has been reported to worsen SMEI,140 and occasionally, to precipitate seizure aggravation and de novo myoclonic status epilepticus if administered at high doses in other conditions.141–145 LTG has also been reported to aggravate seizures in patients with JME.143 In Angelman syndrome worsening of myoclonic and absence seizures may be produced by carbamazepine or oxcarbazepine,146,147,148 phenytoin,146 or vigabatrin.149
Conclusion
Epileptic myoclonus can be defined as an elementary electroclinical manifestations of epilepsy involving descending neurons, whose spatial (spread) or temporal (self-sustained repetition) amplification can trigger overt epileptic activity6 and can be classified as cortical (positive and negative), secondarily generalized, thalamocortical, and reticular.10 Cortical epileptic myoclonus represents a fragment of partial or symptomatic generalized epilepsy; thalamocortical epileptic myoclonus is a fragment of idiopathic generalized epilepsy.5 Reflex reticular myoclonus represents the clinical counterpart of fragments of hypersynchronous epileptic activity of neurons in the brainstem reticular formation. EM, in the setting of an epilepsy syndrome, can be only one component of a seizure (i.e., myoclonic buildup in the clonic-tonic-clonic seizures of juvenile myoclonic epilepsy), the only seizure manifestations (myoclonic seizures of benign myoclonic epilepsy), one of the multiple seizure types (myoclonic astatic seizures in childhood epileptic encephalopathies), or a more stable condition that is manifested in a nonparoxysmal fashion and mimics a movement disorder (i.e., the continuous jerking of cortical tremor or of epilepsia partialis continua or the movement-activated jerks of progressive myoclonus epilepsy that can translate into a myoclonic cascade and a full-blown generalized tonic-clonic seizure). This complex correlation is more obvious in patients with epilepsia partialis continua in which cortical myoclonus (which underlies recurring focal jerks) and overt focal motor seizures usually start in the same somatic (and cortical) region. In patients with cortical tremor, this correlation is less obvious and requires neurophysiological studies to be demonstrated.
1. Marsden CD, Hallett M, Fahn S. The nosology and pathophysiology of myoclonus. In: Marsden CD, Fahn S, editors. Movement Disorders. London: Butterworths Scientific; 1982:196-249.
2. Fahn S, Marsden CD, Van Woert MH. Definition and classification of myoclonus. Adv Neurol. 1986;43:1-5.
3. Caviness JN, Brown P. Myoclonus: current concepts and recent advances. Lancet Neurol. 2006;3:598-607.
4. Patel VM, Jankovic J. Myoclonus. In: Appel SH, ed. Current Neurology, vol 8. Chicago: Year Book Medical Publishers; 1988:109-156.
5. Hallett M. Myoclonus: relation to epilepsy. Epilepsia. 1985;26(Suppl 1):S67-77.
6. Guerrini R, Bonanni P, Rothwell J, Hallet M. Myoclonus and epilepsy. In: Guerrini R, Aicardi J, Andermann F, Hallett M, editors. Epilepsy and Movement Disorders. Cambridge: Cambridge University Press; 2002:165-210.
7. Brown P, Farmer SF, Halliday DM, Marsden J, Rosenberg JR. Coherent cortical and muscle discharge in cortical myoclonus. Brain. 1999;122:461-472.
8. Grosse P, Guerrini R, Parmeggiani L, Bonanni P, Pogosyan A, Brown P. Abnormal corticomuscular and intermuscular coupling in high-frequency rhythmic myoclonus. Brain. 2003;126:326-342.
9. Van Rootselaar AF, Maurits NM, Koelman JHTM, et al. Coherence analysis differentiates between cortical myoclonic tremor and essential tremor. Mov Disord. 2006;21:215-222.
10. Guerrini R, Bonanni P, Parmeggiani L, Hallet M, Oguni H. Pathophysiology of myoclonic epilepsies. Adv Neurol. 2005;95:23-46.
11. Deuschl G, Ebner A, Hammers R, Lucking CH. Differences of cortical activation in spontaneous and reflex myoclonias. Electroencephalogr Clin Neurophysiol. 1991;80:326-328.
12. Kanouchi T, Yakota T, Kamata T, Ishii K, Senda M. Central pathway of photic reflex myoclonus. J Neurol Neurosurg Psychiatry. 1997;62:414-417.
13. Shibasaki H, Kuroiwa Y. Electroencephalographic correlates of myoclonus. Electroencephalogr Clin Neurophysiol. 1975;39:455-463.
14. Hallett M, Chadwick D, Marsden CD. Cortical reflex myoclonus. Neurology. 1979;29:1107-1125.
15. Shibasaki H, Kakigi R, Ikeda A. Scalp topography of giant SEP and pre-myoclonus spike in cortical reflex myoclonus. Electroencephalogr Clin Neurophysiol. 1991;81:31-37.
16. Mima T, Nagamine T, Ikeda A, Yazawa S, Kimura J, Shibasaki H. Pathogenesis of cortical myoclonus studied by magnetoencephalography. Ann Neurol. 1998;43:598-607.
17. Brown P, Day BL, Rothwell JC, Thompson PD, Marsden CD. Intrahemispheric and interhemispheric spread of cerebral cortical myoclonic activity and its relevance to epilepsy. Brain. 1991;114:2333-2355.
18. Sutton GG, Mayer RF. Focal reflex myoclonus. J Neurol Neurosurg Psychiatry. 1974;7:207-217.
19. Shibasaki H, Yamashita Y, Neshige R, Tobimatsu S, Fukui R. Pathogenesis of giant somatosensory evoked potentials in progressive myoclonic epilepsy. Brain. 1985;108:225-240.
20. Rothwell JC, Obeso JA, Marsden CD. Electrophysiology of somatosensory reflex myoclonus. Adv Neurol. 1986;43:385-398.
21. Ikeda A, Shibasaki H, Nagamine T, et al. Peri-rolandic and fronto-parietal components of scalp-recorded giant SEPs in cortical myoclonus. Electroencephalogr Clin Neurophysiol. 1995;96:300-309.
22. Hitomi T, Ikeda A, Matsumoto R, et al. Generators and temporal succession of giant somatosensory evoked potentials in cortical reflex myoclonus: epicortical recording from sensorimotor cortex. Clin Neurophysiol. 2006;117:1481-1486.
23. Thompson PD, Day BL, Rothwell JC, Brown P, Britton TC, Marsden CD. The myoclonus in corticobasal degeneration. Evidence for two forms of cortical reflex myoclonus. Brain. 1994;117:1197-1207.
24. Chadwick D, Hallett M, Harris R, Jenner P, Reynolds EH, Marsden CD. Clinical, biochemical, and physiologic features distinguishing myoclonus responsive to 5-hydroxy-tryptophan, tryptophan with a monoamine oxidase inhibitor, and clonazepam. Brain. 1977;100:455-487.
25. Obeso J, Rothwell JC, Marsden CD. The spectrum of cortical myoclonus: from focal reflex jerks to spontaneous motor epilepsy. Brain. 1985;108:193-224.
26. Chen R, Ashby P, Lang AE. Stimulus-sensitive myoclonus in akinetic-rigid syndromes. Brain. 1992;115:1875-1888.
27. Rodriguez ME, Artieda AJ, Zubieta JL, Obeso JA. Reflex myoclonus in olivopontocerebellar atrophy. J Neurol Neurosurg Psychiatry. 1994;57:316-319.
28. Young RR, Shahani BT. Clinical neurophysiological aspects of post-hypoxic intention myoclonus. Adv Neurol. 1979;26:85-105.
29. Wilkins DE, Hallett M, Berardelli A, Walshe T, Alvarez N. Physiologic analysis of the myoclonus of Alzheimer disease. Neurology. 1984;34:898-903.
30. Ugawa Y, Kkohara N, Hirasawa H, Kuzuhara S, Iwata M, Mannen T. Myoclonus in Alzheimer disease. J Neurol. 1987;235:90-94.
31. Rosing HS, Hopkins LC, Wallace DC, Epstein CM, Weidenheim K. Maternally inherited mitochondrial myopathy and myoclonic epilepsy. Ann Neurol. 1985;17:228-237.
32. So N, Berkovic S, Andermann F, Kuzniecky R, Gendron D, Quesney LF. Myoclonus epilepsy and ragged-red fibres (MERRF). Electrophysiological studies and comparison with other progressive myoclonus epilepsies. Brain. 1989;112:1261-1272.
33. Thompson PD, Bathia KP, Brown P, et al. Cortical myoclonus in Huntington’s disease. Mov Disord. 1994;9:633-641.
34. Guerrini R, Dravet C, Genton P, et al. Epileptic negative myoclonus. Neurology. 1993;43:1078-1083.
35. Shibasaki H. Pathophysiology of negative myoclonus and asterixis. Adv Neurol. 1995;67:199-210.
36. Guerrini R, Parmeggiani L, Shewmon A, Rubboli G, Tassinari CA. Motor dysfunction resulting from epileptic activity involving the sensori-motor cortex. In: Guerrini R, Aicardi J, Andermann F, Hallett M, editors. Epilepsy and Movement Disorder. Cambridge: Cambridge University Press; 2002:77-96.
37. Guerrini R, Belmonte A, Genton P. Antiepileptic drug-induced worsening of seizures in children. Epilepsia. 1998;39(Suppl. 3):S2-S10.
38. Parmeggiani L, Seri S, Bonanni P, Guerrini R. Electrophysiological characterization of spontaneous and carbamazepine-induced epileptic negative myoclonus in benign childhood epilepsy with centro-temporal spikes. Clin Neurophysiol. 2004;115:50-58.
39. Cerminara C, Montanaro ML, Curatolo P, Seri S. Lamotrigine-induced seizure aggravation and negative myoclonus in idiopathic rolandic epilepsy. Neurology. 2004;63:373-375.
40. Noachtar S, Holthausen H, Lüders HO. Epileptic negative myoclonus. Subdural EEG recording indicates a postcentral generator. Neurology. 1997;49:1534-1537.
41. Ikeda A, Ohara S, Matsumoto R, et al. Role of primary sensorimotor cortices in generating inhibitory motor response in humans. Brain. 2000;123:1710-1721.
42. Rubboli G, Parmeggiani L, Tassinari CA. Frontal inhibitory spike component associated with epileptic negative myoclonus. Electroencephalogr Clin Neurophysiol. 1995;95:201-205.
43. Baumgartner C, Podreka I, Olbrich A, et al. Epileptic negative myoclonus: an EEG-single-photon emission CT study indicating involvement of premotor cortex. Neurology. 1996;46:753-758.
44. Meletti S, Tinuper P, Bisulli F, Santucci M. Epileptic negative myoclonus and brief asymmetric tonic seizures. A supplementary sensorimotor area involvement for both negative and positive motor phenomena. Epileptic Disord. 2000;2:163-168.
45. Tassinari CA, Rubboli G, Parmeggiani L, et al. Epileptic negative myoclonus. Adv Neurol. 1995;67:181-197.
46. Lüders HO, Dinner DS, Morris HH, Wyllie E, Comair YG. Cortical electrical stimulation in humans. The negative motor areas. Adv Neurol. 1995;67:115-129.
47. Rubboli G, Mai R, Meletti S, et al. Negative myoclonus induced by cortical electrical stimulation in epileptic patients. Brain. 2006;129:65-81.
48. Commission on classification and terminology of the international league against epilepsy. proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia. 1989;30:389-399.
49. Genton P, Malafosse A, Moulard B, et al. The progressive myoclonus epilepsies. In: Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, editors. Epileptic Syndromes in Infancy and Adolescence. 4th. Montrouge: John Libbey Eurotext; 2005:424-441.
50. Badhwar A, Berkovic SF, Dowling JP, et al. Action myoclonus-renal failure syndrome: characterization of a unique cerebro-renal disorder. Brain. 2004;127:2173-2182.
51. Delgado-Escueta AV, Ganesh S, Yamakawa K. Advances in the genetics of progressive myoclonus epilepsy. Am J Med Genet. 2001;106:129-138.
52. Guerrini R, Bonanni P, Marini C, Parmeggiani L. The myoclonic epilepsies. In: Willie E, Gupta A, Lachhwani DK, editors. The Treatment of Epilepsy. 4th. Philadelphia: Lippincott Williams and Wilkins; 2006:407-427.
53. Roger J, Genton P, Bureau M, Dravet C. Progressive myoclonus epilepsies in childhood and adolescence. In: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, editors. Epileptic Syndromes in Infancy, Childhood and Adolescence. 2nd. London and Paris: John Libbey Eurotext Ltd.; 1992:381-400.
54. Guerrini R, Parmeggiani L, Volzone A. Cortical myoclonus in early childhood epilepsy. In: Majkowski J, Owczarek K, Zwolinski P, editors. Third European Congress of Epileptology. Bologna: Monduzzi Editore; 1998:99-105.
55. Cantello R, Gianelli M, Civardi C, Mutani R. Focal subcortical reflex myoclonus. A clinical and neurophysiological study. Arch Neurol. 1997;54:187-196.
56. Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185-188.
57. Auranen M, Vanhala R, Vosman M, et al. MECP2 gene analysis in classical Rett syndrome and in patients with Rett-like features. Neurology. 2001;56:611-617.
58. Scala E, Longo I, Ottimo F, et al. MECP2 deletions and genotype-phenotype correlation in Rett syndrome. Am J Med Genet A. 2007;143:2775-2784.
59. Guerrini R, Bonanni P, Parmeggiani L, Santucci M, Parmeggiani A, Sartucci F. Cortical reflex myoclonus in Rett syndrome. Ann Neurol. 1998;43:1-8.
60. Armstrong DD. Neuropathology of Rett syndrome. Ment Retard Dev Disabil Res Rev. 2002;8:72-76.
61. Carella F, Scaioli V, Ciano C, Binelli S, Oliva D, Girotti F. Adult onset myoclonic Huntington’s disease. Mov Disord. 1993;8:210-215.
62. Gambardella A, Muglia M, Labate A, etal: Juvenile Huntington disease presenting as progressive myoclonic epilepsy. Neurology. 2001;57:708-711.
63. Lance JW, Adams RD. The syndrome of intention or action myoclonus as a sequel to hypoxic encephalopathy. Brain. 1963;86:111-136.
64. Hallett M, Chadwick D, Adam J, Marsden CD. Reticular reflex myoclonus: a physiologic type of human post-hypoxic myoclonus. J Neurol Neurosurg Psychiatry. 1977;40:253-264.
65. Brown P, Thompson PD, Rothwell JC, Day BL, Marsden CD. A case of postanoxic encephalopathy with cortical action and brainstem reticular reflex myoclonus. Mov Disord. 1991;6:139-144.
66. Kojewnikow AY. Eine besondere Form von corticaler epilepsie. Neurologisches Zentralblatt. 1895;14:47-48.
67. Bancaud J. Kojewnikow’s syndrome (epilepsia partialis continua) in children. In: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, editors. Epileptic Syndromes in Infancy, Childhood and Adolescence. 2nd. London and Paris: John Libbey Eurotext Ltd.; 1992:363-369.
68. Thomas IE, Raegan TJ, Klass DW. Epilepsia partialis continua. A review of 32 cases. Arch Neurol. 1977;34:266-275.
69. Fusco L, Bertini E, Vigevano F. Epilepsia partialis continua and neuronal migration anomalies. Brain Dev. 1992;14:323-328.
70. Kuzniecky R, Powers R. Epilepsia partialis continua due to cortical dysplasia. J Child Neurol. 1993;8:386-388.
71. Andrews PI, McNamara JO, Lewis DV. Clinical and electroencephalographic correlates in Rasmussen’s encephalitis. Epilepsia. 1997;38:189-194.
72. Rogers SW, Andrews PI, Gahring LC, et al. Autoantibodies to glutamate receptor GluR3 in Rasmussen’s encephalitis. Science. 1994;265:648-651.
73. Hart Y. Rasmussen’s encephalitis. Epileptic Disord. 2004;6:133-144.
74. Watson R, Jiang Y, Bermudez I, et al. Absence of antibodies to glutamate receptor type 3 (GluR3) in Rasmussen encephalitis. Neurology. 2004;63:43-50.
75. Veggiotti P, Colamaria V, Dalla Bernardina B, Martelli A, Mangione D, Lanzi G. Epilepsia partialis continua in a case of MELAS: clinical and neurophysiological study. Neurophysiol Clin. 1995;25:158-166.
76. Ikeda A, Kakigi R, Funai N. Cortical tremor: a variant of cortical reflex myoclonus. Neurology. 1990;40:1561-1565.
77. Toro C, Pascual-Leone A, Deuschl G, Tate E, Pranzatelli MR, Hallett M. Cortical tremor: a common manifestation of cortical myoclonus. Neurology. 1993;43:2346-2353.
78. Terada K, Ikeda A, Mima T, et al. Familial cortical tremor as a unique form of cortical reflex myoclonus. Mov Disord. 1997;12:370-377.
79. Okuma Y, Shimo Y, Shimura H, et al. Familial cortical tremor with epilepsy: an under-recognized familial tremor. Clin Neurol Neurosurg. 1998;100:75-78.
80. Elia M, Musumeci SA, Ferri R, et al. Familial cortical tremor, epilepsy, and mental retardation: a distinct clinical entity? Arch Neurol. 1998;55:1569-1573.
81. Guerrini R, Bonanni P, Patrignani A, et al. Autosomal dominant cortical myoclonus and epilepsy (ADCME) with complex partial and generalized seizures: a newly recognized epilepsy syndrome with linkage to chromosome 2p11.1-q12.2. Brain. 2001;124:2459-2475.
82. Gardella E, Tinuper P, Marini C, et al. Autosomal dominant early-onset cortical myoclonus, photic-induced myoclonus, and epilepsy in a large pedigree. Epilepsia. 2006;47:1643-1649.
83. Carr JA, van der Walt PE, Nakayama J, et al. FAME 3: a novel form of progressive myoclonus and epilepsy. Neurology. 2007;68:1382-1389.
84. Guerrini R, De Lorey TM, Bonanni P, et al. Cortical myoclonus in Angelman syndrome. Ann Neurol. 1996;40:39-48.
85. Labauge P, Amer LO, Simonetta-Moreau M, et al. Absence of linkage to 8q24 in a European family with familial adult myoclonic epilepsy (FAME). Neurology. 2002;58:941-944.
86. Mikami M, Yasuda T, Terao A, et al. Localization of a gene for benign adult familial myoclonic epilepsy to chromosome 8q23.3-q24.1. Am J Hum Genet. 1999;65:745-751.
87. van Rootselaar AF, Callenbach PM, Hottenga JJ, et al. A Dutch family with “familial cortical tremor with epilepsy.” Clinical characteristics and exclusion of linkage to chromosome 8q23.3-q24.1. J Neurol. 2002;249:829-834.
88. Striano P, Chifari R, Striano S, et al. A new benign adult familial myoclonic epilepsy (BAFME) pedigree suggesting linkage to chromosome 2p11.1-q12.2. Epilepsia. 2004;45:190-192.
89. De Falco FA, Striano P, de Falco A, et al. Benign adult familial myoclonic epilepsy: Genetic heterogeneity and allelism with ADCME. Neurology. 2003;60:1381-1385.
90. Dravet C, Bureau M, Oguni H, et al. Severe myoclonic epilepsy in infancy (Dravet syndrome). In: Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, editors. Epileptic Syndromes in Infancy, Childhood and Adolescence. 3rd. London and Paris: John Libbey; 2002:81-103.
91. Oguni H, Hayashi K, Awaya Y, Fukuyama Y, Osawa M. Severe myoclonic epilepsy in infants—a review based on the Tokyo Women’s Medical University series of 84 cases. Brain Dev. 2001;23:736-748.
92. Chiron C. Prognosis of severe myoclonic epilepsy in infancy (Dravet syndrome). In: Jallon P, Berg A, Dulac O, Hauser A, editors. Prognosis of Epilepsies. Paris: John Libbey Eurotext; 2003:239-248.
93. Marini C, Mei D, Temudo T, et al. Idiopathic epilepsies with seizures precipitated by fever and SCN1A abnormalities. Epilepsia. 2007;48:1678-1685.
94. Beaumanoir A, Dravet C. The Lennox-Gastaut syndrome. In: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, editors. Epileptic Syndromes in Infancy, Childhood and Adolescence. 2nd ed. London and Paris: John Libbey Eurotext Ltd; 1992:115-132.
95. Gastaut H. The Lennox-Gastaut syndrome: comments on the syndrome’s terminology and nosological position amongst the secondary generalized epilepsies of childhood. Electroencephalogr Clin Neurophysiol. 1982;35(Suppl):S71-S84.
96. Bonanni P, Parmeggiani L, Guerrini R. Different neurophysiologic patterns of myoclonus characterize Lennox-Gastaut syndrome and myoclonic astatic epilepsy. Epilepsia. 2002;43:609-615.
97. Wilkins DE, Hallett M, Erba G. Primary generalized epileptic myoclonus: a frequent manifestation of minipolymyoclonus of central origin. J Neurol Neurosurg Psychiatry. 1985;48:506-516.
98. Gloor P. Generalized epilepsy with spike-and-wave discharge: a reinterpretation of its electrographic and clinical manifestations. Epilepsia. 1979;20:571-588.
99. Dalla Bernardina B, Colamaria V, Capovilla G. Nosological classification of epilepsies in the first three years of life. In: Nisticò G, Di Perri R, Meinardi H, editors. Epilepsy: an Update on Research and Therapy. New York: Alan Liss; 1983:165-183.
100. Dravet C, Bureau M, Roger J. Benign myoclonic epilepsy in infants. In: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, editors. Epileptic Syndromes in Infancy, Childhood and Adolescence. 2nd. London and Paris: John Libbey Eurotext Ltd.; 1992:67-74.
101. Guerrini R, Dravet C, Gobbi G, et al. Idiopathic generalized epilepsies with myoclonus in infancy and childhood. In: Malafosse A, Genton P, Hirsch E, Marescaux C, Broglin D, Bernasconi R, editors. Idiopathic Generalized Epilepsies: Clinical, Experimental and Genetics Aspects. London: John Libbey and Company; 1994:267-280.
102. Dravet C. Les épilepsies myocloniques bénignes du nourrisson. Epilepsies. 1990;2:95-101.
103. Dravet C, Bureau M. Benign myoclonic epilepsy in infancy. Adv Neurol. 2005;95:127-137.
104. Guerrini R, Aicardi J. Epileptic encephalopathies with myoclonic seizures in infants and children (severe myoclonic epilepsy and myoclonic-astatic epilepsy). Journal of Clin Neurophysiol. 2003;20:449-461.
105. Engel JJr. A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: report of the ILAE Task Force on Classification and Terminology. Epilepsia. 2001;42:796-803.
106. Ricci S, Cusmai R, Fusco L, et al. Reflex myoclonic epilepsy in infancy: a new age-dependent idiopathic epileptic syndrome related to startle reaction. Epilepsia. 1995;36:342-348.
107. Genton P, Salas-Puig X, Tunon A, Lahoz C, Del Soccorro M. Juvenile myoclonic epilepsy and related syndromes: clinical and neurophysiological aspects. In: Malafosse A, Genton P, Hirsch E, Marescaux C, Broglin D, Bernasconi R, editors. Idiopathic Generalized Epilepsies: Clinical, Experimental and Genetics Aspects. London: John Libbey and Company; 1994:253-266.
108. Cossette P, Liu L, Brisebois K, et al. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet. 2002;31:184-189.
109. Haug K, Warnstedt M, Alekov AK, et al. Mutations in CLCN2 encoding a voltage-gated chloride channel are associated with idiopathic generalized epilepsies. Nat Genet. 2003;33:527-532.
110. Annesi F, Gambardella A, Michelucci R, et al. Mutational analysis of EFHC1 gene in Italian families with juvenile myoclonic epilepsy. Epilepsia. 2007;48:1686-1690.
111. Suzuki T, Delgado-Escueta AV, Aguan K, et al. Mutations in EFHC1 cause juvenile myoclonic epilepsy. Nat Genet. 2004;36:842-849.
112. Wolf P, Mayer T. Juvenile myoclonic epilepsy: a syndrome challenging syndromic concepts? In: Schmitz B, Sander T, editors. Juvenile Myoclonic Epilepsy: The Janz Syndrome. Petersfield: Wrightson Biomedical Publishing; 2000:33-39.
113. Asconape J, Penry JK. Some clinical and EEG aspects of benign juvenile myoclonic epilepsy. Epilepsia. 1984;25:108-114.
114. Salas-Puig X, Camara da, Silva AM, Dravet C. L’épilepsie myoclonique juveénile dans la population du Centre Saint Paul. Epilepsies. 1990;2:108-113.
115. Thomas P, Genton P, Gelisse P, Wolf P. Juvenile myoclonic epilepsy. In: Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, editors. Epileptic Syndromes in Infancy, Childhood and Adolescence. 3rd. London: John Libbey & Co Ltd; 2002:335-356.
116. Panzica F, Rubboli G, Franceschetti S, et al. Cortical myoclonus in Janz syndrome. Clin Neurophysiol. 2001;112:1803-1809.
117. Doose H. Myoclonic astatic epilepsy of early childhood. In: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, editors. Epileptic Syndromes in Infancy, Childhood and Adolescence. 2nd ed. London and Paris: John Libbey Eurotext Ltd.; 1992:103-114.
118. Kaminska A, Ickowicz A, Plouin P, Bru MF, Dellatolas G, Dulac O. Delineation of cryptogenic Lennox-Gastaut syndrome and myoclonic astatic epilepsy using multiple correspondence analysis. Epilepsy Res. 1999;36:15-29.
119. Oguni H, Tanaka T, Hayashi K, et al. Treatment and long-term prognosis of myoclonic-astatic epilepsy of early childhood. Neuropediatrics. 2002;33:122-132.
120. Guerrini R, Parmeggiani L, Bonnanni P, et al. Myoclonic astatic epilepsy. In: Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, editors. Epileptic Syndromes in Infancy, Childhood and Adolescence. 4th. London and Paris: John Libbey & Co Ltd.; 2005:115-124.
121. Meisler MH, Kearney J, Ottman R, Escayg A. Identification of epilepsy genes in human and mouse. Annu Rev Genet. 2001;35:567-588.
122. Aicardi J. Early myoclonic encephalopathy (neonatal myoclonic encephalopathy). In: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, editors. Epileptic Syndromes in Infancy, Childhood and Adolescence. 2nd. London and Paris: John Libbey Eurotext Ltd; 1992:13-23.
123. Dalla Bernardina B, Fontana E, Darra F. Myoclonic status in nonprogressive encephalopathies. Adv Neurol. 2005;95:59-70.
124. Tassinari CA, Bureau M, Thomas P. Epilepsy with myoclonic absences. In: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, editors. Epileptic Syndromes in Infancy, Childhood and Adolescence. 2nd. London and Paris: John Libbey Eurotext Ltd; 1992:151-160.
125. Tassinari CA, Michelucci R, Rubboli G. Myoclonic absence epilepsy. In: Duncan JS, Panayiotopoulos CP, editors. Typical Absences and Related Epileptic Syndromes. London: Churchill Communications Europe; 1995:187-195.
126. Jeavons PM. Harding GFA. Photosensitive Epilepsy. London: Heinemann, 1975.
127. Jeavons P. Myoclonic epilepsies: therapy and prognosis. In: Akimoto H, Kazamatsuri H, Seino M, Ward AA, editors. Advances in Epileptology: XIIIth Epilepsy International Symposium. New York: Raven Press; 1982:141-144.
128. Kasteleijn-Nolst Trenite DG, Guerrini R, Binnie CD, et al. Visual sensitivity and epilepsy: a proposed terminology and classification for clinical and EEG phenomenology. Epilepsia. 2001;42:692-701.
129. Gastaut H, Broughton R. Epileptic Seizures. Springfield, IL: Charles C. Thomas, 1972.
130. Reilly EL, Peters JF. Relationship of some varieties of electroencephalographic photosensitivity to clinical convulsive disorders. Neurology. 1973;23:1050-1057.
131. Binnie CD, Darby CE, De Korte RA, et al. Self-induction of epileptic seizures by eye closure: incidence and recognition. J Neurol Neurosurg Psychiatry. 1980;43:386-389.
132. Tassinari CA, Rubboli G, Michelucci R. Reflex epilepsy. In: Dam M, Gram L, editors. Comprehensive Epileptology. New York: Raven Press; 1990:233-243.
133. Zuckermann EG, Glaser GH. Urea induced myoclonic seizures. Arch Neurol. 1972;27:14-28.
134. Talwar D, Arora MS, Sher PK. EEG changes and seizure exacerbation in young children treated with carbamazepine. Epilepsia. 1994;35:1154-1159.
135. Viani F, Romeo A, Viri M. Seizure and EEG patterns in Angelman’s syndrome. J Child Neurol. 1995;10:467-471.
136. Lortie A, Chiron C, Mumford J. The potential for increasing seizure frequency, relapse, and appearance of new seizure types with vigabatrin. Neurology. 1993;43:24-27.
137. Marciani MG, Gigli GL, Maschio M. Vigabatrin-induced myoclonus in four cases of partial epilepsy. Epilepsia. 1995;36:107.
138. Marini C, Parmeggiani L, Masi G, D’Arcangelo G, Guerrini R. Nonconvulsive status epilepticus precipitated by carbamazepine presenting as dissociative and affective disorders in adolescents. J Child Neurol. 2005;20:693-696.
139. Dulac O, Kaminska A. Use of lamotrigine in Lennox-Gastaut and related epilepsy syndromes. J Child Neurol. 1997;12(Suppl 1):S23-S28.
140. Guerrini R, Dravet C, Genton P, Belmonte A, Kaminska A, Dulac O. Lamotrigine and seizure aggravation in severe myoclonic epilepsy. Epilepsia. 1998;39:508-512.
141. Briassoulis G, Kalabalikis P, Tamiolaki M, Hatzis T. Lamotrigine childhood overdose. Pediatr Neurol. 1998;19:239-242.
142. Guerrini R, Belmonte A, Parmeggiani L, Perucca E. Myoclonic status epilepticus following high dosage lamotrigine therapy. Brain Dev. 1999;21:420-424.
143. Biraben A, Allain H, Scarabin JM, Schück S, Edan G. Exacerbation of juvenile myoclonic epilepsy with lamotrigine. Neurology. 2000;55:1758.
144. Janszky J, Rásonyi G, Halász P, et al. Disabling erratic myoclonus during lamotrigine therapy with high serum level: report of two cases. Clin Neuropharmacol. 2000;23:86-89.
145. Carrazzana EJ, Wheeler SD. Exacerbation of juvenile myoclonic epilepsy with lamotrigine. Neurology. 2001;56:1424-1425.
146. Minassian BA, DeLorey TM, Olsen RW, et al. Angelman syndrome: correlations between epilepsy phenotypes and genotypes. Ann Neurol. 1998;43:485-493.
147. Laan LA, Renier WO, Arts WF, et al. Evolution of epilepsy and EEG findings in Angelman syndrome. Epilepsia. 1997;38:195-199.
148. Vendrame M, Khurana DS, Cruz M, Melvin J, Valencia I, Legido A, Kothare SV. Aggravation of seizures and/or EEG features in children treated with oxcarbazepine monotherapy. Epilepsia. 2007;48:2116-2120.
149. Kuenzle C, Steinlin M, Wohlrab G, Boltshauser E, Schmitt B. Adverse effects of vigabatrin in Angelman syndrome. Epilepsia. 1998;39:1213-1215.