Pulmonary Hypertension
PATHOPHYSIOLOGY OF PULMONARY HYPERTENSION AND RIGHT VENTRICULAR FAILURE
CLINICAL CLASSIFICATION OF PULMONARY HYPERTENSION
INTEGRATED APPROACH TO THE DIAGNOSIS OF PULMONARY HYPERTENSION
MANAGEMENT OF ARTERIAL PULMONARY HYPERTENSION AND RIGHT VENTRICULAR FAILURE IN THE INTENSIVE CARE UNIT
Pulmonary hypertension (PH) is a complex and heterogeneous pulmonary vascular disorder that leads to elevated pulmonary vascular resistance (PVR) and right ventricular failure. During recent years, multiple advances in the therapy and management of PH have been made; however, this disorder continues to cause significant morbidity and mortality. PH is defined by a resting mean pulmonary arterial pressure (mPAP) greater than 25 mm Hg, associated with PVR greater than 240 dynes ⋅ second ⋅ cm−5 (or >3 Wood units) measured by right-sided heart catheterization. It is very important for the intensivist to distinguish those patients who have chronic PH secondary to any of the five groups included in the clinical classification described in Box 45.1 from other conditions that are usually faced in the intensive care unit (ICU) that can cause an acute elevation in the PVR. The acute elevation of pulmonary artery (PA) pressure observed in critically ill patients can develop on top of preexisting chronic PH (acute on chronic). It can be transient without consequences, or it can be prolonged and progress to severe acute PH, leading to life-threatening complications that include refractory systemic arterial hypotension, severe hypoxemia, right ventricular dysfunction and failure, and ultimately cardiogenic or obstructive shock and death. The most common acute elevation of PA pressures in the ICU is seen in the setting of left-sided heart disease (elevated pulmonary venous pressure) or in patients with preexisting pulmonary vascular disease. It is also well recognized after cardiothoracic surgery, during sepsis, after pulmonary embolism (PE), and in acute respiratory distress syndrome (ARDS). Unfortunately, in most cases acute PH remains underdiagnosed and its treatment begins only after serious complications have been developed.
Pathophysiology of Pulmonary Hypertension and Right Ventricular Failure
Development of Pulmonary Hypertension
In order to understand the pathophysiology of PH and before reviewing its current clinical classification, it is important to identify where the vascular insult originates. Conditions that raise the postcapillary pressure (pulmonary venous pressures) such as left-sided heart failure or mitral stenosis differ significantly from conditions that primarily affect the pulmonary arteries and arterioles such as idiopathic pulmonary artery hypertension (IPAH). The former causes a gradient between the PA diastolic and pulmonary capillary wedge pressure (PCWP) that is relatively small, with histopathologic changes in the arterial vessels that consist of mild medial hypertrophy and reversible intimal changes. In the latter, there is an increased pulmonary arteriovenous pressure gradient, and histologic changes on the pulmonary vasculature are more marked, including significant intimal hypertrophy with fibrosis, marked smooth muscle hypertrophy, vasoconstriction, adventitial proliferation, and thrombosis in situ.1 These changes cause vascular flow obstruction and eventually lead to abnormal angiogenesis and formation of plexiform arteriopathy. Endothelial dysfunction also develops with an imbalance between vasodilation and vasoconstriction and between apoptosis and proliferation, mechanisms that are thought to play the most important role in the development of chronic progressive PH. Hypoxemic pulmonary vasoconstriction is an important determinant of arterial PH in patients with respiratory disorders.1 In many types of PH, production of endogenous vasodilators (nitric oxide [NO] and prostacyclin) is impaired and production of vasoconstrictors (endothelin-1, thromboxane A) is increased.1 That is why the common treatment strategy for PH is to achieve the balance in key molecular pathways by increasing available NO and prostacyclin, or reducing the effects of endothelin-1. Acute cases are characterized by sudden increase in pulmonary arterial pressure (PAP) as seen when mechanical obstruction with subsequent vasoconstriction develops during an acute PE. In ARDS, both hypoxemia and the accumulation of intravascular fibrin and cellular debris contribute to subsequent vascular obliteration and PH.2 Endotoxin and vasoactive mediators related to pulmonary vasoconstriction also play significant roles in development of the PH during sepsis. Several animal studies have shown that endotoxin may cause not only systemic hypotension but also pulmonary biphasic hypertension, decrease in compliance, and increase in resistance of the respiratory system.3 Those endotoxin-dependent hemodynamic and respiratory effects are mediated by excessive release of inflammatory mediators and imbalance in production of NO, prostanoids, and endothelin-1.3,4 PH in endotoxemia is characterized by a constriction of proximal pulmonary arteries during the early phase followed by decreased compliance of distal pulmonary vasculature.5 Endotoxin infusion can also dramatically affect right ventricular function: in the very early phase of endotoxemic shock, right ventricular-vascular coupling is preserved by an increase in right ventricular contractility. Later, myocardial oxygen consumption and the energy cost of right ventricular contractility are increased, which along with progressive endotoxin-induced PH lead to right ventricular dysfunction and failure.6
Right Ventricular Failure
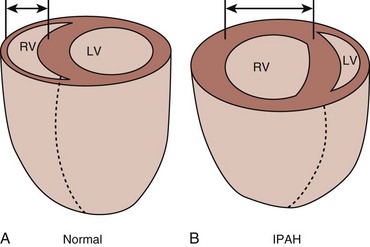
The right ventricle (RV) differs from the left ventricle (LV) in morphologic appearance and functionality.7 Despite the requirement for a similar cardiac output between the RV and LV, the bioenergetic requirement for right ventricular function is approximately 20% of the LV. The RV is thinner than the LV and its shape differs from that of the LV, having a crescent-shaped morphologic appearance. These differences reflect the low resistance, low impedance, and high compliance of the pulmonary circulation.7 The high compliance allows quick adaptations to changes in preload; however, unlike the LV, the RV tolerates poorly the acute increases in afterload, which could lead to hemodynamic collapse.8 It is important to emphasize these differences between the ability of the RV to adapt to sudden (acute) versus gradual (chronic) elevation of PAP. A normal RV can acutely adapt to high flow, but is barely able to tolerate any but very short acute high-pressure load.9 The normal RV cannot acutely increase the mPAP to more than 40 mm Hg.10 In chronic sustained elevation of afterload as seen in PH, the RV increases its wall thickness by hypertrophy of the muscle mass and assumes a more rounded shape (Fig. 45.1). Eventually and despite the compensatory right ventricular hypertrophy to the sustained long-term pressure overload, the RV dilates. Neurohormonal activation develops during the right ventricular dilatation and is an important mechanism in both acute and chronic right ventricular failure. The consequence of sympathetic hyperactivity is an increase in PVR with impedance of flow, causing right ventricular strain that impairs filling and causes right ventricular volume and pressure overload. Furthermore, the RV dilatation increases oxygen consumption and reduces contractility, which is going to decrease right ventricular perfusion, and a vicious circle develops that ultimately leads to death. Tricuspid regurgitation develops as a result of right ventricular dysfunction and defines a poor prognosis.11 It is important also to mention the concept of functional interdependence between the RV and the LV. Anatomically, the superficial myocardial fibers encircle both ventricles, and both chambers are contained within the pericardium, sharing the interventricular septum.12 During elevation of right-sided heart pressures, the interventricular septum shifts progressively to the left with subsequent development of left ventricular diastolic dysfunction that reduces the LV’s cardiac output and coronary perfusion pressure.8,13 A downstream adverse effect of right-sided heart failure is the development of systemic venous hypertension leading to concomitant visceral organ congestion and dysfunction. Regardless of the underlying cause of PH, the final common pathway for hemodynamic deterioration and death is right ventricular failure.
Clinical Classification of Pulmonary Hypertension
The classification of PH has presented different modifications since its first classification made in 1973 at a conference endorsed by the World Health Organization. The most recent classification of PH, described in Box 45.1, is based on causative diseases and was updated during the Fourth World Symposium on PH held in Dana Point, California.14 For the intensivist, it is important to recognize two different scenarios: patients with chronic PH admitted to the ICU for an acute process that may or may not worsen the underlying PH and patients with no history of chronic PH who develop acute PH during their ICU stay secondary to various conditions. It is also important to distinguish between pulmonary arterial hypertension as seen in IPAH and pulmonary venous hypertension as seen in left ventricular failure. A classification of PH in the ICU is described in Box 45.2.
Acute on Chronic Pulmonary Hypertension
Individuals with preexisting PH (i.e., IPAH or portopulmonary hypertension [group 1]) are particularly vulnerable to acute illnesses, which commonly result in rapid clinical deterioration and even death.15 Besides the entities described in group 1 of the Dana Point classification, several other conditions that are associated with chronic PH and are more commonly encountered in the ICU include left ventricular heart failure (with or without preserved ejection fraction causing pulmonary venous hypertension; group 2); interstitial lung diseases, chronic obstructive pulmonary disease (COPD), chronic hypoventilation syndromes, and sleep disorder breathing (group 3); and chronic pulmonary thromboembolic disease (group 4). Several clinical factors faced during intercurrent critical illness can aggravate or unmask the hemodynamics of patients with preexisting PH and are outlined in Table 45.1. These patients with chronic PH can rapidly deteriorate and usually die from progressive right ventricular failure (49%), progressive respiratory failure (18%), or sudden cardiac death (17%). Cardiopulmonary resuscitation (CPR), even when attempted in the hospital setting, is rarely successful. Only 6% of patients survived for more than 90 days and most of the survivors had identifiable causes of circulatory arrest that were rapidly reversible. The pulmonary blood flow is virtually absent in these patients during CPR. In 54%, cardiorespiratory arrest was associated with an intercurrent illness,15 illustrating how preexisting PH adversely affects patients’ compensatory capacity and ability to survive an acute illness.

Acute Pulmonary Hypertension
Acute PH is caused by an abrupt increase in PVR. The prototype of this process is an acute PE; however, other conditions frequently seen in the ICU can also be associated with acute increase in PVR such as acute decompensated left ventricular failure, post cardiac surgery, ARDS, and sepsis.16 Acute right ventricular failure develops in 61% of patients who present with massive PE that involves at least two lobar arteries. The mortality rate ranges from 3% in hemodynamically stable patients to 59% in unstable ones.17,18 Hemodynamic instability in the setting of PE is defined as systolic blood pressure (SBP) less than 90 mm Hg or a drop in SBP greater than 40 mm Hg from baseline for more than 15 minutes that is not otherwise explained by hypovolemia, sepsis, or new arrhythmia.18 The degree of shock inferred from the presence of metabolic acidosis, but not transthoracic echocardiography (TTE) findings, is the most powerful predictor of death in these patients.17,19
Acute Respiratory Distress Syndrome and Sepsis
Right ventricular dysfunction as a complication of ARDS is more gradual than in patients with massive PE, usually occurring at least 48 hours after the beginning of respiratory support.16 Evaluation of right ventricular function by TTE in a group of 75 ARDS patients submitted to protective ventilation demonstrated 25% incidence of acute right ventricular failure, resulting in detrimental hemodynamic consequences associated with tachycardia. However, those changes in heart function were reversible in patients who recovered; furthermore, it did not increase mortality rate.16 Although the initial magnitude of PH was not an indicator of mortality rate, mPAP increased in nonsurvivors, but not in survivors when followed for 7 days.20 Thus, development of PH in ARDS patients seems to be a sign of poor prognosis. In another cohort of 352 ARDS patients, both mortality rate and incidence of right ventricular failure were related to the level of plateau pressure during mechanical ventilation. In patients without acute cor pulmonale, the odds ratio of mortality for an increase in plateau pressure from 18–26 to 27–35 cm H2O was 1.15 (p = 0.635); however, for patients with acute cor pulmonale, the odds ratio of mortality for an increase in plateau pressure from 18–26 to 27–35 cm H2O was 3.32 (p = 0.034), suggesting that the threshold for a safe plateau pressure depends on the presence or not of acute cor pulmonale.21 Importantly, the implementation of low tidal volume ventilation in patients with ARDS has significantly lowered not only mortality rates but also incidence of acute right ventricular failure in this patient population.16 In addition to being the major risk factor for ARDS development, sepsis itself can sometimes lead to severe acute arterial PH.22
Postsurgical Pulmonary Hypertension
Some surgical interventions, in particular vascular, cardiac, and thoracic surgery, may cause acute elevation of mPAP either during the surgery or shortly after the intervention has been completed. This is particularly dangerous in patients with preexisting PH, because even short-lasting increased pressure overload to the RV could lead to profound decompensation with all downstream negative hemodynamic consequences. Preexisting PH is one of the major risk factors for morbidity and death in cardiothoracic surgery patients.23 PH is a major determinant of perioperative morbidity and mortality rate in special situations such as heart and lung transplantation, pneumonectomy, and ventricular assist device placement.24 The elevated PAP during and after surgery is thought to develop secondary to acute left-sided heart failure/dysfunction, or it can also be a consequence of pulmonary parenchymal and endothelial injury with activation of the systemic and pulmonary inflammatory response to cardiopulmonary bypass circulation or ischemia-reperfusion.25 Protamine-mediated acute PH and right ventricular failure in the perioperative period are common complications of cardiopulmonary bypass circulation during open-heart operations.26 PH can also develop later as a result of ARDS27 or other complications (sepsis, PE, etc.) not directly related to either surgery or anesthesia.
Integrated Approach to the Diagnosis of Pulmonary Hypertension
When PH is suspected based on presentation, examination, and risk factors, a comprehensive and structured evaluation should be performed. Physical examination is usually variable and nonspecific. The presence of an accentuated pulmonary component of S2, an early systolic click, and a midsystolic ejection murmur from turbulent pulmonary outflow should raise the suspicion. Left parasternal lift and an S4 are signs of right ventricular hypertrophy. Distended jugular veins and hepatojugular reflux indicate high central venous pressure. Right ventricular S3, hepatomegaly, ascites, systemic hypotension, peripheral edema, and cool extremities are all signs of right ventricular failure. A high level of suspicion is paramount in establishing a timely diagnosis of PH in order to initiate therapy. The diagnostic endeavor is aimed at making the diagnosis of PH and also in finding its cause. General guidelines for the evaluation of PH are described in detail in Figure 45.2. Chest radiographic findings are usually nonspecific but enlarged main and hilar PA shadows could be seen. RV enlargement best seen in the lateral views could also suggest PH. Moreover, the chest radiograph can also present findings of underlying primary lung disease such as emphysema or pulmonary fibrosis. Electrocardiography has a low sensitivity and specificity for the diagnosis of PH; however, evidence of right atrial enlargement, right axis deviation, and RV enlargement is suggestive of the disease. In many cases, PH remains undiagnosed and its treatment begins only after serious complications have developed. Some serologic markers, such as troponin and natriuretic peptides, are important for the evaluation of right ventricular dysfunction. Serum troponin may be elevated in patients with PH and has been associated with right ventricular overdistention and ischemia. Troponin I leak due to acute right ventricular strain from PE has been well studied and may predict mortality rate.28,29 Elevated B-natriuretic peptide (BNP) is an important prognostic indicator and correlates strongly with PVR, cardiac output, and functional status in patients with PH.30 A high level of plasma BNP, and in particular, a further increase in plasma BNP during follow-up, may have a strong independent association with increased mortality rates in patients with PH.31 However, the significance of measuring BNP level in patients with PH in the acute setting remains unclear.
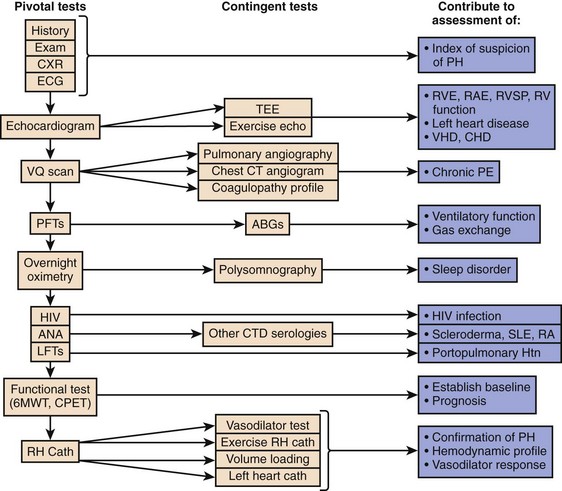
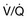 scan and pulmonary angiogram) is contributing or predominant. Contingent tests are recommended to elucidate or confirm results of the pivotal tests and need only be performed in the appropriate clinical context. The combination of pivotal and appropriate contingent tests contributes to assessment of the differential diagnoses in the right-hand column. It should be recognized that definitive diagnosis may require additional specific evaluations not necessarily included in this general guideline. 6MWT indicates a 6-minute walk test. ABGs, arterial blood gases; ANA, antinuclear antibody serologic test; CHD, congenital heart disease; CPET, cardiopulmonary exercise test; CT, computed tomography; CTD, connective tissue disease; CXR, chest x-ray; ECG, electrocardiogram; HIV, human immunodeficiency virus screening; Htn, hypertension; IPAH, idiopathic pulmonary artery hypertension; LFT, liver function test; PAH, pulmonary artery hypertension; PE, pulmonary embolism; PFT, pulmonary function test; PH, pulmonary hypertension; RA, rheumatoid arthritis; RAE, right atrial enlargement; RHC, right-sided heart catheterization; RVE, right ventricular enlargement; RVSP, right ventricular systolic pressure; SLE, systemic lupus erythematosus; TEE, transesophageal echocardiography; VHD, valvular heart disease;
scan and pulmonary angiogram) is contributing or predominant. Contingent tests are recommended to elucidate or confirm results of the pivotal tests and need only be performed in the appropriate clinical context. The combination of pivotal and appropriate contingent tests contributes to assessment of the differential diagnoses in the right-hand column. It should be recognized that definitive diagnosis may require additional specific evaluations not necessarily included in this general guideline. 6MWT indicates a 6-minute walk test. ABGs, arterial blood gases; ANA, antinuclear antibody serologic test; CHD, congenital heart disease; CPET, cardiopulmonary exercise test; CT, computed tomography; CTD, connective tissue disease; CXR, chest x-ray; ECG, electrocardiogram; HIV, human immunodeficiency virus screening; Htn, hypertension; IPAH, idiopathic pulmonary artery hypertension; LFT, liver function test; PAH, pulmonary artery hypertension; PE, pulmonary embolism; PFT, pulmonary function test; PH, pulmonary hypertension; RA, rheumatoid arthritis; RAE, right atrial enlargement; RHC, right-sided heart catheterization; RVE, right ventricular enlargement; RVSP, right ventricular systolic pressure; SLE, systemic lupus erythematosus; TEE, transesophageal echocardiography; VHD, valvular heart disease; 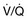 scan, ventilation-perfusion scintigram.
scan, ventilation-perfusion scintigram. Echocardiography
Echocardiography is the most important and useful noninvasive study for screening of PH. It is very important for diagnosing and determining the degree and clinical significance of PH in critically ill patients. It can noninvasively visualize cardiac anatomy and certain intracardiac shunts and valvular abnormalities, estimate right atrial and pulmonary arterial pressures, determine the severity of right and left ventricular dysfunction and wall motion abnormalities, and reveal other potential causes of PH. In the absence of pulmonary outflow obstruction, PA systolic pressure is equivalent to right ventricular systolic pressure (RVSP), which can be calculated from measured systolic regurgitant tricuspid flow velocity and estimated right atrial pressure. PH by TTE is usually defined as RVSP greater than 35 mm Hg with the expected upper normal limit up to 40 mm Hg in older or obese subjects.32 However, it has limitations and echocardiography has a 45% false-positive rate of diagnosis when patients subsequently undergo right-sided heart catheterization.33
Among 3790 healthy people who underwent TTE, RVSP was highly variable in the range of 15 to 57 mm Hg and was associated with age, body mass index (BMI), gender, wall thickness, and ejection fraction. An RVSP greater than 40 mm Hg was found in 6% of those older than 50 years and 5% of those with a BMI greater than 30 kg/m2.32 Therefore, not every elevation of RVSP indicates the presence of a pathologic condition. Possible explanations for mildly elevated RVSP detected by TTE include34 (1) overestimation of the RVSP in a patient with true normal pulmonary pressure; (2) serendipitous observation of a transient pressure elevation in an otherwise healthy individual; (3) discovery of stable mild PH, and (4) discovery of early progressive PH.
Echocardiographic signs of significant PH include right ventricular dilation (D-shaped RV) and its hypertrophy (in sustained cases), septal dyskinesia and bowing into the LV during late systole to early diastole, RV hypokinesis, tricuspid regurgitation, right atrial enlargement, and a dilated inferior vena cava.19,35,36 In patients with chronic PH, predictors of poor outcome include right atrial enlargement, septal bowing, and the development of a pericardial effusion.37 Increased RV size combined with increased outflow resistance and reduced ejection fraction have been also described in acute right ventricular failure.19 A specific pattern of right ventricular dysfunction in acute PE has been characterized by a severe hypokinesia of the RV mid-free wall, with normal contractions of the apical segment.38
Images may be suboptimal in critically ill patients because of limitations related to the patient’s general condition, limited positioning, attached monitoring devices, wound dressings, or ventilatory support. Transesophageal echocardiography (TEE) may be more accurate and sensitive in critically ill patients than TTE, especially in acute diseases such as PE when acute PH is highly suspected.39 Newly developed handheld ultrasound devices capable of TEE may sufficiently replace a standard cart-based TEE system in unstable critically ill patients.40 Advanced Doppler echocardiographic techniques allow for comprehensive hemodynamic assessment of the patients with PH. A high correlation between PA catheter and Doppler echocardiography evaluations of cardiac output, transpulmonary gradient, and PVR were observed in patients with severe PH.41
Right-Sided Heart Catheterization
Invasive hemodynamic assessment using right-sided heart catheterization is considered the gold standard for the diagnosis of PH35; however, this procedure must be performed thoroughly and accurately. Besides direct measurement of the hemodynamic parameters, it also provides useful information regarding response to vasodilator therapy. Analysis of mixed venous oxygen saturations during passage of the PA on its way through the cardiac chambers can allow diagnosis of intracardiac shunts. A PCWP measurement reflects left ventricular end-diastolic (filling) pressure. Values less than 15 mm Hg rule out left ventricular, valvular, and pulmonary venous diseases as possible causes of the PH.35 It is important to emphasize that misinterpretation of the PCWP is a common pitfall during right-sided heart catheterization and it should be measured at the end of expiration and in several segments of the pulmonary vasculature because pulmonary veno-occlusive disease can cause elevated wedge pressure only in affected segments.42 In the ICU, placement of a PA catheter for diagnosis and monitoring is highly desirable in patients with severe PH and in patients with progressive heart failure.43 Although there is a little doubt that the hemodynamic data are valuable in the care of critically ill patients with acute conditions complicated by PH, there are no data available on how PA pressure monitoring could affect management and outcome of these patients. Indeed, placing a PA catheter could be a challenging and dangerous procedure in such patient populations. Technical difficulties could be related to severe tricuspid regurgitation, right ventricular dilatation, elevated PAP, and decreased cardiac output. Complications of PA catheterization are particularly dangerous in patients with PH and right ventricular dysfunction/failure. Arrhythmias in response to PA catheterization can have potentially life-threatening consequences by decreasing cardiac output, or converting into fatal ventricular arrhythmias. Obtaining a PCWP may be technically difficult in patients with markedly elevated PAP and also carries a high risk of sometimes fatal pulmonary arterial rupture. Finally, the presence of tricuspid regurgitation can significantly decrease accuracy of cardiac output calculations by thermodilution. Theoretically, the Fick method may be more accurate, but in critically ill patients with increased pulmonary metabolism and high or very low cardiac output, its accuracy may not be optimal.44
Management of Arterial Pulmonary Hypertension and Right Ventricular Failure in the Intensive Care Unit
General Management Principles
In patients with preexisting PH, an acute illness can lead to significant hemodynamic changes with profound and refractory systemic arterial hypotension, mainly secondary to increased PVR associated with decreased cardiac output and decrease systemic vascular resistance (SVR). This would require tight hemodynamic monitoring and aggressive treatment with combinations of pulmonary vasodilators, inotropic agents, and systemic arterial vasoconstrictors to manage acute right ventricular dysfunction and failure and also to maintain coronary and end-organ perfusion.45 The first step is to optimize the volume status and avoid volume overload. Then efforts should be made to achieve the following: improve cardiac output, reduce PVR (by using vasodilators with fewer systemic effects and 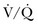 mismatch); treat reversible factors such as hypoxemia, acidosis, anemia; and maintain adequate SVR to ensure end-organ perfusion and adequate coronary filling pressures.46 A general therapeutic approach to pulmonary hypertension in the ICU is outlined in Figure 45-3.
mismatch); treat reversible factors such as hypoxemia, acidosis, anemia; and maintain adequate SVR to ensure end-organ perfusion and adequate coronary filling pressures.46 A general therapeutic approach to pulmonary hypertension in the ICU is outlined in Figure 45-3.
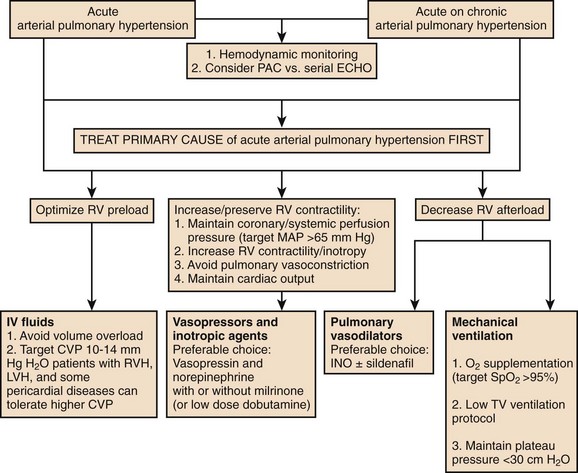
Monitoring
Previous studies have raised concerns and controversy regarding the use of PA catheters and their utility for invasive measurement of cardiac function in critically ill subjects with shock.47,48 However, individuals with severe PH and right ventricular failure are probably best monitored by an invasive method to allow continuous measurement of key hemodynamic values such as cardiac index, mPAP, PVR, and SvO2. General markers of tissue perfusion such as lactate levels and neurohormonal/myocardial markers such as natriuretic peptides and troponin can also be utilized for monitoring.
Fluid Management
Fluid management is a difficult task in these patients because both hypervolemia and hypovolemia can have severe effects on the overall hemodynamic status. With a normal RV, the right ventricular ejection fraction is usually dependent on right ventricular preload.49 However, when right ventricular afterload is increased, even a relatively small increase in blood volume may result in right ventricular dysfunction. Right ventricular dysfunction can occur with volume expansion, despite constant PVR and a decrease in mPAP. Further observations have drawn the conclusion that high right ventricular filling pressure restores normal hemodynamics only if PVR is normal and right ventricular contractility is not markedly reduced.50 In patients with PH, right ventricular dysfunction/failure can reduce left ventricular filling and lead to severe cardiogenic shock. Patients with cardiogenic shock secondary to right ventricular dysfunction usually have a very high (>20 mm Hg) right ventricular filling pressure.51 In addition to decreased right ventricular contractility and cardiac output, right ventricular dilatation can further limit left ventricular filling via ventricular interdependence shifting of the interventricular septum toward the left ventricular cavity. Traditional practice with aggressive fluid resuscitation can thus worsen the patient’s condition. The challenge in fluid management in those patients is to find the optimal right ventricular preload to avoid the detrimental effects of ventricular interdependence on left ventricular function. In the majority of cases (but not in all), right ventricular failure is generally associated with fluid overload and measures should be made to achieve a negative fluid balance.52 Hemodynamic monitoring in patients with right ventricular failure due to acute right ventricular myocardial infarction showed that the cardiac and stroke indexes increased and the RV reached its maximum stroke work index when the filling pressure was 10 to 14 mm Hg. These values may be regarded as the optimal level of RV filling pressure in patients with right ventricular infarction.53 There are no data on optimal right ventricular filling pressure in patients with right ventricular dysfunction secondary to acute PH.
Oxygenation and Ventilatory Support
Adequate oxygenation is a key intervention, as pulmonary arterial hypoxic-induced vasoconstriction is common in these individuals, and might be aggravated also by acidosis as seen in acute and chronic hypercapnia or in severe shock with lactic acidosis. Optimal supplemental oxygen management is an integral component of PH therapy in the ICU.54 One hundred percent oxygen is a selective pulmonary vasodilator in patients with sustained PH, regardless of primary diagnosis, baseline oxygenation, or right ventricular function.55 In patients with ARDS, the vascular response to oxygen was different, and administration of 100% O2 caused the intrapulmonary shunt to deteriorate owing to the collapse of unstable alveolar units with very low ventilation-perfusion ratios. This is in contrast to administration of 100% O2 to patients with COPD, in whom only the dispersion of the blood flow distribution was changed, suggesting release of hypoxic pulmonary vasoconstriction.56
Hypercapnia has been shown to induce PH in animal models. There are no data on how it could affect acute PH in humans. However, a study on healthy volunteers revealed that human pulmonary vascular responses to hypercapnia and hypocapnia consist, respectively, of constriction and dilatation that take 1.5 to 2 hours to reach a steady level. The time courses for recovery in eucapnia are similar. Hypercapnia generated a rise in cardiac output by changing heart rate; hypocapnia produced a fall in cardiac output by changing stroke volume. The finding of marked vasodilatation in response to hypocapnia demonstrates that there is normally a substantial vascular tone in the human pulmonary circulation.57
The management of mechanical ventilation in patients with PH is often challenging due to the effects of positive airway pressure, in addition to the side effects of sedatives. Noninvasive ventilation should initially be considered; however, if intubation is required, careful attention must be paid to the effects of the sedatives on the hemodynamics and to the interaction of the patient with the ventilator. Controlled ventilation alters right ventricular function primarily by increasing right ventricular afterload during the lung inflation period.58 Transpulmonary pressure (and related tidal volume), but not airway pressure itself, was the main determinant factor of right ventricular afterload during mechanical ventilation.59 This supports low-volume strategy in ARDS, recommended as a protective measure for lung parenchyma, which might also represent a protective measure for the RV and pulmonary circulation.59 Frequency of acute right ventricular failure in ARDS patients declined from 61% to 25% over the last 15 to 30 years, which could be explained in part by fundamental alterations in respiratory support and implementation of low tidal volume ventilation.16 Lower incidence of acute right ventricular failure in ARDS patients was associated with lower (<27-30 cm H2O) plateau pressures.21 Right ventricular systolic function was generally negatively affected by positive end-expiratory pressure (PEEP) in ARDS patients undergoing mechanical ventilation. In those patients, PEEP titration significantly affected right ventricular outflow impedance, the lowest values of which were associated with the achieved better total quasi-static lung compliance (calculated by dividing tidal volume by the difference between plateau and end-expiratory airway pressures).60 This suggests that lung hyperinflation along with either inadequate or excessive PEEP can significantly reduce right ventricular systolic function and cardiac output. On other hand, in an experimental study on healthy animals, the open lung concept ventilation resulted in significantly improved lung aeration with no negative effect on right ventricular afterload or left ventricular afterload. This is possibly explained by a loss of hypoxic pulmonary vasoconstriction due to alveolar recruitment. The reductions in the cardiac output and in the mPAP were the consequences of a reduced preload.61 A clinical study in patients after cardiac surgery also found no evidence that ventilation, according to the open lung concept, affects right ventricular afterload.62
Vasopressors and Inotropic Agents
Dobutamine is the most commonly used inotropic agent in patients with PH. It augments myocardial contractility and reduces left ventricular afterload via peripheral vasodilatory effects. In low doses (up to 5 µg/kg/minute) dobutamine decreased PVR, lowered mean systemic arterial pressure, and slightly increased cardiac output63; at doses of 5 to 10 µg/kg/minute, dobutamine caused significant tachycardia and systemic hypotension without improving PVR.63,64 In cases of systemic hypotension patients may require concomitant use of norepinephrine or other peripheral vasoconstrictors to maintain appropriate systemic perfusion pressures. A combination of inhaled nitric oxide (INO) and dobutamine infusion produced an additive effect on pulmonary circulation63 with increased cardiac performance and improved oxygenation65 and had no adverse effects on systemic hemodynamics. In an animal model of acute right ventricular failure secondary to acute PH, dobutamine was superior to norepinephrine in improving right ventricular function by optimizing pulmonary vasodilation and improving right ventricular contractility.63
Norepinephrine (NE) is widely used in critical care settings to treat hemodynamically unstable patients. It exerts significant inotropic effects via β1-receptor agonism and α1-receptor-mediated vasoconstriction. In addition to positive effects on cardiac output and systemic arterial pressure, NE is able to improve the right ventricular oxygen supply/demand ratio,66 although it increases PVR and could worsen PH.67 However, at a lower dose (<7 µg/minute), NE exerts more vasoconstrictive effect in systemic than in pulmonary circulation.67 NE was superior to phenylephrine in restoring systemic arterial pressure, decreasing PVR, augmenting right ventricular myocardial blood flow, and improving cardiac output in animal models of acute PE.68 In patients with chronic PH who developed systemic hypotension following induction of anesthesia, NE in contrast to phenylephrine decreased the ratio of PAP to systemic blood pressure without a change in cardiac index.67 Experimental data also showed that α-adrenergic stimulation can cause a disproportionate rise in PVR,69 which is implicated in the development of acute PH in critical illnesses. Besides this, phenylephrine also causes bradycardia with further adverse consequences on pulmonary and systemic hemodynamics.70 These findings all make NE preferable over phenylephrine for the treatment of hypotension in patients with chronic PH.67 Moreover, NE can be used successfully in combination with selective pulmonary vasodilators for the treatment of patients with acute and chronic PH.45,71
Dopamine produces dose-dependent dopaminergic, β- and α-adrenergic effects on cardiac output and vascular tone. In patients with chronic PH, dopamine infusion led to increased heart rate, mean pulmonary and systemic pressure, and cardiac index with concomitant fall of SVR.72 However, administration of dopamine, similar to epinephrine, is associated with high risk of tachyarrhythmia, with potentially dramatic hemodynamic consequences in patients with severe PH.70 In a small animal study of sepsis-induced PH, epinephrine infusion increased SVR and cardiac output and lowered PVR.73 Isoproterenol is another agent that has positive inotropic and chronotropic effect, which in therapeutic doses increases cardiac output and produces pulmonary and peripheral vasodilation. In animals with acute PH, administration of isoproterenol did not reduce PAP, and instead produced significant tachycardia and was associated with arrhythmias.74
Vasopressin is an endogenous hormone with a weak noradrenergic vasopressor effect on the systemic vasculature and an ability to produce NO-mediated selective pulmonary vasodilation.75 In healthy animals, a linear relationship was observed between vasopressin levels and SVR without significantly affecting PVR or any vascular compliance.74 It was very effective (at a dose of 0.1 U/minute) in treating refractory low SVR hypotension concomitant with PH in postoperative patients.76 Experimental data on the use of vasopressin in acute PH is controversial. In one setting, vasopressin infusion produced significant pulmonary vasodilation,77 although at a higher dose (1.16 U/kg/hour) in another animal model it caused increased PVR and decreased cardiac output with a decrease in right ventricular contractility.78 Studies of the coronary vasoconstriction and the inotropic effect of vasopressin are also controversial, and the effects appear to depend on the dose used and the model studied.79 However, the use of vasopressin at a low dose (0.04 U/minute) produces favorable hemodynamic effects without substantial decline in cardiac output and pulmonary vasoconstriction,79 which makes it a preferable agent for patients with PH.
Levosimendan (a myocardial and vascular calcium sensitizer) besides positive systemic hemodynamic effects also has pulmonary vasodilatory properties and positive inotropic effects on the RV. This makes it particularly effective in treatment of right ventricular dysfunction related to PH.80,81 In this setting, it restored right ventricular function even better than dobutamine, mainly because of more efficient pulmonary vasodilation on top of similar inotropic effects. A pilot prospective randomized controlled study on patients with ARDS in association with septic shock showed that levosimendan improves right ventricular performance mainly through pulmonary vasodilator effects.82 It was also effective in restoring right ventricular function in patients with cardiogenic shock following myocardial infarction.83
Milrinone is a selective phosphodiesterase-3 inhibitor with significant inotropic and vasodilatory effects in both pulmonary and systemic circulation. It is widely used alone or in combinations in patients with decompensated heart failure, particularly in those with nonischemic left ventricular systolic dysfunction,84,85 and those after cardiac surgery.1,86 In animal models of both acute and chronic PH, milrinone significantly reduced PVR and improved right ventricular function.87,88 In combination with INO, milrinone produced additive pulmonary vasodilatation in acute PH.89 In another animal model of PH, addition of sildenafil to milrinone produced more effective pulmonary vasodilation and increased right ventricular contractility without significant systemic hypotension. Cardiac output and right ventricular performance were significantly improved after milrinone alone or in combination with sildenafil, but not with sildenafil alone.90 The systemic vasodilatory effects of milrinone along with its relatively long half-life are the major limiting factors against its utilization in conditions associated with systemic hypotension or hemodynamic instability. Systemic vasopressors (norepinephrine and low-dose vasopressin) were effective in the treatment of the milrinone-induced systemic hypotension. However, a milrinone-vasopressin combination was associated with better hemodynamics than milrinone-norepinephrine during the management of right ventricular failure.91 In a volume-resuscitated pediatric patient with septic shock, a combination of catecholamines with milrinone improved cardiovascular function.92 In animal models, nebulization of milrinone predominantly dilated pulmonary blood vessels, resulting in a reduced pulmonary-to-systemic vascular resistance ratio, improvement in hemodynamic and oxygenation profiles, and prevention of the pulmonary endothelial dysfunction.93,94 Inhaled milrinone was superior to intravenous milrinone in patients with PH after mitral valve replacement surgery; demonstrating less systemic hypotension, more reduction in intrapulmonary shunt fraction, and improvement in PaO2/FIO2 ratio.86
Pulmonary Vasodilators
Reduction of right ventricular afterload is an important part in the management of a patient with PH. Most pulmonary vasodilators can be subdivided according to their action on the pulmonary vasculature via either cyclic guanosine monophosphate, prostacyclin, or endothelin pathways.1 Used in stable patients either as monotherapy or in combination, they were able to reduce mortality rates and improve multiple other clinical and hemodynamic outcomes.95,96 Not all of them can be utilized in a critical care setting: the route of administration, half-life, and systemic effects could significantly affect the choice of the medication. Pulmonary vasodilators with longer half-lives or significant systemic hemodynamic effects can decrease organ perfusion pressure and reduce right coronary blood flow with further deterioration of right ventricular performance.
Inhaled Nitric Oxide
INO is a potent vasodilator able to lower PAP and right ventricular oxygen demand.88 This improves cardiac performance without altering right ventricular contractility and cardiac output in hemodynamically stable patients with a variety of causes of PH.87,89,97 INO also dilates pulmonary vasculature in ventilated lung areas, thereby improving 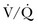 match and oxygenation.91 It has a very short half-life and almost no systemic vasodilatory effects, which is very important in the treatment of hemodynamically unstable patients. Multiple studies on INO utilization in ARDS patients showed improvement in oxygenation for 24 to 48 hours and variable improvement in mPAP.92 A combination of INO and other interventions, such as PEEP and prone positioning, also had beneficial and additive effects on arterial oxygenation.93 However, INO had no favorable impact on the duration of ventilatory support or mortality rate in this patient population.93,98 In cardiac surgery patients with a history of PH, INO was associated with lower heart rates, higher RV ejection fraction, and a lower requirement for vasopressor agents compared to milrinone.99 INO has been successfully used and it was associated with improved outcomes in critically ill postoperative patients who developed severe PH or right ventricular failure.95,100 A combination treatment of INO with other pulmonary vasodilators (milrinone, sildenafil, etc.) has been successfully used in management of acute PH in different conditions.89,101 Besides its high applicability in many acute conditions, INO could be delivered in spontaneously breathing individuals by face mask or even nasal cannula as it was shown in pediatric practice.102 INO treatment is associated with some side effects including methemoglobinemia91,96 and NO2 formation.103 Abrupt withdrawal of INO has been associated with rebound PH, a significant drop in PaO2, and life-threatening hemodynamic deterioration.96,104 Close monitoring and gradual discontinuation are important to prevent and detect rebound PH.
match and oxygenation.91 It has a very short half-life and almost no systemic vasodilatory effects, which is very important in the treatment of hemodynamically unstable patients. Multiple studies on INO utilization in ARDS patients showed improvement in oxygenation for 24 to 48 hours and variable improvement in mPAP.92 A combination of INO and other interventions, such as PEEP and prone positioning, also had beneficial and additive effects on arterial oxygenation.93 However, INO had no favorable impact on the duration of ventilatory support or mortality rate in this patient population.93,98 In cardiac surgery patients with a history of PH, INO was associated with lower heart rates, higher RV ejection fraction, and a lower requirement for vasopressor agents compared to milrinone.99 INO has been successfully used and it was associated with improved outcomes in critically ill postoperative patients who developed severe PH or right ventricular failure.95,100 A combination treatment of INO with other pulmonary vasodilators (milrinone, sildenafil, etc.) has been successfully used in management of acute PH in different conditions.89,101 Besides its high applicability in many acute conditions, INO could be delivered in spontaneously breathing individuals by face mask or even nasal cannula as it was shown in pediatric practice.102 INO treatment is associated with some side effects including methemoglobinemia91,96 and NO2 formation.103 Abrupt withdrawal of INO has been associated with rebound PH, a significant drop in PaO2, and life-threatening hemodynamic deterioration.96,104 Close monitoring and gradual discontinuation are important to prevent and detect rebound PH.
Prostanoids
Prostaglandin E1 and prostacyclin are able to produce significant pulmonary vasodilatation and lower mPAP. They also possess antithrombotic, antiproliferative, and anti-inflammatory properties. Intravenous prostacyclin is highly effective in cases of severe PH and is also associated with a significant survival benefit.105,106 Prostacyclin and its analogs (iloprost, epoprostenol, and treprostinil) in inhaled forms are as effective as intravenous forms in patients with chronic PH in different clinical settings.107,108 All prostanoids have relatively long half-lives and can significantly affect systemic hemodynamics, which limits their use in critically ill or hemodynamically unstable patients.
In the ICU setting, inhaled prostaglandins have been used to treat severe sustained PH and intractable hypoxia, but were associated with systemic hypotension. Inhaled iloprost effectively decreased mPAP and improved RV performance immediately after separation from cardiopulmonary bypass.109 Inhaled iloprost was also successfully used in the treatment of acute right ventricular dysfunction in the setting of preexisting PH in heart transplant recipients during weaning from cardiopulmonary bypass circulation and was not associated with significant systemic side effects. Intravenous prostacyclin (prostaglandin I2 [PGI2]) in combination with norepinephrine and dopamine was effective in the treatment of protamine-mediated acute PH and right ventricular failure in the setting of open heart surgery.110 Inhaled prostacyclin showed different effects on oxygenation and pulmonary hemodynamics in patients presenting with primary pulmonary ARDS (reduction in PaO2/FIO2) compared with an extrapulmonary cause of ARDS (increase in PaO2/FIO2 along with a decrease in mPAP). Despite the observation that RV ejection fraction increased on INO, but not with PGI2, both INO and PGI2-aerosol showed beneficial effects on RV performance and may prove helpful in the treatment of acute PH.104
Phosphodiesterase-5 Inhibitors
Sildenafil is a specific phosphodiesterase-5 inhibitor with sustained pulmonary vasodilatory effect and the ability to lower PVR and mPAP and to increase cardiac output in patients with different forms of chronic PH,111,112 including patients with PH secondary to congestive heart failure.101 In the chronic setting, it is highly effective alone or in combination with other pulmonary vasodilators: epoprostenol,113 iloprost,114,115 BNP,116 and INO.117 It was able to improve central hemodynamics and right ventricular function in ventilated patients with PH who required dobutamine administration.118 Its ability to augment and prolong the hemodynamic effects of other pulmonary vasodilators has been successfully used to minimize rebound PH after INO discontinuation,119 in weaning from intravenous vasodilators in patients after cardiac surgery101 as well as in chronic PH therapy,103 and in severe right ventricular dysfunction related to chronic PH.119 In an animal model of another specific phosphodiesterase-5 inhibitor, zaprinast was more effective than milrinone in the management of acute PH, causing dose-dependent pulmonary vasodilation without significant systemic hypotension.120 Intravenous zaprinast may also increase the efficacy and prolong the duration of action of INO; however, its efficacy in lung injury models was uncertain: nonselective vasodilation induced by intravenously administered zaprinast not only worsens gas exchange but also abolishes the beneficial effects of INO.120 Subthreshold doses (which did not reduce mPAP) of zaprinast and sildenafil in patients with acute lung injury associated with PH improved responsiveness to INO.121
Calcium channel blockers have been used in hemodynamically stable patients with PAH who demonstrated a pulmonary vascular response to acute vasodilator challenge.42 There are limited data on utilization of calcium channel blockers in critically ill patients with acute PH. Acute administration of nifedipine did not cause pulmonary vasodilatation, and in contrast led to increased right ventricular end-diastolic pressure and decreased right ventricular contractility.106 Prolonged half-life and negative inotropic effects, which may precipitate fatal worsening of right ventricular failure, limit the use of calcium channel blockers in treatment of acute PH.
Endothelin receptor antagonists appear to be highly effective in the treatment of PH in the outpatient settings.42 They have not been adequately studied in acute care settings. In an animal model, pretreatment with bosentan completely abolished endotoxin-induced acute PH and changes in pulmonary compliance and resistance.3 Currently, the only possible implication in critically ill patients would be weaning or conversion from inhaled or intravenous pulmonary vasodilators to oral medication (bosentan and ambrisentan). There also seems to be a role for ambrisentan in the management of portopulmonary hypertension patients awaiting liver transplantation.122
Natriuretic peptides (atrial natriuretic peptide [ANP] and BNP) have diuretic and vasorelaxing properties and are able to counteract the renin-angiotensin system.123 In patients with right ventricular failure, they produced dose-related pulmonary vasodilatation without worsening oxygen saturation or systemic hemodynamic and exerted favorable neurohormonal effects by suppressing aldosterone. BNP significantly attenuated the mPAP and acute hypoxic pulmonary vasoconstriction in otherwise healthy individuals,124 but had no effect on mPAP or PVR in patients with PH. Use of nesiritide in hemodynamically unstable and hypotensive patients is limited, mainly because of reduction of SVR and resulting profound systemic hypotension.125
Nitroprusside and nitroglycerin both can cause significant pulmonary vasodilation similar to that of INO. However, nitroprusside has a longer half-life and can cause acute reduction of SVR leading to systemic hypotension, limiting its use in hemodynamically unstable and hypotensive patients.87 A significant venodilator effect of nitroglycerin decreases right ventricular preload leading to adverse consequences in patients with right ventricular failure. Inhaled nitroglycerin may be a safer therapeutic option without adverse effect on systemic hemodynamic parameters.126
Intravenous adenosine is a pulmonary vasodilator with a very short half-life (6-10 seconds) and can be effective for short-term lowering of PVR.105 In the setting of acute PH, adenosine infusion may help lower mPAP without systemic hypotension, which can reverse the clinical state of shock by achieving pulmonary vasodilatation.127 At higher doses (70 to 100 mg/kg/minute) it caused systemic vasodilation.113 Adenosine is also successfully used to treat persistent PH of the newborn.128
Dipyridamole inhibits phosphodiesterase-5 and thromboxane synthase, which explains its strong vasodilatory effect on pulmonary circulation. Dipyridamole can lower PVR, augment INO-induced pulmonary vasodilation,114 acutely attenuate the adverse hemodynamic effects of rapid withdrawal of INO therapy, and attenuate excessive hypoxic pulmonary vasoreactivity. In pediatric patients with PH, dipyridamole was as effective as INO, but caused significant systemic vasodilatation.115 Intravenous dipyridamole was used in acute management of PH in combination either with INO or intravenous nitroglycerin,129 as well as for diagnostic purposes to identify reversibility of PH in potential cardiac transplant recipients with heart failure in whom a pulmonary vasodilator response to inhalation of INO alone was not observed.130
Other Therapies and Mechanical Cardiovascular Support
The observations of survival advantage in patients with PH and a patent foramen ovale suggested that an intra-atrial right-to-left shunt could decompress the RV and increase left ventricular preload, thereby increasing systemic blood flow and improving systemic oxygen transport despite arterial oxygen desaturation. Atrial septostomy has been developed as an alternative/bridge treatment and applied in patients with lack of response to medical therapy in the absence of other surgical treatment options. It has substantial morbidity and mortality rates in critically ill patients with severe right ventricular failure.131 With growing experience, procedure-related death rates have been reduced to 5.4%, and the most suitable patient group has been identified among patients with a mean right atrial pressure between 10 and 20 mm Hg.132 Acute right ventricular failure after orthotopic heart transplantation was successfully managed by decompression of the RV through the patent foramen ovale of the donor heart and inhalation of iloprost.133 Both pericardiectomy and creation of atrial septal defects have been used in extreme cases of acute right ventricular failure secondary to acute myocardial infarction.134 Decompression of the RV through the septostomy may potentially be an effective alternative in the management of severe acute PH in the ICU setting. The defect could be subsequently closed using a transcatheter septal occlusion device after the patient’s condition has been stabilized.
The treatment of choice for PH secondary to chronic thromboembolic disease is pulmonary thromboendarterectomy, which can be performed when organized thrombus is in the proximal vessels135; significant hemodynamic improvement has been reported after such intervention.
Mechanical support for the RV with RV-assist devices (RVAD) could be a reasonable option for reversible cases or as a bridge to final treatment (transplantation). In cases of acute right ventricular failure after heart transplantation, use of RVADs, extracorporeal membrane oxygenation (ECMO), femoral vein–to–femoral artery roller, or centrifugal pumps may facilitate hemodynamic stability until the transplanted heart has recovered or until a new heart has been found for retransplantation.136 These devices could be successfully applied in other cases of potentially reversible acute PH and right ventricular failure. Intra-aortic balloon counterpulsation (IABP) has long been the mainstay of mechanical therapy for cardiogenic shock; however, not every patient has a hemodynamic response to IABP.134 In patients with acute PH and right ventricular failure associated with systemic hypotension, IABP could improve coronary and peripheral perfusion and augment left ventricular performance with an acute decrease in afterload.137
The use of extracorporeal life support, such as venoarterial ECMO, and pumpless lung assist devices have become the preferred bridging strategy for patients with severe right ventricular failure52 who are candidates for transplantation. Heart-lung and lung transplantation has been an option for the therapy of select patients with end-stage PH for the past 25 years. Currently, approximately 4% of the approximately 1700 single lung, double lung, and combined heart and lung transplants annually performed worldwide in adults are for the primary indication of PH.138 Their long-term outcomes are comparable with patients with other primary indications for transplant.138
Prognosis
Despite significant advances made in the therapy of PH, this condition continues to have a poor survival both in the chronic and in the acute setting. The final common pathway is development of right ventricular failure and subsequent multiorgan failure. The natural history of chronic PH was well documented by the National Institutes of Health (NIH) Registry in which 194 patients with IPAH were enrolled in a multicenter observational study from 1981 through 1985.139 The estimated median survival in this registry was 2.8 years with 1-, 3-, and 5-year survival rates of 68%, 48%, and 34%, respectively. Studies from other countries, including Japan, India, and Mexico, have shown similar results, with a median survival estimate of 2 to 3 years. More recently, two large registries have provided more contemporary data regarding prognosis of patients with chronic PH. The French Registry recently characterized survival and important prognostic indicators in PH patients. This registry demonstrated that the survival of PH patients has improved compared to the predicted survival based on the NIH Registry, although it still remains suboptimal with 1-, 2-, and 3-year survival rates of 85.7%, 69.5%, and 54.9% for incident cases.140 In a recent large United States–based registry (REVEAL registry), important prognostic variables found to be predictors of outcome in this study included cause of PH, functional class, gender, exercise tolerance, and hemodynamics that reflect right ventricular function.141 Prognosis is also influenced by an underlying cause; for example, in PH associated with scleroderma, its prognosis appears to be worse than for IPAH, and the untreated 2-year survival rate may be as low as 40%.142 The presence of PH and associated right ventricular failure in the ICU is associated with worse outcomes. In the settings of both COPD and ARDS, the presence of right ventricular failure contributes significantly to shortened survival.143 The 3-year survival rate in patients with severe airflow obstruction and a PVR three to four times normal is less than 10% to 15%.143,144 Death from PH depends mainly on the state of the RV, and patients with symptoms of severe right-sided heart dysfunction, such as syncope, and hemodynamic evidence of impaired right ventricular function, such as reduced cardiac output or mixed venous saturation and elevated right atrial pressure, usually surrender to the disease within 1 to 2 years.139 It is very important to discuss the prognosis and the possibility of transplantation in addition to end-of-life care with these patients and their family members before they present to the ICU. It is also very important to take into account the limitations of current therapies and the complexity of this lethal disease.
References
1. Humbert, M, Morrell, NW, Archer, SL, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004; 43:13S–24S.
2. Moloney, ED, Evans, TW. Pathophysiology and pharmacological treatment of pulmonary hypertension in acute respiratory distress syndrome. Eur Respir J. 2003; 21:720–727.
3. Albertini, M, Ciminaghi, B, Mazzola, S, Clement, MG. Improvement of respiratory function by bosentan during endotoxic shock in the pig. Prostaglandins Leukot Essent Fatty Acids. 2001; 65:103–108.
4. Wort, SJ, Evans, TW. The role of the endothelium in modulating vascular control in sepsis and related conditions. Br Med Bull. 1999; 55:30–48.
5. Lambermont, B, Kolh, P, Detry, O, et al. Analysis of endotoxin effects on the intact pulmonary circulation. Cardiovasc Res. 1999; 41:275–281.
6. Lambermont, B, Ghuysen, A, Kolh, P, et al. Effects of endotoxic shock on right ventricular systolic function and mechanical efficiency. Cardiovasc Res. 2003; 59:412–418.
7. Greyson, CR. The right ventricle and pulmonary circulation: Basic concepts. Rev Esp Cardiol. 2010; 63:81–95.
8. Bronicki, RA, Baden, HP. Pathophysiology of right ventricular failure in pulmonary hypertension. Pediatr Crit Care Med. 2010; 11:S15–S22.
9. Blaise, G, Langleben, D, Hubert, B. Pulmonary arterial hypertension: Pathophysiology and anesthetic approach. Anesthesiology. 2003; 99:1415–1432.
10. Chin, KM, Kim, NH, Rubin, LJ. The right ventricle in pulmonary hypertension. Coron Artery Dis. 2005; 16:13–18.
11. Nath, J, Foster, E, Heidenreich, PA. Impact of tricuspid regurgitation on long-term survival. J Am Coll Cardiol. 2004; 43:405–409.
12. Pinsky, MR. Recent advances in the clinical application of heart-lung interactions. Curr Opin Crit Care. 2002; 8:26–31.
13. Hoffman, D, Sisto, D, Frater, RW, Nikolic, SD. Left-to-right ventricular interaction with a noncontracting right ventricle. J Thorac Cardiovasc Surg. 1994; 107:1496–1502.
14. Simonneau, G, Robbins, IM, Beghetti, M, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009; 54:S43–S54.
15. Hoeper, MM, Galie, N, Murali, S, et al. Outcome after cardiopulmonary resuscitation in patients with pulmonary arterial hypertension. Am J Respir Crit Care Med. 2002; 165:341–344.
16. Vieillard-Baron, A, Schmitt, JM, Augarde, R, et al. Acute cor pulmonale in acute respiratory distress syndrome submitted to protective ventilation: Incidence, clinical implications, and prognosis. Crit Care Med. 2001; 29:1551–1555.
17. Vieillard-Baron, A, Page, B, Augarde, R, et al. Acute cor pulmonale in massive pulmonary embolism: Incidence, echocardiographic pattern, clinical implications and recovery rate. Intensive Care Med. 2001; 27:1481–1486.
18. Kasper, W, Konstantinides, S, Geibel, A, et al. Management strategies and determinants of outcome in acute major pulmonary embolism: Results of a multicenter registry. J Am Coll Cardiol. 1997; 30:1165–1171.
19. Vieillard-Baron, A, Prin, S, Chergui, K, et al. Echo-Doppler demonstration of acute cor pulmonale at the bedside in the medical intensive care unit. Am J Respir Crit Care Med. 2002; 166:1310–1319.
20. Leeman, M. Pulmonary hypertension in acute respiratory distress syndrome. Monaldi Arch Chest Dis. 1999; 54:146–149.
21. Jardin, F, Vieillard-Baron, A. Is there a safe plateau pressure in ARDS? The right heart only knows. Intensive Care Med. 2007; 33:444–447.
22. Sibbald, WJ, Paterson, NA, Holliday, RL, et al. Pulmonary hypertension in sepsis: Measurement by the pulmonary arterial diastolic-pulmonary wedge pressure gradient and the influence of passive and active factors. Chest. 1978; 73:583–591.
23. Bernstein, AD, Parsonnet, V. Bedside estimation of risk as an aid for decision-making in cardiac surgery. Ann Thorac Surg. 2000; 69:823–828.
24. Subramaniam, K, Yared, JP. Management of pulmonary hypertension in the operating room. Semin Cardiothorac Vasc Anesth. 2007; 11:119–136.
25. Wynne, R, Botti, M. Postoperative pulmonary dysfunction in adults after cardiac surgery with cardiopulmonary bypass: Clinical significance and implications for practice. Am J Crit Care. 2004; 13:384–393.
26. Ocal, A, Kiris, I, Erdinc, M, et al. Efficiency of prostacyclin in the treatment of protamine-mediated right ventricular failure and acute pulmonary hypertension. Tohoku J Exp Med. 2005; 207:51–58.
27. Milot, J, Perron, J, Lacasse, Y, et al. Incidence and predictors of ARDS after cardiac surgery. Chest. 2001; 119:884–888.
28. Mehta, NJ, Jani, K, Khan, IA. Clinical usefulness and prognostic value of elevated cardiac troponin I levels in acute pulmonary embolism. Am Heart J. 2003; 145:821–825.
29. La Vecchia, L, Ottani, F, Favero, L, et al. Increased cardiac troponin I on admission predicts in-hospital mortality in acute pulmonary embolism. Heart. 2004; 90:633–637.
30. Leuchte, HH, Holzapfel, M, Baumgartner, RA, et al. Clinical significance of brain natriuretic peptide in primary pulmonary hypertension. J Am Coll Cardiol. 2004; 43:764–770.
31. Nagaya, N, Nishikimi, T, Uematsu, M, et al. Plasma brain natriuretic peptide as a prognostic indicator in patients with primary pulmonary hypertension. Circulation. 2000; 102:865–870.
32. McQuillan, BM, Picard, MH, Leavitt, M, Weyman, AE. Clinical correlates and reference intervals for pulmonary artery systolic pressure among echocardiographically normal subjects. Circulation. 2001; 104:2797–2802.
33. Hachulla, E, Gressin, V, Guillevin, L, et al. Early detection of pulmonary arterial hypertension in systemic sclerosis: A French nationwide prospective multicenter study. Arthritis Rheum. 2005; 52:3792–3800.
34. Barst, RJ, McGoon, M, Torbicki, A, et al. Diagnosis and differential assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2004; 43:40S–47S.
35. Chemla, D, Castelain, V, Herve, P, et al. Haemodynamic evaluation of pulmonary hypertension. Eur Respir J. 2002; 20:1314–1331.
36. Bossone, E, Bodini, BD, Mazza, A, Allegra, L. Pulmonary arterial hypertension: The key role of echocardiography. Chest. 2005; 127:1836–1843.
37. Raymond, RJ, Hinderliter, AL, Willis, PW, et al. Echocardiographic predictors of adverse outcomes in primary pulmonary hypertension. J Am Coll Cardiol. 2002; 39:1214–1219.
38. McConnell, MV, Solomon, SD, Rayan, ME, et al. Regional right ventricular dysfunction detected by echocardiography in acute pulmonary embolism. Am J Cardiol. 1996; 78:469–473.
39. Karski, JM. Transesophageal echocardiography in the intensive care unit. Semin Cardiothorac Vasc Anesth. 2006; 10:162–166.
40. Schneider, C, Schwemmer, U, Kredel, M, et al. Handheld vs. conventional transesophageal echocardiography in non-cardiac surgical intensive care unit patients. Ultraschall Med. 2008; 29(5):531–534.
41. Selimovic, N, Rundqvist, B, Bergh, CH, et al. Assessment of pulmonary vascular resistance by Doppler echocardiography in patients with pulmonary arterial hypertension. J Heart Lung Transplant. 2007; 26:927–934.
42. McLaughlin, VV, Davis, M, Cornwell, W. Pulmonary arterial hypertension. Curr Probl Cardiol. 2011; 36:461–517.
43. Fincke, R, Hochman, JS, Lowe, AM, et al. Cardiac power is the strongest hemodynamic correlate of mortality in cardiogenic shock: A report from the SHOCK trial registry. J Am Coll Cardio. 2004; 44:340–348.
44. Caruso, LJ, Layon, AJ, Gabrielli, A. What is the best way to measure cardiac output? Who cares, anyway? Chest. 2002; 122:771–774.
45. Tsapenko, MV, Tsapenko, AV, Comfere, TB, et al. Arterial pulmonary hypertension in noncardiac intensive care unit. Vasc Health Risk Manage. 2008; 4:1043–1060.
46. Price, LC, Wort, SJ, Finney, SJ, et al. Pulmonary vascular and right ventricular dysfunction in adult critical care: Current and emerging options for management: A systematic literature review. Crit Care. 2010; 14:R169.
47. Harvey, S, Harrison, DA, Singer, M, et al. Assessment of the clinical effectiveness of pulmonary artery catheters in management of patients in intensive care (PAC-Man): A randomised controlled trial. Lancet. 2005; 366:472–477.
48. Sandham, JD, Hull, RD, Brant, RF, et al. A randomized, controlled trial of the use of pulmonary-artery catheters in high-risk surgical patients. N Engl J Med. 2003; 348:5–14.
49. Reuse, C, Vincent, JL, Pinsky, MR. Measurements of right ventricular volumes during fluid challenge. Chest. 1990; 98:1450–1454.
50. Via, G, Braschi, A. Pathophysiology of severe pulmonary hypertension in the critically ill patient. Minerva Anestesiol. 2004; 70:233–237.
51. Jacobs, AK, Leopold, JA, Bates, E, et al. Cardiogenic shock caused by right ventricular infarction: A report from the SHOCK registry. J Am Coll Cardiol. 2003; 41:1273–1279.
52. Hoeper, MM, Granton, J. Intensive care unit management of patients with severe pulmonary hypertension and right heart failure. Am J Respir Crit Care Med. 2011; 184:1114–1124.
53. Berisha, S, Kastrati, A, Goda, A, Popa, Y. Optimal value of filling pressure in the right side of the heart in acute right ventricular infarction. Br Heart J. 1990; 63:98–102.
54. Zamanian, RT, Haddad, F, Doyle, RL, Weinacker, AB. Management strategies for patients with pulmonary hypertension in the intensive care unit. Crit Care Med. 2007; 35:2037–2050.
55. Roberts, DH, Lepore, JJ, Maroo, A, et al. Oxygen therapy improves cardiac index and pulmonary vascular resistance in patients with pulmonary hypertension. Chest. 2001; 120:1547–1555.
56. Santos, C, Ferrer, M, Roca, J, et al. Pulmonary gas exchange response to oxygen breathing in acute lung injury. Am J Respir Crit Care Med. 2000; 161:26–31.
57. Balanos, GM, Talbot, NP, Dorrington, KL, Robbins, PA. Human pulmonary vascular response to 4 h of hypercapnia and hypocapnia measured using Doppler echocardiography. J Appl Physiol. 2003; 94:1543–1551.
58. Jardin, F, Delorme, G, Hardy, A, et al. Reevaluation of hemodynamic consequences of positive pressure ventilation: Emphasis on cyclic right ventricular afterloading by mechanical lung inflation. Anesthesiology. 1990; 72:966–970.
59. Vieillard-Baron, A, Loubieres, Y, Schmitt, JM, et al. Cyclic changes in right ventricular output impedance during mechanical ventilation. J Appl Physiol. 1999; 87:1644–1650.
60. Schmitt, JM, Vieillard-Baron, A, Augarde, R, et al. Positive end-expiratory pressure titration in acute respiratory distress syndrome patients: Impact on right ventricular outflow impedance evaluated by pulmonary artery Doppler flow velocity measurements. Crit Care Med. 2001; 29:1154–1158.
61. Miranda, DR, Klompe, L, Cademartiri, F, et al. The effect of open lung ventilation on right ventricular and left ventricular function in lung-lavaged pigs. Crit Care (London, England). 2006; 10:R86.
62. Reis Miranda, D, Gommers, D, Struijs, A, et al. The open lung concept: Effects on right ventricular afterload after cardiac surgery. Br J Anaesth. 2004; 93:327–332.
63. Kerbaul, F, Rondelet, B, Motte, S, et al. Effects of norepinephrine and dobutamine on pressure load-induced right ventricular failure. Crit Care Med. 2004; 32:1035–1040.
64. Bradford, KK, Deb, B, Pearl, RG. Combination therapy with inhaled nitric oxide and intravenous dobutamine during pulmonary hypertension in the rabbit. J Cardiovasc Pharmacol. 2000; 36:146–151.
65. Vizza, CD, Rocca, GD, Roma, AD, et al. Acute hemodynamic effects of inhaled nitric oxide, dobutamine and a combination of the two in patients with mild to moderate secondary pulmonary hypertension. Crit Care (London, England). 2001; 5:355–361.
66. Schreuder, WO, Schneider, AJ, Groeneveld, AB, Thijs, LG. Effect of dopamine vs norepinephrine on hemodynamics in septic shock. Emphasis on right ventricular performance. Chest. 1989; 95:1282–1288.
67. Kwak, YL, Lee, CS, Park, YH, Hong, YW. The effect of phenylephrine and norepinephrine in patients with chronic pulmonary hypertension. Anaesthesia. 2002; 57:9–14.
68. Hirsch, LJ, Rooney, MW, Wat, SS, et al. Norepinephrine and phenylephrine effects on right ventricular function in experimental canine pulmonary embolism. Chest. 1991; 100:796–801.
69. Brimioulle, S, Vachiery, JL, Brichant, JF, et al. Sympathetic modulation of hypoxic pulmonary vasoconstriction in intact dogs. Cardiovasc Res. 1997; 34:384–392.
70. Tisdale, JE, Patel, RV, Webb, CR, et al. Proarrhythmic effects of intravenous vasopressors. Ann Pharmacother. 1995; 29:269–281.
71. Tritapepe, L, Voci, P, Cogliati, AA, et al. Successful weaning from cardiopulmonary bypass with central venous prostaglandin E1 and left atrial norepinephrine infusion in patients with acute pulmonary hypertension. Crit Care Med. 1999; 27:2180–2183.
72. Holloway, EL, Polumbo, RA, Harrison, DC. Acute circulatory effects of dopamine in patients with pulmonary hypertension. Br Heart J. 1975; 37:482–485.
73. Meadow, WL, Rudinsky, BF, Strates, E. Selective elevation of systemic blood pressure by epinephrine during sepsis-induced pulmonary hypertension in piglets. Pediatr Res. 1986; 20:872–875.
74. Wallace, AW, Tunin, CM, Shoukas, AA. Effects of vasopressin on pulmonary and systemic vascular mechanics. Am J Physiol. 1989; 257:H1228–H1234.
75. Evora, PR, Pearson, PJ, Schaff, HV. Arginine vasopressin induces endothelium-dependent vasodilatation of the pulmonary artery. V1-receptor-mediated production of nitric oxide. Chest. 1993; 103:1241–1245.
76. Tayama, E, Ueda, T, Shojima, T, et al. Arginine vasopressin is an ideal drug after cardiac surgery for the management of low systemic vascular resistant hypotension concomitant with pulmonary hypertension. Interact Cardiovasc Thorac Surg. 2007; 6:715–719.
77. Trempy, GA, Nyhan, DP, Murray, PA. Pulmonary vasoregulation by arginine vasopressin in conscious, halothane-anesthetized, and pentobarbital-anesthetized dogs with increased vasomotor tone. Anesthesiology. 1994; 81:632–640.
78. Leather, HA, Segers, P, Berends, N, et al. Effects of vasopressin on right ventricular function in an experimental model of acute pulmonary hypertension. Crit Care Med. 2002; 30:2548–2552.
79. Holmes, CL, Landry, DW, Granton, JT. Science review: Vasopressin and the cardiovascular system. Part 2—Clinical physiology. Crit Care (London, England). 2004; 8:15–23.
80. Kerbaul, F, Gariboldi, V, Giorgi, R, et al. Effects of levosimendan on acute pulmonary embolism-induced right ventricular failure. Crit Care Med. 2007; 35:1948–1954.
81. Missant, C, Rex, S, Segers, P, Wouters, PF. Levosimendan improves right ventriculovascular coupling in a porcine model of right ventricular dysfunction. Crit Care Med. 2007; 35:707–715.
82. Morelli, A, Teboul, JL, Maggiore, SM, et al. Effects of levosimendan on right ventricular afterload in patients with acute respiratory distress syndrome: A pilot study. Crit Care Med. 2006; 34:2287–2293.
83. Russ, MA, Prondzinsky, R, Carter, JM, et al. Right ventricular function in myocardial infarction complicated by cardiogenic shock: Improvement with levosimendan. Crit Care Med. 2009; 37:3017–3023.
84. Felker, GM, Benza, RL, Chandler, AB, et al. Heart failure etiology and response to milrinone in decompensated heart failure: Results from the OPTIME-CHF study. J Am Coll Cardiol. 2003; 41:997–1003.
85. Mehra, MR, Ventura, HO, Kapoor, C, et al. Safety and clinical utility of long-term intravenous milrinone in advanced heart failure. Am J Cardiol. 1997; 80:61–64.
86. Wang, H, Gong, M, Zhou, B, Dai, A. Comparison of inhaled and intravenous milrinone in patients with pulmonary hypertension undergoing mitral valve surgery. Adv Ther. 2009; 26:462–468.
87. Cockrill, BA, Kacmarek, RM, Fifer, MA, et al. Comparison of the effects of nitric oxide, nitroprusside, and nifedipine on hemodynamics and right ventricular contractility in patients with chronic pulmonary hypertension. Chest. 2001; 119:128–136.
88. Zwissler, B, Welte, M, Messmer, K. Effects of inhaled prostacyclin as compared with inhaled nitric oxide on right ventricular performance in hypoxic pulmonary vasoconstriction. J Cardiothorac Vasc Anesth. 1995; 9:283–289.
89. Bhorade, S, Christenson, J, O’Connor, M, et al. Response to inhaled nitric oxide in patients with acute right heart syndrome. Am J Respir Crit Care Med. 1999; 159:571–579.
90. Lobato, EB, Beaver, T, Muehlschlegel, J, et al. Treatment with phosphodiesterase inhibitors type III and V: Milrinone and sildenafil is an effective combination during thromboxane-induced acute pulmonary hypertension. Br J Anaesth. 2006; 96:317–322.
91. Pepke-Zaba, J, Higenbottam, TW, Dinh-Xuan, AT, et al. Inhaled nitric oxide as a cause of selective pulmonary vasodilatation in pulmonary hypertension. Lancet. 1991; 338:1173–1174.
92. McIntyre, RC, Jr., Moore, FA, Moore, EE, et al. Inhaled nitric oxide variably improves oxygenation and pulmonary hypertension in patients with acute respiratory distress syndrome. J Trauma. 1995; 39:418–425.
93. Kaisers, U, Busch, T, Deja, M, et al. Selective pulmonary vasodilation in acute respiratory distress syndrome. Crit Care Med. 2003; 31:S337–S342.
94. Lamarche, Y, Malo, O, Thorin, E, et al. Inhaled but not intravenous milrinone prevents pulmonary endothelial dysfunction after cardiopulmonary bypass. J Thorac Cardiovasc Surg. 2005; 130:83–92.
95. George, I, Xydas, S, Topkara, VK, et al. Clinical indication for use and outcomes after inhaled nitric oxide therapy. Ann Thorac Surg. 2006; 82:2161–2169.
96. Troncy, E, Francoeur, M, Blaise, G. Inhaled nitric oxide: Clinical applications, indications, and toxicology. Can J Anaesth. 1997; 44:973–988.
97. Fierobe, L, Brunet, F, Dhainaut, JF, et al. Effect of inhaled nitric oxide on right ventricular function in adult respiratory distress syndrome. Am J Respir Crit Care Med. 1995; 151:1414–1419.
98. Taylor, RW, Zimmerman, JL, Dellinger, RP, et al. Low-dose inhaled nitric oxide in patients with acute lung injury: A randomized controlled trial. JAMA. 2004; 291:1603–1609.
99. Solina, A, Papp, D, Ginsberg, S, et al. A comparison of inhaled nitric oxide and milrinone for the treatment of pulmonary hypertension in adult cardiac surgery patients. J Cardiothorac Vasc Anesth. 2000; 14:12–17.
100. Della Rocca, G, Coccia, C. Nitric oxide in thoracic surgery. Minerva Anestesiol. 2005; 71:313–318.
101. Trachte, AL, Lobato, EB, Urdaneta, F, et al. Oral sildenafil reduces pulmonary hypertension after cardiac surgery. Ann Thorac Surg. 2005; 79:194–197.
102. Scheurer, M, Bandisode, V, Atz, AM. Simplified pulmonary vasodilatory testing in the cardiac catheterization laboratory with nasal cannula nitric oxide. Pediatr Cardiol. 2006; 27:84–86.
103. Johnson, RF, Loyd, JE, Mullican, AL, et al. Long-term follow-up after conversion from intravenous epoprostenol to oral therapy with bosentan or sildenafil in 13 patients with pulmonary arterial hypertension. J Heart Lung Transplant. 2007; 26:363–369.
104. Christenson, J, Lavoie, A, O’Connor, M, et al. The incidence and pathogenesis of cardiopulmonary deterioration after abrupt withdrawal of inhaled nitric oxide. Am J Respir Crit Care Med. 2000; 161:1443–1449.
105. McLaughlin, VV, Genthner, DE, Panella, MM, Rich, S. Reduction in pulmonary vascular resistance with long-term epoprostenol (prostacyclin) therapy in primary pulmonary hypertension. N Engl J Med. 1998; 338:273–277.
106. McLaughlin, VV, Shillington, A, Rich, S. Survival in primary pulmonary hypertension: The impact of epoprostenol therapy. Circulation. 2002; 106:1477–1482.
107. Gomberg-Maitland, M, Olschewski, H. Prostacyclin therapies for the treatment of pulmonary arterial hypertension. Eur Respir J. 2008; 31:891–901.
108. Buckley, MS, Feldman, JP. Inhaled epoprostenol for the treatment of pulmonary arterial hypertension in critically ill adults. Pharmacotherapy. 2010; 30:728–740.
109. Theodoraki, K, Rellia, P, Thanopoulos, A, et al. Inhaled iloprost controls pulmonary hypertension after cardiopulmonary bypass. Can J Anaesth. 2002; 49:963–967.
110. Michaels, AD, Chatterjee, K, De Marco, T. Effects of intravenous nesiritide on pulmonary vascular hemodynamics in pulmonary hypertension. J Card Fail. 2005; 11:425–431.
111. Michelakis, E, Tymchak, W, Lien, D, et al. Oral sildenafil is an effective and specific pulmonary vasodilator in patients with pulmonary arterial hypertension: Comparison with inhaled nitric oxide. Circulation. 2002; 105:2398–2403.
112. Mikhail, GW, Prasad, SK, Li, W, et al. Clinical and haemodynamic effects of sildenafil in pulmonary hypertension: Acute and mid-term effects. Eur Heart J. 2004; 25:431–436.
113. Stobierska-Dzierzek, B, Awad, H, Michler, RE. The evolving management of acute right-sided heart failure in cardiac transplant recipients. J Am Coll Cardiol. 2001; 38:923–931.
114. Foubert, L, De Wolf, D, Reyntjens, K, et al. Intermittent nitric oxide combined with intravenous dipyridamole in a piglet model of acute pulmonary hypertension. Anesth Analg. 2003; 97:1497–1500.
115. Ziegler, JW, Ivy, DD, Wiggins, JW, et al. Effects of dipyridamole and inhaled nitric oxide in pediatric patients with pulmonary hypertension. Am J Respir Crit Care Med. 1998; 158:1388–1395.
116. Klinger, JR, Thaker, S, Houtchens, J, et al. Pulmonary hemodynamic responses to brain natriuretic peptide and sildenafil in patients with pulmonary arterial hypertension. Chest. 2006; 129:417–425.
117. Lepore, JJ, Maroo, A, Bigatello, LM, et al. Hemodynamic effects of sildenafil in patients with congestive heart failure and pulmonary hypertension: Combined administration with inhaled nitric oxide. Chest. 2005; 127:1647–1653.
118. Karakitsos, D, Papanikolaou, J, Karabinis, A, et al. Acute effect of sildenafil on central hemodynamics in mechanically ventilated patients with WHO group III pulmonary hypertension and right ventricular failure necessitating administration of dobutamine. Int J Cardiol. 2012.
119. Ng, J, Finney, SJ, Shulman, R, et al. Treatment of pulmonary hypertension in the general adult intensive care unit: A role for oral sildenafil? Br J Anaesth. 2005; 94:774–777.
120. Adrie, C, Holzmann, A, Hirani, WM, et al. Effects of intravenous zaprinast and inhaled nitric oxide on pulmonary hemodynamics and gas exchange in an ovine model of acute respiratory distress syndrome. Anesthesiology. 2000; 93:422–430.
121. Klein, A, Zils, U, Bopp, C, et al. Low-dose phosphodiesterase inhibition improves responsiveness to inhaled nitric oxide in isolated lungs from endotoxemic rats. J Surg Res. 2007; 138:224–230.
122. Cartin-Ceba, R, Swanson, K, Iyer, V, et al. Safety and efficacy of ambrisentan for the treatment of portopulmonary hypertension. Chest. 2011; 139:109–114.
123. Krupicka, J, Janota, T, Kasalova, Z, Hradec, J. Natriuretic peptides—Physiology, pathophysiology and clinical use in heart failure. Physiol Res. 2009; 58(2):171–177.
124. Cargill, RI, Lipworth, BJ. Acute effects of ANP and BNP on hypoxic pulmonary vasoconstriction in humans. Br J Clin Pharmacol. 1995; 40:585–590.
125. Yancy, CW, Krum, H, Massie, BM, et al. The Second Follow-up Serial Infusions of Nesiritide (FUSION II) trial for advanced heart failure: Study rationale and design. Am Heart J. 2007; 153:478–484.
126. Yurtseven, N, Karaca, P, Kaplan, M, et al. Effect of nitroglycerin inhalation on patients with pulmonary hypertension undergoing mitral valve replacement surgery. Anesthesiology. 2003; 99:855–858.
127. Fullerton, DA, Jaggers, J, Jones, SD, et al. Adenosine for refractory pulmonary hypertension. Ann Thorac Surg. 1996; 62:874–877.
128. Motti, A, Tissot, C, Rimensberger, PC, et al. Intravenous adenosine for refractory pulmonary hypertension in a low-weight premature newborn: A potential new drug for rescue therapy. Pediatr Crit Care Med. 2006; 7:380–382.
129. Sulica, R, Dinh, HV, Dunsky, K, et al. The acute hemodynamic effect of IV nitroglycerin and dipyridamole in patients with pulmonary arterial hypertension: Comparison with IV epoprostenol. Congest Heart Fail. 2005; 11:139–144.
130. Lepore, JJ, Dec, GW, Zapol, WM, et al. Combined administration of intravenous dipyridamole and inhaled nitric oxide to assess reversibility of pulmonary arterial hypertension in potential cardiac transplant recipients. J Heart Lung Transplant. 2005; 24:1950–1956.
131. Sandoval, J, Rothman, A, Pulido, T. Atrial septostomy for pulmonary hypertension. Clin Chest Med. 2001; 22:547–560.
132. Klepetko, W, Mayer, E, Sandoval, J, et al. Interventional and surgical modalities of treatment for pulmonary arterial hypertension. J Am Coll Cardiol. 2004; 43:73S–80S.
133. Ozdogan, ME, Erer, D, Iriz, E, et al. Right-to-left shunt through a patent foramen ovale left open in the management of acute right heart failure after heart transplantation. J Heart Lung Transplant. 2008; 27:135–137.
134. Reynolds, HR, Hochman, JS. Cardiogenic shock: Current concepts and improving outcomes. Circulation. 2008; 117:686–697.
135. Hoeper, MM, Mayer, E, Simonneau, G, Rubin, LJ. Chronic thromboembolic pulmonary hypertension. Circulation. 2006; 113:2011–2020.
136. Aubert, S, Leprince, P, Bonnet, N, et al. Limited mechanical circulatory support following orthotopic heart transplantation. Interact Cardiovasc Thorac Surg. 2006; 5:88–89.
137. Arafa, OE, Geiran, OR, Andersen, K, et al. Intraaortic balloon pumping for predominantly right ventricular failure after heart transplantation. Ann Thorac Surg. 2000; 70:1587–1593.
138. Trulock, EP, Edwards, LB, Taylor, DO, et al. Registry of the International Society for Heart and Lung Transplantation: Twenty-third official adult lung and heart-lung transplantation report—2006. J Heart Lung Transplant. 2006; 25:880–892.
139. D’Alonzo, GE, Barst, RJ, Ayres, SM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991; 115:343–349.
140. Humbert, M, Sitbon, O, Chaouat, A, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010; 122:156–163.
141. Benza, RL, Miller, DP, Gomberg-Maitland, M, et al. Predicting survival in pulmonary arterial hypertension: Insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation. 2010; 122:164–172.
142. Kuhn, KP, Byrne, DW, Arbogast, PG, et al. Outcome in 91 consecutive patients with pulmonary arterial hypertension receiving epoprostenol. Am J Respir Crit Care Med. 2003; 167:580–586.
143. Burrows, B, Kettel, LJ, Niden, AH, et al. Patterns of cardiovascular dysfunction in chronic obstructive lung disease. N Engl J Med. 1972; 286:912–918.
144. Traver, GA, Cline, MG, Burrows, B. Predictors of mortality in chronic obstructive pulmonary disease. A 15-year follow-up study. Am Rev Respir Dis. 1979; 119:895–902.