[level-membership-for-pulmolory-and-respiratory-category]13
Pulmonary Embolism
Pathophysiology
When a vessel is occluded by an embolus and forward blood flow through the vessel stops, perfusion of pulmonary capillaries normally supplied by that vessel ceases. If ventilation to the corresponding alveoli continues, it is wasted and the region of lung serves as dead space. As discussed in Chapter 1, assuming that minute ventilation remains constant, increasing the dead space automatically decreases alveolar ventilation and hence CO2 excretion. However, despite the potential for CO2 retention in pulmonary embolic disease, hypercapnia is an unusual consequence of pulmonary embolism, mainly because patients routinely increase their minute ventilation after an embolism occurs and more than compensate for the increase in dead space. In fact, the usual consequence of a pulmonary embolus is hyperventilation and hypocapnia, not hypercapnia. However, if minute ventilation is fixed (e.g., in an unconscious or anesthetized patient whose ventilation is controlled by a mechanical ventilator), a PCO2 rise may result from the increase in dead space caused by a relatively large pulmonary embolus.
In addition to creating an area of dead space, another potential consequence of mechanical occlusion of one or more vessels is an increase in pulmonary vascular resistance. As discussed in Chapter 12, the pulmonary vascular bed is capable of recruitment and distention of vessels. Not surprisingly, experimental evidence indicates no increase in resistance or pressure in the pulmonary vasculature until approximately 50% to 70% of the vascular bed is occluded. The experimental model is somewhat different from the clinical setting, however, because release of chemical mediators may cause vasoconstriction and additional compromise of the pulmonary vasculature.
A variety of bioactive substances are inactivated in the lung (see Chapter 12). Whether pulmonary embolism disturbs some of these nonrespiratory metabolic functions of the lung is not clear, and whether clinical consequences might ensue from such a potential disturbance is unknown.
Diagnostic Evaluation
Recommendations for the best radiologic test to diagnose pulmonary embolism have been shifting in the last decade. Traditionally, the major screening test for pulmonary embolism has been the perfusion lung scan (described in Chapter 3), but contrast computed tomographic angiography (CTA) is increasingly used either instead of or in addition to perfusion lung scanning. Evaluation of the large veins in the lower extremities, typically using ultrasound techniques, is another commonly used diagnostic strategy. Identification of a clot in a vein above the popliteal fossa warrants the same treatment as a documented pulmonary embolus and often obviates the need for further evaluation.
A perfusion lung scan is performed by injecting radiolabeled macroaggregated albumin particles into a peripheral vein. In areas of normal blood flow in the lungs, the albumin particles lodge in a fraction of the small vessels that have been perfused. When blood flow is obstructed by a clot within the pulmonary arterial system, perfusion lung scanning demonstrates no labeled albumin and therefore absence of perfusion to the region of lung supplied by the occluded vessel (Fig. 13-1). If results of the scan are normal, pulmonary embolism is, for all practical purposes, excluded. However, abnormalities do not automatically indicate the presence of embolic disease. False-positive lung scans are common because local decreases in blood flow may result from primary disease of the parenchyma or the airways. A ventilation scan, which involves inhalation of a xenon radioisotope, is often added because if regions of decreased blood flow are secondary to airway disease, corresponding abnormalities should be seen on the ventilation scan. If a defect in perfusion is due to a pulmonary embolism, ventilation still will be present in the area, and the perfusion defect will be mismatched (i.e., will not have a corresponding ventilation defect). If parenchymal disease (e.g., pneumonia) is the cause of a perfusion defect, the corresponding abnormality should be seen on the chest radiograph.
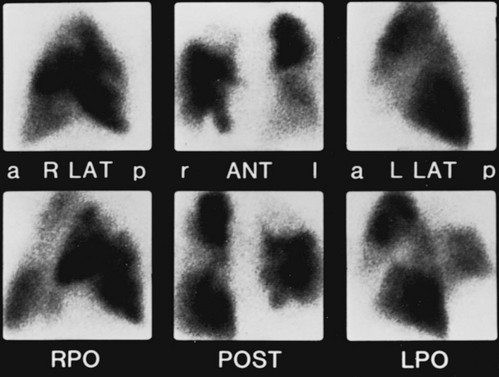
Figure 13-1 Positive perfusion scan shows multiple perfusion defects in a patient with pulmonary emboli. Six views of complete scan are shown: right lateral (R LAT), anterior (ANT), left lateral (L LAT), right posterior oblique (RPO), posterior (POST), and left posterior oblique (LPO). Compare with normal scan results in Figure 3-11. a = Anterior; l = left; p = posterior; r = right. (Courtesy Dr. Henry Royal.)
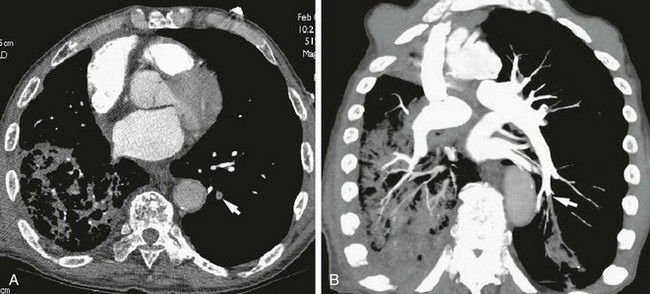
The technique of CTA (discussed in Chapter 3) has become the most commonly used modality in the diagnosis of pulmonary embolism (Fig. 13-2). CTA offers the significant advantage of high-quality visualization of the lung parenchyma, which is helpful in considering the likelihood of competing diagnoses. In addition, in many centers, CTA can be performed quickly and is more readily available than ventilation-perfusion scanning. However, the radiation dose associated with CTA, especially to the breast and chest, is significantly higher than with ventilation-perfusion scanning, so despite its advantages, CTA has not completely replaced perfusion scanning as a diagnostic modality. The clinician must weigh the risk and benefits for the individual patient as well as the practical issues of test availability and interpretation for each patient in whom the diagnosis of pulmonary embolism is being considered.
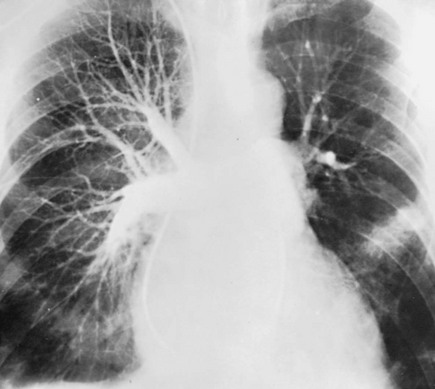
Long thought to be the gold standard for confirmation of pulmonary embolism, conventional pulmonary angiography is now rarely used for this indication (Fig. 13-3). In this technique, direct evaluation of the pulmonary arterial system to identify intraluminal thrombus is accomplished invasively by advancing a catheter through a jugular or femoral vein through the right heart and injecting radiographic contrast material directly into the pulmonary arteries. Compared with CTA, pulmonary angiography is less sensitive for pulmonary emboli, is invasive and more expensive, and is therefore not recommended in a standard evaluation. Although its lack of radiation or iodinated contrast exposure makes magnetic resonance imaging (MRI) seem like a potentially attractive option for diagnosing pulmonary emboli, technical aspects of the study make it currently much less useful and thus rarely considered for evaluation of pulmonary emboli.
Blann, AD, Lip, GY. Venous thromboembolism. BMJ. 2006;332:215–219.
Goldhaber, SZ. Pulmonary embolism. Lancet. 2004;363:1295–1305.
Hunt, JM, Bull, TM. Clinical review of pulmonary embolism: diagnosis, prognosis, and treatment. Med Clin North Am. 2011;95:1203–1222.
Khashper, A, Discepola, F, Kosiuk, J, et al. Nonthrombotic pulmonary embolism. AJR Am J Roentgenol. 2012;198:W152–W159.
Ageno, W, Gallus, AS, Wittkowsky, A, et al. American College of Chest Physicians: Oral anticoagulant therapy: Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141(Suppl 2):e44S–e88S.
Bell, WR, Simon, TL, DeMets, DL. The clinical features of submassive and massive pulmonary emboli. Am J Med. 1977;62:355–360.
Ceriani, E, Combescure, C, Le Gal, G, et al. Clinical prediction rules for pulmonary embolism: a systematic review and meta-analysis. J Thromb Haemost. 2010;8:957–970.
Dalen, JE. Pulmonary embolism: what have we learned since Virchow? Natural history, pathophysiology and diagnosis. Chest. 2004;122:1440–1456.
Dentali, F, Ageno, W, Bozzato, S, et al. Role of factor V Leiden or G20210A prothrombin mutation in patients with symptomatic pulmonary embolism and deep vein thrombosis: a meta-analysis of the literature. J Thromb Haemost. 2012;10:732–737.
Elliott, CG. Pulmonary physiology during pulmonary embolism. Chest. 1992;101:163S–171S.
Garcia, DA, Baglin, TP, Weitz, JI, et al. American College of Chest Physicians: Parenteral anticoagulants: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141(Suppl 2):e24S–e43S.
Kahn, SR, Lim, W, Dunn, AS, et al. American College of Chest Physicians: Prevention of VTE in nonsurgical patients: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141(Suppl 2):e195S–e226S.
Kanne, JP, Lalani, TA. Role of computed tomography and magnetic resonance imaging for deep venous thrombosis and pulmonary embolism. Circulation. 2004;109(12 Suppl 1):I15–I21.
Leung, AN, Bull, TM, Jaeschke, R, et al. An official American Thoracic Society/Society of Thoracic Radiology clinical practice guideline: evaluation of suspected pulmonary embolism in pregnancy. Am J Respir Crit Care Med. 2011;184:1200–1208.
Lucassen, W, Geersing, GJ, Erkens, PM, et al. Clinical decision rules for excluding pulmonary embolism: a meta-analysis. Ann Intern Med. 2011;155:448–460.
PIOPED (Prospective Investigation of Pulmonary Embolism Diagnosis) Investigators. Value of the ventilation/perfusion scan in acute pulmonary embolism: results of the Prospective Investigation of Pulmonary Embolism Diagnosis (PIOPED). JAMA. 1990;263:2753–2759.
PIOPED II Investigators. Diagnostic pathways in acute pulmonary embolism: recommendations of the PIOPED II Investigators. Am J Med. 2006;119:1048–1055.
PIOPED III Investigators. Gadolinium-enhanced magnetic resonance angiography for pulmonary embolism: a multicenter prospective study (PIOPED III). Ann Intern Med. 2010;152:434–443.
Seligsohn, U, Lubetsky, A. Genetic susceptibility to venous thrombosis. N Engl J Med. 2001;344:1222–1231.
Squizzato, A, Donadini, MP, Galli, L, et al. Prognostic clinical prediction rules to identify low-risk pulmonary embolism: a systematic review and meta-analysis. J Thromb Haemost. 2012;10:1276–1290.
Stein, PD, Beemath, A, Matta, F, et al. Clinical characteristics of patients with acute pulmonary embolism: data from PIOPED II. Am J Med. 2007;120:871–879.
Stein, PD, Hull, RD, Patel, KC, et al. D-dimer for the exclusion of acute venous thrombosis and pulmonary embolism: a systematic review. Ann Intern Med. 2004;140:589–602.
Tapson, VF. Treatment of pulmonary embolism: anticoagulation, thrombolytic therapy, and complications of therapy. Crit Care Clin. 2011;27:825–839.
Weichman, K, Ansell, JE. Inferior vena cava filters in venous thromboembolism. Prog Cardiovasc Dis. 2006;49:98–105.
Weitz, JI, Eikelboom, JW, Samama, MM. American College of Chest Physicians: New antithrombotic drugs: antithrombotic therapy and prevention of thrombosis, ed 9, American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141(Suppl 2):e120S–e151S.
[/level-membership-for-pulmolory-and-respiratory-category][not-level-membership-for-pulmolory-and-respiratory-category]13
Pulmonary Embolism
Pathophysiology
When a vessel is occluded by an embolus and forward blood flow through the vessel stops, perfusion of pulmonary capillaries normally supplied by that vessel ceases. If ventilation to the corresponding alveoli continues, it is wasted and the region of lung serves as dead space. As discussed in Chapter 1, assuming that minute ventilation remains constant, increasing the dead space automatically decreases alveolar ventilation and hence CO2 excretion. However, despite the potential for CO2 retention in pulmonary embolic disease, hypercapnia is an unusual consequence of pulmonary embolism, mainly because patients routinely increase their minute ventilation after an embolism occurs and more than compensate for the increase in dead space. In fact, the usual consequence of a pulmonary embolus is hyperventilation and hypocapnia, not hypercapnia. However, if minute ventilation is fixed (e.g., in an unconscious or anesthetized patient whose ventilation is controlled by a mechanical ventilator), a PCO2 rise may result from the increase in dead space caused by a relatively large pulmonary embolus.
In addition to creating an area of dead space, another potential consequence of mechanical occlusion of one or more vessels is an increase in pulmonary vascular resistance. As discussed in Chapter 12, the pulmonary vascular bed is capable of recruitment and distention of vessels. Not surprisingly, experimental evidence indicates no increase in resistance or pressure in the pulmonary vasculature until approximately 50% to 70% of the vascular bed is occluded. The experimental model is somewhat different from the clinical setting, however, because release of chemical mediators may cause vasoconstriction and additional compromise of the pulmonary vasculature.
[/not-level-membership-for-pulmolory-and-respiratory-category]
