[level-membership-for-anesthesiology-category]
Pulmonary arterial hypertension
Diagnosis
< ?xml:namespace prefix = "mml" />
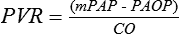
The normal resting mPAP value is 14 ± 3.3 mm Hg; the World Health Organization defines PAH as a sustained elevation of mPAP to more than 25 mm Hg at rest or to more than 30 mm Hg with exercise, with a mean PAOP or left ventricular end-diastolic pressure of less than 15 mm Hg. The World Health Organization classifies PAH as primary or secondary, with recognition that considerable overlap exists between groups, and further divides secondary PAH into left-sided heart disease, PAH from hypoxemia, PAH from pulmonary thrombotic disease, and PAH from miscellaneous causes (Box 228-1). The distinguishing feature between PAH caused by left-sided heart disease and other causes of PAH is that the latter diagnoses require a PVR greater than 3 Wood units (1 Wood unit = 80 dyn·sec−1·cm−5). If left atrial (PAOP) pressure is high (e.g., in patients with mitral valve insufficiency), then PVR is unlikely to be greater than 3 Wood units, and the PAH is secondary to left-sided heart disease.
Anesthetic management of the patient with pulmonary arterial hypertension
Intraoperative management
Both regional and general anesthetic techniques have been used successfully to anesthetize patients with PAH. More important than the technique used is the avoidance or prompt treatment of hypoxia, hypercapnia, acidosis, hypothermia, and pain because all increase PVR and right ventricular afterload. A decreased right ventricular stroke volume leads to decreased left ventricular preload, which is further compromised by the septal displacement from the enlarged right ventricle into the left ventricle. If left ventricular stroke volume decreases (and along with it, aortic root perfusion pressure), coronary blood flow to the right ventricle is decreased, further compromising the performance of the right ventricle. Goals for these patients then include maintaining normal sinus rhythm, with a heart rate of approximately 80 to 90 beats/min, to optimize cardiac output. Right ventricular function is sensitive to both intravascular volume depletion and excess; therefore, fluids should be administered slowly and in small volumes, with a goal of maintaining a central venous pressure of 12 mm Hg or less (Box 228-2).
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
Pulmonary arterial hypertension
Diagnosis
< ?xml:namespace prefix = "mml" />
The normal resting mPAP value is 14 ± 3.3 mm Hg; the World Health Organization defines PAH as a sustained elevation of mPAP to more than 25 mm Hg at rest or to more than 30 mm Hg with exercise, with a mean PAOP or left ventricular end-diastolic pressure of less than 15 mm Hg. The World Health Organization classifies PAH as primary or secondary, with recognition that considerable overlap exists between groups, and further divides secondary PAH into left-sided heart disease, PAH from hypoxemia, PAH from pulmonary thrombotic disease, and PAH from miscellaneous causes (Box 228-1). The distinguishing feature between PAH caused by left-sided heart disease and other causes of PAH is that the latter diagnoses require a PVR greater than 3 Wood units (1 Wood unit = 80 dyn·sec−1·cm−5). If left atrial (PAOP) pressure is high (e.g., in patients with mitral valve insufficiency), then PVR is unlikely to be greater than 3 Wood units, and the PAH is secondary to left-sided heart disease.
