[level-membership-for-basic-science-category]
CHAPTER 46 Programmed Cell Death
The Necessity for Cell Death in Multicellular Organisms
The ability to undergo programmed cell death (Box 46-1) is a built-in latent capacity in virtually all cells of multicellular organisms. Cell death is important for embryonic development, maintenance of tissue homeostasis, establishment of immune self-tolerance, killing by immune effector cells, and regulation of cell viability by hormones and growth factors. It has been proposed that most metazoan cells will die if they fail to receive survival signals from other cells. Abnormalities of the cell death program contribute to a number of diseases, including cancer, Alzheimer’s disease, and acquired immune deficiency syndrome (AIDS).
Programmed Cell Death versus Accidental Cell Death: Apoptosis versus Necrosis
Although cells die in many ways, it is useful to focus on the two poles of this spectrum: apoptosis and necrosis. Apoptosis is the most commonly described pathway for programmed cell death, which is cellular suicide resulting from activation of a dedicated intracellular program (Fig. 46-1). Often, these cells appear completely healthy prior to committing suicide. At the other end of the spectrum is necrosis, also called accidental cell death, which occurs when cells receive a structural or chemical insult that kills them outright (Fig. 46-2). Examples of such insults include extremes of temperature and physical trauma. The cell itself can also initiate necrosis in response to certain stimuli, particularly when induction of apoptosis is inhibited. In contrast to the orderly biochemical pathways of apoptosis, which involve the action of enzyme cascades and the consumption of ATP, necrosis typically involves a collapse of normal cell physiology as a result of ATP depletion.
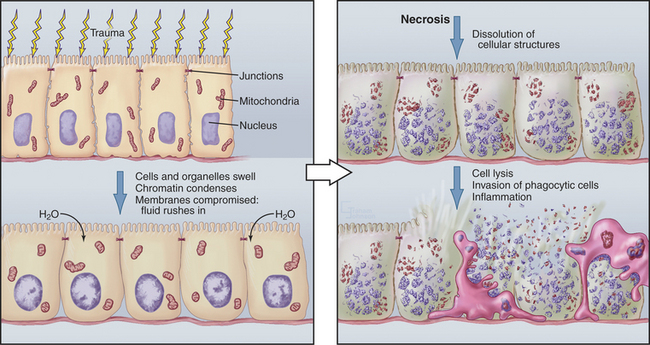
Necrosis corresponds to what most of us naively imagine cell death would be like. Owing to lack of cellular homeostasis, water rushes into the dying cell, causing it to swell greatly so that the plasma and organelle membranes burst. As a result, the cell undergoes a generalized process of autodigestion and dissolution, culminating in the spilling of the cytoplasmic contents out into the surroundings (Fig. 46-2). This, in turn, produces local inflammation as phagocytic cells are activated, flock to the site, and ingest the debris (see Chapter 22). Because agents that damage cells act over areas that are large in comparison to the size of a single cell, necrosis often involves large groups of neighboring cells.
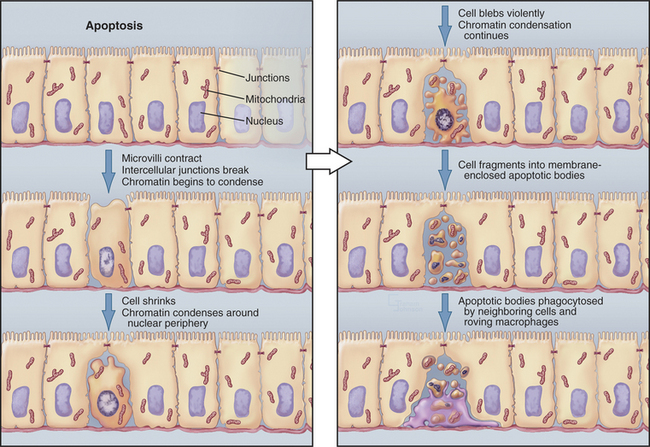
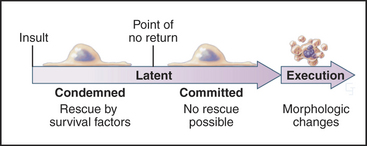
In contrast to necrosis, apoptotic cells shrink rather than swelling, as part of a reproducible pattern of structural alterations of both the nucleus and cytoplasm (Fig. 46-1). Apoptosis is a two-stage process. On receipt of the pro-apoptotic signal that triggers the pathway to death, cells enter a latent phase of apoptosis (Fig. 46-3). Although committed to a pathway that leads to their inevitable demise at some later time, cells in the latent phase look as healthy as their neighbors. The duration of the latent phase of apoptosis is extremely variable, ranging from a few hours to several days. The reason for this variability is not known.
Ultimately, the cells enter the execution phase of apoptosis, lasting about an hour, during which they undergo dramatic morphologic and physiological changes. These include (1) loss of microvilli and intercellular junctions (Fig. 46-4); (2) shrinkage of the cytoplasm; (3) dramatic changes in cytoplasmic motility with activation of violent blebbing (Fig. 46-5); (4) loss of plasma membrane asymmetry, with the distribution of phosphatidylserine being randomized so that it appears in the outer membrane leaflet; (5) hypercondensation of the chromatin and its collapse against the nuclear periphery; and (6) the “explosive” fragmentation of the cell into membrane-enclosed apoptotic bodies that contain remnants of the nucleus, mitochondria, and other organelles. The plasma membrane retains its integrity throughout the entire process. All of these changes are instigated by the action of a specific set of death-inducing proteases, discussed at length later.
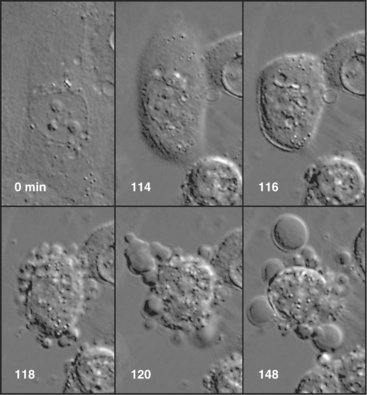
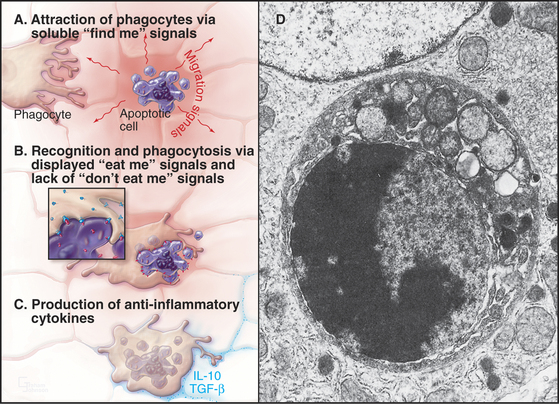
In tissues, apoptotic bodies are rapidly phagocytosed by surrounding cells that recognize the phosphatidylserine and other markers exposed on their surface (Fig. 46-6). Apoptosis can thus be considered to be the disassembly of the cell into “bite-sized” vesicles. Because these vesicles remain membrane bound, the cellular contents are not released into the environment. It is important to note that surface markers on apoptotic bodies cause cells that ingest them to secrete anti-inflammatory cytokines. As a result, apoptotic death does not lead to an inflammatory response.
Nonapoptotic Programmed Cell Death
The terms apoptosis and programmed cell death are sometimes viewed as synonymous. However, in a number of well-documented systems, cells undergo programmed cell death without the dramatic structural changes that classically define apoptosis. Thus, all apoptosis is programmed cell death, but the converse is not necessarily true.
When the adult tobacco hawkmoth emerges from its cocoon, its intersegmental muscles undergo programmed cell death that differs in several ways from apoptosis as described earlier. The chromatin does not condense; DNA is not digested; and cytoplasm does not “boil.” Instead, a polyubiquitin gene is induced and plays an important role in intracellular protein degradation (see Chapter 23). Thus, although these muscle cells unquestionably undergo programmed cell death, they apparently do not use the apoptosis pathway.
Classes of Cells That Undergo Programmed Cell Death
At least six distinct classes of cells undergo programmed cell death (examples are given in Fig. 46-7).
Developmentally Defective Cells
During molecular maturation of T-lymphocyte antigen receptors (see Figs. 27-8 and 28-8), immature T cells in the thymus (known as thymocytes) rearrange the genes encoding the receptor a and b chains. Many newly created receptors bind to foreign antigens, but others interact with self-antigens. Cells with receptors recognizing self-antigens are potentially harmful and are eliminated through apoptosis in a process known as negative selection (Fig. 46-8). The drug cyclosporin A, which inhibits apoptosis in thymocytes, can cause autoimmune disease.
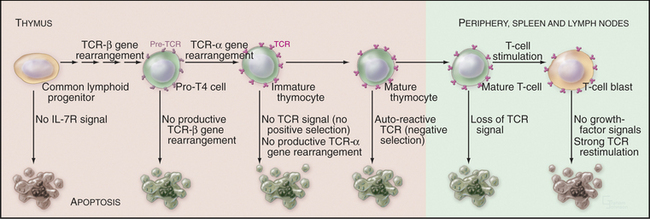
To function properly, the T-cell receptor must recognize major histocompatibility complex (MHC) glycoproteins on other cells during antigen presentation (see Fig. 27-8). T lymphocytes whose T-cell receptors cannot interact with the spectrum of MHC glycoproteins expressed in a given individual are ineffective in the immune response. These cells die by apoptosis in a process known as positive selection (Fig. 46-8). Overall, defects in T-cell receptor assembly are extremely common, and up to 95% of immature T cells die by apoptosis without leaving the thymus.
Similar positive and negative selection steps occur during the maturation of B lymphocytes (see Fig. 28-8), which is accomplished by a combination of gene rearrangements and facilitated mutagenesis. B lymphocytes expressing antibodies directed against self-antigens or producing antibodies whose affinity for antigen is below a critical threshold are eliminated through apoptosis.
Excess Cells
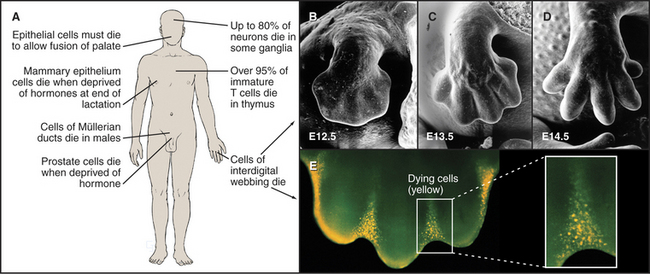
The use of programmed cell death for quality control during development is not limited to the immune system but is also extremely important during brain development. Embryonic ganglia often have many more neurons than are required to enervate their target muscles. Production of excess cells is part of a Darwinian strategy to ensure that a sufficient number of axons reach their targets. Excess neurons that fail to make appropriate connections have no function and are eliminated by programmed cell death. Up to 80% of neurons in certain developing ganglia die in this way. Because of the importance of apoptosis during its development, the brain is often seriously affected in mice that are engineered to lack components of the apoptotic pathway.
Cells That Serve No Function
Mammals also use programmed cell death to eliminate obsolete tissues during development. For example, in humans, the digits of hands and feet are connected by a tissue webbing during embryogenesis. Cells in this webbing serve no purpose in the adult and are eliminated by programmed cell death (Fig. 46-7).
Cells Whose Cell Cycle Is Perturbed
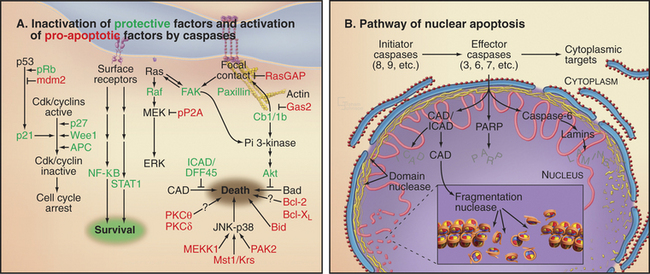
Chapters 40 to 43 describe how biochemical circuits called checkpoints regulate the cell cycle. If DNA is damaged, checkpoint activation blocks cell-cycle progression while repair processes operate. An important downstream effector of checkpoints, the p53 transcription factor, induces the expression of genes encoding proteins that arrest the cell cycle as well as genes encoding proteins that induce cell death. It is generally thought that if the damage cannot be repaired quickly, the pro-death factors win out, and the outcome is apoptosis. Types of DNA damage that commonly trigger cell death are double-strand breaks induced by ionizing radiation and DNA breaks or other damage induced by chemotherapeutic agents.
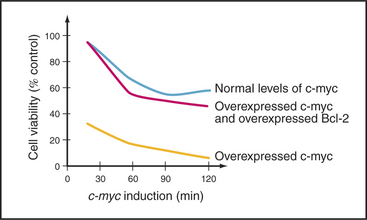
A second important cell-cycle checkpoint regulates the transition from the G1 phase to the S phase. Passage of the restriction point (see Fig. 41-7) represents the commitment of the cell to undergo another cycle of DNA replication and division. Restriction point control centers on the regulation of the E2F family of transcription factors. However, E2F not only regulates genes that promote cell-cycle progression; it also induces the expression of genes that promote apoptosis. It is now thought that if E2F is activated too strongly, as, for example, where restriction point control has broken down (see Fig. 41-10), its function as a death inducer takes over, and the cells undergo apoptosis. Cells that die in response to inappropriate signals to proliferate include those that are infected by certain viruses or overexpress genes involved in cell proliferation (such as c-myc and c-fos [Fig. 46-14]). This ability to recognize an inappropriate stimulus to proliferate and respond to it by undergoing apoptosis may be an important defense against cancer.
Virus-Infected Cells
At least part of the loss of mature CD4+ T helper cells (see Fig. 28-8) in people who are infected with HIV-1 results from programmed cell death. When exposed to agents that normally stimulate cell proliferation, these cells instead undergo apoptosis. Paradoxically, it ap-pears that many of these dying cells are not themselves infected with HIV.
Genetic Analysis of Programmed Cell Death
Several key components that are involved in the apoptotic execution of mammalian cells were first identified by a genetic analysis of the nematode worm Caenorhabditis elegans. Because C. elegans is optically clear, it is possible to see every cell in a developing worm by using differential interference contrast optics (see Fig. 6-2). This enabled investigators to develop a complete fate map for C. elegans that traces the lineage of each cell in an adult worm back to the fertilized egg. These studies led to the surprising discovery that programmed cell death is one of the most common fates for newborn C. elegans cells. Of the 1090 somatic cells that are produced during embryogenesis of the C. elegans hermaphrodite, 131 undergo programmed cell death at reproducible locations and times.
Mutations in at least 14 C. elegans genes affect programmed cell death (Fig. 46-9). These may be divided into three classes: (1) genes that mark cells for subsequent programmed death, (2) genes that are involved in cell killing and its regulation, and (3) genes that are involved in the phagocytosis and subsequent processing of the cell corpses. These mutants are collectively known as “cell death abnormal” (ced) mutants.
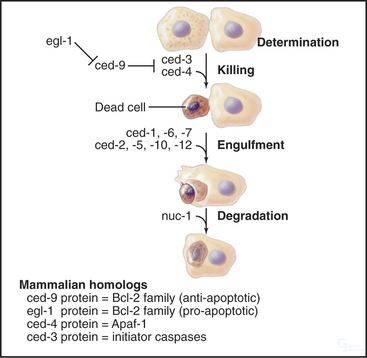
The three best-known cell death genes are ced-3, ced-4, and ced-9. Ced-3 and ced-4 are required for cells to undergo apoptotic programmed cell death. If either gene is inactivated, all cells throughout the organism that should die by apoptosis are reprieved. These cells remain alive and are apparently functional. Interestingly, these worms have normal life spans. This suggests that programmed cell death is not involved in the normal aging process, at least not in C. elegans. Ced-9 regulates ced-3 and ced-4. In ced-9 loss-of-function mutants, many cells die that should normally stay alive. This is deleterious for the organism, and ced-9 mutants die.
Signals and Pathways of Apoptosis
Two principal pathways lead to cell death by apoptosis. These are introduced only briefly here. The intrinsic pathway (Fig. 46-16) is activated by internal surveillance mechanisms or signals sent (or not sent) by other cells. Signals that induce this pathway include DNA damage, exposure to chemicals that interfere with a variety of cellular pathways, excessive activation of factors that promote cell-cycle progression, and receipt of certain pro-apoptotic stimuli from the surrounding medium. Withdrawal of nutrients or of nurturing signals from the environment also activates the intrinsic pathway. Survival signals include lymphokines, such as interleukin-2 and interleukin-3, which are essential for survival of thymocytes; nerve growth factor, which is required for survival of many neurons; and extracellular matrix, which is required for survival of epithelial cells. Signals that activate the intrinsic pathway converge on mitochondria, which release key factors that drive the apoptotic response.
Signals from other cells are the primary triggers of the extrinsic pathway (Fig. 46-17). Direct contact with the target cell activates specific receptors that initiate this pathway, starting on the inner surface of the plasma membrane. Activation of the extrinsic pathway is one strategy that cytotoxic T lymphocytes use to kill cells that are recognized as foreign (or as harboring foreign pathogens). This pathway is also widely used to control cell populations in the immune system.
Protein Regulators and Effectors of Apoptosis
Caspases
C. elegans has three caspases, one of which (Ced-3) is essential for cell death. In contrast, mammals have at least 13 caspase genes (Fig. 46-10A). Analysis based on sequence comparisons divides caspases into two major subfamilies. The caspase 1 subfamily encodes enzymes that process pro-interleukin-1b to yield mature interleukin-1b. Macrophages secrete this cytokine, which is involved in causing inflammation. In contrast, the caspase 3 subfamily of enzymes participates almost exclusively in apoptotic cell death.
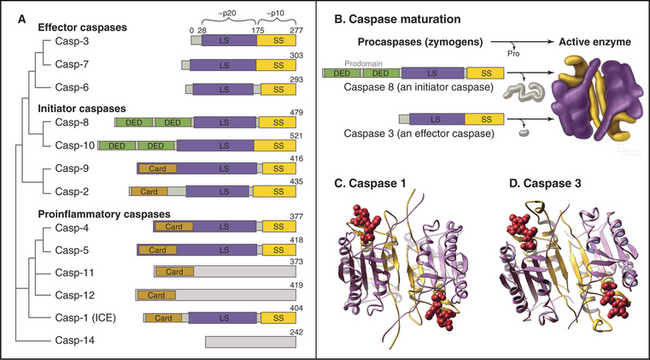
Like many proteases, caspases are synthesized as inactive zymogens. All living vertebrate cells apparently synthesize these zymogens constitutively. The caspase zymogen consists of three domains and an N-terminal prodomain followed by the large and small subunits of the mature enzyme (Fig. 46-10A-B). These three domains are separated by aspartate residues, the cleavage target for caspases. Caspase zymogens are usually activated by zymogen cleavage and release of the prodomains. Following cleavage, the two large and two small subunits associate in a compact, block-like heterotetrameric molecule (Fig. 46-10B-D). Cleavage of the zymogens permits a major conformational change in the polypeptide, creating two stable active site pockets between the large and small subunits.
Two classes of caspases are involved in cell death. Initiator caspases have long prodomains (Fig. 46-10A). These zymogens exist as monomers in cells and become autoactivated when scaffolding cofactors promote their aggregation. Activation is thought to involve dimerization of the zymogens and might not necessarily require zymogen cleavage. Sequences within the extended prodomains are involved in targeting the initiator caspase zymogens to the appropriate cellular locations and in interactions with scaffolding factors.
Scaffolding proteins and adapters play an essential role in the activation of initiator caspases. For the intrinsic pathway of cell death, factors released from mitochondria (discussed later) activate the scaffold protein apoptotic protease activating factor 1 (Apaf-1). Active Apaf-1 forms a seven-spoked ring-like structure called the apoptosome (Fig. 46-16). Binding of the procaspase 9 zymogen to this structure promotes dimerization and activation of the enzyme, which appears to achieve full activity without the necessity of zymogen cleavage. The scaffold proteins for the extrinsic death pathway are cytoplasmic domains of cell surface receptors. When these receptors bind their ligands on the surface of other cells, they form stable trimeric complexes that recruit adapter proteins, which have multiple protein-protein interaction motifs and link the procaspase 8 zymogen to the receptor complex (Box 46-2). This leads to the dimerization, activation, self-cleavage, and release of active caspase 8, thereby starting the apoptotic cascade.
BOX 46-2 Matchmaking for Cell Death: The Key Is in the Domains
Caspases are selective enzymes that cleave a relatively limited subset of cellular proteins (Fig. 46-11). Some targets are structural proteins, but many are involved in cellular signaling. For example, caspases cleave several protein kinases. Many kinases have autoregulatory domains that enable them to be switched on and off in response to physiological stimuli (see Fig. 25-4). Caspase cleavage often neatly removes these regulatory domains, thereby producing constitutively active enzymes. Presumably, these unregulated kinases then activate factors that promote cell death. Caspases also cleave and inactivate a number of proteins that normally function in the detection and repair of DNA damage.
Natural Caspase Inhibitors
Because most healthy cells express initiator procaspases with the potential to oligomerize by mistake and kill the cell, it is important to have a mechanism that dampens this “noise” in the pro-apoptotic pathway. The inhibitor of apoptosis protein (IAP) family is defined by the presence of a motif of approximately 80 amino acids known as a baculovirus IAP repeat domain. This is a type of Zn2+ finger (Fig. 15-17) that mediates protein-protein interactions. IAP proteins inhibit caspases in two ways. First, they bind the caspase and invade the active site, thereby blocking its access to substrates. Second, several IAPs are also E3 ubiquitin ligases (see Fig. 23-8). When they bind caspases, they ubiquitinate them, thereby tagging them for destruction by proteasomes.
IAPs were discovered in studies of the mechanisms viruses use to avoid being eliminated by cell death. When viruses infect cells and disassemble their capsids, they become vulnerable to suicide defense mechanisms: If cells can kill themselves before the virus has had time to complete its life cycle, they will take the virus with them, and the organism will survive. To defend against this, viruses pilfer cellular proteins and adapt them for their own means. For example, insect baculoviruses make two proteins that inhibit apoptosis, keeping the cell alive long enough for the virus to reproduce. One of these, IAP, was derived from a cellular gene. The origin of the second, p35, is less clear. p35 is a broad-spectrum caspase inhibitor that is thought to work by a serpin-like mechanism. Serpins are special protease substrates that, on cleavage, form a tight complex with the enzyme, thereby inactivating it. Several mammalian pox viruses also make a serpin-like inhibitor of certain caspases called CrmA.
CAD Nuclease and Its Chaperone ICAD
During apoptosis, the chromosomal DNA is destroyed. The many nucleases involved in cleaving the cellular DNA during (and after) apoptotic cell death fall into two classes. Cell autonomous nucleases degrade the DNA from within the dying cell (Fig. 46-12A). The best known is the caspase-activated DNase (CAD; see later discussion). In some cell types, a mitochondrial nuclease known as endonuclease G may also be involved. Cell autonomous nucleases are dispensable for cell death and for the life of the organism. They might have evolved to eliminate viral DNA as part of the suicide defense response described in the previous section.
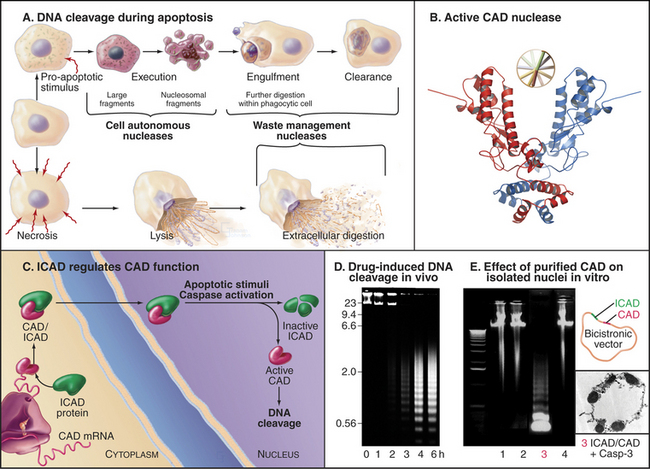
Figure 46-12 the nucleases that digest the cellular dna during apoptosis. A, In apoptosis, the DNA is digested first to large fragments and later to nucleosome-sized pieces (see Fig. 13-1) by cell autonomous nucleases expressed within the dying cell. Waste management nucleases made by other cells also have an essential role in cleaning up apoptotic and necrotic debris. B, The predominant cell autonomous nuclease (CAD) has a scissors-like structure. C, ICAD is an inhibitory chaperone for CAD, promoting its folding on the ribosome and continuing as an inhibitor when CAD is stored in the nucleus. ICAD cleavage leads to CAD activation. D, Cleavage of the chromosomal DNA by CAD during chemotherapy-induced apoptosis of a leukemia cell line. DNA separated according to size by electrophoresis on an agarose gel was stained with ethidium bromide.
(A, Based on Samejima K, Earnshaw WC: Trashing the genome: The role of nucleases during apoptosis. Nat Rev Mol Cell Biol 6:677–688, 2005. B, PDB file: 1V0D. Structure described in Woo EJ, Kim YG, Kim MS, et al: Structural mechanism for inactivation and activation of CAD/DFF40 in the apoptotic pathway. Mol Cell 14:531–539, 2004. D, From Kaufmann SH: Induction of endonucleolytic DNA cleavage in human acute myelogenous leukemia cells by etoposide, camptothecin, and other cytotoxic anticancer drugs: A cautionary note. Cancer Res 49:5870–5878, 1989. E, Courtesy of K. Samejima, Wellcome Trust Institute for Cell Biology, University of Edinburgh, Scotland.)
Cell autonomous nucleases act in two stages. After an initial cleavage of the chromosomes into fragments of roughly 50,000 base pairs, DNA is usually (but not always) cleaved between nucleosomes, producing a characteristic “ladder” of DNA fragments with a periodicity of about 200 base pairs. This ladder is seen when DNA isolated from apoptotic cells is subjected to gel electrophoresis. The responsible nuclease is CAD. CAD is normally present in a complex with ICAD (inhibitor of CAD [Fig. 46-12C]). The complex of CAD and ICAD is also known as DNA fragmentation factor (DFF). ICAD is a chaperone that must be present for CAD to fold into an active conformation as it is being translated on the ribosome. However, ICAD also inhibits the nuclease activity of CAD. This dual function of ICAD guarantees that only inactive CAD can be synthesized in healthy cells. During apoptosis, caspase 3 cleaves ICAD and releases active CAD nuclease.
Bcl-2 Proteins and the Intrinsic Pathway of Apoptotic Cell Death
As was mentioned previously, mitochondria are key players in a pathway to cell death that is triggered by a variety of toxic insults (Fig. 46-16). These mitochondrial events are regulated by the Bcl-2 family of proteins. The following sections describe this important protein family and their regulation of the intrinsic pathway of apoptosis.
Bcl-2 Proteins
Bcl-2 proteins can be grouped into three subfamilies (Fig. 46-13). Bcl-2 protectors protect cells against apoptosis. Bcl-2 killers (e.g., Bax and Bak) are pro-apoptotic proteins that actively kill cells. Bcl-2 regulators promote cell killing by either interfering with the protectors or activating the killers. These proteins primarily regulate the release of death-promoting factors from mitochondria when cells receive signals that activate the intrinsic pathway.
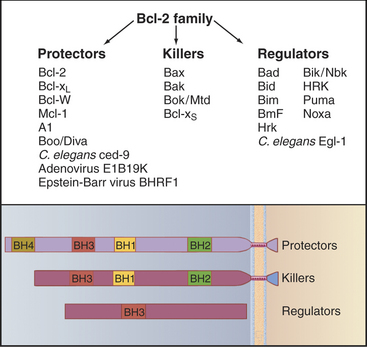
Bcl-2 family members are defined by the presence of one to four short blocks of conserved protein sequence called BH (Bcl-2 homology) domains. Anti-apoptotic Bcl-2 protectors typically have four of the domains. Pro-apoptotic Bcl-2 killers typically have three of these domains, while the Bcl-2 pro-apoptotic regulators have only the BH3 domain. The BH3 domain is a short segment of helix that fits into a groove on the surface of both Bcl-2 protectors and killers, forming a complex that regulates their activity. It is now believed that the Bcl-2 protectors regulate the behavior of Bcl-2 killers by a similar interaction. For example, Bcl-2 protein forms a complex with a pro-apoptotic Bcl-2 killer called Bax, thereby interfering with the ability of Bax to kill cells.
Bcl-2 Family Members and Cancer
A gene that prevents cells from dying poses a potential danger in multicellular organisms, in which rates of cell proliferation and death must be balanced carefully. In fact, the name Bcl-2 comes from the discovery that this gene is the culprit responsible for certain types of B-cell lymphoma. These particular lymphomas arise when a chromosome translocation, involving chromosomes 14 and 18, moves the Bcl-2 gene into the immunoglobulin heavy-chain gene cluster, a site of very active gene transcription in B lymphocytes. Elevated transcription of Bcl-2 is thought to be directly responsible for the cancerous phenotype in these patients, making Bcl-2 a cancer-promoting oncogene (see Chapter 41).
Unlike many other oncogenes, Bcl-2 overexpression does not cause cell proliferation. Instead, it disrupts the balance of regulation between life and death of the affected cells. Cells that overexpress Bcl-2 protein actually grow, if anything, more slowly than do their normal counterparts. However, Bcl-2 overexpressers are highly resistant to many stimuli that normally promote cell death. The net result is an accumulation of B cells: a lymphoma. Figure 46-14 shows an example of Bcl-2 conferring resistance to death when the cell cycle is perturbed by expression of an oncogene.
The Intrinsic Pathway of Apoptotic Death
In addition to their role in energy production, mitochondria have an essential role as sensors of the health of the cell. If cells sense insults from which they cannot recover, mitochondria trigger the intrinsic pathway of cell death (Fig. 46-16). This pathway is regulated by Bcl-2 family members. The regulation seems straightforward in C. elegans, in which the protector CED-9 (Bcl-2–like) binds to the CED-4 scaffolding protein (Apaf-1-like) and interferes with its activation of the CED-3 caspase. Apoptosis is induced when the regulator BH3-only protein EGL-1 binds to CED-9 and blocks it from inactivating CED-4.
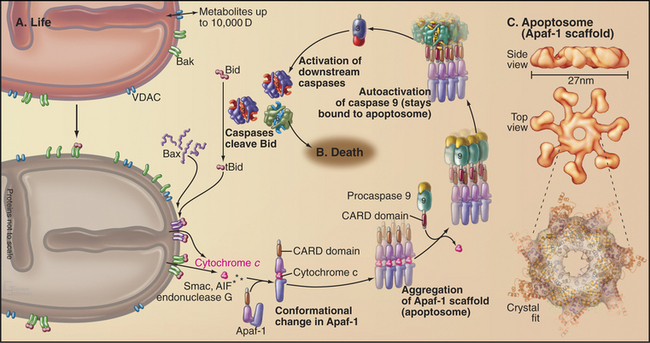
In mammals, the situation is more complex, partly because Bcl-2 family members are more numerous and partly because they do not interact in such a straightforward fashion. In mammals, two of the killer proteins, Bax and Bak, are essential for activation of the intrinsic pathway. In healthy cells, Bak is loosely associated with the mitochondrial outer membrane, and Bax is in the cytoplasm (Fig. 46-16). On receipt of a pro-apoptotic stimulus, Bax and Bak insert deeply into the mitochondrial outer membrane, form oligomers, and somehow (not yet known but possibly involving the formation of membrane pores) cause the release of pro-apoptotic factors from the mitochondrial intermembrane space. Binding of anti-apoptotic Bcl-2 family members to Bax/Bak somehow prevents the release of pro-apoptotic factors from mitochondria. Various BH3-only family members either facilitate Bax/Bak oligomerization or bind and neutralize anti-apoptotic Bcl-2 family members.
The pro-apoptotic factors that are released from the mitochondrial intermembrane space by Bax and Bak include the electron transport protein cytochrome c (see Fig. 19-5), Smac, and endonuclease G (Fig. 46-15). These mitochondrial proteins actively promote apoptotic cell death. In the cytoplasm, cytochrome c binds to the scaffolding protein Apaf-1, a mammalian homologue of C. elegans CED-4 protein, causing it to form a seven-spoked wheel-like structure called the apoptosome (Fig. 46-16). Apaf-1 in the apoptosome binds caspase 9 through an N-terminal caspase recruitment domain.
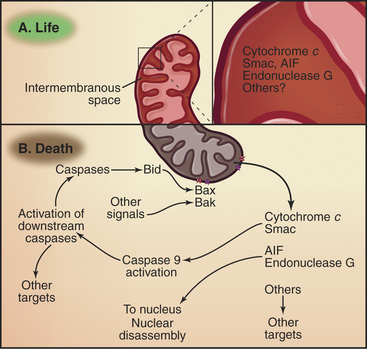
The Extrinsic Pathway of Apoptotic Death
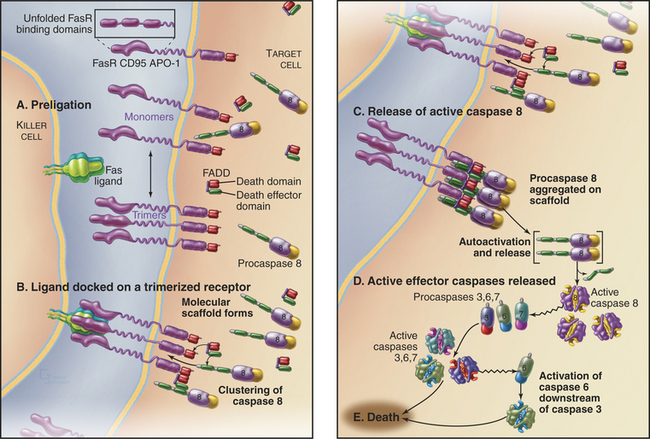
One well-characterized death receptor is called Fas (also known as Apo1 or CD95), a member of the tumor necrosis factor receptor family (see Fig. 24-10). Fas is a type I membrane protein whose extracellular domain consists of three cysteine-rich domains (see Fig. 24-11, which shows the atomic structure of the related trimeric tumor necrosis factor receptor with bound ligand). The cytoplasmic domain of Fas contains a death domain of about 80 residues, which is shared by all of the death receptors (Box 46-2).
The Fas ligand is a trimeric 40-kD intrinsic membrane protein found on the surface of cells. Cytotoxic T lymphocytes use Fas ligand to rid the body of virally infected cells. When a cytotoxic T lymphocyte contacts a target cell, the Fas ligand on the lymphocyte surface binds to Fas on the target cell and initiates the extrinsic pathway of apoptotic death (Fig. 46-17). Ligand binding activates signaling from the intracellular death domain of Fas, possibly by stabilizing Fas trimers or by altering their conformation. Activated Fas binds an adapter protein called FADD (Fas-associated protein with a death domain). The Fas-FADD complex binds procaspase 8 through interactions involving another type of motif called the death effector domain, which is present on both FADD and the prodomain of procaspase 8. On this molecular scaffold, procaspase 8 monomers dimerize and acquire catalytic activity. These dimers can cleave neighboring dimers, creating and releasing heterotetrameric active caspase 8, which initiates the caspase cascade by activating downstream effector caspases.
Role of the Fas Death Receptor in Normal and Diseased Cells
Mouse mutants provide clear evidence for an important role of Fas in regulation of the immune system. Mice with mutated Fas (the lpr mutation) or Fas ligand (the gld mutation) accumulate excessive lymphocytes. In the appropriate genetic background, these mice tend to develop autoimmune disorders that, in some cases, resemble the human disease systemic lupus erythematosus. Evidence that Fas is involved in human systemic lupus erythematosus is still scant.
Linking Apoptosis to the Cell Cycle by p53
No obligate link exists between particular cell-cycle phases and apoptosis. Noncycling G0 cells can undergo apoptosis, and cycling cells appear able to do so from any cell-cycle phase. However, one link between apoptosis and the cell-cycle machinery has now been firmly established. This involves the p53 tumor suppresser and DNA damage.
The p53 transcription factor is one of the downstream effectors of the DNA damage response pathway (see Fig. 40-4). When cells sense DNA damage induced by agents such as ionizing radiation, levels of p53 rise dramatically (Fig. 46-18). When stabilized and activated by phosphorylation (see Fig. 41-13) p53 upregulates the expression of a number of genes, including the Cdk inhibitor p21, which blocks the entry into the S and M phases. p53 also can trigger an apoptotic response in instances in which the DNA damage is too severe to repair. This tumor suppressor protein is very important in the body’s defense against cancer. Mutations in the p53 gene/protein are found in about 50% of all human cancers.
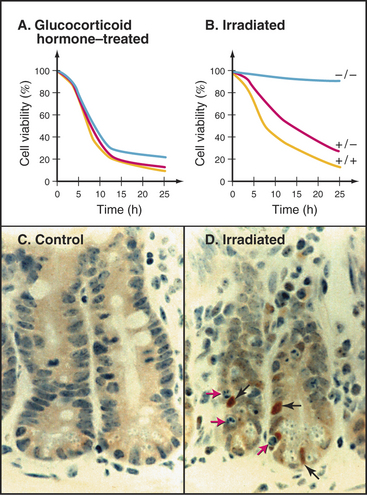
The role of p53 in apoptosis was confirmed in transgenic mice lacking a functional p53 gene (p53 knockout mice). These mice develop normally but are extremely prone to cancer at a very young age. Thus, although mice do not require p53 for programmed cell death during embryogenesis, p53 is critical for apoptosis of certain cells. Thymocytes isolated from p53 knockout mice are extremely resistant to the induction of apoptosis by ionizing radiation and other agents that cause DNA breaks (Fig. 46-18B). However, p53 is not involved in all types of apoptosis. For example, even thymocytes isolated from p53 knockout mice show normal induction of apoptosis following exposure to glucocorticoid hormone (Fig. 46-18).
Importance of Apoptosis in Human Disease
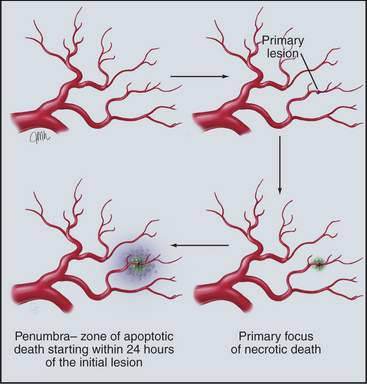
Studies of apoptosis now account for a substantial fraction of cell biology research. Why has this field so caught the scientific eye? The most likely answer is that apoptosis is a point of intersection between cell signaling pathways, cell structure, the cell cycle, and, of course, human disease. This chapter has mentioned the roles that aberrations in apoptosis play in the etiology of autoimmunity, AIDS, and cancer. Apoptosis is also emerging as a key factor in neurodegenerative diseases, such as Huntington’s disease and Alzheimer’s disease, as well as in myocardial infarction and stroke (Fig. 46-19). At a practical level, the realization that many successful chemotherapeutic agents act by inducing cancer cells to undergo apoptosis has motivated searches for newer and better drugs that elicit this response. One promising approach is to combine reagents that either directly promote apoptosis or weaken cellular resistance to apoptosis with cancer chemotherapy. The goal is to find combinations that increase cell killing synergistically. It is hoped that this strategy will improve outcomes in cases where tumors are resistant to current chemotherapy. Conversely, the realization that a large fraction of the cell deaths in stroke are attributable to a wave of apoptosis that radiates outward from the original focus of ischemic death has led to the hunt for molecules that will prevent apoptosis during the critical period following the stroke or infarct. With such important practical problems to be solved, apoptosis will continue to occupy a prominent position in cell biology research over the coming years.
ACKNOWLEDGMENTS
Thanks go to Scott Kaufmann and Yuri Lazebnik for their suggestions on revisions to this chapter.
Abraham MC, Shaham S. Death without caspases, caspases without death. Trends Cell Biol. 2004;14:184-193.
Breckenridge DG, Xue D. Regulation of mitochondrial membrane permeabilization by BCL-2 family proteins and caspases. Curr Opin Cell Biol. 2004;16:647-652.
Cory S, Adams JM. The BCL2 family: Regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647-656.
Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: Structure, activation, substrates and functions during apoptosis. Annu Rev Biochem. 1999;68:383-424.
Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626-629.
Lauber K, Blumenthal SG, Waibel M, Wesselborg S. Clearance of apoptotic cells: Getting rid of the corpses. Mol Cell. 2004;14:277-287.
Lockshin RA, Zakeri Z. Programmed cell death and apoptosis: Origins of the theory. Nat Rev Mol Cell Biol. 2001;2:545-550.
Meier P, Finch A, Evan G. Apoptosis in development. Nature. 2000;407:796-801.
Metzstein MM, Stanfield GM, Horvitz HR. Genetics of programmed cell death in C. elegans: Past, present and future. Trends Genet. 1998;14:410-416.
Nagata S. Apoptosis by death factor. Cell. 1997;88:355-365.
Raff MC. Social control on cell survival and cell death. Nature. 1992;356:397-400.
Reddien PW, Horvitz HR. The engulfment process of programmed cell death in Caenorhabditis elegans. Annu Rev Cell Dev Biol. 2004;20:193-221.
Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. 2004;5:897-907.
Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature. 2000;407:784-788.
Shi Y. Caspase activation: Revisiting the induced proximity model. Cell. 2004;117:855-858.
Strasser A. The role of BH3-only proteins in the immune system. Nat Rev Immunol. 2005;3:189-200.
Vaux DL. Apoptosis timeline. Cell Death Differ. 2002;9:349-354.
Wyllie AH, Kerr JFR, Currie AR. Cell death: The significance of apoptosis. Int Rev Cytol. 1980;68:251-305.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
CHAPTER 46 Programmed Cell Death
The Necessity for Cell Death in Multicellular Organisms
The ability to undergo programmed cell death (Box 46-1) is a built-in latent capacity in virtually all cells of multicellular organisms. Cell death is important for embryonic development, maintenance of tissue homeostasis, establishment of immune self-tolerance, killing by immune effector cells, and regulation of cell viability by hormones and growth factors. It has been proposed that most metazoan cells will die if they fail to receive survival signals from other cells. Abnormalities of the cell death program contribute to a number of diseases, including cancer, Alzheimer’s disease, and acquired immune deficiency syndrome (AIDS).
Programmed Cell Death versus Accidental Cell Death: Apoptosis versus Necrosis
Although cells die in many ways, it is useful to focus on the two poles of this spectrum: apoptosis and necrosis. Apoptosis is the most commonly described pathway for programmed cell death, which is cellular suicide resulting from activation of a dedicated intracellular program (Fig. 46-1). Often, these cells appear completely healthy prior to committing suicide. At the other end of the spectrum is necrosis, also called accidental cell death, which occurs when cells receive a structural or chemical insult that kills them outright (Fig. 46-2). Examples of such insults include extremes of temperature and physical trauma. The cell itself can also initiate necrosis in response to certain stimuli, particularly when induction of apoptosis is inhibited. In contrast to the orderly biochemical pathways of apoptosis, which involve the action of enzyme cascades and the consumption of ATP, necrosis typically involves a collapse of normal cell physiology as a result of ATP depletion.
Necrosis corresponds to what most of us naively imagine cell death would be like. Owing to lack of cellular homeostasis, water rushes into the dying cell, causing it to swell greatly so that the plasma and organelle membranes burst. As a result, the cell undergoes a generalized process of autodigestion and dissolution, culminating in the spilling of the cytoplasmic contents out into the surroundings (Fig. 46-2). This, in turn, produces local inflammation as phagocytic cells are activated, flock to the site, and ingest the debris (see Chapter 22). Because agents that damage cells act over areas that are large in comparison to the size of a single cell, necrosis often involves large groups of neighboring cells.
In contrast to necrosis, apoptotic cells shrink rather than swelling, as part of a reproducible pattern of structural alterations of both the nucleus and cytoplasm (Fig. 46-1). Apoptosis is a two-stage process. On receipt of the pro-apoptotic signal that triggers the pathway to death, cells enter a latent phase of apoptosis (Fig. 46-3). Although committed to a pathway that leads to their inevitable demise at some later time, cells in the latent phase look as healthy as their neighbors. The duration of the latent phase of apoptosis is extremely variable, ranging from a few hours to several days. The reason for this variability is not known.
Ultimately, the cells enter the execution phase of apoptosis, lasting about an hour, during which they undergo dramatic morphologic and physiological changes. These include (1) loss of microvilli and intercellular junctions (Fig. 46-4); (2) shrinkage of the cytoplasm; (3) dramatic changes in cytoplasmic motility with activation of violent blebbing (Fig. 46-5); (4) loss of plasma membrane asymmetry, with the distribution of phosphatidylserine being randomized so that it appears in the outer membrane leaflet; (5) hypercondensation of the chromatin and its collapse against the nuclear periphery; and (6) the “explosive” fragmentation of the cell into membrane-enclosed apoptotic bodies that contain remnants of the nucleus, mitochondria, and other organelles. The plasma membrane retains its integrity throughout the entire process. All of these changes are instigated by the action of a specific set of death-inducing proteases, discussed at length later.
In tissues, apoptotic bodies are rapidly phagocytosed by surrounding cells that recognize the phosphatidylserine and other markers exposed on their surface (Fig. 46-6). Apoptosis can thus be considered to be the disassembly of the cell into “bite-sized” vesicles. Because these vesicles remain membrane bound, the cellular contents are not released into the environment. It is important to note that surface markers on apoptotic bodies cause cells that ingest them to secrete anti-inflammatory cytokines. As a result, apoptotic death does not lead to an inflammatory response.
Nonapoptotic Programmed Cell Death
The terms apoptosis and programmed cell death are sometimes viewed as synonymous. However, in a number of well-documented systems, cells undergo programmed cell death without the dramatic structural changes that classically define apoptosis. Thus, all apoptosis is programmed cell death, but the converse is not necessarily true.
When the adult tobacco hawkmoth emerges from its cocoon, its intersegmental muscles undergo programmed cell death that differs in several ways from apoptosis as described earlier. The chromatin does not condense; DNA is not digested; and cytoplasm does not “boil.” Instead, a polyubiquitin gene is induced and plays an important role in intracellular protein degradation (see Chapter 23). Thus, although these muscle cells unquestionably undergo programmed cell death, they apparently do not use the apoptosis pathway.
Classes of Cells That Undergo Programmed Cell Death
At least six distinct classes of cells undergo programmed cell death (examples are given in Fig. 46-7).
Developmentally Defective Cells
During molecular maturation of T-lymphocyte antigen receptors (see Figs. 27-8 and 28-8), immature T cells in the thymus (known as thymocytes) rearrange the genes encoding the receptor a and b chains. Many newly created receptors bind to foreign antigens, but others interact with self-antigens. Cells with receptors recognizing self-antigens are potentially harmful and are eliminated through apoptosis in a process known as negative selection (Fig. 46-8). The drug cyclosporin A, which inhibits apoptosis in thymocytes, can cause autoimmune disease.
To function properly, the T-cell receptor must recognize major histocompatibility complex (MHC) glycoproteins on other cells during antigen presentation (see Fig. 27-8). T lymphocytes whose T-cell receptors cannot interact with the spectrum of MHC glycoproteins expressed in a given individual are ineffective in the immune response. These cells die by apoptosis in a process known as positive selection (Fig. 46-8). Overall, defects in T-cell receptor assembly are extremely common, and up to 95% of immature T cells die by apoptosis without leaving the thymus.
Similar positive and negative selection steps occur during the maturation of B lymphocytes (see Fig. 28-8), which is accomplished by a combination of gene rearrangements and facilitated mutagenesis. B lymphocytes expressing antibodies directed against self-antigens or producing antibodies whose affinity for antigen is below a critical threshold are eliminated through apoptosis.
Excess Cells
The use of programmed cell death for quality control during development is not limited to the immune system but is also extremely important during brain development. Embryonic ganglia often have many more neurons than are required to enervate their target muscles. Production of excess cells is part of a Darwinian strategy to ensure that a sufficient number of axons reach their targets. Excess neurons that fail to make appropriate connections have no function and are eliminated by programmed cell death. Up to 80% of neurons in certain developing ganglia die in this way. Because of the importance of apoptosis during its development, the brain is often seriously affected in mice that are engineered to lack components of the apoptotic pathway.
Cells That Serve No Function
Mammals also use programmed cell death to eliminate obsolete tissues during development. For example, in humans, the digits of hands and feet are connected by a tissue webbing during embryogenesis. Cells in this webbing serve no purpose in the adult and are eliminated by programmed cell death (Fig. 46-7).
Cells Whose Cell Cycle Is Perturbed
Chapters 40 to 43 describe how biochemical circuits called checkpoints regulate the cell cycle. If DNA is damaged, checkpoint activation blocks cell-cycle progression while repair processes operate. An important downstream effector of checkpoints, the p53 transcription factor, induces the expression of genes encoding proteins that arrest the cell cycle as well as genes encoding proteins that induce cell death. It is generally thought that if the damage cannot be repaired quickly, the pro-death factors win out, and the outcome is apoptosis. Types of DNA damage that commonly trigger cell death are double-strand breaks induced by ionizing radiation and DNA breaks or other damage induced by chemotherapeutic agents.
A second important cell-cycle checkpoint regulates the transition from the G1 phase to the S phase. Passage of the restriction point (see Fig. 41-7) represents the commitment of the cell to undergo another cycle of DNA replication and division. Restriction point control centers on the regulation of the E2F family of transcription factors. However, E2F not only regulates genes that promote cell-cycle progression; it also induces the expression of genes that promote apoptosis. It is now thought that if E2F is activated too strongly, as, for example, where restriction point control has broken down (see Fig. 41-10), its function as a death inducer takes over, and the cells undergo apoptosis. Cells that die in response to inappropriate signals to proliferate include those that are infected by certain viruses or overexpress genes involved in cell proliferation (such as c-myc and c-fos [Fig. 46-14]). This ability to recognize an inappropriate stimulus to proliferate and respond to it by undergoing apoptosis may be an important defense against cancer.
Virus-Infected Cells
At least part of the loss of mature CD4+ T helper cells (see Fig. 28-8) in people who are infected with HIV-1 results from programmed cell death. When exposed to agents that normally stimulate cell proliferation, these cells instead undergo apoptosis. Paradoxically, it ap-pears that many of these dying cells are not themselves infected with HIV.
Genetic Analysis of Programmed Cell Death
Several key components that are involved in the apoptotic execution of mammalian cells were first identified by a genetic analysis of the nematode worm Caenorhabditis elegans. Because C. elegans is optically clear, it is possible to see every cell in a developing worm by using differential interference contrast optics (see Fig. 6-2). This enabled investigators to develop a complete fate map for C. elegans that traces the lineage of each cell in an adult worm back to the fertilized egg. These studies led to the surprising discovery that programmed cell death is one of the most common fates for newborn C. elegans cells. Of the 1090 somatic cells that are produced during embryogenesis of the C. elegans hermaphrodite, 131 undergo programmed cell death at reproducible locations and times.
Mutations in at least 14 C. elegans genes affect programmed cell death (Fig. 46-9
[/not-level-membership-for-basic-science-category]
