CHAPTER 95 PRION DISEASES
Although the human forms of prion disease, including Creutzfeldt-Jakob disease (CJD), are rare, this group of fatal disorders of the central nervous system has become the subject of extensive scientific and public interest. Prion diseases affect both humans and animals (Table 95-1); are caused by infectious agents, which may consist only of a host-encoded protein; and share properties that pose a major challenge to animal and public health (Table 95-2). The critical difference between prion diseases and other neurodegenerative conditions is that they are transmissible experimentally and, sometimes, naturally. The hypothesis that bovine spongiform encephalopathy (BSE) has been transmitted from cattle to the human population as variant CJD (vCJD)1 and the possibility of secondary transmission through blood transfusion2,3 have increased the level of interest in and concern about these diseases.
| Human | Animal |
|---|---|
| Sporadic | Infectious |
| Sporadic CJD | Scrapie in sheep and goats |
| Genetic | Transmissible mink encephalopathy |
| Genetic CJD | BSE |
| Gerstmann-Sträussler-Scheinker disease | |
| Fatal familial insomnia | |
| Infectious | |
| Kuru | |
| Iatrogenic CJD | |
| Variant CJD* |
BSE, bovine spongiform encephalopathy; CJD, Creutzfeldt-Jakob disease.
TABLE 95-2 Characteristics of Prion Diseases
ETIOLOGY AND PATHOGENESIS
According to the prion hypothesis, PrPSc is the infectious agent and acts as a template for its own replication without additional informational material such as DNA or RNA.4 Evidence of the synthetic production of infectious PrP provides strong support to this hypothesis,5 although whether this theory adequately explains the existence of multiple strains of infectious agent remains controversial. After exposure to infection, there is a prolonged incubation period, extending to years or even decades in natural disease, during which there is no evidence of clinical disease, despite the accumulation of PrPSc and infection in peripheral tissues, mainly in the lymphoreticular system. There is no host immune response and no available serological test to identify the presence of infection; this implies that infected animals may enter the human food chain and infected humans may potentially act as blood donors.
Prion diseases are unique in that they occur in sporadic, genetic, and infectious forms.4 According to the prion theory, this is explained by spontaneous conversion of the normal protein to the abnormal form in sporadic disease, by instability in protein structure in cases associated with mutations of the prion protein gene (PRNP), and by transmission of self-replicating PrPSc in infectious forms. The absence of detectable PrPSc in some types of experimental prion disease has led to the hypothesis that it is intermediate forms of PrP, rather than the end-stage protein (PrPSc or PrP27–30), identified in postmortem brain samples, that are infectious. Whatever the structure of infectious PrP, these agents are remarkably resistant to normal sterilizing techniques,6 which adds to the concerns about onward transmission of infection.
HUMAN PRION DISEASES
Epidemiology
Sporadic Creutzfeldt-Jakob Disease
This rare disease occurs worldwide with mortality rates of 1 to 2 cases per million population per year in systematic surveys.7 The cause of sporadic CJD (sCJD) is unknown, and there is no good evidence of clustering of cases to indicate an environmental source of infection and no consistent risk factors identified in case-control studies.8 sCJD occurs predominantly in the older age groups, with a mean age at death of about 66 years, but younger patients can be affected, and a handful of teenagers with sCJD have been described.
Genetic Human Prion Diseases
Genetic Creutzfeldt-Jakob disease, Gerstmann-Sträussler-Scheinker disease, and fatal familial insomnia are all associated with, and perhaps caused by, mutations in PRNP, with variations in clinical and pathological phenotypes that are related to the specific mutation.9 Genetic human prion diseases have been found in most countries, but the frequency of these cases varies; some countries such as Slovakia, Israel, and Italy have high localized incidences.10 Overall, in genetic cases, age at death is approximately 10 years younger than that in sCJD.
Iatrogenic Creutzfeldt-Jakob Disease
This form of CJD is caused by the transmission of infection from person to person in the course of medical or surgical treatment.11 Numerically, the most frequent means of transmission have been through human dura mater grafts (168 cases worldwide) and human pituitary growth hormone (180 cases), although, in rare cases, other vectors have been implicated, including neurosurgical instruments, corneal grafts, and human pituitary gonadotrophin. Incubation periods range from 1.5 to more than 30 years and vary according to the route of inoculation.
Kuru
This human prion disease occurred in the Fore region of Papua New Guinea and was caused by transmission of infection through ritual cannibalism.12 Kuru has now largely disappearedsince the cessation of cannibalism in the late 1950s, although there are still some new cases, with incubation periods exceeding 40 years.
Variant Creutzfeldt-Jakob Disease
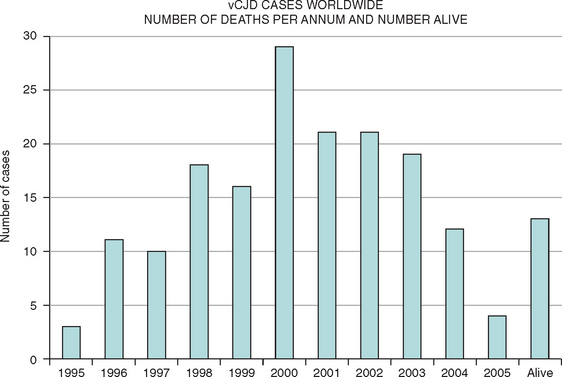
This new disease was identified in 1996, and there is now compelling evidence in support of the hypothesis that vCJD is caused by transmission of BSE to the human population, probably via the human food chain. There have been more than 150 cases of vCJD in the United Kingdom, but the annual mortality rate has dropped since 2000,13 and concerns about a massive epidemic have lessened. Small numbers of cases of vCJD have also been identified in other countries and may be related to export of bovine material from the United Kingdom, travel to the United Kingdom, or indigenous BSE (Fig. 95-1). The mean age at death in vCJD is only 30 years (range, 14 to 74 years). Possible explanations for the young age of patients with vCJD include age-related patterns of dietary exposure, age-related susceptibility, or a combination of these factors.
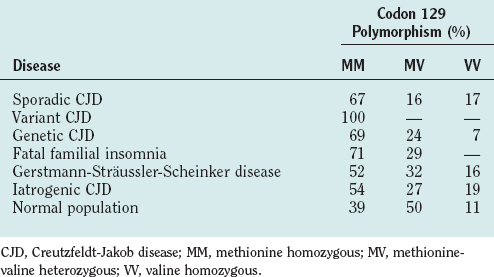
Codon 129 Polymorphism
There is a variable region at codon 129 of human PRNP, which encodes either methionine or valine, leading to three possible genotypes: methionine homozygous (MM), methionine-valine heterozygous (MV), or valine homozygous (VV). Variations in genotype at this locus have a profound effect on susceptibility to disease (Table 95-3) and to disease expression.14 In sCJD, there is an excess of individuals with an MM genotype and, to date, all tested clinical cases of vCJD have been MM. In iatrogenic CJD homozygosity, either MM or VV genotype is a relative risk factor, and in genetic human prion disease, the clinical and pathological phenotype may be influenced by the codon 129 genotype. One example is the codon 178 mutation of PRNP, in which a VV genotype is associated with a disease similar to sCJD, whereas with MM there is an illness (fatal familial insomnia), characterized by early profound autonomic disturbance, particularly affecting sleep.15
Clinical Features of Human Prion Disease
Sporadic Creutzfeldt-Jakob Disease
sCJD typically manifests with a rapidly progressive dementia and myoclonus, often with associated focal neurological features, including ataxia, dysphasia, and pyramidal signs (Table 95-4).16 The median duration of illness is only 5 months from first symptom to death, although a small minority of patients, usually in the younger age groups, survive for more than 1 year. Atypical forms include those with a pure cerebellar onset, with early cortical blindness or with a manifestation mimicking stroke. sCJD has been divided into six subtypes according to the three codon 129 genotypes and the two types of PrP deposited in the brain, as determined by Western blot.17 Patients with the MM type 1 protein and MV type 1 protein subtypes exhibit thetypical features of sCJD, whereas the other four subtypes are often atypical in terms of age, clinical features, and neuropathology.
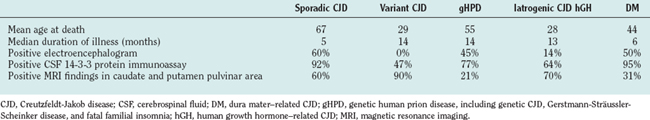
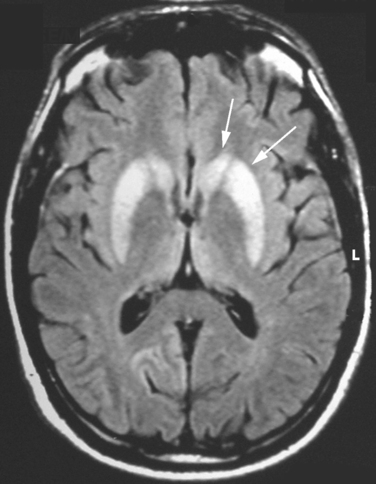
Although the clinical diagnosis of sCJD is usually apparent in the typical forms, investigations are essential for ruling out other conditions, particularly those that are potentially treatable. The electroencephalogram shows pseudo-periodic triphasic complexes in about 60% of cases and, although cerebrospinal fluid (CSF) parameters are usually normal (except for a raised protein level in a minority of cases), the CSF 14-3-3 protein immunoassay has a high sensitivity and specificity for the diagnosis of sCJD in the appropriate clinical context.18 There is increasing evidence that the magnetic resonance imaging (MRI) brain scan may be diagnostically useful; high-intensity signal in the caudate and putamen are evident in the majority of cases on T2-weighted imaging, fluid-attenuated inversion recovery imaging, and diffusion-weighted imaging sequences (Fig. 95-2). Brain biopsy can allow confirmation of the diagnosis during the patient’s life, but this procedure has risks and can yield negative findings as a result of sampling error.
Genetic Human Prion Disease
The clinical features of genetic forms of human prion disease vary according to the mutation in PRNP, although there is also variability both within and between families.19 With some mutations, the clinical features are similar to those of sCJD—for example, with the codon 200 mutation—whereas other mutations are associated with atypical features for CJD, such as a progressive cerebellar syndrome and late cognitive impairment with the codon 102 mutation in Gerstmann-Sträussler-Scheinker disease. Overall, the duration of clinical illness is more protracted in genetic cases than in sCJD, and with some mutations, the illness can last for several years. Although these are dominantly inherited conditions, a significant proportion of patients with genetic cases do not have a family history of a similar disorder, perhaps because of premature death from other causes, nonpaternity, or de novo mutations.
Iatrogenic Creutzfeldt-Jakob Disease
This type of CJD manifests either with features similar to those of sCJD or with a progressive cerebellar syndrome and late cognitive impairment, if this develops at all.11 The factor determining the clinical manifestation is the route of exposure to infection. The sCJD phenotype occurs with effective exposure directly in the central nervous system—for example, with exposure to contaminated neurosurgical instruments—whereas a cerebellar syndrome occurs with a peripheral route of inoculation of infection in human pituitary hormone recipients. The duration of illness is similar to that of sCJD with central infection, but it averages about 12 months with peripheral exposure.
Variant Creutzfeldt-Jakob Disease
The clinical features of vCJD are relatively stereotyped, consistent with a single strain of infectious agent. The early stages are dominated by nonspecific psychiatric symptoms such as depression and anxiety, and in about one third of cases, there are early sensory symptoms, which are usually painful and persistent. This is followed after about 6 months by the development of cerebellar features, often affecting gait; involuntary movements, mainly myoclonus, chorea, or dystonia; and progressive cognitive impairment.20 The median survival length of 14 months is more prolonged than in sCJD.
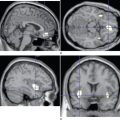
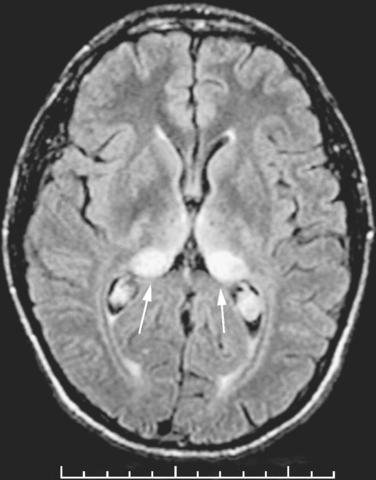
The diagnosis of vCJD is often suspected because of the development of a progressive neuropsychiatric syndrome in a relatively young individual. The electroencephalogram and CSF 14-3-3 protein immunoassay are not diagnostically helpful, but the MRI brain scan reveals a relatively specific abnormality in the form of high-intensity signal in the posterior thalamus (the pulvinar sign) in about 90% of cases (Fig. 95-3). These abnormalities are present on T2-weighted images but are more prominent in fluid-attenuated inversion recovery imaging and diffusion-weighted imaging sequences. Brain and tonsil biopsies can lead to a tissue diagnosis, but both procedures entail risks, and any surgical instruments used in these operations must be destroyed afterward.
Secondary Transmission of Variant Creutzfeldt-Jakob Disease
The level of infectivity in the lymphoreticular system in vCJD is greater than in sCJD, and there is evidence that there may be subclinical vCJD infection in a proportion of the U.K. population, some of whom may act as blood donors. In one case, vCJD was identified in a patient who received a blood donation from an individual who later developed vCJD2; in a second reported case, a recipient of a blood donation from a patient with vCJD was found at postmortem study to have positive PrP staining in the lymphoreticular system but not in the brain.3 This suggests that vCJD may be transmissible through blood transfusion, which is consistent with the demonstration in animal studies of transfusion transmission of experimental BSE in sheep. One of the cases linked to blood transfusion involved a person with heterozygous codon 129 polymorphism,3 which indicates that individuals with this genotype, who make up about 50% of the white population, may be at risk for infection. Numerous measures have been taken to minimize the risk from blood transfusion or blood products in the United Kingdom and many other countries. There is, as yet, no evidence of transmission of vCJD though contaminated surgical instruments.
CONCLUSION
Brown P, Preece M, Brandel J-P, et al. Iatrogenic Creutzfeldt-Jakob disease at the millennium. Neurology. 2000;55:1075-1081.
Collinge J. Variant Creutzfeldt-Jakob disease [Review]. Lancet. 1999;354:317-323.
Gajdusek DC. Le Kuru. Washington, DC: U.S. Department of Health and Human Services, 1981;25-57.
Prusiner SB. Prion diseases of humans and animals. J R Coll Physicians Lond. 1994;28(2, Suppl):1-30.
Will RG, Alpers MP, Dormont D, et al. Infectious and sporadic prion diseases. In: Prusiner SB, editor. Prion Biology and Diseases. 2nd ed. New York: Cold Spring Harbor Laboratory Press; 2004:629-671.
1 Will RG, Ironside JW, Zeidler M, et al. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet. 1996;347:921-925.
2 Llewelyn CA, Hewitt PA, Knight RSG, et al. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet. 2004;363:417-421.
3 Peden AH, Head MW, Ritchie DL, et al. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet. 2004;364:527-529.
4 Prusiner SB. Prion Diseases of Humans and Animals. J R Coll Physicians Lond. 1994;28(2, Suppl):1-30.
5 Legname G, Baskakov IV, Nguyen H-OB, et al. Synthetic mammalian prions. Science. 2004;305:673-676.
6 Taylor DM, McConnell I, Brown DA, et al. New inactivation data for the agents of bovine spongiform encephalopathy (BSE) and scrapie. Neuropathol Appl Neurobiol. 1994;20:510.
7 Ladogana A, Puopolo M, Croes EA, et al. Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology. 2005;64:1586-1591.
8 Wientjens DPWM, Davanipour Z, Hofman A, et al. Risk factors for Creutzfeldt-Jakob disease: a reanalysis of case-control studies. Neurology. 1996;46:1287-1291.
9 Pocchiari M. Prions and related neurological diseases. Mol Aspects Med. 1994;15:195-291.
10 Ladogana A, Puopolo M, Poleggi A, et al. High incidence of genetic human transmissible spongiform encephalopathies in Italy. Neurology. 2005;64:1592-1597.
11 Brown P, Preece M, Brandel J-P, et al. Iatrogenic Creutzfeldt-Jakob disease at the millennium. Neurology. 2000;55:1075-1081.
12 Alpers M. Epidemiology and clinical aspects of kuru. In: Prusiner SB, McKinley MP, editors. Prions. New York: Academic Press; 1987:451-465.
13 Andrews NJ, Farrington CP, Ward HJT, et al. Deaths from variant Creutzfeldt-Jakob disease in the UK. Lancet. 2003;361:751-752.
14 Parchi P, Castellani R, Capellari S, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1996;39:767-778.
15 Montagna P, Cortelli P, Avoni P, et al. Clinical features of fatal familial insomnia: phenotypic variability in relation to a polymorphism at codon 129 of the prion protein gene. Brain Pathol. 1998;8:515-520.
16 Knight R, Collins S. Human prion diseases: cause, clinical and diagnostic aspects. In: Rabenau HF, Cinatl J, Doerr HW, editors. Prions. A challenge for science, medicine and public health system. Basel, Switzerland: Karger; 2001:68-92.
17 Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224-233.
18 Green AJE. Use of 14–3–3 in the diagnosis of Creutzfeldt-Jakob disease. Biochem Soc Trans. 2002;30:382-386.
19 Gambetti P, Petersen RB, Parchi P, et al. Inherited prion diseases. In: Prusiner SB, editor. Prion Biology and Diseases. New York: Cold Spring Harbor Laboratory Press; 1999:509-583.
20 Will RG, Zeidler M, Stewart GE, et al. Diagnosis of new variant Creutzfeldt-Jakob disease. Ann Neurol. 2000;47:575-582.