CHAPTER 18 SCHIZOPHRENIA AND SCHIZOPHRENIA-LIKE PSYCHOSIS
The first part of this chapter deals with schizophrenia as a primary illness, and the second describes schizophrenia-like features that appear in the context of other neurological illness.
SCHIZOPHRENIA
Schizophrenia is a brain disease as common as multiple sclerosis that impairs the ability to work, independent living, and interpersonal relationships. The effect of schizophrenia on health care budgets is substantial and accounts for 1.5% to 3% of total national health care expenditure.1
Kraeplin2 introduced the term dementia praecox to refer to a cluster of symptoms that included catatonia and paranoia and carried a poor prognosis. The term schizophrenia was first used by Bleuler,3 who believed that certain “fundamental symptoms” were present in all affected patients. Current diagnostic classifications, such as the International Classification of Mental and Behavioural Disorders, 10th revision (ICD-10)4 and the Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM-IV),5 still require the presence of a cluster of symptoms, in the absence of drug abuse or other organic brain disease, to establish the diagnosis (Table 18-1).
TABLE 18-1 Key Symptoms of Schizophrenia with Exclusion Criteria
Rights were not granted to include this table in electronic media. Please refer to the printed book.
Epidemiology
The incidence of schizophrenia is rather similar across different countries, with rates between 0.16 to 0.42 per 1000 and with a prevalence around 1% when narrow diagnostic criteria are used.6 There are some remote populations with an increased incidence and prevalence, such as the Afro-Caribbean population in the United Kingdom (incidence ratios above 7),7 and others with reduced rates, such as the Hutterites of South Dakota (ratio of observed to predicted mean rates, 0.48),8 and genetic and environmental factors probably contribute to this variability. Schizophrenia is common in men and women equally, and its onset may occur at any age, although it often starts between the ages of 15 and 45, with an earlier onset in men.9
Clinical Features and Natural History
Schizophrenia is characterized by a multitude of symptoms that vary between patients and encompass a variety of mental functions such as perception, emotion, and language (see Table 18-1). The symptoms of schizophrenia are often categorized as positive and negative. Positive symptoms include delusions, passivity phenomena, and hallucinations. Negative symptoms include apathy and social withdrawal. Functional imaging studies (positron emission tomography) have suggested that auditory hallucinations are accompanied by increased blood flow10 in subcortical nuclei, limbic structures, and paralimbic regions, and functional magnetic resonance imaging (MRI) demonstrates increased activation in the inferior frontal and temporal cortex.11 The neural correlates of other symptoms are less well understood.
Psychotic symptoms usually start in late adolescence or early adulthood,12 tend to persist throughout the illness,13 and are often associated with poor psychosocial functioning.14
For many patients, schizophrenia starts with a prodromal period lasting from months to years. The symptoms of the prodromal stage may include depression, anxiety, dysphoria, social withdrawal, and cognitive underfunctioning, as well as attenuated psychotic symptoms.15
The outcome of schizophrenia is variable. Harrison and Eastwood16 found that one third of patients had recovered at follow-up 15 or 25 years later and that for many patients, schizophrenia is a relapsing-remitting disorder. The study also showed that lack of improvement early in the illness is predictive of persistence of symptoms and long-term disability. Other studies have revealed that an early onset of psychosis is associated with a more severe illness, irrespective of duration of illness.17
Cognitive Deficits
Cognitive impairment is an integral feature of schizophrenia. Attention, executive function, and memory are most commonly impaired.18 Cognitive deficits are already present at the onset of psychosis19,20 and are present irrespective of medication.21 Some patients appear to undergo a decline in general intellectual function in the prodromal stages of the illness or during the onset of psychosis.22,23 After the onset of psychosis, cognitive impairments do not generally deteriorate further, which suggests that they are independent of clinical symptoms and the effects of medication.24 Large-scale studies have found that schizophrenic patients perform worse than healthy controls across a range of cognitive tasks,25,26 that cognitive impairment in a given function is predictive of impairment in all others,27 and that this impairment is global, variation between patients being a matter of degree. Other studies have found evidence for a subgroup of patients with isolated executive dysfunction.22,28 Deficits in executive function may be related to an increased genetic susceptibility to schizophrenia29 and may be part of the schizophrenia endophenotype.30 Similarly episodic memory deficits may be associated with an earlier age at onset,29 and this may suggest that early brain insults (e.g., hypoxia) that constitute a risk factor for young age at onset may, through their action on the amygdala and hippocampus, may also be responsible for the memory deficits.
Functional imaging studies have shown reduced blood flow (positron emission tomography31,32) and reduced activation (functional MRI33) in the dorsolateral and other prefrontal areas of the cortex in schizophrenic patients in comparison with normal controls in response to executive function tasks. The neural correlates of other cognitive deficits are less well understood.
The effect of typical neuroleptics on cognition is still controversial, although the blockade of dopamine D2 receptors achieved by these drugs may have a negative effect on cognition.34 In contrast, atypical neuroleptics, with antipsychotic effects not mediated by D2 blockade, may preserve or enhance cognition.35
Genetics
Twin studies in schizophrenia have shown concordance rates of 41% to 65% in monozygotic pairs and 0% to 28% in dizygotic pairs and a heritability rate of 80% of 85%,36 which are suggestive of an important genetic contribution. The genetic risk for an individual increases with the degree of relation to the affected relative: 40.8% if there is an affected monozygotic twin, 5.3% in siblings of affected patients,37 and a lifetime risk ranging from 3.1% to 16.9% in first-degree relatives of schizophrenic probands.38 A younger age at onset is associated with a higher familial risk for schizophrenia.39
Of the susceptibility genes possibly associated with schizophrenia, catechol-O-methyl-transferase (COMT) is the most likely. COMT is predominantly expressed in prefrontal and hippocampal neurons and implicated in interneuronal monoaminergic signaling, especially dopamine. Hemideletion of chromosome 22q11, where COMT maps, results in the velocardiofacial syndrome40 with a greatly increased risk (24% in a study sample of patients with velocardiofacial syndrome) of schizophrenia-like psychosis.41
Other possible susceptibility genes, suggested by association studies, are DISC1 (“disrupted in schizophrenia”), a complex gene with effects on cytoskeletal proteins, cell migration, and membrane trafficking of receptors likely to influence hippocampal structure and function42; neuregulin 1 (NRG1) with effects in signaling and hence in neuronal development and plasticity; dysbindin (DTNBP1), widely expressed in neurons, including those in the dorsolateral prefrontal cortex, hippocampus, and substantia nigra, with effects on trafficking and tethering of receptors N-methyl-d-aspartate [NMDA], nicotinic acid, and γ-amino butyric acid A [GABAA]), and likely to contribute to glutamatergic hippocampal pathology; the regulator of G protein signaling 4, expressed in the dorsolateral prefrontal cortex involved in signaling; and the metabotropic glutamate receptor gene (GRM3), expressed presynaptically in neurons, astrocytes, and oligodendrocytes and likely to affect glutamatergic neurotransmission in the hippocampus and prefrontal cortex. Other genes associated with glutamatergic transmission and implicated in schizophrenia include G72 and d-amino acid oxidase (DAAO), which appear to directly affect NMDA receptors, and proline dehydrogenase (PRODH), which affects glutamatergic synapses by several mechanisms (Fig. 18-1).43,44
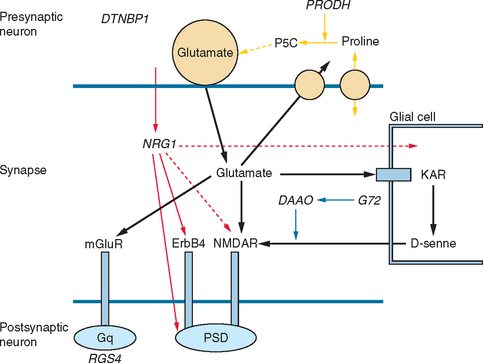
Most of the susceptibility genes so far identified have an effect on the molecular biology of the synapse, particularly glutamatergic synapses, but also influence the dopaminergic and GABAergic systems, thus causing malfunction of cortical microcircuits, which probably explains the pattern of symptoms and cognitive deficits that characterize schizophrenia.44
Neuropathology
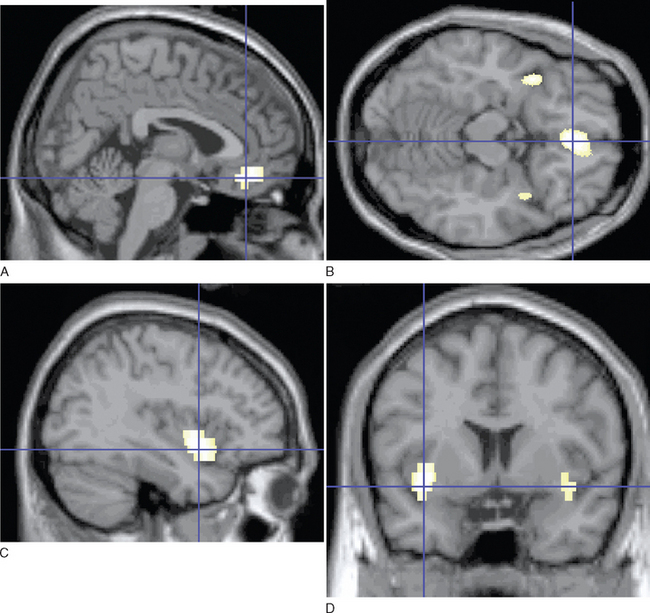
Imaging studies have demonstrated loss of brain volume in schizophrenic patients in comparison with controls45,46 and have highlighted pathological changes in the hippocampus and prefrontal cortext47 and in the superior temporal cortex and thalamus.46 More recent studies with magnetization transfer imaging, a technique sensitive to subtle neuropathological changes (e.g., changes in cell membranes and myelin) have demonstrated diffuse cortical abnormalities in patients with chronic schizophrenia.48 In patients with first episodes,49 these abnormalities are limited to the medial prefrontal cortex, insula, and fasciculus uncinatus in the absence of atrophy. Imaging abnormalities are already evident before or at the time of the first episode of illness (Fig. 18-2)50 and may be present in unaffected relatives,51 which suggests that these abnormalities may be related to the genetic predisposition or early environmental factors, rather than to the illness itself. These changes, modest and often nonspecific, are not diagnostic of schizophrenia and may be present in other psychoses.
The most consistent histological findings are decreases in neuronal size in the hippocampus and neocortex with reduced dendritic arborization and synaptic abnormalities.52 Levels of N-acetyl aspartate, a marker of neuronal integrity, measured in vivo with magnetic resonance spectroscopy, are reduced in the hippocampus53 and prefrontal cortex,54 which is in keeping with these findings. Neuronal loss in the dorsomedial nucleus of the thalamus and pulvinar have been less consistently reported.55 Reduction in the number of oligodendrocytes, important in myelination and synaptic integrity, whether primary or secondary to these neuronal changes, have also been reported.56 These quantitative alterations of the normal neural circuitry may result in subtle loss of cortical volume and thickness.16
Longitudinal imaging studies have not provided clear evidence of progression of brain abnormalities (see Shenton et al [2001]46 for a review), although loss of cortical volume may occur in the early stages of the illness in subgroups of patients with early onset and severe symptoms.57 Other investigators, using diffusion tensor imaging, have described axonal and myelin abnormalities in the corpus callosum of patients with chronic schizophrenia,58 which are absent at the onset of schizophrenia.59 Neuropathologically, astrogliosis and neurodegenerative changes, including those of Alzheimer’s disease, are not overrepresented in schizophrenia, which suggests that apparent clinical deterioration may be difficult to explain as a result of a neurodegenerative process.60–62 In contrast, abnormalities of neuronal migration, evidenced by aberrantly located neurons in the lamina II of the entorhinal cortex and neocortical white matter, are strongly suggestive of disruption of normal brain development (for a review, see Harrison [1999]63).
Further evidence for schizophrenia as a disorder of normal brain development is found in epidemiological studies that suggest early environmental factors that increase the risk for schizophrenia in later life. Evidence points toward a small winter-spring excess of births among patients with schizophrenia,64 as well as exposure to the influenza virus prenatally.65,66 Obstetrical complications are also linked to this risk, although the mechanisms are uncertain.67
Neurochemistry
The dopamine hypothesis has been the chief neurochemical hypothesis in schizophrenia since the early 1960s68 and is supported by observations that dopamine D2 receptor blocking is common to all antipsychotic drugs.69 The mechanism and exact location of dopaminergic abnormalities in schizophrenia still remain unclear, and the dopamine hypothesis has undergone some revision since its initial inception. According to the hypothesis as it stands now, there exists a dopaminergic imbalance between the hyperactive subcortical, mesolimbic dopamine pathways (resulting in positive symptoms), and the hypoactive mesocortical dopaminergic connections to the prefrontal cortex (resulting in negative symptoms and cognitive impairment).70 The alternative glutamate hypothesis is based on the observation that the NMDA glutamate receptor antagonist phencyclidine causes psychosis that resembles both the positive and negative symptoms of schizophrenia.71 The glutamate hypothesis in its most simplified form is that a reduction in glutamate neurotransmission at the NMDA receptor results in symptoms of schizophrenia.72 However, the dopamine and glutamate hypotheses are not mutually exclusive, inasmuch as reciprocal synaptic relations between forebrain dopaminergic projections and glutamatergic systems have been described.73
There is also evidence of dysfunction of the GABAergic system in schizophrenia (see Benes and Berretta74): namely, the reduction of specific GABAergic interneurons (paralbumin-immunoreactive cells) in the prefrontal cortex and hippocampus.75 Various subtypes of GABA neurons provide both inhibitory and disinhibitory modulation of cortical and hippocampal circuits believed to be involved in schizophrenia. The evidence for the role of the serotonergic system in schizophrenia is unclear, although there are serotonergic hallucinogens that block 5-hydroxytryptamine 2 receptors,76 and 5-hydroxytryptamine 2A receptor antagonism may contribute to the efficacy of atypical neuroleptics.77
The Pathophysiology of Schizophrenia
One of the greatest challenges facing psychiatry today is to propose an explanatory theory that encompasses the often disparate facts known about schizophrenia and is able to accommodate emerging knowledge. There is evidence for a genetic predisposition, and a number of possible candidate genes with effects on the molecular biology of the synapse, as well as on the dopaminergic and GABAergic systems, have been identified.44 Histological findings such as aberrant neuronal clusters in the entorhinal cortex and an absence of gliosis also imply a neurodevelopmental etiology. Early environmental insults are additionally implicated, and these include complications of pregnancy and delivery.67 Abnormalities in cortical circuitry, induced by developmental and environmental factors, may limit neuronal information-processing capacity, and demands made on this malfunctioning system later in life may result in the emergence of psychotic symptoms and cognitive deterioration. The neurodevelopmental theory presupposes that pathological changes are not progressive and that changes in brain volume detected during the illness may be the consequence of disease-related changes in neuroplasticity (e.g., unstimulating environments, medication).78
Treatment of Schizophrenia
Drug Treatment
Neuroleptics have been used in the treatment of acute and chronic psychosis since the 1950s. The antipsychotic effect of the first-generation typical neuroleptics such as haloperidol and chlorpromazine depends on their action on the dopamine D2 receptors, and hallucinations are blocked when about 70% of the D2 receptors are occupied by neuroleptic drugs.79 Dopamine D3 and D4 receptor antagonism does not appear to be as important for antipsychotic effects.79 Antipsychotic agents may also affect brain structure directly: There are reports, albeit with a small sample size, of reversal of the superior temporal gyrus volume loss with neuroleptic treatment in a 1-year follow-up.80
Although typical antipsychotics have beneficial effects on positive symptoms, they are less effective in treating negative symptoms and cognitive impairment.81 They also have serious unwanted effects, such as extrapyramidal side effects (EPSEs), tardive dyskinesia, and neuroleptic malignant syndrome (NMS).
Clinical EPSEs include acute dystonia, subjective feelings of restlessness (akathisia), and parkinsonism. Positron emission tomographic studies suggest that EPSEs are related to dopamine D2 occupancy in the range of 75% to 80%,82 but D1 antagonism has also been implicated.83 Although traditional neuroleptics do not necessarily lead to EPSEs, the therapeutic window between therapeutic effect and EPSE is small, and thus many patients receiving these medications have EPSEs.
Tardive dyskinesia is a potentially irreversible side effect of long-term treatment with neuroleptic drugs and is characterized by abnormal involuntary hyperkinetic movements such as grimacing, lip smacking, tongue protrusion, and rapid eye blinking. Involuntary rapid movements of the fingers, arms, legs, and trunk may also occur. Various hypotheses, including overactivity in the striatal dopamine system,84 abnormal GABA-related striatal neurons,85 and free radical production73 have been proposed as pathophysiological mechanisms. Epidemiological data indicate that increasing age86 and female gender are risk factors for tardive dyskinesia.87 The outcome of tardive dyskinesia is more favorable in younger patients.87 Discontinuation of neuroleptic drugs or the use of a drug with fewer EPSEs (e.g., clozapine) is the first action of treatment.
NMS is a life-threatening syndrome characterized by fever, muscular rigidity, and raised serum creatine kinase concentration. The incidence has been estimated to be between 0.07 and 0.9,88,89 and onset can occur within hours but is usually 4 to 14 days after starting neuroleptic therapy. The mechanism of NMS is uncertain, but the most widely accepted mechanism is of blockage of dopamine receptors in the nigrostriatal tracts.90 An alternative hypothesis suggests an imbalance between serotonin and dopamine.91 Risk factors for developing NMS include dehydration, male gender, the presence of organic brain disease or mental retardation, and rapid escalation of ingestion of neuroleptic drugs.92,93 The mortality rate has been reported at 8%,94 but most patients recover within 14 days.93 In NMS, antipsychotics should be discontinued, and, in general, intensive care treatment is required. Drugs that have been used in the treatment of NMS include dopamine agonists such as bromocriptine and apomorphine. If restarting a neuroleptic is deemed necessary, it is worth switching to a neuroleptic in a different chemical class and with a lower D2 affinity than the drug that produced the NMS.95
Maintenance treatment with antipsychotic medication decreases relapse rates96; however, a substantial proportion of patients suffer relapse despite taking medication, and poor compliance is also a problem.
A second generation of so-called atypical neuroleptics (e.g., clozapine, risperidone, quetiapine, olanzapine) has been developed with a spectrum of receptor effects different from those of typical neuroleptics and less severe side effects, resulting in better compliance and improved therapeutic outcome.97
Clozapine is the prototype of this class of neuroleptic. Kane and colleagues98 established the antipsychotic efficacy of clozapine in previously treatment-resistant patients without side effects, and this has been confirmed in other studies.99,100 Clozapine also has an effect on negative symptoms and cognitive functioning.100 One of the properties of clozapine is its high dissociation constant at the D2 receptor, which results in fewer EPSEs.101 Clozapine also has affinities for other nondopamine receptors such as 5-hydroxytryptamine 2, and its α2-adrenoceptor antagonism is believed to contribute to the freedom from EPSEs (see Reynolds102 for review). Clozapine may cause potentially fatal agranulocytosis, and regular blood monitoring is required. Other unwanted effects include hypersalivation, weight gain, and a lowering of the seizure threshold. The increased risk of stroke in older adults with dementia has also been linked to atypical antipsychotics.103
Recent developments in understanding the mechanism of action of antipsychotic medications has led to the development of the partial D2 and 5-hydroxytryptamine 1A receptor agonist aripiprazole,104 which is antipsychotic and possibly without EPSEs.105 Future drug treatments may continue to focus on partial agonism to improve antipsychotic symptoms or focus on NMDA-glutamatergic modulators.106
In the United Kingdom, the guidelines of the National Institute of Clinical Excellence recommend the use of atypical neuroleptics as first-line treatment, because of fewer side effects and efficacy comparable with those of typical neuroleptics, and the use of clozapine in treatment-resistant patients. Early treatment is recommended because of the possible association between a longer duration of untreated psychosis and poor outcome.107
Psychological Treatments
The use of psychological treatments has been shown to improve the outcome of schizophrenia when they are integrated with pharmacological treatments.
Promising results have been reported with cognitive-behavioral therapy. A meta-analysis by Gould and associates108 showed it to be effective for positive symptoms. By challenging the patient’s interpretation of psychotic phenomena, this therapy reduces the frequency of relapse and the degree of distress. There is also evidence to suggest that cognitive-behavioral therapy may ameliorate negative symptoms.109 Cognitive-behavioral therapy has also been used in the prodromal phase of the illness in an attempt to delay or avoid the development of florid symptoms, with promising results.110
Family therapy has also been helpful as an adjuvant to pharmacological treatment in patients exposed to home environments with high levels of criticism, hostility, or emotional over-involvement.111,112 Supported employment programs are also promising.113
SCHIZOPHRENIA-LIKE PSYCHOSIS IN NEUROLOGICAL ILLNESS
The association of epilepsy with schizophrenia-like symptoms was described in the 1960s,114 and similar symptoms have been described in association with other neurological diseases. These psychoses are included in the DSM-IV under the category of “Psychotic Disorder due to a General Medical Condition.” Initially, diseases involving the temporal lobes were believed to be more likely to cause psychotic symptoms,115 but more recent studies have described this association with extratemporal or diffuse pathology,116 and it has been suggested that damage to the dopaminergic limbic projections may be the common mechanism.117
The symptoms are similar to those of schizophrenia, although visual hallucinations, flat affect, passivity feelings, and catatonia may be commoner in patients with neurological disease,118,119 as are complex and specific delusional symptoms (e.g., morbid jealousy, erotomania, delusional parasitosis). Treatment follows the same protocol as that of schizophrenia, but the choice of medication may be dictated by the underlying neurological disease (e.g., danger of aggravating symptoms in patients with Parkinson’s disease).
Epilepsy
The classification of psychotic symptoms in epilepsy is based on their temporal relationship to seizures: ictal, postictal, and interictal (see Sachdev120 for review). The combined prevalence of these syndromes is around 2% to 7%.121 In ictal psychosis, the psychotic episode may be an expression of nonconvulsive status epilepticus (i.e., absence status epilepticus, simple partial status epilepticus, and partial complex status epilepticus). Patients may experience delusions and hallucinations while in a state of altered consciousness.122 Ictal psychoses usually last hours or days, and electroencephalographic (EEG) abnormalities help establish the diagnosis.120
Postictal psychoses account for 25% of psychotic episodes of epilepsy.123 They are usually brief and follow clusters of seizures or an increase in seizure frequency, usually after a lucid period of up to 1 week.124 EEG abnormalities may persist during the psychosis, and consciousness may be normal or impaired.121 The psychotic symptoms include paranoid delusions and hallucinations, and mood abnormalities are common.125 When psychotic symptoms develop gradually and in parallel to increases in seizure activity, the term peri-ictal psychosis may be used, in distinction to postictal psychosis.121 In peri-ictal psychosis, consciousness is usually impaired, and the EEG recording reflects increased epileptic activity.121
Interictal psychoses are not related to ictal activity and can develop when seizures are infrequent or fully controlled.120 They are the commonest psychoses and occur in 4% to 18% of epileptic patients, mostly in those with temporal lobe epilepsy.121 They can last from days to weeks but, once established, may follow a chronic course. Paranoid delusions, auditory hallucinations, and affective symptoms are common,120 and cognitive deficits are similar to those of schizophrenia.126 In some patients, the emergence of psychotic symptoms is accompanied by a normalization of the EEG recording127 or coincides with the use of some anticonvulsants such as vigabatrin and zonisamide, and recurrence of seizure activity ameliorates the psychosis.128
Schizophrenia-like psychosis can also appear after temporal lobe surgery for epilepsy, and it remains to be determined whether surgery is incidental and psychosis would have eventually emerged or whether surgery is a risk factor. If the latter is correct, a mechanism similar to forced normalization127 may also operate in these patients. Of postsurgical psychoses, 85% follow right temporal lobectomies and are usually short-lived, although they may follow a chronic course.129
The pathophysiology of the interictal psychosis is uncertain, but the lack of correlation with seizure frequency and the increased incidence in patients with mesial temporal sclerosis suggest that the epilepsy and the psychotic symptoms may be manifestations of a common pathology, rather than that the epileptic activity causes psychosis, although it is possible that stimulation of temporolimbic circuits may predispose to psychosis.120
Interictal psychoses have been considered a contraindication to temporal lobe surgery for the control of epilepsy, but this opinion has been revised,130 and some patients appear to benefit from surgery.131 Antipsychotic medication is usually needed to treat interictal psychosis, and atypical neuroleptics at low doses to avoid increased seizure activity are the best choice.
Parkinson’s Disease and Diffuse Lewy Body Disease
Psychotic symptoms occur in 50% of patients at some time during the illness and increase with age and severity of the disease. Paranoid ideas and visual hallucinations are the most common psychiatric symptoms, occurring in 10% to 40% of patients.132 Visual hallucinations are usually complex and may be accompanied by auditory and tactile hallucinations.133 Vivid nightmares and reduction of rapid eye movement sleep have been observed in 80% of those who experienced psychotic symptoms.132
The pathophysiology of psychosis in Parkinson’s disease is complex. Dopaminergic medication plays a role in treatment, although psychotic symptoms are not related to the dose of medication,134 and they have even been reported in Parkinson’s disease before the introduction of levodopa therapy.134 In young patients, receptor hypersensitivity in the mesolimbic system may be relevant to psychiatric symptoms whereas in elderly patients, serotonergic hyperactivity caused by dopaminergic agonists (i.e., bromocriptine, lisuride, pergolide, cabergoline, ropinirole, and pramipexole) may be more relevant.135 In elderly patients, diffuse Lewy body disease or coexisting pathological processes such as cerebrovascular and Alzheimer’s disease may also play a role.
In Parkinson’s disease, treatment strategies involve fostering good sleep habits and avoiding sensory overload.135 Progressive reduction of anticholinergic medication and dopamine agonists should be attempted, followed by reduction of the levodopa dosage, but in many patients, mobility reduction may become intolerable before psychotic symptoms are controlled, and antipsychotic medication is often required. Atypical antipsychotics should be tried; clozapine, with its low incidence of EPSEs may be the drug of choice.136 Other atypical neuroleptics (quetiapine, olanzapine, risperidone, and aripiprazole) can also be used in these patients, and the treatment may need to be long term.
In diffuse Lewy body disease, reduction of dopaminergic medication should also be attempted. Frequent adverse reactions to neuroleptics has made their use problematic. Anticholinesterase inhibitors may be the drugs of choice, and their use can also be extended to patients with Parkinson’s disease.135
Psychosis in Traumatic Brain Injury
The prevalence of schizophrenia-like symptoms is increased with traumatic brain injury.115,137 In head injuries of moderate severity, damage to the left temporal lobe appears to be a risk factor for psychosis.138 In subjects with a genetic vulnerability, minor trauma may be the trigger, rather than the cause of psychosis.139
Psychotic symptoms tend to appear 4 or 5 years after the injury, but their appearance could be delayed by many years. Psychotic symptoms that appear immediately after the injury are usually part of a confusional state and tend to be mild and transient. As in other schizophrenia-like psychosis, symptoms may follow a prodrome characterized by social isolation and nonspecific symptoms. Mood changes and agitation are common, and negative symptoms infrequent. Diffuse cognitive deterioration often accompanies psychotic symptoms in these patients.138 Chronicity is the rule, and treatment with neuroleptics is often required, although comprehensive studies of outcome are not available.
Demyelinating Diseases
Schizophrenia-like psychosis is uncommon in multiple sclerosis. Psychotic symptoms tend to appear when muscular sclerosis is well established, usually in patients with lesions in temporal lobe white matter, and usually respond to antipsychotic medication.140 In contrast, psychotic symptoms are far more frequent in metachromatic leukodystrophy, an autosomal recessive disease resulting from a mutation on chromosome 22q that leads to a deficit of the enzyme arylsulfatase A and accumulation of metachromatic material (sulfatides) in the brain and peripheral nervous system, which causes demyelination.141 Patients with the juvenile- and adult-onset forms of metachromatic leukodystrophy have higher arylsulfatase A activity and have a more protracted course than do those with the late infantile forms, and they often present with a schizophrenia-like illness.141 Usually it is only when neurological symptoms, progressive cognitive decline leading to dementia, or white matter MRI abnormalities become apparent that metachromatic leukodystrophy is diagnosed.142 Myelin abnormalities in the prefrontal white matter and frontotemporal connections, detectable by MRI, are likely to play a role in the emergence of psychosis. The high prevalence of psychosis in this age group is in contrast to the low prevalence in multiple sclerosis,143 which suggests that age may determine the psychiatric manifestations of demyelination.31 Treatment of symptoms is based on isolated case reports that indicate that psychosis in patients with metachromatic leukodystrophy improves with neuroleptic treatment.31
Harrison PJ. The neuropathology of schizophrenia. A critical review of the data and their interpretation. Brain. 1999;122:593-624.
Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10:40-68.
Joyce E, Huddy V. Defining the cognitive impairment in schizophrenia. Psychol Med. 2004;34:1151-1155.
Shenton ME, Dickey CC, Frumin M, et al. A review of MRI findings in schizophrenia. Schizophr Res. 2001;49:1-52.
Rapoport JL, Giedd JN, Blumenthal J, et al. Progressive cortical change during adolescence in childhood-onset schizophrenia. A longitudinal magnetic resonance imaging study. Arch Gen Psychiatry. 1999;56:649-654.
1 Knapp M, Mangalore R, Simon J. The global costs of schizophrenia. Schizophr Bull. 2004;30:279-293.
2 Kraeplin E. Psychiatrie. Ein Lehrbuch für Studirende und Aerzte. Fünfte, vollständig umgearbeitete Auflage. Leipzig: Barth Verlag, 1896.
3 Bleuler E. Dementia praecox oder die Gruppe der Schizofrenien. In: Aschaffenburg G, editor. Handbuch der Psychiatrie. Leipzig: G. Aschaffenburg, 1911–1928.
4 World Health Organization. The ICD-10 Classification of Mental and Behavioural Disorders: Clinical Descriptions and Diagnostic Guidelines. Geneva: World Health Organization, 1992.
5 American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 4th ed. Washington, DC: American Psychiatric Association, 1994.
6 Jablensky A, Sartorius N, Ernberg G, et al. Schizophrenia: manifestations, incidence and course in different cultures. A World Health Organization ten-country study. Psychol Med Monogr Suppl. 1992;20:1-97.
7 Harrison G, Glazebrook C, Brewin J, et al. Increased incidence of psychotic disorders in migrants from the Caribbean to the United Kingdom. Psychol Med. 1997;27:799-806.
8 Nimgaonkar VL, Fujiwara TM, Dutta M, et al. Low prevalence of psychoses among the Hutterites, an isolated religious community. Am J Psychiatry. 2000;157:1065-1070.
9 Hafner H, Hambrecht M, Loffler W, et al. Is schizophrenia a disorder of all ages? A comparison of first episodes and early course across the life-cycle. Psychol Med. 1998;28:351-365.
10 Silbersweig DA, Stern E, Frith C, et al. A functional neuroanatomy of hallucinations in schizophrenia. Nature. 1995;378:176-179.
11 Shergill SS, Brammer MJ, Williams SC, et al. Mapping auditory hallucinations in schizophrenia using functional magnetic resonance imaging. Arch Gen Psychiatry. 2000;57:1033-1038.
12 Hafner H, Maurer K, Loffler W, et al. The influence of age and sex on the onset and early course of schizophrenia. Br J Psychiatry. 1993;162:80-86.
13 Arndt S, Andreasen NC, Flaum M, et al. A longitudinal study of symptom dimensions in schizophrenia. Prediction and patterns of change. Arch Gen Psychiatry. 1995;52:352-360.
14 Fenton WS, McGlashan TH. Testing systems for assessment of negative symptoms in schizophrenia. Arch Gen Psychiatry. 1992;49:179-184.
15 Davidson L, McGlashan TH. The varied outcomes of schizophrenia. Can J Psychiatry. 1997;42:34-43.
16 Harrison PJ, Eastwood SL. Neuropathological studies of synaptic connectivity in the hippocampal formation in schizophrenia. Hippocampus. 2001;11:508-519.
17 Suvisaari JM, Haukka J, Tanskanen A, et al. Age at onset and outcome in schizophrenia are related to the degree of familial loading. Br J Psychiatry. 1998;173:494-500.
18 Gold S, Arndt S, Nopoulos P, et al. Longitudinal study of cognitive function in first-episode and recent-onset schizophrenia. Am J Psychiatry. 1999;156:1342-1348.
19 Bilder RM, Lipschutz-Broch L, Reiter G, et al. Intellectual deficits in first-episode schizophrenia: evidence for progressive deterioration. Schizophr Bull. 1992;18:437-448.
20 Hoff AL, Riordan H, O’Donnell DW, et al. Neuropsychological functioning of first-episode schizophreniform patients. Am J Psychiatry. 1992;149:898-903.
21 Joyce E, Hutton S, Mutsatsa S, et al. Executive dysfunction in first-episode schizophrenia and relationship to duration of untreated psychosis: the West London Study. Br J Psychiatry Suppl. 2002;43:s38-s44.
22 Weickert TW, Goldberg TE, Gold JM, et al. Cognitive impairments in patients with schizophrenia displaying preserved and compromised intellect. Arch Gen Psychiatry. 2000;57:907-913.
23 Fuller R, Nopoulos P, Arndt S, et al. Longitudinal assessment of premorbid cognitive functioning in patients with schizophrenia through examination of standardized scholastic test performance. Am J Psychiatry. 2002;159:1183-1189.
24 Joyce E, Huddy V. Defining the cognitive impairment in schizophrenia. Psychol Med. 2004;34:1151-1155.
25 Townsend LA, Malla AK, Norman RM. Cognitive functioning in stabilized first-episode psychosis patients. Psychiatry Res. 2001;104:119-131.
26 Addington J, Brooks BL, Addington D. Cognitive functioning in first episode psychosis: initial presentation. Schizophr Res. 2003;62:59-64.
27 Dickinson D, Iannone VN, Wilk CM, et al. General and specific cognitive deficits in schizophrenia. Biol Psychiatry. 2004;55:826-833.
28 Goldstein G, Shemansky WJ. Influences on cognitive heterogeneity in schizophrenia. Schizophr Res. 1995;18:59-69.
29 Tuulio-Henriksson A, Partonen T, Suvisaari J, et al. Age at onset and cognitive functioning in schizophrenia. Br J Psychiatry. 2004;185:215-219.
30 Glahn DC, Therman S, Manninen M, et al. Spatial working memory as an endophenotype for schizophrenia. Biol Psychiatry. 2003;53:624-626.
31 Weinberger DR, Lipska BK. Cortical maldevelopment, antipsychotic drugs, and schizophrenia: a search for common ground. Schizophr Res. 1995;16:87-110.
32 Meyer-Lindenberg A, Miletich RS, Kohn PD, et al. Reduced prefrontal activity predicts exaggerated striatal dopaminergic function in schizophrenia. Nat Neurosci. 2002;5:267-271.
33 Volz HP, Gaser C, Hager F, et al. Brain activation during cognitive stimulation with the Wisconsin Card Sorting Test—a functional MRI study on healthy volunteers and schizophrenics. Psychiatry Res. 1997;75:145-157.
34 Gallhofer B, Lis S, Meyer-Lindenberg A, et al. Cognitive dysfunction in schizophrenia: a new set of tools for the assessment of cognition and drug effects. Acta Psychiatr Scand Suppl. 1999;395:118-128.
35 Keefe RS, Silva SG, Perkins DO, et al. The effects of atypical antipsychotic drugs on neurocognitive impairment in schizophrenia: a review and meta-analysis. Schizophr Bull. 1999;25:201-222.
36 Cardno AG, Gottesman II. Twin studies of schizophrenia: from bow-and-arrow concordances to Star Wars Mx and functional genomics. Am J Med Genet. 2000;97:12-17.
37 Cardno AG, Marshall EJ, Coid B, et al. Heritability estimates for psychotic disorders: the Maudsley twin psychosis series. Arch Gen Psychiatry. 1999;56:162-168.
38 Gershon ES, DeLisi LE, Hamovit J, et al. A controlled family study of chronic psychoses. Schizophrenia and schizoaffective disorder. Arch Gen Psychiatry. 1988;45:328-336.
39 Kendler KS, MacLean CJ. Estimating familial effects on age at onset and liability to schizophrenia. I. Results of a large sample family study. Genet Epidemiol. 1990;7:409-417.
40 Driscoll DA, Spinner NB, Budarf ML, et al. Deletions and microdeletions of 22q11.2 in velocardiofacial syndrome. Am J Med Genet. 1992;44:261-268.
41 Murphy KC, Jones LA, Owen MJ. High rates of schizophrenia in adults with velocardiofacial syndrome. Arch Gen Psychiatry. 1999;56:940-945.
42 Hodgkinson CA, Goldman D, Jaeger J, et al. Disrupted in schizophrenia 1: association with schizophrenia, schizoaffective disorder, and bipolar disorder. Am J Hum Genet. 2004;75:862-872.
43 Harrison PJ, Owen MJ. Genes for schizophrenia? Recent findings and their pathophysiological implications. Lancet. 2003;361:417-419.
44 Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10:40-68.
45 Lawrie SM, Whalley H, Kestelman JN, et al. Magnetic resonance imaging of brain in people at high risk of developing schizophrenia. Lancet. 1999;353:30-33.
46 Shenton ME, Dickey CC, Frumin M, et al. A review of MRI findings in schizophrenia. Schizophr Res. 2001;49:1-52.
47 Wright IC, Rabe-Hesketh S, Woodruff PW, et al. Meta-analysis of regional brain volumes in schizophrenia. Am J Psychiatry. 2000;157:16-25.
48 Foong J, Symms MR, Barker GJ, et al. Neuropathological abnormalities in schizophrenia: evidence from magnetization transfer imaging. Brain. 2001;124:882-892.
49 Bagary MS, Symms MR, Barker GJ, et al. Gray and white matter brain abnormalities in first-episode schizophrenia inferred from magnetization transfer imaging. Arch Gen Psychiatry. 2003;60:779-788.
50 Gur RE, Turetsky BI, Cowell PE, et al. Temporolimbic volume reductions in schizophrenia. Arch Gen Psychiatry. 2000;57:769-775.
51 McIntosh AM, Job DE, Moorhead TW, et al. Voxel-based morphometry of patients with schizophrenia or bipolar disorder and their unaffected relatives. Biol Psychiatry. 2004;56:544-552.
52 Blennow K, Davidsson P, Gottfries C-G, et al. Synaptic degeneration in thalamus in schizophrenia [Letter]. Lancet. 1996;348:692-693.
53 Maier M, Ron MA, Barker GJ, et al. Proton magnetic resonance spectroscopy: an in vivo method of estimating hippocampal neuronal depletion in schizophrenia. Psychol Med. 1995;25:1201-1209.
54 Bertolino A, Roffman JL, Lipska BK, et al. Reduced N-acetylaspartate in prefrontal cortex of adult rats with neonatal hippocampal damage. Cereb Cortex. 2002;12:983-990.
55 Byne W, Buchsbaum MS, Kemether E, et al. Magnetic resonance imaging of the thalamic mediodorsal nucleus and pulvinar in schizophrenia and schizotypal personality disorder. Arch Gen Psychiatry. 2001;58:133-140.
56 Uranova NA, Vostrikov VM, Orlovskaya DD, et al. Oligodendroglial density in the prefrontal cortex in schizophrenia and mood disorders: a study from the Stanley Neuropathology Consortium. Schizophr Res. 2004;67:269-275.
57 Rapoport JL, Giedd JN, Blumenthal J, et al. Progressive cortical change during adolescence in childhood-onset schizophrenia. A longitudinal magnetic resonance imaging study. Arch Gen Psychiatry. 1999;56:649-654.
58 Foong J, Maier M, Clark CA, et al. Neuropathological abnormalities of the corpus callosum in schizophrenia: a diffusion tensor imaging study. J Neurol Neurosurg Psychiatry. 2000;68:242-244.
59 Price G, Bagary MS, Cercignani M, et al. The corpus callosum in first episode schizophrenia: a diffusion tensor imaging study. J Neurol Neurosurg Psychiatry. 2005;76:585-587.
60 Arnold SE, Trojanowski JQ, Gur RE, et al. Absence of neurodegeneration and neural injury in the cerebral cortex in a sample of elderly patients with schizophrenia. Arch Gen Psychiatry. 1998;55:225-232.
61 Purohit DP, Perl DP, Haroutunian V, et al. Alzheimer disease and related neurodegenerative diseases in elderly patients with schizophrenia: a postmortem neuropathologic study of 100 cases. Arch Gen Psychiatry. 1998;55:205-211.
62 Falke E, Han LY, Arnold SE. Absence of neurodegeneration in the thalamus and caudate of elderly patients with schizophrenia. Psychiatry Res. 2000;93:103-110.
63 Harrison PJ. The neuropathology of schizophrenia. A critical review of the data and their interpretation. Brain. 1999;122:593-624.
64 Torrey EF, Miller J, Rawlings R, et al. Seasonality of births in schizophrenia and bipolar disorder: a review of the literature. Schizophr Res. 1997;28:1-38.
65 Wright P, Takei N, Rifkin L, et al. Maternal influenza, obstetric complications, and schizophrenia. Am J Psychiatry. 1995;152:1714-1720.
66 Brown AS, Begg MD, Gravenstein S, et al. Serologic evidence of prenatal influenza in the etiology of schizophrenia. Obstet Gynecol Surv. 2005;60:77-78.
67 Cannon M, Jones PB, Murray RM. Obstetric complications and schizophrenia: historical and meta-analytic review. Am J Psychiatry. 2002;159:1080-1092.
68 Carlsson A, Lindqvist M. Effect of chlorpromazine or haloperidol on formation of 3-methoxytyramine and normetanephrine in mouse brain. Acta Pharmacol Toxicol (Copenh). 1963;20:140-144.
69 Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science. 1976;192:481-483.
70 Davis KL, Kahn RS, Ko G, et al. Dopamine in schizophrenia: a review and reconceptualization. Am J Psychiatry. 1991;148:1474-1486.
71 Luby ED, Cohen BD, Rosenbaum G, et al. Study of a new schizophrenomimetic drug: sernyl. AMA Arch Neurol Psychiatry. 1959;81:363-369.
72 Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia [Review]. Am J Psychiatry. 1991;148:1301-1308.
73 Carlsson M, Carlsson A. Interactions between glutamatergic and monoaminergic systems within the basal ganglia—implications for schizophrenia and Parkinson’s disease. Trends Neurosci. 1990;13:272-274.
74 Benes FM, Berretta S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology. 2001;25:1-27.
75 Reynolds GP, Beasley CL, Zhang ZJ. Understanding the neurotransmitter pathology of schizophrenia: selective deficits of subtypes of cortical GABAergic neurons. J Neural Transm. 2002;109:881-889.
76 Gouzoulis-Mayfrank E, Hermle L, Thelen B, et al. History, rationale and potential of human experimental hallucinogenic drug research in psychiatry. Pharmacopsychiatry. 1998;31(Suppl 2):63-68.
77 Meltzer HY, Matsubara S, Lee JC. Classification of typical and atypical antipsychotic drugs on the basis of dopamine D-1, D-2 and serotonin 2 pKi values. J Pharmacol Exp Ther. 1989;251:238-246.
78 Weinberger DR, McClure RK. Neurotoxicity, neuroplasticity, and magnetic resonance imaging morphometry: what is happening in the schizophrenic brain? Arch Gen Psychiatry. 2002;59:553-558.
79 Seeman P. Dopamine receptor sequences. Therapeutic levels of neuroleptics occupy D2 receptors, clozapine occupies D4. Neuropsychopharmacology. 1992;7:261-284.
80 Keshavan MS, Haas GL, Kahn CE, et al. Superior temporal gyrus and the course of early schizophrenia: progressive, static, or reversible? J Psychiatr Res. 1998;32:161-167.
81 Hawkins KA, Mohamed S, Woods SW. Will the novel antipsychotics significantly ameliorate neuropsychological deficits and improve adaptive functioning in schizophrenia? Psychol Med. 1999;29:1-8.
82 Farde L, Nordstrom AL, Wiesel FA, et al. Positron emission tomographic analysis of central D1 and D2 dopamine receptor occupancy in patients treated with classical neuroleptics and clozapine. Relation to extrapyramidal side effects. Arch Gen Psychiatry. 1992;49:538-544.
83 Casey DE. Dopamine D1 (and D2 [haloperidol]) antagonists in drug-naïve monkeys. Psychopharmacology (Berl). 1992;107:18-22.
84 Klawans HLJr, Rubovits R. An experimental model of tardive dyskinesia. J Neural Transm. 1972;33:235-246.
85 Gunne LM, Haggstrom JE. Pathophysiology of tardive dyskinesia. Psychopharmacology Suppl. 1985;2:191-193.
86 Jeste DV, Caligiuri MP, Paulsen JS, et al. Risk of tardive dyskinesia in older patients. A prospective longitudinal study of 266 outpatients. Arch Gen Psychiatry. 1995;52:756-765.
87 Kane JM, Jeste DV, Barnes TRE, et al. Tardive Dyskinesia: A Task Force Report of the American Psychiatric Association. Washington, DC: American Psychiatric Association, 1992.
88 Gelenberg AJ, Bellinghausen B, Wojcik JD, et al. A prospective survey of neuroleptic malignant syndrome in a short-term psychiatric hospital. Am J Psychiatry. 1988;145:517-518.
89 Keck PE, Sebastianelli J, Pope HG, et al. Frequency and presentation of neuroleptic malignant syndrome in a state psychiatric hospital. J Clin Psychiatry. 1989;50:352-355.
90 Adnet P, Lesteval P, Krivosic-Horber R. Neuroleptic malignant syndrome. Br J Anaesth. 2000;85:129-135.
91 Ames D, Wirshing W. Ecstasy, the serotonin syndrome and neuroleptic malignant syndrome: a possible link? JAMA. 1993;269:869.
92 Sachdev P, Mason C, Hadzi-Pavlovic D. Case-control study of neuroleptic malignant syndrome. Am J Psychiatry. 1997;154:1156-1158.
93 Addonizio G, Susman VL, Roth SD. Neuroleptic malignant syndrome: review and analysis of 115 cases. Biol Psychiatry. 1987;22:1004-1020.
94 Levinson DF, Simpson GM. Neuroleptic-induced extrapyramidal symptoms with fever: heterogeneity of the “neuroleptic malignant syndrome.”. Arch Gen Psychiatry. 1986;43:839-848.
95 Pelonero AL, Levenson JL, Pandurangi AK. Neuroleptic malignant syndrome: a review. Psychiatr Serv. 1998;49:1163-1172.
96 Gilbert P, Harris MJ, McAdams LA. Neuroleptic withdrawal in schizophrenic patients: a review of the literature. Arch Gen Psychiatry. 1995;52:173-188.
97 Leucht S, Barnes TR, Kissling W, et al. Relapse prevention in schizophrenia with new-generation antipsychotics: a systematic review and exploratory meta-analysis of randomized, controlled trials. Am J Psychiatry. 2003;160:1209-1222.
98 Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45:789-796.
99 Simpson GM, Josiassen RC, Stanilla JK, et al. Double-blind study of clozapine dose response in chronic schizophrenia. Am J Psychiatry. 1999;156:1744-1750.
100 Wahlbeck K, Cheine M, Essali A, et al. Evidence of clozapine’s effectiveness in schizophrenia: a systematic review and meta-analysis of randomized trials. Am J Psychiatry. 1999;156:990-999.
101 Seeman P, Tallerico T. Antipsychotic drugs which elicit little or no parkinsonism bind more loosely than dopamine to brain D2 receptors, yet occupy high levels of these receptors. Mol Psychiatry. 1998;3:123-134.
102 Reynolds GP. Receptor mechanisms in the treatment of schizophrenia. J Psychopharmacol. 2004;18:340-345.
103 Wooltorton E. Risperidone (Risperdal): increased rate of cerebrovascular events in dementia trials. CMAJ. 2002;167:1269-1270.
104 Lawler CP, Prioleau C, Lewis MM, et al. Interactions of the novel antipsychotic aripiprazole (OPC-14597) with dopamine and serotonin receptor subtypes. Neuropsychopharmacology. 1999;20:612-627.
105 Taylor DM. Aripiprazole: a review of its pharmacology and clinical use. Int J Clin Pract. 2003;57:49-54.
106 Heresco-Levy U. N-Methyl-D-aspartate (NMDA) receptor-based treatment approaches in schizophrenia: the first decade. Int J Neuropsychopharmacol. 2000;3:243-258.
107 Wyatt RJ, Henter ID. The effects of early and sustained intervention on the long-term morbidity of schizophrenia. J Psychiatr Res. 1998;3:169-177.
108 Gould RA, Mueser KT, Bolton E, et al. Cognitive therapy for psychosis in schizophrenia: an effect size analysis. Schizophr Res. 2001;48:335-342.
109 Sensky T, Turkington D, Kingdon D, et al. A randomized controlled trial of cognitive-behavioral therapy for persistent symptoms in schizophrenia resistant to medication. Arch Gen Psychiatry. 2000;57:165-172.
110 McGorry P, Yung AR, Phillips LJ, et al. Randomized controlled trial of interventions designed to reduce the risk of progression to first episode psychosis in a clinical sample with sub-threshold symptoms. Arch Gen Psychiatry. 2002;59:921-928.
111 Vaughn CE, Leff JP. The influence of family and social factors on the course of psychiatric illness. A comparison of schizophrenic and depressed neurotic patients. Br J Psychiatry. 1976;129:125-137.
112 Pitschel-Walz G, Leucht S, Bauml J, et al. The effect of family interventions on relapse and rehospitalization in schizophrenia—a meta-analysis. Schizophr Bull. 2001;27:73-92.
113 Bond GR, Becker DR, Drake RE, et al. Implementing supported employment as an evidence-based practice. Psychiatr Serv. 2001;52:313-322.
114 Slater E, Beard AW, Glithero E. The schizophrenia-like psychosis of epilepsy. Br J Psychiatry. 1963;109:95-105.
115 Davidson K, Bagley CR. Schizophrenia-like psychosis associated with organic disorders of the central nervous system: a review of the literature. In: Herrington RN, editor. Current Problems in Neuropsychiatry. British Journal of Psychiatry Special Publication No. 4. Ashford, Kent, UK: Hedley Brothers, 1969.
116 Feinstein A, Ron MA. Psychosis associated with demonstrable brain disease. Psychol Med. 1990;20:793-803.
117 Cummings JL. Organic psychosis [Review]. Psychosomatics. 1988;29:16-26.
118 Johnstone EC, Cooling NJ, Frith CD, et al. Phenomenology of organic and functional psychoses and the overlap between them. Br J Psychiatry. 1988;153:770-776.
119 Toone BK, Garralda EM, Ron MA. The psychosis of epilepsy and the functional psychosis. A clinical and phenomenological evaluation. Br J Psychiatry. 1982;141:256-261.
120 Sachdev P. Schizophrenia-like psychosis and epilepsy: the status of the association. Am J Psychiatry. 1998;155:325-336.
121 Gaitatzis A, Trimble MR, Sander JW. The psychiatric comorbidity of epilepsy. Acta Neurol Scand. 2004;110:207-220.
122 Wolf P, Trimble MR. Biological antagonism and epileptic psychosis. Br J Psychiatry. 1985;146:272-276.
123 Dongier S. Statistical study of clinical and electroencephalographic manifestations of 536 psychotic episodes occurring in 516 epileptics between clinical seizures. Epilepsia. 1959;1:117-142.
124 Torta R, Keller R. Behavioral, psychotic, and anxiety disorders in epilepsy: etiology, clinical features, and therapeutic implications. Epilepsia. 1999;40(Suppl 10):S2-S20.
125 Logsdail SJ, Toone BK. Postictal psychoses. A clinical and phenomenological description. Br J Psychiatry. 1988;152:246-252.
126 Nathaniel-James DA, Brown RG, Maier M, et al. Cognitive abnormalities in schizophrenia and schizophrenia-like psychosis of epilepsy. J Neuropsychiatry Clin Neurosci. 2004;16:472-479.
127 Landolt H. Some clinical EEG correlations in epileptic psychoses (twilight states). EEG Clin Neurophysiol. 1953;5:121.
128 Kanner AM. Psychosis of epilepsy: a neurologist’s perspective. Epilepsy Behav. 2000;1:219-227.
129 Trimble MR. The Psychoses of Epilepsy. New York: Raven Press, 1991.
130 Toone BK. The psychoses of epilepsy. J Neurol Neurosurg Psychiatry. 2000;69:1-4.
131 Reutens DC, Savard G, Andermann F, et al. Results of surgical treatment in temporal lobe epilepsy with chronic psychosis. Brain. 1997;120:1929-1936.
132 Sánchez-Ramos JR, Ortoll R, Paulson GW. Visual hallucinations associated with Parkinson’s disease. Arch Neurol. 1996;53:1265-1268.
133 Fenelon G, Mahieux F, Huon R, et al. Hallucinations in Parkinson’s disease: prevalence, phenomenology and risk factors. Brain. 2000;123:733-745.
134 Burn DJ, Troster AI. Neuropsychiatric complications of medical and surgical therapies for Parkinson’s disease. J Geriatr Psychiatry Neurol. 2004;17:172-180.
135 Wint DP, Okun MS, Fernandez HH. Psychosis in Parkinson’s disease. J Geriatr Psychiatry Neurol. 2004;17:127-136.
136 Friedman JH, Factor SA. Atypical antipsychotics in the treatment of drug-induced psychosis in Parkinson’s disease. Mov Disord. 2000;15:201-211.
137 Wilcox JA, Nasrallah HA. Childhood head trauma and psychosis. Psychiatry Res. 1987;21:303-306.
138 Sachdev P, Smith JS, Cathcart S. Schizophrenia-like psychosis following traumatic brain injury: a chart-based descriptive and case-control study. Psychol Med. 2001;31:231-239.
139 Fujii DE, Ahmed I. Risk factors in psychosis secondary to traumatic brain injury. J Neuropsychiatry Clin Neurosci. 2001;13:61-69.
140 Feinstein A, du Boulay G, Ron MA. Psychotic illness in multiple sclerosis. A clinical and magnetic resonance imaging study. Br J Psychiatry. 1992;161:680-685.
141 Black DN, Taber KH, Hurley RA. Metachromatic leukodystrophy: a model for the study of psychosis. J Neuropsychiatry Clin Neurosci. 2003;15:289-293.
142 Hyde TM, Ziegler JC, Weinberger DR. Psychiatric disturbances in metachromatic leukodystrophy. Insights into the neurobiology of psychosis. Arch Neurol. 1992;49:401-406.
143 Ron MA, Feinstein A. Multiple sclerosis and the mind. J Neurol Neurosurg Psychiatry. 1992;55:1-3.