Prion diseases
 Creutzfeldt–Jakob disease (CJD) and variant Creutzfeldt–Jakob disease (vCJD).
Creutzfeldt–Jakob disease (CJD) and variant Creutzfeldt–Jakob disease (vCJD).
 Gerstmann–Sträussler–Scheinker disease (GSS).
Gerstmann–Sträussler–Scheinker disease (GSS).


Common to all of these diseases are:
 The accumulation of an abnormal form of a cellular protein (prion protein).
The accumulation of an abnormal form of a cellular protein (prion protein).
 Neuronal death, synaptic loss, and microvacuolation (spongiform change) in the brain.
Neuronal death, synaptic loss, and microvacuolation (spongiform change) in the brain.

 CELL BIOLOGY OF PRION DISEASE
CELL BIOLOGY OF PRION DISEASEThe prion diseases have a common molecular pathology that involves the conversion of a normal cellular protein called prion protein (PrP) into an abnormal isoform (Fig. 32.1). Most evidence implicates the abnormal protein as the transmissible factor; evidence is lacking for the involvement of DNA or RNA in the infective process.
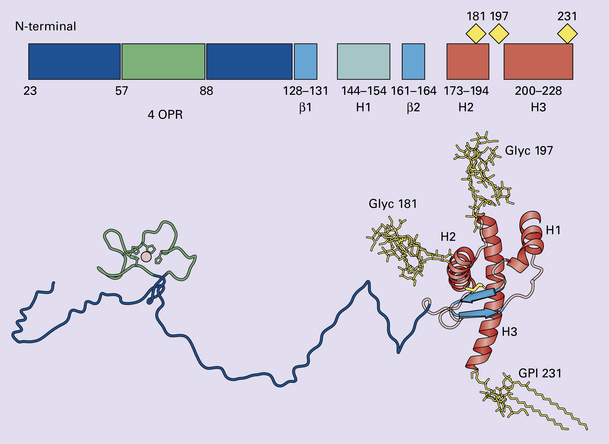
32.1 N-terminal domain: contains a large unstructured region (blue) and the octapeptide repeat domain (green), containing a copper binding site (tan-colored sphere representing the copper ion). The structured region of PrP encompasses approximately residues 124–231 and contains: (1) three α-helices (red: residues 144–154, 173–194, and 200–228); (2) a small two-strand antiparallel β-sheet (light blue: residues 128–131 and 161–164); (3) three unstructured residues at the carboxyl terminus. Glycosylation trees (yellow diamonds) are attached to residues 181 and 197 (there are un-, mono-and di-glycosylated forms.) A GPI anchor is attached to residue 231 (yellow, thick outline).
The naturally occurring form of the mature PRNP gene product. Its presence in a given cell type is necessary, but not sufficient, for replication of the prion. The terms PrPC (cellular PrP) and PrPsen (proteinase-sensitive) are both used (Fig. 32.2).
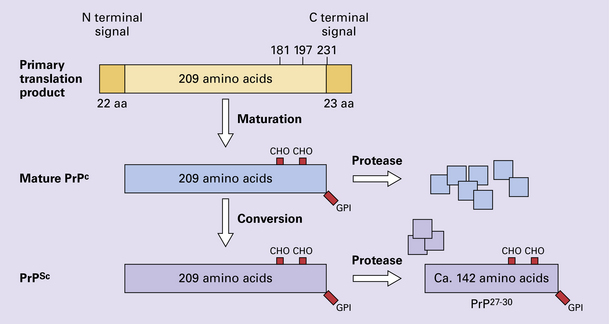
32.2 PrPC and its isoforms. The primary translation product contains amino (N) terminal and carboxyl (C) terminal signal sequences, which are cleaved off during maturation. Amino acid residues 181 and/or 197 may undergo glycosylation, with the attachment of glycosylation trees (CHO, also known as glycans) at either, both or neither of these residues (i.e. resulting in PrPC which is monoglycosylated, diglycosylated or unglycosylated), and a glycosyl phosphoinositol (GPI) anchor is added to attach the PrP molecule to the cell surface.
PrPSc (other terms PrP res or PrPCJD)
‘Abnormal’ form of the mature Prnp gene product (Fig. 32.3). Partly resistant to digestion by proteinase K. Believed to differ from PrPC conformationally. Often considered to be the transmissible agent or prion. The designations PrPSc (from scrapie, the spongiform encephalopathy of sheep); PrPCJD (CJD-associated PrP); PrPres (protease resistant); PrP*, and PrPP have all been used for this form of PrP.
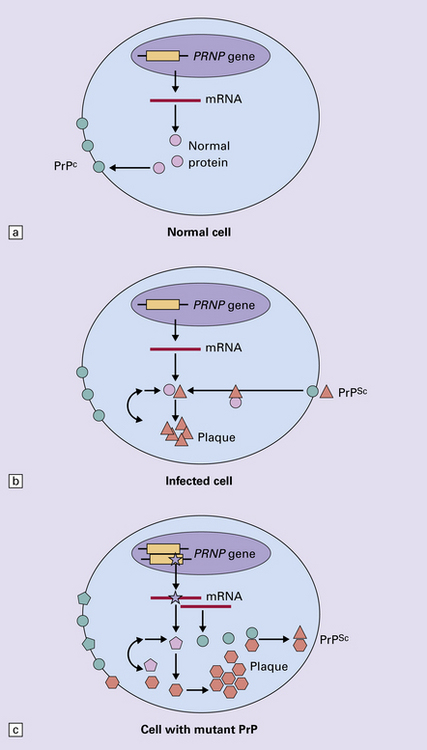
32.3 (a,b) Conversion of normal to abnormal PrP. Endogenous or exogenous abnormal PrP can interact with normal PrP and induce it to adopt an abnormal pathogenic conformation. The abnormal form accumulates within cells, interacting with and converting more of the normal cell protein. In this way, normal PrPC is converted to PrPSc. The circles symbolize wild type PrP (mauve for immature, and blue for processed PrP), while the red triangles represent PrPSc molecules. (c) Pathogenesis of inherited prion diseases. Cells contain both mutant and non-mutant (wild type) PrP, encoded by mutant (?) and wild type alleles of the PRNP gene. Mutant PrP has a reduced threshold for conversion into pathogenic PrP. In this diagram, the blue circles represent processed wild type PrP, the mauve pentagons mutant, non-pathogenic PrP molecules, and the red hexagons mutant PrP molecules that have adopted a pathogenic conformation. The mutant, pathogenic PrP (red hexagons) is capable of converting non-mutant, non-pathogenic PrPC (blue circles) into non-mutant, pathogenic PrPSc (red triangles).
PrP in normal function and disease



 The accumulated PrPSc is abnormal in that it is relatively resistant to degradation in vitro by proteinase K – this property is the basis of detection of abnormal PrP by immunohistochemical techniques.
The accumulated PrPSc is abnormal in that it is relatively resistant to degradation in vitro by proteinase K – this property is the basis of detection of abnormal PrP by immunohistochemical techniques.
 According to the template-directed refolding hypothesis (Fig. 32.4), exogenously added PrPSc serves as a template for the conversion of PrPC into more PrPSc. This conformational change is kinetically controlled in that a high activation-energy barrier prevents spontaneous conversion at detectable rates.
According to the template-directed refolding hypothesis (Fig. 32.4), exogenously added PrPSc serves as a template for the conversion of PrPC into more PrPSc. This conformational change is kinetically controlled in that a high activation-energy barrier prevents spontaneous conversion at detectable rates.
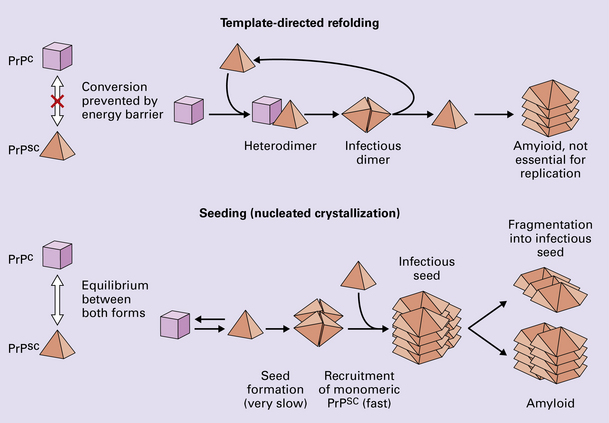
32.4 Mechanisms believed to give rise to PrPSc. (1) According to the template-directed refolding hypothesis, exogenously added PrPSc serves as a template and lowers the energy barrier for the conversion of PrPC into more PrPSc. In contrast, a high activation-energy barrier prevents spontaneous conversion at detectable rates. (2) The seeding (or nucleated crystallization) model hypothesizes that the conversion is reversible. However, PrPSc is stabilized by the formation of a crystal-like ‘infectious’ seed, and once this has formed, there is rapid accumulation and stabilization of further monomers.
 The seeding (or nucleated crystallization) model (Fig. 32.4) hypothesizes that the conversion is reversible. PrPSc stabilizes when it form a crystal-like seed. Once a seed is formed, further monomers add rapidly.
The seeding (or nucleated crystallization) model (Fig. 32.4) hypothesizes that the conversion is reversible. PrPSc stabilizes when it form a crystal-like seed. Once a seed is formed, further monomers add rapidly.

Distinct prion strains can be identified with characteristic patterns of CNS pathology and distinct incubation times. Such strains can be stably propagated in experimental animals that are homozygous for their PrP genes (Fig. 32.5).
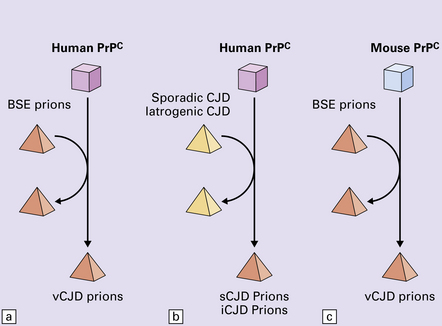
32.5 Strains in prion diseases: a characteristic, but still unexplained feature of these diseases. The principle of strain preservation applies to transmission within one species as well as across species. (a) Transmission of BSE prions into humans has caused vCJD. vCJD prions have identical properties to those of BSE. (b) Human prions transmitted into humans generate prions that are indistinguishable from each other. Characteristically however, they generate a type 3 banding pattern and produce characteristic histological features – small or medium sized plaques and perineuronal networks located in deep cortical layers. (c) Transmission of BSE prions into mice produces mouse BSE prions that are identical to vCJD prions.
Differences in the pattern of PrP glycosylation
Research has revealed variations in the pattern of PrP glycosylation in CJD and in spongiform encephalopathies in animals. Western blot analysis of PrP from affected brain tissue shows three bands, corresponding to protein with two, one, or no attached polysaccharide chains (Fig. 32.6).
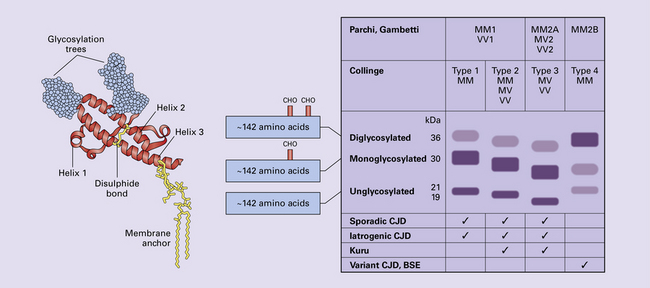
32.6 Glycoforms of PrP and electrophoretic patterns of prion protein in western blots. Left: PrP has two glycosylation sites and can exist in diglycosylated, monoglycosylated and unglycosylated forms. During the conversion to PrPSc, the glycans (glycosylation trees) are preserved. Right: To detect PrPSc in a Western blot, the tissue homogenate (which contains both PrPC and PrPSc) needs to be treated with proteinase K to digest the protease-sensitive PrPC but retain the partially protease resistant PrPSc, leaving a fragment of approximately 142 amino acids. Separation of the ~142-AA fragment by western blotting reveals three bands. The types of PrPSc can be further characterized according to the amino acid – methionine (M) or valine (V) – that is encoded at position 129 in PrP by each of the patient’s two PRPN alleles. The combination of electrophoretic mobility and codon 129 genotyping allows discrimination between different types human of CJD: types 1, 2, 3 or MM1/VV1 and MM2/MV2/VV2 are seen in sporadic CJD, Kuru and iatrogenic CJD, while a distinct, highly characteristic diglycosylation-dominant banding pattern is seen in vCJD, in BSE and other forms where BSE prions were transmitted accidentally or experimentally (type 4 or type MM2B). This diglycosylation-dominant banding pattern is regarded as strong evidence that BSE is the origin of vCJD. Variable glycotype patterns can be found in a single patient. A large, detailed study of 4200 samples from 200 brains showed that two types of PrP coexist in about 35% of sCJD cases. PrPSc types 1 and 2 co-occur more frequently in the MM than in the MV or VV genotypes. These molecular findings correlate to some extent to the histological phenotype. (Adapted from Parchi P, Strammiello R, Notari S, et al. Incidence and spectrum of sporadic Creutzfeldt–Jakob disease variants with mixed phenotype and co-occurrence of PrPSc types: an updated classification. Acta Neuropathol 2009; 118:659–671)
The commonest phenotype of prion disease is CJD. Patients typically present with subtle motor signs, which herald severe cerebellar ataxia, and progress to global dementia in under 1 year. Criteria for the clinical diagnosis of CJD have been proposed and widely adopted (Table 32.1).
Table 32.1
Clinical criteria for the diagnosis of CJD

aDepression, anxiety, apathy, withdrawal, delusions.
bThis includes both frank pain and/or unpleasant dysesthesia.
cGeneralized triphasic periodic complexes at approximately one per second.
dSpongiform change and extensive PrP deposition with florid plaques, throughout the cerebrum and cerebellum.
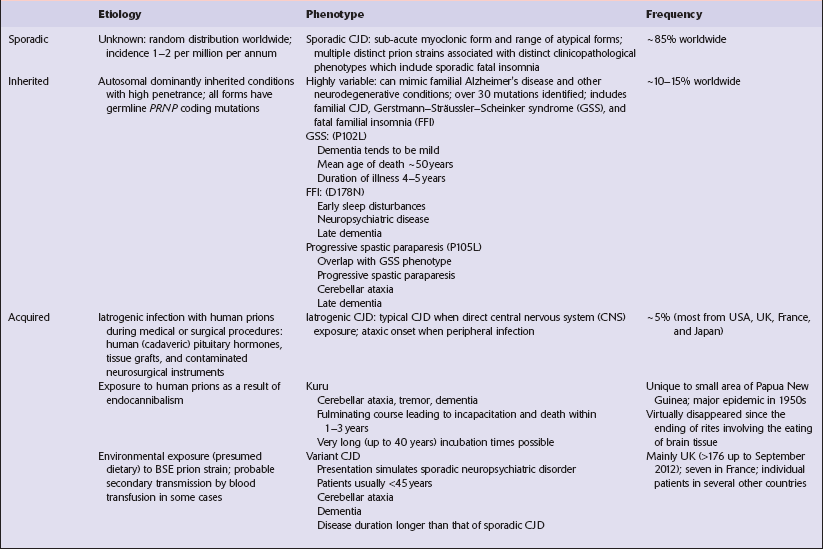
Several phenotypes of prion disease other than CJD have been identified (Table 32.2). In all of these, the mainstay of diagnosis is clinical examination supplemented by additional radiologic, electrophysiologic and neuropathologic investigations (Table 32.3).

 PRP GENE AND PATHOGENESIS OF PRION DISEASE
PRP GENE AND PATHOGENESIS OF PRION DISEASE
Several point mutations and insertions have been identified in the PrP gene that increase the susceptibility of PrP to assume a pathologic conformation (Fig. 32.7).
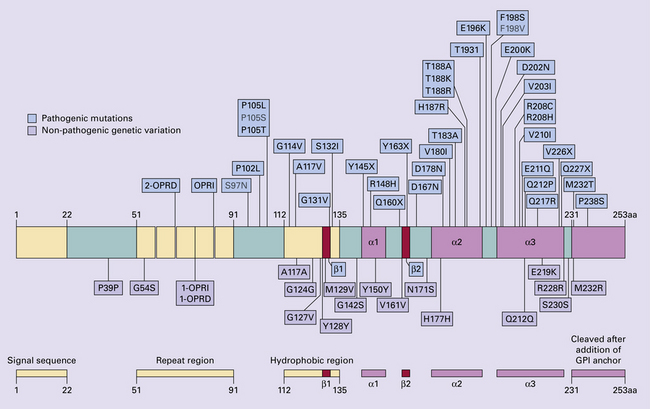
32.7 Schematic diagram of PRNP. The domains and tertiary protein structures are shown in the lower part of the figure. The upper part indicates pathogenic mutations (red) and non-pathogenic genetic polymorphisms (green). The gray text indicates PRNP variations found in cases of neurodegenerative disease in which the diagnosis was not prion disease (From Beck JA, Poulter M, Campbell TA, et al. PRNP allelic series from 19 years of prion protein gene sequencing at the MRC Prion Unit. Hum Mutat 2010; 31(7):E1551–E1563)
Nomenclature of PrP gene mutations
This takes the form of disease phenotype (original amino acid, codon position, substituted amino acid), for example GSS (P102L).
Polymorphism at codon 129 acting as a susceptibility factor
In addition to these pathogenic mutations, a polymorphism at codon 129 (which codes for either valine or methionine) acts as a susceptibility factor, modulating the facility with which PrPC assumes an abnormal conformation when interacting with exogenous abnormal PrPP (see below). The frequency of this polymorphism in Caucasian populations is:






 EPIDEMIOLOGY OF CJD
EPIDEMIOLOGY OF CJD CJD occurs throughout the world and has an annual incidence of 1–2 cases/million population.
CJD occurs throughout the world and has an annual incidence of 1–2 cases/million population.

 Approximately equal numbers of men and women are affected.
Approximately equal numbers of men and women are affected.
 About 10% of cases are familial.
About 10% of cases are familial.
 The age of onset is usually 55–75 years with a peak incidence in the 7th decade.
The age of onset is usually 55–75 years with a peak incidence in the 7th decade.
 Rare cases with an onset as young as 16 years or as old as 85 years have been reported.
Rare cases with an onset as young as 16 years or as old as 85 years have been reported.

PATHOLOGY
MACROSCOPIC APPEARANCES
The brain may appear macroscopically normal, even in cases with long clinical histories. Most cases, however, show some atrophy (Fig. 32.8) and this may be severe, with a reduction in brain weight to as low as 850 g. In such cases, ventricular enlargement is marked and the atrophy often includes the caudate nucleus and thalamus. The hippocampus may be relatively spared, even in cases with severe atrophy elsewhere.
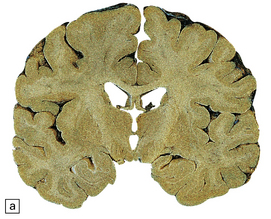
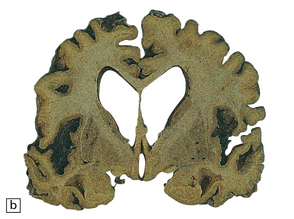
MICROSCOPIC APPEARANCES
The histologic features of prion diseases are:
 Spongiform change (intraneuronal vacuolation).
Spongiform change (intraneuronal vacuolation).
 Loss of synapses and subsequently of neurons.
Loss of synapses and subsequently of neurons.


 Paucity or absence of lymphocytic inflammation.
Paucity or absence of lymphocytic inflammation.
 Hyperphosphorylation of tau (immunohistochemical detection only).
Hyperphosphorylation of tau (immunohistochemical detection only).

Spongiform change
Microscopic examination of affected regions of the brain reveals vacuolation of the neuropil, an appearance termed spongiform change (Fig. 32.9). The vacuoles are typically round, relatively small (20–50 μm in diameter), and quite evenly distributed, but some can be large and irregular. The vacuoles are intracellular. Electron microscopy of early lesions shows that most of the vacuoles are within neuronal processes (Fig. 32.10). The vacuolation occurs mainly in gray matter. The distribution of pathology varies greatly between cases. The most consistently affected regions are the cerebral and cerebellar cortices, but the basal ganglia and thalamus are often involved. The distribution of lesions in the cerebral and cerebellar cortices is often patchy, affected regions alternating with seemingly unaffected areas, but may be confluent.
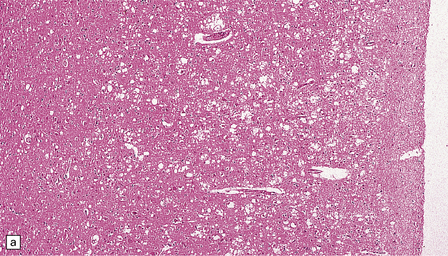
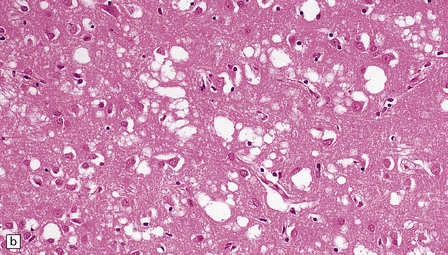
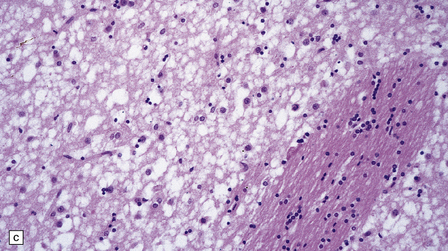
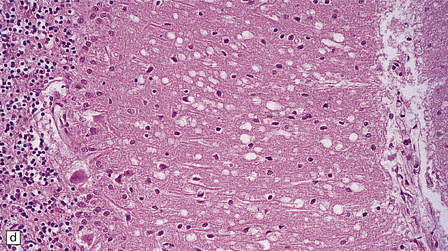
32.9 Spongiform change. There is fine vacuolation of the neuropil, even well away from blood vessels and neuronal somata (where tissue shrinkage during processing may give an artifactual appearance of vacuolation). The fine round vacuoles in the affected cortex and deep gray matter are interspersed with coarser vacuoles, some of which appear to be formed by the coalescence of smaller ones. (a) Spongiform change affecting all layers of the cerebral cortex. (b) Some of the larger vacuoles appear to result from the coalescence of smaller ones. (c) Spongiform change involving the putamen. (d) Spongiform change involving the cerebellum in which there are numerous small vacuoles in the molecular layer. The cerebellar cortex is commonly, but not invariably, affected in prion disease.
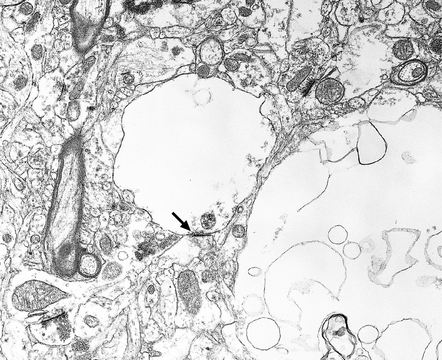
32.10 Electron microscopic appearance of spongiform change. Dendrites and axons contain vacuoles, which appear empty apart from fragments of membrane and occasional wisps of amorphous material. Note the synaptic density (arrow). Small vacuoles may be enclosed by a membrane. Larger vacuoles are usually incompletely enclosed or lack any discernible surrounding membrane.
 DIFFERENTIAL DIAGNOSIS OF SPONGIFORM CHANGE IN PRION DISEASES
DIFFERENTIAL DIAGNOSIS OF SPONGIFORM CHANGE IN PRION DISEASES
 Microvacuolation may occur in other diseases and can be confused with the spongiform change of prion disease.
Microvacuolation may occur in other diseases and can be confused with the spongiform change of prion disease.
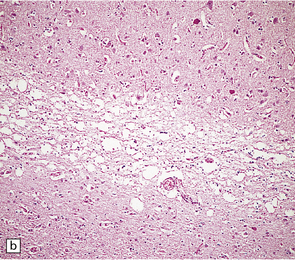
 Status spongiosus (Fig. 32.11) refers to the coarse microvacuolation that accompanies astrocytosis in association with severe neuronal loss. It is a nonspecific finding in several neurodegenerative diseases. It may be part of the pathology of prion disease in some cases, but is not a diagnostic feature.
Status spongiosus (Fig. 32.11) refers to the coarse microvacuolation that accompanies astrocytosis in association with severe neuronal loss. It is a nonspecific finding in several neurodegenerative diseases. It may be part of the pathology of prion disease in some cases, but is not a diagnostic feature.
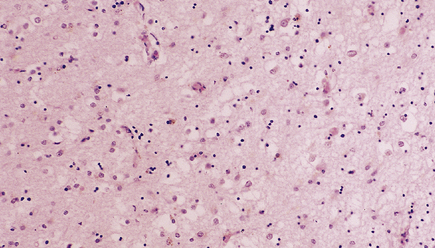
32.11 Status spongiosus. Irregular vacuolation of gliotic cerebral cortex can be a feature of several degenerative disorders.
 Superficial microvacuolation involving layers II and III of the frontal and temporal cortices is a feature of neurodegenerative diseases that present as frontotemporal dementias, especially dementia of frontal type, but also Pick’s disease, dementia with motor neuron disease inclusions, and dementia associated with corticobasal degeneration (Fig. 32.12).
Superficial microvacuolation involving layers II and III of the frontal and temporal cortices is a feature of neurodegenerative diseases that present as frontotemporal dementias, especially dementia of frontal type, but also Pick’s disease, dementia with motor neuron disease inclusions, and dementia associated with corticobasal degeneration (Fig. 32.12).
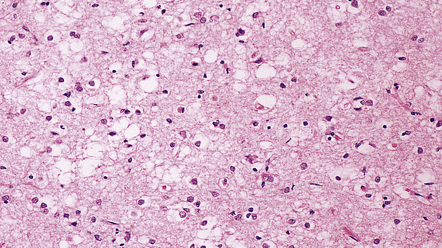
32.12 Superficial microvacuolation in frontal dementia. Microvacuolation limited to the outer cortical laminae in the frontal and temporal lobes is a feature of several forms of neurodegenerative disorders (see Chapter 31) not related to prion disease. In contrast to prion disease, the vacuolation does not involve the full thickness of the cortex.
 Dementia with Lewy bodies may be associated with transcortical microvacuolation very similar to that of prion disease (Fig. 32.13). This is localized to medial temporal lobe structures and is not associated with accumulation of PrP.
Dementia with Lewy bodies may be associated with transcortical microvacuolation very similar to that of prion disease (Fig. 32.13). This is localized to medial temporal lobe structures and is not associated with accumulation of PrP.
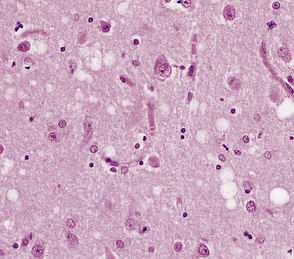
32.13 Microvacuolation in dementia with Lewy bodies. Spongiform change, which is transcortical, may be seen in dementia with Lewy bodies, but the distribution is restricted to medial temporal lobe structures.
 Spongiform change in gray and white matter can occur in several metabolic encephalopathies, including aminoaciduria syndromes (Fig. 32.14a), Alpers’ disease, and chronic hepatocerebral degeneration (Fig. 32.14b).
Spongiform change in gray and white matter can occur in several metabolic encephalopathies, including aminoaciduria syndromes (Fig. 32.14a), Alpers’ disease, and chronic hepatocerebral degeneration (Fig. 32.14b).
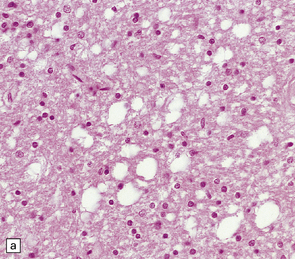
32.14 Spongiform vacuolation in disorders other than prion disease. (a) Cortical vacuolation in aminoaciduria. (b) Coarse vacuolation of the subcortical white matter in liver failure.
 Microvacuolation can occur in acute hypoxic–ischemic encephalopathy.
Microvacuolation can occur in acute hypoxic–ischemic encephalopathy.

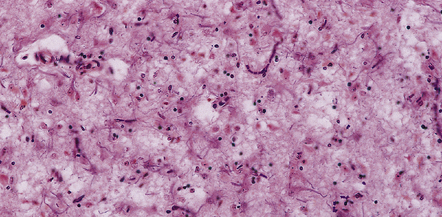
Neuronal loss
In most cases there is a marked loss of neurons. This loss is greatest in cortical layers III–V and in focal regions of the caudate nucleus and thalamus. The pattern of loss is variable. There may be destruction of almost all neurons over large areas, causing a loss of lamination and coarse vacuolation (status spongiosus) (Fig. 32.15). Examination may reveal scattered shrunken remnants of neurons in the midst of apparently unaffected cells. In some cases residual neurons swell to form ballooned cells.
Astrocytosis
Loss of neurons is accompanied by massive activation and proliferation of astrocytes (Fig. 32.16). In severe cases, the neuronal component of cortical areas may have been replaced by reactive gliosis.
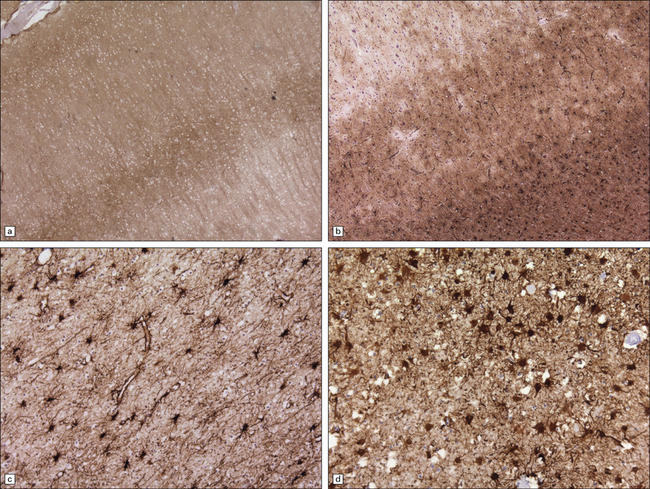
32.16 Gliosis in prion diseases. (a) Immunolabeling for abnormal PrP shows diffuse (synaptic) staining, with accentuation in cortical layers 3 and 4. (b) Immunolabeling of an adjacent section for GFAP reveals marked gliosis, particularly towards the junction with the white matter (bottom right of panel). Labeling for GFAP also highlights gliosis of the white matter (c) in sporadic CJD, and of the gray matter (d) in vCJD.
Synaptic loss
Electron microscopy has shown that axons and dendrites are damaged early in the disease. Quantitative studies of synaptic density in cases with obvious spongiform change have estimated synaptic loss at about 30% in the cerebral cortex. A synaptic loss of approximately 20% has been shown in atypical cases lacking obvious pathology at the light microscopic level (Fig. 32.17).
Hyperphosphorylation of tau
This can occur in response to the cerebral accumulation of amyloid (Aβ in Alzheimer’s disease, ABri and ADan in familial British dementia and familial Danish dementia), and in prion disease. This is particularly prominent in areas of plaque formation but can also be seen in the context of synaptic PrPSc. The deposits are distinct from those seen in Alzheimer’s disease or British dementia, in that neurofibrillary tangles or threads are not formed. PrP-associated tau forms short stubs or rods (Fig. 32.18).
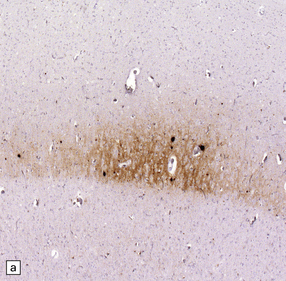
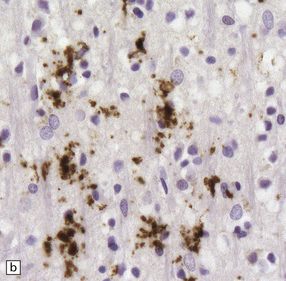
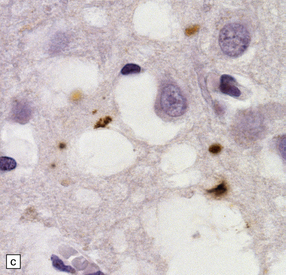
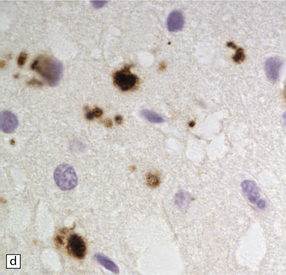
32.18 Deposition of hyperphosphorylated tau in prion disease. (a) sCJD with deposition of abnormal PrP in the deep cortical layer with formation of occasional small plaques. (b) Detection of dense granular or rod-shaped hyperphosphorylated tau in the vicinity of plaques (same area shown in the box in ‘a’). (c) Tau deposits in the cortex of sCJD near large vacuoles or (d) in the cerebellum in vCJD. (Courtesy of Reiniger L, Lukic A, Linehan J, Rudge P, Collinge J, Mead S, Brandner S. Tau, prions and Ab: the triad of neurodegeneration. Acta Neuropathol. 2010 Jan;121(1):5-20)
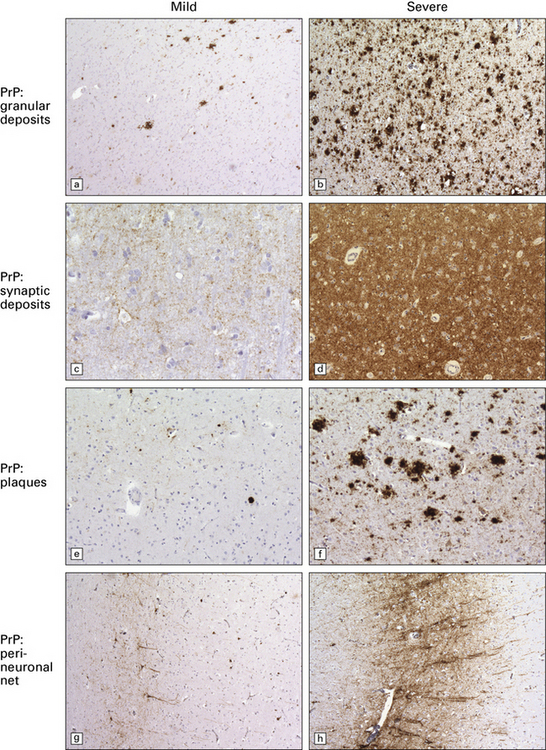
Accumulation of PrP: PrP accumulates in the brain in prion diseases and can be detected by immunohistochemistry. There are several patterns of accumulation:
 a diffuse synaptic pattern (Fig. 32.17)
a diffuse synaptic pattern (Fig. 32.17)
 perivacuolar deposits (Fig. 32.17)
perivacuolar deposits (Fig. 32.17)
 larger deposits or plaques (Fig. 32.19) in which some of the PrP may be in the form of amyloid. There are five main types of plaque in prion disease:
larger deposits or plaques (Fig. 32.19) in which some of the PrP may be in the form of amyloid. There are five main types of plaque in prion disease:
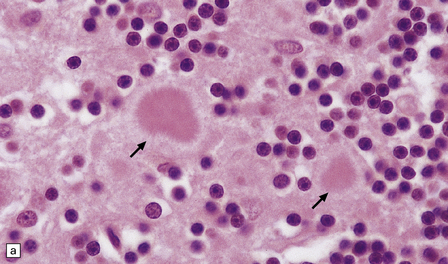
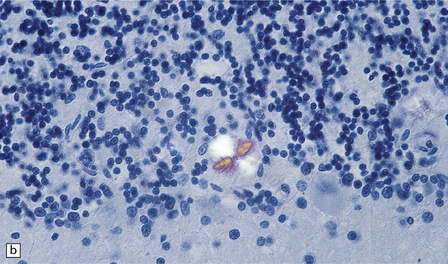
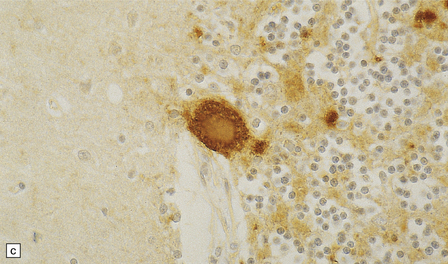
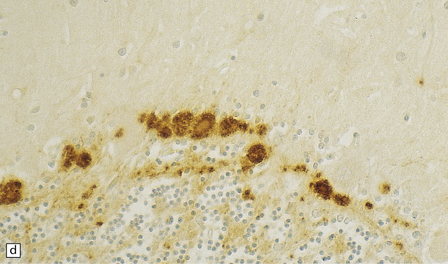
32.19 Plaques in prion disease. About 15% of brains from patients with prion disease contain amyloid plaques, usually restricted to the cerebellum. The plaques occur within the molecular layer, the granule cell layer, and the Purkinje cell layer. (a) They appear as homogeneous round or oval eosinophilic structures, and show birefringence when stained with Congo red and viewed under polarized light (b). (c) The plaques show immunoreactivity for PrP. (d) Plaques within the Purkinje cell layer.
• Unicentric plaques, consisting of an amyloid core and radiating spicules of amyloid, which together form a spherical deposit. The amyloid is periodic acid–Schiff (PAS)-positive. This type is often called a kuru plaque.
• Multicentric plaques, consisting of several dense core regions of amyloid with radiating spicules, which form multilobed structures as if made by the fusion of many unicentric plaques. The amyloid is PAS-positive. This form is characteristic of GSS.
• Unicentric plaques, consisting of a dense amyloid core that lacks surrounding radiating spicules of amyloid material. The amyloid is PAS-positive.
• Florid plaques, appearing as unicentric amyloid deposits with radiating spicules of amyloid and a surrounding rim of spongiform change. The amyloid is PAS-positive. This type is characteristic of vCJD.
• Diffuse plaques consisting of large (100–200 μm diameter) ill-defined deposits of PrP that are visible on immunostaining. The deposits are not PAS-positive.
SPORADIC CJD






 129MM homozygote and 129MV heterozygote with PrPP type 1 (CJDMM1 and CJDMV1) are common, between them accounting for about 70% of sporadic cases, and characterized by a rapid clinical course, with early dementia, myoclonus, and periodic sharp waves on the EEG. Histologically, there is mild to moderate spongiosis and gliosis in the cerebral cortex, striatum, thalamus, and cerebellar cortex, with general sparing of the brain stem, hippocampus, and hypothalamus. PrP deposition is mainly of the synaptic pattern, in areas of spongiosis.
129MM homozygote and 129MV heterozygote with PrPP type 1 (CJDMM1 and CJDMV1) are common, between them accounting for about 70% of sporadic cases, and characterized by a rapid clinical course, with early dementia, myoclonus, and periodic sharp waves on the EEG. Histologically, there is mild to moderate spongiosis and gliosis in the cerebral cortex, striatum, thalamus, and cerebellar cortex, with general sparing of the brain stem, hippocampus, and hypothalamus. PrP deposition is mainly of the synaptic pattern, in areas of spongiosis.




GERSTMANN–STRÄUSSLER–SCHEINKER DISEASE (GSS)
Several clinical conditions are encompassed by the designation GSS, as defined by the presence of multicentric PrP amyloid plaques in the brain (Fig. 32.20):
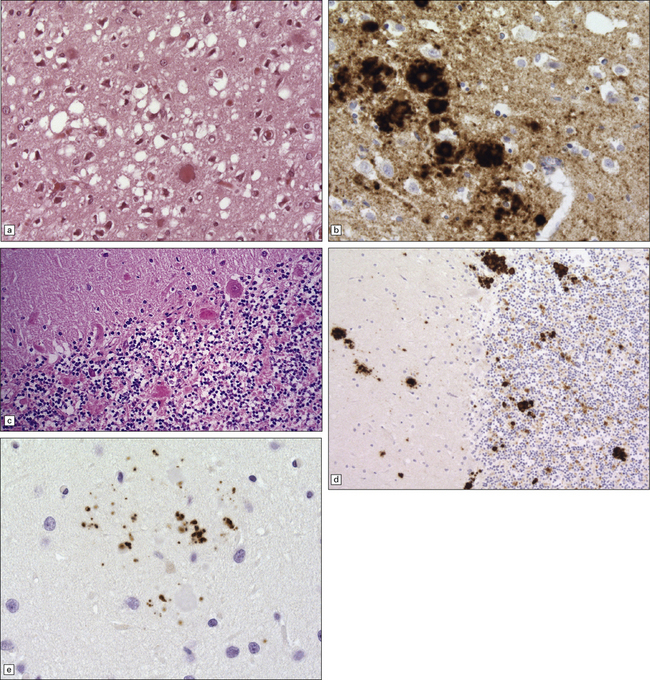
32.20 Cortical and cerebellar plaques in GSS (P102L). (a) The cerebral cortex contains dense core amyloid plaques and very significant spongiform change with formation of large vacuoles. Often vacuolation is less prominent but the extent can vary considerably within a single brain. (b) Immunohistochemistry for abnormal PrP highlights numerous, multicentric amyloid plaques but also synaptic PrP, which can be a feature in GSS. (c) Amyloid plaques in the cerebellar cortex. (d) Labeling of cerebellar plaques with PrP antibody. (e) Hyperphosphorylated tau forms small rod-shaped deposits in the area of a cerebellar plaque.
 Ataxic GSS (PRNP P102L) in which ataxia is combined with varying degrees of dementia. Unicentric and multicentric amyloid plaques are present in the molecular layer of the cerebellar cortex and in smaller numbers in the cerebral cortex. Usually spongiform change is not prominent. The severity of neuronal loss from cerebral and cerebellar cortices is variable.
Ataxic GSS (PRNP P102L) in which ataxia is combined with varying degrees of dementia. Unicentric and multicentric amyloid plaques are present in the molecular layer of the cerebellar cortex and in smaller numbers in the cerebral cortex. Usually spongiform change is not prominent. The severity of neuronal loss from cerebral and cerebellar cortices is variable.





FATAL FAMILIAL INSOMNIA (FFI)
 marked neuronal loss and astrocytic gliosis in the thalamus
marked neuronal loss and astrocytic gliosis in the thalamus
 inconsistent and mild neuronal loss and gliosis in the cerebral cortex
inconsistent and mild neuronal loss and gliosis in the cerebral cortex



VARIANT CJD (VCJD)
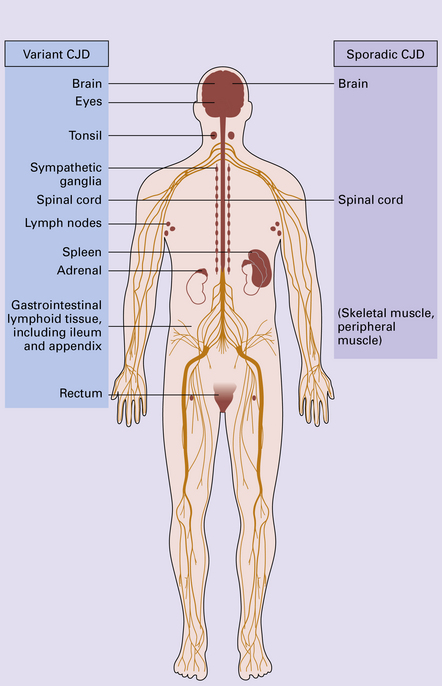
vCJD has a distinct neuropathological profile. The cerebral cortex and cerebellum typically contain numerous amyloid plaques which are distinct from those in Kuru or inherited forms of prion disease (Fig. 32.21). Many plaques are surrounded by vacuoles and have been termed florid plaques (Fig. 32.22). Spongiform change, neuronal loss, and astrocytic gliosis are generally very severe and are most evident in the thalamus and basal ganglia. Within the CNS, immunohistochemistry typically shows an abundant accumulation of PrP (Fig. 32.23). Additionally, and in remarkable contrast to all other forms of prion diseases, there is accumulation of PrPSc in lymphoreticular tissues such as the tonsil, lymph nodes, spleen, and appendix (Fig. 32.23). This finding is unique to vCJD and has not been observed in sporadic, inherited or, importantly, in acquired prion diseases in which human prions were transmitted (i.e. iatrogenic forms and Kuru). This unique property of the vCJD strain allows pre-mortem diagnosis of vCJD by tonsil biopsy. PrPSc can be demonstrated in follicular dendritic cells of lymphoid tissue by immunohistochemistry.
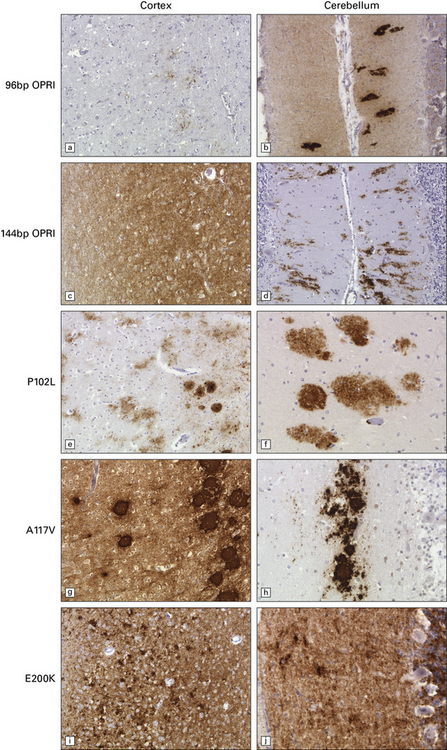
32.21 Phenotypic variability of genetic (inherited) prion disease. (a-j) The very variable appearance of the PrPSc deposits associated with PRNP mutations is shown in these panels. OPRI = octapeptide repeat insert. The other PRNP mutations are indicated using the conventional abbreviation ‘A number B’, where the number is the position of the mutant codon, A is the single-letter code for the amino acid at that position in wild-type PrP, and B is the code for the amino acid at that position in the mutant PrP.
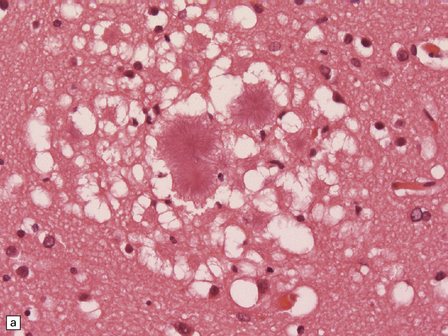
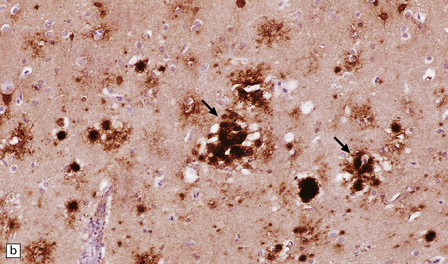
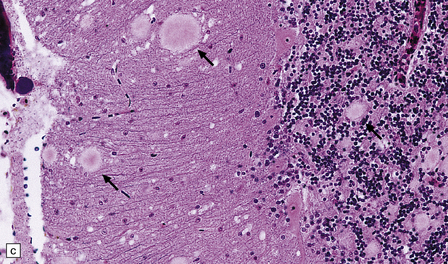
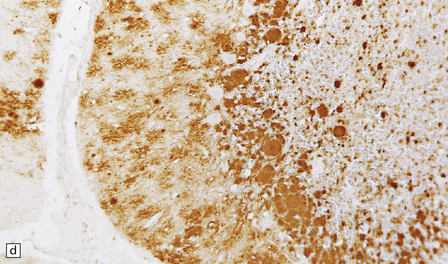
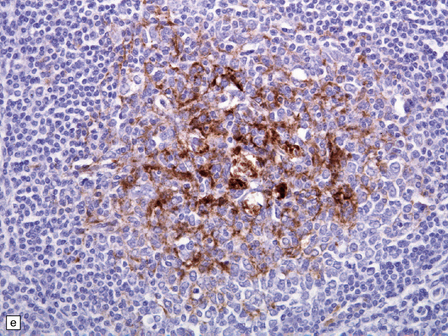
32.22 vCJD. (a) The plaques of vCJD are typically of ‘florid’ type (arrows), consisting of an amyloid core with relatively coarse surrounding vacuoles in a pattern that has been likened to the petals of a daisy. (b) The coarsely granular amyloid cores are strongly immunoreactive for PrP (arrows). (c) Large numbers of plaques (arrows) are usually present in the cerebellum. (d) The cerebellum shows abundant deposition of PrP on immunostaining. (e) Germinal center from tonsil showing positive immunostaining for PrP. Staining is concentrated in follicular dendritic cells.
IATROGENIC CJD
There are typically widespread neuronal loss and spongiform change in the cerebellar cortex and to a lesser degree in the cerebral cortex (Fig. 32.24). Plaques are common. PrP immunohistochemistry reveals heavy deposits in the cerebellum and a linear pattern of labeling involving the deeper laminae of the cerebral cortex.
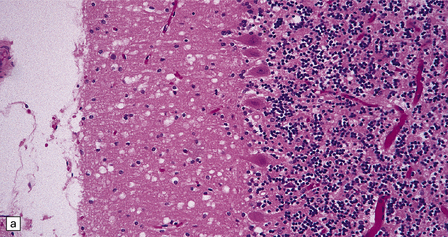
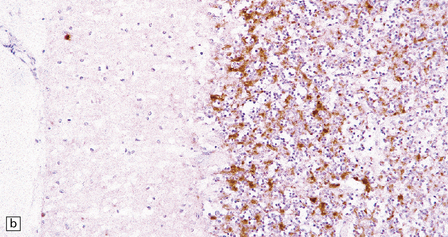
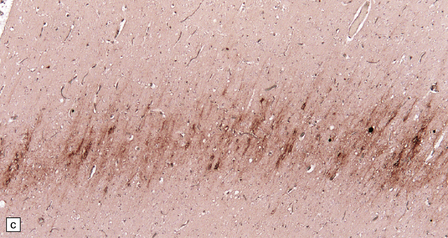
32.24 Iatrogenic CJD. A recipient of human pituitary derived growth hormone, this patient presented with ataxia and later developed dementia. (a) Spongiform change is present in the molecular layer of the cerebellum. (b) Immunohistochemistry for PrP reveals scattered deposits in the cerebellum and (c) a linear pattern of immunoreactivity involving the deeper laminae in the cerebral cortex.
PANENCEPHALOPATHIC CJD
The panencephalopathic form of CJD is characterized by extensive involvement of white matter as well as cerebral cortex (Fig. 32.25). It has been suggested that this is an end-stage pattern that reflects relatively prolonged disease.
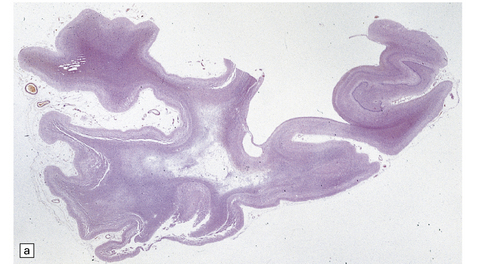
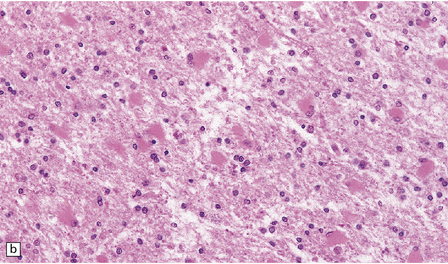
32.25 Panencephalopathic CJD. (a) Severe white matter rarefaction and focal cavitation in panencephalopathic CJD. This patient had typical spongiform change in the basal ganglia and thalamus. (b) Marked reactive astrocytosis in the cerebral white matter. (c) Elsewhere, the white matter is severely rarefied and contains scattered lipid-laden macrophages as well as reactive astrocytes.
The brain generally shows severe atrophy. Marked neuronal loss and spongiform change in the cerebral cortex are associated with an intense astrocytosis. There is diffuse myelin pallor in the hemispheric white matter, which appears spongy and may become cavitated. These changes are accompanied by moderate astrocytosis and an accumulation of lipid in macrophages (Fig. 32.25).
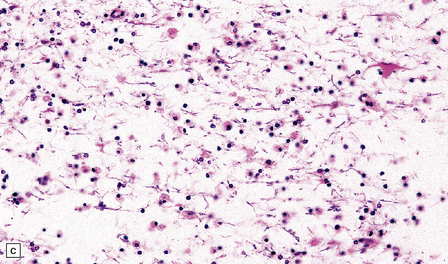
KURU
Clinical presentation and the neuropathology of Kuru are distinct from the majority of patients with sporadic CJD, in that Kuru presents with progressive cerebellar ataxia. Dementia is a late and less prominent feature. In contrast to vCJD there is no accumulation of abnormal PrP in lymphoreticular tissue (Fig. 32.26).
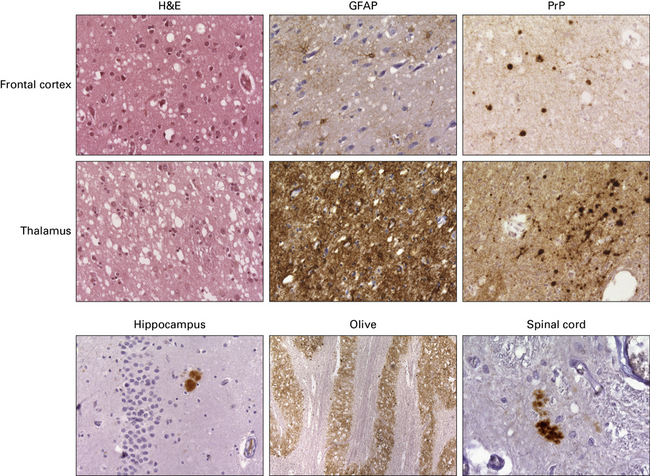
32.26 Neuropathology of Kuru. These images are from a patient in whom there was a 40-year latent interval between infection and the development of the first symptoms. The time from onset of symptoms to death was 2 years. Spongiform changes are highly variable in Kuru, as are the degree of gliosis and intensity and type of PrP deposition. Note that PrP accumulates both in small, spiculated, so-called Kuru plaques, but that many regions also show synaptic deposition of abnormal PrP. The codon 129 genotype in this case was 129MV and the pattern on western blotting type 3/type 2MV.
 NECROPSY AND DECONTAMINATION PROCEDURES
NECROPSY AND DECONTAMINATION PROCEDURES
 CJD is usually suspected ante-mortem, and so the necropsy is undertaken with an appreciation of the potential risks.
CJD is usually suspected ante-mortem, and so the necropsy is undertaken with an appreciation of the potential risks.












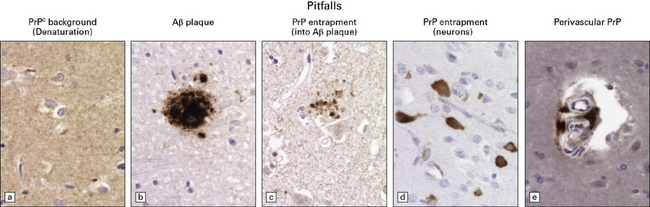
PITFALLS IN THE HISTOPATHOLOGICAL DIAGNOSIS OF PRION DISEASE
The diagnosis of prion diseases by examination of brain biopsies can be complicated by a number of pitfalls, some related to technical difficulties associated with PrP immunostaining, others to the biochemical properties of PrPC. See Figure 32.27 for details.
REFERENCES
Aguzzi, A., Weissmann, C. Prion research: the next frontiers. Nature. 1997;389:795–798.
Baldwin, M.A., Stahl, N., Reinders, L.G., et al. Permethylation and tandem mass spectrometry of oligosaccharides having free hexosamine: analysis of the glycoinositol phospholipid anchor glycan from the scrapie prion protein. Anal Biochem.. 1990;191:174–182.
Beck, J.A., Poulter, M., Campbell, T.A., et al. PRNP allelic series from 19 years of prion protein gene sequencing at the MRC Prion Unit. Hum Mutat.. 2010;31(7):E1551–E1563.
Bell, J.E., Ironside, J.W. How to tackle a possible Creutzfeldt–Jakob disease necropsy. J Clin Pathol.. 1993;46(3):193–197.
Brandner, S., Whitfield, J., Boone, K., et al. Central and peripheral pathology of kuru: pathological analysis of a recent case and comparison with other forms of human prion disease. Philos Trans R Soc Lond B Biol Sci.. 2008;363(1510):3755–3763.
Budka, H., Aguzzi, A., Brown, P., et al. Neuropathological diagnostic criteria for Creutzfeldt–Jakob disease (CJD) and other human spongiform encephalopathies (prion diseases). Brain Pathol.. 1995;5(4):459–466.
Budka, H., Aguzzi, A., Brown, P., et al. Tissue handling in suspected Creutzfeldt–Jakob disease (CJD) and other human spongiform encephalopathies (prion diseases). Brain Pathol.. 1995;5(3):319–322.
Collinge, J. Molecular neurology of prion disease. J Neurol Neurosurg Psychiatry.. 2005;76:906–919.
Collinge, J., Rossor, M. A new variant of prion disease. Lancet.. 1996;347(9006):916–917.
Collinge, J., Whitfield, J., McKintosh, E., et al. A clinical study of kuru patients with long incubation periods at the end of the epidemic in Papua New Guinea. Philos Trans R Soc Lond B Biol Sci.. 2008;363(1510):3725–3739.
Collinge, J., Sidle, K.C., Meads, J., et al. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature.. 1996;383(6602):685–690.
Gambetti, P., Cali, I., Notari, S., et al. Molecular biology and pathology of prion strains in sporadic human prion diseases. Acta Neuropathol.. 2011;121(1):79–90.
Hill, A.F., Zeidler, M., Ironside, J., et al. Diagnosis of new variant Creutzfeldt–Jakob disease by tonsil biopsy. Lancet.. 1997;349:99.
Hill, A.F., Desbruslais, M., Joiner, S., et al. The same prion strain causes vCJD and BSE [letter]. Nature.. 1997;389(6650):448–450.
Hilton, D.A., Ghani, A.C., Conyers, L., et al. Accumulation of prion protein in tonsil and appendix: review of tissue samples. BMJ.. 2002;325:633–634.
Ironside, J.W., Bell, J.E. Florid plaques and new variant Creutzfeldt–Jakob disease [letter]. Lancet.. 1997;350(9089):1475.
Ironside, J.W., Bell, J.E. The ‘high-risk’ neuropathological autopsy in AIDS and Creutzfeldt–Jakob disease: principles and practice. Neuropathol Appl Neurobiol.. 1996;22(5):388–393.
Kovacs, G.G., Seguin, J., Quadrio, I., et al. Genetic Creutzfeldt–Jakob disease associated with the E200K mutation: characterization of a complex proteinopathy. Acta Neuropathol (Berl).. 2011;121(1):39–57.
Medori, R., Montagna, P., Tritschler, H.J., et al. Fatal familial insomnia: a second kindred with mutation of prion protein gene at codon 178. Neurology.. 1992;42(3 Pt 1):669–670.
Palmer, M.S., Dryden, A.J., Hughes, J.T., et al. Homozygous prion protein genotype predisposes to sporadic Creutzfeldt–Jakob disease. Nature.. 1991;352(6333):340–342.
Parchi, P., Castellani, R., Capellari, S., et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt–Jakob disease. Ann Neurol.. 1996;39(6):767–778.
Parchi, P., Strammiello, R., Notari, S., et al. Incidence and spectrum of sporadic Creutzfeldt–Jakob disease variants with mixed phenotype and co-occurrence of PrPSc types: an updated classification. Acta Neuropathol (Berl).. 2009;118:659–671.
Reiniger, L., Lukic, A., Linehan, J., et al. Tau, prions and Ab: the triad of neurodegeneration. Acta Neuropathol (Berl).. 2011;121(1):5–20.
Wadsworth, J.D., Hill, A.F., Beck, J.A., et al. Molecular and clinical classification of human prion disease. Br Med Bull.. 2003;66:241–254.
Wadsworth, J.D., Joiner, S., Linehan, J.M., et al. Phenotypic heterogeneity in inherited prion disease (P102L) is associated with differential propagation of protease-resistant wild-type and mutant prion protein. Brain.. 2006;129(Part 6):1557–1569.
Weber, T., Tumani, H., Holdorff, B., et al. Transmission of Creutzfeldt–Jakob disease by handling of dura mater [letter]. Lancet.. 1993;341(8837):123–124.
Whittington, M.A., Sidle, K.C., Gowland, I., et al. Rescue of neurophysiological phenotype seen in PrP null mice by transgene encoding human prion protein. Nat Genet.. 1995;9:197–201.
Will, R.G., Ironside, J.W., Zeidler, M., et al. A new variant of Creutzfeldt–Jakob disease in the UK. Lancet.. 1996;347(9006):921–925.
Young, K., Clark, H.B., Piccardo, P., et al. Gerstmann–Sträussler–Scheinker disease with the PRNP P102l mutation and valine at codon 129. Mol Brain Res.. 1997;44(1):147–150.
Zerr, I., Giese, A., Windl, O., et al. Phenotypic variability in fatal familial insomnia (D178N-129M) genotype. Neurology.. 1998;51(5):1398–1405.
Zou, W.Q., Puoti, G., Xiao, X., et al. Variably protease-sensitive prionopathy: a new sporadic disease of the prion protein. Ann Neurol.. 2010;68(2):162–172.