CHAPTER 24 Principles of Neurocritical Care
Medical care of a patient with an acutely deteriorating neurological or neurosurgical disorder is vastly different from that for patients with other medical or surgical disorders. Management is distinguished by recognition of the potential for further harmful injury to the central nervous system (CNS), identification of the causes of neurological deterioration, and recognition of the urgent need for neurosurgical procedures. Admission to a dedicated neurosciences intensive care unit (NICU) is preferred for any patient with severe traumatic head injury, aneurysmal subarachnoid hemorrhage, large ischemic stroke, expanding intracerebral hematoma, and CNS infections. Any “unstable” patient with an acute neurological or neurosurgical disorder, however, is better served in an NICU and, depending on the underlying cause, may require specific interventions. This chapter discusses the general principles of the initial assessment and care of critically ill neurological and neurosurgical patients. More detailed information on the complex care of these patients can be found in two monographs.1,2
Assessment of Impaired Consciousness
Neurologists and neurosurgeons have a singular aptitude for the assessment and correct interpretation of impaired consciousness. Because consciousness is often decreased in patients with acute CNS lesions,1 a brief review of the processes underlying abnormal consciousness is warranted. One of the critical anatomic elements that maintain or, more strictly speaking, activate consciousness has been defined under the term ascending reticular activating system. This neuronal system, located in the caudal brainstem, connects to the thalamus. These synapsing fibers extend from the entry of the trigeminal nerve in the midpons to the thalamus, which in turn loops fibers to the cortex and back, thereby creating a thalamic-cortical circuitry. Coma can be expected when destructive or compressive lesions interrupt these synapsing fibers. Therefore, a structural lesion in the pons or mesencephalon can interrupt cortical stimulation and generate an altered state of consciousness. Common lesions are occlusion of the basilar artery, which causes ischemia of major portions of the brainstem; pontine hemorrhage involving the tegmentum; and compression of the brainstem from acute, mostly hemorrhagic cerebellar lesions.
Thus, disturbances in arousal lead to diminished alertness. Altered arousal involves an altered state of awareness, and these two components are interrelated but sometimes dissociated. One can be awake and aware (normal vigilance), awake but not aware (persistent vegetative state), and not awake and not aware (coma or brain death).1
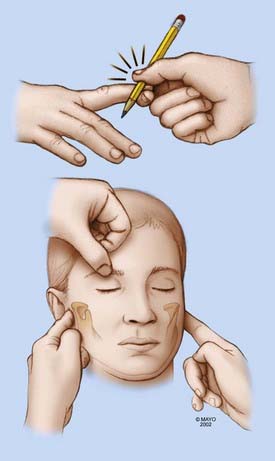
Neurological examination starts with assessment of responsiveness of the patient, and the simple reaction to painful stimuli is often one of the most important tests. Absent eye opening, vertical eye movements, or blinking (locked-in syndrome) and absent motor response to a voice command lead to the application of noxious stimuli. These stimuli should be limited to three known and reliable stimuli (compression of the supraorbital nerve, nail bed, and temporomandibular joints). The response of the patient to these stimuli is the first step in determining the depth of coma (Fig. 24-1).
We have developed and validated a new coma scale, the FOUR score, and tested it for accuracy when used by neurologists, emergency physicians, and medical intensivists.3–5 We compared ratings by residents and nursing staff specialists from all intensive care units (ICUs) and found a high degree of agreement. In this scale (Fig. 24-2) there is no assessment of verbal response, which at least in the GCS, is more a measure of orientation than alertness. The FOUR score is more useful in intubated, critically ill patients. This scale identifies different levels of coma but also locked-in syndrome, uncal herniation, and brain death. The FOUR score can be summed, and the summed scores reasonably correlate with certain degrees of impaired consciousness (alert, 16; drowsy, 12; stupor, 8; coma, 4; brain death, 0). However, it is better to describe the separate components when communicating (e.g., “this patient with traumatic brain injury has his eyes open but does not track a finger, has intact pupil and corneal reflexes, localizes to pain, is intubated, and triggers the ventilator”).
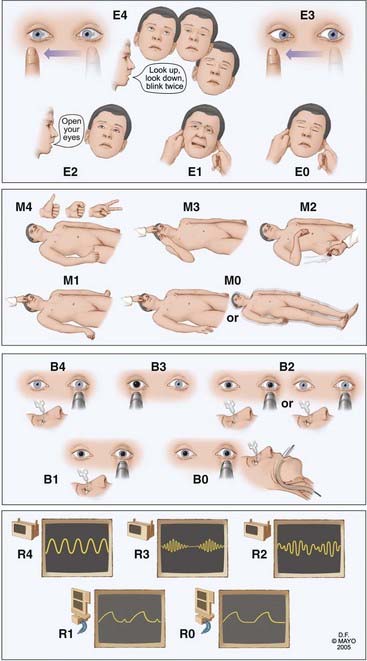
The FOUR score has been received well and is currently used or being piloted in ICUs. After initial assessment of the depth of coma, a more detailed examination follows, including a comprehensive assessment of brainstem reflexes, eye movements in particular. The eyes should be evaluated for spontaneous movement, position of the eyes with eyelid opening, and movement after turning the head or irrigation of the ear with cold water. Abnormal motor movements, pathologic reflexes, and tone are noted. With these neurological findings it should be possible to localize the lesion to three main parts of the brain—both hemispheres (or thalamus), the brainstem, or the cerebellum; key findings are shown in Table 24-1. These findings are then combined with findings on computed tomography (CT) and lead to a tailored differential diagnosis. Causes of impaired consciousness in patients in the NICU are mostly structural, secondary to postictal stupor, or, less commonly, due to acute metabolic derangements or the use of toxic substances, drugs, alcohol, or sedatives.
TABLE 24-1 Main Areas of Lesion Localization in Coma with Common Clinical Pointers
| LOCATION | CLINICAL POINTERS |
|---|---|
| Bihemispheric |
Assessment of the Airway and Need for Mechanical Ventilation
Depression of the level of consciousness is not an absolute indication for mechanical ventilation. Intubated patients with acute CNS disease are generally able to maintain efficient gas exchange. However, patients with abnormal breathing patterns that result in inadequate oxygen delivery and hypercapnia need to be mechanically ventilated. Noninvasive mechanical ventilation (BiPAP) is a reasonable option in many postoperative patients with marginal oxygenation.6 In any patient on mechanical ventilation, a “ventilator bundle” is ordered (head elevation 30 degrees; peptic ulcer and venous thromboembolism prophylaxis).7 Intubation after a seizure is not usually necessary if the airway is not obstructed.
Assessment of Volume Status and Blood Pressure
Monitoring of volume status should include laboratory values (Table 24-2), with serial body weight and fluid balance being the most practical indicators. Patient with adequate fluid balance should have a hematocrit of less than 55%, an osmolality of less than 350 mOsm, and a serum sodium concentration of less than 150 mEq/L. Any higher values should signal dehydration.
TABLE 24-2 Indicators of Volume Status in Patients with Acute Neurologic Illness
| Basic principles |
From Wijdicks EFM. The Practice of Emergency and Critical Care Neurology. New York: Oxford University Press; 2010.
Hypertension has been arbitrarily defined as systolic blood pressure of 180 mm Hg or greater and mean arterial blood pressure of 120 mm Hg or greater. There are very few data on the need for blood pressure control and the best pharmaceutical agents in patients with acute neurological illness. In most patients in the NICU, acute hypertension is treated with a labetalol bolus of 20 to 40 mg and hydralazine, 20 mg (when patients have significant bradycardia). Recalcitrant hypertension may be treated with nicardipine by titrating up from a starting dose of 5 mg/hr while closely monitoring for hours via an arterial line. Recently, a study in China suggested that aggressive lowering of blood pressure with urapidil and furosemide to a systolic pressure of 140 mm Hg is safe and may possibly reduce expansion of intracerebral hemorrhage.8 It remains very uncertain whether outcome is affected by this measure, however.
How to best manage blood pressure in patients with aneurysmal subarachnoid hemorrhage is not known. Most neurosurgeons feel comfortable only if systolic blood pressure is maintained at less than 180 mm Hg, particularly in patients with an unsecured aneurysm. The relationship between blood pressure and rebleeding has not been established. A recent study suggested that systolic blood pressure above 160 mm Hg is associated with an increased risk for rebleeding, but many of these studies have poorly defined rebleeding.9
Postoperative hypertension is best managed by treatment of pain (mostly fentanyl), hypoxemia, and volume overload, if present.10
Assessment of Infection Potential
Fever is a key clinical sign that mostly signals infection. Fever is also associated with poor outcome after ischemic stroke,11 intracerebral hemorrhage,12 and aneurysmal subarachnoid hemorrhage,13 but the pathophysiologic effects of increased brain temperature on neuronal function are not well understood.
Fever develops in 25% to 50% of NICU patients. Fifty-two percent of fevers were explained by an infectious etiology, with pulmonary pathology being most predominant.14 Infectious causes should be aggressively sought (chest radiograph; blood, urine, and sputum cultures), but noninfectious causes of fever may occur and include reaction to blood products, deep venous thrombosis (DVT), drug fever, postsurgical local tissue injury, pulmonary embolism (PE), and central fever with its extreme autonomic storms (episodes of profuse sweating, tachycardia, tachypnea, and bronchial hypersecretion). Subarachnoid hemorrhage, intraventricular hemorrhage, and posterior fossa surgery can lead to a sterile inflammatory meningitis characterized by a progressive increase in cerebrospinal fluid (CSF) white blood cell counts and hypoglycorrhachia. Penicillins and phenytoin are the most common causes of drug-induced fever.15 DVT occurs in 9% of NICU patients and should be monitored with lower extremity ultrasonography.
The systemic inflammatory response syndrome may be diagnosed when fever is associated with tachypnea, tachycardia, or leukocytosis in the absence of detectable infection.16
Pneumonia is the most common nosocomial infection in critically ill neurological patients, and approximately half of all nosocomial infections are associated with mechanical ventilation. The rate of pneumonia increases 5- to 20-fold in intubated patients and with the duration of mechanical ventilation.17 Empirical antibiotic therapy is typically used at the first signs of infection. In general, early infections (<3 days) require coverage for Staphylococcus aureus, Haemophilus influenzae, Streptococcus pneumoniae, and nonpseudomonal gram-negative rods. Patients with late infections require double coverage for resistant Pseudomonas or Acinetobacter and treatment with vancomycin for methicillin-resistant S. aureus. Treatment should be modified after culture and sensitivity results are available. The duration of treatment with an effective agent should range from 7 to 14 days.
Urinary tract infections commonly develop after chronic indwelling bladder catheterization. After 10 days, nearly 50% of all catheterized patients have significant bacteriuria.18 Because asymptomatic bacterial colonization is extremely common, treatment should be reserved for patients with pyuria (>10 cells/mm3) and fever and leukocytosis. Early uncomplicated urinary tract infection can be treated with sulfamethoxazole/trimethoprim or fluoroquinolone for 7 days. Treatment should be guided by the results of culture and sensitivity testing. Changing the catheter before treatment of infection may reduce the rate of secondary infection.19
Ventriculostomy-related infection is rare, with the incidence of ventriculostomy-related meningitis or ventriculitis being approximately 8%.20 A combination of ceftazidime and vancomycin is the preferred regimen for empirical coverage. The infected catheter should be removed and treatment given for a minimum of 14 days.
Initial management of fever includes the administration of acetaminophen, but even high doses may not be effective. Currently, fever is aggressively treated with water-circulating cooling pads, and a feedback mechanism in the device secures a constant core temperature. A clinical trial comparing a simple cooling blanket with administration of acetaminophen for controlling fever found a 30% failure rate with both modes of treatment.21 The use of cooling pads with constant temperature control was shown to be superior to simple cooling blankets for the management of fever.22
Assessment of Seizures
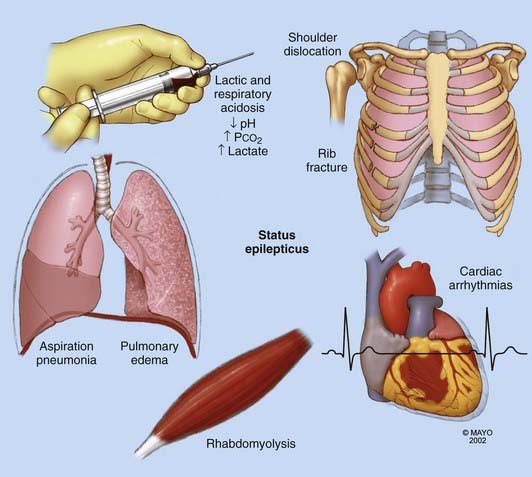
Seizures may affect other systems and cause cardiac and pulmonary complications. Rhabdomyolysis may potentially lead to acute renal failure if not recognized. Metabolic (lactic) acidosis occurs because of excessive muscular contraction, which results in depletion of glycogen and anaerobic glycolysis and thus the promotion of lactic acid formation from pyruvic acid (Fig. 24-3).
Benzodiazepines—in particular, lorazepam—are the preferred first-line drugs for acute control of tonic-clonic seizures. Lorazepam terminates seizures in more than 80% of patients.23
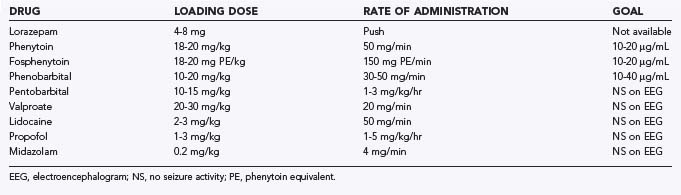
Failure of lorazepam and fosphenytoin in adequate doses to control seizures indicates transition to refractory status epilepticus. The different approaches for refractory status epilepticus have seldom been compared in a prospective controlled study. At this point, either increasing doses of barbiturates or midazolam should be used for treatment.24 Propofol is another alternative, but high doses are needed. Propofol infusion syndrome, or sudden cardiovascular collapse with metabolic acidosis, is a serious complication that limits the routine use of this otherwise very effective medication.25 Drugs used for seizures and status epilepticus are presented in Table 24-3.
Assessment of Nutritional Needs
Early feeding of the gut to ensure adequate nutrition is part of daily care. Outcome studies in patients with severe strokes and head injury suggest that early nutritional support reduces mortality and the incidence of nosocomial infections.26–28 The main goal of nutrition should be to preserve muscle mass and provide adequate fluids, minerals, and fats.29,30
The integrity of the gut is maintained by enteral feeding and greatly challenged by parenteral nutrition. It is prudent to consider postpyloric feeding in patients with neurological catastrophes because gastric atony increases the risk for aspiration. However, drug absorption in the jejunum may be unreliable. Enteral feeding should preferably be carried out by continuous infusion with a volumetric pump. Current protocols recommend that feeding start at 25 mL/hr and the volume be increased by 25 mL/hr every 4 hours until the goal of nutrition is achieved.31 In patients with a gastric residual volume of greater than 250 mL, feeding should be withheld for 4 hours and then restarted at the same rate but with a more gradual increase. Problems with enteral feeding are frequent and include diarrhea, nausea, and abdominal distention.
Assessment of Prophylaxis and Need for Anticoagulation
Patients with acute brain injury and hemiparesis have a higher incidence of DVT because of lack of mobility of the affected limb. Clinically apparent DVT was reported in 2% to 5% of patients with ischemic stroke,30,32 whereas subclinical DVT occurred in 28% to 73%, mostly in the paralyzed extremity.33
No observational studies are available to clarify the incidence of DVT in patients with intracerebral hemorrhage. One study demonstrated that DVT was detected at day 10 in 16% of patients wearing elastic stockings alone.34 In another study, 5% of patients with intracranial hemorrhage died of PE within the first 30 days.35
Only general recommendations can be made for postoperative neurosurgical patients. The most recent consensus statement published by the American College of Chest Physicians in 2004 recommended mechanical methods (intermittent pneumatic compression with or without elastic stockings) as the standard of care.36 The use of unfractionated heparin was left to the discretion of the physician, but the use of low-molecular-weight heparin was discouraged.36 Only one randomized clinical trial compared low-dose unfractionated heparin with no prophylaxis in craniotomy patients and found an 85% reduction in DVT in heparinized patients.37
The use of DVT prophylaxis in patients with traumatic head injury is another challenging situation. Several studies have tried to address the safety of anticoagulation in patients with recent hemorrhagic contusions. One study demonstrated no increased risk for hemorrhage in patients treated with unfractionated heparin within 72 hours.38 Another comprehensive study of 150 patients with hemorrhagic traumatic brain injury tested enoxaparin, 30 mg twice daily subcutaneously, starting 26.5 ± 11.5 hours after head injury. Overall, 23% of the patients demonstrated progression of the hemorrhagic lesion on CT. Nineteen percent had enlarging lesions before anticoagulation, but only 4% demonstrated an increase after initiation of anticoagulation. In 8% of patients who underwent craniotomy, hemorrhagic complications developed and required reoperation.39 These studies emphasize the potential dangers with the use of unfractionated heparin, and no definitive recommendation can be made.
Full anticoagulation is indicated for patients with cardioembolic strokes, acute DVT, or PE. It might be considered in patients with critical stenosis or acute occlusion of the carotid artery.40 Anticoagulation is used frequently in patients with high-grade basilar artery stenosis or recent basilar artery occlusion despite a virtual lack of any evidence of effectiveness. Anticoagulation is resumed in patients with a mechanical heart valve and recent cerebral hematoma, usually after a 10- to 14-day interval.
The weight-based nomogram for the use of intravenous heparin has been shown to be more effective than the standard-care nomogram.41 Heparin boluses are not recommended in neurological patients because of the increased incidence of intracerebral hemorrhage. The activated partial thromboplastin time (aPTT) is used to monitor heparin therapy closely, with values ranging from 1.5 to 2.0 times the baseline aPTT (50 to 75 seconds) being recommended. Bleeding is possible at any location with heparin but frequently occurs in the gastrointestinal tract as a result of stress ulcers. Bilateral adrenal hemorrhage might be manifested as sudden shock without any evidence of overt blood loss.42 Retroperitoneal hemorrhage should be considered in patients with a rapid decrease in hematocrit, in those with labile blood pressure, and especially when a recent cerebral angiogram or endovascular procedure has been performed. Thrombocytopenia secondary to the development of heparin-induced antibodies is another well-established complication. It is usually delayed by 5 to 10 days after the initiation of heparin therapy and might be associated with hemorrhagic and ischemic complications.43,44
Assessment of Intracranial Pressure
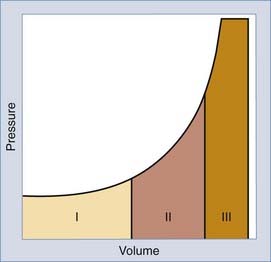
Intracranial pressure (ICP) is tightly controlled. According to the Monro-Kellie model, overall intracranial volume is constant because of the rigidity of skull, which does not permit any expansion. An increase in the volume of any component leads to a compensatory decrease in other components or eventual displacement of brain tissue through the tentorial opening or foramen of Magnum and possibly brainstem displacement and loss of brainstem function. Several compensatory mechanisms may prevent this complication. First, CSF may shift from the ventricular or subarachnoid space into the spinal compartment. Second, reduction of intracranial blood volume is achieved by collapse of veins and dural sinuses and by changes in the diameter of cerebral vessels. If the limits of the compensatory mechanisms are exceeded, a minimal increase in intracranial volume will lead to a precipitous rise in ICP (Fig. 24-4).
Monitoring of ICP is an integral function of the NICU. Indications for placement of ICP monitors include a coma from severe traumatic brain injury, and massive cerebral edema from infarction. ICP can be reduced by decreasing total intracranial volume, which is achieved by withdrawal of CSF via ventricular drainage, reduction of cerebral tissue volume (osmotic dehydration), or removal or decompression of an intracranial mass.45
Head position should be neutral to reduce any possible compression of the jugular veins. Head elevation of 30 degrees is considered standard.46 Patients should be made comfortable, and pain, bladder distention, and agitation should be avoided because these factors might increase ICP.
Hyperventilation is one method of reducing ICP. Hypocapnia causes cerebral vasoconstriction, which in turn reduces cerebral blood flow. This effect is maintained for several hours, after which it becomes ineffective because of compensatory rapid buffering of alkalotic CSF.47 Moreover, monitoring of brain tissue oxygenation has demonstrated that even a small reduction in PCO2 causes a decrease in cerebral oxygenation in comatose patients.48 Aggressive hyperventilation might thus decrease cerebral blood flow to levels approaching ischemia. Consequently, hyperventilation should be used only as a bridging measure while other means of ICP control are being instituted.
Osmotic diuresis is the mainstay of medical therapy for elevated ICP. The most widely used diuretic agents for the treatment of increased ICP are mannitol and hypertonic saline. The basic mechanism of this intervention is transport of extracellular water to the intravascular space.49 Mannitol not only facilitates the movement of extracellular water but also might increase CSF absorption. This osmotic gradient remains the overriding mechanism, but other mechanisms of action are increased cerebral blood flow from transient hypervolemia and hemodilution resulting in a decrease in blood viscosity.50 Cerebral blood volume, however, remains much the same as the result of a compensatory reflex vasoconstriction of the cerebral arterioles. Mannitol is typically used in a 20% solution, and the agent is excreted through the kidneys. The initial dose is 1 g/kg, which is then tapered to 0.25 to 0.5 g/kg. Rarely, higher doses (up to 2 g/kg) are needed to reduce ICP. The effect is apparent within 15 minutes, and failure to respond to mannitol is a sign of poor compliance and failing compensatory mechanisms. Not infrequently, it is a reason for decompressive surgery to allow more volume expansion within the skull. The maximal effect lasts about 60 minutes. The adverse reactions of mannitol, including congestive heart failure and profound pulmonary edema, are a result of rapid intravascular expansion.
Failure of mannitol to control ICP should lead to a trial of hypertonic saline. In a recent retrospective study, hypertonic saline was effective in 75% of patients deteriorating from increased ICP.51 Hypertonic saline can be administered only by central venous access, especially when given in more concentrated solutions. Brief hypotension, caused by a sudden reduction in peripheral vascular resistance in reaction to a sudden osmotic load, is the most common side effect of administration of hypertonic saline and can be avoided by slow administration lasting 10 to 15 minutes. No cases of central pontine myelinolysis were documented after treatment with a 23.4% solution.51 Cardiac arrhythmia is another potential complication of this treatment.
Monitoring of Deterioration After Acute Brain Injury
Deterioration in a patient with an acute brain injury is disease specific but predictable. Examples are shown in Table 24-4. In many instances, neurological deterioration is due to further displacement of brain tissue and, eventually, brainstem displacement. A lateral shift in brain tissue distorts the thalamus and mesencephalon. These patients have a decline in consciousness (thalamus-mesencephalon). A unilateral fixed dilated (varying from a difference of 2 to 5 mm) pupil is seen early and can be followed by bilateral fixed pupils. The pontine reflexes remain intact. This course, however, can be mimicked by acute lesions in the thalamus that suddenly extend asymmetrically to the mesencephalon (e.g., a thalamic hemorrhage). Lesions in the cerebellum may produce compression of the brainstem, but more often at the pontine level. Most notable is a predominance of pontine signs with possible bilateral miosis and loss of both corneal reflexes and the oculocephalic reflex. Frequent episodes of bradycardia with or without hypertension may occur (Cushing’s sign). A mass located more centrally will distort the thalamus and mesencephalon in a vertical plane and cause fixed midposition (4 to 6 mm) pupils initially. Asymmetric compression of the mesencephalon with anisocoria and a larger pupil or an oval-shaped pupil on the side with the lesion may be seen. Motor responses vary from decorticate to extensor responses, sometimes even with variation throughout the day and no evidence of other signs of deterioration. In patients with a gaze preference toward the expanding mass, gaze may reverse as a result of thalamic compression. Brief episodes of periodic lateral gaze may occur. Further vertical displacement of the entire thalamus-mesencephalon pontine structure may take place, but only after the upper brainstem has been destroyed directly from compression. It may occur with bilateral thalamic compression as a result of diffuse brain edema. Patients who lose all brainstem reflexes generally lose their pontomesencephalic reflexes at onset and medulla function later. A common progression is the appearance of flaccidity and no motor response with loss of the pontomesencephalic reflexes and, finally, failure to trigger the ventilator indicative of brain death.
TABLE 24-4 Causes of Clinical Deterioration in Selected Disorders
| DISORDER | CAUSES OF CLINICAL DETERIORATION | CLINICAL SIGNS OF DETERIORATION |
|---|---|---|
, Becker PS, Miller VT. Heparin-induced thrombocytopenia. Stroke. 1989;20:1449-1459.
, Carmona Suazo JA, Maas AI, van den Brink WA, et al. CO2 reactivity and brain oxygen pressure monitoring in severe head injury. Crit Care Med. 2000;28:3268-3274.
, Kirby DF. As the gut churns: feeding challenges in the head-injured patient. JPEN J Parenter Enteral Nutr. 1996;20:1-2.
, Lacut K, Bressollette L, Le Gal G, et al. Prevention of venous thrombosis in patients with acute intracerebral hemorrhage. Neurology. 2005;65:865-869.
, Norton B, Homer-Ward M, Donnelly MT, et al. A randomized prospective comparison of percutaneous endoscopic gastrostomy and nasogastric tube feeding after acute dysphagic stroke. BMJ. 1996;312:13-16.
, Norwood SH, McAuley CE, Berne JD, et al. Prospective evaluation of the safety of enoxaparin prophylaxis for venous thromboembolism in patients with intracranial hemorrhagic injuries. Arch Surg. 2002;137:696-702.
, Ropper A, Rockoff M. Physiology and Clinical Aspects of Raised Intracranial Pressure. New York: McGraw-Hill; 1993.
, Wijdicks EFM. The Comatose Patient. Oxford University Press; 2008.
, Wijdicks EFM. The Clinical Practice of Critical Care Neurology, 2nd ed. Oxford University Press; 2003.
, Wijdicks EFM, Bamlet WR, Maramattom BV, et al. Validation of a new coma scale: the FOUR score. Ann Neurol. 2005;58:585-593.
1 Wijdicks EFM. The Comatose Patient. Oxford, UK: Oxford University Press; 2008.
2 Wijdicks EFM. The Clinical Practice of Critical Care Neurology, 2nd ed. Oxford, UK, Oxford University Press; 2003.
3 Wijdicks EFM, Bamlet WR, Maramattom BV, et al. Validation of a new coma scale: the FOUR score. Ann Neurol. 2005;58:585-593.
4 Stead LG, Wijdicks EF, Bhagra A, et al. The FOUR score in the emergency department. Neurocrit Care. 2009;10:50-54.
5 Wolf CA, Wijdicks EFM, Bamlet WR, et al. Further validation of the FOUR score coma scale by intensive care nurses. Mayo Clin Proc. 2007;82:435-438.
6 Chatburn RL. Which ventilators and modes can be used to deliver noninvasive ventilation? Respir Care. 2009;54:85-101.
7 Wip C, Napolitano L. Bundles to prevent ventilation-associated pneumonia: how valuable are they? Curr Opin Infect Dis. 2009;22:159-166.
8 Anderson CS, Huang Y, Wang JG, et al. Intensive blood pressure reduction in acute cerebral hemorrhage trial (INTERACT): a randomized pilot trial. Lancet Neurol. 2008;7:391-399.
9 Ohkuma H, Tsurutani H, Suzuki S. Incidence and significance of early aneurysmal rebleeding before neurosurgical or neurological management. Stroke. 2001;32:1176-1180.
10 Van Aken H, Cottrell JE, Anger C, et al. Treatment of intraoperative hypertensive emergencies in patients with intracranial disease. Am J Cardiol. 1989;63:43C-47C.
11 Hajat C, Hajat S, Sharma P. Effects of poststroke pyrexia on stroke outcome: a meta-analysis of studies in patients. Stroke. 2000;31:410-414.
12 Schwarz S, Hafner K, Aschoff A, et al. Incidence and prognostic significance of fever following intracerebral hemorrhage. Neurology. 2000;54:354-361.
13 Oliveira-Filho J, Ezzeddine MA, Segal AZ, et al. Fever in subarachnoid hemorrhage: relationship to vasospasm and outcome. Neurology. 2001;56:1299-1304.
14 Georgilis K, Plomaritoglou A, Dafni U, et al. Etiology of fever in patients with acute stroke. J Intern Med. 1999;246:203-209.
15 Mackowiak PA, LeMaistre CF. Drug fever: a critical appraisal of conventional concepts. An analysis of 51 episodes in two Dallas hospitals and 97 episodes reported in the English literature. Ann Intern Med. 1987;106:728-733.
16 Yoshimoto Y, Tanaka Y, Hoya K. Acute systemic inflammatory response syndrome in subarachnoid hemorrhage. Stroke. 2001;32:1989-1993.
17 George DL. Epidemiology of nosocomial ventilator-associated pneumonia. Infect Control Hosp Epidemiol. 1993;14:163-169.
18 Garibaldi RA, Burke JP, Dickman ML, Smith CB. Factors predisposing to bacteriuria during indwelling urethral catheterization. N Engl J Med. 1974;291:215-219.
19 Nickel JC, Ruseska I, Wright JB, et al. Tobramycin resistance of Pseudomonas aeruginosa cells growing as a biofilm on urinary catheter material. Antimicrob Agents Chemother. 1985;27:619-624.
20 Lozier AP, Sciacca RR, Romagnoli MF, et al. Ventriculostomy-related infections: a critical review of the literature. Neurosurgery. 2002;51:170-181.
21 Mayer SA, Commichau C, Scarmeas N, et al. Clinical trial of an air-circulating cooling blanket for fever control in critically ill neurologic patients. Neurology. 2001;56:292-298.
22 Mayer SA, Kowalski RG, Presciutti M, et al. Clinical trial of a novel surface cooling system for fever control in neurocritical care patients. Crit Care Med. 2004;32:2508-2515.
23 Leppik IE, Derivan AT, Homan RW, et al. Double-blind study of lorazepam and diazepam in status epilepticus. JAMA. 1983;249:1452-1454.
24 Kumar A, Bleck TP. Intravenous midazolam for the treatment of refractory status epilepticus. Crit Care Med. 1992;20:483-488.
25 Fong JJ, Sylvia L, Ruthazer R, et al. Predictors of mortality in patients with suspected propofol infusion syndrome. Crit Care Med. 2008;36:2281-2287.
26 Kirby DF. As the gut churns: feeding challenges in the head-injured patient. JPEN J Parenter Enteral Nutr. 1996;20:1-2.
27 Norton B, Homer-Ward M, Donnelly MT, et al. A randomized prospective comparison of percutaneous endoscopic gastrostomy and nasogastric tube feeding after acute dysphagic stroke. BMJ. 1996;312:13-16.
28 Yanagawa T, Bunn F, Roberts I, et al. Nutritional support for head-injured patients. Cochrane Database Syst Rev. 2000;3:CD001530.
29 Bower RH. Nutritional and metabolic support of critically ill patients. JPEN J Parenter Enteral Nutr. 1990;14:257S-259S.
30 Koretz RL. Nutritional supplementation in the ICU. How critical is nutrition for the critically ill? Am J Respir Crit Care Med. 1995;151:570-573.
32 Pinilla JC, Samphire J, Arnold C, et al. Comparison of gastrointestinal tolerance to two enteral feeding protocols in critically ill patients: a prospective, randomized controlled trial. JPEN J Parenter Enteral Nutr. 2001;25:81-86.
32 The International Stroke Trial (IST): a randomized trial of aspirin, subcutaneous heparin, both, or neither among 19435 patients with acute Ischemic stroke. Lancet. 1997;349:1569-1581.
33 Skaf E, Stein PD, Beemath A, et al. Venous thromboembolism in patients with ischemic and hemorrhagic stroke. Am J Cardiol. 2005;96:1731-1733.
34 Lacut K, Bressollette L, Le Gal G, et al. Prevention of venous thrombosis in patients with acute intracerebral hemorrhage. Neurology. 2005;65:865-869.
35 Counsella C, Boonyakarnkula S, Dennis M, et al. Primary intracerebral hemorrhage in the Oxfordshire Community Stroke Project. 2. Prognosis. Cerebrovasc Dis. 1995;5:26-34.
36 Geerts WH, Pineo GF, Heit JA, et al. Prevention of venous thromboembolism: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126:338S-400S.
37 Cerrato D, Ariano C, Fiacchino F. Deep vein thrombosis and low-dose heparin prophylaxis in neurosurgical patients. J Neurosurg. 1978;49:378-381.
38 Kim J, Gearhart MM, Zurick A, et al. Preliminary report on the safety of heparin for deep venous thrombosis prophylaxis after severe head injury. J Trauma. 2002;53:38-42.
39 Norwood SH, McAuley CE, Berne JD, et al. Prospective evaluation of the safety of enoxaparin prophylaxis for venous thromboembolism in patients with intracranial hemorrhagic injuries. Arch Surg. 2002;137:696-702.
40 Pessin MS, Hinton RC, Davis KR, et al. Mechanisms of acute carotid stroke. Ann Neurol. 1979;6:245-252.
41 Raschke RA, Reilly BM, Guidry JR, et al. The weight-based heparin dosing nomogram compared with a “standard care” nomogram. A randomized controlled trial. Ann Intern Med. 1993;119:874-881.
42 Hardwicke MB, Kisly A. Prophylactic subcutaneous heparin therapy as a cause of bilateral adrenal hemorrhage. Arch Intern Med. 1992;152:845-847.
43 Alberio L. Heparin-induced thrombocytopenia: some working hypothesis on pathogenesis, diagnostic strategies and treatment. Curr Opin Hematol. 2008;15:456-464.
44 Becker PS, Miller VT. Heparin-induced thrombocytopenia. Stroke. 1989;20:1449-1459.
45 Aarabi B, Hesdorffer DC, Simard JM, et al. Comparative study of decompressive craniectomy after mass lesion evacuation in severe head injury. Neurosurgery. 2009;64:927-939.
46 Feldman Z, Kanter MJ, Robertson CS, et al. Effect of head elevation on intracranial pressure, cerebral perfusion pressure, and cerebral blood flow in head-injured patients. J Neurosurg. 1992;76:207-211.
47 Ropper A, Rockoff M. Physiology and Clinical Aspects of Raised Intracranial Pressure. New York: McGraw-Hill; 1993.
48 Carmona Suazo JA, Maas AI, van den Brink WA, et al. CO2 reactivity and brain oxygen pressure monitoring in severe head injury. Crit Care Med. 2000;28:3268-3274.
49 Muizelaar JP, Wei EP, Kontos HA, et al. Mannitol causes compensatory cerebral vasoconstriction and vasodilation in response to blood viscosity changes. J Neurosurg. 1983;59:822-828.
50 Muizelaar JP, Lutz HAIII, Becker DP. Effect of mannitol on ICP and CBF and correlation with pressure autoregulation in severely head-injured patients. J Neurosurg. 1984;61:700-706.
51 Koenig MA, Bryan M, Lewin JL3rd, et al. Reversal of transtentorial herniation with hypertonic saline. Neurology. 2008;70:1023-1029.