Chapter 11 Principles of Electropharmacology
Long before actual medicines were available for arrhythmias, much had been written about the theoretical mechanisms of arrhythmias.1,2 Today, these studies still provide a rational argument for why a certain drug with certain properties should be an effective antiarrhythmic agent. For example, as discussed in Chapter 2, re-entrant excitation involves a circulating excitatory wavefront along a certain path and has a certain conduction velocity (CV) and refractoriness (effective refractory period [ERP]); thus we have the concept that wavelength (λ) = CV × ERP. Depending on path length and the value of λ, the re-entrant circuit will have an excitable gap. Any change in λ would be expected to determine the inducibility and stability of re-entrant excitation. On the basis of this concept, antiarrhythmic drugs are known to affect properties of cardiac excitability (class I and IV drugs) and refractoriness (class III drugs). Class II drugs remain specific for blocking β-adrenergic receptors. Thus what has emerged is a classification of drugs that depends on a drug’s effect on certain ionic channels or receptors of the cardiac cell sarcolemma (Vaughan Williams classification).3 However, as a result of the Cardiac Arrhythmia Suppression Trial (CAST), which was designed to suppress premature ventricular depolarizations, some of these drugs lost favor, particularly for treatment of ventricular arrhythmias.
Thus most drugs currently used interrupt the direct mediators of electrogenesis, cardiac ion channels, by affecting the channel pore, the channel’s gating mechanism, or both. In 2001, the members of the Sicilian Gambit, while not disposing of the Vaughan Williams drug classification, emphasized the important and emerging role of new drug targets for pharmacological therapy and/or prevention (Figure 11-1).4 These suggestions were based on the concept that most hearts that need antiarrhythmic drugs are “remodeled,” which means that drugs are not working at all or not working well because the fundamental nature of the ion channel “pore” or channel gating mechanism has been altered by an underlying disease. A clear example here is the effect of flecainide in patients following myocardial infarction (CAST) and its effect on a re-entrant circuit and the remodeled sodium channels of cells surviving in the infarcted heart.5,6
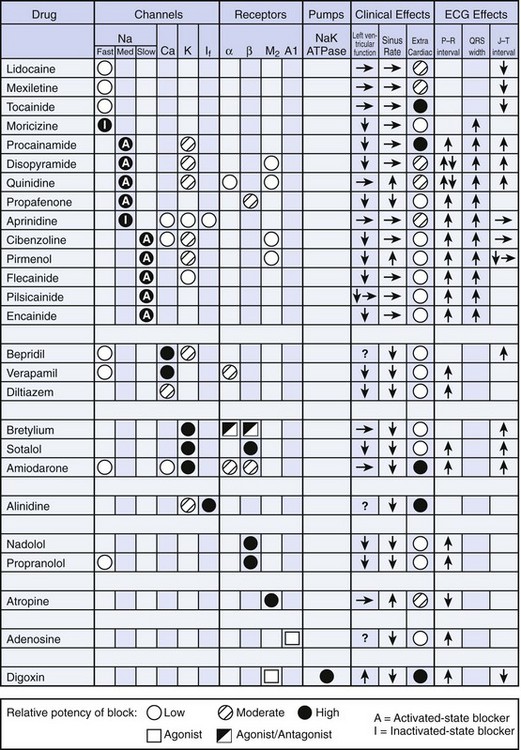
FIGURE 11-1 Drug targets for pharmacological therapy and intervention.
(From New approaches to antiarrhythmic therapy, part I: Emerging therapeutic applications of the cell biology of cardiac arrhythmias, Circulation 104:2865–2873, 2001.)
Biology and Biophysics of Cardiac Ion Channels
A drug that blocks inward plateau currents would be considered an anti–class III drug in that it should shorten APD. An antianginal drug, ranolazine, is being investigated for its antiarrhythmic properties, since it strongly inhibits the late INa current with little or no effect on peak INa.7 Thus ranolazine would shorten APD rather than reduce excitability in cardiac tissues. More interestingly, this drug, by blocking sodium (Na+) influx into the cardiac cell, would be expected to reduce intracellular Na+ and therefore would indirectly affect intracellular Ca2+ and all its sequelae (see below).
One form of re-entrant excitation is anisotropic re-entry, which was first considered in atrial samples.8 In this type of re-entry, conduction in the longitudinal direction is faster than that in the transverse direction. Spach and his colleagues suggested that a component of conduction slowing in the transverse direction in these cardiac tissues was caused by a change in gap junctional conductance. Thus slowing of conduction was brought about by the uncoupling of cells at the level of the gap junctional proteins, for example, connexins 43 and 40 (Cx43 and Cx40). While experiments have clearly shown that gap junctional uncouplers (e.g., heptanol) are, in fact, arrhythmogenic, a preferential effect appears to occur on transverse conduction velocities in various animal models. Thus a corollary would be that in re-entrant circuits in highly remodeled substrates such as those affected by cell uncoupling caused by ischemia, one would expect drugs that enhance gap junctional conductance to be antiarrhythmic. Rotigaptide is now under study as a gap junctional coupler. In experimental arrhythmia models, this drug improves conduction and abolishes lines of block that perpetuate circuits and therefore has been deemed antiarrhythmic. This experimental drug is thought to affect coupling by preventing dephosphorylation of Cx43, a ventricular gap junctional protein, or by maintaining phosphorylation of the protein.
Factors Contributing to Cardiac Remodeling
Altered Mediators of Electrogenesis
Cardiac remodeling is an all-encompassing term that can refer to both structural and functional changes in cardiac cells in response to a disease. It is an adaptive response of the heart that, when over-compensated, can be maladaptive.4
The cardiac cellular action potential changes differ in several types of disease, from chronic reduction in excitability (post-MI hearts), to APD prolongation in heart failure, to APD shortening in chronic AF. As such, the cellular ionic channel changes in different acquired diseases are varied, from a loss in Na+ current function in cells of the epicardial border zone after MI to the enhancement of the constitutively active IKAch in atrial cells in AF. In some cases, drugs have been developed to specifically target the remodeled channels of the diseased cell. The best example is the development of tertiapin-Q–type drugs that block IKAch to prolong atrial APDs shortened by muscarinic activation of IKAch. Tertiapin-Q is a highly selective blocker of Kir3, the ion channel subunit that underlies IKAch. It can significantly prolong the duration of the AF-remodeled atrial AP, where IKach activity is upregulated.9
Gene-Based Arrhythmias
For loss of function of the Na+ channel, no specific Na+ channel agonists would be useful in restoring excitability to the mutant channel cardiac cells. While trafficking defective mutants have been identified in Na+ channels and are associated with BrS, they can be “rescued” using mexilitene. However, Na+ channel–rescuing agents are not practical, since, at this time, most candidates also have Na+ channel–blocking effects. Some have suggested that by blocking K currents (e.g., Ito), the altered plateau of action potentials in BrS can be overcome, restoring normal electrical function and preventing the initiation of arrhythmia in BrS patients. To this end, quinidine (class I) and tedisamil, a new agent, have been tested. Interestingly, isoproterenol has also been reported to be antiarrhythmic (see www.BrugadaDrugs.org).
Intracellular Calcium and Targets
Under normal conditions (see Chapter 2), during systole, Ca2+ is released from the sarcoplasmic reticulum (SR) through a channel known as the ryanodine receptor (RyR). The important property of RyR protein is its open probability that is increased by the elevation of cytoplasmic Ca2+ concentration [Ca2+]i. Thus Ca2+ entry into the cell via the L-type Ca2+ current produces a small increase of Ca2+, which leads to an opening of the RyR and the release of a much greater amount of Ca2+ from the SR. This process is known as calcium-induced calcium release (CICR).
When a cell is “overloaded” with calcium, Ca2+ leaks out of the SR and waves of CICR propagate along the cell. It appears that Ca2+ waves occur when the SR Ca2+ content is elevated above a threshold value.10,11 Some of the Ca2+ in the wave is pumped out of the cell by the electrogenic Na+-Ca2+ exchange (NCX). The resulting current depolarizes the membrane and can initiate an action potential (Figure 11-2).
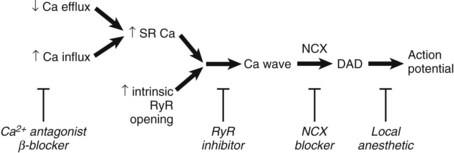
The arrhythmias mentioned above result when SR Ca2+ content is increased above the threshold level at which waves are produced. Recent work has suggested that a decrease of threshold may also produce waves. One example relates to arrhythmias seen in heart failure, where the involvement of DADs in some ventricular arrhythmias has been shown.12,13 However, studies on heart failure have found that the SR Ca2+ content is actually decreased, which suggests that the threshold for Ca2+ release may be lower such that Ca2+ waves occur at a lower SR Ca2+ content. This may be a consequence of the increased leakiness of the RyR during diastole such that Ca2+ efflux is increased at a given SR Ca2+ content. The exact molecular mechanisms responsible for this are still being debated, but it may be associated with the increased phosphorylation of the RyR caused by protein kinase A or CaMKII.14–16
An example of the occurrence of DADs in the absence of increased SR Ca2+ content is provided by catecholaminergic polymorphic ventricular tachycardia (CPVT). This arrhythmia is seen in patients during exercise or other stress. The similarity of the abnormalities in the electrocardiogram (ECG) to those observed in digitalis toxicity led to the suggestion of similarities in the underlying mechanisms. Genetic studies have shown that many patients with CPVT have a mutation in RyR or the intrasarcoplasmic protein calsequestrin. The current hypothesis is that the mutated protein causes an increased leak of Ca2+ from the SR. Thus Ca2+ waves and DADs occur at a lower SR Ca2+ content than in controls.17
Potential Therapies for Delayed After-Depolarization–Related Arrhythmias
In principle, as indicated in Figure 11-2, arrhythmias can be treated in several ways: (1) by preventing DAD, (2) by preventing DAD from producing an action potential, or (3) by both. The latter can potentially be achieved by Na+ channel blockers. A better solution, however, would be to remove the underlying DAD directly. Again, several potential approaches to this are possible. In the case of arrhythmias resulting from Ca2+ overload, it may be possible to remove the underlying “overload.” Local anesthetics reduce intracellular Na+ concentration as a consequence of decreasing Na+ entry; therefore, via NCX, this will decrease the Ca2+ load. β-Blockers (class II) are the mainstay of therapy for patients with CPVT, but even with this therapy, the recurrence rate is about 30%. β-Blockers decrease the cellular Ca2+ load by decreasing phosphorylation of the L-type Ca2+ channel and phospholamban, the latter leading to a decrease of SERCA2 activity and thus SR Ca2+ content. It would also be possible to modulate Ca2+ by affecting the membrane transports or channels involved in Ca2+ homeostasis. L-type Ca2+ channel pore blockers obviously decrease Ca2+ influx and, in so doing, would be expected to eventually reduce the SR load and [Ca2+]i and diminish force. Thus, Ca2+ channel pore blockers will affect Ca2+ but at the expense of force generation. Alternatively, one might target the molecular mechanism involved in the inactivation of the Ca2+ channel proteins or the Ca2+-dependent processes known to affect the Ca2+ channel function (e.g., CaMKII) or the small proteins (e.g., Gem) that are known to affect Ca2+ channel subunit assembly.
An alternative approach would be to stop the Ca2+ wave from developing. One caution is required here. Although the Ca2+ efflux during the wave is proarrhythmogenic, it does have the useful effect of removing Ca2+ from the cell. Abolishing the wave may result in an increase of diastolic [Ca2+]i and thus impair relaxation. Many drugs target the RyR. The local anesthetic tetracaine decreases RyR opening and thereby increases the SR threshold. In experimental studies, tetracaine was shown to abolish Ca2+ waves.18 Tetracaine is not used clinically for this purpose, since at concentrations at which it affects the RyR, it also blocks sarcolemmal Na+ channels. Very recent work has shown that flecainide suppresses CPVT arrhythmias both in humans and in a murine model.19 This appears to be caused by a combination of a direct effect to decrease RyR opening that decreases the occurrence of Ca2+ waves and an effect to inhibit Na+ channels.
Another compound is JTV519 (K201), which has been shown to decrease arrhythmias in animal models. JTV519 (K201) and its sister drug S107 appear to affect SR Ca2+ leak.20–22 This drug is a 1,4 benzothiazepine and thus can also decrease the L-type Ca2+ current. However, in isolated SR vesicle systems, it has been shown to reduce Ca2+ leak by restoring the normal FKBP12.6 stabilization of the RyR complex and improving defective gating.20,23,24
Would modulation of the Na/Ca exchanger activity be antiarrhythmic? NCX serves to maintain Ca2+ homeostasis and in several acquired diseases appears to be “upregulated.” Therefore reducing Na/Ca forward mode function would be expected to reduce the size of the current enhanced for any given change in Ca2+. Therefore there would be a decrease in the depolarization produced by the Ca2+ waves of the triggering beats. However the consequences of nonselective inhibitors of this transporter are complex. KB-R7943 is a well-characterized inhibitor but its specificity is open to question. A newer agent, SN-6, holds promise for reducing cell injury in the presence of ischemia but there are no data on its antiarrhythmic effects. There are also other agents such as SEA0400 that inhibit the exchanger.25 A recent study using the XIP peptide in normal and failing cells in fact shows a positive inotropic effect of the peptide.26 Indirect effects of such XIP effects would be expected to reduce Ca2+-dependent activation of Iti and XIP-induced SR Ca2+ release would shorten the APD prolonged by aberrant Ca2+. Finally, at least in principle, it is clearly possible to partially inhibit NCX and thereby decrease the depolarizing current generated by a Ca2+ wave. The problem with this approach, however, is that it will increase [Ca2+]i and lead to other undesirable consequences.
Would modulation of SR Ca2+ adenosine triphosphatase (ATPase; SERCA2) function be antiarrhythmic? Overexpression of SERCA would increase Ca2+ uptake into the SR at the expense of Ca2+ efflux via the Na+-Ca2+ exchanger. Therefore, decreased Na+-Ca2+ exchanger current would be evident immediately. In this way, on the one hand, cytosolic Ca2+ would (should) decrease and the SR fill, increasing the amplitude of the stimulated Ca2+ transient. On the other hand, the increase of SR Ca2+ content might make Ca2+ waves more likely. Gene transfer techniques to treat arrhythmias are still far from being used in practice, but some studies have offered proof-of-principle results. SERCA overexpression via gene transfer techniques has been shown to do just this, but no reports of an antiarrhythmic effect exist, although it decreases aftercontractions and also accelerates APDs.27,28 In a more recent report, SERCA2a overexpression previous to ligation of the left anterior descending coronary artery greatly reduced episodes of ventricular tachycardia plus VF.29 This effect was aligned with a decrease in Ca2+. Pharmacologic enhancement of SERCA pump activity is possible; however, it is not known if these agents are antiarrhythmic.
Acknowledgment
Key References
Bers DM, Eisner DA, Valdivia HH. Sarcoplasmic reticulum Ca2+ and heart failure: Roles of diastolic leak and Ca2+ transport. Circ Res. 2003;93:487-490.
Chen-Izu Y, Ward CW, Stark WJr, et al. Phosphorylation of RyR2 and shortening of RyR2 cluster spacing in spontaneously hypertensive rat with heart failure. Am J Physiol Heart Circ Physiol. 2007;293:H2409-H2417.
Diaz ME, Trafford AW, O’Neill CL, Eisner DA. A measurable reduction of SR Ca content follows spontaneous Ca release in rat ventricular myocytes. Pfluegers Arch. 1997;434:852-854.
Hirose M, Stuyvers BD, Dun W, et al. Function of Ca2+ release channels in Purkinje cells that survive in the infarcted canine heart: A mechanism for triggered Purkinje ectopy. Circ Arrhythmia Electrophysiol. 2008;1:387-395.
Kohno M, Yano M, Kobayashi S, et al. A new cardioprotective agent, JTV519, improves defective channel gating of ryanodine receptor in heart failure. Am J Physiol. 2003;284:H1035-H1042.
Janse MJ. Electrophysiological changes in heart failure and their relationship to arrhythmogenesis. Cardiovasc Res. 2004;61:208-217.
Liu N, Colombi B, Memmi M, et al. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: Insights from a RyR2 R4496C knock-in mouse model. Circ Res. 2006;99:292-298.
2001 Members of the Sicilian Gambit: New approaches to antiarrhythmic therapy, Part I: Emerging therapeutic applications of the cell biology of cardiac arrhythmias. Circulation. 2001;104:2865-2873.
Pogwizd SM, McKenzie JP, Cain ME. Mechanisms underlying spontaneous and induced ventricular arrhythmias in patients with idiopathic dilated cardiomyopathy. Circulation. 1998;98:2404-2414.
Venetucci LA, Trafford AW, Diaz ME, et al. Reducing ryanodine receptor open probability as a means to abolish spontaneous Ca2+ release and increase Ca2+ transient amplitude in adult ventricular myocytes. Circ Res. 2006;98:1299-1305.
Yano M, Ono K, Ohkusa T, et al. Altered stoichiometry of FKBP12.6 versus ryanodine receptor as a cause of abnormal Ca2+ leak through ryanodine receptor in heart failure. Circulation. 2000;102:2131-2136.
Zaza A, Belardinelli L, Shryock JC. Pathophysiology and pharmacology of the cardiac “late sodium current,”. Pharmacol Ther. 2008;119:326-339.
1 Mines GR. On dynamic equilibrium in the heart. J Physiol. 1913;46:349-383.
2 Lewis T. The mechanism and graphic registration of the heart beat, ed 3. London, UK: Shaw and Sons; 1925.
3 Vaughan Williams EM. Classification of antidysrhythmic drugs. PharmacolTher. 1975;1:115-138.
4 Members of the Sicilian Gambit: New approaches to antiarrhythmic therapy, part I: Emerging therapeutic applications of the cell biology of cardiac arrhythmias. Circulation. 2001;104:2865-2873.
5 Echt DS, Liebson PR, Mitchell LB, et al. Mortality and morbidity in patients receiving encainide, flecainide or placebo: The Cardiac Arrhythmia Suppression Trial. N Engl J Med. 1991;324:781-788.
6 Pu J, Balser J, Boyden PA. Lidocaine action on sodium currents of ventricular myocytes from the epicardial border zone of the infarcted heart. Circ Res. 1998;83:431-440.
7 Zaza A, Belardinelli L, Shryock JC. Pathophysiology and pharmacology of the cardiac “late sodium current,”. Pharmacol Ther. 2008;119:326-339.
8 Spach MS, Miller WTI, Miller-Jones E, et al. Extracellular potentials related to intracellular action potentials during impulse conduction in anisotropic canine cardiac muscle. Circ Res. 1979;45:188-204.
9 Cha TJ, Ehrlich JR, Chartier D, et al. Kir3-based inward rectifier potassium current: Potential role in atrial tachycardia remodeling effects on atrial repolarization and arrhythmias. Circulation. 2006;113:1730-1737.
10 Diaz ME, Trafford AW, O’Neill CL, Eisner DA. A measurable reduction of SR Ca content follows spontaneous Ca release in rat ventricular myocytes. Pfluegers Arch. 1997;434:852-854.
11 Chen-Izu Y, Ward CW, Stark WJr, et al. Phosphorylation of RyR2 and shortening of RyR2 cluster spacing in spontaneously hypertensive rat with heart failure. Am J Physiol Heart Circ Physiol. 2007;293:H2409-H2417.
12 Pogwizd SM, McKenzie JP, Cain ME. Mechanisms underlying spontaneous and induced ventricular arrhythmias in patients with idiopathic dilated cardiomyopathy. Circulation. 1998;98:2404-2414.
13 Janse MJ. Electrophysiological changes in heart failure and their relationship to arrhythmogenesis. Cardiovasc Res. 2004;61:208-217.
14 Bers DM, Eisner DA, Valdivia HH. Sarcoplasmic reticulum Ca2+ and heart failure: Roles of diastolic leak and Ca2+ transport. Circ Res. 2003;93:487-490.
15 Wehrens XHT, Lehnart SE, Reiken S, et al. Enhancing calstabin binding to ryanodine receptors improves cardiac and skeletal muscle function in heart failure. PNAS. 2005;102:9607-9612.
16 Ai X, Curran JW, Shannon TR, et al. Ca2+/Calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314-1322.
17 Liu N, Colombi B, Memmi M, et al. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: Insights from a RyR2 R4496C knock-in mouse model. Circ Res. 2006;99:292-298.
18 Venetucci LA, Trafford AW, Diaz ME, et al. Reducing ryanodine receptor open probability as a means to abolish spontaneous Ca2+ release and increase Ca2+ transient amplitude in adult ventricular myocytes. Circ Res. 2006;98:1299-1305.
19 Watanabe H, Chopra N, Laver D, et al. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med. 2009;15:380-383.
20 Yano M, Ono K, Ohkusa T, et al. Altered stoichiometry of FKBP12.6 versus ryanodine receptor as a cause of abnormal Ca2+ leak through ryanodine receptor in heart failure. Circulation. 2000;102:2131-2136.
21 Hirose M, Stuyvers BD, Dun W, et al. Function of Ca2+ release channels in Purkinje cells that survive in the infarcted canine heart: A mechanism for triggered Purkinje ectopy. Arrhythmia Electrophysiol. 2008;1:387-395.
22 Lehnart SE, Mongillo M, Bellinger A, et al. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J Clin Invest. 2008;118:2230-2245.
23 Yano M, Kobayashi S, Kohno M, et al. FKBP12.6-mediated stabilization of calcium-release channel (ryanodine receptor) as a novel therapeutic strategy against heart failure. Circulation. 2003;107:477-484.
24 Kohno M, Yano M, Kobayashi S, et al. A new cardioprotective agent, JTV519, improves defective channel gating of ryanodine receptor in heart failure. Am J Physiol. 2003;284:H1035-H1042.
25 Tanaka H, Nishimaru K, Aikawa T, et al. Effect of SEA0400, a novel inhibitor of sodium-calcium exchanger on myocardial ionic currents. Br J Pharmacol. 2002;135:1096-1100.
26 Hobai IA, Maack C, O’Rourke B. Partial inhibition of sodium/calcium exchange restores cellular calcium handling in canine heart failure. Circ Res. 2004;95:292-299.
27 Davia K, Bernobich E, Ranu HK, et al. SERCA2A overexpression decreases the incidence of aftercontractions in adult rabbit ventricular myocytes. J Mol Cell Cardiol. 2001;33:1005-1015.
28 Terraccianco CM, Hajjar RJ, Harding SE. Overexpression of SERCA2a accelerates repolarization in rabbit ventricular myocytes. Cell Calcium. 2002;31:299-305.
29 del Monte F, Lebeche D, Guerrero JL, et al. Abrogation of ventricular arrhythmias in a model of ischemia and reperfusion by targeting myocardial calcium cycling. Proc Natl Acad Sci U S A. 2004;101:5622-5627.