Chapter 46 Ventricular Fibrillation
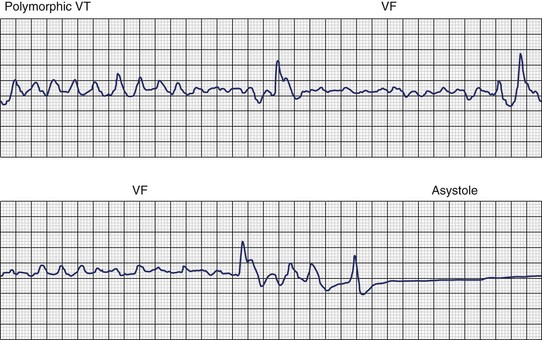
Ventricular fibrillation (VF) is the most serious cardiac arrhythmia and has a primary role in mediating sudden cardiac death (SCD). It leads to immediate circulatory arrest with cardiovascular collapse. A variable period may elapse, but cardiac asystole usually supervenes (Figure 46-1). Spontaneous termination of VF, which is seen in animal experiments, is rare in humans. VF is often preceded by an organized, rapid ventricular tachycardia (VT) of variable duration; recordings from implantable devices have now substantiated this. Ischemic injury may trigger VF. If untreated, this leads to irreversible end-organ damage, including cerebral and myocardial damage after 5 to 7 minutes of VF. Even with optimally performed basic cardiopulmonary resuscitation (CPR), the mortality rate is greater than 95% if defibrillation is delayed by more than 10 minutes. The rule of survival from out-of-hospital VF is that survival decreases by 10% for each minute before defibrillation. The sine qua non of effective resuscitation from VF is thus prompt defibrillation, delivered as early as possible. Importantly, however, a brief period of CPR before defibrillation in out-of-hospital cardiac arrest, especially if the arrest is more than 4 minutes in duration, may increase survival rates.
Etiology
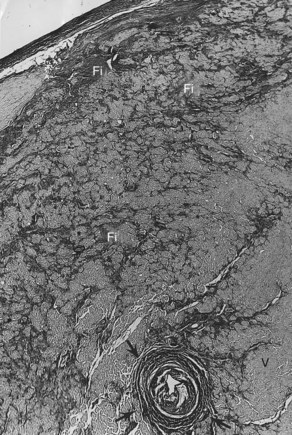
VF occurs in many disease states of the myocardium and the conduction system and can be broadly grouped under two categories: (1) genetic/familial and (2) acquired. Genetically based abnormalities of the myocardium or the specialized conduction fibers or both may give rise to the clinical manifestations of familial occurrence of VT and VF. In most cases, the initial arrhythmia is one of various forms of VT (monomorphic or polymorphic VT, which then degenerates to VF). However, acquired cardiac diseases such as coronary artery disease (CAD), hypertensive heart disease, cardiomyopathy of any etiology with or without heart failure, and other miscellaneous disease states (see Chapter 47) are the most common causes of VF and SCD.1–3 VT leading to VF may occur in acute myocarditis of any etiology. However, intractable VT with recurrent cardiac arrest refractory to medications or defibrillator device implantation may be related to chronic myocarditis (Figure 46-2).4–6
Pathology
The anatomic basis for VF in acquired heart diseases is similar to that of VT. In this chapter, emphasis is given to the morphologic findings for VF in athletes or healthy youths who were seemingly living a “normal, asymptomatic” life and died suddenly. In addition, familial occurrences of SCD caused by recurrent VT degenerating to VF is briefly discussed.1–3,5,6
Sudden Cardiac Death in Athletes or the Young and Healthy
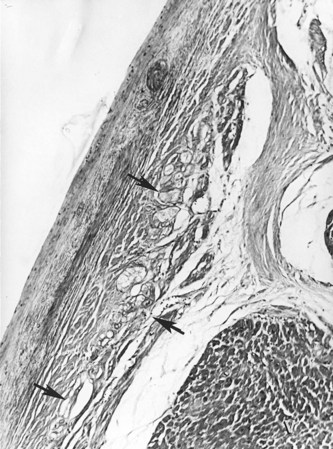
The conduction system has been carefully studied in several young persons who were living a “normal” life and died suddenly. Routine autopsies are often unremarkable. In the majority, the heart is hypertrophied and enlarged to a mild, moderate, or marked extent; however, at the gross level, no significant abnormalities were seen.3 A serial section examination of the conduction system and the surrounding myocardium reveals varying types of abnormalities, either of a congenital or acquired nature. Congenital abnormalities can exist in the sinoatrial (SA) node, atrioventricular (AV) node, or the AV bundle and the bundle branches (Figure 46-3). Abnormal formation of the SA or AV nodes, such as a double SA node, a double AV node, or the abnormal location of the SA and AV nodes in unexpected areas, can be seen pathologically. The AV bundle may be considerably fragmented into several components, abnormally located, or both. In addition, acquired pathologic findings exist in the form of focal myocardial disarray, fat, or fibrosis to varying degrees, disrupting or replacing parts of the specialized conduction fibers and the surrounding myocardium (see Figure 46-3) with arteriolosclerosis (small vessel disease) of the ventricular septum. In general, the findings in the conduction system are accompanied by pathologic findings in the surrounding myocardium. Mononuclear cell infiltration in the approaches to the SA node and in the SA node itself is present.3
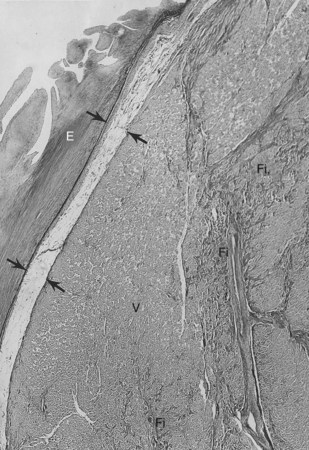
Despite such pathologic findings in and around the conduction system, these persons were considered to be asymptomatic, and SCD was the first manifestation of the disorder. Clinically, in almost all, VF was the only common denominator observed by the paramedics at the time of resuscitation. It can be hypothesized that varying types of congenital or acquired pathologic findings in and around the conduction system may remain “silent,” and these individuals are asymptomatic for long periods. Other disorders (e.g., right ventricular cardiomyopathy, hypertrophic cardiomyopathy, idiopathic dilated cardiomyopathy) can lead to VT degenerating to VF, the first evident manifestation of the disease. During an altered physiological or metabolic state, anatomic findings, pathologic findings, or both may trigger an arrhythmia that progresses to VT, VF, and SCD.3
Familial Sudden Cardiac Death
A genetic tendency for the development of an abnormal conduction system, the surrounding myocardium, or both may also lead to VT, VF, and SCD (Figure 46-4).5,6 Familial occurrence of ventricular arrhythmias may be related to the many genetic mutations associated with congenital long QT syndrome (LQTS) or Brugada syndrome. These disorders of ion channel function may also lead to cellular dysfunction and pathologic changes such as fatty infiltration, fibrosis, and disruption of the conduction system and the adjoining myocardium.7,8 These disorders are discussed in other chapters in this text and are alluded to in subsequent sections of this chapter.
Epidemiology
Ventricular Fibrillation and Sudden Cardiac Death
The epidemiology of VF intertwines with the available data on SCD (or cardiac arrest) and its documentation by emergency medical system (EMS) personnel.9 In 1998, an epidemiologic prospective study from northeast Italy reported an incidence of cardiac arrest of 95 of 100,000 persons per year.10 In this study, VF accounted for 30.2% of the initially recorded rhythms, asystole for 48.3%, and pulseless electrical activity for 21.5%. A similar study of out-of-hospital cardiac arrest confirmed VT or VF as the initial rhythm in 59 (30%) of 197 patients of a patient cohort from the Los Angeles area.11 Although VT and VF were consistently more likely to be associated with return of spontaneous circulation in these two studies, the time dependency of the initial recorded rhythm is still debated. The Ontario Prehospital Advanced Life Support (OPALS) study quoted an annual incidence of 58 out-of-hospital cardiac arrests per 100,000 persons, and in the subgroup of EMS-witnessed cardiac arrests, VT and VF accounted for 34.2% of cases.12 However, a recent analysis from Sweden estimated that patients with an electrocardiogram (ECG) taken within the first 10 minutes from a witnessed cardiac arrest have an incidence of VF of 50% to 60%. Linear regression further estimated that ECGs taken within the first 4 minutes should have an incidence of 75% to 80%.13
Ventricular Fibrillation and Population Considerations
Despite the debate on the true proportion and incidence of VF in prehospital cardiac arrest, event rates range from 250,000 to more than 450,000 per year in the United States.14 This absolute number is heavily influenced by population dynamics and the defined subpopulation being discussed.
The median incidence of cardiac arrest in 10 large Canadian and U.S. communities is 52 cases per 10,000 annually.15 The well-documented high-risk subgroups (ischemic heart disease with low left ventricular ejection fraction [LVEF], complex ventricular ectopy, prior hospital admission for congestive heart failure [CHF], or previous cardiac arrest) contribute a minority of the total number of cardiac arrests, although the subgroup percent incidence per year is the highest (Figure 46-5).
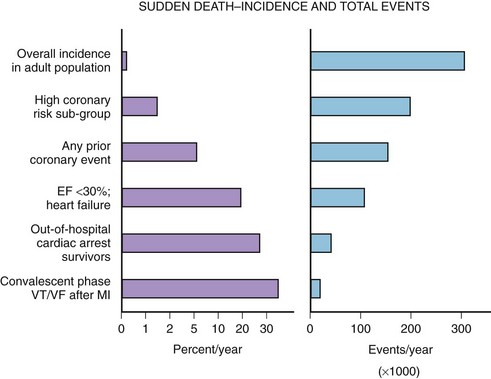
A screening tool or preventative intervention would need to be applied to 999 of 1000 persons to influence the 1 of 1000 previously unidentified persons destined for a cardiac arrest. The Framingham study demonstrated a large disparity from the highest to lowest decile (14-fold increase in risk) for SCD measuring the well-known risk factors such as age, family history, gender, tobacco use, hyperlipidemia, and hypertension.16 Electron beam computed tomography for the detection of coronary artery calcification has been correlated to coronary event risk.17 A recent publication compared the Framingham risk index and coronary artery calcification in an autopsy series of SCD victims.18 The two risk assessment methods had modest correlation with each other (63%). Of the cases, 83.5% had a coronary artery calcification score or Framingham risk index above average for age. The remaining 17.5% highlight the need for exploration of new cardiac risk factors (e.g., fibrinogen, homocysteine, infectious agents, or ion channel abnormalities) that could be applied to the population at large.
The high-risk subpopulations have been targeted in recent publications for epidemiologic analysis. The Worcester Heart Attack Study described a relatively fixed incidence rate of 4.7% for VF in hospitalized patients with validated acute myocardial infarction (MI) over a 22-year period (1975 to 1997).19 The Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico (GISSI-2) database reported an incidence of early-onset VF (<4 hours after onset of acute MI) and late VF (>4 to 48 hours) of 3.1% and 0.6%, respectively. In-hospital prognosis was worse in patients with VF than without VF, but the postdischarge to 6-month mortality rate was similar for both VF subgroups and controls.20 The patient with prior cardiac arrest has a similar “time dependence” of risk of recurrent events, as was presented by Furukawa et al (Figure 46-6).21
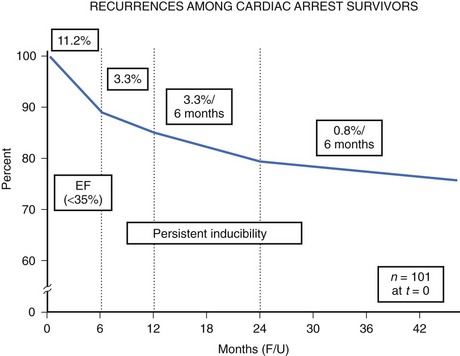
Low LVEF was a strong predictor of the highest risk of recurrence (11% in the first 6 months). Persistent inducibility at electrophysiological study (EPS) was the better predictor for recurrent events from 6 to 42 months, but the subsequent risk fell to 0.8% over the final 6 months of the study period. In the patients who entered the Antiarrhythmics Versus Implantable Defibrillators (AVID) study with VF, the survival curve shows the same early recurrence (<6 months) in the patients treated with antiarrhythmic drugs as those who did not receive this therapy (Figure 46-7).22
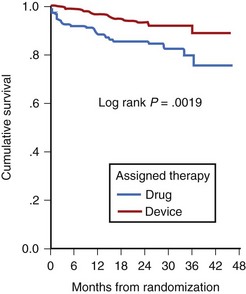
Clinical trials are assessing the “static” risk of cardiac events outside the setting of acute hospitalization.23 The Multicenter Automatic Defibrillator Implantation Trial (MADIT-II) trial tested implantable cardioverter-defibrillator (ICD) therapy versus standard medical therapy in patients with ischemic heart disease, an LVEF less than 30%, and previous MI. It showed a 31% relative risk reduction with ICD therapy. The Sudden Cardiac Death in Heart Failure Trial (SCD-HeFT) trial compared three arms of therapy in patients with congestive heart failure and LVEF less than 30%: best conventional heart failure therapy with placebo, best conventional heart failure therapy with amiodarone, and best conventional heart failure therapy with ICD.23 In a preliminary report, there was a 23% relative reduction in all-cause mortality with the ICD compared with placebo (7.2% to 5.8% per year), and no reduction in mortality rate with amiodarone compared with placebo. The most recent developments in assessing risk of cardiac arrest have been in the blossoming technology of molecular genetics. The familial types of LQTS and their respective genetic bases have been well described elsewhere. An increasingly recognized syndrome of right bundle branch block (RBBB), ST segment elevation, and aborted SCD (Brugada syndrome) has also been ascribed to a genetic mutation in the gene encoding the cardiac sodium channel. Its prevalence in a European study by ECG screening was estimated at 0.1% of the general population.24 If the only effective therapy for the syndrome is ICD implantation, the economic impact on the public health budget is obvious.
A trial of the automated external defibrillator (AED) to be used in the home by trained spouses or cohabitants of individuals with prior anterior MI (the Home Automated External Defibrillator Trial [HAT]) was not able to show a reduction in mortality rate, in part because of the unexpectedly low incidence of VF as the initial recorded arrhythmia in these patients.25
Basic Electrophysiology
Nature of Fibrillatory Wavefronts
Re-entrant excitation is the fundamental mechanism responsible for VF. The precise nature of these wavefronts, their formation, and sustainability are key to understanding VF. Various theories exist regarding the nature of re-entrant waves. One such theory is the leading circle hypothesis, originally put forward by Allessie and coworkers to explain atrial fibrillation (AF).26 This hypothesis states that a re-entrant circuit is the smallest pathway in which the impulse can continue to circulate and that the core is kept permanently refractory (i.e., there is no excitable gap) (Figure 46-8). However, Janse questioned whether the core is truly refractory during re-entry, since membrane potential is relatively normal in this region and diastolic intervals are relatively long.27 Furthermore, the presence of an excitable gap allows wavefronts (spontaneous or induced) to invade the area and terminate or entrain the re-entrant circuit; Allessie et al were able to terminate tachycardia with premature beats (i.e., anti-tachycardia pacing).26 It now appears that in most cases of VF, an excitable gap does exist; however, there may still be room for leading circle re-entry in a subset of arrhythmic phenomena, particularly with regard to AF.28
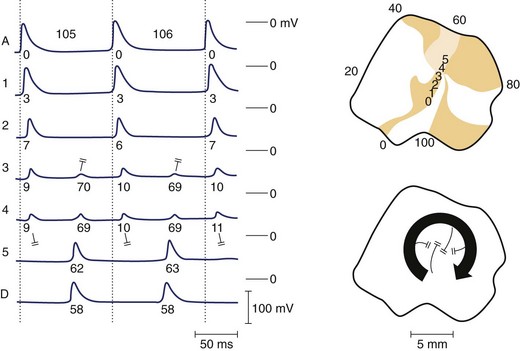
An alternative mechanism for VF is spiral wave re-entry. This theory suggests that the wavefront curves, or forms a spiral, because curvature is negatively related to conduction velocity (because of the source-sink relationship). As a result, the wave is highly curved near the core and moves slowly, but at the distal end the wave speed increases, resulting in a spiral shape. Two-dimensional spiral waves may theoretically exist in surviving two-dimensional layers overlying the healed myocardial infarcts, although three-dimensional waves (i.e., scroll waves) are clearly more relevant to cardiac arrhythmias. A scroll wave can be thought of as a stack of spiral waves in which the cores line up to form a “filament” at the center. Simple scroll waves, in which the filament forms a straight line, have been demonstrated during VF.29 A scroll wave with a core that is perpendicular to the recording surface will appear as a two-dimensional spiral wave, whereas a filament that is parallel to the recording surface will appear as a plane wave. It has been suggested that during VF, scroll waves are usually oriented parallel to the surface, which may explain why mapping studies sometimes fail to detect re-entry during VT or VF. A scroll wave with a ring-shaped filament is called a scroll ring. It is interesting to note that the cross-section of such a wave would represent classic figure-of-eight re-entry. Only indirect experimental evidence is available for the existence of scroll rings, probably because of the difficulty of measuring them, because they manifest only transmurally, never on the surface.30 The shape of a scroll as it is initiated strictly depends on the shape of the inexcitable obstacle encountered by the wavefront. Thus, filaments of varying shape can readily develop in the myocardium. Scroll rings ultimately collapse on themselves, but simple scroll waves can be stabilized and maintained.31
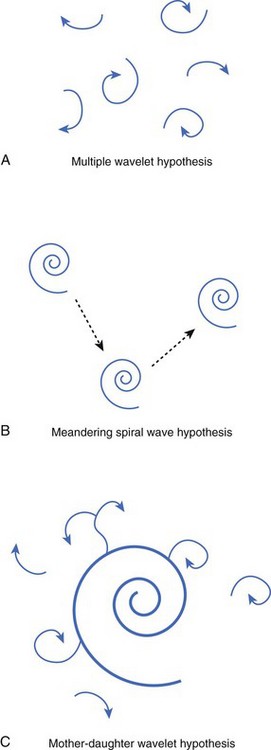
Numerous hypotheses have been offered regarding the nature of fibrillatory waves that underlie VF. Three of these theories are illustrated in Figure 46-9: (1) the multiple wavelet hypothesis, (2) the meandering spiral wave hypothesis, and (3) the mother-daughter wavelet hypothesis. The multiple wavelet hypothesis was put forward in the early 1960s by Moe and describes fibrillation as consisting of multiple, nonstationary wavefronts that continuously form, fractionate, and reform (Figure 46-9, A).32 Spatial dispersion of repolarization was an important condition for initiating multiple wavelet VF. Others have suggested that VF is caused by spiral re-entrant waves that meander around the myocardium (see Figure 46-9, B).33 According to this theory, the chaotic appearance of the ECG in VF is attributable to the meandering of a single spiral wave rotor throughout the ventricular myocardium. Jalife suggested that the mechanism of fibrillation may consist of a stable, periodic re-entrant wave that gives off “daughter wavelets” that meander and fractionate (see Figure 46-9, C).34 According to this hypothesis, unstable daughter wavelets form because of local gradients of refractoriness and give rise to the fibrillatory patterns of the ECG.
Substrates for Ventricular Fibrillation: Heterogeneities of Repolarization and Refractoriness
Various anatomic and functional substrates can lead to the development of the fibrillatory dynamics previously described. Early studies of AF suggested that the heterogeneity of refractoriness was a necessary substrate for fibrillation in the heart.32 Although it was later shown that VF can occur in hearts without large dispersions of refractoriness (i.e., in normal hearts), or in modeling studies using simulations of electrically homogenous tissue, these studies may not be clinically relevant.35 It is more difficult to initiate VF in a normal heart, and the vast majority of fibrillatory episodes occur in diseased hearts, in which the gradients of repolarization kinetics (and therefore refractoriness) are almost certainly abnormal. The important role of repolarization in the development of re-entrant substrates has recently been emphasized by studies demonstrating the inherent heterogeneities in repolarization properties that exist transmurally.36 Antzelevitch and others have demonstrated that trans-mural heterogeneities of repolarization are critical in the development of the substrates for re-entry under various drug-induced or disease-induced conditions.37,38
Rosenbaum et al recently developed a hypothesis that explains how the critical heterogeneities of repolarization that provide a substrate for re-entry may occur.39 Beat-by-beat alternation of cardiac action potential duration (APD) has been shown to underlie T-wave alternans, an electrocardiographic indicator that correlates well with the risk of SCD.39 We have demonstrated that when alternans occurs in various regions of the myocardium in a “discordant” manner (i.e., some regions are on a long-short-long cycle, and others are on a short-long-short cycle), steep gradients of repolarization develop and reverse the direction on a beat-by-beat basis, which can lead to conduction block, re-entry, and VF.40
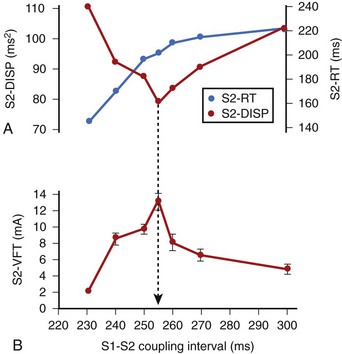
In related work, Laurita et al demonstrated that premature beats modulate the dispersion of repolarization in a manner that has direct effects on vulnerability to VF.41 As the S1-S2 interval is shortened, gradients of repolarization decrease concomitant to a decrease in vulnerability to VF, but then increase with a concomitant increase in VF vulnerability as S1-S2 is further shortened (Figure 46-10). Such biphasic modulation of dispersion relates back to the concept of the “vulnerable period,” which arose from critical studies by Moe, who described the window of time during the ECG T wave when vulnerability to VF is greatest.32 Thus, dispersion of repolarization and refractoriness appears to play a major role in the mechanism of re-entrant VF.
Restitution Hypothesis
Restitution is a property of cardiac myocytes that dictates the APDs of a premature action potential after a period of steady-state rhythm. The APD of the extrasystolic beat is determined not only by the S1-S2 interval but also by the APD of the pre-extrasystolic beat. Furthermore, gradients of restitution kinetics are known to exist across the epicardial surface such that cells in different regions of the heart having equal baseline APDs could have different extrasystolic APDs following the same S1-S2 interval.42 In theory, if gradients of restitution kinetics are steep enough, underlying heterogeneities of repolarization would not be necessary to create dispersions of repolarization following a premature beat.42 However, cardiac tissue is known to exhibit repolarization heterogeneities across epicardial and trans-mural surfaces, so the role of restitution may be to enhance existing dispersion of repolarization and promote VF.
One possible mechanism to describe the role of restitution in VF is the restitution hypothesis, which states that when the restitution curve (a plot of APD vs. diastolic interval) has a slope greater than 1, VF may be initiated. For example, a wavefront encroaching on the tail end of a previous wave creates a gradient of diastolic interval between the two (assuming the degree of curvature is not identical); in regions of tissue where restitution is steep, this will create large dispersions of APD along the second wavefront. Some areas of the wave may have such a short diastolic interval that they fail to propagate, resulting in wavefront fractionation. This was demonstrated by Garfinkel et al, who also used pharmacologic agents to flatten the restitution curve slope and decrease vulnerability to VF. Koller et al showed that elevating extracellular potassium decreases the portion of the restitution curve where the slope is 1 or more and converts VF to a periodic rhythm.43,44 Other investigators have shown that the biphasic nature of a restitution curve, demonstrated in a number of cardiac tissue types, is also an important determinant of vulnerability to VF.45 More recently, the restitution hypothesis has been applied to restitution curves for both APD and conduction velocity.46
Myocardial Ischemia
VF is commonly caused by acute myocardial ischemia. Under experimental conditions, it is often difficult to provoke VF in normal tissue using premature stimuli; however, in ischemic tissue, premature beats exacerbate dispersion of refractoriness and readily lead to tachyarrhythmias.47 This is because of the various electrical alterations that occur during ischemia and the consequent formation of various substrates that promote re-entry and VF.
At the cellular level, acute ischemia results in depolarization of the resting membrane potential, decreased maximum rise rate and amplitude of the action potential, and decreased APD, as well as reduced intercellular coupling.48–53 Depolarization of the membrane is attributed largely to accumulation of extracellular potassium, as is post-repolarization refractoriness.54,55 Acidosis can produce a small depolarization of resting membrane potential and also decreases gap junction conductance and cell-to-cell coupling.56 The net result of the electrical changes that occur during ischemia is the formation of various substrates that can lead to re-entry and wavebreak, such as slowed conduction velocity, increased dispersion of refractoriness, and alternans.
Healed Myocardial Infarcts
The process of tissue healing and scar formation after an episode of acute myocardial ischemia involves necrosis of the infarcted region as well as swelling and hypertrophy of the noninfarcted region as it attempts to compensate for the loss of cardiac muscle.57 Heterogeneity of repolarization may result from the uneven prolongation of APD in the hypertrophied post-MI ventricle, changes in expression of gap junction proteins, or both.58,59 A healed infarct may also provide an anatomic substrate for arrhythmias. Infarcts vary from simple surviving muscle bundles that form accessory pathways to complex subendocardial sheetlike structures, which are linked to the surrounding myocardium by multiple connecting bundles, creating complex matrices of conductive tissue that may promote multiple re-entrant circuits. In particular, the combined effects of slowed conduction and the presence of structural anomalies is significant because in tissue where excitability is low, a wavefront breaking past an obstacle will curl at the inner ends (where the break occurred) to create figure-of-eight re-entry, whereas in a normally excitable medium, the wave tends to reform on the other side of the obstacle.60
Autonomic Modulation of Ventricular Fibrillation
It has been well established that increases in sympathetic tone increase the risk for SCD, whereas vagal reflexes have the opposite effect.61–63 Studies in dogs have shown that left cardiac sympathetic denervation, or left stellectomy, increases survival and decreases arrhythmias during acute MI.64 In contrast, unilateral blockade of the right stellate ganglion by cooling increases the number of arrhythmias after coronary occlusion.65 Many clinical trials have shown that β-adrenergic receptor blocking drugs have a potential benefit in preventing SCD in patients with MI, whereas anti–α-adrenergic agents have not been shown to be effective.66–69
It has been suggested that during acute MI, sympathetic nerves that traverse the myocardial wall are damaged, leading to denervation hypersensitivity.70 This, in turn, leads to spatially inhomogeneous responses to β-adrenergic stimulation across the ventricle, forming potential substrates for re-entrant excitation. Further investigation is required to elucidate fully the mechanistic role of β-adrenergic stimulation and arrhythmogenesis.
Genetic Substrates for Ventricular Fibrillation
Brugada syndrome is a cardiac disease characterized by an elevated ST segment that is unrelated to ischemia, Q-T interval prolongation, electrolyte abnormalities, or structural heart disease.71 Patients with Brugada syndrome are at increased risk of SCD from VF. The syndrome is caused by a mutation in the gene encoding the cardiac sodium channel (SCN5A) and is common in Southeastern Asia, where it is the second highest cause of death among young men.72 In Europe, gender specificity is less distinct, although affected women tend to be less symptomatic than affected men. Patients with Brugada syndrome often die in their sleep, and a possible relationship to sudden infant death syndrome has been suggested.73
It is believed that the mechanism of VF in patients with Brugada syndrome is related to reduced function of the mutant late inward sodium current, forcing the balance of plateau currents in favor of repolarization. This is why any further impairment of sodium channel function (e.g., by sodium-blocking drugs) can exacerbate the electrocardiographic and arrhythmogenic phenotype in this disorder.74 Because epicardial, not endocardial, cells possess the transient outward current (Ito), they are most susceptible to the repolarizing effect of the sodium channel mutation in Brugada syndrome. Marked and selective shortening of epicardial action potentials relative to endocardial action potentials produces a transmural voltage gradient during the plateau that, in turn, is believed to account for the characteristic pattern of ST segment elevation seen in these patients.75 Transmural action potential gradients also account for the presumed mechanism of VF (i.e., phase 2 re-entry).
In a recent provocative study, Haisaguerre et al identified a select group of patients with recurrent idiopathic VF that seemed to originate with premature beats emanating from the distal His-Pukinje system; these patients were successfully treated with radiofrequency ablation targeting the originating premature beats, with a low rate of VF recurrence.76
Clinical Electrocardiography
The role of electrocardiography has been increasing in patients presenting with VF. Certain ECG features may identify high-risk patients or transient events that can result in VF, which may be preceded by VT (see Chapter 47).
Electrocardiographic Features of Patients at High Risk for Ventricular Fibrillation
Electrocardiographic recordings obtained at the onset of VF provide insight into events that precipitate SCD. Documentation of these events has been obtained by continuous monitoring in hospital telemetry units and by ambulatory monitoring. The patients involved in these studies had a high incidence of CAD, and most had frequent or complex ventricular ectopy. The terminal events were associated with sinus arrest, complete heart block, or ventricular asystole in approximately 10% of patients; in 90%, VF was preceded by VT or ventricular flutter of variable duration.77–80 ST segment and T-wave changes indicative of ischemia related to acute MI or coronary spasm have also been reported to precede terminal ventricular arrhythmias.77–80 Other studies indicate that bradycardia and electromechanical dissociation are important causes of SCD in patients with advanced heart failure and nonischemic cardiomyopathy.81 These observations have significant implications for strategies to reduce mortality rates in patients at risk for SCD.
The diagnostic role of the ECG in identifying patients with genetic disorders associated with SCD from ventricular arrhythmias has continued to evolve over the past decade. The electrocardiographic characteristics of LQTS, Brugada syndrome, and arrhythmogenic right ventricular cardiomyopathy (ARVC) have gained widespread attention. This is vital to the prevention of SCD in young patients with these disorders, particularly if these patients participate in athletics. A recent study has suggested a higher incidence of early repolarization changes in the inferolateral leads (J-point elevation and notching in the terminal portion of the QRS complex) among survivors of cardiac arrest caused by idiopathic VF when compared with control subjects.82
The pattern of early repolarization associated with highest risk seems to be global or inferolateral early repolarization with prominent J waves on the resting ECG.83 It is not clear if a single disorder of repolarization causes at least some of the cases previously labeled “idiopathic VF,” or, as seems more likely, a spectrum of disorders is associated with abnormal and heterogeneous repolarization that predisposes otherwise normal individuals to VF. These disorders have been collectively named early repolarization disease.84
Long QT Syndrome
The electrocardiographic manifestations of LQTS include QT prolongation, abnormalities in T-wave morphology, increases in QT dispersion, T-wave alternans, and a relative degree of bradycardia in children (Figure 46-11). The upper limits of normal for the QTc values are 460 to 470 ms for females and 440 to 460 ms for males.85,86 Longer QT values may be observed in normal women after puberty.87 The degree of QT prolongation does not directly correspond with the risk of syncope, but malignant ventricular arrhythmias are more frequent when the QTc exceeds 600 ms.87 A diagnostic dilemma is that the Q-T interval shows temporal variations in patients with this syndrome, and the QTc may fall within the normal range on a random recording.88 Garson reported that 6% of LQTS patients had a normal Q-T interval, and data from the International Registry showed that 10% of family members with a QTc less than 440 ms had a cardiac arrest.89,90 Thus, an ECG with a normal QTc does not exclude the diagnosis if strong suspicion exists that a patient has the syndrome, especially if the QTc is on the border of normal.
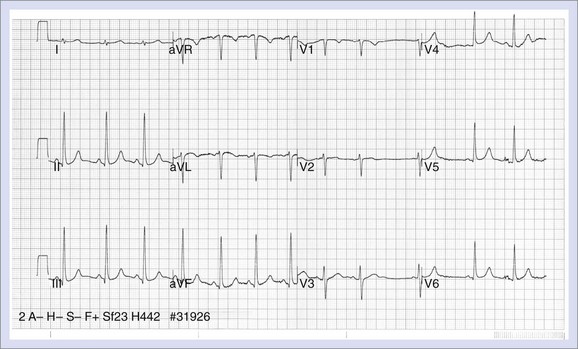
The effect of exercise increases the QTc in patients with LQTS, but this effect is less apparent in patients with LQT3 than in those with other genotypes.91 Approximately 62% of patients with LQTS exhibit T waves that are biphasic or notched, and a higher incidence of these abnormalities is seen in patients with cardiac events.92 The characteristic features are most pronounced in precordial leads V2 to V5. The appearance of notched T waves may be provoked by exercise. The degree of QT dispersion is measured by the difference between the longest and shortest Q-T intervals on the 12-lead ECG and is prolonged in patients with LQTS.93 It is thought to represent increased dispersion of repolarization. Patients who show no change in the degree of QT dispersion when they are treated with β-blockers appear to be at increased risk for cardiac events.90,94 T-Wave alternans is a beat-to-beat alternation in the amplitude or polarity of the T wave. It appears to be a marker of electrical instability that may precede torsades de pointes.94 Children with LQTS often have resting heart rates that are lower than normal and may exhibit a blunted chronotropic response to exercise.87,95 Torsades de pointes, which is the ventricular arrhythmia associated with LQTS, is characterized by the undulating amplitude of the QRS complex, which gives the appearance of twisting about its axis. The onset is frequently associated with pause-dependent ventricular ectopy that falls on the T wave.96–98
Brugada Syndrome
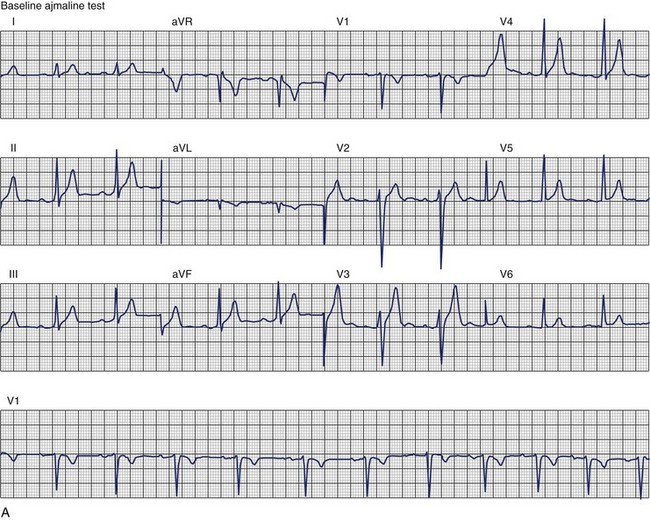
The ECG patterns associated with Brugada syndrome are (1) a terminal R′ in lead V1 with complete or incomplete RBBB; (2) convex downward (coved) ST segment elevation equal to 0.1 mV in lead V1 or leads V1 and V2; convex upward (saddle-shaped) ST-segment elevation equal to 0.1 mV; and (3) J-point elevation followed by a downsloping ST segment ending in a negative deflection (triangular shape).98 Serial ECGs performed on the same patient may show variation from one pattern of ST-segment elevation to another, normalization, and progressive development of RBBB. Figure 46-12 shows examples of variable Brugada ECG patterns that occurred during an ajmaline test in the same patient.99,100 The prevalence of the Brugada ECG pattern is reported to be 0.07% to 0.7%, and there is a male predominance that is especially marked in Asians.101–106 The range in prevalence appears to depend on the criteria used to make the diagnosis. The saddleback ST-segment elevation is more common. The typical coved pattern was found in 0.1% to 0.26% of community-based populations in Japan and Europe.104–106 In a Japanese study population that underwent ECGs during health examinations, the prevalence of all types of Brugada ECG patterns was 0.7%.103 The coved-type ST-segment elevation was found in 38% of subjects with the Brugada pattern, and the saddleback-type ST-segment elevation was seen in 62%. In the same study, the rsR′ pattern in lead V1 was observed in 41%, and the Rsr′ pattern was recorded in 59%. The prevalence of the coved pattern was 0.26%, and the typical Brugada ECG pattern with coved ST-segment elevation and the rsR′ pattern in lead V1 was 0.12%. If only male subjects were considered, the criteria for a Brugada ECG pattern was met in 2.14% of the population, but the typical pattern in males was 0.38%.104 In a European population studied by Hermida, the prevalence of ST segment elevation was 6.1%; however, only 1 (0.1%) of the 61 subjects who met the study’s criteria for the Brugada pattern had the coved pattern. All of the others had the saddleback pattern.103
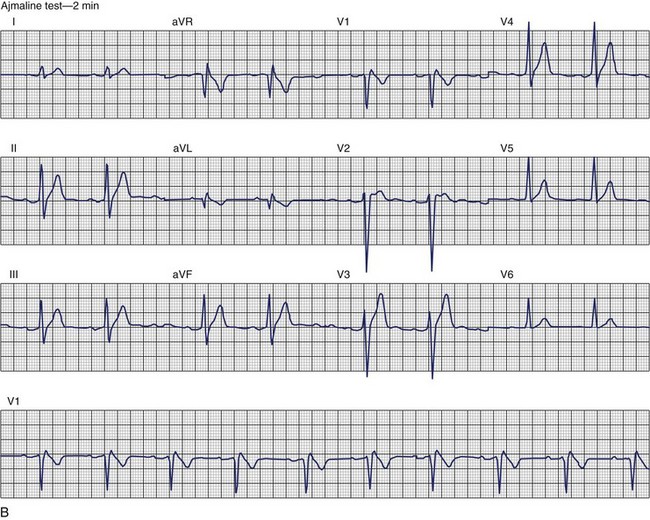
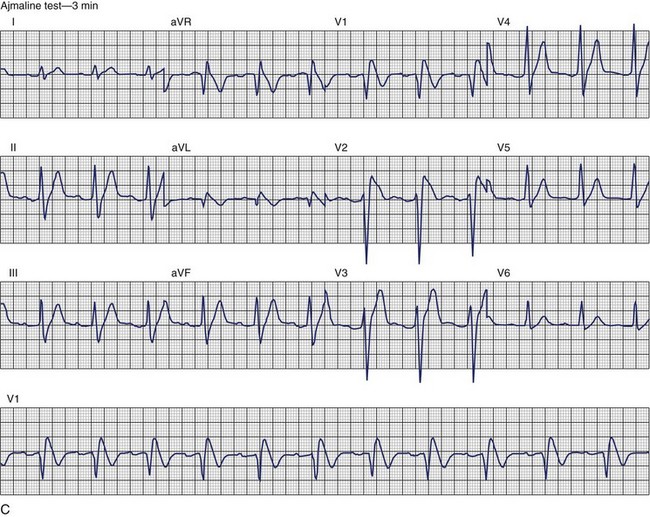
The prognostic significance of the Brugada ECG pattern is difficult to assess. Brugada reported an 8% incidence of arrhythmic events in an asymptomatic hospital-based population.106 The degree and type of ST segment elevation require further study for risk stratification of asymptomatic individuals in community-based populations. Matsuo evaluated mortality in patients younger than 50 years who had ECGs recorded during biannual examinations from 1958 through 1999.107 A total of 32 patients were identified with the Brugada ECG pattern. Seven of these patients died suddenly or died of an unexplained accident. Although total mortality was not increased in patients with the Brugada ECG pattern, the mortality rate from unexpected death was significantly higher. No increase in mortality was observed in studies by Miyasaka, Takenaka, or Priori.104,105,107 In the Osaka population, one SCD occurred among the 98 subjects with the Brugada ECG pattern during a mean follow-up of 2.6 years.104 A 3-year follow-up reported by Atarashi of patients with a Brugada ECG pattern found cardiac event–free rates of 67.6% in symptomatic patients and 93.4% in an asymptomatic group.108 Coved-typed ST segment elevation appeared to be related to cardiac events. The higher incidence in hospital-based studies may reflect referral patterns based on a family history of SCD. Differences in criteria or ECG interpretation affect the diagnosis of this pattern, and the follow-up in most studies is too short to draw definitive conclusions about the long-term prognosis.
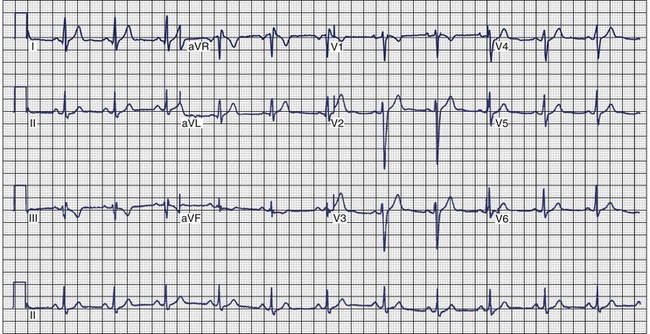
Arrhythmogenic Right Ventricular Cardiomyopathy
ECG recordings during sinus rhythm in patients with ARVC have several distinctive features (Figure 46-13).109 The QRS may be prolonged in the right precordial leads to a greater extent than in leads I or V6. The QRS is often greater than 110 ms in lead V1 (sensitivity, 55%), and a pattern of incomplete RBBB is observed.110 In 30% of cases, the delay in conduction over the right ventricle results in a small potential at the terminal portion of the QRS in lead V1 that has been termed an epsilon (ε) wave, which can be amplified with bipolar recordings over the inferior and superior aspects of the sternum.109 This can be achieved by repositioning the left arm lead over the xiphoid process, positioning the right arm lead over the manubrium sternum, and applying the left leg lead at the position customarily used for lead V4 or V5.111 The other major feature of ECGs recorded from patients with ARVC is inversion of the T waves in the precordial leads, which is observed in 42% to 54% of patients.111,112 Metzger assessed the value of serial 12-lead ECGs in 20 patients to recognize progression of ARVC over a mean of 71 ± 48 months.113 Abnormalities were detected in 90% of the patients. The most frequent abnormality was T-wave inversion in the precordial leads. No correlation was demonstrated between the ECG and the extent of disease detected by echocardiography. In the 14 patients who had several ECGs recorded over time, no clear progression of electrocardiographic abnormalities was observed.
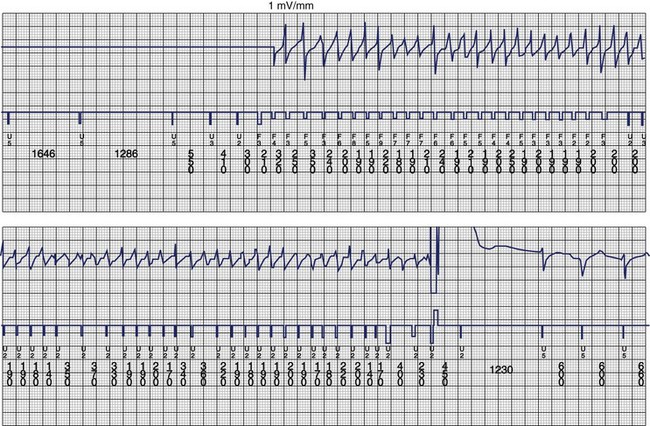
The ventricular arrhythmias associated with ARVC typically show a morphology resembling left bundle branch block in lead V1 with a variable axis. Nava published a clinical profile and long-term follow-up of 37 families with ARVC that demonstrated a correlation between the severity of echocardiographic findings and the severity of ventricular arrhythmias, which were seen in all patients with the severe form of the disease, in 82% with moderate disease, and in 23% in those with mild disease.112 Overall, 60 (45%) of 132 affected living members had ventricular arrhythmias. These included VF in 1, sustained VT in 14, nonsustained VT in 8, ventricular couplets and triplets in 16, and frequent ventricular premature depolarizations in 8. Exercise-induced polymorphic VT was observed in 13 patients with no other documented arrhythmias. Only one patient who was judged to have mild disease had a sustained ventricular arrhythmia. Although the data from this study showed a low incidence of VF, the incidence may be higher. In 19 of the 37 families, the proband died at a young age, and the diagnosis was made at autopsy. One may speculate that some of these subjects had VF. Figure 46-14 shows electrograms recorded from an ICD implanted in a teenager (male) with ARVC and frequent nonsustained ventricular arrhythmias. The recording shows the sudden onset and successful termination of VF, which occurred at night while he was asleep. He had no prior history of syncope or sustained ventricular arrhythmias.
Diagnostic Evaluation
Evaluation of Transient or Reversible Causes
VF that occurs secondary to a reversible or transient cause may be adequately treated by correction of the reversible cause or by short-term therapy or close observation while awaiting spontaneous resolution of the transient cause.114 The causes of VF with these characteristics are usually readily identified with a few focused investigations. Electrolyte abnormalities are identified with serum electrolyte testing performed as soon as possible after resuscitation. The most common electrolyte abnormalities leading to VF are hypokalemia, hypomagnesemia, or both. When considering the temptation to ascribe an episode of VF wholly to hypokalemia, hypomagnesemia, or both, one must recall that the adrenergic discharge state during resuscitated VF results in redistribution of extracellular potassium and magnesium into the intracellular compartment. Accordingly, relative hypokalemia, hypomagnesemia, or a combination is very common after VF.115 Only marked hypokalemia, hypomagnesemia, or both and confidence that future episodes can be prevented should prompt the belief that the episode of VF had a reversible cause.
The early performance of a 12-lead ECG and serum markers of myocardial necrosis (creatine kinase [muscle or brain type], troponin) will permit identification of the patient whose VF has occurred in the acute phase (first 48 hours) of an MI. Evidence that acute MI produces an environment that constitutes only a transient risk of VF is most convincing for a Q-wave MI.116 Nevertheless, the risk of VF may also be transient in the setting of a non–Q-wave MI.117 However, the practitioner must avoid the temptation to ascribe an episode of VF wholly to an acute MI if the only evidence of acute myocardial necrosis is a marginal elevation in a serum marker of necrosis; such elevations may be secondary to the VF-induced cardiac arrest rather than represent its cause. The diagnosis of MI in the setting of out-of-hospital cardiac arrest from VF is made difficult by the usual absence of a history of chest pain, frequent nondiagnostic ECG abnormalities that may be a consequence of the myocardial ischemia resulting from the global “no-flow or low-flow state” during VF, and the absence of a specific cut-off for the degree of cardiac enzyme elevation that separates MI causing VF from VF causing myocardial necrosis. In general, the presence of a culprit lesion (coronary thrombus or ulcerated plaque) on angiography early after cardiac arrest and well-preserved or normal left ventricular function suggests an ischemic or infarction-related cause.
The other common reversible or transient cause of VF is the use of a proarrhythmic drug: a classic antiarrhythmic drug, other drugs with electrophysiological effects, and recreational drugs, particularly cocaine.118 Use of these agents is determined from the history. However, if the practitioner believes that a report of such drug use will not be forthcoming from a high-risk individual, a toxicology screen is advised. When therapeutic drug use is reported, early determination of a serum concentration of the agent may be important when toxicity related to that agent has a relationship to serum concentrations (i.e., digitalis).
Identification of Structural Heart Disease
Exercise testing of the patient who has been resuscitated from an episode of VF provides information about the inducibility of both exercise-related myocardial ischemia and exercise-related arrhythmias.119 The sensitivity, specificity, and spatial localization of reversible myocardial ischemia can be enhanced by coupling the exercise test with radionuclide or echocardiographic imaging techniques.
Despite the availability of these noninvasive diagnostic procedures, the critical importance of accurate and complete anatomic and functional diagnosis of structural heart disease in a patient who has been resuscitated from VF usually necessitates cardiac catheterization and coronary angiography. Occasionally, the combination of information available from the history of events surrounding the spontaneous episode of VF (especially when preceded by physical or emotional stress accompanied by angina), evidence of reversible myocardial ischemia on exercise testing (especially when associated with evidence of ventricular electrical instability), and documentation of CAD (especially when not associated with severe left ventricular dysfunction) will suggest the possibility that the episode of VF was caused by reversible myocardial ischemia that can be corrected. Although coronary revascularization in this setting has been reported to prevent further episodes of VF in some patients, the effect is not necessarily predictable.120 Accordingly, it has become customary to either assume that the revascularization procedure was insufficient for the prevention of VF recurrences or document that VT or VF is not inducible by programmed stimulation performed during a transvenous catheter EPS study after the revascularization procedure—preferably after a preoperative catheter EPS documented the inducibility of sustained VT or VF.
Documentation of the Mechanism of Ventricular Fibrillation
The debate on the advisability of offering a baseline transvenous catheter EPS to all patients who have had an episode of VF in the absence of a reversible or transient cause is still ongoing.121 The major potential advantage of an EPS for patients with VF is the possibility of demonstrating that the VF was caused by another arrhythmia that would be treated in another way. Of course, VF may result from the degeneration of other tachyarrhythmias best treated by trans-catheter ablation procedures such as supraventricular tachyarrhythmias (including AF in the setting of ventricular pre-excitation) and certain VTs (including bundle branch re-entrant VT). EPS may also help distinguish patients with a permanent VT substrate (typically those with a myocardial scar who have inducible VT) from those without a permanent VT substrate (typically those without a myocardial scar who do not have inducible monomorphic VT). This distinction may assist the practitioner in the selection of treatment modalities. For example, revascularization of CAD is rarely, if ever, useful in the former circumstance. Finally, EPS may also provide information regarding optimal programming of a subsequently implanted ICD. Nevertheless, each of these potential advantages is more likely to be had by the patient who presents with VT.
Assessment and Monitoring of Selected Antiarrhythmic Therapy
The assessment and monitoring of some forms of antiarrhythmic therapy require other selected investigations. Although now infrequently used, the selection of antiarrhythmic drug therapy by suppressing ventricular premature beats requires a baseline EPS as well as antiarrhythmic drug–free, and drug assessment 24-hour ambulatory ECG examinations along with exercise tolerance tests.119,122 Similarly, the selection of antiarrhythmic drug therapy using the approach of suppression of ventricular tachyarrhythmias induced by programmed stimulation requires a baseline EPS as well as antiarrhythmic drug–free and drug assessment EPSs.123,124 Of course, the follow-up of patients with a treated propensity to VF usually requires long-term surveillance—most commonly with repeated 24-hour ambulatory ECG examinations. However, no direct evidence suggests that such surveillance is of value to the patient with VF.
Electrophysiological Study
The role of EPS in the patient with VF depends on the etiology of the arrhythmia. Secondary VF is associated with acute reversible derangement such as ischemia, electrolyte imbalance, or cardiac trauma. In contrast, primary VF is not associated with any acute precipitant. In secondary VF, the best approach is to treat the specific precipitant responsible for VF. The role of EPS is often limited in secondary VF. Therefore, the focus of this section is on the approach to primary VF in the electrophysiology laboratory. In the setting of primary VF, it is imperative to first define the anatomic substrate. Because healed MI is the most common cause of primary VF, echocardiography, stress testing, and cardiac catheterization are all important diagnostic tools. If myocardial ischemia is demonstrated, revascularization (percutaneous or surgical) can improve long-term survival.125,126
Induction of Ventricular Tachycardia Versus Ventricular Fibrillation
After defining the underlying heart disease, EPS is important for risk stratification for primary VF. It is believed that VF is often preceded by VT, which degenerates into VF, so the induction of VT in the electrophysiological laboratory is indicative of the clinical rhythm (Figure 46-15). Therefore, the principal goal at EPS is the induction of sustained monomorphic VT. In the setting of a healed MI, VT can be induced at EPS in 20% to 45% of patients. The anatomic and electrophysiological substrate of VT is well described. Areas of healed MI, regions of slow conduction, and inhomogeneities of refractoriness all contribute to the specific responses to programmed stimulation and the induction of VT. As the induction of VT at EPS is already well described (see Chapters 25, 26, and 47), the focus of this section is induction of VF.
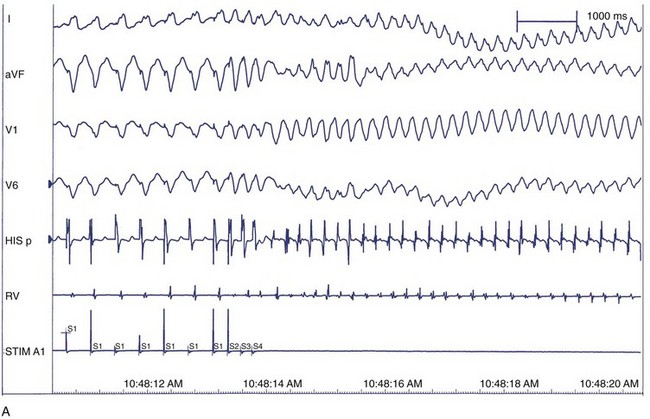
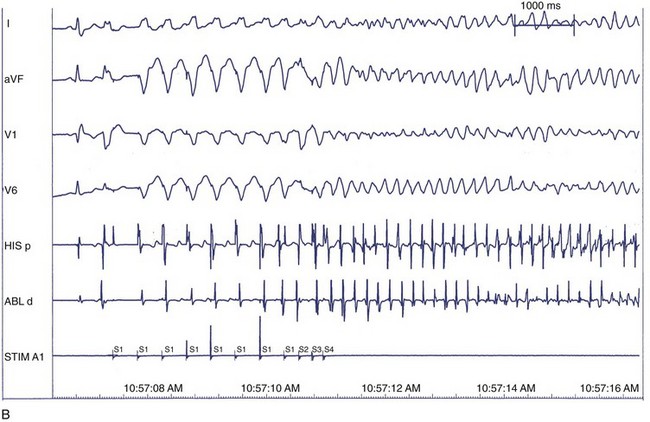
The correlation between the induction of VF in the electrophysiology laboratory and the incidence of VF as the presenting arrhythmia is limited, perhaps because of transient factors or prior VT in either situation.127 Among patients with previous cardiac arrest, 20% to 50% do not have VF induced at EPS. VF is inducible in up to 25% of cardiac arrest survivors compared with 3% of patients who presented with monomorphic VT. This suggests that induction of VF in patients who present clinically with VF may be specifically predictive of spontaneous VF episodes, whereas induction of VF in patients who presented with VT may represent a nonspecific response to programmed electrical stimulation.128
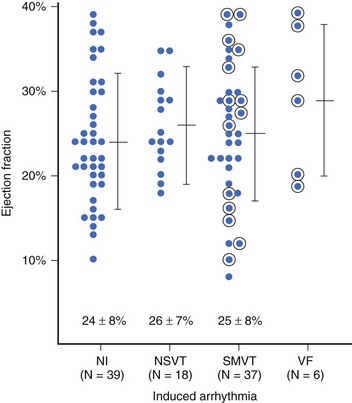
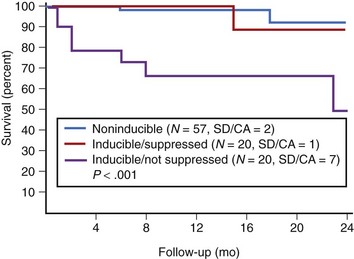
In patients with asymptomatic nonsustained VT, CAD, and an ejection fraction less than 40%, VF or polymorphic VT is induced in up to 6% of cases.129 Clinical variables often fail to distinguish patients with inducible arrhythmias from those without inducible arrhythmias. Ejection fraction is not significantly different in patients with no inducible arrhythmia, inducible nonsustained VT, inducible sustained monomorphic VT, or inducible VF (Figure 46-16). Of the 6% of patients in whom VF or polymorphic VT was induced, 17% died suddenly during follow-up.126 The presence of inducible sustained ventricular arrhythmias and the persistence of inducible sustained ventricular arrhythmias on therapy were significant univariate predictors of SCD. However, only the persistence of inducible sustained arrhythmias on therapy was an independent predictor of SCD (Figure 46-17).
Electrophysiological Characteristics Associated with Induction of Ventricular Fibrillation
It is not well understood why VF is induced in some patients in the clinical electrophysiology laboratory and not in others. Because VF can be induced in perfectly normal hearts by using either multiple closely coupled premature stimuli or by shocks applied on the T wave, induction of VF by catheter stimulation is not necessarily associated with a poor prognosis. Several potential reasons exist for the nonspecific response of induction of VF at EPS, including local graded response in normal muscle or decremental conduction block caused by short coupling intervals. This appears to have less to do with dispersion of refractoriness or propagation and more to do with local graded responses in normal muscle. For this reason, premature coupling intervals are usually not shortened below 180 ms to avoid inducing VF that has no diagnostic value.129 Other causes of nonspecific responses to programmed stimulation and induction of VF include increased conduction latency and prolongation of local activation time in proximity of the stimulus electrode. Induction of VF is preceded by increased latency (Figure 46-18).130
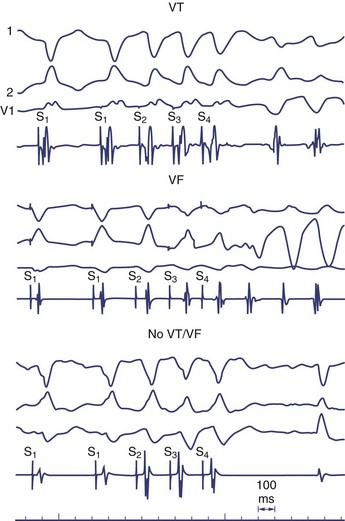
Another hypothesis suggesting why VF is induced with closely coupled premature beats during programmed stimulation is delayed conduction. Conduction slowing by itself causes dispersion of activation time between sites near the stimulating electrode and distant sites. Also, conduction slowing of a premature beat allows distant sites to have a longer coupling interval of the premature beat compared with that from the pacing site. These factors result in dispersion of refractoriness, with the refractory periods following the premature beat being longer at distant sites than at the pacing site. This, in turn, allows additional premature beats to induce VF. Therefore, the measurement technique itself alters what we are attempting to measure (Table 46-1).131 In addition, indirect evidence suggests that the mechanism of VF in the setting of a healed MI may be related to areas of slow conduction and stable re-entrant circuits. Occasionally, polymorphic VT or VF can transform to monomorphic VT by type I antiarrhythmic medications.132 These tachycardias have been mapped in the operating room and have been cured by endocardial resection. These findings have not been observed in patients with a normal heart and inducible polymorphic VT or VF. Therefore, use of a type I antiarrhythmic agent may be helpful in differentiating between a nonspecific finding from a significant one in the electrophysiology laboratory.
Prognosis and Clinical Relevance
The prognostic value of electrophysiological evaluation has been extensively studied in survivors of cardiac arrest. The reproducibility of inducible VF is strongly predictive of recurrent cardiac arrest. However, the failure to induce arrhythmias with programmed stimulation may not necessarily be associated with a benign prognosis. In a specific subset of patients with CAD, LVEF less than 40%, asymptomatic nonsustained VT, and no inducible arrhythmias, the relative risk of death was 1.8.125
In addition, several secondary prevention trials have supported the efficacy of defibrillator implantation in out-of-hospital VF survivors irrespective of the outcome of EPS. Results of the Cardiac Arrest Study Hamburg (CASH), AVID, and the Canadian Implant Defibrillator Study (CIDS) support defibrillator placement in cardiac arrest survivors who have no reversible cause of VF.133,134
Evidence-Based Therapy
Rescue of the Patient with Ventricular Fibrillation
Optimal resuscitation from VF requires restoring a perfusing cardiac rhythm as early as possible as well as providing the maximum possible cerebral and coronary blood flow during the resuscitation process. Mechanical aids, which improve cardiac output during CPR, have consistently shown improved outcomes in experimental settings and have shown, in limited human clinical trials, improved early resuscitation and improved survival to hospital admission.135 However, definitive clinical trials with respect to improving survival to discharge from hospital are lacking although such interventions may be able to improve long-term outcomes in patients with VF, particularly if the interventions are begun relatively early (e.g., at 5 to 10 minutes) after the onset of cardiac arrest from VF.
Mechanical interventions of this type include the active compression-decompression CPR device, interposed abdominal and chest compression devices, the impedance threshold valve, mechanical or pneumatically driven chest compression devices, or devices inserted into the chest cavity to assist in direct cardiac compression.136 All these devices increase cardiac output by increasing intrathoracic pressure during the compression phase of CPR or by increasing venous return during the decompression phase, either by positive abdominal pressure or by negative intrathoracic pressure during the decompression phase of CPR. The recent Prehospital Resuscitation Using an Impedance Valve and Early vs. Delayed Analysis Impedance Threshold Device (ROC PRIMED ITD) study was stopped at the recommendation of the Data and Safety Monitoring Board; the ITD did not improve survival to hospital discharge (see www.nih.gov/news/health/nov2009/nhlbi-06.htm).
Drug Therapy in Acute Management of Ventricular Fibrillation
The benefit of pharmacologic therapy as an adjunct to defibrillation in out-of-hospital VF is incompletely established. Despite many decades of use and ample laboratory experimental evidence of benefit, controlled clinical trials have not provided any clear evidence that either low-dose or high-dose epinephrine is beneficial in the treatment of patients with out-of-hospital VF.137 Vasopressin, which appears to be superior to epinephrine in experimental models of cardiac arrest in improving survival from experimental VF, appears to be superior to epinephrine in some studies but not in others.138–140 However, vasopressin has also not been proven superior to placebo in randomized controlled trials.
Similarly, the evidence base regarding antiarrhythmic therapy is incomplete. The standard therapy used to assist electrical defibrillation has been lidocaine; no evidence from controlled clinical trials is available to suggest that lidocaine is superior to placebo or any other agent in improving survival to hospital admission or survival to hospital discharge.141 In a meta-analysis of prophylactic lidocaine used in the post-infarction period, lidocaine may reduce the incidence of primary VF but appears to increase mortality rate.142 Lidocaine likely increases defibrillation thresholds and may increase the incidence of asystole. Magnesium is not superior to placebo in in-hospital cardiac arrest caused by VF.143–145 Bretylium, although studied in VF, is no longer available.
Intravenous (IV) amiodarone for VF resistant to three defibrillation shocks was compared with placebo in a blinded, randomized clinical trial by Kudenchuk et al.146 The 246 patients with shock-resistant, out-of-hospital VF were randomized to 300 mg amiodarone by IV bolus versus placebo; the primary study endpoint was survival to hospital admission, achieved in 44% of amiodarone-treated patients versus 34% of placebo-treated patients. In a related study, IV amiodarone was compared with IV lidocaine in a blinded, randomized trial of patients with VF persisting after three shocks, IV epinephrine, and a further defibrillation shock. Survival to hospital admission was achieved in 23% of amiodarone-treated patients versus 12% of lidocaine-treated patients (P = .009).147 Although neither of these studies demonstrated statistically significant improvements in survival to hospital discharge, if any antiarrhythmic drug is to be used in out-of-hospital VF, on the basis of these studies it seems reasonable to consider amiodarone as the drug of choice.
It is important to note that resuscitation from VF is a dynamic clinical situation, and many patients will have multiple recurrences of VF in the seconds to minutes after initial defibrillation and also undergo multiple transitions among VF, asystole, pulseless electrical activity, and a perfusing organized rhythm during the course of a protracted cardiac arrest. The use of adjunctive drug therapy can therefore be considered a means to prevent recurrences of VF as much as to assist in defibrillation. In particular, patients with frequent recurrences of VF may benefit from intensive antiadrenergic therapy, IV amiodarone therapy, and occasionally from anti-ischemic therapy, revascularization therapy, or therapy with an intra-aortic balloon pump.148,149
“Non-antiarrhythmic” Drugs that Prevent Sudden Cardiac Death
In the setting of heart failure, a cascade of neurohumoral activation initially supports perfusion.150 Over time, however, the biologically active molecules released in the neurohumoral activation, including the sympathetic nervous system and the renin-angiotensin-aldosterone system, result in progressive left ventricular dysfunction. Pharmacologic antagonism of these two systems has proven to be an effective treatment paradigm.
β-Blockers
Strong evidence for a beneficial effect of β-blocker treatment in patients following an MI was reported in 1981 by the Norwegian Multicenter Study Group.68 In their study, 1884 patients were randomly assigned to treatment with timolol (10 mg twice a day) or to placebo. Mortality in rate the control group was 16% compared with 10% in the timolol group (P = .0001), and a reduction in SCD from 13.9% to 7.7% (P = .0001) was observed. In the following year, the Beta-Blocker Heart Attack Trial (BHAT) reported a comparison of propranolol and placebo in patients with prior MI.66 In that study, 3837 patients were randomized to treatment with propranolol or placebo 5 to 10 days after an MI. The trial was stopped 9 months early after an average follow-up of 25 months because of a highly significant difference in mortality in favor of propranolol (7.2% in the propranolol group vs. 9.8% in the placebo group). These trials, along with a series of subsequent studies, were combined in a meta-analysis confirming that β-blockers reduce both total mortality and SCD.153
More recently, two studies investigating treatment of patients with heart failure have suggested the benefit of β-blockers with respect to reduction of total mortality and prevention of SCD.154,155 The Cardiac Insufficiency Bisoprolol Study II (CIBIS II) trial randomized 2647 patients with an LVEF of 35% or less and New York Heart Association (NYHA) functional class III or IV heart failure to the β1-selective agent bisoprolol or placebo.156 All-cause mortality was reduced from 17.3% in the placebo group to 11.8% in the bisoprolol group (P < .0001). Significantly fewer SCDs occurred in bisoprolol-treated patients compared with placebo-treated patients (3.6% vs. 6.3%; P = .0011). The Metoprolol Randomized Intervention Trial in Congestive Heart Failure (MERIT-HF) randomized 3991 patients with LVEF of 40% or less in NYHA functional class II to IV heart failure to treatment with the β1-selective agent metoprolol, in its controlled release or extended release form, or placebo.155 All-cause mortality was 7.2% per year in the metoprolol group and 11% in the placebo group (P = .00009). A reduction in SCD was seen in metoprolol-treated patients (odds ratio [OR], 0.59; confidence interval [CI], 0.45 to 0.78).
Angiotensin-Converting Enzyme Inhibitors
The survival advantage conferred by angiotensin-converting enzyme (ACE) inhibitors in patients with heart failure has been demonstrated in a number of studies.156–159 However, only in the Tandolapril Cardiac Evaluation Trial (TRACE) did the reduction in SCD reach statistical significance.160 A recently published meta-analysis has now clearly demonstrated that ACE inhibitors reduce SCD.160 In this study, 15,104 patients from 15 trials were studied. The relative risk of SCD in ACE inhibitor–treated patients was 0.80 (95% CI, 0.70 to 0.92).
Aldosterone Antagonism
The effect of spironolactone on survival was studied in the Randomized Aldactone Evaluation Study (RALES).161 A total of 822 patients on an ACE inhibitor with an LVEF of 35% or less and severe heart failure symptoms were randomly assigned to treatment with spironolactone or placebo. After a mean follow-up of 24 months, the mortality rate was 46% in the placebo group and 35% in the spironolactone group (P < .001). The relative risk of SCD in spironolactone-treated patients was 0.71 (CI, 0.54 to 0.95).
Recent studies have confirmed these findings and extended the benefit to patients with mild heart failure, in whom eplerenone, a selective aldosterone inhibitor, reduces all-cause mortality. A total of 12.5% of patients receiving eplerenone and 15.5% of those receiving placebo died (hazard ratio [HR], 0.76; 95% CI, 0.62 to 0.93; P = .008).162 Taken together, the data confirm that antagonizing the various components of the neuroendocrine activation that accompanies heart failure results in a reduction in SCD.
Anti-thrombotic and Anticoagulant Therapy
Accumulated data have suggested that ischemia caused by thrombus formation in stenotic coronary arteries may result in SCD. On the basis of pathologic examinations, greater than 80% of SCD cases in patients with ischemic heart disease may be associated with thrombus formation or plaque fissuring.163,164 In the Second International Study of Infarct Survival (ISIS-2), aspirin use was associated with a reduction in total mortality and SCD.165 A multivariate analysis of the 6797 participants in the Studies of Left Ventricular Dysfunction (SOLVD) demonstrated that both anti-platelet and anticoagulant therapies were associated with a reduction in the risk of SCD.166 This provides a rationale for anti-thrombotic therapy, anti-platelet therapy, or a combination of both in patients with left ventricular dysfunction.
Antiarrhythmic Drugs
Class I Agents
The Cardiac Arrhythmia Suppression Trial (CAST) studied patients with a history of MI and frequent PVCs that were suppressible with encainide, flecainide, or moricizine (type IC agents that are sodium channel blockers).167 More than 90 days after MI, patients were also required to have an LVEF of 40% or less. Despite suppression of PVCs, the encainide and flecainide arms of the trial were stopped early because of increased mortality in patients treated with antiarrhythmic drugs. The moricizine arm was subsequently stopped because it became clear there would be no benefit and because of concern about an apparent early increase in mortality.168
New Class III Drugs
In the Survival with Oral D-Sotalol (SWORD) study, the impact on SCD of d-sotalol, a potassium channel blocker without β-blocker properties, was studied.3,127,169 Patients with a history of MI and LVEF less than 40% were randomly assigned to d-sotalol or placebo. The study was terminated because of an excess risk of mortality in d-sotalol–treated patients (relative risk [RR], 1.65; P = .006). Dofetilide was also evaluated in patients with CHF.170 In a study of 1518 patients with symptomatic CHF and severe left ventricular dysfunction randomized to dofetilide or placebo, no difference in mortality rate was seen, although dofetilide was successful in converting AF to sinus rhythm. Similar results were obtained in patients with prior MI but without CHF.171 These data suggest that although dofetilide has no role in the prevention of SCD, it may be a useful treatment option in patients with AF who are also at risk for SCD because it did not increase mortality rate in these patients. A meta-analysis performed by Teo et al of antiarrhythmic trials was reported in 1993. Data were drawn from 138 trials and 98,000 patients.172 The mortality rate of patients randomized to receive class I agents was significantly higher than that of patients receiving placebo (OR, 1.14; 95% CI, 1.01 to 1.28; P = .03).
Given the data presented, it is reasonable to ask whether the reason for the failure of these antiarrhythmic drugs to prevent SCD is the intrinsic ineffectiveness of drug action or the method of guiding selection of the drug. If the problem were the method of guiding selection of potentially effective drugs, it would be expected that although the drugs might not prevent an arrhythmia, at least they would not be proarrhythmic. However, most antiarrhythmic drugs other than amiodarone were shown to provoke SCD in clinical trials. This suggests that the failure to prevent SCD is based on the molecule rather than the selection paradigm. The results of the Electrophysiologic Study Versus Electrocardiographic Monitoring (ESVEM) study provide support for this view.122 In this study, 486 patients with sustained monomorphic VT, inducible with programmed electrical stimulation (PES) and 10 PVCs or more per hour on Holter monitoring, were randomly assigned to guidance by demonstrating suppression of PES-stimulated VT or Holter recording suppression of ventricular ectopy. Although the PES protocol used in ESVEM was limited, the results in ESVEM were no different in either arm, indicating that PES guidance was not better than Holter guidance. In contrast, the Calgary study, using a standard PES protocol, showed superior results with PES-guided therapy for the prevention of recurrent VT or VF in less drug-refractory patients.123,124
Amiodarone
A number of early studies suggested that amiodarone, unlike other antiarrhythmics, may have a role in suppressing VTs in the post-MI population. The Basel Antiarrhythmic Study of Infarct Survival (BASIS) randomized 312 patients with previous MI and complex ventricular ectopy to amiodarone, individualized drug therapy starting with procainamide, or no antiarrhythmic drug therapy.173 After 1 year of follow-up, amiodarone-treated patients had a significantly greater survival compared with patients treated with no antiarrhythmic therapy (95% vs. 87%; P = .048). Amiodarone-treated patients also had better survival compared with patients who received individualized therapy, but this did not reach statistical significance. Ceremuzynski et al randomized 613 patients with previous MI who were ineligible to receive β-blockers to amiodarone or placebo.174 A statistically significant (P = .048) reduction in cardiac mortality was seen in amiodarone-treated patients, although the reduction in total mortality did not achieve statistical significance. The meta-analysis by Teo et al, discussed earlier, showed a significant mortality reduction in amiodarone-treated patients with previous MI.172 Taken together, these studies suggested that amiodarone may reduce arrhythmic death.
This picture was further clarified by the European Myocardial Infarction Amiodarone Trial (EMIAT) and the Canadian Amiodarone Myocardial Infarction Trial (CAMIAT).175,176 EMIAT randomized 1486 patients who had an LVEF of 40% or less, 5 to 21 days after an MI, to treatment with amiodarone or placebo. No difference was observed in the primary endpoint of total mortality or in cardiac mortality. Arrhythmic death was reduced from 7% in the placebo group to 4% in the amiodarone group (P = .05). All the mortality benefit occurred in amiodarone-treated patients, who were also treated with a β-blocker. In CAMIAT, 1202 patients with a history of MI and frequent ventricular ectopy (>10 PVCs per hour or at least one episode of VT) were randomized to receive amiodarone or placebo. A reduction was seen in the endpoint of resuscitated VF or arrhythmic death from 6% in the placebo group to 3.3% in the amiodarone group (P = .03). No difference in total mortality was observed. As in EMIAT, only amiodarone-treated patients who were also on a β-blocker appeared to derive any benefit. Two recent meta-analyses that provide a quantitative overview of the available randomized trials have been published. The Amiodarone Trials Meta-Analysis Investigators examined 13 randomized trials involving 6553 patients.177 Five trials of patients with CHF and eight trials involving patients with previous MI were included. The mean LVEF in this population was 0.31. They found a 29% reduction in SCD (OR, 0.71; 95% CI, 0.59 to 0.85). No difference was seen between the post-MI and CHF populations. Sim et al studied 15 randomized trials of amiodarone to prevent SCD using the random effects model.178 Amiodarone was found to significantly reduce the risk of total mortality (OR, 0.77; 95% CI, 0.66 to 0.89; P < .001) and SCD (OR, 0.70; 95% CI, 0.58 to 0.85; P < .001).
In the Optimal Pharmacological Therapy in Cardioverter Defibrillator Patients (OPTIC) trial, amiodarone was more effective that β-blockers alone or sotalol in preventing ICD discharges for ventricular arrhyhthmias.179 However, amiodarone is rarely recommended as the sole therapy for the secondary prevention of SCD in patients with a history of VF not caused by a reversible cause, the ICD being the therapy of choice. The fact that the reduction in arrhythmic death is not easily translated into an improvement in total mortality is likely attributable to the limited protective effect of amiodarone. SCD-HeFT did not show any mortality benefit from amiodarone versus placebo; the HR for mortality in 845 amiodarone-treated patients followed up to 5 years versus the 847 patients treated with placebo was 1.06 (97.5% CI, 0.86 to 1.30).
Nonpharmacologic Therapy
Revascularization
As previously discussed, ischemia appears to be a precipitating event in SCD and is frequently caused by plaque disruption and coronary thrombus formation. Data supporting the usefulness of anti-thrombotic agents suggest a potential antiarrhythmic role for revascularization. Interestingly, trial-derived data bearing directly on the issue of revascularization are rare. In the Coronary Artery Surgery Study (CASS) registry, revascularization was independently associated with improved survival free of SCD (SCD occurred in 4.9% of patients assigned to medical therapy and 1.6% of patients assigned to surgical therapy).180 Garan et al showed that myocardial revascularization can result in elimination of PES-inducible ventricular tachyarrhythmias.181 Hii et al found that a patent infarct-related artery was associated with the effective drug suppression of inducible VT at PES.182 In sum, data supporting a role for ischemia in the production of ventricular arrhythmias and studies suggesting a positive treatment effect of eliminating the ischemia lead to the conclusion that elimination of inducible ischemia should be a component of treatment to prevent SCD.
Implantable Cardioverter-Defibrillator
Since the first implantation of an ICD by Mirowski in 1980, these systems have continued to evolve.183 Today, they are generally placed transvenously, with an implantation-related mortality rate of less than 1%.184 Current systems are capable of defibrillation of VF, tiered therapy of VT (competitive pacing, synchronized cardioversion, defibrillation), and antibradycardia pacing. Increasingly sophisticated electrogram storage and telemetry are useful for rhythm diagnosis.
A number of randomized, controlled clinical trials in patients with malignant ventricular arrhythmias, including SCD, have been performed and have provided an increasingly clear picture of the appropriate role of and considerations for the ICD (Box 46-1). The AVID trial was a secondary prevention study that randomized patients with a history of symptomatic VT or VF to ICD implantation or to antiarrhythmic therapy.131 Randomized patients must have been resuscitated from VF or have had an episode of VT that was hemodynamically compromising or that was associated with an LVEF less than 40%. A total of 1016 patients were randomized to immediate ICD implantation or antiarrhythmic drug therapy. Although sotalol or amiodarone was permitted in the antiarrhythmic drug arm, only 2.6% of patients randomized to antiarrhythmic drug were discharged on sotalol. Thus, for all practical purposes, this was an amiodarone versus ICD trial. The trial was stopped early because of significantly better survival in ICD-treated patients (75.4% vs. 64.1% at 3 years; P < .02). Similar patient populations were studied in CIDS and CASH, with results similar to AVID.134,135
Box 46-1 Indications for Implantable Cardioverter-Defibrillator Therapy for Patients at Risk of Ventricular Fibrillation
Class I
Class IIa
Class IIb
Class III
Adapted from Epstein, AE, DiMarco, JP, Ellenbogen, KA, et al: ACC/AHA/HRS 2008 Guidelines for Device-Based Therapy of Cardiac Rhythm Abnormalities: American College of Cardiology/American Heart Association Task Force on Practice Guidelines Developed in Collaboration With the American Association for Thoracic Surgery and Society of Thoracic Surgeons, J Am Coll Cardiol 51:1–62, 2008.
MADIT was a primary prevention trial that studied 196 patients with previous MI, nonsustained VT, and an LVEF of 35% or less who were inducible to a sustained monomorphic VT at PES not suppressible by procainamide.185 This population was different from the AVID patients in that they did not have a spontaneous sustained ventricular arrhythmia but had shown that VT could be induced by PES. These patients were randomly assigned to immediate ICD implantation or antiarrhythmic drug therapy. At 1-month follow-up, 74% of the patients in the antiarrhythmic drug therapy group were on amiodarone. Survival was significantly better in the ICD group than in patients treated with an antiarrhythmic drug (mostly amiodarone) (HR for overall mortality, 0.46; 95% CI, 0.26 to 0.82; P = .009).
All the ICD trials (AVID, MADIT, CIDS, CASH) presented thus far entered patients known to be at high risk for a fatal arrhythmia because they had a history of prior VT, either as a presenting problem or at PES. The Coronary Artery Bypass Graft PATCH (CABG-PATCH) trial examined patients at risk of SCD because of the presence of coronary disease for which they were to undergo coronary artery bypass grafting (CABG), who had an LVEF of 35% or less, and who had an abnormal signal-averaged ECG.186 In contrast to the patients presented in the other studies, these patients had no history of a sustained VT that occurred spontaneously or at PES. This study randomly assigned 900 patients to ICD implantation or no implantation at the time of CABG. During an average follow-up of 32 ± 16 months, no significant difference was seen in mortality between patients assigned to ICD and those assigned to no ICD. The HR for total mortality with ICD placement was 1.07 (95% CI, 0.81 to 1.42). In CABG-PATCH, 71% of the deaths were nonarrhythmic, which accounts for the absence of benefit from the ICD.187 It is possible that the revascularization reduced the frequency of VTs by removing the contributing role of inducible ischemia. The results of this trial do not provide support for the use of the signal-averaged ECG in risk stratification.
The Multicenter Unsustained Tachycardia Trial (MUSTT) studied patients with CAD, an LVEF of 40% or less, and nonsustained VT on Holter recording and who had inducible, sustained monomorphic VT or VF.188 Patients were randomized to standard therapy for coronary disease or standard therapy with a PES-guided attempt to suppress inducible VT or VF. Patients in whom inducible VT or VF was suppressed or whose VT was hemodynamically tolerated were treated with the drug that suppressed the VT (or rendered it tolerable). Patients who continued to be inducible to a sustained VT at PES that was hemodynamically intolerable had an ICD placed. The primary endpoint of the study was cardiac arrest or death. A total of 704 patients were randomized. The patients who received the ICD because of nonsuppressibility would be expected to be the highest risk group, but they had a better survival compared with patients not receiving antiarrhythmic therapy and those who were suppressible with PES-guided antiarrhythmic therapy (RR, 0.24; 95% CI, 0.13 to 0.45; P < .001). No difference was seen in mortality rate or cardiac arrest between patients who received no antiarrhythmic therapy and those with antiarrhythmic drug suppression of inducibility. Interestingly, unlike the other ICD trials, it is possible to compare ICDs with standard treatment without an antiarrhythmic drug in patients who have had a demonstrated VT (in this case, at PES). These data suggest that the ICD is the most effective approach to preventing SCD in these patients. Also, they proved an important additional demonstration of the ineffectiveness of PES-guided antiarrhythmic drug selection. The conclusions drawn from MUSTT suggest that the potentially large group of coronary patients with depressed ejection fraction and nonsustained VT who are inducible into a sustained monomorphic VT or VF by PES should have an ICD inserted. MADIT-II randomized patients with previous MI and an LVEF of 30% or less to ICD or standard therapy.189 The primary endpoint was total mortality. Most patients were in NYHA class I or II. The ICD arm had a 31% relapse risk reduction in total mortality, suggesting that an important role for SCD prevention exists in this population.189 However, most of the benefit was seen in patients with a prolonged QRS complex on the resting ECG. In summary, the accumulated data suggest that the ICD reduces mortality in patients at high risk for a fatal VT and that it is more effective than amiodarone in doing so.
A consideration of these facts leads to the current management approach to patients at risk for SCD:
Primary Prevention of Sudden Cardiac Death
At this point, no doubt exists about the capacity of the ICD to prevent arrhythmic death. This means that any patient destined to have a potentially fatal ventricular arrhythmia (in spite of otherwise appropriate medical therapy) could potentially benefit from having an ICD in place. A central issue for future investigation is the identification of patients at sufficiently high risk of SCD to justify the morbidity and cost of placing a device. Some subpopulations of patients described earlier may be effectively treated with amiodarone. For instance, a post hoc analysis of the AVID database suggests that ICDs have no benefit over amiodarone for patients with LVEF greater than 35%.190 A randomized trial in such patients would be needed to make definitive treatment recommendations.
Certain populations of patients are known to be at risk for SCD, but because of competing risks of death, it is not clear whether the ICD will confer a benefit to the population as a whole. The Defibrillator in Acute Myocardial Infarction Trial (DINAMIT) study used a risk factor representing autonomic dysfunction as a risk stratifier to allow defibrillation therapy to more effectively focus on the highest-risk patients. A reduction in heart rate variability (HRV) has been shown to portend a worse prognosis in patients with CAD.191–193 Patients in DINAMIT had low HRV and an ejection fraction of 35% or less. They were within 40 days of an acute MI. No mortality benefit was seen in patients randomized to the ICD (n = 332) compared with control patients (n = 342), with an HR for all-cause death of 1.08 (95% CI, 0.76 to 1.55) in the ICD versus control groups, over 30 ± 13 months of follow-up.194
In the Defibrillators In Non-Ischemic Cardiomyopathy Treatment Evaluation (DEFINITE) study, 458 patients with nonischemic cardiomyopathy, an LVEF of 35% or less, and more than 10 PVCs per hour or any episode of unsustained VT were randomized to standard medical therapy versus standard therapy plus ICD. The HR for all-cause mortality after 29 ± 14.4 months of follow-up in the ICD versus control groups was 0.65 (95% CI, 0.4 to 1.06; P = .08).195
The entire discussion to this point has focused on populations known to be at high risk for SCD. Myerberg and others have emphasized the fact that the majority of cardiac arrests occur in the low-risk, but very large, population of patients whose increased risk has not come to clinical recognition.196 To make major inroads into the prevention of SCD, learning how to screen the asymptomatic population inexpensively, but safely and effectively, will be necessary. Identifying high-risk patients in the low-risk, asymptomatic population and identifying members of known high-risk populations who are not at high enough risk to justify ICD placement is the next frontier in SCD prevention.
Key References
The Antiarrhythmics versus Implantable Defibrillators (AVID) Investigators. A comparison of antiarrhythmic drug therapy with implantable defibrillators in patients resuscitated from near fatal ventricular arrhythmias. N Engl J Med. 1997;337:1576-1583.
BHAT investigators. A randomized trial of propranolol in patients with acute myocardial infarction: I. Mortality results. JAMA. 1982;247:1707-1714.
Brugada P, Brugada J, Brugada R. The Brugada syndrome. Cardiovasc Drugs Ther. 2001;15:15-17.
Buxton AE, Lee KL, Fisher JD, et al. A randomized study of the prevention of sudden death in patients with coronary artery disease. N Engl J Med. 1999;341:1882-1890.
The Cardiac Arrhythmia Suppression Trial II Investigators. Effect of antiarrhythmic agent moricizine on survival after myocardial infarction. N Engl J Med. 1992;327:227-233.
CIBIS-II Investigators and Committees. The Cardiac Insufficiency Bisoprolol Study II (CIBIS II): A randomized trial. Lancet. 1999;353:9-13.
Cobb LA, Baum RS, Alvarez HIII, et al. Resuscitation from out-of-hospital ventricular fibrillation: 4 years follow-up. Circulation. 1975;51(Suppl III):III223-III228.
Cobb LA, Fahrenbruch CE, Walsh TR, et al. Influence of cardiopulmonary resuscitation prior to defibrillation in patients with out-of-hospital ventricular fibrillation. JAMA. 1999;281:1182-1188.
Connolly SJ, Dorian P, Roberts RS, et al. Comparison of beta-blockers, amiodarone plus beta-blockers, or sotalol for prevention of shocks from implantable cardioverter defibrillators: The OPTIC Study. A randomized trial. JAMA. 2006;295:165-171.
Domanski M, Saksena S, Epstien A, et al. Relative effectiveness of the implantable cardioverter-defibrillator and antiarrhythmic drugs in patients with varying degrees of left ventricular dysfunction who have survived malignant ventricular arrhythmias, for the AVID Investigators. J Am Coll Cardiol. 1999;34:1090-1095.
Dorian P, Cass D, Schwartz B, et al. Amiodarone as compared with lidocaine for shock-resistant ventricular fibrillation. N Engl J Med. 2002;346:884-890.
Furukawa T, Rozanski J, Nogami A, et al. Time-dependent risk of and predictors for cardiac arrest recurrence in survivors of out-of-hospital cardiac arrest with chronic coronary artery disease. Circulation. 1989;80:599-608.
Haissaguerre M, Derval N, Sacher F, et al. Sudden cardiac arrest associated with early repolarization. N Engl J Med. 2008;358(19):2016-2023.
Hohnloser SH, Kuck KH, Dorian P, Roberts RS, Hampton JR, Hatala R, Fain E, Gent M, Connolly SJ. Prophylactic use of an implantable cardioverter-defibrillator after acute myocardial infarction. N Engl J Med. 2004;351:2481-2488.
Kannel W, Thomas H. Sudden coronary death: The Framingham study. Ann N Y Acad Sci. 1982;382:3-21.
Kober L, Torp-Pederson C, Carlsen J, et al. for the Trandolapril Cardiac Evaluation (TRACE) Study Group: A clinical trial of the ACE inhibitor Trandolapril in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 1995;333:1670-1676.
Kowey PR, Levine JH, Herre JM, et al. Randomized, double-blind comparison of intravenous amiodarone and bretylium in the treatment of patients with recurrent, hemodynamically destabilizing ventricular tachycardia or fibrillation. The Intravenous Amiodarone Multicenter Investigators Group. Circulation. 1995;92:3255-3263.
Luu M, Stevenson WG, Stevenson LW, et al. Diverse mechanisms of unexpected sudden cardiac arrest in advanced heart failure. Circulation. 1989;80:1675-1680.
Mason J for the Electrophysiologic Study Versus Electro cardiographic Monitoring Investigators. A comparison of electrophysiologic testing with Holter monitoring to predict antiarrhythmic drug efficacy for ventricular tachyarrhythmias. N Engl J Med. 1993;329:445-451.
Mitchell LB, Duff HJ, Manyari DE, Wyse DG. A randomized clinical trial of the noninvasive and invasive approaches to drug therapy ventricular tachycardia. N Engl J Med. 1987;317:1681-1687.
Moss A, Hall J, Cannom D, et al. for the Multicenter Automatic Defibrillator Implantation Trial Investigators: Improved survival with an implanted defibrillator in patients with coronary disease at high risk for ventricular arrhythmia. N Engl J Med. 1996;335:1933-1940.
Moss AJ, Schwartz PJ, Crampton RS, et al. The long QT syndrome: Prospective longitudinal study of 328 families. Circulation. 1991;84:1136-1144.
Moss AJ, Zareba W, Hall WJ, et al. Multicenter Automatic Defibrillator Implantation Trial II investigators: Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med. 2002;346:877-883.
Pfeffer M, Braunwald E, et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction: Results of the Survival and Ventricular Enlargement Trial. N Engl J Med. 1992;327:669-677.
Pitt B, Zannad F, Remme W, et al. for the Randomized Aldactone Evaluation Study Investigators: The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med. 1999;341:709-717.
Podrid PJ, Graboys TB. Exercise stress testing in the management of cardiac rhythm disorders. Med Clin North Am. 1984;68:1139-1152.
Saksena S. The PCD investigators Group: Clinical outcome of patients with malignant ventricular tachyarrhythmias and a multiprogrammable cardioverter-defibrillator implanted with or without thoracotomy: An international multicenter study. J Am Coll Cardiol. 1994;23:1521-1530.
Teo K, Yusef S, Furberg C. Effects of prophylactic antiarrhythmic drug therapy in acute myocardial infarction: An overview of results from randomized controlled trials. JAMA. 1993;270:1589-1595.
1981 Timolol-induced reduction in mortality and reinfarction in patients surviving acute myocardial infarction. N Engl J Med. 1981;304:801-807.
Volpi A, Cavalli A, Santoro L, et al. Incidence and prognosis of early primary ventricular fibrillation in acute myocardial infarction—results of the Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico (GISSI-2) database. Am J Cardiol. 1998;82:265-271.
Waldo A, Camm J, deRuyter H, et al. Survival with oral d-sotalol in patients with left ventricular dysfunction after myocardial infarction: Rationale, design and methods (the SWORD Trial. J Am Coll Cardiol. 1995;75:1023-1027.
1 Bharati S. Pathology of the conduction system. In: Silver MD, Gotlieb AI, Schoen FJ, editors. Cardiovascular pathology. New York: Churchill Livingston, 2001.
2 Bharati S, Lev M. The pathologic aspects of ventricular tachycardia. In: Iwa T, Fontaine G, editors. Cardiac arrhythmias: Recent investigation and management. New York: Elsevier, 1988.
3 Bharati S. Pathology of the conduction system. In: Silver MD, Gotlieb AI, Schoen FJ, editors. Cardiovascular pathology. New York: Churchill Livingston; 2001:607-628. 20
4 Bharati S, Lev M. The pathologic aspects of ventricular tachycardia. In: Fontaine G, editor. Cardiac arrhythmias: Recent investigation and management. New York: Elsevier, 1988.
5 Bharati S, Lev M. The cardiac conduction system in unexplained sudden death. Mt. Kisco, NY: Futura Publishing; 1990.
6 Bharati S, Olshansky B, Lev M. Pathological study of an explanted heart due to intractable ventricular fibrillation. J Cardiovasc Electrophysiol. 1992;3:437-441.
7 Gault JH, Cantwell J, Lev M, Braunwald E. Fatal familial cardiac arrhythmias: Histologic observation in the cardiac conduction system. Am J Cardiol. 1972;29:548-553.
8 Husson GS, Blackman MS, Rogers MC, et al. Familial congenital bundle branch system disease. Am J Cardiol. 1973;32:365-369.
9 Cobb LA, Fahrenbruch CE, Walsh TR, et al. Influence of cardiopulmonary resuscitation prior to defibrillation in patients with out-of-hospital ventricular fibrillation. JAMA. 1999;281:1182-1188.
10 Kette F, Sbrojavacca R, Rellini G, et al. Epidemiology and survival rate of out-of-hospital cardiac arrest in northeast Italy: The FACS study. Resuscitation. 1998;36:153-159.
11 Stratton S, Niemann J. Outcome from out-of-hospital cardiac arrest caused by nonventricular arrhythmias: Contribution of successful resuscitation to overall survivorship supports the current practice of initiating out-of-hospital ACLS. Ann Emerg Med. 1998;32:448-453.
12 De Maio V, Stiell I, Wells G. Spaite D for the OPALS study group: Cardiac arrest witnessed by emergency medical services personnel: Descriptive epidemiology, prodromal symptoms, and predictors of survival. Ann Emerg Med. 2000;35:138-146.
13 Holmberg M, Holmberg S, Herlitz J. Incidence, duration and survival of ventricular fibrillation in out-of-hospital cardiac arrest patients in Sweden. Resuscitation. 2000;44:7-17.
14 Myerburg RJ. Sudden cardiac death: Exploring the limits of our knowledge. J Cardiovasc Electrophysiol. 2001;12:369-381.
15 Nichol G, Thomas E, Callaway CW, et al. T1 regional variation in out-of-hospital cardiac arrest incidence and outcome. JAMA. 2008;300(12):1423-1431.
16 Kannel W, Thomas H. Sudden coronary death: The Framingham study. Ann N Y Acad Sci. 1982;382:3-21.
17 Secci A, Wong N, Tang W, et al. Electron beam computed tomographic coronary calcium as a predictor of coronary events: Comparison of two protocols. Circulation. 1997;96:122-129.
18 Taylor A, Burke A, O’Malley P, et al. A comparison of the Framingham risk index, coronary artery calcification and culprit plaque morphology in sudden cardiac death. Circulation. 2000;101:1243-1248.
19 Thompson C, Yarzebski J, Goldberg R, et al. Changes over time in the incidence and case-fatality rates of primary ventricular fibrillation complicating acute myocardial infarction: Perspectives from the Worcester heart attack study. Am Heart J. 2000;139:1014-1021.
20 Volpi A, Cavalli A, Santoro L, Negri E. Incidence and prognosis of early primary ventricular fibrillation in acute myocardial infarction. Results of the Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico (GISSI-2) Database. Am J Cardiol. 1998;82:265-271.
21 Furukawa T, Rozanski J, Nogami A, et al. Time-dependent risk of and predictors for cardiac arrest recurrence in survivors of out-of-hospital cardiac arrest with chronic coronary artery disease. Circulation. 1989;80:599-608.
22 The AVID investigators. Causes of death in the Antiarrhythmics versus Implantable Defibrillators trial. J Am Coll Cardiol. 1999;34:1552-1559.
23 Klein H, Auricchi A, Reek S, Geller C. New primary prevention trials of sudden cardiac death in patients with left ventricular dysfunction: SCD-HeFT and MADIT-II. Am J Cardiol. 1999;83:91d-97d.
24 Hermida J, Lemoine J, Aoun F, et al. Prevalence of the Brugada syndrome in an apparently healthy population. Am J Cardiol. 2000;86:91-94.
25 Bardy GH, Lee KL, Mark DB, et al. Home use of automated external defibrillators for sudden cardiac arrest. N Engl J Med. 2008;358(17):1793-1804.
26 Allessie MA. Circus movement in rabbit atrial muscle as a mechanism of tachycardia. III. The “leading circle” concept: A new model of circus movement in cardiac tissue without the involvement of an anatomic obstacle. Circ Res. 1977;41:9-18.
27 Janse MJ. Functional reentry: Leading circle or spiral wave? J Cardiovasc Electrophysiol. 1999;10:621-622.
28 Ravelli F, Disertori M, Cozzi F, et al. Ventricular beats induce variations in cycle length of rapid (type II) atrial flutter in humans: Evidence of leading circle reentry. Circulation. 1994;89:2107-2116.
29 Frazier DW, Wolf PD, Wharton JM, et al. Stimulus-induced critical point mechanism for electrical initiation of reentry in normal canine myocardium. J Clin Invest. 1988;83:1039-1052.
30 Krinsky V. Qualitative theory of reentry. In Zipes D, Jalife J, editors: Cardiac electrophysiology. From cell to bedside, ed 3, Philadelphia: WB Saunders, 2000.
31 Panfilov A, Persov A. Vortex rings in a three-dimensional medium described by reaction-diffusion equations. Dokl Biophys. 1984;274:58-60.
32 Moe GK. On the multiple wavelet hypothesis of atrial fibrillation. Arch Int Pharmacodyn. 1962;140:183-188.
33 Perstov AM, Davidenko JM, Salomonsz R, et al. Spiral waves of excitation underlie reentrant activity in isolated cardiac muscle. Circ Res. 1993;72:631-650.
34 Jalife J, Berenfeld O, Skanes A, Mandapati R. Mechanisms of atrial fibrillation: Mother rotors or multiple daughter wavelets, or both? J Cardiovasc Electrophysiol. 1993;9(Suppl):S2-S12. 1998
35 Chen P, Wolf PD, Dixon EG, et al. Mechanism of ventricular vulnerability to single premature stimuli in open-chest dogs. Circ Res. 1988;62:1191-1209.
36 Antzelevitch C, Sicouri S, Litovsky SH, et al. Heterogeneity within the ventricular wall: Electrophysiology and pharmacology of epicardial, endocardial and M cells. Circ Res. 1991;69:1427-1449.
37 Antzelevitch C, Nesterenko VV, Yan G-X. Role of M cells in acquired long QT syndrome, U wakes, and torsade de pointes. J Electrocardiol. 1991;28(Suppl):131-138. 1996
38 Akar FG, Laurita KR, Rosenbaum DS. Cellular basis for dispersion of repolarization underlying reentrant arrhythmias. J Electrocardiol. 2000;33(Suppl):23-31.
39 Rosenbaum DS, Jackson LE, Smith JM, et al. Electrical alternans to the genesis of cardiac fibrillation. Circulation. 1999;99:1385-1394.
40 Pastore JM, Girouard SD, Laurita KR, et al. Mechanism linking T-wave alternans to the genesis of cardiac fibrillation. Circulation. 1999;99:1385-1394.
41 Laurita KR, Girouard SD, Akar FG, et al. Modulated dispersion explains changes in arrhythmia vulnerability during premature stimulation of the heart. Circulation. 1998;98:2774-2780.
42 Watanabe MA, Fenton FH, Evans SJ, et al. Mechanisms for discordant alternans. J Cardiovasc Electrophysiol. 2001;12:196-206.
43 Garfinkel A, Kim YH, Voroshilovsky O, et al. Preventing ventricular fibrillation by flattening cardiac restitution. Proc Natl Acad Sci USA. 2000;97:6061-6066.
44 Koller M, Riccio M, Gilmour RJ. Effects of [K+]0 on electrical restitution and activation dynamics during ventricular fibrillation. Am J Physiol Heart Circ Physiol. 2000;279:H2665-H2672.
45 Watanabe M, Otani NF, Gilmour FRJr. Biphasic restitution of action potential duration and complex dynamics in ventricular myocardium. Circ Res. 1995;76:915-921.
46 Dekker L, Coronel R, VanBavel E, et al. Intracellular Ca2+ and the delay of ischemia-induced electrical uncoupling in preconditioned rabbit ventricular myocardium. Cardiovasc Res. 1999;44:101-112.
47 Han J, Moe GK. Nonuniform recovery of excitability of ventricular muscle. Circ Res. 1964;14:44-60.
48 Franz MR, Flaherty JT, Platia EV, et al. Localization of regional myocardial ischemia by recording of monophasic action potentials. Circulation. 1984;69:593-604.
49 Mohabir R, Franz MR, Clusin WT. In vivo electrophysiological detection of myocardial ischemia through monophasic action potential recording. Prog Cardiovasc Dis. 1991;34:15-28.
50 Dilly SG, Lab MJ. Changes in monophasic action potential duration during the first hour of regional myocardial ischemia in the anaesthetized pig. Cardiovasc Res. 1987;21:908-915.
51 Jongsma HJ. Gap junctions in cardiovascular disease. Circ Res. 2000;86:1193-1197.
52 Owens LM, Fralix TA, Murphy E, et al. Correlation of ischemia-induced extracellular and intracellular ion changes to cell-to-cell electrical uncoupling in isolated blood-perfused rabbit hearts. Experimental Working Group. Circulation. 1996;94:10-13.
53 Owens L, Murphy E, Fralix TA. Relationship of cellular electrical uncoupling to changes of CA, pH, ATP, and contracture in ischemic rabbit myocardium [abstract. Circulation. 1993;88:1373.
54 Coronel R, Fiolet JW, Wilms-Schopman FJ, et al. Distribution of extracellular potassium and its relation to electrophysiologic changes during acute myocardial ischemia in the isolated perfused porcine heart. Circulation. 1988;77:1125-1138.
55 Cascio WE, Yan GX, Kleber AG. Passive electrical properties, mechanical activity, and extracellular potassium in arterially perfused and ischemic rabbit ventricular muscle. Effects of calcium entry blockade or hypocalcemia. Circ Res. 1990;66:1461-1473.
56 Kagiyama Y, Hill JL, Gettes LS. Interaction of acidosis and increased extracellular potassium on action potential characteristics and conduction in guinea pig ventricular muscle. Circ Res. 1982;51:614-623.
57 Anversa P, Olivetti G, Meggs LG, et al. Cardiac anatomy and ventricular loading after myocardial infarction. Circulation. 1993;87(Suppl):VII22-VII27.
58 Qin D, Zhang ZH, Caref EB, et al. Cellular and ionic basis of arrhythmias in post-infarction remodeled ventricular myocardium. Circ Res. 1996;79:461-473.
59 Peters NS, Green CR, Poole-Wilson PA, Severs NJ. Reduced content of connexin43 gap junctions in ventricular myocardium from hypertrophied and ischemic human hearts. Circulation. 1993;88:864-875.
60 Starobin JM, Zilberter YI, Rusnak EM, Starmer CF. Wavelet formation in excitable cardiac tissue: The role of wavefront obstacle interactions in initiating high-frequency fibrillatory-like arrhythmias. Biophys J. 1996;70:581-594.
61 Stramba-Badiale M, Vanoli EDFG, et al. Sympathetic- parasympathetic interaction and accentuated antagonism in conscious dogs. Am J Physiol. 1991;260:H335-H340.
62 Vanoli E, De Ferrari G, Stramba-Badiale M, et al. Vagal stimulation and prevention of sudden death in conscious dogs with a healed myocardial infarction. Circ Res. 1991;68:1471-1481.
63 De Ferrari G, Vanoli E, Stramba-Badiale M, et al. Vagal reflexes and survival during acute myocardial ischemia in conscious dogs with a healed myocardial infarction. Am J Physiol. 1991;261:H63-H69.
64 Schwartz PJ, Stone HL. Left stellectomy in the prevention of ventricular fibrillation caused by acute myocardial ischemia in conscious dogs with anterior myocardial infarction. Circulation. 1980;62:1256-1265.
65 Schwartz PJ, Stone HL, Brown AM. Effects of unilateral stellate ganglion blockade on the arrhythmias associated with coronary occlusion. Am Heart J. 1976;92:589-599.
66 BHAT investigators. A randomized trial of propranolol in patients with acute myocardial infarction: I. Mortality results. JAMA. 1982;247:1707-1714.
67 Hjalmarsen A. Effects of beta blockade on sudden cardiac death during acute myocardial infarction and the postinfarction period. Am J Cardiol. 1997;80:35J-39J.
68 Timolol-induced reduction in mortality and reinfarction in patients surviving acute myocardial infarction. N Engl J Med. 1981;304:801-807.
69 Vanoli E, Hull SJ, Foreman R, et al. Alpha-1 adrenergic blockade and sudden cardiac death. J Cardiovasc Electrophysiol. 1994;5:76-89.
70 Kammerling JJ, Green FJ, Watanabe AM, et al. Denervation supersensitivity of refractoriness in noninfarcted areas apical to transmural myocardial infarction. Circulation. 1987;76:383-393.
71 Brugada P, Brugada J, Brugada R. The Brugada syndrome. Cardiovasc Drugs Ther. 2001;15:15-17.
72 Chen QY, Kirsch GE, Zhang DM, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293-296.
73 Priori SG, Napolitano C, Giordano U, et al. Brugada syndrome and sudden cardiac death in children. Lancet. 2000;355:808-809.
74 Antzelevitch C. The Brugada syndrome. J Cardiovasc Electrophysiol. 1998;9:513-516.
75 Lukas A, Antzelevitch C. Phase 2 reentry as a mechanism of initiation of circus movement reentry in canine epicardium exposed to simulated ischemia. Cardiovasc Res. 1996;32:593-603.
76 Haïssaguerre M, Shoda M, Jaïs P, et al. Mapping and ablation of idiopathic ventricular fibrillation. Circulation. 2002;106:962-967.
77 Pandis IP, Morganroth J. Sudden death in hospitalized patients: Cardiac rhythm disturbances detected by ambulatory electrocardiographic monitoring. J Am Coll Cardiol. 1983;2:798-805.
78 Pandis IP, Morganroth J. Holter monitoring and sudden cardiac death. Cardiovasc Rev Rep. 1984;5:283-304.
79 Pratt CM, Francis MJ, Luck JC. Observations on sudden cardiac death recorded during ambulatory electrocardiographic monitoring. Circulation. 1982;66(Suppl):2. 26
80 Savage DD, Dasteli WP, Anderson SJ. Sudden unexpected death during ambulatory electrocardiographic monitoring: The Framingham study. Am J Med. 1983;74:148-152.
81 Luu M, Stevenson WG, Stevenson LW, et al. Diverse mechanisms of unexpected sudden cardiac arrest in advanced heart failure. Circulation. 1989;80:1675-1680.
82 Haissaguerre M, Derval N, Sacher F, et al. Sudden cardiac arrest associated with early repolarization. N Engl J Med. 2008;358(19):2016-2023.
83 Antzelevitch C, Yan GX. J wave syndromes. Heart Rhythm. 2010;7(4):549-558.
84 Shah AJ, Sacher F, Chatel S, et al. Early repolarization disease. Card Electrophysiol Clin. 2010;2(4):559-569.
85 Merri M, Benhorin J, Alberti M, et al. Electrocardiographic quantitation of ventricular repolarization. Circulation. 1989;80:1301-1308.
86 Surawicz B, Knoebel SB. Long QT: Good, bad, or indifferent. J Am Coll Cardiol. 1984;4:398-413.
87 Priori S, Bloise R, Crotti L. The long QT syndrome. Europace. 2001;3:16-27.
88 Schwartz PJ. The long QT syndrome. In: Kulbertus HE, Wellens HJJ, editors. Sudden death. The Hague: M Nijhoff, 1980.
89 Garson AJr, Dick MII, Fournier A, et al. The long QT syndrome in children: An international study of 287 patients. Circulation. 1993;87:11866-11872.
90 Moss AJ, Schwartz PJ, Crampton RS, et al. The long QT syndrome: Prospective longitudinal study of 328 families. Circulation. 1991;84:1136-1144.
91 Schwartz PJ, Priori SG, Locati EH, et al. Long QT syndrome patients with mutations on the SCN5A and HERG genes have differential responses to Na channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation. 1995;92:3381-3386.
92 Malfatto G, Beria G, Sala S, et al. Quantitative analysis of T wave abnormalities and their prognostic implications in the idiopathic long QT syndrome. J Am Coll Cardiol. 1994;23:296-301.
93 De Ambroggi L, Negroni MS, Monza E, et al. Dispersion of ventricular repolarization in the long QT syndrome. Am J Cardiol. 1991;68:614-620.
94 Priori SG, Napolitano C, Diehl L, Schwartz PJ. Dispersion of the QT interval: A marker of therapeutic efficacy in the idiopathic long QT syndrome. Am Heart J. 1975;89:1681-1689.
95 Schwartz PJ, Periti M, Maliani A. The long QT syndrome. Am Heart J. 1975:378-390.
96 Kay GN, Plumb VJ, Arciniegas JG, et al. Torsade de pointes: The long-short initiating sequence and other clinical features: Observations in 32 patients. Am J Cardiol. 1983:806-817.
97 Jackman WM, Friday KJ, Anderson JL, et al. The long QT syndrome: A critical review, new clinical observations, and a unifying hypothesis. Prog Cardiovasc Dis. 1986;31:115-172.
98 Guzak I, Antzelevitch C, Bjerregaard P, et al. The Brugada syndrome: Clinical, electrophysiologic and genetic aspects. J Am Coll Cardiol. 1999;33:5-15.
99 Atararashi J, Ogawa W, Harumi K, et al. Characteristics of patients with right bundle branch block and ST-segment elevation in right precordial leads. Am J Cardiol. 1996;78:581-583.
100 Brugada P, Brugada R, Brugada J. Sudden death in high-risk family members. Brugada syndrome. Am J Cardiol. 2000;86(9A):40K-43K.
101 Matsuo K, Akahoshi M, Nakashima E, et al. The prevalence, incidence and prognostic value of the Brugada-type electrocardiogram. A population-based study of incidence and prognostic value of the Brugada-type electrocardiogram. A population-based study of four decades. J Am Coll Cardiol. 2001;38:765-770.
102 Monroe MH, Littman L. Two-year case collection of the Brugada syndrome electrocardiogram pattern at a large teaching hospital. Clin Cardiol. 2002;23:849-851.
103 Hermida JS, Lemoine JL, Aoun FB, et al. Prevalence of the Brugada syndrome in an apparently healthy population. Am J Cardiol. 2000;86:91-94.
104 Myasaka Y, Tsuji H, Yamada K, et al. Prevalence and mortality of the Brugada-type electrocardiogram in one city in Japan. Am J Cardiol. 2001;38:771-774.
105 Takenaka S, Hisamatsu K, Nagase S, et al. Relatively benign clinical course in asymptomatic patients with Brugada-type electrocardiogram without family history of sudden death. J Cardiovasc Electrophysiol. 2001;12:2-6.
106 Brugada J, Brugada R, Brugada P. Asymptomatic patients with Brugada electrocardiogram: Are they at risk? J Cardiovasc Electrophysiol. 2001;12:7-8.
107 Priori SG, Napolitano C, Gasparini M, et al. Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome. A prospective evaluation of 52 families. Circulation. 2000;102:2509-2515.
108 Atarachi H, Ogawa S, Harumi K, et al. Three year follow-up of patients with right bundle branch block and ST segment elevation in the right precordial leads. Japanese registry of Brugada syndrome. Idiopathic ventricular fibrillation investigators. J Am Coll Cardiol. 2001;37:1916-1920.
109 Macus FI, Fontaine GH, Guiradon G, et al. Right ventricular dysplasia: A report of 24 adult cases. Circulation. 1982;65:384-398.
110 Fontaine G, Umemura J, Di Donna P, et al. Duration of the QRS complexes in arrhythmogenic right ventricular dysplasia. A new non-invasive diagnostic marker. Ann Cardiol Angeiol (Paris). 1993;42:399-405.
111 Fontaine G, Fontaliran F, Hebert JL, et al. Arrhythmogenic right ventricular dysplasia. Annu Rev Med. 1999;50:17-35.
112 Nava A, Bauce B, Basso C, et al. Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2000;36:2226-2233.
113 Metzger JT, de Chillou C, Cheriex E, et al. Value of the 12-lead electrocardiogram in arrhythmogenic right ventricular dysplasia and absence of correlation with echocardiographic findings. Am J Cardiol. 1993;72:964-967.
114 Mitchell LB. Drug therapy of sustained ventricular tachyarrhythmias: Is there still a role? Cardiol Clin. 2000;18:357-373.
115 Salerno DM, Katz A, Dunbar DN, et al. Serum electrolytes and catecholamines after cardioversion from ventricular tachycardia and atrial fibrillation. PACE. 1993;16:1862-1871.
116 Cobb LA, Baum RS, Alvarez HIII, et al. Resuscitation from out-of-hospital ventricular fibrillation: 4 years follow-up. Circulation. 1975;51(Suppl III):III223-III228.
117 Volpi A, Cavalli A, Santoro L, et al. Incidence and prognosis of early primary ventricular fibrillation in acute myocardial infarction—results of the Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico (GISSI-2) database. Am J Cardiol. 1998;82:265-271.
118 Kerin NZ, Somberg J. Proarrhythmia: Definition, risk factors, causes, treatment, and controversies. Am Heart J. 1994;128:575-585.
119 Podrid PJ, Graboys TB. Exercise stress testing in the management of cardiac rhythm disorders. Med Clin North Am. 1984;68:1139-1152.
120 Kelly P, Ruskin JN, Vlahakes GJ, et al. Surgical coronary artery revascularization in survivors of pre-hospital cardiac arrest: Its effect on inducible ventricular arrhythmias and long-term survival. J Am Coll Cardiol. 1990;15:267-273.
121 Mitchell LB, Gettes LS. Is a baseline electrophysiologic study mandatory for the management of patients with spontaneous, sustained, ventricular tachyarrhythmias? Prog Cardiovasc Dis. 1996;38:385-392.
122 Mason J for the Electrophysiologic Study Versus Electro cardiographic Monitoring Investigators. A comparison of electrophysiologic testing with Holter monitoring to predict antiarrhythmic drug efficacy for ventricular tachyarrhythmias. N Engl J Med. 1993;329:445-451.
123 Mitchell LB, Duff HJ, Manyari DE, Wyse DG. A randomized clinical trial of the noninvasive and invasive approaches to drug therapy ventricular tachycardia. N Engl J Med. 1987;317:1681-1687.
124 Mitchell LB, Duff HJ, Miller CE, et al. Drug therapy of ventricular tachycardia: A cost comparison of randomized noninvasive/ invasive approaches. Can J Cardiol. 1992;8:487-494.
125 Buxton AE, Lee KL, Fisher JD, et al. A randomized study of the prevention of sudden death in patients with coronary artery disease. N Engl J Med. 1999;341:1882-1890.
126 Wilber DJ, Olshansky B, Moran JF, Scanlong PJ. Electro physiological testing and nonsustained ventricular tachycardia. Circulation. 1990;82:350-358.
127 Swerdlow CD, Bardy GH, McAnulty J, et al. Determinants of induced ventricular arrhythmias in survivor of out-of -hospital VF. Circulation. 1987;76:1053-1060.
128 Adhar GC, Larson LW, Bardy GH, et al. Sustained ventricular arrhythmias, differences between survivors of cardiac arrest and patients with recurrent sustained ventricular tachycardia. J Am Coll Cardiol. 1988;12:159-165.
129 Morady F, DiCarlo LAJr, Baerman JM, DeBuitleir M. Comparison of coupling intervals that induce clinical and nonclinical forms of ventricular tachycardia during programmed stimulation. Am J Cardiol. 1986;57:1269-1273.
130 Avitall B, McKinnie J, Jazayeri M, et al. Induction of VF versus monomorphic ventricular tachycardia during programmed stimulation. Circulation. 1992;85:1271-1278.
131 Avitall B, Levine HJ, Naimi S, et al. Local effects of electrical and mechanical stimulation in the recovery properties of the canine ventricle. Am J Cardiol. 1982;50:263-270.
132 Horowitz LN, Greenspan AM, Speilman SR, Josephson ME. Torsades de pointes: Electrophysiologic studies in patients without transient pharmacologic of metabolic abnormalities. Circulation. 1981;63:1120.
133 The Antiarrhythmics versus Implantable Defibrillators (AVID) Investigators. A comparison of antiarrhythmic drug therapy with implantable defibrillators in patients resuscitated from near fatal ventricular arrhythmias. N Engl J Med. 1997;337:1576-1583.
134 Cappato R, Siebels J, Ruppel R, et al. For the Cardiac Arrest Study Hamburg (CASH) Investigators. PACE. 2000;23:568.
135 Connolly SJ, Gent M, Roberts RS, et al. Canadian Implantation Defibrillator Study (CIDS) Investigators. Circulation. 2000;101:1297-1302.
136 Voelckel WG, Lurie KG, Sweeney M, et al. Effects of active compression-decompression cardiopulmonary resuscitation with the inspiratory threshold valve in a young porcine model of cardiac arrest. Pediatr Res. 2002;51:523-527.
137 Gueugniaud PY, Mols P, Goldstein P, et al. A comparison of repeated high doses and repeated standard doses of epinephrine for cardiac arrest outside the hospital. European Epinephrine Study Group. N Engl J Med. 1998;339:1595-1601.
138 Wenzel V, Lindner KH, Krismer AC, et al. Survival with full neurologic recovery and no cerebral pathology after prolonged cardiopulmonary resuscitation with vasopressin in pigs. J Am Coll Cardiol. 2000;35:527-533.
139 Lindner KH, Dirks B, Strohmenger HU, et al. Randomized comparison of epinephrine and vasopressin in patients with out- of-hospital ventricular fibrillation. Lancet. 1997;349:535-537.
140 Stiell IG, Hebert PC, Wells GA, et al. Vasopressin versus epinephrine for inhospital cardiac arrest: A randomized controlled trial. Lancet. 2001;358:105-109.
141 Weaver WD, Fahrenbruch CE, Johnson DD, et al. Effect of epinephrine and lidocaine therapy on outcome after cardiac arrest due to ventricular fibrillation. Circulation. 1990;82:2027-2034.
142 Hine LK, Laird N, Hewitt P, Chalmers TC. Meta-analytic evidence against prophylactic use of lidocaine in acute myocardial infarction. Arch Intern Med. 1989;149:2694-2698.
143 Echt DS, Gremillion ST, Lee JT, et al. Effects of procainamide and lidocaine on defibrillation energy requirements in patients receiving implantable cardioverter defibrillator devices. J Cardiovasc Electrophysiol. 1994;5:752-760.
144 Dorian P, Fain ES, Davy JM, Winkle RA. Lidocaine causes a reversible, concentration-dependent increase in defibrillation energy requirements. J Am Coll Cardiol. 1986;8:327-332.
145 Thel MC, Armstrong AL, McNulty SE, et al. Randomized trial of magnesium in in-hospital cardiac arrest. Duke Internal Medicine Housestaff. Lancet. 1997;350:1272-1276.
146 Kudenchuk PJ, Cobb LA, Copass MK, et al. Amiodarone for resuscitation after out-of-hospital cardiac arrest due to ventricular fibrillation. N Engl J Med. 1999;341:871-878.
147 Dorian P, Cass D, Schwartz B, et al. Amiodarone as compared with lidocaine for shock-resistant ventricular fibrillation. N Engl J Med. 2002;346:884-890.
148 Nademanee K, Taylor R, Bailey WE, et al. Treating electrical storm: Sympathetic blockade versus advanced cardiac life support-guided therapy. Circulation. 2000;102:742-747.
149 Kowey PR, Levine JH, Herre JM, et al. Randomized, double-blind comparison of intravenous amiodarone and bretylium in the treatment of patients with recurrent, hemodynamically destabilizing ventricular tachycardia or fibrillation. The Intravenous Amiodarone Multicenter Investigators Group. Circulation. 1995;92:3255-3263.
150 Mann D. Mechanisms and models in heart failure. Circulation. 1999;100:999-1008.
151 Reference deleted in proofs.
152 Reference deleted in proofs.
153 Teo K, Yusef S, Furberg C. Effects of prophylactic antiarrhythmic drug therapy in acute myocardial infarction: An overview of results from randomized, controlled trials. JAMA. 1993;270:1589-1595.
154 CIBIS-II Investigators and Committees. The Cardiac Insufficiency Bisoprolol Study II (CIBIS II): A randomized trial. Lancet. 1999;353:9-13.
155 The MERIT-HF Study Group. Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomized Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet. 1999;353:2001-2007.
156 Pfeffer M, Braunwald E, Moye LA, et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction: Results of the Survival and Ventricular Enlargement Trial. N Engl J Med. 1992;327:669-677.
157 The Consensus Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure: Results of the cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N Engl J Med. 1987;316:1429-1435.
158 The Studies of Left Ventricular Dysfunction Investigators. Effect of enalapril in survival in patients with reduced left ventricular ejection fraction and congestive heart failure. N Engl J Med. 1991;325:293-302.
159 Kober L, Torp-Pederson C, Carlsen J, et al. for the Trandolapril Cardiac Evaluation (TRACE) Study Group: A clinical trial of the ACE inhibitor Trandolapril in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 1995;333:1670-1676.
160 Domanski M, Exner D, Borkowf C, et al. Effect of angiotensin converting enzyme inhibition on sudden cardiac death in patients following a myocardial infarction: A meta-analysis of randomized clinical trials. J Am Coll Cardiol. 1999;33:598-604.
161 Pitt B, Zannad F, Remme W, et al. for the Randomized Aldactone Evaluation Study Investigators: The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med. 1999;341:709-717.
162 Zannad F, McMurray JJV, Krum H, et al. for the EMPHASIS-HF Study Group : Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med. 2011;364:11-21.
163 Davies M. Anatomic features in victims of sudden coronary death: Coronary artery pathology. Circulation. 1992;85(Suppl I):I19-I24.
164 Farb A, Tang A, Burke A, et al. Sudden coronary death: Frequency of active coronary lesions, inactive coronary lesions and myocardial infarction. Circulation. 1995;92:1701-1709.
165 ISIS-2 (Second International Study of Infarction Survival) Collaborative Group. Randomized trial of intravenous streptokinase, oral aspirin, both or neither among 17,187 cases of suspected myocardial infarction. Lancet. 1988;ii:349-360.
166 Dries D, Domanski M, Waclawiw M, Gersh B. Effect of antithrombotic therapy on risk of sudden coronary death in patients with congestive heart failure. Am J Cardiol. 1997;79:909-913.
167 Echt D, Liebson P, Mitchell B, et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo: The Cardiac Arrhythmia Suppression Trial (CAST I). N Engl J Med. 1992;327:227-233.
168 The Cardiac Arrhythmia Suppression Trial II Investigators. Effect of antiarrhythmic agent moricizine on survival after myocardial infarction. N Engl J Med. 1992;327:227-233.
169 Waldo A, Camm J, deRuyter H, et al. Survival with oral d-sotalol in patients with left ventricular dysfunction after myocardial infarction: Rationale, design and methods (the SWORD Trial. J Am Coll Cardiol. 1995;75:1023-1027.
170 Torp-Pedersen C, Moller M, Bloch-Thomsen P, et al. Dofetilide in patients with congestive heart failure and left ventricular dysfunction. N Engl J Med. 1999;341:857-865.
171 Kober L, Bloch Thomsen PE, Moller M, et al. Danish Investigations of Arrhythmia and Mortality on Dofetilide (DIAMOND) Study Group: Effect of dofetilide in patients with recent myocardial infarction and left-ventricular dysfunction: A randomised trial. Lancet. 2000;356:2052-2058.
172 Teo K, Yusef S, Furberg C. Effects of prophylactic antiarrhythmic drug therapy in acute myocardial infarction: An overview of results from randomized controlled trials. JAMA. 1993;270:1589-1595.
173 Burkart F, Pfisterer M, Kiowski W, et al. Effect of antiarrhythmic therapy on mortality on survivors of myocardial infarction with asymptomatic complex ventricular arrhythmias: Basel Antiarrhythmic Study of Infarct Survival (BASIS). J Am Cardiol. 1992;20:1056-1062.
174 Ceremuzynski L, Kleczar K, Krzeminska-Pakula M, et al. Effect of amiodarone on mortality after myocardial infarction: A double-blind, placebo-controlled, pilot study. J Am Coll Cardiol. 1992;20:1056-1062.
175 Julian D, Camm A, Frangin G, et al. for the European Myocardial Infarction Amiodarone Trial Investigators: Randomized trial of effect of amiodarone on mortality in patients with left ventricular dysfunction after recent myocardial infarction: EMIAT. Lancet. 1997;349:667-674.
176 Cairns J, Connolly S, Roberts R. Gent M for the Canadian Amiodarone myocardial Infarction Arrhythmia Trial Investigators: Randomized trial of outcome after myocardial infarction in patients with frequent of repetitive premature depolarizations: CAMIAT. Lancet. 1997;349:675-682.
177 Amiodarone Trials Meta-Analysis Investigators. Effect of prophylactic amiodarone in mortality after acute myocardial infarction and in congestive heart failure: Meta-analysis of individual data from 6500 patients in randomized trials. Lancet. 1997;350:1417-1424.
178 Sim I, McDonald K, Lavori P, Norbutas B. Quantitative overview of randomized trials of amiodarone to prevent sudden cardiac death. Circulation. 1997;96:2823-2829.
179 Connolly SJ, Dorian P, Roberts RS, et al. Comparison of beta-blockers, amiodarone plus beta-blockers, or sotalol for prevention of shocks from implantable cardioverter defibrillators: The OPTIC Study. A randomized trial. JAMA. 2006;295:165-171.
180 Holmes D, Davis K, Gersh B, et al. and participants in the Coronary Artery Surgery Study: Risk factor profiles of patients with sudden death and from other cardiac causes: A report of the Coronary Artery Surgery Study (CASS. J Am Cardiol. 1989;13:524-530.
181 Garan H, Ruskin J, DiMarco J, et al. Electrophysiologic studies before and after myocardial revascularization in patients with life-threatening ventricular arrhythmias. Am J Cardiol. 1983;51:519-524.
182 Hii J, Traboulsi M, Mitchell L, et al. Infarct artery patency predicts outcome of electropharmacological studies in patients with malignant tachyarrhythmias. Circulation. 1993;87:764-772.
183 Mirowski M, Reid P, Mower M, et al. Termination of malignant ventricular arrhythmias with an implanted automatic defibrillator in human beings. N Engl J Med. 1980;303:322-324.
184 Saksena S. The PCD investigators Group: Clinical outcome of patients with malignant ventricular tachyarrhythmias and a multiprogrammable cardioverter-defibrillator implanted with or without thoracotomy: An international multicenter study. J Am Coll Cardiol. 1994;23:1521-1530.
185 Moss A, Hall J, Cannom D, et al. for the Multicenter Automatic Defibrillator Implantation Trial Investigators: Improved survival with an implanted defibrillator in patients with coronary disease at high risk for ventricular arrhythmia. N Engl J Med. 1996;335:1933-1940.
186 Bigger J for the Coronary Artery Bypass Graft (CABG) Patch Trial Investigators. Prophylactic use of implanted cardiac defibrillators in patients at high risk for ventricular arrhythmias after coronary artery bypass graft surgery. N Engl J Med. 1997;337:1569-1575.
187 Bigger J, Whang W, Rottman J, et al. Mechanisms of death in the CABG Patch Trial. A randomized trial of implantable cardiac defibrillator prophylaxis in patients at high risk of death after coronary artery bypass graft surgery. Circulation. 1999;99:1416-1421.
188 Buxton A, Lee K, Fisher J, et al. A randomized study of the prevention of sudden death in patients with coronary artery disease, for the Multicenter Unsustained Tachycardia Trial (MUSTT) Investigators. N Engl J Med. 1999;341:1882-1890.
189 Moss AJ, Zareba W, Hall WJ, et al. Multicenter Automatic Defibrillator Implantation Trial II investigators: Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med. 2002;346:877-883.
190 Domanski M, Saksena S, Epstien A, et al. Relative effectiveness of the implantable cardioverter-defibrillator and antiarrhythmic drugs in patients with varying degrees of left ventricular dysfunction who have survived malignant ventricular arrhythmias, for the AVID Investigators. J Am Coll Cardiol. 1999;34:1090-1095.
191 Hohnloser S, Klingengeben T, Zabel M, Li Y. Heart rate variability used as an arrhythmia risk stratifier after myocardial infarction. Pacing Clin Electrophysiol. 1997;20:2594-2601.
192 LaRovere M, Bigger J, Marcus F, et al. Baroreflex sensitivity and heart-rate variability in prediction of total cardiac mortality after myocardial infarction. ATRAMI (Autonomic Tone and Reflexes After Myocardial Infarction) Investigator. Lancet. 1998;351:478-484.
193 Nolan J, Batin P, Andrews R, et al. Prospective study of heart rate variability and mortality in chronic heart failure: Results in the United Kingdom heart failure evaluation and assessment of risk trial (UK-HEART. Circulation. 1998;98:1510-1516.
194 Hohnloser SH, Kuck KH, Dorian P, Roberts RS, Hampton JR, Hatala R, Fain E, Gent M, Connolly SJ. Prophylactic use of an implantable cardioverter-defibrillator after acute myocardial infarction. N Engl J Med. 2004;351:2481-2488.
195 Kadish A, Dyer A, Daubert JP, et al. Prophylactic defibrillator implantation in patients with non-ischemic dilated cardio myopathy. N Engl J Med. 2004;350:2151-2158.
196 Huikuri HV, Makikallio TH, Raatikainen MJ, et al. Prediction of sudden cardiac death: Appraisal of the studies and methods assessing the risk of sudden arrhythmic death. Circulation. 2003;108:110-115.
Related posts:
 Mechanisms of Re-entrant Arrhythmias
Mechanisms of Re-entrant Arrhythmias
 Current Indications for Temporary and Permanent Cardiac Pacing
Current Indications for Temporary and Permanent Cardiac Pacing
 Surgery for Ventricular Arrhythmias
Surgery for Ventricular Arrhythmias
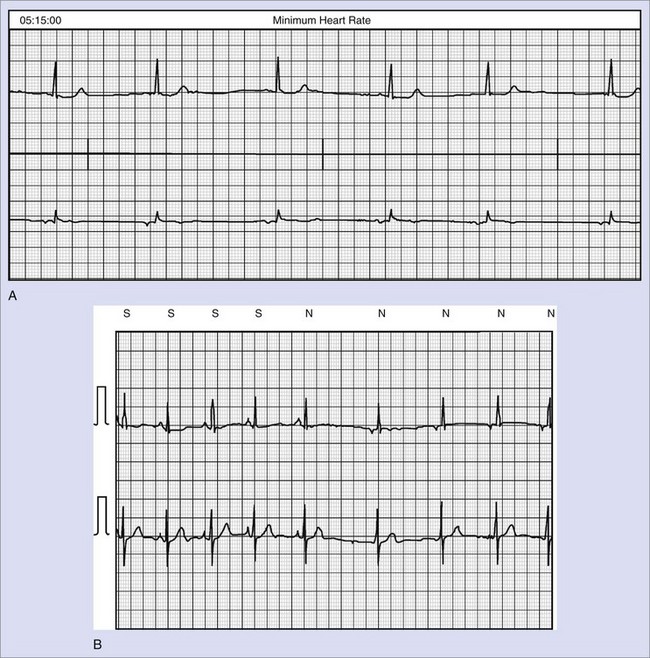 Sinus Node Dysfunction
Sinus Node Dysfunction
 Molecular and Cellular Basis of Cardiac Electrophysiology
Molecular and Cellular Basis of Cardiac Electrophysiology
 Implantable Cardioverter-Defibrillators: Device Technology and Implantation Techniques
Implantable Cardioverter-Defibrillators: Device Technology and Implantation Techniques
