[level-membership-for-cardiovascular-category]
Chapter 19 Principles of Clinical Trials
Introduction
In items 1 and 2, the intervention, niacin use, is not under the control of the investigators. Clinical factors such as age, smoking status, and other comorbidities as well as HDL levels may affect the physician’s choice to prescribe niacin, so the statement that can be made on the basis of these studies is that niacin use is associated with changes in HDL. In contrast, when the intervention is randomized, factors that could be related to HDL and clinical characteristics that influence the decision to prescribe niacin are ignored. As a result, the statement that niacin use causes changes in HDL is valid. The causal relationship between a test treatment and an outcome is called efficacy when it is measured in clinical trials and effectiveness when it is measured in a routine care setting. RCTs are the gold standard for evidence of the efficacy of a drug or other intervention, and this evidence is used by the U.S. Food and Drug Administration (FDA) to determine whether new medical treatments should be approved.1
Hypothesis Testing
Niacin use raises HDL by at least 10% on average
Niacin use raises HDL by less than 10% or lowers HDL on average
Types of Comparisons
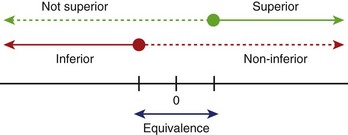
Hypotheses usually involve demonstrating that one treatment is significantly better than another (superiority) or that one treatment is not meaningfully worse than another treatment (non-inferiority, equivalence) (Figure 19-1). When testing an active drug or treatment compared with placebo, the study should be designed to detect the smallest clinically meaningful improvement as well as the cost and side effects of the drug. Placebo-controlled trials should always be superiority trials if the goal is to test the efficacy of the new drug. Sometimes non-inferiority placebo-controlled trials are used to establish the safety of a new drug.
Primary Outcome
The human phase of development for a drug or intervention is divided into four parts, and each part has unique objectives (Table 19-1). The choice of the outcome measure should be based on the goals of the development phase.
Table 19-1 Human Phase of Development for a Drug or Intervention
| PHASE | GOALS | EXAMPLES OF OUTCOMES |
|---|---|---|
| I | Maximum tolerated dose, toxicity, and safety profile | Pharmacokinetic parameters, adverse events |
| II | Evidence of biological activity | Surrogate markers* such as CRP, blood pressure, cholesterol, arterial plaque measure by intravascular ultrasound, hospitalization for CHF |
| III | Evidence of impact on hard clinical endpoints | Time to death, myocardial infarction, stroke, hospitalization for CHF, some combination of these events |
| IV | Postmarketing studies collecting additional information from widespread use of drugs or devices | Death, myocardial infarction, lead fracture, stroke, heart failure, liver failure |
CRP, C-reactive protein; CHF, congestive heart failure.
* A surrogate marker is an event that precedes the clinical event, which is (ideally) in the causal pathway to the clinical event. For example, if a drug is thought to decrease risk of myocardial infarction (MI) by reducing arterial calcification, changes in arterial calcification might be used as a surrogate endpoint because these changes should occur sooner than MI, leading to a reduction in trial time. Hospitalization is a challenging surrogate marker because it is not clearly in the causal pathway; hospitalization does not cause MI, but heart failure worsening to the degree that hospitalization is required is in the causal pathway. When hospitalization is used as a measure of new or worsening disease, it is important that the change in disease status and not just the hospitalization event is captured.
Randomization
Why Randomize?
Randomization, then, tends to even out any differences between the study participants assigned to one treatment arm compared with those to the other. The Coronary Artery Surgery Study (CASS) randomly assigned patients with stable class III angina to initial treatment with bypass surgery or medical therapy.2 If this trial had not been randomized and treatment assignment had been left to the discretion of the enrolling physician, it is likely that these physicians would have selected patients who they believed would be “good surgical candidates” for the bypass surgery arm. This would have led to a comparison of patients who were good surgical candidates receiving bypass surgery with a group of patients who, for one reason or another, were not good surgical candidates receiving medical treatment. It is likely that the medically selected patients would have been sicker, with more comorbidities than the patient selected for surgery. This design would not result in a fair comparison of the two treatment strategies. Randomization levels the playing field.
Intention to Treat
To make randomization work, analysis of RCT data needs to adhere to the principle of “intention to treat.” In its purest form, this means that data from each study participant are analyzed according to the treatment arm to which they were randomized, period. In the case of the Antiarrhythmics Versus Implantable Defibrillators (AVID) trial, this meant that data from patients randomly assigned to the implantable defibrillator arm were analyzed in that arm, whether or not they ever received the device.3 Data from patients randomly assigned to the antiarrhythmic drug treatment arm were analyzed with that arm, even if they had received a defibrillator. This may not make obvious sense, and many trial sponsors have argued that their new treatment just could not show an effect if the patient never received the new treatment. However, the principle of “intention to treat” protects the integrity of the trial by removing a large source of potential bias. In a trial to examine the efficacy of a new antiarrhythmic drug for preventing sudden cardiac death (SCD), for example, a sponsor might be tempted to count events only while the patient was still taking the drug. How, they would argue, could the drug have a benefit if the patient was not taking it? But, if, as has happened, the drug exacerbates congestive heart failure (CHF), then patients assigned to the experimental drug would be likely to discontinue taking the drug. And any subsequent SCD or cardiac arrests would not be attributed to the drug. In fact, it could be argued that the drugs led to a situation where the patient was more likely to die of an arrhythmia.
Stratification and Site Effect
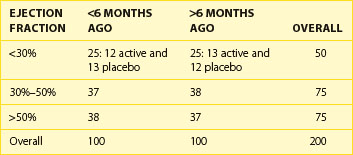
Some investigators get carried away with the concept of stratification to try to design randomization in such a way that all possible factors are balanced. It is easy to design a trial with too many strata. Take, for example, a trial stratifying on the basis of ejection fraction at baseline (<30%, 30% to 50%, and >50%) and a required history of myocardial infarction (MI) being “recent,” that is, within the past 6 months, or distant, that is, more than 6 months ago. With this, six strata have been created, a reasonable number for a sample size of, say, 200 or more subjects, randomized to one of two treatment options (Table 19-2). But if the decision is made to stratify by site, with 10 sites, for example, more strata than expected patients would be created. It has been shown that as the number of strata in a conventional randomization design is increased, the probability of imbalances between treatment groups is, in fact, increased as well.4
Types of Randomization Designs
Permuted block designs can lead to the same problems as with too many strata. For this reason, adaptive randomization is sometimes used. Several types of adaptive designs exist. Basically, the next randomization assignment is dependent, in some way, on the characteristics of the patients who have been randomized so far. Baseline adaptive techniques adjust the probabilities of assigning the next patient to one treatment arm or the other on the basis of the baseline characteristics of the patient compared with other patients already randomized.5 In the study design described above where randomization will be stratified by the class of angina, recent or distant history of MI, and ejection fraction, the objective is to keep a balance of treatment assignments within each stratum. So, as each patient is randomized, the randomization algorithm will look at the existing balance in that stratum and assign treatment on the basis of a biased coin toss.
Accuracy of Measurement and Event Ascertainment
Principles of Outcome Selection
The Outcome Should Be Unambiguously Defined
The American College of Cardiology (ACC) has devoted much effort to standardizing the definition for MI. Whereas earlier trials such as CASS had to define MI for their trials, current studies refer to the ACC definition.6
The Outcome Should Be Important to the Patient Population
For a patient with AF, what is important? Clearly, avoiding death or disability caused by a stroke or other cardiovascular cause is most important. Thus, some of the most critical trials in patients with AF were the Atrial Fibrillation Follow-up Investigation of Rhythm Management trial (AFFIRM), which examined the effect of two treatment strategies, heart rate control, and restoration of sinus rhythm, to prevent overall mortality in patients with atrial fibrillation,8 and the Stroke Prevention in Atrial Fibrillation (SPAF),9 Aspirin vs. Warfarin Standard Dose (AFASAK),10 and other trials of anticoagulation to prevent stroke.11 Then, is functional status important? The AFFIRM study attempted to measure change in functional status using the New York Heart Association (NYHA) functional class scale, Canadian Cardiovascular Society Angina Classification, a Mini-Mental State Examination, and a 6-minute walk test.12 The investigators in the study were able to detect a difference in functional status related to the presence or absence of AF. In contrast, inflammatory measures such as C-reactive protein and degree of stenosis may be good measures of disease burden, but changes in these measures may not be identifiable to patients.
Overreads and Clinical Endpoint Adjudication
As clinical trials have to reach further and include many more sites to recruit enough patients, the study is dependent on the clinical judgment of a large number of physicians to assess study endpoints. In the AFFIRM trial, for example, the more than 200 clinical sites were located primarily in cardiology practices. One of the important endpoints was cardioembolic stroke. Although the patients were, of course, treated by neurologists, the study physicians were, in almost all cases, not neurologists. To make sure that all of the strokes included in the study-defined endpoint met the protocol definition for cardioembolic stroke, the investigators used a clinical events committee (CEC) to evaluate each potential study endpoint by reviewing collected medical records. These records were masked to anything that could reveal the patient’s assigned treatment arm (rhythm or rate control). Two separate CEC neurologists examined records that included imaging reports, hospital discharge summaries, and a narrative discussion provided by the local physician. Concordance was required between two CEC neurologists or consensus by the committee in order for an event to be “ruled in” (E. Nasco, personal communication). In the final analysis, only events confirmed by the CEC were included. In the AFFIRM trial, only 171 of 247 reported strokes were ruled in. In the Trial With Dronedarone to Prevent Hospitalization or Death in Patients with Atrial Fibrillation (ATHENA), an events adjudication committee categorized the causes of death using a modification to a previously published classification scheme that standardized the criteria for determining arrhythmic, nonarrhythmic cardiac, noncardiac vascular, or noncardiovascular death.13
Interim Analyses and Adaptive Designs
In the simplest approach, a clinical trial would be designed, the trial would be launched, and participants would be enrolled, treated, and followed up. Follow-up would continue up to a prespecified endpoint, such as 6 months after the last patient was treated. While the trial is ongoing, data would be collected, but no one would look at the results. At the end of the trial, the data would be completed, closed, and analyzed. However, for many trials conducted these days, investigators have valid reasons to look at the data even while the trial is ongoing. In the case of experimental treatments, the sponsor may want an independent group monitoring the trial to make sure that participants are not exposed to undue risk or harm from the adverse effects of the treatment. In the case of trials with long-term follow-ups, it may be possible to reach a reasonable conclusion about the treatment that is being evaluated without continuing the trial to its planned conclusion. For these reasons, many trials are planned with “interim analyses.” Further, regulatory agencies have, in some situations, strongly recommended the use of external, independent monitoring committees, commonly referred to as Data Monitoring Committees (DMCs) or Data and Safety Monitoring Boards.1,14 Although the specific charge of DMCs will vary from one trial to another, in general, DMCs are charged with monitoring the trial for the following reasons:
Much has been written about DMCs, which are being used more frequently for late phase II and phase III trials. Ellenburg and colleagues make the following recommendations regarding when a DMC should be used15:
During the CAST I study, the DSMB was concerned that the reports contained information about all of the deaths that had occurred. For this reason, the coordinating center organized and performed an “events sweep.” During a 2-week period, research coordinators at each of the clinical sites were instructed to call all of their participants to find out, primarily, if they were still alive.16 This ensured that the DMC had current (within the past 4 weeks) follow-up data on all randomized participants. This information supplemented the data from a normal database freeze.
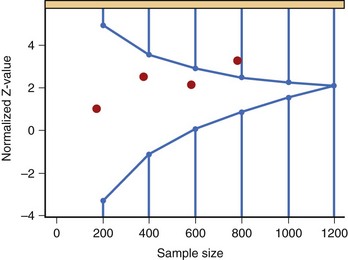
The various approaches to a group sequential design can best be illustrated by Figure 19-2, which depicts a typical O’Brien-Fleming boundary. The O’Brien-Fleming approach is very conservative at early looks at the data and is less conservative as more information is available. The Pocock approach assigns the same alpha level at each look. The actual shape of the O’Brien-Fleming boundary can be adjusted based on the risk tolerance and objectives of the sponsor. The decisions regarding the number of formal interim analyses and the shape of the boundary are important for management to consider carefully.
Issues Related to the Conduct of Clinical Trials
Recruitment
In designing a trial, delineating the population to be studied requires careful thought. These definitions will ultimately define the conclusions that can be drawn from the trial. For example, the Physicians Health Study studied 22,017 male physicians in the United States.17 By excluding women from their study, the investigators were limited in any conclusions that could be drawn from their data as they related to women.
The inclusion criteria define the disease condition to be studied. For example, in CAST, the inclusion criteria stated that potential participants should have had an MI 6 months to 2 years before enrollment and should have demonstrated sufficient ventricular arrhythmia to warrant treatment (≥6 VPDs per hour on a 24-hour Holter monitor). The investigators gave considerable thought to setting a cutoff of 6 or more VPDs per hour. Would 10 or more VPDs have defined a population that would have better benefited from the treatment? Would a cutoff of 10 VPDs per hour have excluded too many patients? The Cardiac Arrhythmia Pilot Study (CAPS) was designed as a feasibility study to see if a long-term trial could be conducted to test the hypothesis that suppression of ventricular arrhythmias in a post-MI population would decrease the rate of arrhythmic and overall death.18 Data from CAPS provided needed information about the proportion of patients likely to be excluded if a cutoff of 10 VPDs per hour was used.
It has been said that as soon as a trial begins, the prevalence of the disease decreases. In some respects, this is true about trials in cardiology. Over the past 20 years, the rate of death from cardiovascular cause has decreased to the point that it is expected that cancer will exceed cardiovascular disease as the primary cause of death in the United States.19 The number of clinical trials being conducted in the United States has increased in the past 20 years to such an extent that in any community more competition occurs for the pool of available patients. Nearly all large clinical trials have struggled with recruitment.
The ATHENA trial was a multicenter randomized trial of dronedarone in patients with AF.20 Patients with paroxysmal or persistent AF were eligible if they were older than 70 years or had at least one risk factor for cardiac complications (arterial hypertension, diabetes mellitus, previous stroke, transient ischemic attack or systemic embolism, enlarged left atrium, or depressed ejection fraction). As the trial proceeded, overall mortality rates were lower than expected. For this reason, the investigators modified the entry criteria to increase the risk profile of patients enrolled. Patients between ages 70 and 75 years were eligible only if they had one or more of the above risk factors for cardiac death. Patients younger than 70 years were no longer eligible.
Conclusion
Medical journals publishing the results of clinical trials have set certain standards for reporting these results. This is been done to prevent the publication of false or misleading reports.21 Consolidated Standards of Reporting Trials (CONSORT) is an organization that has examined the quality of reporting of medical studies.22 CONSORT was formed by a group of clinical trialists and journal editors who originally met in 1993 to set a scale for evaluating published results from clinical trials. As a result of their examination, CONSORT has published standards for results manuscripts, which include the following key points:
The real result of clinical trials research is improvement in the way that diseases are treated and prevented. If the results of a clinical trial are not published or disseminated in a trustworthy manner, it is unlikely that the practice of health care professionals will change on the basis of trial results. It has been estimated that it takes, on average, 17 years for the results of a clinical trial to bring about an effect in general practice.23 The Beta-blocker Heart Attack Trial (BHAT) in 1982 showed that treatment with β-blockers after an MI would significantly decrease mortality in this at-risk population.24 And yet, the widespread use of β-blockers did not become the standard of care until the AHA/ACC published guidelines for the use of β-blockers in 1996 that this class of drug became standard of care.25,26 The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack (ALLHAT) trial was a National Heart, Lung, and Blood Institute sponsored trial of antihypertensives and lipid-lowering treatment to reduce the rate of cardiac death and nonfatal MI in participants with hypertension and at least one other risk factor for cardiovascular disease.27,28 The trial showed that aggressive treatment with antihypertensives could significantly decrease the mortality rate in this population. Following the conclusion of the trial and the publication of the initial results, the study group for ALLHAT embarked on a project to focus on the dissemination of the study results in such a way as to maximize the impact of the trial’s results.29 Their dissemination plan included the usual press releases, presentations at national medical meetings, and refereed articles in major medical journals. In addition, they targeted the “determinants of clinician behavior” to communicate more directly with clinicians about the implications of the ALLHAT results. They took their message to places where physicians routinely went to get information and conducted numerous in-person, interactive sessions. Their approach included, among other strategies, Web forums, online continuing medical education sources, and other more immediately available media.
1 International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. Statistical principles for clinical trials (E9). February 5, 1998: Available at http://www.ich.org/fileadmin/Public_Web_site/ICH_Products/Guidelines/Efficacy/E9/Step4/E9_Guideline.pdf Accessed October 19, 2009
2 CASS Principal Investigators. National Heart, Lung, and Blood Institute Coronary Artery Surgery Study. Circulation. 1981;63(II):6.
3 AVID Investigators. Antiarrhythmics Versus Implantable Defibrillators (AVID)—rationale, design, and methods. Am J Cardiol. 1995;75:470-475.
4 Hallstrom AP, Davis K. Imbalance in treatment assignments in stratified blocked randomization. Control Clin Trials. 1988;9:375-382.
5 Efron B: Forcing a sequential experiment to be balanced, Biometrika. 1971;58:403-417.
6 The Joint European Society of Cardiology/American College of Cardiology Committee. Myocardial infarction redefined—a consensus document of the Joint European Society of Cardiology/American College of Cardiology committee for the redefinition of myocardial infarction. J Am Coll Cardiol. 2000;36:959-969.
7 Epstein AE, Hallstrom AP, Rogers WJ, et alfor the CAST Investigators. Mortality following ventricular arrhythmia suppression by encainide, flecainide, and moricizine after myocardial infarction: The original design concept of the Cardiac Arrhythmia Suppression Trial (CAST). JAMA. 1993;270(20):2451-2455.
8 The AFFIRM Investigators. A comparison of rate control and rhythm control in patients with atrial fibrillation. N Engl J Med. 2002;347(23):1825-1833.
9 The Stroke Prevention in Atrial Fibrillation Investigators. Stroke prevention in atrial fibrillation study, final results. Circulation. 1991;84:527-539.
10 Petersen P, Boyson G, Godtfredsen J, et al. Placebo-controlled, randomised trial of warfarin and aspirin for prevention of thromboembolic complications in chronic atrial fibrillation. The Copenhagen AFASAK study. Lancet. 1989;i:175-179.
11 Atrial Fibrillation Investigators. Risk factors for stroke and efficacy of antithrombotic therapy in atrial fibrillation. Arch Intern Med. 1994;154:1449-1457.
12 Chung MK, Shemanski LR, Sherman DG, et aland AFFIRM Investigators. Functional status in rate- versus rhythm-control strategies for atrial fibrillation: Results of the Atrial Fibrillation Follow-Up Investigation of Rhythm Management (AFFIRM) Functional Status substudy. J Am Coll Cardiol. 2005;46(10):1891-1899.
13 Hinkle LE, Thaler HT. Clinical classification of cardiac deaths. Circulation. 1982;65:457-464.
14 U.S. Food and Drug Administration. Guidance for clinical trial sponsors: Establishment and operation of clinical trial data monitoring committees. March 2006. Available online at http://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM127073.pdf Accessed October 19, 2009
15 Ellenberg SS, Fleming TR, DeMets DL. Data monitoring committees in clinical trials: A practical perspective. New York: John Wiley & Sons; 2002.
16 Hallstrom AP, McBride R, Moore R. Toward vital status sweeps: A case history in sequential monitoring. Stat Med. 1995;14:1927-1931.
17 Steering Committee of the Physicians’ Health Study Research Group. Final report on the aspirin component of the ongoing Physicians’ Health Study. N Engl J Med. 1989;321(3):129-135.
18 The CAPS Investigators. The cardiac arrrhythmia pilot study. Am J Cardiol. 1986;57:91-95.
19 Davis DL, Dinse GE, Hoel DG. Decreasing cardiovascular disease and increasing cancer among whites in the United States from 1973 through 1987. JAMA. 1994;271:431-437.
20 Hohnloser SH, Crijris HJGM, van Eickels M, et alfor the ATHEN Investigators. Effect of dronedarone on cardiovascular events in atrial fibrillation. N Engl J Med. 2009;360:668-678.
21 Lancet fighting plagiarism [editorial]. Lancet. 2008;371:2146.
22 Consolidated Standards of Reporting Trials. CONSORT home page. Available online at http://www.consort-statement.org Accessed October 5, 2009
23 Balas EA, Boren SA. Managing clinical knowledge for health care improvement. Yearbook Med Informat. 2000:65-70.
24 BHAT Investigators. A randomized trial of propranolol in patients with acute myocardial infraction. I. Mortality results. JAMA. 1982;247:1707-1714.
25 Howard PA, Ellerbeck EF. Optimizing beta-blocker use after myocardial infarction. Am Fam Physician. 2000;62(8):1853-1860. 1865–1866
26 Ryan TJ, Anderson JL, Antman EM, et al. ACC/AHA guidelines for the management of patients with acute myocardial infarction: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee on Management of Acute Myocardial Infarction). J Am Coll Cardiol. 1996;28:1328-1428.
27 The ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic. JAMA. 2002;288:2981-2997.
28 National Heart, Lung, and Blood Institute. ALLHAT information for health professionals: Quick reference for health care providers. Available online at http://www.nhlbi.nih.gov/health/allhat/qckref.htm Accessed October 5, 2009
29 Bartholomew LK, Cushman WC, Cutler JA, et alfor the ALLHAT Collaborative Research Group. Getting clinical trial results into practice: Design, implementation, and process evaluation of the ALLHAT Dissemination Project. Clin Trials. 2009;6:329-6343.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
Chapter 19 Principles of Clinical Trials
Introduction
In items 1 and 2, the intervention, niacin use, is not under the control of the investigators. Clinical factors such as age, smoking status, and other comorbidities as well as HDL levels may affect the physician’s choice to prescribe niacin, so the statement that can be made on the basis of these studies is that niacin use is associated with changes in HDL. In contrast, when the intervention is randomized, factors that could be related to HDL and clinical characteristics that influence the decision to prescribe niacin are ignored. As a result, the statement that niacin use causes changes in HDL is valid. The causal relationship between a test treatment and an outcome is called efficacy when it is measured in clinical trials and effectiveness when it is measured in a routine care setting. RCTs are the gold standard for evidence of the efficacy of a drug or other intervention, and this evidence is used by the U.S. Food and Drug Administration (FDA) to determine whether new medical treatments should be approved.1
Hypothesis Testing
Niacin use raises HDL by at least 10% on average
Niacin use raises HDL by less than 10% or lowers HDL on average
Types of Comparisons
Hypotheses usually involve demonstrating that one treatment is significantly better than another (superiority) or that one treatment is not meaningfully worse than another treatment (non-inferiority, equivalence) (Figure 19-1). When testing an active drug or treatment compared with placebo, the study should be designed to detect the smallest clinically meaningful improvement as well as the cost and side effects of the drug. Placebo-controlled trials should always be superiority trials if the goal is to test the efficacy of the new drug. Sometimes non-inferiority placebo-controlled trials are used to establish the safety of a new drug.
Primary Outcome
The human phase of development for a drug or intervention is divided into four parts, and each part has unique objectives (Table 19-1). The choice of the outcome measure should be based on the goals of the development phase.
Table 19-1 Human Phase of Development for a Drug or Intervention
| PHASE | GOALS | EXAMPLES OF OUTCOMES |
|---|---|---|
| I | Maximum tolerated dose, toxicity, and safety profile | Pharmacokinetic parameters, adverse events |
| II | Evidence of biological activity | Surrogate markers* such as CRP, blood pressure, cholesterol, arterial plaque measure by intravascular ultrasound, hospitalization for CHF |
| III | Evidence of impact on hard clinical endpoints | Time to death, myocardial infarction, stroke, hospitalization for CHF, some combination of these events |
| IV | Postmarketing studies collecting additional information from widespread use of drugs or devices | Death, myocardial infarction, lead fracture, stroke, heart failure, liver failure |
CRP, C-reactive protein; CHF, congestive heart failure.
* A surrogate marker is an event that precedes the clinical event, which is (ideally) in the causal pathway to the clinical event. For example, if a drug is thought to decrease risk of myocardial infarction (MI) by reducing arterial calcification, changes in arterial calcification might be used as a surrogate endpoint because these changes should occur sooner than MI, leading to a reduction in trial time. Hospitalization is a challenging surrogate marker because it is not clearly in the causal pathway; hospitalization does not cause MI, but heart failure worsening to the degree that hospitalization is required is in the causal pathway. When hospitalization is used as a measure of new or worsening disease, it is important that the change in disease status and not just the hospitalization event is captured.
Randomization
Why Randomize?
Randomization, then, tends to even out any differences between the study participants assigned to one treatment arm compared with those to the other. The Coronary Artery Surgery Study (CASS) randomly assigned patients with stable class III angina to initial treatment with bypass surgery or medical therapy.2 If this trial had not been randomized and treatment assignment had been left to the discretion of the enrolling physician, it is likely that these physicians would have selected patients who they believed would be “good surgical candidates” for the bypass surgery arm. This would have led to a comparison of patients who were good surgical candidates receiving bypass surgery with a group of patients who, for one reason or another, were not good surgical candidates receiving medical treatment. It is likely that the medically selected patients would have been sicker, with more comorbidities than the patient selected for surgery. This design would not result in a fair comparison of the two treatment strategies. Randomization levels the playing field.
Intention to Treat
To make randomization work, analysis of RCT data needs to adhere to the principle of “intention to treat.” In its purest form, this means that data from each study participant are analyzed according to the treatment arm to which they were randomized, period. In the case of the Antiarrhythmics Versus Implantable Defibrillators (AVID) trial, this meant that data from patients randomly assigned to the implantable defibrillator arm were analyzed in that arm, whether or not they ever received the device.3 Data from patients randomly assigned to the antiarrhythmic drug treatment arm were analyzed with that arm, even if they had received a defibrillator. This may not make obvious sense, and many trial sponsors have argued that their new treatment just could not show an effect if the patient never received the new treatment. However, the principle of “intention to treat” protects the integrity of the trial by removing a large source of potential bias. In a trial to examine the efficacy of a new antiarrhythmic drug for preventing sudden cardiac death (SCD), for example, a sponsor might be tempted to count events only while the patient was still taking the drug. How, they would argue, could the drug have a benefit if the patient was not taking it? But, if, as has happened, the drug exacerbates congestive heart failure (CHF), then patients assigned to the experimental drug would be likely to discontinue taking the drug. And any subsequent SCD or cardiac arrests would not be attributed to the drug. In fact, it could be argued that the drugs led to a situation where the patient was more likely to die of an arrhythmia.
Stratification and Site Effect
Some investigators get carried away with the concept of stratification to try to design randomization in such a way that all possible factors are balanced. It is easy to design a trial with too many strata. Take, for example, a trial stratifying on the basis of ejection fraction at baseline (<30%, 30% to 50%, and >50%) and a required history of myocardial infarction (MI) being “recent,” that is, within the past 6 months, or distant, that is, more than 6 months ago. With this, six strata have been created, a reasonable number for a sample size of, say, 200 or more subjects, randomized to one of two treatment options (Table 19-2). But if the decision is made to stratify by site, with 10 sites, for example, more strata than expected patients would be created. It has been shown that as the number of strata in a conventional randomization design is increased, the probability of imbalances between treatment groups is, in fact, increased as well.4
Types of Randomization Designs
Permuted block designs can lead to the same problems as with too many strata. For this reason, adaptive randomization is sometimes used. Several types of adaptive designs exist. Basically, the next randomization assignment is dependent, in some way, on the characteristics of the patients who have been randomized so far. Baseline adaptive techniques adjust the probabilities of assigning the next patient to one treatment arm or the other on the basis of the baseline characteristics of the patient compared with other patients already randomized.5 In the study design described above where randomization will be stratified by the class of angina, recent or distant history of MI, and ejection fraction, the objective is to keep a balance of treatment assignments within each stratum. So, as each patient is randomized, the randomization algorithm will look at the existing balance in that stratum and assign treatment on the basis of a biased coin toss.
