[level-membership-for-cardiovascular-category]
Chapter 2 Principles of Cellular Architecture and Physiology with Applications in Electrophysiology
Specialized Excitable Cells Tightly Regulate Cardiac Depolarization
The highly coordinated and efficient propagation of electrical activity through the heart is maintained by the combined activities of a diverse set of specialized excitable cardiac cells, each with its own structural, electrical, and molecular signature. The cardiac sinoatrial (SA) node, a small group of spontaneously active cells in the right atria, is the primary initiation site of cardiac electrical activity because of its relatively positive threshold potential.1 Once generated by the sinus node, the cardiac action potential propagates through the atria to the atrioventricular (AV) node (the maximum diastolic potential of the AV node is only slightly more negative than that of the SA node), a second small but critical group of specialized cells that display slow conduction properties preventing inappropriate depolarization of the ventricles. In fact, the slow conduction of the AV node is a critical safeguard against the development of ventricular arrhythmias from pathologic atrial pacing defects (i.e., atrial flutter/fibrillation). After the AV node, the cardiac action potential propagates through the cardiac conduction system comprising the AV bundle (bundle of His), the left and right bundle branches, and the cardiac Purkinje system. Interestingly, this conduction system, particularly the cardiac Purkinje fibers, has evolved to rapidly propagate cardiac electrical activity at up to 2 to 4 m/s for the nearly instantaneous spread of depolarization through the sub-endocardium of the left and right ventricles.2 In comparison with the rapid conduction pathways of the Purkinje system, left ventricular tissue conduction velocity is significantly slower (0.3 to 1 m/s).2 Importantly, Purkinje fibers communicate with the ventricular mass at well-defined discrete loci (Purkinje-muscle junctions).
Form Fits Function: The Ventricular Cardiomyocyte and Excitation-Contraction Coupling
As is discussed in detail in Chapter 3, the electrical activity of cardiac cells (the action potential) is primarily modulated by the coordinated movement of sodium (Na+), calcium (Ca2+), and potassium (K+) across the external plasma membrane (sarcolemma) and the internal sarcoplasmic reticulum (SR) membrane. The specialized function of each cardiac cell type is the result of the evolution of specific molecular and structural components that regulate ion flux across the membrane and dictate specific cell properties.3 In this section, the primary structural and molecular components of different cardiac cells is discussed in relation to cell type–specific action potentials. Because of its central role in cardiac excitability, the ventricular cardiomyocyte will be used as the central point of comparison for other excitable cardiac cell types.
The ventricular action potential is notable for its hyperpolarized resting membrane potential, rapid upstroke, and prolonged plateau (Figure 2-1). The resting membrane potential of the ventricular cardiomyocyte (held at ~–90 mV, roughly 30 mV more negative than the human sinus node) is the most negative of all excitable cell types (hence the final cell type to depolarize), primarily because of a large inwardly rectifying K+ current, IK1, prominently expressed in these cells.4 The rapid upstroke, because of the presence of rapidly activating voltage-gated Na+ channels, allows for rapid propagation of the electrical signal through the ventricles, which is required for synchronized muscle contraction. Finally, the extended plateau allows sufficient time for Ca to enter the cell and signal contraction.3
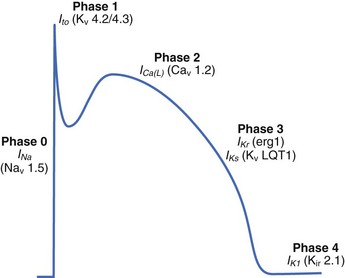
Once the excitation reaches the ventricles through the cardiac conduction system (see above), the electrical signal passes from cell to cell as a flux of ions through specialized intercellular ion channels called gap junctions (discussed in detail below). This flow of ions into the cell from neighboring activated cells depolarizes the membrane potential. If the membrane reaches a threshold potential (~–60 mV), a large population of voltage-gated Na+ channels (primarily Nav1.5, encoded by SCN5A) is activated, which results in a large inward influx of Na+ across the membrane into the myocyte. Na+ channels are functionally well suited to this task as they undergo rapid activation (activation time constant <0.5 ms) in response to membrane depolarization. Importantly, these channels also experience rapid voltage-dependent inactivation, which prevents reactivation until the membrane has returned to rest. The inward flux of Na+ ions carried by voltage-gated Na+ channels produces the initial rapid spike of the ventricular action potential (phase 0; see Figure 2-1).3 Depolarization of the cardiac membrane by rapidly activating voltage-gated Na+ channels activates higher threshold, more slowly activating voltage-gated Ca2+ channels (primarily CACNA1C-encoded Cav1.2 in the ventricle; see Figure 2-1).2 Ca2+ influx through voltage-gated Ca2+ channels serves two major purposes: (1) maintaining the action potential plateau (important for controlling heart rhythm), and (2) triggering Ca2+ release from internal stores for the purpose of triggering the mechanical contraction of the heart (excitation-contraction coupling).2 During excitation-contraction (EC) coupling, a small inward Ca2+ current across the plasma membrane is sensed by functional SR ryanodine receptor (RyR2 in the ventricle, encoded by RYR2) clusters, which subsequently release large quantities of Ca2+ from the internal SR stores into the cytosol, giving rise to a dramatic increase (order of magnitude) in intracellular Ca2+ (the Ca2+ transient).2 In this process, termed Ca2+-induced Ca2+ release, a small increase in local Ca2+ via Cav1.2 produces a relatively large release of SR Ca2+ (high gain function).
While voltage-gated Na+ and Ca2+ channels are responsible for myocyte depolarization and contraction, a host of plasma membrane–associated K+ channels regulate ventricular cardiomyocyte repolarization during phases 1, 2, and 3, as well as the rest potential.4 Specifically, the characteristic repolarization “notch” of phase 1 is modulated by the outward flux of K+ carried by the transient outward K+ current, Ito (primarily Kv4.2/Kv4.3 channels; see Figure 2-1). The duration of the action potential plateau (phase 2) is determined by a delicate balance between the inward Ca2+ current and the outward delayed rectifier K+ currents IKr and IKs (erg1/MiRP1 and KvLQT1/MinK channels encoded by KCNH2/KCNE2 and KCNQ1/KCNE1, respectively; see Figure 2-1).4 As the action potential proceeds, the Ca2+ current decreases because of deactivation as well as inactivation, and the K+ currents IKr and IKs increase, ultimately tilting the balance in favor of the late repolarization phase (phase 3; see Figure 2-1).4 The cell eventually returns to a resting potential maintained primarily by the time-independent inward rectifier current IK1 (see Figure 2-1). Other currents such as IKATP, while not central to the healthy control action potential, may have key roles in disease. Finally, the Na+-K+ adenotriphosphatase (ATPase) (which uses ATP to remove 3 Na+ from the cell and bring in 2 K+) is a key feature for generating and maintaining the myocyte electrochemical gradient.
Cell Membrane Architecture Defines Myocyte Local Electrical Activity
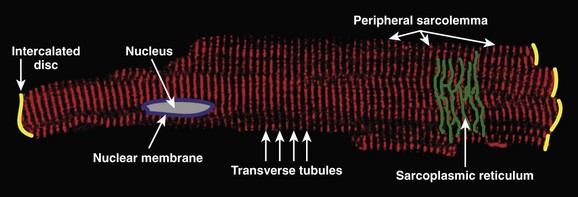
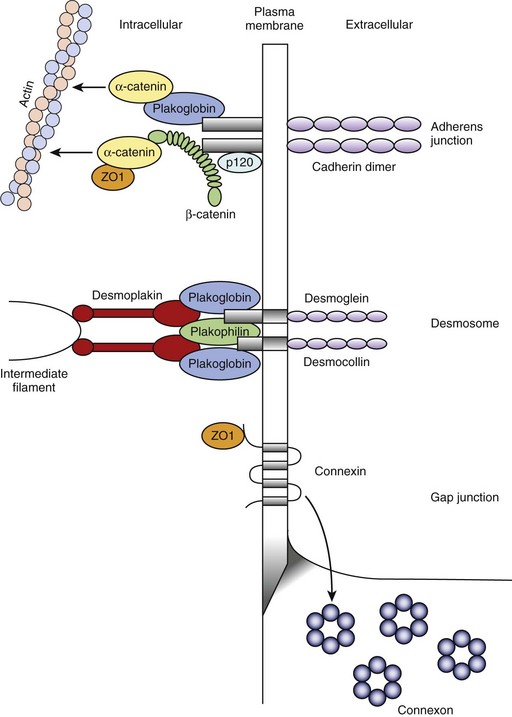
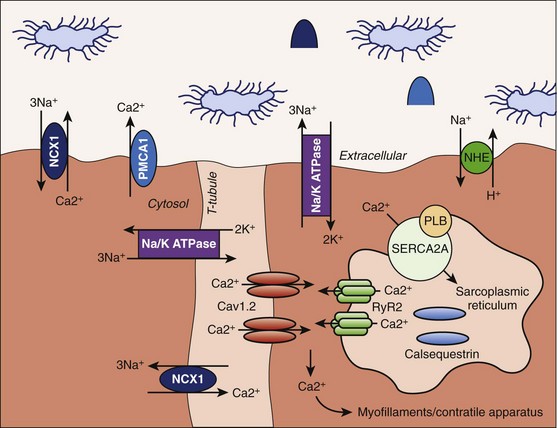
The ventricular cardiomyocyte plasma membrane, or sarcolemma, is divided into multiple and unique membrane structures (Figure 2-2). In addition to the external sarcolemma (resident proteins include the Na+-Ca2+ exchanger (NCX1) and plasma membrane Ca2+-ATPase [PMCA1]), the ventricular cardiomyocyte contains a large array of regularly spaced (~1.8 μm) plasma membrane invaginations, termed transverse tubules, or T-tubules. This membrane system, instrumental in myocyte EC coupling, evolved to facilitate coordinated EC coupling in the relatively large ventricular cardiomyocyte (system not present in smaller atrial and sinoatrial node cells). T-tubule–resident proteins include the L-type Ca2+ channel Cav1.2, the Na+-Ca2+ exchanger, and Na+-K+-ATPase.2 Finally, a highly specialized domain is present where the ventricular cardiomyocyte plasma membrane lies in close apposition to the plasma membrane of a neighboring cell. This complex membrane system, termed the intercalated disc, is required for myocyte cell-cell adhesion as well as intercellular action potential propagation and comprises three subdomains: (1) the gap junction, (2) the adherens junction, and (3) the desmosome (Figure 2-3).2 The gap junction comprises hundreds of hemi-channels (connexons) that span the lipid bilayer and allow electrical and metabolic coupling when docked with hemi-channels from a neighboring cell. At least four different connexin proteins (functional units of the connexon) with distinct biophysical properties are expressed throughout the heart. Gap junctions in ventricular tissue consist primarily of connexin43, which forms large conductance channels to allow rapid conduction. In contrast, gap junctions in the sinus node contain mostly connexin45, which forms lower conductance channels ideal for slow but safe conduction. The adherens junction is maintained by the function of a cadherin-catenin complex that provides a stabilizing link from the intercalated disc to the actin cytoskeleton (see Figure 2-3). Finally, the desmosome also supports cell adhesion through a complex, including plakoglobin (γ-catenin, also found in the adherens junction), desmoplakin, and plakophilin, that interacts with intermediate filaments (see Figure 2-3). Interestingly, defects in desmosomal proteins have been linked to cardiomyopathies and arrhythmias, including arrhythmogenic right ventricular cardiomyopathy (ARVC). In fact, loss of plakoglobin immunostaining in human heart biopsies has been recently developed into a diagnostic marker for Naxos disease, a cardiocutaneous syndrome characterized by wooly hair, palmoplantar keratoderma, and severe cardiomyopathy.5
In addition to the external plasma membrane, the cardiac myocyte (as well as many other excitable cells, including neurons and skeletal muscle) contains SR, the specialized endoplasmic reticulum that has key roles in the regulation of intracellular Ca2+.2 The cardiac SR is an extensive network of tubules linking key signaling networks, including the plasma membrane, nuclear envelope, nucleus, and mitochondria, to mediate a host of diverse functions. The pre-eminent role of the cardiac SR is to regulate myocyte EC coupling. Specifically, the vertebrate SR sequesters a large pool of releasable Ca2+ (1 mM inside SR vs. 100 nM in cytosol2) that serves as the primary source of Ca2+ for troponin-C (TnC) activation and muscle contraction (discussed in detail below).2 The majority of this Ca2+ is internally buffered by the Ca2+-binding protein calsequestrin, which forms a critical RyR2 regulatory complex with triadin and junctin. However, Ca2+ entry into the cell during the action potential plateau through voltage-gated Ca2+ channels activates SR ryanodine receptor Ca2+ channels. These RyR2 channels then rapidly release sequestered SR Ca2+ into the cytosol (see below) to signal contraction. During diastole, SR Ca2+-ATPase (SERCA2) and its regulatory protein phospholamban play central roles in the reuptake of released Ca2+ from the myocyte cytosol into the SR. Interestingly, inappropriate regulation of SR Ca2+ because of defects in either Ca2+ buffering (i.e., human calsequestrin-2 gene mutations) or Ca2+ release (human RyR2 gene mutations) has been linked with potentially fatal human arrhythmia (catecholaminergic polymorphic ventricular tachyarrhythmia). SR membrane–resident proteins also include inositol 1,4,5 trisphosphate (InsP3) receptors that have been linked to cardiac hypertrophy and arrhythmia.6
During the ventricular action potential, the rise in cytosolic Ca2+ via the SR membrane–associated RyR2 is rapidly translated into mechanical activity between the thick myosin filaments and the thin actin filaments through the regulatory functions of the troponin-tropomyosin complex.2 This complex consists of four subunits. Tropomyosin is a double-stranded α-helical molecule, which, under basal conditions (low Ca2+), covers myosin-binding sites along a span of seven actin monomers.2 Troponin T (TnT; tropomyosin-binding subunit) connects tropomyosin to the two remaining subunits TnC (Ca2+-binding) and troponin I (TnI; inhibitory subunit). The cardiac isoform of TnC has two high-affinity binding sites for Ca2+ or Mg2+ in the C-terminal domain and a low-affinity regulatory binding site that is Ca2+ specific in the N-terminal domain. During diastole, the C-terminal domain of TnI interacts with actin. During systole, Ca2+ binds to a low-affinity binding site in the TnC N-terminal domain, causing an increased affinity between this domain and the TnI N-terminal domain. As a result, the TnI-actin interaction is destabilized, which ultimately leads to a conformational change in the troponin-tropomyosin complex.2 Specifically, this complex is shifted and exposes myosin binding sites on actin, leading to “cross-bridge” formation between actin and myosin, force production, and cellular shortening.2 On removal of Ca2+ from the cytosol (primarily by the activities of SERCA2A and the plasma membrane Na+-Ca2+ exchanger), these molecular events are rapidly reversed, which results in cellular relaxation.2 Specifically, as cytosolic Ca2+ levels decrease, Ca2+ is removed from the TnC low-affinity binding site, causing TnI to dissociate from TnC and then reassociate with actin, effectively re-establishing the steric hindrance imposed by the troponin-tropomyosin complex.
As discussed in detail below, the dynamic range of cardiac excitation-mechanical coupling is, in part, regulated by phosphoregulation of key ion channels and transporters, which modulate intracellular Ca2+ in addition to altering the Ca2+ sensitivity of contractile proteins. For example, protein kinase A (PKA) phosphorylation of phospholamban at serine 16 relieves inhibition of phospholamban on SERCA2, thereby increasing SR Ca2+ uptake and promoting muscle relaxation.7 In addition to phosphorylating membrane proteins, PKA also phosphorylates contractile proteins to decrease their Ca2+ sensitivity, thereby promoting muscle relaxation. These modifications allow for increased cycle frequency elicited by exercise. For example, the N-terminal domain of cardiac TnI is phosphorylated by cyclic adenosine monophosphate (cAMP)-dependent PKA phosphorylation at serines 22 and 23. Phosphorylation of these residues desensitizes TnI to Ca2+-bound TnC and reduces the Ca2+ affinity of the Ca2+-specific regulatory site on TnC.8 Thus, these findings clearly illustrate the highly collaborative roles of structural, electrical, mechanical, and signaling proteins in the modulation of myocyte EC coupling and cardiac function.
Recent findings demonstrate key cellular roles for nuclear and mitochondrial membranes in the regulation of myocyte transcriptional pathways and in metabolism. The nuclear envelope is a complex structure comprising outer and inner nuclear membranes. The outer nuclear membrane is continuous with the SR, and the inner membrane contains a number of critical membrane proteins involved in nuclear assembly and gene transcription, such as lamins, which create a structural lattice for nuclear envelope integrity, and emerins, which bind directly to actin filaments. Mutations in lamin A/C and emerin have been linked to Emery-Dreifuss muscular dystrophy, a degenerative muscle disease featuring cardiac conduction defects. Human mutations in the nuclear lamina protein emerin (EMD), which are relevant to cardiac arrhythmia, have also been linked to familial atrial fibrillation and sinus node disease.9 Specifically, identified probands display a complex arrhythmia phenotype, including irregular, chaotic atrial rhythm and first-degree atrioventricular block.9 One proband displayed premature atrial complexes with rate variability (30 to 100 beats/min), and sinus arrest with junctional escape rhythm.9 Interestingly, the identified EMD mutation is hypothesized to affect the interaction between the emerin LEM domain and intranuclear binding proteins.9 Moreover, analysis of emerin localization in EMD mutation carriers revealed defects in nuclear emerin localization.9 These findings clearly demonstrate the unexpected link between cardiac atypical cellular architecture, in this case the nuclear lamina, and normal cellular excitability. In addition to having clear roles in orchestrating cellular structure and intermediate filament organization, the nuclear membrane also contains an autonomous system for Ca2+ signaling. InsP3 receptors located on both the inner and outer nuclear membranes allow Ca2+ release from the nuclear membrane lumen into the nucleoplasm and cytosol, respectively.10 In fact, work by Bers and colleagues demonstrated that Ca2+ in the nuclear membrane is tightly regulated by SR Ca2+, and this Ca2+ is central to cardiac excitation-transcription signaling via InsP3 receptor-dependent signaling.11
Cardiac mitochondria play an important role not only in energy production but also in Ca2+ homeostasis and apoptosis. Mitochondria occupy a large percentage of the cell volume (about 30%) and are concentrated near myofilaments, T-tubules, and the SR.12 Mitochondria comprise inner and outer membranes surrounding the mitochondrial matrix, where oxidative phosphorylation drives ATP production. A large potential gradient (ΔΨm about –180 mV) across the inner mitochondrial membrane, together with a proton gradient (ΔpH), is necessary for the conversion of adenosine diphosphate (ADP) to ATP. Respiration is regulated by many factors, including ADP and Ca2+, which activate key enzymes in the tricarboxylic acid (TCA) cycle. Highly selective Ca2+ uniporters expressed in the inner mitochondrial membrane use the electrical gradient across the inner mitochondrial membrane to move Ca2+ from the myoplasm into the mitochondria. The proximity of mitochondria to the Ca2+ channels at T-tubules and to the Ca2+ release sites on the SR create a local Ca2+ domain that provides mitochondria with access to an important resource for respiration and enables mitochondria to serve as an important buffer of intracellular Ca2+. Interestingly, disruption of the normal mitochondrial arrangement within the cell has been linked to mitochondrial dysfunction, apoptosis, and arrhythmias in a murine model of desmin-related cardiomyopathy. The mitochondrial Na+-Ca2+ exchanger helps maintain mitochondrial Ca2+ homeostasis, and the Na+-H+ exchanger and the Na+-K+-ATPase use the proton gradient and ATP, respectively, for maintaining mitochondrial Na+ homeostasis. Ion homeostasis in the myoplasm and in the mitochondrial matrix are coupled such that dysregulation of homeostasis in the myoplasm may alter mitochondrial energetics. Thus, myoplasmic Na+ accumulation in the setting of heart failure may accelerate Ca2+ removal from the mitochondrial matrix via the Na+-Ca2+ exchanger, leading to decreased mitochondrial Ca2+, decreased NADH production via the TCA cycle, and decreased ATP production.13 Conversely, myoplasmic Ca2+ overload (e.g., during myocardial ischemia) may lead to the accumulation of mitochondrial Ca2+ and the opening of the mitochondrial permeability transition pore, a large nonspecific conductance in the inner mitochondrial membrane that discharges the mitochondrial membrane potential, which leads to cell death.13
The Cardiac Dyad
A striking example of the evolution of cardiac molecular and structural components converging on function is the cardiac dyad. Franzini-Armstrong and colleagues identified the dyad using electron microscopy and showed that it represents the pre-eminent cardiac signaling unit for EC coupling.14 Specifically, the cardiac dyad is a key juxtaposition of cardiac external plasma membrane (T-tubules) to internal SR membrane (Figure 2-4).2 Central to the dyad T-tubule membrane is a large population of membrane-associated L-type Ca2+ channels (comprising a central α-subunit pore and multiple regulatory β-subunits).15 Across the tiny dyadic cleft (~15 nm) reside large clusters of ryanodine receptor Ca2+ release channels in the SR membrane.15 Thus, the dyad structure has evolved to provide spatial constraints that, together with the high gain of the Ca2+-induced Ca2+ release process, allows for the activation of internal Ca2+ release by a very small influx of Ca2+ through L-type Ca2+ channels. Since the divalent cation Ca2+ is used for a host of cellular processes, including contraction, transcription, and apoptosis, the spatially privileged environment of the dyad is critical for maintaining the fidelity of intracellular Ca2+ signaling.
A second key component of cardiac architecture for physiology illustrated by the dyad is the convergence of local signaling networks for function. For example, multiple key proteins in the cardiac dyad, including the L-type Ca2+ channel and RyR, are dynamically regulated by phosphorylation and dephosphorylation cascades. In fact, phosphorylation of both Cav1.2 and RyR2 by both protein kinase A and Ca2+-calmodulin–dependent protein kinase II (CaMKII) regulates channel activity. Recent work has illustrated that phosphorylation signaling pathways are tightly regulated at the local level. For example, PKA is directly linked to its target molecule RyR2, by direct interaction of RyR2 with the PKA-anchoring protein mAKAP.16 Moreover, PKA-dependent regulation of RyR2 is directly antagonized by local phosphatase 2A, also directly linked to RyR2 via mAKAP.16 Similarly, PKA-dependent regulation of Cav1.2 activity at the cell membrane occurs via a protein complex that involves AKAP150. Like PKA, CaMKII regulates the activities of proteins on either side of the dyad. CaMKII directly phosphorylates the Cav1.2 channel complex to produce an alternative channel-gating mode characterized by long open times (mode 2) and current facilitation.17 Recent studies have identified the Cav1.2–auxillary subunit β2a as a critical target for CaMKII-mediated current facilitation.18 Moreover, β2a contains a CaMKII-binding site with high homology to established motifs in the NR2B subunit of the NMDA receptor and in the CaMKII association domain.18,19 Thus, in addition to playing an important role in regulating Cav1.2 activity, β2a also serves as a Cav1.2-specific anchoring protein for CaMKII.18 CaMKII co-localizes with and phosphorylates RyR2 to regulate channel activity, although the nature of this association and the functional effects (increase or decrease in activity) remain uncertain.20,21 However, several studies have provided compelling evidence that CaMKII hyperphosphorylation of RyR2 in the setting of heart failure leads to inappropriately active channels that promote diastolic Ca2+ leak from the SR, reduced SR Ca2+ content, and contractile dysfunction.22 Thus, the cardiac dyad, by functionally linking key ion channel components on closely apposed excitable membrane structures and by recruiting key signaling proteins, has evolved into an all-in-one signaling unit for the regulation of cardiac excitability.
The presence of large membrane complexes for local cardiac signaling extends far beyond the dyad (Figure 2-5). As discussed above, ventricular repolarization is regulated by the activity of IKs. The IKs current is the result of a heteromeric channel complex encoded by KCNQ1 (α-subunit) and KCNE1 (β-subunit).4 In fact, the importance of IKs for cardiac repolarization is clearly illustrated by human gene mutations in KCNQ1 and KCNE1 linked with both atrial and ventricular arrhythmias.4 Similar to the cardiac L-type Ca2+ channel and RyR2, IKs is dynamically regulated by PKA-dependent phosphorylation, and this regulation is coordinated by a protein complex involving the AKAP, Yotiao (see Figure 2-5). Yotiao interacts with the KCNQ1 C-terminus as well as with protein phosphatase 1 (PP1) and PKA to create a macromolecular signaling complex for regulating cardiac repolarization. Interestingly, mutations in KCNQ1 that affect the binding of KCNQ1 to Yotiao result in cardiac arrhythmia (long QT syndrome).16,23 Similarly, Yotiao (AKAP9) mutants that block binding to KCNQ1 result in defects in IKs and are associated with human long QT syndrome.24 Thus, in addition to inherent channel biophysical properties, regulation of signaling at the level of the local membrane microdomain is essential for normal cardiac excitability and human physiology.
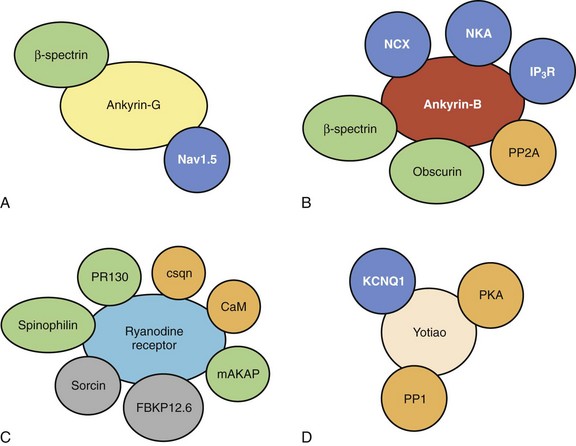
Biogenesis and Maintenance of Local Signaling Domains
Ankyrins, a family of cytoskeletal adapter proteins, were first identified in the erythrocyte in the late 1970s as a structural link between plasma membrane proteins and the actin-spectrin–based cytoskeleton.25 However, recent findings have clearly demonstrated key roles for ankyrin polypeptides in ion channel and transporter expression and localization in diverse cardiac cell types.26 As described above, the voltage-gated Na+ channel Nav1.5 is required for the rapid upstroke of the ventricular action potential (phase 0). As discussed in Chapters 3 and 7, defects in Nav1.5 biophysical activity resulting in aberrant inward INa are associated with sinus node disease, conduction defects, and ventricular arrhythmias. In ventricular cardiomyocytes, Nav1.5 is primarily localized to the cardiac intercalated disc, although it may also be found at T-tubules and the peripheral sarcolemma in less abundance.27,28 In fact, ankyrin-G (encoded by human ANK3) is required for the expression and localization of Nav1.5 at the intercalated disc (see Figure 2-5).28,29 Myocytes lacking ankyrin-G display loss of Nav1.5 expression at the cardiac intercalated disc and corresponding reductions in cellular INa.29 Interestingly, the ankyrin-G membrane–targeting pathway appears specific for Nav1.5 versus other cardiac ion channels and transporters, as the localization, expression, and functioning of Cav1.2 and the Na+-Ca2+ exchanger are unaffected in myocytes lacking ankyrin-G.29 Moreover, the loss of Nav1.5 targeting is rescued by exogenous expression of wild-type ankyrin-G, but not a mutant ankyrin-G lacking key Nav1.5 binding residues.29 Therefore, ankyrin-G is critical for the localization and functioning of Nav1.5 at the cardiac intercalated disc. Consistent with these findings, loss of ankyrin-G in the murine cerebellum results in defects in neuronal Na+ channel targeting, defects in neuronal action potentials, and ataxia.30 Defects in the ankyrin-G–based pathway for Nav1.5 membrane targeting are associated with human arrhythmia. Specifically, key residues in the Nav1.5 domain I-II cytoplasmic domain are required for interaction with ankyrin-G.28 Interestingly, a human Brugada syndrome mutation is located in this Nav1.5 motif and blocks the interaction of Nav1.5 with ankyrin-G.28 Moreover, consistent with the role of ankyrin-G in targeting Nav1.5, the human Nav1.5 Brugada syndrome mutant displays aberrant trafficking to the intercalated disc.28 Instead, the mutant channel is trapped intracellularly in the biosynthetic process.28 Thus, these findings illustrate the importance of membrane-targeting pathways for normal human cardiac excitability. While ankyrin-G is required for Nav1.5 targeting to the intercalated disc, the pathways for Na+ channel targeting to other excitable domains are not yet known. However, alternative pathways are almost certainly required for these domains. Likely protein suspects for peripheral sarcolemmal targeting for Nav1.5 include syntrophin and dystrophin.31
While ankyrin-G is required for protein targeting to the intercalated disc, a second ankyrin-gene product, termed ankyrin-B (human ANK2), is responsible for targeting key cardiac ion channels and transporters to the ventricular cardiomyocyte T-tubule and the SR. Specifically, ankyrin-B directly associates with the T-tubule membrane proteins Na+-Ca2+ exchanger and Na+-K+-ATPase (see Figure 2-5).32,33 Moreover, ankyrin-B interacts with the SR membrane protein InsP3 receptor.33,34 Ventricular cardiomyocytes from mice lacking ankyrin-B expression display striking loss of Na+-K+-ATPase, the Na+-Ca2+ exchanger, and InsP3 receptor expression and function.33–36 Similar to the findings on ankyrin-G, loss of ankyrin-B is specific for Na+-K+-ATPase, the Na+-Ca2+ exchanger, and InsP3 receptor membrane targeting, as ankyrin-B loss does not affect Nav1.5 or Cav1.2 membrane expression.35,37 Consistent with loss of Na+-K+-ATPase and the Na+-Ca2+ exchanger (and similar to the effects of digitalis, which blocks Na+-K+-ATPase activity), ankyrin-B+/− cells display increased SR Ca2+ load and Ca2+ transient amplitudes.35 Moreover, while stable in resting conditions, ankyrin-B+/− ventricular cardiomyocytes display cellular after-depolarizations (oscillations in membrane excitability) in response to catecholaminergic stimulation.35 Consistent with these findings, ankyrin-B+/− mice may display polymorphic arrhythmia and sudden death in response to severe catecholaminergic stimulation (exercise and/or catecholamine injection).35 Finally, consistent with the role of ankyrin-G in human arrhythmia (Brugada syndrome, see above), defects in the ankyrin-B–based pathway for ion channel and transporter targeting in ventricular cardiomyocytes result in arrhythmia in the human heart. Specifically, human loss-of-function mutations in ANK2 (ankyrin-B gene) result in a complex arrhythmia syndrome that includes sinus node disease, atrial fibrillation, conduction block, catecholaminergic polymorphic ventricular tachycardia, and sudden death.35,36,38 In fact, nine ANK2 loss of function variants have been identified in a host of kindred worldwide.35,36,38 While the clinical phenotypes of the probands may differ, depending on the variant and the environment (i.e., sinus node disease plus ventricular arrhythmia versus simple sinus node disease), the cardiac phenotypes present in these patients clearly demonstrate the importance of proper ion channel and transporter targeting for normal human cardiovascular excitability. Interestingly, while the ventricular phenotypes are primarily due to defects in the Na+-Ca2+ exchanger and Na+-K+-ATPase, ANK2-associated defects in sinus node function are due to defects in ankyrin-B–based targeting of Cav1.3, an atrial and sinus node–specific isoform of the L-type Ca2+ channel.39 Specifically, loss of ankyrin in the sinus node affects Cav1.3, but not Cav1.2 expression and targeting, which results in decreased ICa,L and aberrant automaticity, consistent with findings from mice lacking Cav1.3 expression.39–41 Ankyrin-B dysfunction has also been observed in electrically unstable regions of the ventricle following myocardial infarction.42 These data suggest that changes in ankyrin-B expression may play a role in the more common, acquired forms of cardiac arrhythmia.
Following the identification of ankyrins in ion channel and transporter targeting and human arrhythmia, identical membrane components have been linked to human arrhythmia. For example, syntrophin, the large dystrophin-associated protein, is critical for targeting Nav1.5 to the peripheral sarcolemma.31 Mice lacking the dystrophin complex display defects in Nav1.5 expression and aberrant cardiac electrical activity.31 In support of these findings, a human mutation in SNTA1 (encodes α1-syntrophin) was recently linked to long QT syndrome.43,44 Specifically, the novel syntrophin mutation resulted in increased persistent INa. While the mutant does not directly block Nav1.5-syntrophin interactions, the mutation may disrupt association of Nav1.5 with PMCA4b, resulting in defective regulation of nNOS (nitric oxide synthase), S-nitrosylation of Nav1.5, and increased late INa.44
Finally, in addition to defects in integral membrane ion channels, targeting proteins, and structural proteins, defects in membrane coat proteins have been linked to aberrant cardiomyocyte electrical activity and pathophysiology. Caveolae are small invaginations of the cell membrane that have roles in vesicular trafficking and endocytosis. In fact, work in excitable cells strongly implicates caveolae-rich membrane domains in the clustering of ion channel and receptor signaling complexes, including both voltage-gated Na+, K+, and Ca2+ channels, as well as β-adrenergic receptor signaling complexes. One key coat protein for the formation of the cardiac caveolae membrane domain is the ~20 kD protein caveolin-3 (encoded by CAV3). Recent data link CAV3 mutations to human excitable cell disease. Specifically, CAV3 mutations have been linked to long QT syndrome, limb-girdle muscular dystrophy, and sudden infant death syndrome.45–48 Similar to type 3 long QT mutations affecting Nav1.5, cardiac phenotypes associated with human CAV3 mutations include increased late INa and QTc prolongation.45 However, skeletal muscle phenotypes associated with CAV3 mutations include decreased L-type Ca2+ channel current.47,48 Thus, it appears that caveolin-3 and caveolae, in general, are likely to have pleiotropic effects on cardiac and skeletal muscle membrane ion channels, transporters, and receptors, which result in complex electrical phenotypes in response to loss of function. Nonetheless, these data clearly demonstrate the importance of unlikely cellular proteins in the pathogenesis of human cardiac arrhythmias.
1 Dobrzynski H, Boyett MR, Anderson RH. New insights into pacemaker activity: Promoting understanding of sick sinus syndrome. Circulation. 2007;115(14):1921-1932.
2 Bers DM. Excitation-contraction coupling and cardiac contractile force, ed 2. Dordrecht: Kluwer Academic Publishers; 2001.
3 Kleber AG, Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev. 2004;84(2):431-488.
4 Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85(4):1205-1253.
5 Asimaki A, Tandri H, Huang H, et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009;360(11):1075-1084.
6 Roderick HL, Bootman MD. Pacemaking, arrhythmias, inotropy and hypertrophy: The many possible facets of IP3 signalling in cardiac myocytes. J Physiol. 2007;581(Pt 3):883-884.
7 Koss KL, Kranias EG. Phospholamban: A prominent regulator of myocardial contractility. Circ Res. 1996;79(6):1059-1063.
8 Robertson SP, Johnson JD, Holroyde MJ, et al. The effect of troponin I phosphorylation on the Ca2+-binding properties of the Ca2+-regulatory site of bovine cardiac troponin. J Biol Chem. 1982;257(1):260-263.
9 Karst ML, Herron KJ, Olson TM. X-linked nonsyndromic sinus node dysfunction and atrial fibrillation caused by emerin mutation. J Cardiovasc Electrophysiol. 2008;19(5):510-515.
10 Bootman MD, Collins TJ, Peppiatt CM, et al. Calcium signalling—an overview. Semin Cell Dev Biol. 2001;12(1):3-10.
11 Wu X, Zhang T, Bossuyt J, et al. Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J Clin Invest. 2006;116(3):675-682.
12 Maack C, O’Rourke B. Excitation-contraction coupling and mitochondrial energetics. Basic Res Cardiol. 2007;102(5):369-392.
13 Maack C, Cortassa S, Aon MA, et al. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res. 2006;99(2):172-182.
14 Franzini-Armstrong C. Studies of the triad. II. Penetration of tracers into the junctional gap. J Cell Biol. 1971;49(1):196-203.
15 Scriven DR, Dan P, Moore ED. Distribution of proteins implicated in excitation-contraction coupling in rat ventricular myocytes. Biophys J. 2000;79(5):2682-2691.
16 Marx SO, Reiken S, Hisamatsu Y, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101(4):365-376.
17 Dzhura I, Wu Y, Colbran RJ, et al. Calmodulin kinase determines calcium-dependent facilitation of L-type calcium channels. Nat Cell Biol. 2000;2(3):173-177.
18 Grueter CE, Abiria SA, Dzhura I, et al. L-type Ca(2+) channel facilitation mediated by phosphorylation of the beta subunit by CaMKII. Mol Cell. 2006;23(5):641-650.
19 Strack S, McNeill RB, Colbran RJ. Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 2000;275(31):23798-23806.
20 Witcher DR, Kovacs RJ, Schulman H, et al. Unique phosphorylation site on the cardiac ryanodine receptor regulates calcium channel activity. J Biol Chem. 1991;266(17):11144-11152.
21 Wehrens XH, Lehnart SE, Reiken SR, et al. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res. 2004;94(6):e61-e70.
22 Zhang T, Maier LS, Dalton ND, et al. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res. 2003;92(8):912-919.
23 Schwartz PJ, Priori SG, Spazzolini C, et al. Genotype-phenotype correlation in the long-QT syndrome: Gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103(1):89-95.
24 Chen L, Marquardt ML, Tester DJ, et al. Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc Natl Acad Sci U S A. 2007;104(52):20990-20995.
25 Mohler PJ, Gramolini AO, Bennett V. Ankyrins. J Cell Sci. 2002;115(Pt 8):1565-1566.
26 Cunha SR, Mohler PJ. Cardiac ankyrins: Essential components for development and maintenance of excitable membrane domains in heart. Cardiovasc Res. 2006;71(1):22-29.
27 Cohen SA. Immunocytochemical localization of rH1 sodium channel in adult rat heart atria and ventricle. Presence in terminal intercalated disks. Circulation. 1996;94(12):3083-3086.
28 Mohler PJ, Rivolta I, Napolitano C, et al. Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc Natl Acad Sci U S A. 2004;101(50):17533-17538.
29 Lowe JS, Palygin O, Bhasin N, et al. Voltage-gated Nav channel targeting in the heart requires an ankyrin-G dependent cellular pathway. J Cell Biol. 2008;180(1):173-186.
30 Zhou D, Lambert S, Malen PL, et al. AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J Cell Biol. 1998;143(5):1295-1304.
31 Gavillet B, Rougier JS, Domenighetti AA, et al. Cardiac sodium channel Nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ Res. 2006;99(4):407-414.
32 Cunha SR, Bhasin N, Mohler PJ. Targeting and stability of Na/Ca exchanger 1 in cardiomyocytes requires direct interaction with the membrane adaptor ankyrin-B. J Biol Chem. 2007;282(7):4875-4883.
33 Mohler PJ, Davis JQ, Bennett V. Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP(3) receptor in a cardiac T-tubule/SR microdomain. PLoS Biol. 2005;3(12):e423.
34 Mohler PJ, Davis JQ, Davis LH, et al. Inositol 1,4,5-trisphosphate receptor localization and stability in neonatal cardiomyocytes requires interaction with ankyrin-B. J Biol Chem. 2004;279(13):12980-12987.
35 Mohler PJ, Schott JJ, Gramolini AO, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421(6923):634-639.
36 Mohler PJ, Splawski I, Napolitano C, et al. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc Natl Acad Sci U S A. 2004;101(24):9137-9142.
37 Mohler PJ, Gramolini AO, Bennett V. The ankyrin-B C-terminal domain determines activity of ankyrin-B/G chimeras in rescue of abnormal inositol 1,4,5-trisphosphate and ryanodine receptor distribution in ankyrin-B (-/-) neonatal cardiomyocytes. J Biol Chem. 2002;277(12):10599-10607.
38 Mohler PJ, Le Scouarnec S, Denjoy I, et al. Defining the cellular phenotype of “ankyrin-B syndrome” variants: Human ANK2 variants associated with clinical phenotypes display a spectrum of activities in cardiomyocytes. Circulation. 2007;115(4):432-441.
39 Le Scouarnec S, Bhasin N, Vieyres C, et al. Dysfunction in ankyrin-B-dependent ion channel and transporter targeting causes human sinus node disease. Proc Natl Acad Sci U S A. 2008;105(40):15617-15622.
40 Mangoni ME, Couette B, Bourinet E, et al. Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc Natl Acad Sci U S A. 2003;100(9):5543-5548.
41 Zhang Z, Xu Y, Song H, Rodriguez J, et al. Functional roles of Ca(v)1.3 (alpha(1D)) calcium channel in sinoatrial nodes: Insight gained using gene-targeted null mutant mice. Circ Res. 2002;90(9):981-987.
42 Hund TJ, Wright PJ, Dun W, et al. Regulation of the ankyrin-B-based targeting pathway following myocardial infarction. Cardiovasc Res. 2009;81(4):742-749.
43 Wu G, Ai T, Kim JJ, et al. Alpha1 syntrophin mutation and the long QT syndrome. Circ Arrhythmia Electrophysiol. 2008;1:193-201.
44 Ueda K, Valdivia C, Medeiros-Domingo A, et al. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci U S A. 2008;105(27):9355-9360.
45 Vatta M, Ackerman MJ, Ye B, et al. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006;114(20):2104-2112.
46 Cronk LB, Ye B, Kaku T, Tester DJ, et al. Novel mechanism for sudden infant death syndrome: Persistent late sodium current secondary to mutations in caveolin-3. Heart Rhythm. 2007;4(2):161-166.
47 Couchoux H, Allard B, Legrand C, et al. Loss of caveolin-3 induced by the dystrophy-associated P104L mutation impairs L-type calcium channel function in mouse skeletal muscle cells. J Physiol. 2007;580(Pt.3):745-754.
48 Weiss N, Couchoux H, Legrand C, et al. Expression of the muscular dystrophy-associated caveolin-3(P104L) mutant in adult mouse skeletal muscle specifically alters the Ca(2+) channel function of the dihydropyridine receptor. Pflugers Arch. 2008;457(2):361-375.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
Chapter 2 Principles of Cellular Architecture and Physiology with Applications in Electrophysiology
Specialized Excitable Cells Tightly Regulate Cardiac Depolarization
The highly coordinated and efficient propagation of electrical activity through the heart is maintained by the combined activities of a diverse set of specialized excitable cardiac cells, each with its own structural, electrical, and molecular signature. The cardiac sinoatrial (SA) node, a small group of spontaneously active cells in the right atria, is the primary initiation site of cardiac electrical activity because of its relatively positive threshold potential.1 Once generated by the sinus node, the cardiac action potential propagates through the atria to the atrioventricular (AV) node (the maximum diastolic potential of the AV node is only slightly more negative than that of the SA node), a second small but critical group of specialized cells that display slow conduction properties preventing inappropriate depolarization of the ventricles. In fact, the slow conduction of the AV node is a critical safeguard against the development of ventricular arrhythmias from pathologic atrial pacing defects (i.e., atrial flutter/fibrillation). After the AV node, the cardiac action potential propagates through the cardiac conduction system comprising the AV bundle (bundle of His), the left and right bundle branches, and the cardiac Purkinje system. Interestingly, this conduction system, particularly the cardiac Purkinje fibers, has evolved to rapidly propagate cardiac electrical activity at up to 2 to 4 m/s for the nearly instantaneous spread of depolarization through the sub-endocardium of the left and right ventricles.2 In comparison with the rapid conduction pathways of the Purkinje system, left ventricular tissue conduction velocity is significantly slower (0.3 to 1 m/s).2 Importantly, Purkinje fibers communicate with the ventricular mass at well-defined discrete loci (Purkinje-muscle junctions).
Form Fits Function: The Ventricular Cardiomyocyte and Excitation-Contraction Coupling
As is discussed in detail in Chapter 3, the electrical activity of cardiac cells (the action potential) is primarily modulated by the coordinated movement of sodium (Na+), calcium (Ca2+), and potassium (K+) across the external plasma membrane (sarcolemma) and the internal sarcoplasmic reticulum (SR) membrane. The specialized function of each cardiac cell type is the result of the evolution of specific molecular and structural components that regulate ion flux across the membrane and dictate specific cell properties.3 In this section, the primary structural and molecular components of different cardiac cells is discussed in relation to cell type–specific action potentials. Because of its central role in cardiac excitability, the ventricular cardiomyocyte will be used as the central point of comparison for other excitable cardiac cell types.
The ventricular action potential is notable for its hyperpolarized resting membrane potential, rapid upstroke, and prolonged plateau (Figure 2-1). The resting membrane potential of the ventricular cardiomyocyte (held at ~–90 mV, roughly 30 mV more negative than the human sinus node) is the most negative of all excitable cell types (hence the final cell type to depolarize), primarily because of a large inwardly rectifying K+ current, IK1, prominently expressed in these cells.4 The rapid upstroke, because of the presence of rapidly activating voltage-gated Na+ channels, allows for rapid propagation of the electrical signal through the ventricles, which is required for synchronized muscle contraction. Finally, the extended plateau allows sufficient time for Ca to enter the cell and signal contraction.3
Once the excitation reaches the ventricles through the cardiac conduction system (see above), the electrical signal passes from cell to cell as a flux of ions through specialized intercellular ion channels called gap junctions (discussed in detail below). This flow of ions into the cell from neighboring activated cells depolarizes the membrane potential. If the membrane reaches a threshold potential (~–60 mV), a large population of voltage-gated Na+ channels (primarily Nav1.5, encoded by SCN5A) is activated, which results in a large inward influx of Na+ across the membrane into the myocyte. Na+ channels are functionally well suited to this task as they undergo rapid activation (activation time constant <0.5 ms) in response to membrane depolarization. Importantly, these channels also experience rapid voltage-dependent inactivation, which prevents reactivation until the membrane has returned to rest. The inward flux of Na+ ions carried by voltage-gated Na+ channels produces the initial rapid spike of the ventricular action potential (phase 0; see Figure 2-1).3 Depolarization of the cardiac membrane by rapidly activating voltage-gated Na+ channels activates higher threshold, more slowly activating voltage-gated Ca2+ channels (primarily CACNA1C-encoded Cav1.2 in the ventricle; see Figure 2-1).2 Ca2+ influx through voltage-gated Ca2+ channels serves two major purposes: (1) maintaining the action potential plateau (important for controlling heart rhythm), and (2) triggering Ca2+ release from internal stores for the purpose of triggering the mechanical contraction of the heart (excitation-contraction coupling).2 During excitation-contraction (EC) coupling, a small inward Ca2+ current across the plasma membrane is sensed by functional SR ryanodine receptor (RyR2 in the ventricle, encoded by RYR2) clusters, which subsequently release large quantities of Ca2+ from the internal SR stores into the cytosol, giving rise to a dramatic increase (order of magnitude) in intracellular Ca2+ (the Ca2+ transient).2 In this process, termed Ca2+-induced Ca2+ release, a small increase in local Ca2+ via Cav1.2 produces a relatively large release of SR Ca2+ (high gain function).
While voltage-gated Na+ and Ca2+ channels are responsible for myocyte depolarization and contraction, a host of plasma membrane–associated K+ channels regulate ventricular cardiomyocyte repolarization during phases 1, 2, and 3, as well as the rest potential.4 Specifically, the characteristic repolarization “notch” of phase 1 is modulated by the outward flux of K+ carried by the transient outward K+ current, Ito (primarily Kv4.2/Kv4.3 channels; see Figure 2-1). The duration of the action potential plateau (phase 2) is determined by a delicate balance between the inward Ca2+ current and the outward delayed rectifier K+ currents IKr and IKs (erg1/MiRP1 and KvLQT1/MinK channels encoded by KCNH2/KCNE2 and KCNQ1/KCNE1, respectively; see Figure 2-1).4 As the action potential proceeds, the Ca2+ current decreases because of deactivation as well as inactivation, and the K+ currents IKr and IKs increase, ultimately tilting the balance in favor of the late repolarization phase (phase 3; see Figure 2-1).4 The cell eventually returns to a resting potential maintained primarily by the time-independent inward rectifier current IK1 (see Figure 2-1). Other currents such as IKATP, while not central to the healthy control action potential, may have key roles in disease. Finally, the Na+-K+ adenotriphosphatase (ATPase) (which uses ATP to remove 3 Na+ from the cell and bring in 2 K+) is a key feature for generating and maintaining the myocyte electrochemical gradient.
Cell Membrane Architecture Defines Myocyte Local Electrical Activity
The ventricular cardiomyocyte plasma membrane, or sarcolemma, is divided into multiple and unique membrane structures (Figure 2-2). In addition to the external sarcolemma (resident proteins include the Na+-Ca2+ exchanger (NCX1) and plasma membrane Ca2+-ATPase [PMCA1]), the ventricular cardiomyocyte contains a large array of regularly spaced (~1.8 μm) plasma membrane invaginations, termed transverse tubules, or T-tubules. This membrane system, instrumental in myocyte EC coupling, evolved to facilitate coordinated EC coupling in the relatively large ventricular cardiomyocyte (system not present in smaller atrial and sinoatrial node cells). T-tubule–resident proteins include the L-type Ca2+ channel Cav1.2, the Na+-Ca2+ exchanger, and Na+-K+-ATPase.2 Finally, a highly specialized domain is present where the ventricular cardiomyocyte plasma membrane lies in close apposition to the plasma membrane of a neighboring cell. This complex membrane system, termed the intercalated disc, is required for myocyte cell-cell adhesion as well as intercellular action potential propagation and comprises three subdomains: (1) the gap junction, (2) the adherens junction, and (3) the desmosome (Figure 2-3).2 The gap junction comprises hundreds of hemi-channels (connexons) that span the lipid bilayer and allow electrical and metabolic coupling when docked with hemi-channels from a neighboring cell. At least four different connexin proteins (functional units of the connexon) with distinct biophysical properties are expressed throughout the heart. Gap junctions in ventricular tissue consist primarily of connexin43, which forms large conductance channels to allow rapid conduction. In contrast, gap junctions in the sinus node contain mostly connexin45, which forms lower conductance channels ideal for slow but safe conduction. The adherens junction is maintained by the function of a cadherin-catenin complex that provides a stabilizing link from the intercalated disc to the actin cytoskeleton (see Figure 2-3). Finally, the desmosome also supports cell adhesion through a complex, including plakoglobin (γ-catenin, also found in the adherens junction), desmoplakin, and plakophilin, that interacts with intermediate filaments (see Figure 2-3). Interestingly, defects in desmosomal proteins have been linked to cardiomyopathies and arrhythmias, including arrhythmogenic right ventricular cardiomyopathy (ARVC). In fact, loss of plakoglobin immunostaining in human heart biopsies has been recently developed into a diagnostic marker for Naxos disease, a cardiocutaneous syndrome characterized by wooly hair, palmoplantar keratoderma, and severe cardiomyopathy.5
In addition to the external plasma membrane, the cardiac myocyte (as well as many other excitable cells, including neurons and skeletal muscle) contains SR, the specialized endoplasmic reticulum that has key roles in the regulation of intracellular Ca2+.2 The cardiac SR is an extensive network of tubules linking key signaling networks, including the plasma membrane, nuclear envelope, nucleus, and mitochondria, to mediate a host of diverse functions. The pre-eminent role of the cardiac SR is to regulate myocyte EC coupling. Specifically, the vertebrate SR sequesters a large pool of releasable Ca2+ (1 mM inside SR vs. 100 nM in cytosol2) that serves as the primary source of Ca2+ for troponin-C (TnC) activation and muscle contraction (discussed in detail below).2 The majority of this Ca2+ is internally buffered by the Ca2+-binding protein calsequestrin, which forms a critical RyR2 regulatory complex with triadin and junctin. However, Ca2+ entry into the cell during the action potential plateau through voltage-gated Ca2+ channels activates SR ryanodine receptor Ca2+ channels. These RyR2 channels then rapidly release sequestered SR Ca2+ into the cytosol (see below) to signal contraction. During diastole, SR Ca2+-ATPase (SERCA2) and its regulatory protein phospholamban play central roles in the reuptake of released Ca2+ from the myocyte cytosol into the SR. Interestingly, inappropriate regulation of SR Ca2+ because of defects in either Ca2+ buffering (i.e., human calsequestrin-2 gene mutations) or Ca2+ release (human RyR2 gene mutations) has been linked with potentially fatal human arrhythmia (catecholaminergic polymorphic ventricular tachyarrhythmia). SR membrane–resident proteins also include inositol 1,4,5 trisphosphate (InsP3) receptors that have been linked to cardiac hypertrophy and arrhythmia.6
During the ventricular action potential, the rise in cytosolic Ca2+ via the SR membrane–associated RyR2 is rapidly translated into mechanical activity between the thick myosin filaments and the thin actin filaments through the regulatory functions of the troponin-tropomyosin complex.2 This complex consists of four subunits. Tropomyosin is a double-stranded α-helical molecule, which, under basal conditions (low Ca2+), covers myosin-binding sites along a span of seven actin monomers.2 Troponin T (TnT; tropomyosin-binding subunit) connects tropomyosin to the two remaining subunits TnC (Ca2+-binding) and troponin I (TnI; inhibitory subunit). The cardiac isoform of TnC has two high-affinity binding sites for Ca2+ or Mg2+ in the C-terminal domain and a low-affinity regulatory binding site that is Ca2+ specific in the N-terminal domain. During diastole, the C-terminal domain of TnI interacts with actin. During systole, Ca2+ binds to a low-affinity binding site in the TnC N-terminal domain, causing an increased affinity between this domain and the TnI N-terminal domain. As a result, the TnI-actin interaction is destabilized, which ultimately leads to a conformational change in the troponin-tropomyosin complex.2
[/not-level-membership-for-cardiovascular-category]
